

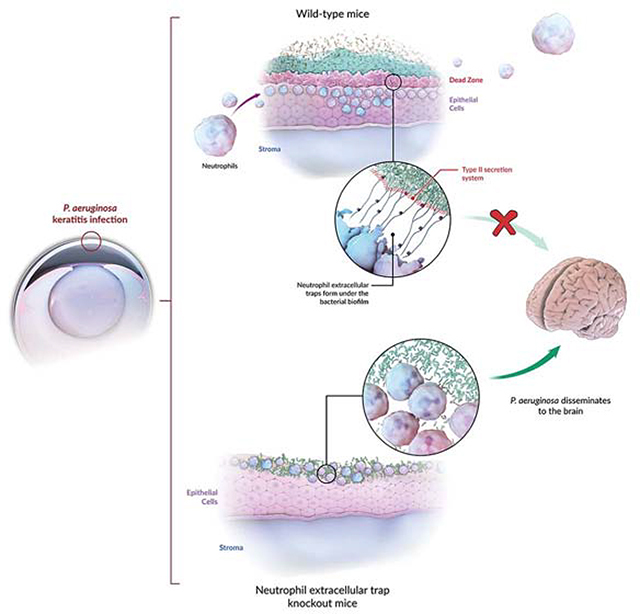
Summary
Bacterial biofilm infections are difficult to eradicate due to antibiotic insusceptibility and high recurrence rates. Biofilm formation by Pseudomonas aeruginosa, a leading cause of bacterial keratitis, is facilitated by the bacterial Psl exopolysaccharide and associated with heightened virulence. Using intravital microscopy, we observe that neutrophilic recruitment to corneal infections limits P. aeruginosa biofilms to the outer eye surface, preventing bacterial dissemination. Neutrophils moved to the base of forming biofilms, where they underwent neutrophil extracellular trap formation (NETosis) in response to high expression of the bacterial type-3 secretion system (T3S). NETs formed a barrier “dead-zone”, confining bacteria to the external corneal environment, and inhibiting bacterial dissemination into the brain. Once formed, ocular biofilms were resistant to antibiotics and neutrophil killing, advancing eye pathology. However, blocking both Psl and T3S together with antibiotic treatment broke down the biofilm and reversed keratitis, suggesting future therapeutic strategies for this intractable infection.
eTOC abstract
Thanabalasuriar et. al. characterize the interplay between neutrophils and Pseudomonas biofilms on the cornea. At the base of the biofilm, neutrophils encounter expression of the type-three secretion system, leading to an expansive deposition of neutrophil extracellular traps (NETs). NET formation impeded bacterial dissemination into the brain, yet promoted antibiotic resistance.
Graphical Abstract
Introduction
Neutrophils are the first-line of defense deployed against an invading pathogen (Rosales et al., 2016). They overcome pathogens by 1) rapid and robust neutrophilic recruitment 2) intracellular killing by oxidants and proteases like elastase after phagocytosis (Okada et al., 2006) and/or 3) via a PAD4 and elastase dependent, extracellular release of neutrophil extracellular traps (NETs) (Papayannopoulos, 2018). The latter has been highlighted by release of DNA decorated with proteases and other antimicrobial peptides (Yang et al., 2016). Recent work has proposed that NET release may be in part caused by non-specific frustrated phagocytosis wherein the pathogen or foreign particles are too large to be engulfed (Branzk et al., 2014). While individual planktonic bacteria are small and easily phagocytosed, biofilms are large impenetrable masses, that could potentially induce NET formation. These bacterial biofilm infections are difficult to eradicate because of antibiotic insusceptibility and, even with “apparent” eradication, recurrence is a common feature (Lebeaux et al., 2014). How neutrophils handle bacterial biofilms is poorly understood because visualizing growing biofilms and the immune response in vivo is very challenging. Bacterial keratitis represents a clinically relevant and more accessible opportunity to directly visualize the progression of a bacterial biofilm and the interactions with the host immune response with intravital imaging technologies.
Much is still unknown about ocular surface immunity despite its direct contact with the external environment and its critical role in vision (Barabino et al., 2012). For many years, eyes were thought to be immune privileged much like the brain (Benhar et al., 2012). However, there is growing evidence that the anti-microbial property of tears and the unique surface of the eye, combined with the nightly vacuuming by neutrophils and help from resident populations of other immune cells like macrophages, prevent infections (Liyanage et al., 2016; Liu et al., 2017). While the eye has also been reported to have a microbiome (Lu and Liu, 2016), pathogen growth is infrequent unless there is foreign body involvement (i.e. contact lenses) or an injury (i.e. abrasion) that exposes the underlying surface (Metruccio et al., 2018). In addition, when the host is immunosuppressed and neutropenic, following organ transplantation or cancer treatment for example (Ozkan et al., 2017; Nanda et al., 1991), the eye may rapidly succumb to opportunistic bacteria such as Pseudomonas aeruginosa, a leading cause of bacterial keratitis (Sy et al., 2012). P. aeruginosa employs virulence factors such as the type-3-secretion (T3S) system to infuse toxins into host cells, as well as immune evasion and persistence determinants such as the Psl exopolysaccharide to form tenacious biofilms (Sousa and Pereira, 2014; Ma et al., 2006; Ma et al., 2012). Current understanding of P. aeruginosa is based predominately on in vitro systems and advocate that the T3S injects various virulence effectors (ExoS, ExoT, ExoY, and ExoU) into host cells (Gellatly and Hancock, 2013). Recent studies suggest that T3S may be activated on the surface of P. aeruginosa in low pH environments such as those found in the phagolysosome (Thanabalasuriar et al., 2017). ExoS and ExoU are rarely seen to be expressed by the same strain of P. aeruginosa, while ExoT is expressed by almost all strains. ExoS and ExoT are closely related and have been shown to block reactive oxygen species (ROS) production, to induce neutrophil apoptosis, and to inhibit neutrophil phagocytosis (Vareechon et al., 2017; Sun et al., 2012; Kaminski et al., 2018; Rangel et al., 2014). The survival benefit of having a T3S has been attributed to these two effector proteins (Angus et al., 2010). Little is known about ExoY’s role in T3S virulence, however it was recently shown to play a role in acute lung infections (Kloth et al., 2018). To date, very few studies have shown a role for the T3S in biofilm formation, and at least in vitro on glass surfaces it was not detectable in formed biofilms (Ray et al., 2018; Tran et al., 2014). However, the in vivo environment varies in many ways that is difficult to recapitulate in vitro. In vivo, neutrophils emigrate out of blood vessels, which is known to prime their activity and trigger NETosis (Yipp et al., 2012). When biofilms are studied in vitro, they are often grown in the absence of many tissue factors and, only once formed, are biofilms exposed to isolated circulating neutrophils (Ray et al. 2017). Thus, this begs the question: how do bacteria ever survive in the harsh in vivo environment?
Herein we tracked neutrophil recruitment to a developing biofilm on the corneal surface. We observed that neutrophils move into the eye after P. aeruginosa corneal infection and strategically migrate underneath to the base of the bacterial layer. Interestingly, the basolateral area of the biofilm expressed high levels of T3S and, in response, neutrophils released NETs. These NETS created a dead-zone of DNA and degraded collagen thereby walling off the P. aeruginosa biofilm to the outer surface of the eye. While this prevented further bacterial dissemination, the massive biofilm and neutrophil-mediated pathology also resulted in severe local ulcerative damage to the eye. Preventing NET formation reduced eye pathology yet resulted in bacterial dissemination to the brain. Blocking the exopolysaccharide Psl matrix of the biofilm and the T3S PcrV protein enabled specific neutrophil penetration into the bacterial area and enhanced the killing and clearance of the pathogen. This treatment combined with topical antibiotics was the only strategy that optimally cleared a pre-existing bacterial biofilm.
Results
Neutrophils are unable to penetrate P. aeruginosa biofilm on the surface of the cornea
The corneas of naïve mice are devoid of neutrophils (Figure 1A). Inoculating the eye with as much as 107 CFU of P. aeruginosa, failed to induce: 1) bacterial adhesion to the ocular surface, 2) persistent infection or 3) inflammation in the form of neutrophil recruitment (Figure S1B). In other words, the intact surface of the cornea resists colonization of even large amounts of pathogens consistent with previous observations (Augustin et al., 2011). Abrasion of corneas (Figure S1C) induced an inflammatory response that was undetectable by a crude myeloperoxidase measurement (Figure S1D). However, corneal abrasion did induce subtle neutrophil recruitment detectable by intravital imaging and flow cytometry over the first 24 hours (Figure 1B,C; S1E). The recruited neutrophils were dispersed across the cornea (Figure 1B) and were extremely motile, migrating randomly over the surface of the cornea (Video S1). The neutrophils did not show characteristic signs of responding to the injury however (Video S1), such as swarming behaviour reported in liver and skin (Lammermann et al., 2013; McDonald et al., 2010). Despite this neutrophil recruitment, there was no overt eye pathology including opacification or cytokine production including IL-1β release (Figure S1F,G).
