

Abstract
Objective: Multiple myeloma (MM), a plasma cell neoplasm, afflicts elder individuals accounting for 10% of hematologic malignancies. The MM plasma cells largely reside within the bone marrow niche and are accessible through an invasive bone marrow biopsy, which is challenging during serial monitoring of patients. In this setting, cell free DNA (cfDNA) may have a role to ascertain the molecular aberrations at diagnosis and in assessment of residual disease during therapy. The aim of this review was to explore the utility and current status of cfDNA in MM. Method: PubMed was searched with terms including cell-free DNA, circulating-tumor DNA, Multiple Myeloma, diagnosis, genomic profiling, Minimal Residual Disease individually or in combination to shortlist the relevant studies. Result: cfDNA serves as a non-invasive source of tumor-specific molecular biomarker, ctDNA that has immense potential in facilitating management of cancer patients. The mutation detection platforms for ctDNA include hybrid capture and ultra-deep sequencing. Hybrid capture allows full length gene sequencing for mutation and CNV detection. The disease progression can be monitored by profiling prognostic somatic copy number alterations by ultra-low pass whole genome sequencing of ctDNA cost-effectively. Evolution of both the laboratory protocols and bioinformatics tools may further improve the sensitivity of ctDNA detection for better disease management. Only a limited number of studies were available in MM exploring the potential utility of cfDNA. Conclusion: In this review, we discuss the nuances and challenges associated with molecular evaluation of cfDNA and its potential role in diagnosis and monitoring of treatment response in MM.
Keywords: Cell free DNA, cfDNA, circulating tumor DNA, ctDNA, multiple myeloma, genomic profiling, deep sequencing, minimal residual disease, MRD
Introduction
Pathobiology of multiple myeloma (MM) has come a long way since its advent in terms of diagnosis, prognosis, risk stratification and disease monitoring. Multiple myeloma is primarily a hematological malignancy characterized by proliferation of neoplastic plasma cells, which are largely confined to the bone marrow but is also frequently associated with extra-medullary disease. The pattern of distribution of malignant plasma cells in bone marrow compartment is essentially patchy in over 80% of the cases and thus a bone marrow aspirate and/or biopsy from a single site, as is the current practice, may not give complete information about the entire disease burden [1-4]. Recently, Rasche L et al [5] performed sequencing from multiple sites, including bone marrow, focal bone and soft tissue lesions from MM patients and demonstrated that the spatial heterogeneity is positively associated with regional outgrowth of tumor-subclones in over two-third of the patients. Also, the mutations in TP53 gene were primarily restricted to the focal lesions with the disease progression [1,5]. Besides, in extramedullary disease, the myeloma subclones occupy niches outside the bone marrow and are not sampled by a conventional bone marrow biopsy and may lead to sampling bias [2,3].
The residual tumor cells commonly referred to as minimal residual disease (MRD), may be present in a micro environmental niche that is not well sampled during the collection and testing of marrow aspirate. Both the spatial heterogeneity and variegated nature of MM pose a challenge in MRD detection in the BM. This might lead to false negative results by missing the extra-medullary tumor cells disease burden, which may account for relapse in substantial proportion of MRD negative patients [6-8]. Another limitation of bone marrow-based approach is invasive nature of the procedure which further pose challenge in serial evaluation of patients at multiple time-points of MRD monitoring. All these factors question the utility of bone marrow based studies to diagnose and monitor disease activity in multiple myeloma warranting the need for newer strategies for comprehensive disease characterization, tumor burden and MRD monitoring in MM. Exploring circulating tumor cells (CTCs) by multiparametricflowcytometry (MFC) or circulating DNA (ctDNA) by next generation sequencing (NGS) and imaging techniques, such as MRI and PET are few of the techniques to capture temporal and spatial genetic heterogeneity in MM [9-14].
Here, we systematically review the available data on the current status of ctDNA for assessing disease burden and MRD monitoring in Multiple Myeloma (MM). This includes current status of ctDNA analysis over repeated, invasive, and often uninformative tissue biopsies in MM with a future potential of response assessment and MRD monitoring.
Biology of cell free DNA
Circulating free tumor derived DNA (ctDNA) constitutes a fraction of the total cell free DNA (cfDNA) that is released by neoplastic cells in the blood and other body fluids [15,16]. The ctDNA can be distinguished from cfDNA as the former contain cancer-specific somatic mutations, chromosomal abnormalities, copy-number alterations and epigenetic modifications [17]. These degraded short fragments of DNA harbor altered gene signatures present in both primary tumors and metastases. The first evidence of cfDNA was given by Mandel and Metais and was initially found in immune complexes derived from systemic lupus erythematosus (SLE) patients [18,19]. Almost three decades later, tumor specific NRAS mutations were shown to be present in the plasma derived from patients with myeloid malignancy [20].
It is widely accepted that cfDNA are released into the bloodstream by both active and passive secretion including cellular degradation, apoptosis, and necrosis [15,21]. Active secretion occurs mainly in metabolically active, live cells, and mainly includes nuclear expulsion from erythroid precursors, neutrophil extracellular trap release (NETosis) in response to various stimuli and excision repair mechanism [22]. Previous studies demonstrated the size of cfDNA in the order of ~166 bp, fragmentation of this length of DNA suggests that apoptosis and DNA methylation may be the primary source of cfDNA release [23-26]. The cfDNA has variable half-life ranging from 15 minutes to several hours and it is continuously cleared from the circulation by liver, spleen and kidney [27,28].
Distribution and levels of cfDNA in health and disease
cfDNA is found to be present in both health and disease conditions, however their level is extremely elevated in malignancy. In normal subjects, cfDNA is pre-dominantly of hematopoietic origin, and derived from turnover of myeloid and erythroid precursors [21]. Endothelial cells, hepatocytes and neurons are other less common sources of cfDNA [22]. Apart from malignancy, there are numerous non-malignant pathological conditions in which increased levels of cfDNA are detected viz any tissue damage, inflammation, sepsis, post-transplant and in pregnancy (physiological) [29,30]. In malignancy, the quantity of cfDNA is found to be correlated with type, grade and stage of tumor as seen in various studies [31,32].
As a reference, 100 g of tumor load in a patient releases 3.3% of ctDNA into the circulation [33]. In healthy individuals, the concentration of cfDNA ranges from 0 to 100 ng/mL of blood (average ~30 ng/mL), whereas in cancer patients, the concentration ranges from 0 to over 1000 ng/mL of blood (average ~180 ng/mL) due to high cellular turnover rate [34,35]. The pathophysiology of cfDNA is summarized in Figure 1.
Figure 1.
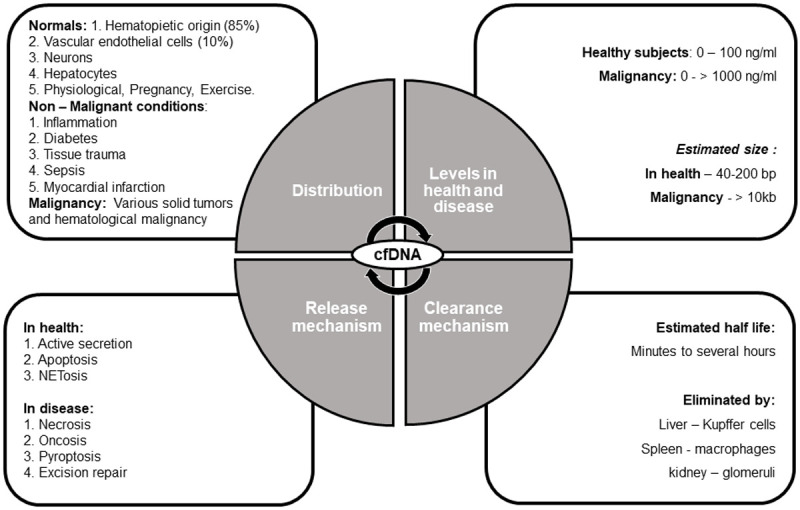
Pathophysiology of cell free DNA in health and disease.
Biological functions of cell free DNA
Cellular homeostasis
cfDNA release is an important mechanism to remove damaged nuclear material from cells as part of normal cellular homeostasis. Damaged DNA is mainly removed from the circulation via exosomes as oxidized mitochondrial DNA (mtDNA) maintaining the normal body homeostasis [21].
Cellular transformation
An important function of cfDNA is transformation as demonstrated experimentally by treating murine cells with serum from cancer patients and tumorigenesis of recipient cells in vivo model [36]. This led to the development of genometastasis hypothesis, where integration of cfDNA from diseased serum into healthy genome causes malignancy [37,38].
Tumor metastasis
Presence of cfDNA in the circulation and microvasculature leads to trapping and immobilization of tumor DNA, contributing to the formation of pre-metastatic niche and spread of tumor [21,23].
Immunomodulation and inflammation
Studies have shown the cytotoxic and pro-inflammatory actions of cfDNA in variable proportion, which regulates aberrant immune responses as well as mediates various inflammatory pathways [22,39,40].
cfDNA as diagnostic tool
In recent years, there has been a drastic increase in the number of publications referring to cfDNA/ctDNA analysis or liquid biopsy in various cancers [41,42]. The first blood cfDNA liquid biopsy for epidermal growth factor receptor (EGFR) genotyping assay was approved by US-FDA for lung cancer patients in 2016 [43]. Since then the analysis of cfDNA is now frequently integrated into clinical trials [44-46]. Moreover, the application of cfDNA for diagnostics is rapidly evolving but is presently limited to betterment of treatment choices in late stage cancers.
Role in disease monitoring and tumor prognostication
The concentration of cfDNA in blood plasma may differ among patients and is shown to increase with increasing stage or type of cancer, and metastasis [47-49]. In fact, the increased levels of cfDNA are associated with poor survival in different cancer types [50-52]. It may also be used for predicting patient risk stratification, therapy selection, prognostication, early relapse, monitoring emergence of resistance to treatment, and metastasis [53-59].
Utility over and above other techniques in disease evaluation
Till date, invasive tissue biopsies and radiological scans that involve radiation exposure have dominated the clinical assessment of cancer. In the current scenario, liquid biopsies come of age and among which, ctDNA is one of the most widely investigated markers in cancer patients. The advantages of cfDNA analysis over conventional tissue biopsies include: (1) Non-invasive technique; (2) Potential for cancer screening in asymptomatic individuals; (3) Early detection of cancer occurrence [60]; (4) Longitudinal study through serial sampling; (5) Monitoring of clonal evolution of malignant cells in real-time [61]; (6) Representative sampling of unreachable and non-resectable cancers [62,63] and (7) Comprehensive characterization of tumor evolution and tumor dynamics [64-66].
