

Abstract
The polo-like kinases (Plks1–5) are emerging as an important class of proteins involved in many facets of cell cycle regulation and response to DNA damage and stress. Here we show that Plk3 phosphorylates the key cell cycle protein phosphatase Cdc25A on two serine residues in its cyclinB/cdk1 docking domain and regulates its stability in response to DNA damage. We generated a Plk3 knock-out mouse and show that Cdc25A protein from Plk3-deficient cells is less susceptible to DNA damage-mediated degradation than cells with functional Plk3. We also show that absence of Plk3 correlates with loss of the G1/S cell cycle checkpoint. However, neither this compromised DNA damage checkpoint nor reduced susceptibility to proteasome-mediated degradation after DNA damage translated into a significant increase in tumor incidence in the Plk3-deficient mice.
Keywords: Polo-like kinases, Cdc25A, DNA damage, Checkpoints, Ubiquitinylation
1. Introduction
Polo-like kinases (Plks) constitute a family of evolutionarily conserved serine/threonine kinases that participate in normal cell cycle progression and in cellular response to DNA damage [1–8]. Subsequent to the original identification of Polo in Drosophila melanogaster [9–11], the Saccharaomyces cerevisiae Cdc5 gene was identified as a polo kinase ortholog [12], as was the Plo1 gene in Schizosaccharomyces pompe [13]. While there is only one polo gene in Drosophila and the yeasts, there is a family comprised of multiple Plk genes in Xenopus laevis, Caenorhabditis elegans and mammals [14]. In mammals there are now five known Plks [14,15] with apparently diverse functions. The polo-like kinases are identified by polo box motifs within the carboxy region of the protein which are responsible for sub-cellular localization and substrate recognition [16–19]. Among the functions attributed to polo-like kinases are the promotion of spindle formation at the G2 to mitosis transition, activation of the anaphase-promoting complex (APC) at the time of mitotic exit, a requirement for cytokinesis, an involvement in Golgi fragmentation, and participation in the DNA damage checkpoint signaling cascade during the transition from G2 to mitosis [1–8,20–24].
Polo-like kinase 3 (Plk3) serves numerous and diverse cellular functions. It participates in cell cycle control [7,25] and is reported to be a tumor suppressor that is down-regulated in several primary human tumor types including lung, stomach, uterus, bladder and head and neck tumors [26]. Following DNA damage due to ionizing radiation (IR) or oxidative stress, Plk3 is phosphorylated in an ATM-dependent manner [7,27–29]. It is also phosphorylated in an ATM-independent manner following disruption of the mitotic spindle [7]. It phosphorylates p53 on serine 20, which also is a target phosphorylation site for Chk2, and which contributes to the release of p53 from MDM2 and its consequent stabilization [30]. Plk3 appears to have a regulatory function in the DNA damage response pathway. It phosphorylates Chk2 on serine 73 following exposure to IR in a priming capacity [28]. This phosphorylation event is important for the full activation of Chk2 by ATM and the subsequent phosphorylation of Cdc25A by Chk2, targeting it for proteasome-mediated degradation. Prior to the entry of cells into mitosis, Plk3 also phosphorylates Cdc25C on two serine residues that appear to regulate the localization of Cdc25C [31].
Cdc25A phosphatase is an essential activator of cell cycle progression and its expression is very tightly regulated at many levels. Cdc25A activity is regulated by transcriptional activation, reversible phosphorylation, protein-protein interaction and ubiquitin-mediated degradation [32–38]. In response to DNA damage or to stalled replication, the activation of the ATM and ATR protein kinases leads to Chk1 and Chk2 activation and to Cdc25A hyperphosphorylation. These events stimulate SCF-mediated ubiquitinylation of Cdc25A and its proteolysis [39,40]. This contributes to delaying cell cycle progression, thereby preventing genomic instability.
The fact that Cdc25A levels are precisely controlled by a multilayered mechanism is underscored by the observation that its overproduction leads to accelerated entry of cells into both S phase [41] and mitosis [40]. Furthermore, failure to regulate Cdc25A during a checkpoint response causes bypass of DNA damage and replication checkpoints, resulting in enhanced DNA damage [33,39,40,42,43]. These defects in maintaining correct levels of Cdc25A translate into an increased capacity for cell proliferation that can lead to cancer. Indeed, Cdc25A is frequently overexpressed in multiple cancer types [44].
Ubiquitinylation-mediated degradation of Cdc25A is associated with phosphorylation of Cdc25A at the β-TrCP docking site (DSG motif). Serines 79, 82 and 88 constitute the Cdc25A DSG docking site and absence of their phosphorylation is sufficient to abolish β-TrCP binding and to inhibit Cdc25A proteolysis [32]. Several kinases have recently been reported to phosphorylate the DSG motif and target Cdc25A for SCFβ-TrCP-mediated degradation. Human tumors show a strong correlation between Cdc25A overproduction and GSK-3β inactivation, consistent with the observation that GSK-3β targets Cdc25A for ubiquitin-mediated proteolysis [45]. It is, therefore, likely that GSK-3β inactivation may account for Cdc25A overproduction in a subset of human tumors [45]. When Cdc25A and Plk3 are co-expressed in cultured cells, phosphorylation of Cdc25A on threonine 80 is enhanced with subsequent β-TrCP-mediated degradation. Furthermore, knockdown of Plk3, but not other Plk family members, with siRNA resulted in the stabilization of Cdc25, consistent with Plk3 serving a priming function for Cdc25A degradation [45]. A separate study, in which an shRNA library was screened to identify new genes involved in the G2/M checkpoint, identified NIMA-related kinase 11 (NEK11) as the kinase that phosphorylates Cdc25A on the DSG motif [46]. This study further showed that depleting cells of NEK11 stabilized Cdc25A, suggesting that phosphorylation of Cdc25A by NEK11 is essential for its SCFβ-TrCP-mediated degradation. Other pathways have also been implicated in the phosphorylation of the DSG domain. When the extracellular signal-regulated kinase (ERK) pathway is activated in Xenopus eggs, Cdc25A serine 85 (serine 88 in human) is phosphorylated and targeted for degradation [47]. This degradation involves the SCFβ-TrCP ubiquitin ligase and requires phosphorylation by both ERK and its downstream kinase p90rsk. An earlier study has also suggested that Smad3 plays a key role in the regulation of Cdc25A ubiquitinylation by SCFβ-TrCP and that Cdc25A stabilization observed in various cancers could be associated with defects in the TGF-β–Smad3 pathway [48].
We now show that Plk3 phosphorylates Cdc25A at its carboxy end and helps regulate its stability. To better understand the roles of Plk3 in cell cycle regulation and DNA damage control, particularly as it relates to Cdc25A, we have produced Plk3 knockout mice by standard gene targeting methods. The availability of these mice has enabled us to directly address whether or not Plk3 participates in cell cycle regulation and in the cellular response to DNA damage. When mouse embryo fibroblasts (MEFs) are deprived of serum to arrest them in G1, with subsequent release by the addition of serum, the distribution of Plk3−/− cells in the cell cycle is altered showing an increased number of cells with a 4 N complement of DNA, a diminished number of cells in G1, and a small polyploidy peak of 8 N or greater. When thymocytes derived from both wildtype and mutant mice were treated with etoposide, a DNA damaging agent that produces double strand breaks, the cell cycle distribution of the mutant cells showed a compromised G1/S checkpoint consistent with a role for Plk3 in DNA damage response. Furthermore, the level of Cdc25A in thymocytes from Plk3-deficient mice is markedly elevated compared with that of wildtype mice following etoposide treatment, indicating that the absence of Plk3 in vivo renders Cdc25A more stable in these cells. Surprisingly, the compromised lability of Cdc25A and G1/S cell cycle checkpoint did not translate into a significant elevation in spontaneous tumor incidence in the Plk3-deficient mice. No statistically significant increase in tumor incidence was observed between any of the genotypes. It was only when the data were fractionated by gender that a slight increase was observed in the heterozygous and homozygous female mice, possibly indicating hormonal involvement.
2. Materials and methods
2.1. Plasmid construction
GST-Cdc25A constructs were produced by subcloning the indicated regions of a human Cdc25A cDNA (a gift from Dr. Yolanda Sanchez, Dartmouth College) into the PGEX2T vector (Amersham) between the BamH1 and EcoR1 sites. The construct for GST-Plk3 expression was provided by Dr. Klaus Strebhardt (Goethe-University, Frankfurt, Germany). To construct the Myc-tagged Cdc25A, a Myc-His-tag was first cloned into the EcoRI site of pcDNA 3.1 myc/his (−) and the human Cdc25A cDNA was cloned into EcoRI and BamHI sites. Each of the serine 513 and serine 519 point mutations of Cdc25A was generated by site-directed mutatagenesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
2.2. Cell culture and transfection
HT1080 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Eurobio), 2 mM glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml) in a humidified atmosphere containing 5% CO2 at 37 °C. Transfections were performed using Lipofectamine 2000 (Invitrogen) following the manufacturer’s recommendations.