Figure 1. Neutrophils move into the eye during P. aeruginosa infection but are unable to stop biofilm development.

A. In vivo microscopy of a naive mouse cornea, outlined in white (green autofluorescence signal of epithelial cells).White arrows point to neutrophils (red) moving quickly through blood vessels in the sclera. B. In vivo microscopy 24 hours post eye abrasion. Neutrophils (red) start to move onto the cornea. C. Flow cytometry analysis of the full corneal abrasion model at 4, 24 hours post-abrasion only as well as abrasion + infection. All samples were gated as singlets, live, CD45+ cells with neutrophils gated as Ly6G+ Cd11b+ cells. D. In vivo microscopy 4 hours post abrasion + infection of cornea with P. aeruginosa strain PAO1 (green), neutrophils (red). E. In vivo microscopy 24 hours post abrasion + infection with P. aeruginosa strain PAO1 (green), neutrophils (red). F. Zoomed in 3D-reconstruction of the biofilm in vivo. P. aeruginosa (green), neutrophils (red). G. Scanning electron microscopy of the corneal surface at 24 hours post P. aeruginosa infection. Yellow arrow defines mushroom-like structures characteristically seen in developed biofilms. Right panel is a zoomed in section 6.00KX magnification of the 650X magnification image on the left. White arrows define single bacteria that are encased in a matrix (biofilm matrix).
In contrast, mice that had corneal abrasions and received a dose of 1×107 CFU of P. aeruginosa developed significant bacterial growth along the abrasion by 4 hours (Figure 1D; Video S2). Rapid and robust neutrophil recruitment in the form of a huge continuous swarm was noted near the abrasion at 4 hours by IVM, flow cytometry and detectable by increased myeloperoxidase (Figure 1C,D; S1D,E, Video S2). Using a series of small abrasion + infection sites on the cornea we found that neutrophils moved onto the cornea through the peripheral limbal vasculature (Figure S1H,I). This was evident by the timing of neutrophil recruitment: when the infection sites were at the corners of the cornea the neutrophils arrived more rapidly than when the abrasion was at the center of the cornea ((Hanlon et al., 2014); Figure S1H,I). It is important to note that the full abrasion as well as the small abrasion model were used throughout this study to examine bacterial growth and neutrophil behaviour respectively. While the results were similar it was much easier to visualize single neutrophil behaviour by intravital microscopy using the small abrasion model and assess clinical pathology scores, bacterial CFU counts, and 3D reconstruction using the full abrasion model.
At 24 hours post-infection, a large film of P. aeruginosa persisted and a mass of neutrophils were recruited with minimal overlap at the border of bacterial growth (Figure 1E,F; Video S3). Using multiphoton microscopy and the 3D reconstruction feature (Figure 1F) in combination with electron microscopy (Figure 1G) and basic Gram-staining techniques of eye biopsies (Figure S2A) revealed that P. aeruginosa formed a thick biofilm on the surface of the cornea. This biofilm seemed to be fully developed by 24 hours, based on the mushroom like structures seen by electron microscopy and Gram-staining of eye biopsies (Figure 1G; S2A; (Miller et al., 2012)). Intravital imaging at 4 and 24 hours further revealed that P. aeruginosa moved in rod-like clusters and grew into a thick sheet of bacteria while the neutrophils all assembled beneath this layer in the upper part of the cornea and formed an apparent shield (Figure 1D,E,F; Video S2,3). These visual observations and molecular measurements (including Psl: see later Figure 4A) were all consistent with the sheet of bacteria being a biofilm. A ΔpslA knockout isogenic strain (Psl-deficient) did not form an organized biofilm nor did it stain well for mannose lectin, consistent with previous observations in other in vitro systems (Mann and Wozniak, 2012) showing Psl to be crucial for biofilm production (Figure 2F,G and S2B).
Figure 4. Prophylactic treatment of mice with bispecific therapeutic antibodies reduces biofilm formation and bacterial load.

A. Schematic and visualization of antibody binding to the bacterial biofilm using in vivo microscopy. Antibodies (anti-Psl, anti-PcrV, bispecific: blue), biofilm (green). B. Schematic of antibody treatment and infection models (using small localized abrasion model for in vivo intravital imaging and classic full abrasion model to assess clinical pathology and CFUs). C,D. In vivo imaging of neutrophils (red) in the cornea and quantification of neutrophil behavior at 4 hours post-infection in the presence of therapeutic antibodies. P. aeruginosa, green. E,F. In vivo imaging of neutrophils (red) in the cornea and quantification of biofilm (green) height at 24 hours post-infection in antibody treated mice. White dotted line defines the dead-zone. Small focal infection point model was used. G. Quantification of bacterial CFUs in eyes at 24 hours post-infection. H,I. Clinical outcome and scores of eyes at 7 days post-infection. J. Quantification of bacterial CFUs in eyes at 7 days post infection. n=4–5 mice per group prophylactically treated with therapeutic antibodies. K. Quantification of bacterial CFUs in brains at 7 days post-infection. L,M. Clinical outcome and scores of eyes at 7 days post-infection in NE−/−, PAD4−/−,and Cstc−/− mice treated with bispecific therapeutic antibody. Data in G, J, and K are represented as median and interquartile range and tested by non-parametric Kruskal-Wallis ANOVA. Data in D, F, I, and M are represented as the mean ± standard error and tested by one-way ANOVA. *p<0.05; **p<0.01, ***p<0.001. n=4–5 per group.
Figure 2. Developed bacterial biofilm and dead-zone is dependent on P. aeruginosa T3S and Psl exopolysaccharide.

A. In vivo microscopy 24 hours post infection. Red (Catchup reporter mouse, dtTomato neutrophils), blue (Ly6G staining, antibody administered intravenously), green (P. aeruginosa biofilm). White dotted line defines the dead-zone. B. In vivo microscopy showing collagen (second harmonic signal, white) around bacterial biofilm (green), neutrophils (red). White dotted line defines the dead-zone. C. Active neutrophil elastase staining (blue) in vivo 24 hours post-infection. P. aeruginosa biofilm (green), neutrophils (red). White dotted line defines the dead-zone. D. Histone H2A staining (blue) in vivo 24 hours post-infection. P. aeruginosa biofilm (green), neutrophils (red). White dotted line defines the dead-zone. E. Sytox Red DNA staining (blue) in vivo 24 hours post-infection. P. aeruginosa biofilm (green), neutrophils (red). White dotted line defines the dead-zone. F. Mice were given ocular infections with wild-type P. aeruginosa and isogenic mutants (Δpsl, Δpcrv, Δexos). Top panel: in vivo microscopy and 3D reconstructions were used to observe and quantify biofilm and dead-zone formation (G, H). Bottom panel: scanning electron microscopy images taken at 6.00kX magnification. White arrows indicate bacteria. I. Quantification of bacterial CFUs in eyes at 24 and 48 hours post-infection. J. Active neutrophil elastase activity (blue) was visualized in vivo and quantified (K) at 24 hours post-infection with P. aeruginosa (Δpsl, Δpcrv, and Δexos; green), neutrophils (red). Data in I are represented as median and interquartile range and tested by non-parametric Kruskal-Wallis ANOVA. Data in G, H, and K are represented as the mean ± standard error and tested by one-way ANOVA. *p<0.05; **p<0.01, ***p<0.001. n=4–5 per group.
Characterization of a dead-zone that separates host from pathogen
A notable chasm (black area) devoid of bacteria or dtTomato neutrophils formed between the host and pathogen (Figure 1E). Fluorescent anti-Ly6G antibody given intravenously also labelled the layer of neutrophils but not the dead-zone (Figure 2A). The dark area also lacked second harmonic signal associated with properly formed collagen matrix composing the corneal structure (Figure 2B; Video S4) potentially suggesting proteolytic destruction of the pre-existing matrix. Consistent with this observation, analysis of eye biopsy cross-sections revealed empty spaces under the bacterial growth (Figure S2A). Indeed, a probe that detects host elastase activity showed profound but localized staining in this area (Figure 2C). The elastase activity was localized to the dead-zone along with histone H2A.X, myeloperoxidase, and DNA indicative of NETs (Figure 2D,E; S2C). DNA (devoid of histones) was also present in the biofilm consistent with the well-known feature of P. aeruginosa releasing its own DNA to form the biofilm matrix (Figure 2D,E).