Techniques for detection of cell free DNA
Since ctDNA is present at a very low variant allelic frequency (VAF; also known as Mutant AF) in the peripheral blood, high-end technologies are needed to detect such low VAFs. In a study done by Underhill et al, it was shown that the size selection of cfDNA fragments in plasma can be used to increase the amount of ctDNA [67]. Advancement of techniques like next generation sequencing (NGS) and droplet digital polymerase chain reaction (ddPCR) have enabled detection of specific mutations present in ctDNA in blood with high sensitivity [68-70]. However, these newer techniques are still limited to detect small number of changes in ctDNA and yet to be validated in large prospective studies. Furthermore, there are certain analytic and post-analytic factors in the detection of cfDNA, that need thorough standardization viz. the experimental design, the choice of cfDNA detection method and analytical platform (e.g., digital PCR or NGS), data quality control methods, raw data processing and management (e.g., molecular bar-coding, in silico error suppression) etc. Nonetheless patient related confounding parameters like therapeutic or interventional protocols, co-morbidities, infections, lifestyle and dietary modifications may also influence the cfDNA concentration. The documented studies so far have reported wide variability in sensitivity and specificity in the assessment of cfDNA as they are based on diverse technical approaches [35,71-74]. Both the pre-analytical and analytical steps involved in cfDNA analysis are summarized in Table 1.
Table 1.
The pre-analytical and analytical steps involved in cfDNA isolation and analysis
| Pre-analytical Considerations | ||
|
| ||
| Plasma Collection (Tubes and Stability) | Plasma Centrifugation Procedure | Isolation of cfDNA (Kit and plasma input volume) |
|
| ||
| BD vacutainer tubes | First centrifugation step, 1200-1600 × g for 10 minutes at 4°C | QIAGEN (QIAamp Circulating Nucleic Acid Kit) |
| Stability: 4-6 hr | Type: Silica column based | |
| Temp.: RT or 4°C | Plasma input volume: 1-5 mL | |
| PAX gene tubes | Second centrifugation step, 16,000 × g for 10 minutes at 4°C | Promega (Maxwell RSC ccfDNA Plasma kit) |
| Stability: 7 days | Type: Paramagnetic particles | |
| Temp.: RT (15-25°C)/24 h at 35°C | Plasma input volume: 1-5 mL | |
| Streck tubes | Thermo Fisher (MagMAX Cell-Free DNA Isolation Kit) | |
| Stability: 14 days | Type: Magnetic bead based | |
| Temp.: RT (6-37°C) | Plasma input volume: 0.5-10 mL | |
| NORGEN tubes | MACHEREY-NAGEL (NucleoSpin Plasma XS) | |
| Stability: 30 days | Type: Silica column based | |
| Temp.: RT (15-25°C)/8 days at 37°C | Plasma input volume: 0.2-0.72 mL | |
| Roche tubes | EPIGENTEK (EpiQuik Circulating Cell-Free DNA Isolation Kit) | |
| Stability: 7 days | Type: Magnetic bead based | |
| Temp.: RT (18-25°C)/16 h at RT (15-30°C) | Plasma input volume: 0.1-1 mL | |
| ZYMO RESEARCH (Quick-cfDNA Serum and Plasma Kit) | ||
| Type: Silica column based | ||
| Plasma input volume: 0.2-10 mL | ||
| NORGEN (Plasma/Serum Cell-Free Circulating DNA Purification) | ||
| Type: Silica column based | ||
| Plasma input volume: 0.01-10 mL | ||
|
| ||
| Analytical Considerations | ||
|
| ||
| Quantification | Detection techniques | |
|
| ||
| NanoDrop 2000 (Thermo Fisher)/2100 Bioanalyzer DNA Quantification Kit (Agilent)/Quant-iT Pico Green dsDNA Assay Kit (Thermo Fisher) | qPCR/Real time qPCR | |
| Digital PCR | ||
| Next Generation Sequencing | ||
The primary molecular based approaches for cfDNA quantitation include digital PCR and NGS (Table 2). Digital PCR is a highly-sensitive technology which can quantitatively monitor ctDNA for the detection of tumor-associated genetic mutations but is limited by the number of single candidate genetic loci [75-77]. On the contrary, to determine tumor mutation profiles or monitor tumor clonal evolution, the NGS-based platforms facilitate the coverage of entire sequence of a vast gene panel and allow identification of novel or epigenetic alteration. Despite the advantages of NGS-based analysis of ctDNA, the major limitation remains the cost-effectiveness and hands-on-time. For this reason, ultra-deep, high-coverage NGS-approach is still far from clinical use for ctDNA-based routine analysis.
Table 2.
Various molecular approaches for the detection of genetic aberrations in cfDNA
| Technique | Sensitivity | Aberrations detected | |
|---|---|---|---|
| qPCR | ARMS-Scorpions PCR | 0.05-0.1% | point mutations |
| Clamping PCR | 0.1-1% | point mutations | |
| TaqMan | 0.1-1% | point mutations | |
| Digital PCR | Beaming | 0.01% | point mutations |
| ddPCR | 0.001% | point mutations | |
| Targeted DNA sequencing | TAm-Seq | >2% | Point mutations and structural alterations in gene |
| SAFE-SeqS | 0.1% | Point mutations and structural alterations in gene | |
| CAPP-Seq | 0.01% | Point mutations and structural alterations in gene | |
| Whole genome sequencing (WES) | Digital karyotyping | 0.001% | Genome-wide copy-number changes |
| PARE | 0.001% | Genome-wide rearrangements | |
Genomic landscape of MM
Multiple myeloma (MM) is characterized by a myriad of cytogenetic and molecular aberrations, which primarily include chromosomal translocations that involve the immunoglobin heavy chain locus and driver and/or secondary mutations in several oncogenic signaling pathways [78-87]. Since the first whole genome sequencing revealed the landscape of non-recurrent somatic alterations in MM [88], several NGS studies of the MM genome and exome have shed light on the spectrum of gene mutations associated with tumor progression [89-92]. Mutations have been reported most commonly in KRAS (23-26%), NRAS (20-24%) and BRAF (4-6%) genes that play a key role in MAPK pathway, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-KB) signaling and FGFR3, TRAF3, and TP53 [88,91-94]. Novel mutations in genes involved in RNA processing (DIS3) and protein homeostasis (FAM46C), regulator of MYC transcription (FUBP1), linker histones HIST1H1B, HIST1H1D, HIST1H1E, and HIST1H2BK have also been reported [89,91,95]. Using whole exome sequencing, Walker and group (2015) identified 15 gene mutations that were either actionable targets or carried prognostic value, which were incorporated into a mutation-based staging system [96]. The most frequently occurring gene mutations in multiple myeloma are listed in the Table 3.
Table 3.
Data from various studies comparing the frequency of recurrent somatic gene mutations in multiple myeloma
| Gene Mutation Frequency (%) | |||
|---|---|---|---|
|
| |||
| Gene | Lohr et al 2014 [91] (n = 203) | Bolli et al 2014 [89] (n = 67) | Walker et al 2015 [96] (n = 463) (NCT01554852) |
| KRAS | 23 | 20 | 21 |
| NRAS | 20 | 20 | 19 |
| FAM46C | 11 | 10 | 6 |
| BRAF | 6 | 12 | 7 |
| TP53 | 8 | 12 | 3 |
| DIS3 | 11 | 1 | 9 |
| SP140 | 4 | 6 | - |
| EGR1 | 4 | 6 | 4 |
| TRAF3 | 5 | 2 | 4 |
| ATM | 4 | 3 | 3 |
| CCND1 | 3 | 4 | 2 |
| LTB | 1 | 4 | 3 |
| IRF4 | 2 | - | 3 |
| FGFR3 | 2 | - | 3 |
Further technological advancement came with the development of a cost-effective genomics assay for myeloma, which was based on a moderate-depth capture sequencing platform, instead of Whole Genome Sequencing or Whole Exome Sequencing, for identification of chromosomal translocations, CNAs, and single-nucleotide variants [97]. In parallel, genomic classification of multiple myeloma was proposed based on comprehensive genotyping of patients by generating innovative clustering algorithms [98]. This study used custom target pulldown (TPD) analysis of a limited fraction of the genome as compared to WES and identified novel prognostic markers and disease subgroups [99]. Majority of the MM cases with hyperdiploidy status were characterized by the least number of gene mutations and CNAs and had a relatively good prognosis. Whereas, non-hyperdiploidy cases containing multiple subchromosomal CNAs and gene mutations showed a worse prognosis. More recently for improved stratification, chromosomal abnormalities detected by Next generation sequencing Seq-FISH and data from the Multiple Myeloma Research Foundation (MMRF) CoMMpass study was validated by R-ISS staging [100]. This study was undertaken because of the heterogeneity in methods and reporting of interphase FISH data in the MMRF CoMMpass study.
Cell free DNA in evaluation of genomic landscape of multiple myeloma
A few studies had recently documented the ctDNA based prediction of disease progression in MM patients by evaluation of the mutational spectrum and real-time monitoring of mutant clones [11-13,55]. These studies demonstrated significantly higher levels of cfDNA in MM patients relative to controls and non-MM cancers. Using a high sensitivity targeted sequencing platform, a proof-of-concept study demonstrated the detection of mutations exclusively in the plasma but not in the bone marrow obtained from a subset of MM patients [13]. The existence of spatial and genetic heterogeneity in progressive MM disease was shown by sequential ctDNA quantitation using ddPCR by longitudinal monitoring of specific clones, which are missed with single-site BM WES studies. The fractional abundance of specific clones correlated with the disease status and provided improved mutational characterization and therapeutic monitoring in MM. Moreover, the detection of plasma-based PIK3CA mutations raised the possibility of their association with extramedullary disease. Oberle A et al (2017) demonstrated an association between response to therapy and the presence of cfDNA clonotypic V(D)J rearrangements in a cohort of 27 MM patients [101].
A hybrid-capture-based liquid biopsy Sequencing (LB-Seq) method was evaluated for sequencing of gene targets including KRAS, NRAS, BRAF, EGFR and PIK3CA. This method included a variant filtering algorithm that enabled the detection of tumor-derived fragments available in cfDNA at very low variant allele frequencies (range 0.25-46%), albeit did not specifically reflect the typical mutational landscape in MM [12]. Another study used a targeted sequencing gene panel, spanning the exons and splice sites of 14 genes (BRAF, KRAS, NRAS, TP53, IRF4, DIS3, CCND1, CYLD, EGR1, FAM46C, PRDM1, SP140, TRAF3, and ZNF462). The CAPP-seq ultra-deep targeted NGS approach was used for genotyping this gene panel that was specifically created to obtain maximum mutation recovery in plasma cell tumors. Also, the mutational profile of cfDNA and tumor genomic DNA (gDNA) of purified PCs from bone marrow aspirates was compared [102]. This proof-of-concept demonstrated the feasibility of ctDNA genotyping in real-time approach that could reliably detect both clonal and subclonal somatic mutations present in at least 5% of alleles in tumor PCs.