2.3. Cell lysis, Western blots and immunoprecipitation
Cells transfected with the Myc-tagged Cdc25A constructs were harvested 24 h after transfection and lysed in ice-cold lysis buffer (50 mM HEPES pH 7.0, 250 mM NaCl, 0.2% Nonidet P-40, 10% (vol/vol) glycerol, 1 mM NaVO4, 1 mM DTT) freshly supplemented with 1 mM PMSF, 10 μg/ml aprotinin (Sigma), pepstatin (Sigma), leupeptin (Sigma), and microcystin LR (Calbiochem). The lysates were boiled for 5 min in sample buffer and loaded on a 10% SDS–PAGE gel. Proteins were analyzed by Western blot using an anti-Myc antibody (Santa Cruz cat# sc-40). Cells were also transfected with a Flag-tagged Cdc25A construct and Cdc25A was immunoprecipitated with mouse antibody to Cdc25A (Neomarkers, Freemont, CA) and protein A/G agarose beads (Santa Cruz Biotechnology, CA).
2.4. Expression and purification of GST fusion proteins
Escherichia coli BL21 was transformed with the following plasmids: pGEX2T (Pharmacia), pGEX2T-Plk3, pGEX2T-Plk3-KD, pGEX-Chk2–KD, pGEX2T-Cdc25C, pGEX2T-hCdc25A, and each of the pGEX2T-hCdc25A derivatives. Cultures were grown at 37 °C to an A600 of 0.8 and IPTG was added to a final concentration of 0.5 mM. After growing for an additional 3 h at 25 °C, cells were pelleted by centrifugation. Cell pellets were washed with PBS and were resuspended in STE buffer (100 mM NaCl, 10 mM Tris HCl, pH 8.0, 1 mM EDTA) supplemented with 2 mM DTT, 1 mg/ml lysozyme and protease inhibitors (2 mM PMSF, 0.15 U/ml aprotinin, 20 μM leupeptin, 20 μM pepstatin). Cells were incubated on ice for 1 h with occasional mixing. Lysates were clarified by centrifugation (13,000 × g for 15 min) and Triton X-100 was added to a final concentration of 2%. Proteins were precipitated with GST-agarose beads (Sigma Chemical Co.), washed three times with STE buffer, and eluted from GST-agarose with 20 mM glutathione and 1 mM DTT in PBS.
2.5. In vitro kinase reactions
Protein kinases were incubated with substrates at 37 °C for 1 h in 1× kinase buffer (30 mM HEPES pH 7.4, 10 mM MgCl2 and 1 mM DTT) supplemented with 10 μM nonradioactive ATP and 10 μCi of [γ−32P] ATP (Amersham Pharmacia Biotech). Kinase reactions were terminated by the addition of 6× SDS loading buffer then were boiled for 10 min and fractionated by SDS–PAGE on 10% polyacrylaminde gel. For Plk3 phosphorylation of GST-Cdc25A, the SDS–PAGE gels were silver stained to demonstrate the relative amounts of substrate.
2.6. Ionizing radiation treatment
HT1080 cells were transfected as previously described with a vector expressing wildtype and mutant Cdc25A. Twenty-four hours after transfection each plate was split into four plates. The cells were used to determine Cdc25A stability before and after treatment with IR. To control for stress induced by time spent outside the incubator, additional plates were taken out and left at room temperature during the time of the treatment, but left untreated. To determine the level of Cdc25A that accumulates in the absence of degradation, cells were treated with the proteasome inhibitor MG132. Transfected cells were either left untreated or treated with 10 Gy of IR using a 137Cs source.
2.7. Cell cycle analysis
MEF cells and thymocytes derived from both Plk3+/+ and Plk3−/− mice were grown to ~80% confluency and subject serum starvation or etopside treatment (10 mM). Cells were then trypsinized, washed three times in cold PBS, and fixed in cold 70% ethanol for 15 min at −20 °C. Cells were then stained with 10 μg/ml propidium iodide (Molecular Probes) and 40 μg/ml RNase A (Sigma) in PBS for 20 min at room temperature. Cells were then washed and resuspended in 1 ml of PBS and analyzed with a BD LSR II flow cytometer system (Becton Dickinson, Franklin Lakes, NJ). Figures were prepared from listmode data with the use of DIVA v6.0 software and quantitation of the cell cycle profile was performed by curve fitting with the use of the ModFitLT program (Becton Dickinson, Franklin Lakes NJ).
2.8. Generation of a Plk3 knock-out mouse and derivation of mouse embryo fibroblasts
In order to generate the Plk3 knock-out targeting construct, two fragments of Plk3 genomic DNA were PCR amplified using Turbo-Taq polymerase. The first fragment covers the 5′ non translated region of the Plk3 gene and does not include the promoter region (2.2 kb). This fragment was amplified using two primers containing the KpnI sites in both ends. The second fragment covers the 3′ part of Plk3 genomic sequence spanning from exon 7 through exon 14. The fragment was amplified using two primers containing BamHI sites in both ends. The two fragments were cloned into KpnI and BamHI sites of PL452 targeting vector flanking a floxed neomycin resistance gene. The construct was checked for orientation by restriction mapping and for potential mutations introduced by PCR by sequencing. Integration of this construct in genomic DNA leads to replacement of the promoter region of the genomic Plk3 as well as the first 6 exons, which encode the kinase domain of the Plk3 protein, with a neomycin resistance gene. The targeting construct was electroporated into ES cells and mice (129/BS background) were generated by conventional methods. Mouse tails were clipped and genomic DNA extracted and analyzed for gene targeting by Southern blot and PCR. The heterozygous mice were crossed and MEFs were derived from E13.5 embryos. DNA and RNA extracted from those MEFs were checked for Plk3 gene inactivation by Southern and Northern blots respectively.
2.9. Identification of phosphorylation sites of Cdc25A
Protein preparations of cdc25A captured by immuno-affinity methods and separated by SDS–PAGE, were excised from the gel, digested with trypsin, and analyzed by MALDI-TOF/TOF as described previously [49]. For those samples digested with chymotrypsin (Promega), the method was the same as described for trypsin but with the addition of 10 mM CaCl2 to the digestion buffer. MALDI-TOF mass spectra were collected in positive ion reflector mode in 4-HCCA matrix on an MDS Analytical Technologies 4800 MALDI-TOF/TOF instrument. For sequence confirmation and mapping sites of phosphorylation, individual peptide masses detected in reflector ion mode were further subjected to isolation and fragmentation in the MALDI-TOF/TOF instrument as described previously [49]. For phosphopeptide enrichment, cdc25A preparations digested with chymotrypsin were captured on TiO2 beads equilibrated in 80% acetonitrile, 1%TFA 1 M glycolic acid then eluted with 1% ammonium hydroxide as described previously [50]. The enriched phosphopeptides were evaluated and sequenced by MALDI-TOF/TOF as described above. Samples of bovine alpha casein (a standard phosphoprotein) digested with trypsin were used as a positive control for TiO2 capture and elution.
3. Results
3.1. Plk3 phosphorylates Cdc25A at discrete sites in vitro
The family of Cdc25 phosphatases regulates the progression of cells through the cell cycle [35]. They in turn are regulated by phosphorylation that promotes their proteasome-mediated degradation [36–39]. Subcellular localization of Cdc25C is partly controlled by Plk3-mediated phosphorylation which helps target Cdc25C to the nucleus in late G2 where it presumably is able to dephosphorylate Cdk1 and promote transit from G2 into mitosis[31]. To explore whether Cdc25A might also serve as a substrate for Plk3, recombinant Cdc25A was incubated with recombinant Plk3 and γ-[32P] ATP in an in vitro kinase assay. As shown in Fig. 1A, Plk3 phosphorylated the full-length Cdc25A similar to Cdc25C and Chk2, two known substrates of Plk3. To better assess the site of Plk3 phosphorylation, a series of overlapping GST-Cdc25A fragments were prepared and tested as substrates for in vitro Plk3 kinase activity, with a focus on the carboxy half of Cdc25A, which contains its catalytic domain. Further analysis showed that Plk3 phosphorylates a fragment that extends from residue 250 to 524 (Cdc25A-CT) but not a fragment that extends from 250 to 501 or from 250 to 508, indicating that the phosphorylated residues reside between 508 and the carboxy terminus at 524 (Fig. 1B). There are two serines within this region which were mutated to a non-phosphorylatable alanine. In contrast to the wildtype sequence, the singly mutated versions, S513A or S519A, were poor substrates for Plk3 phosphorylation (Fig. 1C) indicating that these two residues are the Plk3 phosphorylation target sites. The location of the two phosphorylation sites in Cdc25A is shown in Fig. 1D.
Fig. 1.