Biofilm and dead-zone formation are dependent on P. aeruginosa exopolysaccharide Psl and the T3S system expression
The Psl exopolysaccharide is a key molecule required for P. aeruginosa to develop and maintain biofilms in vitro (Irie et al., 2017; Ray et al., 2017). In the absence of Psl, we observed that the bacteria no longer formed a structured biofilm on the eye and a planktonic mass spread across the corneal surface at 24 hours post-infection (Figure 2F,G; S2B). In addition, evidence of a dead-zone was not observed and the neutrophils were intermingled among small colonies of bacteria (Figure 2F,H; S2B). However, the bacteria still managed to survive, with ample bacteria on the eye surface and very little improvement of eye opacity or keratitis score (Figure 2I; S2D). Upon phagocytosis of P. aeruginosa, neutrophils have been shown to die in a T3S dependent manner (Thanabalasuriar et al., 2017; Sun et al., 2012; Rangel et al., 2014). Surprisingly, in the absence of the T3S (ΔpcrV), bacteria grew in a cluster but did not form into a complete biofilm (Figure 2F,G). This may be due to the fact that neutrophils were also able to move directly into the bacterial cluster and disrupt it with no evidence of a dead-zone (Figure 2F–H). Moreover, there were fewer ΔpcrV bacteria by 48 hours post-infection and the majority were cleared by 7 days (Figure 2I; S2E). In addition, the opacity of the eye was greatly improved and the keratitis score was reduced in comparison to wildtype bacteria (Figure S2D). Interestingly, neutrophil elastase activity was only detected close to the interface between the bacteria and the neutrophils. There was less pronounced staining in the ΔpslA infected mice and even less in the ΔpcrV bacteria (Figure 2J,K). This suggests that if neutrophils are able to phagocytose and perform intracellular killing of P. aeruginosa, there is less need for elastase and presumably NET release.
We further characterized the mechanism by which T3S is required for dead-zone formation. Given that ExoS has previously been shown to manipulate neutrophil function and since the PAO1 strain we are utilizing does not express ExoU, we tested PAO1 Δexos in our in vivo biofilm model. When mice were infected with the ExoS mutant, there was no apparent dead-zone formation and reduced elastase activity (Figure 2F,H,J,K). In fact, in these mice the neutrophils penetrated into the biofilm (Figure 2F). In addition, the bacterial load on the corneal surface in ExoS mutant infected mice was significantly reduced compared to wild-type bacteria (Figure 2I; S2E). It is worth noting that the ExoS mutant was not as compromised in biofilm formation as the complete T3S mutant indicating that another T3S effector may be contributing to the phenotype (Figure 2F,G). A likely candidate would be ExoT which has previously been shown to work in synergy with ExoS. However, the lack of a dead-zone suggests that ExoS but not ExoY or ExoT play a prominent role in NET formation.
The dead-zone is formed by neutrophil extracellular traps (NETs)
To further establish that NET machinery was involved in the response to P. aeruginosa and dead-zone formation, peptidyl arginine deiminase, type IV (PAD4−/−), neutrophil elastase (NE−/−), and cathepsin C (Cstc−/−) knockout mice were systematically examined. All three of these strains of mice have previously been shown to have impaired NET production (Papayannopoulos et al., 2010) (Akk et al., 2016). No dead-zone was detected in any of these mice (Figure 3A,C). The absence of this dead-zone was associated with absence of the striking biofilm seen on the cornea of wild-type mice (Figure 3A,B,D; Video S6) suggesting that the presence of NETs caused the highly replicative planktonic bacteria to form a biofilm. Indeed, there was significantly increased bacterial growth in the serine protease deficient mice (NE−/− and Cstc−/−) compared to wild-type mice (Figure 3E). The PAD4−/− mice had a less significant increase in bacterial growth (Figure 3E). However, unlike the NE−/− and Cstc−/− mice that could not make NETs or release elastase, PAD4−/− mice could still release elastase perhaps limiting bacterial growth (Figure 3F,G). The lack of biofilm production at 24 hours post-infection reduced the opacity of the eye in these mutant mice (Figure 3D). To determine whether NETs were critical as a barrier, bacteria were measured in other compartments in the mouse. Surprisingly, P. aeruginosa was able to disseminate beyond the eye into the brain of all three knockout mouse strains by 7 days (Figure 3H), suggesting that neutrophils release a NET barrier to keep bacteria external to the host and prevent dissemination to the brain while sacrificing the pathology of the eye. This dissemination is likely through a direct portal to the brain, for example through the optic nerve, since we were unable to detect bacteria in the spleen or blood of any of the knockout mouse strains (Figure S3G). This was not due to better killing of P. aeruginosa in the brain by the wild-type versus PAD4−/− mice, as neither mouse strain demonstrated any ability to kill the pathogen when inoculated into the brain. In fact, only 500 CFU of P. aeruginosa injected directly into the brain resulted in massive bacterial growth in the cerebral parenchyma of both wild-type and PAD4−/− mice (Figure S3H). It is worth mentioning that some immune pathways were not involved in this host-pathogen encounter. For example, we examined whether complement C3 was involved in the host response to P. aeruginosa. Despite the many roles complement plays in the host response to P. aeruginosa, it had no readily apparent effect on the ocular surface (Figure 3A–E).
Figure 3. Formation of the dead-zone is dependent on the production of NETs and proteases by host neutrophils.

Mice lacking neutrophil elastase (NE−/−), PAD4 (PAD4−/−), Cathepsin C (Cstc−/−), or complement component 3 (C3−/−) were infected with wild-type P. aeruginosa. Biofilm and dead-zone formation were visualized using in vivo microscopy (A) and quantified (B, C). D. Eye pathology was observed and assessed. E. Bacterial CFUs in eyes were assessed at 24 hours post-infection in mutant mice. F. Active elastase staining (blue) in PAD4−/− and NE−/− mice was visualized in vivo and mean fluorescence intensity was quantified (G). H. Quantification of bacterial CFUs in brains at 7 days post-infection. I. Quantification of bacterial CFUs in eyes at 7 days post-infection. Wild-type C57 and PAD−/− mice were treated with topical eye drops with or without tobramycin two times a day for 7 days. Data in E, H, and I are represented as median and interquartile range and tested by non-parametric Kruskal-Wallis ANOVA. Data in B, C, D, and G are represented as the mean ± standard error and tested by one-way ANOVA. *p<0.05; **p<0.01, ***p<0.001. n=4–5 per group.
We speculated that this spread of bacteria from the eye to the brain was likely due to the bacteria being in an extremely motile planktonic form rather than a more immobile biofilm form. When P. aeruginosa strain PAO1 is grown in planktonic culture it is susceptible to the antibiotic tobramycin (Hill et al., 2005). Therefore, we tested the susceptibility of PAO1 on the surface of wildtype versus PAD4−/− mice to tobramycin (Figure 3I). While tobramycin had no effect on the biofilm growing on corneas of wild-type mice, PAD4−/− mice treated with antibiotics had significantly lower bacterial CFUs (Figure 3I) consistent with the absence of a biofilm in PAD4−/− mice.
P. aeruginosa biofilm formation can be blocked in vivo using a bispecific antibody targeting both T3S and Psl
Having established Psl and T3S to be important for P. aeruginosa biofilm formation on the ocular surface, we next examined the impact of systemically-administered therapeutic antibodies against these components (anti-Psl, anti-PcrV, and a bispecific antibody-MEDI3902; (DiGiandomenico et al., 2014; Warrener et al., 2014; DiGiandomenico et al., 2012)). These antibodies distributed from the bloodstream to the ocular surface very rapidly. In fact, 1.5 hours after intravenous administration, antibodies could be detected in the eye (Figure 4A; S3A). Systemic application of fluorescent antibodies targeting Psl and PcrV of the T3S system showed strikingly different binding patterns. Anti-Psl antibody administration resulted in antibody binding on the outer surface of the bacterial mass, while anti-PcrV antibody localized to the bottom of the biofilm at the host-pathogen interface (Figure 4A). The bispecific antibody incorporating both anti-Psl and anti-PcrV complementarity determining regions, bound to the surface, underneath and throughout the biofilm heterogeneously (Figure 4A). Intriguingly, we could detect only Psl and no PcrV when we grew biofilms in vitro as previously described (Ray et al., 2018). Prophylactic treatment with IgG control antibody had no discernable impact on the bacteria and neutrophil distribution compared to naïve mice, with bacteria growing in a biofilm surrounded by a dead-zone and neutrophils (Figure 4C–I). By contrast, administration of antibodies against Psl or PcrV prophylactically reduced the height of the biofilm and eradicated the dead-zone with neutrophils inter-mixed with the bacteria (Figure 4E,F). Inhibition of PcrV but not Psl also somewhat improved the opacity of the eye (Figure 4H,I; S3C,D). In the case of the bispecific antibody which blocks both effector functions of the bacteria, the opacity of the eye was improved and a dramatic reduction in the ocular bacteria by 7 days post infection was noted (Figure 4G–M; S3B–D).