In a case report, the utility of ctDNA was demonstrated as a non-invasive qualitative (characterization of the tumor genome) and quantitative (tumor burden) biomarker in MM, especially in the setting of Extramedullary-MM patient with high risk oligosecretory disease [14]. In this longitudinal monitoring study of patient undergoing therapy, ctDNA specific mutation was demonstrated by the presence of NRAS Q61H that coincided with disease progression. This observation was further validated by alterations in the spectrum of single nucleotide variants (VAFs) and insertions/deletions in the ctDNA during the disease progression. This study provided evidence for the presence of clonal dynamics and tumor burden concomitant with drug resistance [14]. A recent Phase Ib trial of relapsed/refractory patients revealed a significant correlation with higher mutational fractional abundance in plasma with shorter overall survival whereas decrease in ctDNA levels at day five post-induction correlated with superior progression-free survival. This study claimed to be the first to demonstrate ctDNA in monitoring tumor burden for predicting disease outcome in MM [103].
Residual disease monitoring in MM
The major advancement in the treatment strategies of MM such as proteasome inhibitors and immunomodulatory agents have resulted in improved survival outcomes. According to the IMWG criteria, the minimal residual disease (MRD) negativity in the bone marrow is recommended for response assessment and correlates with improved progression-free survival and overall survival in myeloma [104]. Earlier, the myeloma guidelines included urine assessment for monitoring monoclonal FLCs, with the exception of patients with non-measurable M-protein levels in serum (<10 g/L by SPE) and urine (BJP <200 mg/24 h by UPE) [105-107]. The recent update to the IMWG consensus criteria firmly emphasized on the possibility of novel biomarkers of response in MM with improved sensitivity. Detection of MRD within the BM, either by Multicolor Flow Cytometry for phenotypic markers or by Next Generation Sequencing (NGS) technologies for genotypic aberrations is currently followed. The sensitivity of at least 10-5 is considered as sustained response when confirmed at least one year apart [108]. In a meta-analysis, MRD status emerged as a surrogate for PFS in newly diagnosed MM [109]. The continuous improvement in the methods for MRD testing have been witnessed over the past two decades, which allow sensitive detection as low as one in a million cells. A comparison of various techniques to detect MRD in Multiple myeloma is summarized in Table 4.
Table 4.
Comparison between different techniques for detection of Minimal Residual Disease in Multiple Myeloma (Adapted from [116,137])
| Method | Sample Type | Applicability | Sensitivity | Quantification | Clonal Evolution | Advantage | Limitations |
|---|---|---|---|---|---|---|---|
| NGS | Nucleic acid (DNA) | ~90% | 10-6 | Absolute | Detectable | Provides information on background repertoire | Not standardized, no guidelines for analysis and data interpretation, available in few labs, Expensive |
| Flow Cytometry (Next Gen. Flow) | BMA (Cells) | 90-100% | 3-4 colors-10-3/10-4 | Absolute | Not applicable | Fast, reproducible, Accurate quantification well standardized, cost-effective | Large cell number needed (~6 million), Processing to be done on fresh samples |
| 6-8 colors-10-4 | |||||||
| 8-10 colors-10-5 | |||||||
| ASO-qPCR | Nucleic acid (DNA) | ~80% | 10-5 | Absolute | Not detected | More sensitive and specific than MFC, fresh sample not required, provides information on background repertoire | More time consuming and relatively laborious, requires patient specific primers, need of a bioinformatics analysis, Expensive |
| ddPCR | Nucleic acid (DNA) | Not reported | 10-4/10-5 | Absolute | Not detected | Standard curve Not needed, easy to perform | Dependent on ASO-primer, not standardized, no guidelines for analysis, data interpretation, Expensive |
| RT-qPCR | Nucleic acid (DNA) | 90-95% | 10-4/10-5 | Absolute | Not detected | Well standardized, International guidelines for analysis and data interpretation | Dependent on ASO-primer, laborious, time consuming, affected by clonal evolution, large amount of diagnostic DNA, Expensive |
MRD detection by MFC allows detection and monitoring of neoplastic and normal plasma cells that can be distinguished by identifying aberrant expression of cell surface and intracellular protein markers [6]. Initially, three to four colors MFC was used for MRD detection, which has currently expanded to 8-10 colors MFC in a single tube with monoclonal antibodies in dried formulation with sensitivity of 10-5 [110]. More recently, Next Generation Flow (NGF)-MRD has been optimized as a two-tube eight-color antibody panel. After bulk lysis, acquisition of ≥107 cells/sample is recommended, and novel software tools for automatic plasma cell gating ease MRD detection. NGF-MRD showed better sensitivity than conventional eight-color flow-MRD [111]. Quality of sample and hemodilution impacts MFC based MRD detection results [112]. The sensitivity of NGF can be improved by increasing the number of cells analyzed. The minimum requirement by current consensus guidelines is 2 million and recommend 5 million events to be acquired to achieve a minimum sensitivity of 10-5-10-6 [112]. The BMT CTN Myeloma Intergroup and IMWG flow MRD-negative response criterion was reported to be highly translational and sensitive for the evaluation of treatment efficacy in MM [7,113]. Recently, the utility of a non-invasive approach combining immune-magnetic beads and flow cytometry for enrichment and detection of circulating myeloma cells for monitoring MRD in MM patients was shown [114].
In regard to the molecular approaches for MRD assessment, initially, polymerase chain reaction-based amplification of the V(D)J clonal rearrangements was used for MRD assessment on BM samples for qualitative information. Later, clonal-size based methods (PAGE, Gene Scanning) and allele specific oligonucleotide PCR (ASO-RQ-PCR) were used for quantitation [115]. Minimal residual disease negativity by deep sequencing emerged as a major prognostic factor in MM [116]. In a retrospective study, patients with MM who underwent autologous stem-cell transplantation were evaluated by sequencing of all rearranged immunoglobulin gene segments present in a myeloma clone and the prognostic value of sequencing-based MRD detection became evident. NGS-based approach was more sensitive for low level MRD detection as compared to multiparameter flow cytometry, real-time quantitative ASO-PCR and droplet digital PCR (ddPCR) [77,117]. Digital PCR based MRD detection is still in the phase of standardization and optimization with varying definition of positivity in different studies [75,76]. The sensitivity and accuracy of ddPCR are almost comparable to ASO-PCR [76]. Unlike qASO-PCR, ddPCR does not require a standard curve construction and gives absolute number of copies of the target sequence. Both ASO-PCR and ddPCR cannot detect clonal evolution and therefore have limited applicability.
A limitation of the molecular approach is that consensus primers for clonality identification are used in the diagnostic sample followed by evaluation of follow-up sample for MRD detection. Also, there is an absolute need for the patient specific primers and probes. In NGS approaches, MRD detection can be enabled once the clonality of the sequence in the diagnostic sample is established. In contrast to this, in ASO-PCR even if the clonality is established there are many hurdles that hinder its applicability in MRD detection which includes, sequence mismatch against consensus primer and diversity of the CDR region. NGS can detect almost all clones and subclones and hence can monitor clonal evolution. Unlike ASO-PCR, additional advantage of NGS is that it is independent of a standard curve.
CtDNA in disease monitoring and MRD assessment in myeloma
Recent technological developments in detection of MRD by utilizing peripheral blood instead of bone marrow have emerged [118,119]. Usually cell-free fraction of peripheral blood (serum/plasma) and the cellular part (PBMCs) are used to detect MRD in peripheral blood. In a study by Korde et al (2014), myeloma associated clonotypes by NGS of the immunoglobulin VDJ segments were detected in 13/14 plasma samples (i.e. cfDNA) at diagnosis, while only 1/13 plasma samples after therapy showed myeloma associated clonotype, while monoclonal protein was still detected in 12/13 serum samples by immunofixation and/or serum electrophoresis [118]. Although, detection of myeloma-specific clonotypes in the peripheral blood plasma was shown at diagnosis; however, after two cycles of combination therapy these clonotypes could not be quantified using standard volumes of peripheral blood plasma as the tumor load in the plasma is several folds lower than in the BM [118]. Study of MRD detection in PB using NGS of clonotypic VDJ gene rearrangements by Oberle et al (2017) showed that myeloma clonotypes were absent in PB samples despite the presence of M-protein [101]. Furthermore, this study showed discordance in 30% of the samples in plasma cfDNA and PB leukocytes in MRD-positive cases. Hence, MRD analysis needs to be performed in both the compartments of PB (plasma and leukocytes) [101].
A correlation was shown between ctDNA and tumor burden in non-responders compared with less than half of responder myeloma patients. However, Mazzotti et al (2018) demonstrated no correlation in paired bone marrow and blood samples (ctDNA) for MRD by NGS utilizing only Ig gene rearrangements [120]. In a long-term study on blood-based MRD monitoring in MM, cfDNA detection of VDJ rearrangement by ASO-qPCR was used to demonstrate its utility as a prognostic marker [121]. Also, the length of cfDNA fragments was shown to be associated with treatment response of patients. Overall, myeloma clonotypes are present at a much higher level in BM samples than in PB samples, therefore, MRD testing using PB samples still poses many challenges.
Recently, advancement in the analysis of ctDNA was based on a two-step sequencing approach [122]. The first step, developed by the team at Broad and Dana-Farber Cancer Institute was called “ultra-low pass” whole genome sequencing (ULPS). This method is cost-effective in identification of blood samples with tumor DNA fraction of at least 5-10%, which allows more comprehensive genetic analysis. This was followed by whole exome sequencing (analyzing the protein-coding regions of the genome) of the filtered samples in the second step. As opposed to NGS alone, wherein the emphasis is on increasing sequencing depth, ultra-low pass whole-genome sequencing allows sequencing of the DNA at ultra-low depth as low as 0.1X. This two-step approach is unique and novel technique for screening and monitoring tumor fraction and copy-number variations in patient samples [122]. The ULPS requires low amount of sample to be analyzed as compared to conventional NGS and requires high-end computational analysis to interpret the results. In contrast to traditional array-based technique for getting molecular information, ULPS provides accurate and cost-effective genetic information. Before proceeding with genomic analysis of patient sample, ULPS provides tumor quality and content information to guide for further steps. By this QC step, researchers can identify samples with high purity for further genomic characterization. The current status of various studies for the utility of ctDNA in molecular characterization of MM is summarized in Table 5.