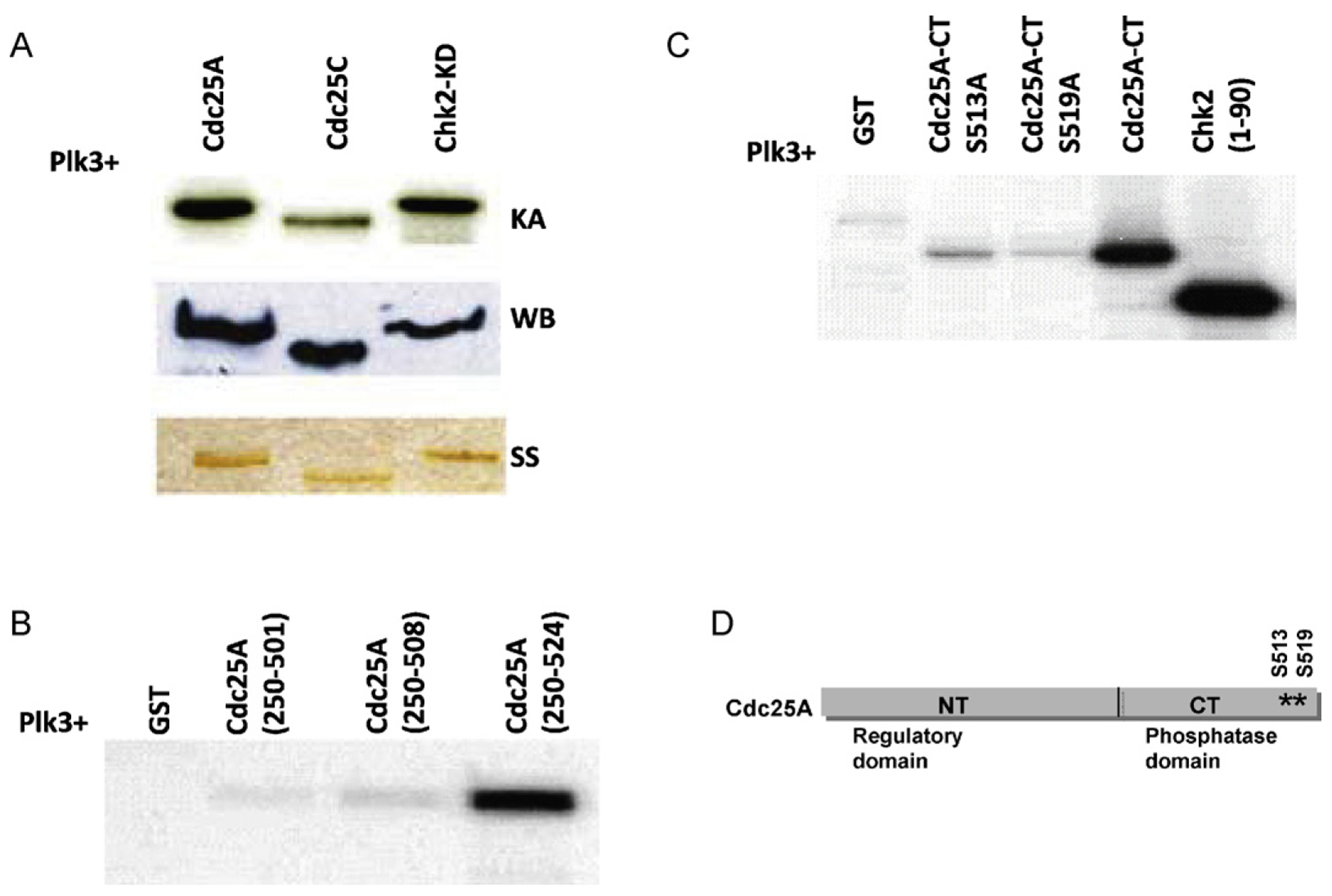
Plk3 phosphorylates Cdc25A on serines 513 and 519 in vitro. (A) GST fusion Plk3 protein was expressed in bacteria and used to phosphorylate either Cdc25A or known substrates of Plk3 (Cdc25C and Chk2) in a kinase assay (KA). The amounts of used substrates were checked by both Western blot (WB) and silver staining (SS). (B) Deletion mutants of Cdc25A show that the phosphorylation sites are within the last 16 amino acids of Cdc25A protein. (C) Mutation of S513 or S519 on Cdc25A abolishes Plk3 phosphorylation activity. (D) A schematic representation of Cdc25A protein showing the location of the two phosphorylated sites on the C-terminal part of the protein.
3.2. Mutation of phosphorylatable residues alter the stability of Cdc25A following IR treatment
To test whether mutation of these serines has physiological consequences, cDNAs encoding myc-tagged wildtype Cdc25A protein or mutant Cdc25A with serines 513 and 519 substituted with alanine or aspartate were transfected into HT1080 cells followed by exposure to 10 Gy IR. Protein expression of the transfected constructs was detected in unchallenged cells in all cases (Fig. 2). Following exposure to IR, however, wildtype Cdc25A and the mutant Cdc25A proteins containing the phosphomimetic S513D and S519D substitutions were rapidly degraded. The proteins with the non-phosphorylatable S513A and S519A substitutions remained stable. After IR treatment, Cdc25A undergoes proteasome-mediated degradation [36–39]. To determine whether or not the degradation of the aspartate-substituted proteins is dependent upon proteasome function, transfected cells were treated with 10 μM MG132, a proteasome inhibitor. Both wildtype and mutant variants were stabilized following IR treatment in the presence of MG132 (Fig. 2) supporting the contention that phosphorylation of serines 513 and 519 promotes proteasome-mediated degradation of Cdc25A. In aggregate, these data suggest that Cdc25A is a substrate for Plk3 kinase activity in vitro, that the target phosphorylation sites reside at the carboxy terminus of Cdc25A, and that phosphorylation of these sites may contribute to proteasome-dependent Cdc25A degradation after IR treatment.
Fig. 2.
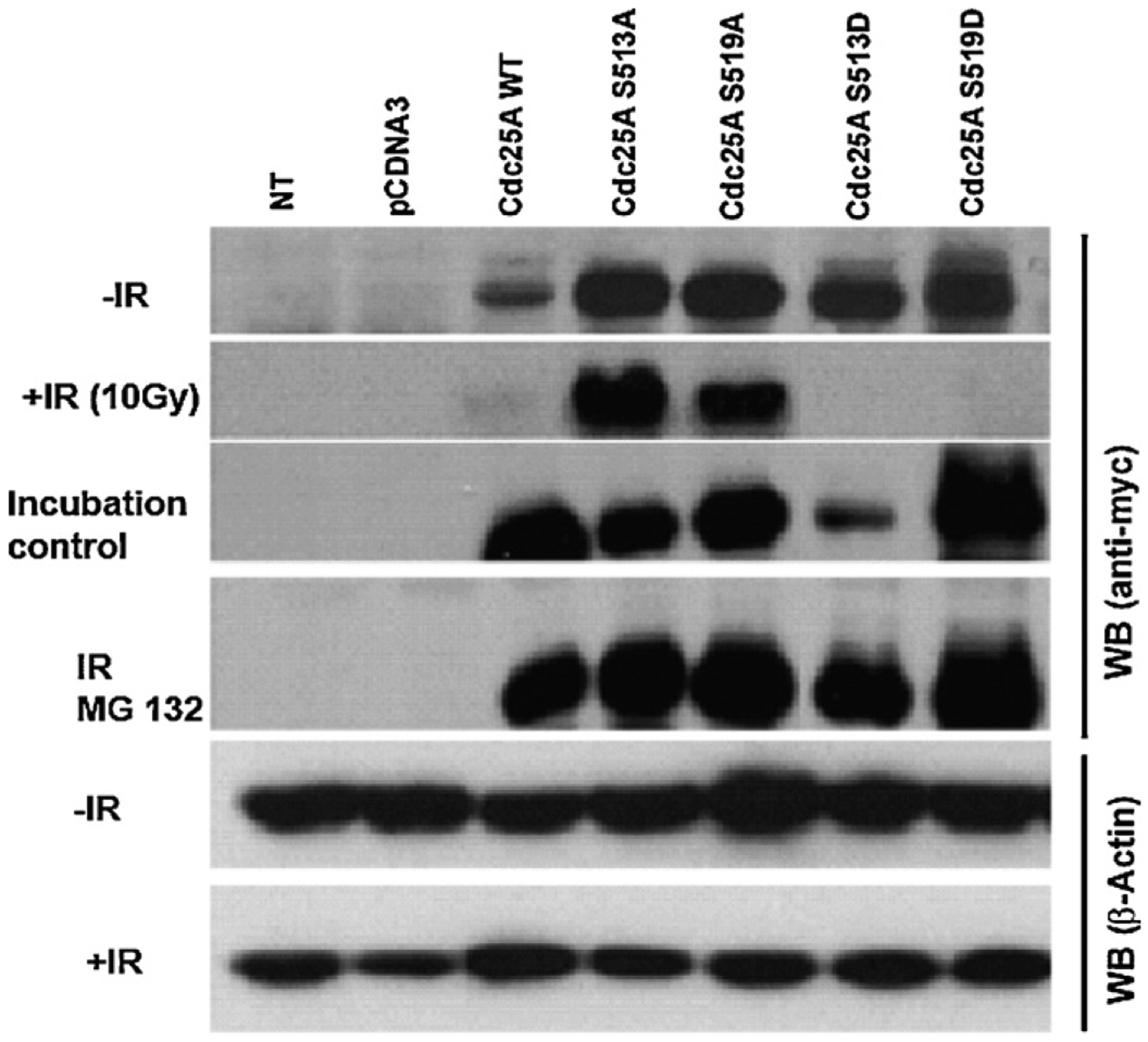
Mutation of serines 513 or 519 to alanine in Cdc25A prevents proteasome-mediated degradation following IR treatment. HT1080 cells were transfected with pCDNA3 plasmid control, pcDNA3-myc-Cdc25A-WT, and with each of the pcDNA3-myc-Cdc25A mutants or left non-transfected (NT), and were subjected to IR treatment or left unchallenged. Cell lysates were probed with antibody to Myc. An incubation control where cells were taken out of the incubator while other cells were being treated with IR was also included. Beta-actin was used as the loading control.