Using the small abrasion site + infection model (Figure 4B) to image the neutrophil-bacterial interactions, we observed an improved immune cell meandering index directly towards the P. aeruginosa in antibody-treated mice (Figure 4D). A further assessment of blocking both Psl and PcrV revealed neutrophils directly engulfing bacteria and disrupting the forming biofilm (Figure 4 C–F; Video S5). In fact, much less extracellular elastase was detected at 24 hours post-infection (Figure S3E,F). This suggested a conversion from extracellular NET formation to intracellular phagocytosis and killing in the absence of both these bacterial virulence factors. By 24 hours post-infection, very few bacteria were seen by IVM but large numbers of neutrophils were still visible in biphasic antibody-treated mice (Figure 4E,G).
While complement was not important in untreated wild-type mice, this was not due to a lack of complement on the corneal surface inasmuch as the bispecific antibody required both functional complement and the Fc receptors. In C3−/− and FcRγ−/− mice, the bispecific and anti-PcrV showed no difference in keratitis compared to isotype control mice whereas both antibodies showed significant benefit in wild-type mice (Figure S4A–D). In addition, elastase was critical for the intracellular neutrophil killing of P. aeruginosa, as bispecific antibody treatment of NE−/− mice provided no protection and bacteria were able to disseminate beyond the neutrophils into the brain (Figure 4J–M). It is interesting to note that functional neutrophils were required for effective antibiotic clearance given that tobramycin treatment was ineffective in NE−/− mice (Figure S4E). By contrast, lacking the ability to make NETs (PAD4−/− mice) while still having normal elastase levels allowed the antibody to induce neutrophil killing of bacteria (Figure 4J–M). Indeed, blocking both Psl and PcrV on P. aeruginosa protected PAD4−/− mice from ocular damage and brain dissemination when prophylactically administered (Figure 4J–M). These data indicate that antibody therapy promoted switching from a NET-mediated bacterial killing program to an intracellular protease-mediated killing mechanism. They also strongly support the view that T3S is key to NET formation.
Adjunctive treatment with bispecific antibody in tandem with antibiotics significantly reduced keratitis
While prophylactic use of these antibodies would be useful in certain patients, such as those receiving organ transplants, their efficacy after biofilms have started to form is unclear. Tobramycin is the antibiotic of choice in P. aeruginosa infections but outcomes are variable (Mulcahy et al., 2014). At 6 hours post-infection, tobramycin was started every day for one week but had little effect on the clearance of the P. aeruginosa biofilm (Figure 5A,B). In fact, the bacterial CFUs were comparable to the isotype control treatment, even though the PAO1 strain used is highly susceptible to tobramycin when grown in planktonic culture in vitro (Tre-Hardy et al., 2010). Clearly it is much less effective in biofilms. Importantly, bispecific antibody administration post-infection only partially rescued the eye (Figure 5B) with significantly less opacity and a reduced clinical score but certainly some pathology remained (Figure 5C,D). Tobramycin given in tandem with the antibody twice a day starting at 6 hours post-infection had by far the best outcome. It completely eradicated P. aeruginosa in at least 80% of mice and also resulted in a profound reduction of proinflammatory cytokines and chemokines (Figure 5B–E). When either Psl or PcrV alone with antibiotics was used, neither showed any efficacy or clearance of the biofilm (Figure S4F). The simultaneous blocking of Psl and PcrV together with antibiotics was the key to breaking down the biofilm, showing the tremendous resistance of biofilms in vivo.
Figure 5. Treatment with the bispecific antibody can breakdown biofilm infections in a neutrophil serine protease mediated manner.

A. Timeline of antibody and tobramycin treatment of mice post-infections. n=5 per group. B. Quantification of bacterial CFUs at 7 days post-infection and treatment. C,D. Clinical outcome and scores of eyes at 7 days post-infection. E. Concentration of TNF-α, IL-1β, and KC in whole eyes at 7 days post-infection and treatment. Classic full abrasion of the cornea model was used here. Data in B are represented as median and interquartile range and tested by non-parametric Kruskal-Wallis ANOVA. Data in D and E are represented as the mean ± standard error and tested by one-way ANOVA. *p<0.05; **p<0.01, ***p<0.001. n=6–8 per group.
Discussion
The impact of innate and adaptive immunity on biofilm formation or eradication has been difficult to study in vivo despite biofilm contribution to numerous diseases including ocular complications. Indeed, bacterial keratitis continues to be a major concern in both healthy and immunocompromised individuals in developed and developing nations (Robertson et al., 2007). In this study, we systematically examined the role of the bacteria as well as the host in the progression of debilitating keratitis caused by P. aeruginosa. Once formed, neutrophils were incapable of penetrating the biofilm, and this was primarily due to high expression of the T3S system. This led to the production of NETs, which formed a formidable barrier against the pathogen and inhibited the spread of the bacteria to the brain. It is intriguing to speculate, that this might be an evolutionarily beneficial mechanism to shield the brain from infections via the ocular route.
In this study we examined the dynamic host-pathogen interplay in the development of biofilms in vivo using intravital microscopy. Indeed, our work provides insights not readily implicit from our previous in vitro studies using a flow cell chamber (Ray et al., 2018). Preformed biofilms grown on a glass slide harboured abundant Psl but completely lacked T3S staining and Psl inhibition was sufficient to significantly affect the biofilm (Ray et al., 2018). In vivo on the corneal surface, neutrophils form a barrier of NETs below the bacteria, T3S was directionally induced towards the amassing neutrophils, keeping them at bay and allowing the biofilm to mature. In vitro, it is impossible to place neutrophils below the biofilm on a coverslip and as such the results are limited. Indeed, neutrophils that had emigrated out of the vasculature formed a barrier by releasing NETs in a directional manner in vivo. Nevertheless, some in vitro data had complementarity to our work. As one example, in vitro Staphylococcus aureus biofilms were shown to stimulate NET release and this facilitated growth of the bacteria (Bhattacharya et al., 2018). Of course, this in vitro study could not have possibly conceived the importance of the NET barrier in preventing dissemination of bacteria to brain. It is also worth noting that in our study we see that in the absence of NETs, the bacteria remained planktonic giving them the capacity to penetrate deeper into tissues. These observations suggest that the neutrophils and their effector mechanisms are an impetus for P. aeruginosa to make biofilms. Finally, whereas in vitro studies use neutrophils that are isolated from blood, in vivo, neutrophils will have emigrated into the tissue. The emigration process itself has been documented to prime neutrophils to do various things including make more NETs (Branzk et al., 2014; Yipp et al., 2012). As such we would submit that there is much value to studying the host-pathogen biofilm interface in vivo, which provided detailed insights from both the bacterial and the neutrophil aspect. These findings of differential virulence factor expression within an in vivo biofilm and the critical neutrophil responses could have significant implications for other biofilm infections including those common to cystic fibrosis patients, for example.
Using a combination of intravital imaging, bacterial mutants, and antibodies that block P. aeruginosa virulence we have found that neutrophils come in from the peripheral corners of the eye. It is intriguing that we did not observe neutrophils moving on top of the biofilm. This could be due to toxins released around the biofilm restricting the proximity of neutrophils to the biofilm, so the neutrophils crawl underneath these structures. High expression of the Psl exopolysaccharide on the surface of the biofilm might cloak P. aeruginosa from neutrophil recognition as was previously described (Mishra et al., 2012; Thanabalasuriar et al., 2017). Indeed, the Psl mutant allowed neutrophils to infiltrate throughout the bacterial clusters. However, this was not sufficient to eradicate the bacteria. In this study, we found that T3S plays a critical role in keeping the neutrophils at bay so that the biofilm could mature. Further work revealed that the ExoS mutant of P. aeruginosa failed to form the T3S-dependent dead-zone, suggesting that this toxin is released in the direction of the amassing neutrophils. Another unique observation that highlights the complex neutrophil-pathogen interplay is that, as the neutrophils moved under the biofilm, they were triggered to form NETs by P. aeruginosa T3S toxin release. While this NET formation effectively blocked bacterial dissemination into the brain, it also resulted in the formation of an impenetrable biofilm. This biofilm formation is drug resistant and difficult to treat with topical antibiotics.
P. aeruginosa is known to be highly virulent, causing eye ulcerations that are more difficult to treat than other bacterial corneal ulcers resulting in poor visual outcomes (Sy et al., 2012). In this study we found that a bispecific antibody targeting two evasion mechanisms in P. aeruginosa can effectively and rapidly reach the eye and together with antibiotics can help to breakdown bacterial biofilms. Blocking the T3S, and therefore the cytotoxic effects of P. aeruginosa, reduced the need for NET formation. In addition, targeting Psl with the antibody either disrupts the development of a biofilm or opsonizes the bacteria (DiGiandomenico et al., 2014) that are trying to form a biofilm, thus promoting neutrophil-mediated clearance. In combination with topical antibiotic treatment we found that blocking both virulence factors was necessary for the effective eradication of infection and reduction of eye inflammation in mice with a developed biofilm.