Table 5.
Current status of various studies on the utility of ctDNA in molecular characterization of MM
| S.No. | Title of Study | Genes Targets by ctDNA | Outcome | Reference |
|---|---|---|---|---|
| 1 | ctDNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates | Five genes (18 kb) (KRAS, NRAS, BRAF, EGFR, PIK3CA) | Ultra-deep sequencing of cfDNA to >20,000 × median coverage. Showed 96% concordance and >98% specificity when compared with matched BMA samples. | [12] |
| 2 | ctDNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma | Four genes (KRAS, NRAS, BRAF, TP53) | Mutations detected by OnTarget Mutation Detection (OMD) and ddPCR. High frequency of Plasma-Only mutations was observed in relapsed/refractory patients than newly diagnosed. | [13] |
| 3 | Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma | All genes and their exons | Screened and monitored tumor fraction and copy-number changes in cfDNA and CTCs using ultra-low pass whole-genome sequencing and whole-genome sequencing of matched CTCs, cfDNA and bone marrow biopsies from MM patients. ~99% concordance in clonal somatic mutations and ~81% concordance in copy number alterations between liquid and tumor biopsies. | [122] |
Challenges and pitfalls in cfDNA analysis in MM
Akin to the technical challenges involved in the isolation and characterization of plasma cells in MM, still several other methodological issues are in the standardization phase and therefore limit the clinical use of cfDNA analysis in the current scenario [123,124]. In regard to the pre-analytical procedures starting from sample collection timing, isolation of the plasma or serum from the blood, cfDNA extraction and quantification, and the storage condition for the cfDNA are critical for the data interpretation. In a study, the stability of cfDNA was compared in standard EDTA and other specialized blood collection tubes (including Streck, Roche, PAXgene, Norgen and CellSave tubes). Ten metastatic cancer patients with KRAS-mutation served as a source of blood samples, which was collected and stored for three days. It was demonstrated that the cfDNA levels were the least stable in EDTA tubes (≤6 hours), whereas cfDNA could be recovered from all the specialized tubes after 48-72 h, irrespective of the temperature [125]. The choice of specialized blood collection tubes is less critical if the samples are processed within a few hours.
In a comparative study, the isolation efficiency of cfDNA using various commercially available kits including QIAamp circulating nucleic acid kit (QIA), Maxwell RSC ccfDNA Plasma Kit (RSC), PME free-circulating DNA Extraction Kit (PME), the EpiQuick Circulating Cell-Free DNA Isolation Kit (EQ), and NEXTprep-Mag cfDNA Isolation Kit (NpMV1/2) was performed [125]. Both the total cfDNA concentration as well as the ctDNA fraction (KRAS mutation) was quantitated by ddPCR. Both QIA and the RSC kits had comparable isolation efficiencies of both non-mutated total cfDNA and KRAS mutated ctDNA, whereas the yield generated by other kits was relatively less. The advantage of RSC kit was its fully automated protocol over the labor-intensive QIA kit.
A multicentric Innovative Medicines Initiative consortium CANCER-ID (http://www.cancer-id.eu) recently documented a comparison of different technologies for cfDNA purification, quantification, and characterization [126]. The suitability of mononucleosomal DNA as a surrogate for cfDNA as a process quality control from nucleic acid extraction to mutation detection was demonstrated. The key challenges of cfDNA based tumor profiling still include the low overall yield (<10 ng/ml plasma) and low percentage of ctDNA fraction (<5%) relative to the total cfDNA.
Apart from the technical challenges, other practical issues in ctDNA analysis include the source of ctDNA origin, signal stability, specificity, sensitivity and limit of detection. The identification of tumor-specific somatic mutations is restricted to the hot-spot regions used to discriminate the tumor DNA from healthy DNA. In addition, the variable amounts of ctDNA detected in the bloodstream poses sensitivity issues in the detection of early-stage cancers. Thus, alternative strategies have been investigated towards improvement of the specificity and sensitivity of ctDNA detection.
The sensitivity of cfDNA analysis has been improved by undertaking several considerations. The size selection approaches of cfDNA, which are believed to reflect the tissue of origin, tumor-derived cfDNA are considerably shorter in size. Either physical methods or using bioinformatics tools, this challenge of short length has been overcome. Many studies focused on physical sorting of cfDNA which includes electrophoresis [127-130] while in-silico analysis of the fragmentation pattern of ctDNA require different size selection algorithms to select for appropriate size, which exhibits preferred end-coordinates [131,132]. When the tumor fraction in the total cfDNA sample is low, targeted approach for detecting point mutations are used such as qPCR, digital PCR and targeted DNA sequencing. These targeted approaches are highly sensitive and enhance the depth of mutational analysis. In contrast, when the tumor fraction is high, whole genome analysis-based approaches (such as whole genome sequencing and whole exome sequencing) are used which are less sensitive than targeted approaches [133]. Therefore, for ctDNA monitoring, targeted approaches may prove to be more beneficial. As tumors continuously evolve during the course of the disease and their heterogeneity pattern change upon exposure to selective influence of different therapies, therefore the sensitivity and efficiency of NGS based technologies are continuously being improved over time.
Future directions and conclusion
The cell-free DNA is a potential non-invasive biomarker for early cancer detection and can facilitate monitoring of myeloma patients. It is proposed that a combi-panel of multiple biomarkers could serve as a promising approach for disease monitoring. Such multi-panels could include the analysis of tumor-specific mutations in cfDNA using NGS, which can be combined with the identification of genetic alterations in cfDNA using ultra-low pass coverage WGS. In addition, the methylation profiling of cfDNA and the concurrent quantification of protein biomarkers can be add-ons for a more comprehensive cancer profiling. Together all these could increase the sensitivity and specificity of liquid biopsy in MM, especially in the early stages of the disease. To overcome the various hurdles in the translational potential of cfDNA in clinical practice, the unresolved issue of primary location of the disease could be achieved by the analysis of epigenetic markers. It is suggested that the highly tissue-specific DNA methylation profile is representative of cfDNA tissue of origin although further targeted studies are warranted [134]. To achieve this goal, multiple research groups may collaborate in a global consortium approach, which may cover a wide population cohort.
The cfDNA genotyping in MM may have an immediate clinical application as it could be incorporated into clinical trials. This may facilitate the identification of patients carrying actionable mutations, their longitudinal genetic monitoring during targeted therapy administration and MRD estimation. To enhance the utility of ctDNA as a routinely applicable biomarker for disease monitoring in MM, analyzing ctDNA epigenetic modifications and immune signatures may boost its potential for early detection in MM [135-137]. The need of the hour is an improved understanding of the kinetics of ctDNA production in myeloma to monitor MM disease burden using peripheral blood in routine clinical practice.
In conclusion, identifying tumor-specific mutations (ctDNA) at diagnosis and by sequential tracking of plasma-only mutations may be useful for prognostication that can help clinicians guide suitable treatment for individual MM patients. Moreover, ctDNA analysis holds great potential in prediction of MM disease progression, treatment outcome monitoring, tumor dynamics monitoring, MRD assessment or early relapse prior to its actual clinical occurrence.
Acknowledgements
This work was supported by grant from Department of Biotechnology, Govt. of India to Prof. Ritu Gupta [Grant: BT/MED/30/SP11006/2015]. AtulBasnal would like to thank University Grants Commission, Govt. of India for CSIR-UGC-Senior Research Fellowship.
Disclosure of conflict of interest
None.