3.3. In vivo assessment of S513 and S519 phosphorylation
Since serines 513 and 519 are phosphorylated in vitro (Fig. 1), we sought to establish whether these residues are also phosphorylated in cells after DNA damage. Since no phosphospecific antibodies are available for these particular residues, the preferred approach was to detect the phosphorylated forms of these serines by mass spectrometry. The approach is somewhat problematic since phosphorylation of these residues, as well as other residues in Cdc25A, triggers its degradation. In an attempt to circumvent this problem, cells were first transfected with flag-tagged Cdc25A and treated with etoposide for 12 h or left untreated as a control. In both cases, the proteasome inhibitor MG 132 was subsequently added for 4 h to prevent Cdc25A degradation. These conditions were optimal for maximal Cdc25A degradation and for its subsequent accumulation following proteasome inhibition (Supplementary Fig. 1). Accumulated Cdc25A was immunoprecipitated using antibody to Cdc25A and protein A/G beads, the immunoprecipitates subjected to SDS PAGE, the separated proteins detected by Coomassie blue staining, and the bands eluted for identification and determination of phosphorylated residues (Supplementary Fig. 2).
Digestion of the protein and isolation of peptides suitable for sequencing by mass spectrometry and characterization of phosphorylation at serine 513 and 519 of cdc25A was problematic due to the sequence context around residues 513 and 519 which produced peptides that were either too small or hydrophilic for sequence mapping of both sites with any single endopeptidase. Digestion of separate samples with trypsin and chymotrypsin produced a peptide corresponding to serine 519 (EMYSR, trypsin digestion) and serine 513 (AGEKSKREMY, chymotrypsin digestion) that could be isolated and sequenced (Supplementary Figs. 3 and 4, respectively). There was, however, no evidence of phosphorylation of serines 513 or 519 in either challenged or control samples. Neither was there evidence of other serine residues being phosphorylated, including S82 and S88 which by antibody staining are known to be phosphorylated after DNA damage [46]. The only residue that was phosphorylated, both in control and challenged samples, was S178 (Supplementary Fig. 4), a CHK1 and CHK2 target (38). The S178 residue is phosphorylated primarily during the G1 and S phases of the cell cycle, and is not dependent on presence of DNA damage [38]. Detection of this phosphorylated serine indicates that the methodology should be sufficient to identify phosphorylated amino acids, particularly after further enrichment by TiO2 capture [50]. The inability to detect phosphorylated S513 and S519, as well as other residues known to be phosphorylated in response to DNA damage and detected by immunocytochemistry, suggests a very low stoichiometry in the cell under the conditions used that limit their detection by mass spectrometry. An alternative approach would be to co-express tagged Cdc25A and Plk3 with and without challenge and to assess the phosphorylation status of the ectopically expressed Cdc25A. Since overexpression of Plk3, however, is toxic to the cells, this approach did not seem feasible.
Fig. 4.
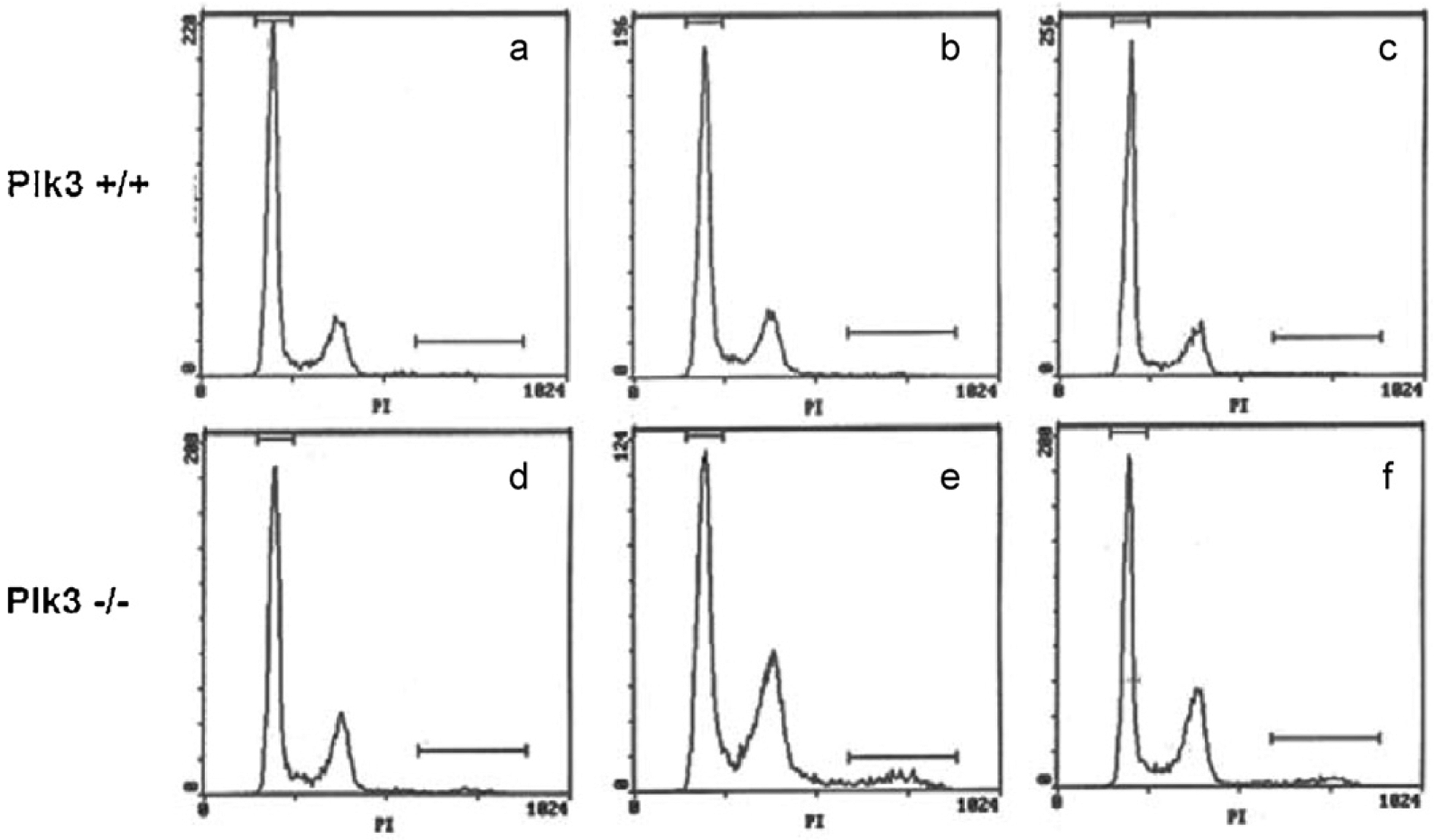
Defect in cell cycle distribution in cells deficient in Plk3 following serum deprivation and release. Cells from wildtype mice and from mice lacking Plk3 were grown on regular medium (a and d) or deprived of serum and subsequently released for 24 h (b and e) or 36 h (c and f). Cell cycle distribution was analyzed by flow cytometry.
3.4. Generation of a Plk3 knockout mouse
To further analyze the physiological role of Plk3 in development and in control of the cell cycle, we have created and exploited a Plk3 knockout mouse that is homozygous for Plk3 null alleles. The murine Plk3 gene is about 12 kb in length and contains 14 exons. The targeting strategy is depicted in Fig. 3A. The targeted allele lacks the Plk3 promoter and the first six exons, which have been replaced with a neo marker. Southern blot analysis confirmed correct targeting using genomic DNA extracted from both mouse tails and MEFs (Fig. 3B). Northern blot analysis with RNA from wildtype, heterozygous and homozygous knockout mice confirmed reduced levels of Plk3 mRNA in mice heterozygous at Plk3 and no detectable Plk3 mRNA in mice homozygous for the null allele (Fig. 3C). Western blots with extracts from cells homozygous for the Plk3 knockout allele, using all available commercial antibodies to Plk3, produced disturbing results. Every antibody tested displayed bands at approximately the expected size, even though it was apparent that Plk3 mRNA was absent (Fig. 3C). Further analysis demonstrated that the cross-reacting band is Plk5, a newly described polo-like kinase family member [15].
Fig. 3.