Initially, neutrophils were thought to perform frustrated phagocytosis in an attempt to engulf either inert or large particles. More recently, it was reported that these types of large particles including pathogens such as hyphae may induce NET release (Branzk et al., 2014). In our study, we saw that neutrophils primarily attempted to phagocytose planktonic P. aeruginosa, but when presented with the same species of bacterium as a large impenetrable biofilm, the neutrophils release NETs that formed a barrier restricting dissemination into the brain. The inability to make NETs resulted in bacteria entering the brain from the corneal surface. While both blood and the optic nerve could function as portals for this dissemination, we speculate that bacteria utilize the optic nerve to enter the brain as no other organ was infected despite the fact that this strain of P. aeruginosa is trophic for the lung and spleen when in blood (Thanabalasuriar et al., 2017). A similar mechanism of dissemination through the optic nerve was previously suggested for viral infections (Olson et al., 1987; Francis et al., 2003). In this study we show an intriguing example of host-response co-evolution in that NET formation seemed to occur hand in hand with biofilm formation. While it is impossible to delineate whether the NETs induced biofilm formation or whether the biofilm induced NET formation, it is clear NETs absolutely serve a barrier function, that seems critical to survival of the host but comes at the expense of the eye. As such, treatment strategies must be designed with this very precarious balance in mind.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paul Kubes (pkubes@ucalgary.ca).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
6–8 week-old C57BL/6 wild-type male and female mice were purchased from The Jackson Laboratory originally and the bred in house. Experiments consisted of a combination of males and females since there were no sex specific effects seen in the data sets. B6.129S4-C3,tm1Crr./J referred to as C3 −/− mice in this paper were purchased from The Jackson Laboratory (3641). FcRγ−/− on the C57 background were purchased from Taconic (583). Catchup mice were a gift from Matthias Gunzer. Neutrophil elastase knockout mice were purchased from Jax strain 6112. Cathepsin C knockout mice (Cstc−/−) were a gift from GlaxoSmithKline. Peptidyl arginine deiminase, type IV, (PAD4−/−) were a generous gift from Kerri A. Mowen, La Jolla California. Mice were housed under pathogen-free conditions and received sterilized rodent feed and water. All mice were used in experiments at the 6–8 weeks of age and combination of males and females were used. Littermates of the same age were allocated to experimental groups. All experimental animal protocols were approved by the University of Calgary Animal Care Committee and followed the Canadian Council for Animal Care Guidelines (protocol nr. AC16–0224 and AC16–0144).
Bacterial Strains
PAO1 bacteria were kindly donated to this work by Dr. Shawn Lewenza. Plasmid CTX-Ptac-GFP was obtained from D. Wozniak (Wyckoff and Wozniak, 2001). Plasmid pPM297 was generated by replacing the GFP coding sequence in CTX-Ptac-GFP with a synthetic gene encoding dsRed2. Both constructs were propagated in E. coli JM109. Tri-parental mating resulting in chromosomal integration of the fluorescent protein expression cassettes were carried out as described previously (Wyckoff and Wozniak, 2001). No difference in viability or in vitro growth kinetics were observed for all PAO1 isogenic mutant strains used in our manuscript when compared to the wild type parental strain (data not shown). For keratitis infections bacteria were grown overnight on blood agar plates at 37 degrees Celsius. Bacteria were scraped off the plate and resuspended in PBS. OD 650 was measured and bacterial volumes were calculated based on the OD.
METHOD DETAILS
Eye intravital microscopy
Mice were placed under anesthetic (ketamine (200mg/kg) and xylazine (10mg/kg)) and a jugular catheter was inserted as previously described (McDonald et al., 2010; Lee et al., 2010). Mice were placed on a heating pad (World precision Instruments), on their side and restrained using umbilical tape by their arms and feet to reduce movement from breathing. Throughout the imaging process the mouse remained on the heating pad with a rectal probe to maintain body temperature. A 25 mm circular coverslip (48380–080 VWR) mounted on a plastic restrainer was used to maintain the position of the head under the upright microscope (Figure S1A). A small drop of saline was placed on the eye before putting the mouse under the restrainer to keep the eye moist. Gentle and uniform pressure was placed on the eye with the coverslip (Figure S1A). Mice were imaged either using spinning disk confocal microscopy (Quorum) or multiphoton microscopy (Leica SP8). Leica SP8 microscope (Leica Microsystems), equipped with white light laser and resonance scanner, a 20X 0.95 NA water objective with a piezo actuator and controller was used. A combination of HyD-detectors and PMT internal (confocal) and external (2-photon) detectors were used to detect with 650–700 nm, 565–620 nm and 500–550 nm. Spinning disk confocal images were acquired with an upright microscope (BX51; Olympus; Quorum) XLUM Plan F1 10X 0.3 NA air and 20X 0.95 NA water immersion objectives. A C9100–13; Hamamatsu camera was used for fluorescence detection (Quorum). Simultaneous observation of bacteria, neutrophils, and therapeutic antibodies was assessed using 3 laser excitation wavelengths (488, 561, and 635 nm; Cobolt AB) in rapid succession and visualized with the appropriate long-pass filters (Semrock).
Neutrophils were labelled using tail vein injection of Ly6G antibody 2–4 hours before imaging or using Catchup transgenic mice (Hasenberg et al., 2015). Experiments with knockout mouse strains we used intravenous inject of Ly6G 1A8 antibody to identify neutrophils, control wild-type mice were similarly stained with Ly6G antibody. Mice were imaged for 30 minutes to1 hour at a rate of 10 frames per second. Z-stacks were performed based on optimal slice size and numbers based on software recommendations. Data were analyzed using Volocity imaging software, Leica image analysis software, and FIJI software. Neutrophil elastase staining was quantified by taking 3D images and compressing them into one 2D stack. Mean fluorescence intensity was quantified in the single stack using Leica software. Lectin staining was performed as previously described (Ma et al., 2009). For lectin staining of the biofilm, the lectin stain (MOA-TRITC) was placed on the eye 20 minutes before imaging. The eye was stained for 10 minutes and then gently washed 3 times PBS.
Keratitis models
The eye was abraded using a 25-gauge needle in the form of a scratch, disturbing the corneal epithelial cells but not causing excessive damage to the eye itself, as previously described (Sun et al., 2012). For all experiments, if a mouse’s eye was considered smaller than the other mice in the group or had any signs of trauma in comparison to other mice in the group this eye or mouse was omitted from the analysis.
For CFU experiments, clinical scores and biofilm 3D reconstruction, animals were anesthetized with isoflurane followed by initiation of three 1-mm abrasions on the cornea using a 25-guage needle (full/traditional abrasion technique). Mice were infected with GFP expressing P. aeruginosa strain PAO1 and isogenic mutants (1×107 CFU/eye) by placing the bacteria directly on the eye in a total volume of 0.05ml. At designated time points post-infection, animals were euthanized by CO2 asphyxiation followed by either: determination of bacterial burden in eye homogenates or determination of corneal pathology scores.
For IVM we redeveloped the traditional keratitis model to make a focal infection point. A 25 Gauge needle was used to disturb approximately 10–30 corneal epithelial cells, making a scarified zone no bigger than the diameter of the 25-gauge needle. For IVM imaging to track neutrophil-bacterial interactions and to further define the dead zone, a localized abrasion (small abrasion technique) was performed at the temporal corner of each eye. This procedure was performed using a dissection microscope and a 25-guage needle. The needle was carefully place onto the eye without the application of pressure. We used the needle to scratch a small area in the region of interest. Bacteria (1×107 CFU/eye) were then dropped directly on the eye in a total volume of 0.05ml. These bacteria only adhered to the area where the small abrasion was made.
CFU experiments. Organ burden analyses and quantification of proinflammatory cytokines/chemokines
6–8 weeks old male or female with ocular infections were sacrificed using CO2 at specified time points. Eyes, spleen, and brains were harvested from animals followed by homogenizing of organs. Homogenized tissues were plated on tryptic soy agar plates for identification of bacterial colony forming units (CFU). The CFU for each eye was determined. The weights of the spleens and brains were measured to determine CFU per gram of tissue. Supernatants from tissues were then analyzed for IL-1β, IL-6, KC/GRO, and TNF-α using a multiplex kit (Meso Scale Diagnostics) according to the manufacturer’s instructions.
Flow cytometry
Mice were euthanized at various time points post infection and perfused with 0.9% saline through the right ventricle of the heart. The eyes were harvested and minced into pieces. Eye tissue was digested in 2mL RPMI-1640 medium containing 0.5 mg/mL collagenase VI (Sigma) for 90 minutes at 37°C with shaking (60 rpm). The digested corneal tissue was then passed through a 100 μm filter cell strainer (Corning) to remove the debris. The cells were then pelleted by centrifugation at 400g for 5 minutes and washed once with 1x PBS. The cells were subjected to antibody staining for flow cytometric analysis. Cell numbers were measured using absolute cell count beads (eBioscience) according to the manufacturers’ instruction. Flow cytometric data were analyzed using FlowJo X software (Tree star). Refer to the Key Resource Table for antibodies used. Cells were gated using FSCA and FSCH to identify singlets, then gated for live cells by taking a population that was negative for live/dead stain. Gating strategy is presented in Figure S1J. Eyes were not pooled together for analysis.