References
- 1.Rasche L, Kortüm KM, Raab MS, Weinhold N. The impact of tumor heterogeneity on diagnostics and novel therapeutic strategies in multiple myeloma. Int J Mol Sci. 2019;20:1248. doi: 10.3390/ijms20051248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Haart SJ, Willems SM, Mutis T, Koudijs MJ, van Blokland MT, Lokhorst HM, de Weger RA, Minnema MC. Comparison of intramedullary myeloma and corresponding extramedullary soft tissue plasmacytomas using genetic mutational panel analyses. Blood Cancer J. 2016;6:e426. doi: 10.1038/bcj.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melchor L, Jones JR, Lenive O, Peterson EA, Murison A, Wardell CP, Kaiser MF, P P, Boyle EM, Begum DB, Pawlyn C, Johnson DC, Rapado I, Cairns DA, Gregory WM, Owen RG, Jackson GH, Drayson MT, Davies FE, Martínez-López J, Houlston RS, Greaves M, Walker BA, Morgan GJ. Spatiotemporal analysis of intraclonal heterogeneity in multiple myeloma: unravelling the impact of treatment and the propagating capacity of subclones using whole exome sequencing. Blood. 2015;126:371–371. [Google Scholar]
- 4.Egan J, Kortuem KM, Kurdoglu A, Izatt T, Aldrich J, Reiman R, Phillips L, Baker A, Shi CX, Schmidt J, Liang WS, Craig DW, Carpten JD, Stewart AK. Extramedullary myeloma whole genome sequencing reveals novel mutations in cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br J Haematol. 2013;161:748–751. doi: 10.1111/bjh.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, Bauer M, Stein C, Deshpande S, Wardell C, Buzder T, Molnar G, Zangari M, van Rhee F, Thanendrarajan S, Schinke C, Epstein J, Davies FE, Walker BA, Meissner T, Barlogie B, Morgan GJ, Weinhold N. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. 2017;8:268. doi: 10.1038/s41467-017-00296-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paiva B, Van Dongen JJ, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015;125:3059–3068. doi: 10.1182/blood-2014-11-568907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holstein S, Al-Kadhimi Z, Costa L, Hahn T, Hari P, Hillengass J, Jacob A, Munshi N, Oliva S, Pasquini M, Shi Q, Stadtmauer E, Waldvogel S, McCarthy P. Summary of the third annual blood and marrow transplant clinical trials network myeloma intergroup workshop on minimal residual disease and immune profiling. Biol Blood Marrow Transplant. 2019;26:e7–e15. doi: 10.1016/j.bbmt.2019.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrot A, Lauwers-Cances V, Corre J, Robillard N, Hulin C, Chretien ML, Dejoie T, Maheo S, Stoppa AM, Pegourie B, Karlin L, Garderet L, Arnulf B, Doyen C, Meuleman N, Royer B, Eveillard JR, Benboubker L, Dib M, Decaux O, Jaccard A, Belhadj K, Brechignac S, Kolb B, Fohrer C, Mohty M, Macro M, Richardson PG, Carlton V, Moorhead M, Willis T, Faham M, Anderson KC, Harousseau JL, Leleu X, Facon T, Moreau P, Attal M, Avet-Loiseau H, Munshi N. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018;132:2456–2464. doi: 10.1182/blood-2018-06-858613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishima Y, Paiva B, Shi J, Park J, Manier S, Takagi S, Massoud M, Perilla-Glen A, Aljawai Y, Huynh D, Roccaro AM, Sacco A, Capelletti M, Detappe A, Alignani D, Anderson KC, Munshi NC, Prosper F, Lohr JG, Ha G, Freeman SS, Van Allen EM, Adalsteinsson VA, Michor F, San Miguel JF, Ghobrial IM. The mutational landscape of circulating tumor cells in multiple myeloma. Cell Rep. 2017;19:218–224. doi: 10.1016/j.celrep.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, Brenton JD, Rosenfeld N. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra168. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 11.Mithraprabhu S, Khong T, Ramachandran M, Chow AWS, Klarica D, Mai L, Walsh S, Broemeling D, Marziali A, Wiggin M, Hocking J, Kalff A, Durie B, Spencer A. Mutational characterisation and tracking disease progression using circulating cell-free tumor DNA in multiple myeloma patients. Blood. 2016;31:1695–1705. [Google Scholar]
- 12.Kis O, Kaedbey R, Chow S, Danesh A, Dowar M, Li T, Li Z, Liu J, Mansour M, Masih-Khan E, Zhang T, Bratman SV, Oza AM, Kamel-Reid S, Trudel S, Pugh TJ. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat Commun. 2017;8:15086. doi: 10.1038/ncomms15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mithraprabhu S, Khong T, Ramachandran M, Chow A, Klarica D, Mai L, Walsh S, Broemeling D, Marziali A, Wiggin M, Hocking J, Kalff A, Durie B, Spencer A. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia. 2017;31:1695–1705. doi: 10.1038/leu.2016.366. [DOI] [PubMed] [Google Scholar]
- 14.Mithraprabhu S, Sirdesai S, Chen M, Khong T, Spencer A. Circulating tumour dna analysis for tumour genome characterisation and monitoring disease burden in extramedullary multiple myeloma. Int J Mol Sci. 2018;19:1858. doi: 10.3390/ijms19071858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- 16.Stroun M, Maurice P, Vasioukhin V, Lyautey J, Lederrey C, Lefort F, Rossier A, Chen XQ, Anker P. The origin and mechanism of circulating DNA. Ann N Y Acad Sci. 2000;906:161–168. doi: 10.1111/j.1749-6632.2000.tb06608.x. [DOI] [PubMed] [Google Scholar]
- 17.Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O’Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B, Diaz LA, Velculescu VE. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4:162ra154. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandel P, Metais P. Les acides nucléiques du plasma sanguin chez l’homme. C R Seances Soc Biol Fil. 1948;142:241–243. [PubMed] [Google Scholar]
- 19.Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest. 1966;45:1732–1740. doi: 10.1172/JCI105479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasioukhin V, Anker P, Maurice P, Lyautey J, Lederrey C, Stroun M. Point mutations of the N-Ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol. 1994;86:774–779. doi: 10.1111/j.1365-2141.1994.tb04828.x. [DOI] [PubMed] [Google Scholar]
- 21.Kustanovich A, Schwartz R, Peretz T, Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol Ther. 2019;20:1057–1067. doi: 10.1080/15384047.2019.1598759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goggs R, Jeffery U, LeVine DN, Ronald HL. Neutrophil-extracellular traps, cell-free DNA, and immunothrombosis in companion animals: a review. Vet Pathol. 2020;57:6–23. doi: 10.1177/0300985819861721. [DOI] [PubMed] [Google Scholar]
- 23.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 24.Zheng YW, Chan KC, Sun H, Jiang P, Su X, Chen EZ, Lun FM, Hung EC, Lee V, Wong J, Lai PB, Li CK, Chiu RW, Lo YM. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: a transplantation model. Clin Chem. 2012;58:549–558. doi: 10.1373/clinchem.2011.169318. [DOI] [PubMed] [Google Scholar]
- 25.Sun K, Jiang P, Wong AIC, Cheng YKY, Cheng SH, Zhang H, Chan KCA, Leung TY, Chiu RWK, Lo YMD. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc Natl Acad Sci U S A. 2018;115:E5106–E5114. doi: 10.1073/pnas.1804134115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T, Kis O, Zhao Z, Spreafico A, Medina TdS, Wang Y, Roulois D, Ettayebi I, Chen Z, Chow S, Murphy T, Arruda A, O’Kane GM, Liu J, Mansour M, McPherson JD, O’Brien C, Leighl N, Bedard PL, Fleshner N, Liu G, Minden MD, Gallinger S, Goldenberg A, Pugh TJ, Hoffman MM, Bratman SV, Hung RJ, De Carvalho DD. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–583. doi: 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 27.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 28.Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer--a survey. Biochim Biophys Acta. 2007;1775:181–232. doi: 10.1016/j.bbcan.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Aucamp J, Bronkhorst AJ, Badenhorst CPS, Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc. 2018;93:1649–1683. doi: 10.1111/brv.12413. [DOI] [PubMed] [Google Scholar]
- 30.Hummel E, Hessas E, Müller S, Beiter T, Fisch M, Eibl A, Wolf O, Giebel B, Platen P, Kumsta R, Moser D. Cell-free DNA release under psychosocial and physical stress conditions. Transl Psychiatry. 2018;8:236. doi: 10.1038/s41398-018-0264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barták B, Nagy Z, Spisák S, Tulassay Z, Dank M, Igaz P, Molnar B. In vivo analysis of circulating cell-free DNA release and degradation. Orv Hetil. 2018;159:223–233. doi: 10.1556/650.2018.30929. [DOI] [PubMed] [Google Scholar]
- 32.Laurent D, Semple F, Starkey Lewis PJ, Rose E, Black HA, Coe J, Forbes SJ, Arends MJ, Dear JW, Aitman TJ. Absolute measurement of the tissue origins of cell-free DNA in the healthy state and following paracetamol overdose. BMC Med Genomics. 2020;13:60. doi: 10.1186/s12920-020-0705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gedvilaitė V, Schveigert D, Cicėnas S. Cell-free DNA in non-small cell lung cancer. Acta Med Litu. 2017;24:138–144. doi: 10.6001/actamedica.v24i2.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esposito A, Criscitiello C, Trapani D, Curigliano G. The emerging role of “liquid biopsies”, circulating tumor cells, and circulating cell-free tumor DNA in lung cancer diagnosis and identification of resistance mutations. Curr Oncol Rep. 2017;19:1. doi: 10.1007/s11912-017-0564-y. [DOI] [PubMed] [Google Scholar]
- 36.Trejo-Becerril C, Pérez-Cárdenas E, Taja-Chayeb L, Anker P, Herrera-Goepfert R, Medina-Velázquez LA, Hidalgo-Miranda A, Pérez-Montiel D, Chávez-Blanco A, Cruz-Velázquez J, Díaz-Chávez J, Gaxiola M, Dueñas-González A. Cancer progression mediated by horizontal gene transfer in an in vivo model. PLoS One. 2012;7:e52754. doi: 10.1371/journal.pone.0052754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.García-Olmo D, García-Olmo DC. Functionality of circulating DNA: the hypothesis of genometastasis. Ann N Y Acad Sci. 2001;945:265–275. doi: 10.1111/j.1749-6632.2001.tb03895.x. [DOI] [PubMed] [Google Scholar]
- 38.García-Casas A, García-Olmo DC, García-Olmo D. Further the liquid biopsy: gathering pieces of the puzzle of genometastasis theory. World J Clin Oncol. 2017;8:378–388. doi: 10.5306/wjco.v8.i5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breitbach S, Tug S, Simon P. Circulating cell-free DNA: an up-coming molecular marker in exercise physiology. Sports Med. 2012;42:565–586. doi: 10.2165/11631380-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 40.Atamaniuk J, Kopecky C, Skoupy S, Säemann MD, Weichhart T. Apoptotic cell-free DNA promotes inflammation in haemodialysis patients. Nephrol Dial Transplant. 2012;27:902–905. doi: 10.1093/ndt/gfr695. [DOI] [PubMed] [Google Scholar]