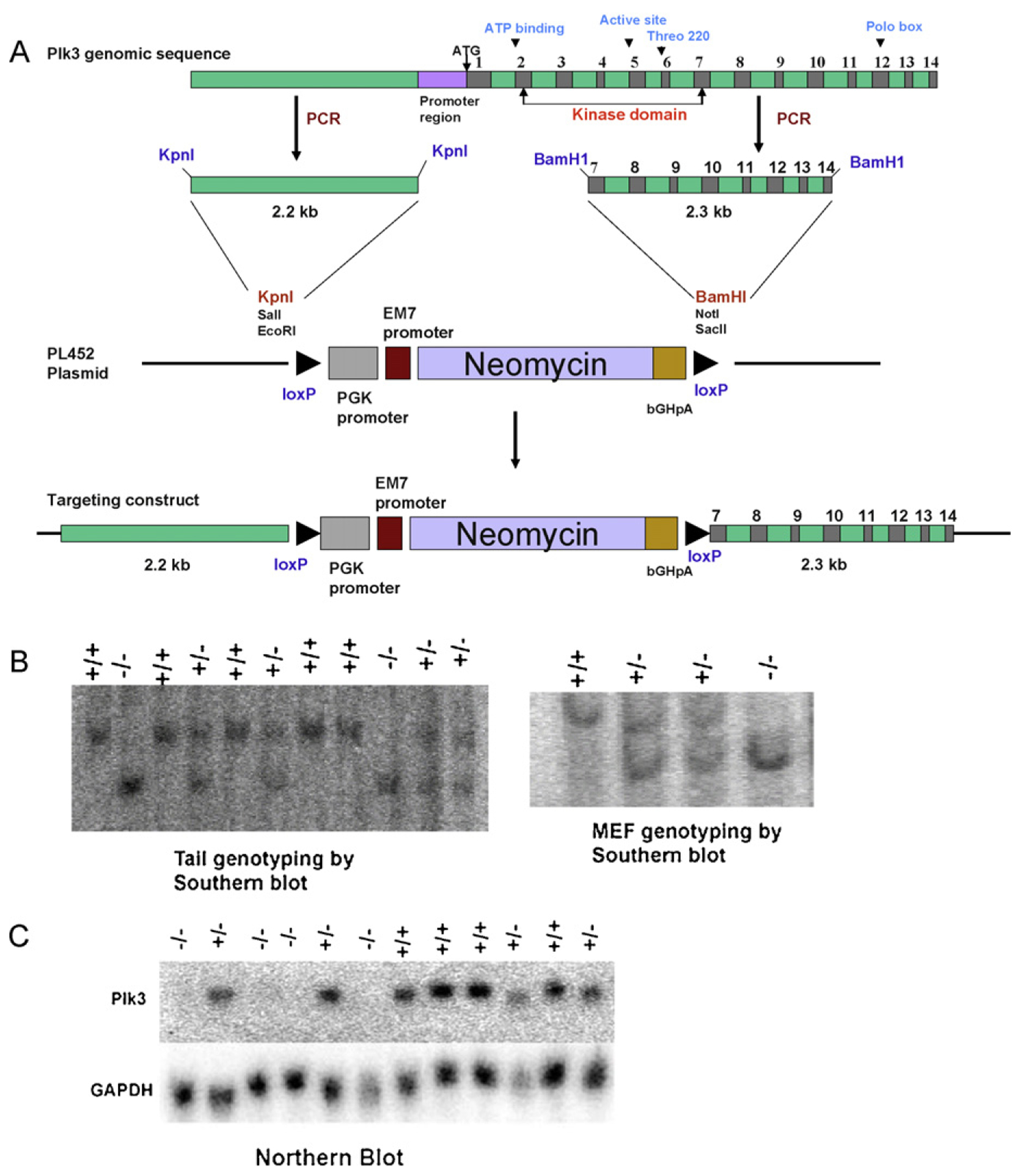
Generation of a Plk3 knockout mouse. (A) A schematic representation of the targeting construct. (B) The targeted clones were identified by Southern blot (tail and MEF genotyping) and (C) Northern blot (mRNA expression).
3.5. MEFs derived from Plk3−/− mice show cell cycle defects following release from serum deprivation
A recent report argued that Plk3 protein is necessary for the G1/S transition [25]. Following shRNA-mediated depletion of Plk3 from serum deprived cells, Plk3 appeared to be required for cyclin E expression and entry into S phase after re-administration of serum. It was suggested that Plk3 may regulate entry into S phase in part through interaction with Cdc25A [25]. To examine this proposition, MEFs derived from embryos homozygous for the Plk3 null allele were examined for cell cycle distribution following release from serum starvation. There was no obvious difference in cell cycle distribution between wildtype and Plk3-deficient MEFs (Fig. 4, panels a and d). However, when cells were arrested by serum deprivation and then released by addition of serum for 24 h (panels b and e), there was a clear accumulation of Plk3-deficient cells in G2/M compared with wildtype control. There was also a clear polyploid population that appeared at 24 h. At 36 h post release from serum starvation, the cell cycle had a more normal profile with fewer cells in G2/M and a smaller polyploid population (panels c and f). Given that Plk3 may play a role in mitosis, accumulation of cells in G2/M and the increase in polyploid population may be the result of a mitotic checkpoint activation due to a defect in spindle formation. The Plk3 protein localizes to centrosomes or spindle pole bodies and undergoes significant subcellular redistribution during the cell cycle. Deregulated activities of Plk3 often result in abnormalities in centrosome duplication, maturation, and/or microtubule dynamics (reviewed in Ref. [51]).
3.6. Thymocytes from Plk3-deficient mice have elevated levels of Cdc25A and a defective G1/S checkpoint in response to DNA damage
A predicted consequence of a lack of Plk3 activity is the stabilization and increased abundance of Cdc25A. This outcome is suggested in part by the altered behavior of Cdc25A in transfected cultured cells when the serine residues in Cdc25A that are phosphorylated by Plk3 were substituted by alanine. This hypothesis was tested by comparing the status of Cdc25A in cells from mice deficient in Plk3 and from mice with the wildtype allele. In thymocytes from Plk3 deficient mice, Cdc25A was minimally diminished, if at all, following treatment with etoposide, whereas Cdc25A degradation was clearly apparent after etoposide treatment in thymocytes in mice with wildtype Plk3 (Fig. 5A). These data suggest that Plk3 facilitates proteasome-mediated degradation of Cdc25A following the introduction of DNA damage, at least in some cell types such as thymocytes. To test whether the absence of Plk3 and consequent stabilization of Cdc25A translates into compromised cell cycle checkpoints following DNA damage, thymocytes from wildtype and mutant mice were either treated with etoposide or left untreated. Following etoposide treatment, the cell cycle distribution of cells from Plk3−/− mice was clearly perturbed with apparent retention of an intra-S phase checkpoint. These data are consistent with our observation that Cdc25A protein in Plk3−/− cells is less susceptible to proteasome-mediated degradation after DNA damage compared with wildtype cells, thereby allowing progression of cells from G1 into S phase (Fig. 5B).
Fig. 5.
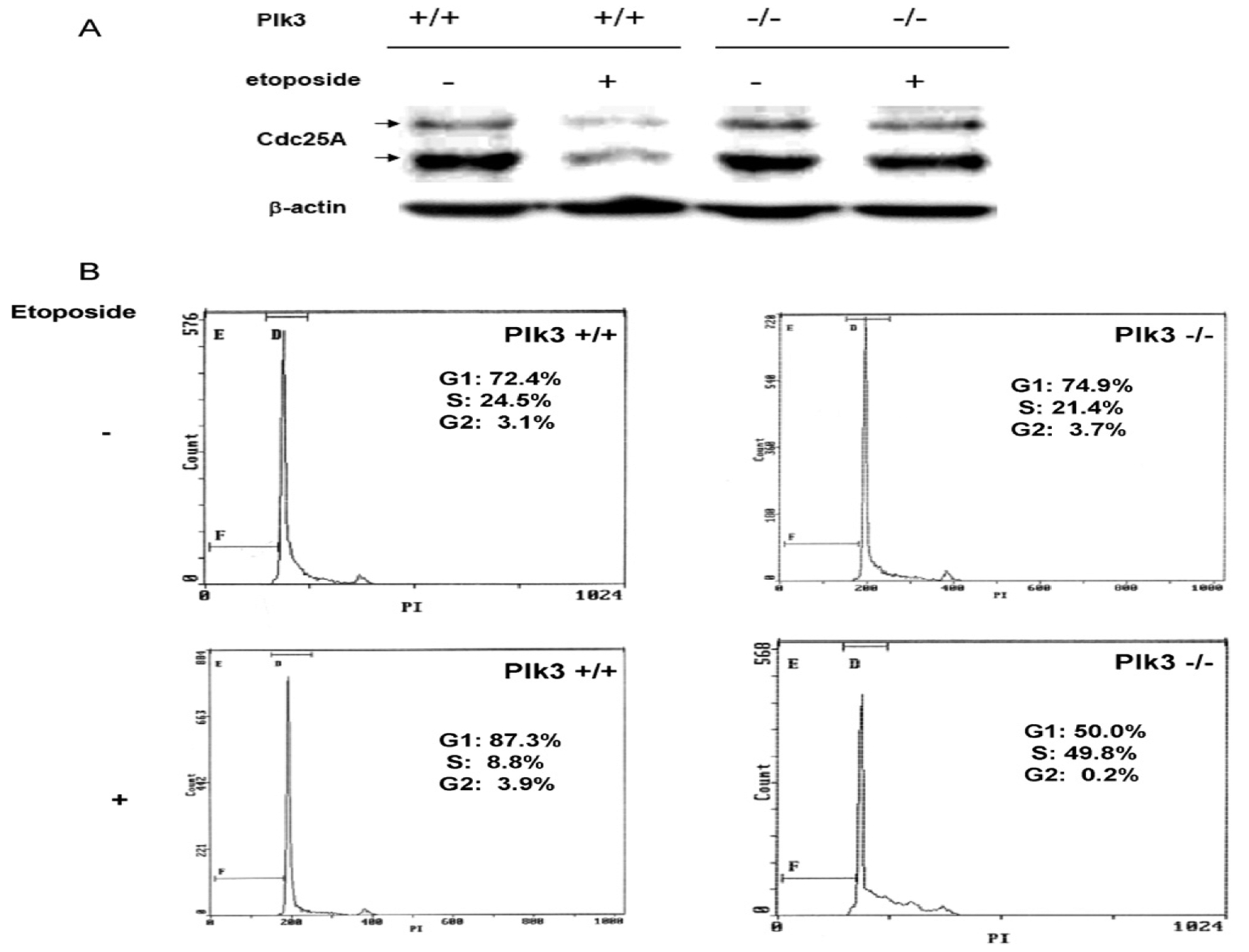
Thymocytes derived from Plk3−/− mice have a compromised G1/S checkpoint and are more resistant to degradation of Cdc25A than wildtype cells following DNA damage. Thymocytes derived from Plk3+/+ and Plk3−/− mice were left untreated or subjected to treatment with etoposide. (A) The Cdc25A level was analyzed by Western blot. (B) Cell cycle profile was determined by flow cytometry.