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ly6G (clone: 1A8) | Biolegend | Cat. # 127609, RRID:AB_1134162 |
| F480 (clone BM8) | Ebioscience | Cat. # 47-4801-82 RRID:AB_2735036 |
| CD11c (N148) | Ebioscience | Cat # 11-0114-82 RRID:AB_464940 |
| CD11b (M1/70) | Ebioscience | Cat # 25-0112-82 RRID:AB_469588 |
| MHCII (M5/114.15.2) | Ebioscience | Cat # 12-5321-82 AB_465928 |
| CD45 (clone 30-F11) | Biolegend | Cat # 103137 RRID:AB_2561392 |
| H2A.X histone antibody | Abcam | Cat#: Ab188819 RRID:AB_10672053 |
| Mouse anti-MPO | Abcam | Cat # Ab9535 RRID:AB_307322 |
| Neutrophil Elastase 680 FAST Fluorescent Imaging Agent | PerkinElmer | Cat # NEV11169 |
| Ghost Red Live/dead | Tonbo | Cat # 13-0871-T100 |
| IgGA control | Abcam | Cat # ab37415 RRID:AB_2631996 |
| MOA (Marasmium oreades agglutinin)-TRITC | EY Labs | Cat # R-9001-1 |
| Anti-Psl (Psl0096) | (DiGiandomenico et al., 2012) | NA |
| Anti-PcrV (V2L2) | (Warrener et al., 2014) | NA |
| Bispecific (MEDI3902) | (DiGiandomenico et al., 2014) | NA |
| Bacterial Strains | ||
| Pseudomonas aeruginosa PAO1 WT GFP | Shawn Lewenza | NA |
| Pseudomonas aeruginosa PAO1 Δpsl GFP | Created in this study | NA |
| Pseudomonas aeruginosa PAO1 ΔpcrV GFP | Created in this study | NA |
| PAO1 dexos GFP | Created in this study | NA |
| E. coli JM109 | Promega | Cat# (P9751) |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 wild-type | Jackson | #664 |
| B6.129S4-C3,tm1Crr./J | Jackson | #3641 |
| Catchup Ly6G reporter mice | Hasenberg et al., 2015 | NA |
| Peptidyl arginine deiminase, type IV (PAD4−/−) | Gift from Kerri A. Mowen, La Jolla California | NA |
| Cathepsin C knockout mice (Cstc−/−) | GalxoSmithKline | NA |
| Neutrophil elastase knockout (NE−/−) | Jackson | #6112 |
| Fc R gamma receptor knockout mice (FcRγ−/−) | Taconic | #583 |
| Critical Commercial Assays | ||
| Meso Scale Multiplex assay Kits | Proinflammatory panel | K152QOD |
| Plasmids | ||
| CTX-Ptac-GFP | Wyckoff and Wozniak, 2001 | NA |
| pPM297 | Wyckoff and Wozniak, 2001 | NA |
| Software and Algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Volocity | Quorum | http://quorumtechnologies.com/index.php/component/content/article/31-volocity-software/31-volocity-quantitation |
| FIJI | ImageJ | https://fiji.sc/ |
| Leica SP8 software | Leica | https://www.leica-microsystems.com/products/ |
| FlowJo | Tree Star | https://www.flowjo.com/ |
| Other. | ||
| 0.3% Tobrex (3mg/mL tobramycin eye ointment) | Canadian distributors of veterinary products | NA |
| Eye ointment (no antibiotics) | VWR | NA |
Intracerebral administration of Pseudomonas aeruginosa
Mice were anesthetized with ketamine (200 mg/kg) and xylazine (10 mg/kg) i.p and placed on a stereotaxic frame. After a midline skin incision, a burr hole (0.5 diameter) was made using a motorized drill 1.0 mm anterior and 2.0 mm lateral to the bregma. A 10 μl microsyringe (Hamilton Company) was loaded with a 2 ul suspension containing 500 CFU Pseudomonas aeruginosa GFP (PAO1) and affixed to the stereotaxic frame. The needle was advanced 4.0 mm into the brain from the surface of the dura and the suspension was slowly administered. The burr hole was then sealed with wax and the incision closed. Mice were recovered and closely monitored. At 24 hours post-infection, animals were euthanized and tissues were collected to determine CFUs in brain and spleen.
Antibiotic treatment
0.3% tobramycin eye drops (Tobrex, sterile ophthalmic ointment) were administered to mice two times daily, directly onto the eye. As a control, mice were administered eye drops with no antibiotics.
Antibody conjugation
Abcam antibodies were conjugated to Alexa-647 using the Biotium conjugation kits.
Therapeutic antibodies
Anti-psl commercial name Psl0096, anti-PCRV commercial name V2L2, bispecific commercial name MEDI3902. Produced by MedImmune/AstraZeneca.
NET staining
Sytox red (ThermoFisher) was diluted 1:500 and gently dripped on the surface of the eye, left for 5 minutes, and then gently washed off with PBS (3x). H2A (10μg of labelled antibody per mouse) and neutrophil elastase probe (100μl as recommended by Perkin Elmer) was injected I.V. into the mouse 2–3 hours prior to imaging. Refer to Extended Table 1 for detailed list of antibodies used. Elastase antibody probe used was to detect activity of mammalian elastase enzyme, as seen by the lack of staining only in neutrophil elastase deficient animals. We did not see reactivity with P. aeruginosa elastase using this probe.
Total human IgG quantification from mouse eye
The total concentration of MEDI3902 in mouse homogenates was determined by enzyme-linked immunosorbent assay (ELISA) as previously described (DiGiandomenico et al., 2014). The procedure is a heterogeneous format in which wash steps follow after each incubation. All plates were washed 3X with PBS supplemented with 0.1% tween 20. A 96 well microtiter plate (NUNC™ MaxiSorp™) was coated with 0.05 ml/well of sheep anti-human IgG (H+L) diluted to 0.5 μg/mL in carbonate/bicarbonate. The standard curve was established using a four parameter logistical curve fit model [y-(A-D)/(1+(x/C)^B+D] without weighting. The nominal range of this assay was 1–1000 ng/mL.
Scanning electron microscopy
Eye samples were collected from mice at 24 hours post-infection. The full eye with the optic nerve was prepared and mounted. The optic nerve was used to orient our imaging. Samples were prepared as previously described using the critical drying point method (Saraswathi and Beuerman, 2015). Images were obtained from John’s Hopkin’s EM facility. All SEM images except for the first panel of Figure 1G were collected at 6.0KX magnification. Panel 1 of Figure 1G was captured at magnification of 650X.
Histology
In order to keep the shape of the eyes intact and not disturb the biofilm on the surface of the cornea, we fixed the whole head of the mouse in 10% buffered formalin after the mice were sacrificed. Eyes were removed from mice 24 hours after fixation and paraffin embedded. Samples were sectioned using a microtome and stained using Gram-stain and hematoxylin and eosin stain as previously described (Brown and Hopps, 1973).
QUANTIFICATION AND STATISICAL ANALYSIS
Statistical analysis
All experiments were repeated three times with n=4–5 mice per group. Both eyes were infected on each mouse and plotted individually (to show variability); however statistics were performed on the average of the two eyes from the same mouse. For example, an experiment with n=4 mice will have 8 points to account for the individual eyes, unless otherwise stated. Data are represented as the mean ± standard error of the mean or median and interquartile range (IQR; 25th and 75th percentiles), as specified in figure legends. Normally distributed data were analyzed using Student’s t-test or one-way analysis of variance (ANOVA) with Holms-Sidak’s multiple comparisons post-hoc test. Non-normally distributed data were analyzed using the Mann Whitney U test or Kruskal-Wallis ANOVA with Dunn’s multiple comparisons post-hoc test. All statistical tests were two-sided and a p-value <0.05 was considered significant. Data were analyzed using GraphPad Prism (Version 7, GraphPad Software Inc., CA, USA).
IVM image quantification
For IVM image quantification, each point in a graph represents one mouse as only one eye per mouse could be imaged using this technique. Leica software TCS SP8 software was used to quantify the height of the biofilm. Images were gathered and 3D projections were made of each z-stack. The z-stacks were then sliced at three regions and the height of the biofilm at three points in each region was measured and averaged. The average of these quantifications is represented in the graphs. Similarly, dead-zone measurements were performed at three points in the 3D image and the average was plotted. Please refer to Figure S1K for schematic of how we made these measurements.