- 41.Shimada H. Biomarkers in cancer therapy liquid biopsy comes of age. Springer Nature Singapore. 2019 [Google Scholar]
- 42.Domínguez-Vigil IG, Moreno-Martínez AK, Wang JY, Roehrl MHA, Barrera-Saldaña HA. The dawn of the liquid biopsy in the fight against cancer. Oncotarget. 2018;9:2912–2922. doi: 10.18632/oncotarget.23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwapisz D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? . Ann Transl Med. 2017;5:46. doi: 10.21037/atm.2017.01.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sellami D, Dharan B, Wilke C, Scherer SJ, Hirawat S. Circulating tumor DNA as a novel tool to shape clinical trial designs with the potential to impact outcomes: a focus on PI3K inhibitors. Ann Oncol. 2017;28:2882–2887. doi: 10.1093/annonc/mdx480. [DOI] [PubMed] [Google Scholar]
- 45.Ulrich BC, Paweletz CP. Cell-free DNA in oncology: gearing up for clinic. Ann Lab Med. 2018;38:1–8. doi: 10.3343/alm.2018.38.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lustberg MB, Stover DG, Chalmers JJ. Implementing liquid biopsies in clinical trials: state of affairs, opportunities, and challenges. Cancer J. 2018;24:61–66. doi: 10.1097/PPO.0000000000000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646–650. [PubMed] [Google Scholar]
- 48.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih lM, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu TL, Zhang D, Chia JH, Tsao KC, Sun CF, Wu JT. Cell-free DNA: measurement in various carcinomas and establishment of normal reference range. Clin Chim Acta. 2002;321:77–87. doi: 10.1016/s0009-8981(02)00091-8. [DOI] [PubMed] [Google Scholar]
- 50.Spindler KL, Appelt AL, Pallisgaard N, Andersen RF, Brandslund I, Jakobsen A. Cell-free DNA in healthy individuals, noncancerous disease and strong prognostic value in colorectal cancer. Int J Cancer. 2014;135:2984–2991. doi: 10.1002/ijc.28946. [DOI] [PubMed] [Google Scholar]
- 51.Singh N, Gupta S, Pandey RM, Chauhan SS, Saraya A. High levels of cell-free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Invest. 2015;33:78–85. doi: 10.3109/07357907.2014.1001894. [DOI] [PubMed] [Google Scholar]
- 52.Shaw JA, Guttery DS, Hills A, Fernandez-Garcia D, Page K, Rosales BM, Goddard KS, Hastings RK, Luo J, Ogle O, Woodley L, Ali S, Stebbing J, Coombes RC. Mutation analysis of cell-free DNA and single circulating tumor cells in metastatic breast cancer patients with high circulating tumor cell counts. Clin Cancer Res. 2017;23:88–96. doi: 10.1158/1078-0432.CCR-16-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, Halsall D, Wallis M, Bentley D, Caldas C, Rosenfeld N. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;369:93–94. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, Cheang M, Osin P, Nerurkar A, Kozarewa I, Garrido JA, Dowsett M, Reis-Filho JS, Smith IE, Turner NC. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 55.Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, Søreide JA, Javle M, Nordgård O, Smaaland R. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016;10:635–643. doi: 10.1016/j.molonc.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, Harding TC, Durkin KA, Otterson GA, Purcell WT, Camidge DR, Goldman JW, Sequist LV, Piotrowska Z, Wakelee HA, Neal JW, Alizadeh AA, Diehn M. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal R, Marafioti T, Kirkizlar E, Watkins TBK, McGranahan N, Ward S, Martinson L, Riley J, Fraioli F, Al Bakir M, Grönroos E, Zambrana F, Endozo R, Bi WL, Fennessy FM, Sponer N, Johnson D, Laycock J, Shafi S, Czyzewska-Khan J, Rowan A, Chambers T, Matthews N, Turajlic S, Hiley C, Lee SM, Forster MD, Ahmad T, Falzon M, Borg E, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Hafez D, Naik A, Ganguly A, Kareht S, Shah R, Joseph L, Marie Quinn A, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Oukrif D, Akarca AU, Hartley JA, Lowe HL, Lock S, Iles N, Bell H, Ngai Y, Elgar G, Szallasi Z, Schwarz RF, Herrero J, Stewart A, Quezada SA, Peggs KS, Van Loo P, Dive C, Lin CJ, Rabinowitz M, Aerts HJWL, Hackshaw A, Shaw JA, Zimmermann BG TRACERx consortium; PEACE consortium. Swanton C. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–451. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rustad EH, Coward E, Skytøen ER, Misund K, Holien T, Standal T, Børset M, Beisvag V, Myklebost O, Meza-Zepeda LA, Dai HY, Sundan A, Waage A. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica. 2017;102:1266–1272. doi: 10.3324/haematol.2016.160564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thierry AR, Pastor B, Jiang ZQ, Katsiampoura AD, Parseghian C, Loree JM, Overman MJ, Sanchez C, El Messaoudi S, Ychou M, Kopetz S. Circulating DNA demonstrates convergent evolution and common resistance mechanisms during treatment of colorectal cancer. Clin Cancer Res. 2017;23:457. doi: 10.1158/1078-0432.CCR-17-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Molparia B, Nichani E, Torkamani A. Assessment of circulating copy number variant detection for cancer screening. PLoS One. 2017;12:e0180647. doi: 10.1371/journal.pone.0180647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khan KH, Cunningham D, Werner B, Vlachogiannis G, Spiteri I, Heide T, Mateos JF, Vatsiou A, Lampis A, Damavandi MD, Lote H, Huntingford IS, Hedayat S, Chau I, Tunariu N, Mentrasti G, Trevisani F, Rao S, Anandappa G, Watkins D, Starling N, Thomas J, Peckitt C, Khan N, Rugge M, Begum R, Hezelova B, Bryant A, Jones T, Proszek P, Fassan M, Hahne JC, Hubank M, Braconi C, Sottoriva A, Valeri N. Longitudinal liquid biopsy and mathematical modeling of clonal evolution forecast time to treatment failure in the prospect-C phase II colorectal cancer clinical trial. Cancer Discov. 2018;8:1270–1285. doi: 10.1158/2159-8290.CD-17-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang TY, Piunti A, Lulla RR, Qi J, Horbinski CM, Tomita T, James CD, Shilatifard A, Saratsis AM. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol Commun. 2017;5:28. doi: 10.1186/s40478-017-0436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, Zheng Y, Skakodub A, Mehta SA, Campos C, Hsieh WY, Selcuklu SD, Ling L, Meng F, Jing X, Samoila A, Bale TA, Tsui DWY, Grommes C, Viale A, Souweidane MM, Tabar V, Brennan CW, Reiner AS, Rosenblum M, Panageas KS, DeAngelis LM, Young RJ, Berger MF, Mellinghoff IK. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019;565:654–658. doi: 10.1038/s41586-019-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Molecular analysis of circulating tumour cells-biology and biomarkers. Nat Rev Clin Oncol. 2014;11:129–144. doi: 10.1038/nrclinonc.2013.253. [DOI] [PubMed] [Google Scholar]
- 65.Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, Hendifar AE, Wang Z, Atreya CE, Kelley RK, Van Loon K, Ko AH, Tempero MA, Bivona TG, Munster PN, Talasaz A, Collisson EA. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov. 2015;5:1040–1048. doi: 10.1158/2159-8290.CD-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coombes RC, Page K, Salari R, Hastings RK, Armstrong A, Ahmed S, Ali S, Cleator S, Kenny L, Stebbing J, Rutherford M, Sethi H, Boydell A, Swenerton R, Fernandez-Garcia D, Gleason KLT, Goddard K, Guttery DS, Assaf ZJ, Wu HT, Natarajan P, Moore DA, Primrose L, Dashner S, Tin AS, Balcioglu M, Srinivasan R, Shchegrova SV, Olson A, Hafez D, Billings P, Aleshin A, Rehman F, Toghill BJ, Hills A, Louie MC, Lin CJ, Zimmermann BG, Shaw JA. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin Cancer Res. 2019;25:4255–4263. doi: 10.1158/1078-0432.CCR-18-3663. [DOI] [PubMed] [Google Scholar]
- 67.Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP, Shendure J. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:e1006162. doi: 10.1371/journal.pgen.1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sykes PJ, Neoh SH, Brisco MJ, Hughes E, Condon J, Morley AA. Quantitation of targets for PCR by use of limiting dilution. Biotechniques. 1992;13:444–449. [PubMed] [Google Scholar]
- 69.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102:16368–16373. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao GJ, Chan KC, Jiang P, Sun H, Leung TY, Chiu RW, Lo YM. Noninvasive prenatal diagnosis of fetal trisomy 21 by allelic ratio analysis using targeted massively parallel sequencing of maternal plasma DNA. PLoS One. 2012;7:e38154. doi: 10.1371/journal.pone.0038154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, Anagnostou V, Fiksel J, Cristiano S, Papp E, Speir S, Reinert T, Orntoft MW, Woodward BD, Murphy D, Parpart-Li S, Riley D, Nesselbush M, Sengamalay N, Georgiadis A, Li QK, Madsen MR, Mortensen FV, Huiskens J, Punt C, van Grieken N, Fijneman R, Meijer G, Husain H, Scharpf RB, Diaz LA Jr, Jones S, Angiuoli S, Ørntoft T, Nielsen HJ, Andersen CL, Velculescu VE. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9:eaan2415. doi: 10.1126/scitranslmed.aan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dello Russo C, Cesta A, Longo S, Barone MA, Cima A, Mesoraca A, Sparacino D, Viola A, Giorlandino C. Validation of extensive next-generation sequencing method for monogenic disorder analysis on cell-free fetal DNA: noninvasive prenatal diagnosis. J Mol Diagn. 2019;21:572–579. doi: 10.1016/j.jmoldx.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 74.Leal A, Cristiano S, Phallen J, Fiksel J, Adleff V, Bruhm DC, Jensen SO, Medina JE, Palsgrove DN, Niknafs N, Anagnostou V, Forde PM, Brahmer JR, Fijneman RJA, Johansen JS, Nielsen HJ, Meijer GA, Andersen CL, Scharpf RB, Velculescu V. Genome-wide cell-free DNA fragmentation profiling for early cancer detection. J. Clin. Oncol. 2019;570:385–389. [Google Scholar]