3.7. Loss of Plk3 and stabilization of Cdc25A does not predispose mutant mice to a significant increase in tumorigenesis.
The elevated abundance of Cdc25A due to its diminished proteasome-mediated degradation, coupled with cell cycle perturbation and increased genomic instability, predicts a higher incidence of spontaneous tumors in Plk3−/− mice. To test this prediction, mice heterozygous and homozygous at Plk3, as well as their wildtype counterparts, were maintained for up to two years and checked twice a week for palpable tumors. Around the 10th month (40 weeks) of age, mice of all genotypes started developing tumors (Fig. 6A) involving several different tissues. When analyzed regardless of gender, there was no statistically significant difference in tumor incidence between the three genotypes (Fig. 6A and C). When analyzed solely by gender, male mice had a higher incidence of tumors regardless of genotype and female wildtype mice had a lower tumor incidence than their female heterozygous and homozygous mutant counterparts. The fact that female mice deficient for Plk3 are slightly more prone to developing tumors than female wildtype mice suggests that spontaneous tumorigenesis in these mice may be partly hormone dependent. Aging mice that lack Plk3 are reported to have accelerated tumor development, larger tumor size, and more pronounced angiogenesis than their wildtype littermates [29]. The differences observed between this study and the previous study may be due to the difference in mouse strain background used in each case.
Fig. 6.
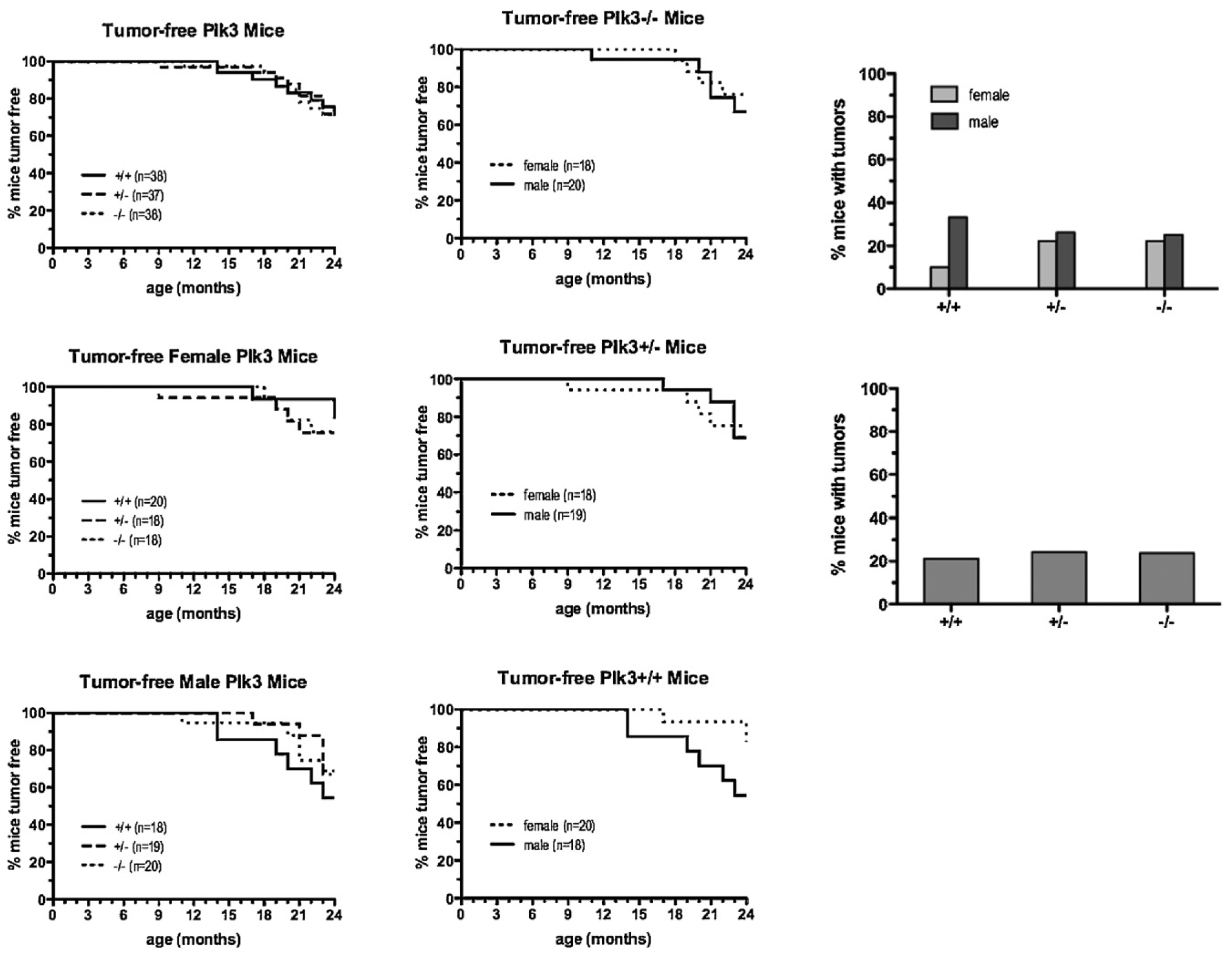
Development of spontaneous tumors in mice with wildtype and mutant Plk3. (A) Wildtype mice and mice heterozygous and homozygous for Plk3 were maintained for 24 months and monitored for palpable tumors twice a week. Mice were killed when tumors reached 1.0 cm in diameter or when mice appeared to be in distress or were found dead. In all cases, necropsy with histology of representative organs was performed, and tumors and tumor types were noted. (B) Comparison by gender of mice of each genotype that had developed spontaneous tumors by 24 months of age. (C) Comparison by gender (upper panel) or regardless of gender (lower panel) of mice of each genotype that had developed spontaneous tumors by 24 months of age.
4. Discussion
Our understanding of the biology of Cdc25A and its functions is expanding from an exclusive role as key regulator of the G1/S transition to broader roles in the cell cycle, with essential functions in mitosis as well as in cellular responses to DNA damage. The controlled degradation of Cdc25A in a cell cycle dependent manner, mainly in the S and G2 phases, as well as in response to DNA damage, is mediated by multiple phosphorylation events. These phosphorylation events lead to SCFβ-TrCP binding to Cdc25A and its proteasome-mediated degradation in late G1 and S phase. During late G2 and exit from mitosis, Cdc25A is degraded by the APC complex [36]. After DNA damage, Cdc25A is phosphorylated by Chk1 on serine 76 or Chk2 on serine 123 and thereby primed for subsequent phosphorylation by other kinases [36]. Several kinases including NEK11, GSK-β, ERK and casein kinase 1 [45–47,52] are among the kinases recently implicated in the phosphorylation within or proximal to the Cdc25A phosphodegron that binds SCFβ-TrCP which directs ubiquitinylation and degradation of Cdc25A [32,53].
Clearly, the regulation of Cdc25A degradation is complex since introduction of mutant non-phosphorylatable Cdc25A proteins (S76A or S76/123A) is not sufficient to overcome the S phase checkpoint [54], suggesting that other kinases or mechanisms, in addition to phosphorylation by Chk1 and Chk2, may contribute to S phase checkpoint activation. Here we present evidence that one such regulatory mechanism may be mediated by Plk3. We show that Plk3 phosphorylates Cdc25A and regulates its stability. The role of Plk3 in the DNA damage response is now well established. It is known that Plk3 is activated in response to DNA damage, phosphorylating and activating key proteins in the DNA damage checkpoint pathways, including p53 and Chk2 [27,28]. The site on p53 is serine 20, which is the same site targeted by Chk2 [27]. The principal site on Chk2 is serine 73, which serves as a priming phosphorylation that facilitates the activation phosphorylation by ATM on Chk2 threonine 68 [28]. Thus, these series of phosphorylation events constitute a regulatory circuit involving Cdc25A, ATM and Chk2.
The data in the current report support a role for Plk3 in the controlled degradation of Cdc25A in either the presence or absence of DNA damage. The Cdc25A protein is phosphorylated by multiple kinases at multiple residues during the cell cycle and in response to DNA damage. In the latter case, mutation of any one of the target serines or threonines can interfere with its proteasome-mediated degradation, which may account, in part, for its elevated level in many tumors [55]. The Plk3 kinase phosphorylates both the N-terminus [45] and C-terminus of Cdc25A in vitro. The present study shows that Plk3 phosphorylates two serine residues in positions 513 and 519 near the Carboxy terminus. Phosphomimetic substitution of S513 and S519 with aspartates promotes IR-mediated Cdc25A proteolysis in asynchronous cells. In support of these results, we have also shown that Cdc25A is more resistant to DNA damage-mediated degradation in Plk3-deficient thymocytes.