Keratitis severity score
Full abrasion model was used for keratitis scores. Keratitis severity score was adapted from Hazlett et al. (Hazlett et al., 1987). We modified the semiquantitative score to include an additional category for eyes without any visual opacity and adjusted the score developed by Hazlett et al. accordingly. Eyes of infected mice were scored by two individuals and the identity of the samples were blinded to the individuals. The average of both individuals was processed and graphed (Hazlett et al., 1987). Scoring format: 1= Clear or slight opacity, partially covering pupil; 2 = Slight opacity fully covering anterior segment; 3 = Dense opacity, partially or fully covering pupil; 4 = Dense opacity, covering entire anterior segment; 5 = Corneal perforation or phthisis.
Supplementary Material
Moderate number of neutrophils move into the eye after corneal abrasion. Related to Figure 1. A. Neutrophil recruitment to the cornea first imaged at the edge of the cornea. B. Neutrophil behaviour after abrasion imaged at the centre of the cornea. Neutrophils (red) in the cornea 24 hours post-abrasion. Classic full abrasion of the cornea model was used here.
A robust number of neutrophils move towards the infection. Related to Figure 1. A. 25-minute spinning-disk confocal video 4 hours post abrasion + infection. Neutrophils (red) and P. aeruginosa (green). B. 3D reconstruction of infection at 4 hours using multiphoton microscopy. Neutrophils (red) and P. aeruginosa (green). Classic full abrasion of the cornea model was used here.
P. aeruginosa forms a layer on the surface of the cornea that neutrophils are unable to penetrate. Related to Figure 1. A. Spinning-disk confocal video 24 hours post-infection. P. aeruginosa (green) and neutrophils (red). B. P. aeruginosa (green) and neutrophils (red), 3D reconstruction using multiphoton microscopy. Classic full abrasion of the cornea model was used here.
There is a break-down of the collagen matrix of the cornea around the site of infection. Related to Figure 3. 3D reconstruction using multiphoton microscopy at 24 hours post abrasion + infection. Second harmonic imaging represents collagen matrix in white, P. aeruginosa (green), neutrophils (red). Small localized abrasion site model was used here.
Prophylactic treatment with a bispecific antibody results in enhanced neutrophil recognition of bacteria at 4 hours post-infection. Related to Figure 4. Twenty-five minute spinning-disk confocal video in mice prophylactically treated with bispecific antibody. P. aeruginosa (green) and neutrophils (red). Small localized abrasion site model was used here.
NET deficient, PAD4−/− mice, show reduced biofilm formation. Related to Figure 3. A. Twenty-minute spinning-disk confocal video in PAD4−/− mice at 24 hours post-infection. B. 3D reconstruction of corneal infection in PAD4−/− mice. P. aeruginosa (green) and neutrophils (red). Classic full abrasion of the cornea model was used here.
Highlights.
P. aeruginosa keratitis infections result in biofilm formation on the cornea
NETs form at the base of the biofilm, triggered by the typethree secretion system (T3S)
NETs stop bacterial dissemination into the brain but promote antibiotic resistance
Blocking exopolysaccharide Psl and the T3S allowed neutrophils to breakdown the biofilm
Acknowledgments
Thank you to Trecia Nussbaumer for breeding and genotyping mice, Karen Poon and the microbial core for use of the Flow cytometry facility. Andrew K. Chojnack for assistance with image analysis. In addition, the authors would like to thank Dr. Shawn Lewenza and Erik van Tilburg Bernardes for sharing the PAO1 bacterial strains. The authors would also like to thank Justin Deniset for critical analysis of the manuscript and assistance with keratitis scores.
This work was funded partially by MT6TYG1 MedImmune/AstraZeneca. AT is funded by Banting Postdoctoral Fellowship, MP is funded by German Research Foundation. PK holds funding from Cystic fibrosis Canada and CIHR foundations grant that fund MEW. ZZ is supported by Alberta Innovative Heath Solutions. BS is supported by Canadian Institute of Health Research. JTK was supported by the Academy of Finland. M.G. was funded by NIH (R01EY022054).
Footnotes
Declaration of conflict of interest.
We would like to confirm no competing interests regarding the work presented in this manuscript. This work was partially funded by MedImmune/AstraZeneca. All members of this manuscript affiliated with MedImmune/AstraZeneca have personal financial interests. MedImmune/AstraZeneca holds patents for anti-Psl (Psl0096), anti-PcrV (V2L2MD), and bispecific (MEDI3902) antibodies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akk A, Springer LE, and Pham CT (2016). Neutrophil Extracellular Traps Enhance Early Inflammatory Response in Sendai Virus-Induced Asthma Phenotype. Front Immunol 7, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus AA, Evans DJ, Barbieri JT, and Fleiszig SM (2010). The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect Immun 78, 4500–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin DK, Heimer SR, Tam C, Li WY, Le Due JM, Evans DJ, and Fleiszig SM (2011). Role of defensins in corneal epithelial barrier function against Pseudomonas aeruginosa traversal. Infect Immun 79, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabino S, Chen Y, Chauhan S, and Dana R (2012). Ocular surface immunity: homeostatic mechanisms and their disruption in dry eye disease. Prog Retin Eye Res 31, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar I, London A, and Schwartz M (2012). The privileged immunity of immune privileged organs: the case of the eye. Front Immunol 3, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, Torres VJ, and Wozniak DJ (2018). Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A 115, 7416–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, and Papayannopoulos V (2014). Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RC, and Hopps HC (1973). Staining of bacteria in tissue sections: a reliable gram stain method. Am J Clin Pathol 60, 234–240. [DOI] [PubMed] [Google Scholar]
- DiGiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, Bonnell J, Fleming R, Bezabeh B, Dimasi N, et al. (2014). A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med 6, 262ra155. [DOI] [PubMed] [Google Scholar]
- DiGiandomenico A, Warrener P, Hamilton M, Guillard S, Ravn P, Minter R, Camara MM, Venkatraman V, Macgill RS, Lin J, et al. (2012). Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J Exp Med 209, 1273–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PJ, Jackson H, Stanford MR, and Graham EM (2003). Inflammatory optic neuropathy as the presenting feature of herpes simplex acute retinal necrosis. Br J Ophthalmol 87, 512–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellatly SL, and Hancock RE (2013). Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis 67, 159–173. [DOI] [PubMed] [Google Scholar]
- Hanlon SD, Smith CW, Sauter MN, and Burns AR (2014). Integrin-dependent neutrophil migration in the injured mouse cornea. Exp Eye Res 120, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, Kraus A, Engel DR, Klingberg A, Seddigh P, et al. (2015). Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods 12, 445–452. [DOI] [PubMed] [Google Scholar]
- Hazlett LD, Moon MM, Strejc M, and Berk RS (1987). Evidence for N-acetylmannosamine as an ocular receptor for P. aeruginosa adherence to scarified cornea. Invest Ophthalmol Vis Sci 28, 1978–1985. [PubMed] [Google Scholar]
- Hill D, Rose B, Pajkos A, Robinson M, Bye P, Bell S, Elkins M, Thompson B, Macleod C, Aaron SD, et al. (2005). Antibiotic susceptabilities of Pseudomonas aeruginosa isolates derived from patients with cystic fibrosis under aerobic, anaerobic, and biofilm conditions. J Clin Microbiol 43, 5085–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie Y, Roberts AEL, Kragh KN, Gordon VD, Hutchison J, Allen RJ, Melaugh G, Bjarnsholt T, West SA, and Diggle SP (2017). The Pseudomonas aeruginosa PSL Polysaccharide Is a Social but Noncheatable Trait in Biofilms. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski A, Gupta KH, Goldufsky JW, Lee HW, Gupta V, and Shafikhani SH (2018). Pseudomonas aeruginosa ExoS Induces Intrinsic Apoptosis in Target Host Cells in a Manner That is Dependent on its GAP Domain Activity. Sci Rep 8, 14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloth C, Schirmer B, Munder A, Stelzer T, Rothschuh J, and Seifert R (2018). The Role of Pseudomonas aeruginosa ExoY in an Acute Mouse Lung Infection Model. Toxins (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, and Germain RN (2013). Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaux D, Ghigo JM, and Beloin C (2014). Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol Mol Biol Rev 78, 510–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, Chaconas G, and Kubes P (2010). An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol 11, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xue Y, Dong D, Xiao C, Lin C, Wang H, Song F, Fu T, Wang Z, Chen J, et al. (2017). CCR2(−) and CCR2(+) corneal macrophages exhibit distinct characteristics and balance inflammatory responses after epithelial abrasion. Mucosal Immunol 10, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage SE, Gardner PJ, Ribeiro J, Cristante E, Sampson RD, Luhmann UF, Ali RR, and Bainbridge JW (2016). Flow cytometric analysis of inflammatory and resident myeloid populations in mouse ocular inflammatory models. Exp Eye Res 151, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LJ, and Liu J (2016). Human Microbiota and Ophthalmic Disease. Yale J Biol Med 89, 325–330. [PMC free article] [PubMed] [Google Scholar]