- 75.Drandi D, Kubiczkova-Besse L, Ferrero S, Dani N, Passera R, Mantoan B, Gambella M, Monitillo L, Saraci E, Ghione P, Genuardi E, Barbero D, Omedè P, Barberio D, Hajek R, Vitolo U, Palumbo A, Cortelazzo S, Boccadoro M, Inghirami G, Ladetto M. Minimal residual disease detection by droplet digital pcr in multiple myeloma, mantle cell lymphoma, and follicular lymphoma: a comparison with real-time PCR. J Mol Diagn. 2015;17:652–660. doi: 10.1016/j.jmoldx.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 76.Della Starza I, Nunes V, Cavalli M, De Novi LA, Ilari C, Apicella V, Vitale A, Testi AM, Del Giudice I, Chiaretti S, Foà R, Guarini A. Comparative analysis between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell receptor gene rearrangements to monitor minimal residual disease in acute lymphoblastic leukaemia. Br J Haematol. 2016;174:541–549. doi: 10.1111/bjh.14082. [DOI] [PubMed] [Google Scholar]
- 77.Takamatsu H. Comparison of minimal residual disease detection by multiparameter flow cytometry, ASO-qPCR, droplet digital PCR, and deep sequencing in patients with multiple myeloma who underwent autologous stem cell transplantation. J Clin Med. 2017;6:91–101. doi: 10.3390/jcm6100091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene. 2001;20:5611–5622. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- 79.Shaughnessy J Jr, Gabrea A, Qi Y, Brents L, Zhan F, Tian E, Sawyer J, Barlogie B, Bergsagel PL, Kuehl M. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood. 2001;98:217–223. doi: 10.1182/blood.v98.1.217. [DOI] [PubMed] [Google Scholar]
- 80.Avet-Loiseau H, Facon T, Grosbois B, Magrangeas F, Rapp MJ, Harousseau JL, Minvielle S, Bataille R. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood. 2002;99:2185–2191. doi: 10.1182/blood.v99.6.2185. [DOI] [PubMed] [Google Scholar]
- 81.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, Morgan G, Van Ness B, Chesi M, Minvielle S, Neri A, Barlogie B, Kuehl WM, Liebisch P, Davies F, Chen-Kiang S, Durie BG, Carrasco R, Sezer O, Reiman T, Pilarski L, Avet-Loiseau H International Myeloma Working Group. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210–2221. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar S, Fonseca R, Ketterling RP, Dispenzieri A, Lacy MQ, Gertz MA, Hayman SR, Buadi FK, Dingli D, Knudson RA, Greenberg A, Russell SJ, Zeldenrust SR, Lust JA, Kyle RA, Bergsagel L, Rajkumar SV. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood. 2012;119:2100–5. doi: 10.1182/blood-2011-11-390658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neben K, Jauch A, Hielscher T, Hillengass J, Lehners N, Seckinger A, Granzow M, Raab MS, Ho AD, Goldschmidt H, Hose D. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. 2013;31:4325–4332. doi: 10.1200/JCO.2012.48.4923. [DOI] [PubMed] [Google Scholar]
- 84.Rajkumar SV, Gupta V, Fonseca R, Dispenzieri A, Gonsalves WI, Larson D, Ketterling RP, Lust JA, Kyle RA, Kumar SK. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27:1738–1744. doi: 10.1038/leu.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, Richardson P, Caltagirone S, Lahuerta JJ, Facon T, Bringhen S, Gay F, Attal M, Passera R, Spencer A, Offidani M, Kumar S, Musto P, Lonial S, Petrucci MT, Orlowski RZ, Zamagni E, Morgan G, Dimopoulos MA, Durie BG, Anderson KC, Sonneveld P, San Miguel J, Cavo M, Rajkumar SV, Moreau P. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J. Clin. Oncol. 2015;33:2863–2869. doi: 10.1200/JCO.2015.61.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Du Pont SR, Cleynen A, Fontan C, Attal M, Munshi N, Corre J, Avet-Loiseau H. Genomics of multiple myeloma. J. Clin. Oncol. 2017;35:963–967. doi: 10.1200/JCO.2016.70.6705. [DOI] [PubMed] [Google Scholar]
- 87.Coffey DG, Wu QV, Towlerton AMH, Ornelas S, Morales AJ, Xu Y, Green DJ, Warren EH. Ultradeep, targeted sequencing reveals distinct mutations in blood compared to matched bone marrow among patients with multiple myeloma. Blood Cancer J. 2019;9:77. doi: 10.1038/s41408-019-0238-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, Baker A, Bergsagel PL, Bernstein BE, Drier Y, Fonseca R, Gabriel SB, Hofmeister CC, Jagannath S, Jakubowiak AJ, Krishnan A, Levy J, Liefeld T, Lonial S, Mahan S, Mfuko B, Monti S, Perkins LM, Onofrio R, Pugh TJ, Rajkumar SV, Ramos AH, Siegel DS, Sivachenko A, Stewart AK, Trudel S, Vij R, Voet D, Winckler W, Zimmerman T, Carpten J, Trent J, Hahn WC, Garraway LA, Meyerson M, Lander ES, Getz G, Golub TR. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, Dawson KJ, Iorio F, Nik-Zainal S, Bignell GR, Hinton JW, Li Y, Tubio JM, McLaren S, O’Meara S, Butler AP, Teague JW, Mudie L, Anderson E, Rashid N, Tai YT, Shammas MA, Sperling AS, Fulciniti M, Richardson PG, Parmigiani G, Magrangeas F, Minvielle S, Moreau P, Attal M, Facon T, Futreal PA, Anderson KC, Campbell PJ, Munshi NC. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC, Kaiser MF, Mirabella F, Lopez-Corral L, Humphray S, Murray L, Ross M, Bentley D, Gutiérrez NC, Garcia-Sanz R, San Miguel J, Davies FE, Gonzalez D, Morgan GJ. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia. 2014;28:384–390. doi: 10.1038/leu.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, Cibulskis K, McKenna A, Chapman MA, Straussman R, Levy J, Perkins LM, Keats JJ, Schumacher SE, Rosenberg M, Getz G, Golub TR. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–113. doi: 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 93.Lionetti M, Barbieri M, Todoerti K, Agnelli L, Marzorati S, Fabris S, Ciceri G, Galletti S, Milesi G, Manzoni M, Mazzoni M, Greco A, Tonon G, Musto P, Baldini L, Neri A. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: implication for MEK-ERK pathway activation. Oncotarget. 2015;6:24205–24217. doi: 10.18632/oncotarget.4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kortüm KM, Mai EK, Hanafiah NH, Shi CX, Zhu YX, Bruins L, Barrio S, Jedlowski P, Merz M, Xu J, Stewart RA, Andrulis M, Jauch A, Hillengass J, Goldschmidt H, Bergsagel PL, Braggio E, Stewart AK, Raab MS. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016;128:1226–1233. doi: 10.1182/blood-2016-02-698092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, Mitchell TJ, Fullam A, Gonzalez S, Szalat R, Abascal F, Rodriguez-Martin B, Samur MK, Glodzik D, Roncador M, Fulciniti M, Tai YT, Minvielle S, Magrangeas F, Moreau P, Corradini P, Anderson KC, Tubio JMC, Wedge DC, Gerstung M, Avet-Loiseau H, Munshi N, Campbell PJ. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun. 2019;10:3835. doi: 10.1038/s41467-019-11680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, Proszek PZ, Johnson DC, Kaiser MF, Melchor L, Aronson LI, Scales M, Pawlyn C, Mirabella F, Jones JR, Brioli A, Mikulasova A, Cairns DA, Gregory WM, Quartilho A, Drayson MT, Russell N, Cook G, Jackson GH, Leleu X, Davies FE, Morgan GJ. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015;33:3911–3920. doi: 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.White BS, Lanc I, O’Neal J, Gupta H, Fulton RS, Schmidt H, Fronick C, Belter EA, Fiala M, King J, Ahmann GJ, Derome M, Mardis ER, Vij R, Dipersio JF, Levy J, Auclair D, Tomasson MH. A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in IGLL5. Blood Cancer J. 2018;8:35. doi: 10.1038/s41408-018-0062-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, Maura F, Sathiaseelan V, Tai YT, Mudie L, O’Meara S, Raine K, Teague JW, Butler AP, Carniti C, Gerstung M, Bagratuni T, Kastritis E, Dimopoulos M, Corradini P, Anderson KC, Moreau P, Minvielle S, Campbell PJ, Papaemmanuil E, Avet-Loiseau H, Munshi NC. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604–2616. doi: 10.1038/s41375-018-0037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bolli N, Li Y, Sathiaseelan V, Raine K, Jones D, Ganly P, Cocito F, Bignell G, Chapman MA, Sperling AS, Anderson KC, Avet-Loiseau H, Minvielle S, Campbell PJ, Munshi NC. A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer J. 2016;6:e467–475. doi: 10.1038/bcj.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goldsmith SR, Fiala MA, Dukeman J, Ghobadi A, Stockerl-Goldstein K, Schroeder MA, Tomasson M, Wildes TM, Vij R. Next generation sequencing-based validation of the revised international staging system for multiple myeloma: an analysis of the MMRF CoMMpass study. Clin Lymphoma Myeloma Leuk. 2019;19:285–289. doi: 10.1016/j.clml.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 101.Oberle A, Brandt A, Voigtlaender M, Thiele B, Radloff J, Schulenkorf A, Alawi M, Akyüz N, März M, Ford CT, Krohn-Grimberghe A, Binder M. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica. 2017;102:1105–1111. doi: 10.3324/haematol.2016.161414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gerber B, Manzoni M, Spina V, Bruscaggin A, Lionetti M, Fabris S, Barbieri M, Ciceri G, Pompa A, Forestieri G, Lerch E, Servida P, Bertoni F, Zucca E, Ghielmini M, Cortelezzi A, Cavalli F, Stussi G, Baldini L, Rossi D, Neri A. Circulating tumor DNA as a liquid biopsy in plasma cell dyscrasias. Haematologica. 2018;103:e245–e248. doi: 10.3324/haematol.2017.184358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mithraprabhu S, Morley R, Khong T, Kalff A, Bergin K, Hocking J, Savvidou I, Bowen KM, Ramachandran M, Choi K, Wong BKL, Reynolds J, Spencer A. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia. 2019;33:2022–2033. doi: 10.1038/s41375-019-0469-x. [DOI] [PubMed] [Google Scholar]
- 104.Manasanch EE. What to do with minimal residual disease testing in myeloma. Hematology Am Soc Hematol Educ Program. 2019;1:137–141. doi: 10.1182/hematology.2019000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alyanakian MA, Abbas A, Delarue R, Arnulf B, Aucouturier P. Free immunoglobulin light-chain serum levels in the follow-up of patients with monoclonal gammopathies: correlation with 24-hr urinary light-chain excretion. Am J Hematol. 2004;75:246–248. doi: 10.1002/ajh.20007. [DOI] [PubMed] [Google Scholar]
- 106.Puig N, Sarasquete ME, Alcoceba M, Balanzategui A, Chillón MC, Sebastián E, Marín LA, Díaz MG, San Miguel JF, Sanz RG. The use of CD138 positively selected marrow samples increases the applicability of minimal residual disease assessment by PCR in patients with multiple myeloma. Ann Hematol. 2013;92:97–100. doi: 10.1007/s00277-012-1566-3. [DOI] [PubMed] [Google Scholar]