Consistent with a role for Plk3 in Cdc25A stability, serum-starved MEFs derived from Plk3-deficient mice had a greater proportion of cells in S phase and mitosis than their wildtype counterparts and displayed a polyploid population. The data suggest an accumulation of Cdc25A in S phase, leading to unscheduled replication, a hallmark of proto-oncogene overexpression [56]. This stabilization resulted in loss of the G1/S checkpoint and activation of an intra-S checkpoint.
It is worth noting that serines 513 and 519 on Cdc25A that are phosphorylated by Plk3 are adjacent to the cyclinB/cdk1 docking domain at residues K514 and R520. These residues are important for Cdc25A/cyclinB/cdk1 complex interaction [38] and are in close proximity to threonine 507, a Chk1 phosphorylation site involved in 14-3-3 binding [38]. Binding of 14-3-3 protein to this region appears to block Cdc25A from functionally interacting with the cyclinB/cdk1 complex and probably with other cyclin/cdk complexes. One scenario for regulatory phosphorylation is that Plk3 phosphorylates Cdc25A in concert with Chk1 to allow subsequent binding of 14-3-3 that will interfere with Cdc25A interaction with cyclin/cdk complexes during the G1 and S phases of the cell cycle. Modulation of such interactions will be important in the “opening/closing” of Cdc25A protein and subsequent binding and release of ubiquitin ligase, which targets Cdc25A for degradation (Fig. 7). The precise involvement of each of the various players in the DNA damage response pathway, however, and the mechanisms by which they interact to regulate controlled proteolysis of Cdc25A following DNA damage, remain unclear.
Fig. 7.
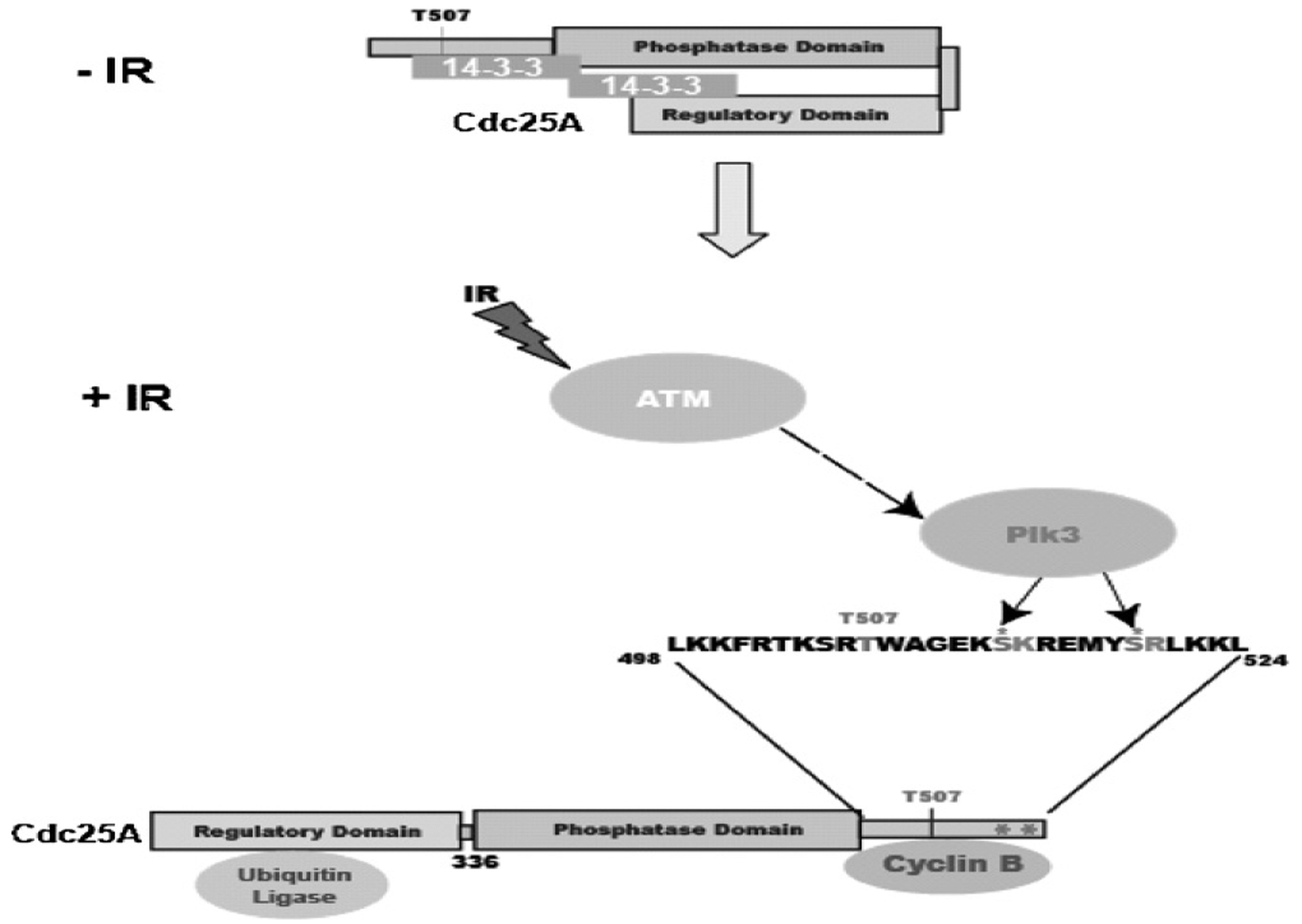
A model depicting the regulation of Cdc25A degradation by Plk3. Plk3 phosphorylates serines 513 and 519 which are adjacent to the cyclinB/cdk1 docking site (K514 and R520). This phosphorylation, together with Chk1 phosphorylation of T507 allows 14–3–3 binding and prevents cyclinB/cdk1 from binding to Cdc25A. 14–3–3 binding leads to an open conformation of Cdc25A that allows binding of the ubiquitin ligase and subsequent degradation of the protein.
A previous report has described a tumorigenic phenotype in Plk3 deficient mice in which the Plk3 gene had been ablated in a manner very similar to that described here [29]. The absence of tumors in the Plk3 knockout mice described in this report, therefore, is somewhat disturbing. There are several explanations that may account for this discrepancy. Firstly, it is not uncommon for mice with the same gene ablated, but of different strain backgrounds, to display a different phenotype. Secondly, an animal’s phenotype can be affected by its intestinal flora, and indirectly by its diet, dictating the propensity for developing tumors or other disease [57,58]. Lastly, it is possible that other kinases within the Plk family may compensate for the absence of Plk3 to reduce the risk of developing tumors. Although it is apparent that under certain circumstances Plk3 deficient mice are at elevated risk for developing tumors, it is clear that this is not always the case.
Supplementary Material
Acknowledgements
This work was supported in part by NIH grants R03 ES015307 to EMB, R01 ES012695 and R01 ES016625 to PJS, and the Center for Environmental Genetics P30-ES006096. DLM was supported in part by training grant T32 ES07250. The authors thank Sandy Schwemberger and Dr. George Babcock, Shriners Hospital for Children, Cincinnati, for their help with flow cytometry.
Footnotes
Conflict of interest
None.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.mrfmmm.2011.02.006.