- Ma L, Conover M, Lu H, Parsek MR, Bayles K, and Wozniak DJ (2009). Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog 5, e1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Jackson KD, Landry RM, Parsek MR, and Wozniak DJ (2006). Analysis of Pseudomonas aeruginosa conditional psl variants reveals roles for the psl polysaccharide in adhesion and maintaining biofilm structure postattachment. J Bacteriol 188, 8213–8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Wang S, Wang D, Parsek MR, and Wozniak DJ (2012). The roles of biofilm matrix polysaccharide Psl in mucoid Pseudomonas aeruginosa biofilms. FEMS Immunol Med Microbiol 65, 377–380. [DOI] [PubMed] [Google Scholar]
- Mann EE, and Wozniak DJ (2012). Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev 36, 893–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, and Kubes P (2010). Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366. [DOI] [PubMed] [Google Scholar]
- Metruccio MME, Wan SJ, Horneman H, Kroken AR, Sullivan AB, Truong TN, Mun JJ, Tam CKP, Frith R, Welsh L, et al. (2018). A novel murine model for contact lens wear reveals clandestine IL-1R dependent corneal parainflammation and susceptibility to microbial keratitis upon inoculation with Pseudomonas aeruginosa. Ocul Surf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JK, Badawy HT, Clemons C, Kreider KL, Wilber P, Milsted A, and Young G (2012). Development of the Pseudomonas aeruginosa mushroom morphology and cavity formation by iron-starvation: a mathematical modeling study. J Theor Biol 308, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, Schlesinger LS, and Wozniak DJ (2012). Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol 14, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy LR, Isabella VM, and Lewis K (2014). Pseudomonas aeruginosa biofilms in disease. Microb Ecol 68, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda M, Pflugfelder SC, and Holland S (1991). Fulminant pseudomonal keratitis and scleritis in human immunodeficiency virus-infected patients. Arch Ophthalmol 109, 503–505. [DOI] [PubMed] [Google Scholar]
- Okada F, Kobayashi M, Tanaka H, Kobayashi T, Tazawa H, Iuchi Y, Onuma K, Hosokawa M, Dinauer MC, and Hunt NH (2006). The role of nicotinamide adenine dinucleotide phosphate oxidase-derived reactive oxygen species in the acquisition of metastatic ability of tumor cells. Am J Pathol 169, 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson RM, Holland GN, Goss SJ, Bowers WD, and Meyers-Elliott RH (1987). Routes of viral spread in the von Szily model of herpes simplex virus retinopathy. Curr Eye Res 6, 59–62. [DOI] [PubMed] [Google Scholar]
- Ozkan J, Nielsen S, Diez-Vives C, Coroneo M, Thomas T, and Willcox M (2017). Temporal Stability and Composition of the Ocular Surface Microbiome. Sci Rep 7, 9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos V (2018). Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18, 134–147. [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V, Metzler KD, Hakkim A, and Zychlinsky A (2010). Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191, 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel SM, Logan LK, and Hauser AR (2014). The ADP-ribosyltransferase domain of the effector protein ExoS inhibits phagocytosis of Pseudomonas aeruginosa during pneumonia. MBio 5, e01080–01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray VA, Hill PJ, Stover CK, Roy S, Sen CK, Yu L, Wozniak DJ, and DiGiandomenico A (2018). Publisher Correction: Anti-Psl Targeting of Pseudomonas aeruginosa Biofilms for Neutrophil-Mediated Disruption. Sci Rep 8, 9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray VA, Hill PJ, Stover KC, Roy S, Sen CK, Yu L, Wozniak DJ, and DiGiandomenico A (2017). Anti-Psl Targeting of Pseudomonas aeruginosa Biofilms for Neutrophil-Mediated Disruption. Sci Rep 7, 16065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DM, Petroll WM, Jester JV, and Cavanagh HD (2007). Current concepts: contact lens related Pseudomonas keratitis. Cont Lens Anterior Eye 30, 94–107. [DOI] [PubMed] [Google Scholar]
- Rosales C, Demaurex N, Lowell CA, and Uribe-Querol E (2016). Neutrophils: Their Role in Innate and Adaptive Immunity. J Immunol Res 2016, 1469780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswathi P, and Beuerman RW (2015). Corneal Biofilms: From Planktonic to Microcolony Formation in an Experimental Keratitis Infection with Pseudomonas Aeruginosa. Ocul Surf 13, 331–345. [DOI] [PubMed] [Google Scholar]
- Sousa AM, and Pereira MO (2014). Pseudomonas aeruginosa Diversification during Infection Development in Cystic Fibrosis Lungs-A Review. Pathogens 3, 680–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Karmakar M, Taylor PR, Rietsch A, and Pearlman E (2012). ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J Immunol 188, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy A, Srinivasan M, Mascarenhas J, Lalitha P, Rajaraman R, Ravindran M, Oldenburg CE, Ray KJ, Glidden D, Zegans ME, et al. (2012). Pseudomonas aeruginosa keratitis: outcomes and response to corticosteroid treatment. Invest Ophthalmol Vis Sci 53, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanabalasuriar A, Surewaard BG, Willson ME, Neupane AS, Stover CK, Warrener P, Wilson G, Keller AE, Sellman BR, DiGiandomenico A, et al. (2017). Bispecific antibody targets multiple Pseudomonas aeruginosa evasion mechanisms in the lung vasculature. J Clin Invest 127, 2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CS, Rangel SM, Almblad H, Kierbel A, Givskov M, Tolker-Nielsen T, Hauser AR, and Engel JN (2014). The Pseudomonas aeruginosa type III translocon is required for biofilm formation at the epithelial barrier. PLoS Pathog 10, e1004479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tre-Hardy M, Nagant C, El Manssouri N, Vanderbist F, Traore H, Vaneechoutte M, and Dehaye JP (2010). Efficacy of the combination of tobramycin and a macrolide in an in vitro Pseudomonas aeruginosa mature biofilm model. Antimicrob Agents Chemother 54, 4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vareechon C, Zmina SE, Karmakar M, Pearlman E, and Rietsch A (2017). Pseudomonas aeruginosa Effector ExoS Inhibits ROS Production in Human Neutrophils. Cell Host Microbe 21, 611–618 e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrener P, Varkey R, Bonnell JC, DiGiandomenico A, Camara M, Cook K, Peng L, Zha J, Chowdury P, Sellman B, et al. (2014). A novel anti-PcrV antibody providing enhanced protection against Pseudomonas aeruginosa in multiple animal infection models. Antimicrob Agents Chemother 58, 4384–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff TJ, and Wozniak DJ (2001). Transcriptional analysis of genes involved in Pseudomonas aeruginosa biofilms. Methods Enzymol 336, 144–151. [DOI] [PubMed] [Google Scholar]
- Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, and Herrmann M (2016). New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol 7, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, et al. (2012). Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18, 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Moderate number of neutrophils move into the eye after corneal abrasion. Related to Figure 1. A. Neutrophil recruitment to the cornea first imaged at the edge of the cornea. B. Neutrophil behaviour after abrasion imaged at the centre of the cornea. Neutrophils (red) in the cornea 24 hours post-abrasion. Classic full abrasion of the cornea model was used here.
A robust number of neutrophils move towards the infection. Related to Figure 1. A. 25-minute spinning-disk confocal video 4 hours post abrasion + infection. Neutrophils (red) and P. aeruginosa (green). B. 3D reconstruction of infection at 4 hours using multiphoton microscopy. Neutrophils (red) and P. aeruginosa (green). Classic full abrasion of the cornea model was used here.
P. aeruginosa forms a layer on the surface of the cornea that neutrophils are unable to penetrate. Related to Figure 1. A. Spinning-disk confocal video 24 hours post-infection. P. aeruginosa (green) and neutrophils (red). B. P. aeruginosa (green) and neutrophils (red), 3D reconstruction using multiphoton microscopy. Classic full abrasion of the cornea model was used here.
There is a break-down of the collagen matrix of the cornea around the site of infection. Related to Figure 3. 3D reconstruction using multiphoton microscopy at 24 hours post abrasion + infection. Second harmonic imaging represents collagen matrix in white, P. aeruginosa (green), neutrophils (red). Small localized abrasion site model was used here.
Prophylactic treatment with a bispecific antibody results in enhanced neutrophil recognition of bacteria at 4 hours post-infection. Related to Figure 4. Twenty-five minute spinning-disk confocal video in mice prophylactically treated with bispecific antibody. P. aeruginosa (green) and neutrophils (red). Small localized abrasion site model was used here.
NET deficient, PAD4−/− mice, show reduced biofilm formation. Related to Figure 3. A. Twenty-minute spinning-disk confocal video in PAD4−/− mice at 24 hours post-infection. B. 3D reconstruction of corneal infection in PAD4−/− mice. P. aeruginosa (green) and neutrophils (red). Classic full abrasion of the cornea model was used here.