- 107.Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93:981–1114. doi: 10.1002/ajh.25117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, Munshi N, Lonial S, Bladé J, Mateos MV, Dimopoulos M, Kastritis E, Boccadoro M, Orlowski R, Goldschmidt H, Spencer A, Hou J, Chng WJ, Usmani SZ, Zamagni E, Shimizu K, Jagannath S, Johnsen HE, Terpos E, Reiman A, Kyle RA, Sonneveld P, Richardson PG, McCarthy P, Ludwig H, Chen W, Cavo M, Harousseau JL, Lentzsch S, Hillengass J, Palumbo A, Orfao A, Rajkumar SV, Miguel JS, Avet-Loiseau H. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e346. doi: 10.1016/S1470-2045(16)30206-6. [DOI] [PubMed] [Google Scholar]
- 109.Avet-Loiseau H, Ludwig H, Landgren O, Paiva B, Morris C, Yang H, Zhou K, Ro S, Mateos MV. Minimal residual disease status as a surrogate endpoint for progression-free survival in newly diagnosed multiple myeloma studies: a meta-analysis. Clin Lymphoma Myeloma Leuk. 2019;20:30–37. doi: 10.1016/j.clml.2019.09.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carulli G, Tarasco A, Sammuri P, Ottaviano V, Domenichini C, Ciancia EM, Petrini M. Assessment of response to therapy in multiple myeloma by multiparameter flow cytometry. Usefulness of an eight-color single tube with monoclonal antibodies in dried formulation. Clin Ter. 2019;170:e352–e356. doi: 10.7417/CT.2019.2159. [DOI] [PubMed] [Google Scholar]
- 111.Flores-Montero J, Sanoja-Flores L, Paiva B, Puig N, García-Sánchez O, Böttcher S, van der Velden VHJ, Pérez-Morán JJ, Vidriales MB, García-Sanz R, Jimenez C, González M, Martínez-López J, Corral-Mateos A, Grigore GE, Fluxá R, Pontes R, Caetano J, Sedek L, Del Cañizo MC, Bladé J, Lahuerta JJ, Aguilar C, Bárez A, García-Mateo A, Labrador J, Leoz P, Aguilera-Sanz C, San-Miguel J, Mateos MV, Durie B, van Dongen JJM, Orfao A. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia. 2017;31:2094–2103. doi: 10.1038/leu.2017.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soh KT, Wallace P. Monitoring of measurable residual disease in multiple myeloma by multiparametric flow cytometry. Curr Protoc Cytom. 2019;90:e63. doi: 10.1002/cpcy.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Paiva B, Puig N, Cedena MT, Rosiñol L, Cordón L, Vidriales MB, Burgos L, Flores-Montero J, Sanoja-Flores L, Lopez-Anglada L, Maldonado R, de la Cruz J, Gutierrez NC, Calasanz MJ, Martin-Ramos ML, Garcia-Sanz R, Martinez-Lopez J, Oriol A, Blanchard MJ, Rios R, Martin J, Martinez-Martinez R, Sureda A, Hernandez MT, de la Rubia J, Krsnik I, Moraleda JM, Palomera L, Bargay J, Van Dongen JJM, Orfao A, Mateos MV, Blade J, San-Miguel JF, Lahuerta JJ GEM (Grupo Español de Mieloma)/PETHEMA (Programa Para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Study Group. Measurable residual disease by next-generation flow cytometry in multiple myeloma. J. Clin. Oncol. 2020;38:784–792. doi: 10.1200/JCO.19.01231. [DOI] [PubMed] [Google Scholar]
- 114.Wang N, Tesfaluul N, Li J, Gao X, Liu S, Yue B. Enrichment of circulating myeloma cells by immunomagnetic beads combined with flow cytometry for monitoring minimal residual disease and relapse in patients with multiple myeloma. Ann Hematol. 2019;98:2769–2780. doi: 10.1007/s00277-019-03833-5. [DOI] [PubMed] [Google Scholar]
- 115.Sarasquete ME, García-Sanz R, González D, Martínez J, Mateo G, Martínez P, Ribera JM, Hernández JM, Lahuerta JJ, Orfão A, González M, San Miguel JF. Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica. 2005;90:1365–1372. [PubMed] [Google Scholar]
- 116.Bai Y, Orfao A, Chim CS. Molecular detection of minimal residual disease in multiple myeloma. Br J Haematol. 2018;181:11–26. doi: 10.1111/bjh.15075. [DOI] [PubMed] [Google Scholar]
- 117.Takamatsu H, Takezako N, Zheng J, Moorhead M, Carlton VEH, Kong KA, Murata R, Ito S, Miyamoto T, Yokoyama K, Matsue K, Sato T, Kurokawa T, Yagi H, Terasaki Y, Ohata K, Matsumoto M, Yoshida T, Faham M, Nakao S. Prognostic value of sequencing-based minimal residual disease detection in patients with multiple myeloma who underwent autologous stem-cell transplantation. Ann Oncol. 2017;28:2503–2510. doi: 10.1093/annonc/mdx340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Korde N, Mailankody S, Roschewski M, Faham M, Kotwaliwale C, Moorhead M, Kwok ML, Manasanch EE, Bhutani M, Tageja N, Kazandjian D, Costello R, Zhang Y, Zingone A, Burton D, Mulquin M, Carpenter A, Zuchlinski D, Lamping E, Carter G, Morrison C, Kurdziel K, Lindenberg M, Kurlander R, Maric I, Calvo KR, Braylan RC, Yuan C, Stetler-Stevenson M, Arthur DC, Steinberg SM, Figg WD, Choyke P, Landgren O. Minimal residual disease (MRD) testing in newly diagnosed multiple myeloma (MM) patients: a prospective head-to-head assessment of cell-based, molecular, and molecular-imaging modalities. Blood. 2014;124:2105. [Google Scholar]
- 119.Vij R, Mazumder A, Klinger M, O’Dea D, Paasch J, Martin T, Weng L, Park J, Fiala M, Faham M, Wolf J. Deep sequencing reveals myeloma cells in peripheral blood in majority of multiple myeloma patients. Clin Lymphoma Myeloma Leuk. 2014;14:131–139. doi: 10.1016/j.clml.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 120.Mazzotti C, Buisson L, Maheo S, Perrot A, Chretien ML, Leleu X, Hulin C, Manier S, Hébraud B, Roussel M, Do Souto L, Attal M, Avet-Loiseau H, Corre J. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018;2:2811–2813. doi: 10.1182/bloodadvances.2018025197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vrabel D, Sedlarikova L, Besse L, Rihova L, Bezdekova R, Almasi M, Kubaczkova V, Brožová L, Jarkovsky J, Plonkova H, Jelinek T, Sandecka V, Stork M, Pour L, Sevcikova S, Hajek R. Dynamics of tumor-specific cfDNA in response to therapy in multiple myeloma patients. Eur J Haematol. 2019;104:190–197. doi: 10.1111/ejh.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Manier S, Park J, Capelletti M, Bustoros M, Freeman SS, Ha G, Rhoades J, Liu CJ, Huynh D, Reed SC, Gydush G, Salem KZ, Rotem D, Freymond C, Yosef A, Perilla-Glen A, Garderet L, Van Allen EM, Kumar S, Love JC, Getz G, Adalsteinsson VA, Ghobrial IM. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat Commun. 2018;9:1691. doi: 10.1038/s41467-018-04001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ignatiadis M, Lee M, Jeffrey SS. Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin Cancer Res. 2015;21:4786–4800. doi: 10.1158/1078-0432.CCR-14-1190. [DOI] [PubMed] [Google Scholar]
- 124.Gorgannezhad L, Umer M, Islam MN, Nguyen NT, Shiddiky MJA. Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip. 2018;18:1174–1196. doi: 10.1039/C8LC00100F. [DOI] [PubMed] [Google Scholar]
- 125.Sorber L, Zwaenepoel K, Deschoolmeester V, Roeyen G, Lardon F, Rolfo C, Pauwels P. A comparison of cell-free DNA isolation kits: isolation and quantification of cell-free DNA in plasma. J Mol Diagn. 2017;19:162–168. doi: 10.1016/j.jmoldx.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 126.Lampignano R, Neumann MHD, Weber S, Kloten V, Herdean A, Voss T, Groelz D, Babayan A, Tibbesma M, Schlumpberger M, Chemi F, Rothwell DG, Wikman H, Galizzi JP, Riise Bergheim I, Russnes H, Mussolin B, Bonin S, Voigt C, Musa H, Pinzani P, Lianidou E, Brady G, Speicher MR, Pantel K, Betsou F, Schuuring E, Kubista M, Ammerlaan W, Sprenger-Haussels M, Schlange T, Heitzer E. Multicenter evaluation of circulating cell-free DNA extraction and downstream analyses for the development of standardized (Pre)analytical work flows. Clin Chem. 2020;66:149–160. doi: 10.1373/clinchem.2019.306837. [DOI] [PubMed] [Google Scholar]
- 127.Li Y, Zimmermann B, Rusterholz C, Kang A, Holzgreve W, Hahn S. Size separation of circulatory DNA in maternal plasma permits ready detection of fetal DNA polymorphisms. Clin Chem. 2004;50:1002–1011. doi: 10.1373/clinchem.2003.029835. [DOI] [PubMed] [Google Scholar]
- 128.Jorgez CJ, Bischoff FZ. Improving enrichment of circulating fetal DNA for genetic testing: size fractionation followed by whole gene amplification. Fetal Diagn Ther. 2009;25:314–319. doi: 10.1159/000235877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hahn T, Drese KS, O’Sullivan CK. Microsystem for isolation of fetal DNA from maternal plasma by preparative size separation. Clin Chem. 2009;55:2144–2152. doi: 10.1373/clinchem.2009.127480. [DOI] [PubMed] [Google Scholar]
- 130.Hellwig S, Nix DA, Gligorich KM, O’Shea JM, Thomas A, Fuertes CL, Bhetariya PJ, Marth GT, Bronner MP, Underhill HR. Automated size selection for short cell-free DNA fragments enriches for circulating tumor DNA and improves error correction during next generation sequencing. PLoS One. 2018;13:e0197333. doi: 10.1371/journal.pone.0197333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yu SC, Chan KC, Zheng YW, Jiang P, Liao GJ, Sun H, Akolekar R, Leung TY, Go AT, van Vugt JM, Minekawa R, Oudejans CB, Nicolaides KH, Chiu RW, Lo YM. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci U S A. 2014;111:8583–8588. doi: 10.1073/pnas.1406103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, Mair R, Goranova T, Marass F, Heider K, Wan JCM, Supernat A, Hudecova I, Gounaris I, Ros S, Jimenez-Linan M, Garcia-Corbacho J, Patel K, Østrup O, Murphy S, Eldridge MD, Gale D, Stewart GD, Burge J, Cooper WN, van der Heijden MS, Massie CE, Watts C, Corrie P, Pacey S, Brindle KM, Baird RD, Mau-Sørensen M, Parkinson CA, Smith CG, Brenton JD, Rosenfeld N. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10:eaat4921. doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61:112–123. doi: 10.1373/clinchem.2014.222679. [DOI] [PubMed] [Google Scholar]
- 134.Huang J, Wang L. Cell-free DNA methylation profiling analysis-technologies and bioinformatics. Cancers (Basel) 2019;11:1741. doi: 10.3390/cancers11111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.van der Pol Y, Mouliere F. Toward the early detection of cancer by decoding the epigenetic and environmental fingerprints of cell-free DNA. Cancer Cell. 2019;36:350–368. doi: 10.1016/j.ccell.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 136.Perakis S, Auer M, Belic J, Heitzer E. Advances in circulating tumor DNA analysis. Adv Clin Chem. 2017;80:73–153. doi: 10.1016/bs.acc.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 137.Romano A, Palumbo GA, Parrinello NL, Conticello C, Martello M, Terragna C. Minimal residual disease assessment within the bone marrow of multiple myeloma: a review of caveats, clinical significance and future perspectives. Front Oncol. 2019;9:699. doi: 10.3389/fonc.2019.00699. [DOI] [PMC free article] [PubMed] [Google Scholar]