References
- [1].Glover DM, Hagan IM, Tavares AAM, Polo-like kinases: a team that plays throughout mitosis, Genes Dev. 12 (1998) 3777–3787. [DOI] [PubMed] [Google Scholar]
- [2].Barr FA, Sillje HH, Nigg EA, Polo-like kinases and the orchestration of cell division, Nat. Rev. Mol. Cell Biol 5 (2004) 429–440. [DOI] [PubMed] [Google Scholar]
- [3].Lane HA, Nigg EA, Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes, J. Cell Biol 135 (1996) 1701–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Qian YW, Erikson E, Li C, Maller JL, Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis, Mol. Cell. Biol 18 (1998) 4262–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moutinho-Santos T, Sampaio P, Amorim I, Costa M, Sunkel CE, In vivo localisation of the mitotic POLO kinase shows a highly dynamic association with the mitotic apparatus during early embryogenesis in Drosophila, Biol. Cell 91 (1999) 585–596. [PubMed] [Google Scholar]
- [6].Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH, Polo-like kinase-1 is a target of the DNA damage checkpoint, Nat. Cell Biol 2 (2000) 672–676. [DOI] [PubMed] [Google Scholar]
- [7].Bahassi EM, Conn CW, Myer DL, Hennigan RF, McGowan CH, Sanchez Y, Stambrook PJ, Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways, Oncogene 21 (2002) 6633–6640. [DOI] [PubMed] [Google Scholar]
- [8].Xie S, Wu H, Wang Q, Kunicki J, Thomas RO, Hollingsworth RE, Cogswell J, Dai W, Genotoxic stress-induced activation of Plk3 is partly mediated by Chk2, Cell Cycle 1 (2002) 424–429. [DOI] [PubMed] [Google Scholar]
- [9].Llamazares S, Moreira A, Tavares A, Girdham C, Spruce BA, Gonzalez C, Karess RE, Glover DM, Sunkel CE, Polo encodes a protein kinase homolog required for mitosis in Drosophila, Genes Dev. 5 (1991) 2153–2165. [DOI] [PubMed] [Google Scholar]
- [10].Sunkel CE, Glover DM, Polo, a mitotic mutant of Drosophila displaying abnormal spindle poles, J. Cell Sci 89 (1988) 25–38. [DOI] [PubMed] [Google Scholar]
- [11].Logarinho E, Sunkel CE, The Drosophila POLO kinase localises to multiple compartments of the mitotic apparatus and is required for the phosphorylation of MPM2 reactive epitopes, J. Cell Sci 111 (1998) 2897–2909. [DOI] [PubMed] [Google Scholar]
- [12].Song S, Grenfell TZ, Garfield S, Erikson RL, Lee KS, Essential function of the polo box of Cdc5 in subcellular localization and induction of cytokinetic structures, Mol. Cell. Biol 20 (2000) 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ohkura H, Hagan IM, Glover DM, The conserved Schizosaccharomyces pombe kinase plo1, required to form a bipolar spindle, the actin ring, and septum, can drive septum formation in G1 and G2 cells, Genes Dev. 9 (1995) 1059–1073. [DOI] [PubMed] [Google Scholar]
- [14].Lowery DM, Lim D, Yaffe MB, Structure and function of Polo-like kinases, Oncogene 24 (2005) 248–259. [DOI] [PubMed] [Google Scholar]
- [15].Andrysik Z, Bernstein WZ, Deng L, Myer DL, Li YQ, Tischfield JA, Stambrook PJ, Bahassi EM, The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus, Nucleic Acids Res. 38 (2010) 2931–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee KS, Grenfell TZ, Yarm FR, Erikson RL, Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk, Proc. Natl. Acad. Sci. U.S.A 95 (1998) 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van de Weerdt BC, Littler DR, Klompmaker R, Huseinovic A, Fish A, Perrakis A, Medema RH, Polo-box domains confer target specificity to the Polo-like kinase family, Biochim. Biophys. Acta 1783 (2008) 1015–1022. [DOI] [PubMed] [Google Scholar]
- [18].Elia AE, Cantley LC, Yaffe MB, Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates, Science 299 (2003) 1228–1231. [DOI] [PubMed] [Google Scholar]
- [19].Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB, The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain, Cell 115 (2003) 83–95. [DOI] [PubMed] [Google Scholar]
- [20].Nigg EA, Polo-like kinases: positive regulators of cell division from start to finish, Curr. Opin. Cell Biol 10 (1998) 776–783. [DOI] [PubMed] [Google Scholar]
- [21].Carmena M, Riparbelli MG, Minestrini G, Tavares AM, Adams R, Callaini G, Glover DM, Drosophila polo kinase is required for cytokinesis, J. Cell Biol 143 (1998) 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lin CY, Madsen ML, Yarm FR, Jang YJ, Liu X, Erikson RL, Peripheral Golgi protein GRASP65 is a target of mitotic polo-like kinase (Plk) and Cdc2, Proc. Natl. Acad. Sci. U.S.A 97 (2000) 12589–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sutterlin C, Lin CY, Feng Y, Ferris DK, Erikson RL, Malhotra V, Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis, Proc. Natl. Acad. Sci. U.S.A 98 (2001) 9128–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shirayama M, Zachariae W, Ciosk R, Nasmyth K, The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae, EMBO J. 17 (1998) 1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zimmerman WC, Erikson RL, Polo-like kinase 3 is required for entry into S phase, Proc. Natl. Acad. Sci. U.S.A 104 (2007) 1847–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, Fukuzawa M, Nakagawara A, Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation, J. Biol. Chem 279 (2004) 25549–25561. [DOI] [PubMed] [Google Scholar]
- [27].Xie S, Wu H, Wang Q, Cogswell JP, Husain I, Conn C, Stambrook P, Jhanwar-Uniyal M, Dai W, Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway, J. Biol. Chem 276 (2001) 43305–43312. [DOI] [PubMed] [Google Scholar]
- [28].Bahassi EM, Myer DL, McKenney RJ, Hennigan RF, Stambrook PJ, Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage, Mutat. Res 596 (2006) 166–176. [DOI] [PubMed] [Google Scholar]
- [29].Yang Y, Bai J, Shen R, Brown SA, Komissarova E, Huang Y, Jiang N, Alberts GF, Costa M, Lu L, Winkles JA, Dai W, Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions, Cancer Res. 68 (2008) 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chehab NH, Malikzay A, Appel M, Halazonetis TD, Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53, Genes Dev. 14 (2000) 278–288. [PMC free article] [PubMed] [Google Scholar]
- [31].Bahassi EM, Hennigan RF, Myer DL, Stambrook PJ, Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation, Oncogene 23 (2004) 2658–2663. [DOI] [PubMed] [Google Scholar]
- [32].Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF, Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage, Nature 426 (2003) 87–91. [DOI] [PubMed] [Google Scholar]
- [33].Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J, The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis, Nature 410 (2001) 842–847. [DOI] [PubMed] [Google Scholar]
- [34].Bernardi R, Lieberman DA, Hoffman B, Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation, Oncogene 19 (2000) 2447–2454. [DOI] [PubMed] [Google Scholar]
- [35].Boutros R, Dozier C, Ducommun B, The when and whereas of CDC25 phosphatases, Curr. Opin. Cell Biol 18 (2006) 185–191. [DOI] [PubMed] [Google Scholar]
- [36].Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF, Dual mode of degradation of Cdc25 A phosphatase, EMBO J. 21 (2002) 4875–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Busino L, Chiesa M, Draetta GF, Donzelli M, Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis, Oncogene 23 (2004) 2050–2056. [DOI] [PubMed] [Google Scholar]
- [38].S Chen M, Ryan CE, Piwnica-Worms H, Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14-3-3 binding, Mol. Cell. Biol 23 (2003) 7488–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J, Rapid destruction of human Cdc25A in response to DNA damage, Science 288 (2000) 1425–1429. [DOI] [PubMed] [Google Scholar]
- [40].Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF, Human Cdc25A inactivation in response to S phase inhibition and its role in preventing premature mitosis, EMBO Rep. 1 (2000) 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Blomberg I, Hoffmann I, Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases, Mol. Cell. Biol 19 (1999) 6183–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bartek J, Lukas J, Pathways governing G1/S transition and their response to DNA damage, FEBS Lett. 490 (2001) 117–122. [DOI] [PubMed] [Google Scholar]
- [43].Zhao H, Watkins JL, Piwnica-Worms H, Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints, Proc. Natl. Acad. Sci. U.S.A 99 (2002) 14795–14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kristjansdottir K, Rudolph J, CDC25 phosphatases and cancer, Chem. Biol 11 (2004) 1043–1051. [DOI] [PubMed] [Google Scholar]
- [45].Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H, GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers, Cancer Cell 13 (2008) 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Melixetian M, Klein DK, Sørensen CS, Helin K, NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint, Nat. Cell Biol 11 (2009) 1247–1253. [DOI] [PubMed] [Google Scholar]
- [47].Isoda M, Kanemori Y, Nakajo N, Uchida S, Yamashita K, Ueno H, Sagata N, The extracellular signal-regulated kinase-mitogen-activated protein kinase pathway phosphorylates and targets Cdc25A for SCF beta-TrCP-dependent degradation for cell cycle arrest, Mol. Biol. Cell 20 (2009) 2186–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, Zou X, Ghosh AK, Varga J, Draetta GF, Kiyokawa H, Transforming growth factor beta facilitates beta-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner, Mol. Cell. Biol 25 (2005) 3338–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Eismann T, Huber N, Shin T, Kuboki S, Galloway E, Wyder M, Edwards MJ, Greis KD, Shertzer HG, Fisher AB, Lensch AB, Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice, Am. J. Physiol. Gastrointest. Liver Physiol 296 (2009) G266–G274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Thingholm TE, Jensen ON, Robinson PJ, Larsen MR, SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides, Cell Proteomics 7 (2008) 661–671. [DOI] [PubMed] [Google Scholar]
- [51].Dai W, Cogswell JP, Polo-like kinases and the microtubule organization center: targets for cancer therapies, Prog. Cell Cycle Res 5 (2003) 327–334. [PubMed] [Google Scholar]
- [52].Honaker Y, Piwnica-Worms H, Casein kinase 1 functions as both penultimate and ultimate kinase in regulating Cdc25A destruction, Oncogene 29 (2010) 3324–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW, SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase, Genes Dev. 17 (2003) 3062–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ Jr., D.V. Bulavin, Regulation of human Cdc25A stability by Serine 75 phosphorylation is not sufficient to activate a S phase checkpoint, Cell Cycle 2 (2003) 473–478. [PubMed] [Google Scholar]
- [55].Boutros R, Lobjois V, Ducommun B, CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 7 (2007) 495–507. [DOI] [PubMed] [Google Scholar]
- [56].Halazonetis TD, Gorgoulis VG, Bartek J, An oncogene-induced DNA damage model for cancer development, Science 319 (2008) 1352–1355. [DOI] [PubMed] [Google Scholar]
- [57].Fox JG, Feng Y, Theve EJ, Raczynski AR, Fiala JLA, Doernte AL, Williams M, et al. , Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens, Gut 59 (2010) 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria, Cell 139 (2009) 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.