

Abstract
Epigenetic mechanisms such as histone modifications play critical roles in adaptive tuning of chromatin structures. Profiling various histone modifications at the genome scale using tissues from animal and human samples is an important step for functional studies of epigenomes and epigenomics-based precision medicine. Because the profile of a histone mark is highly specific to a cell type, cell isolation from tissues is often necessary to generate a homogeneous cell population and such operation tends to yield a low number of cells. In addition, high-throughput processing is often desirable due to the multiplexity of histone marks of interest and the large quantity of samples in a hospital setting. In this protocol, we describe detailed instructions on device fabrication, setup, and operation of microfluidic oscillatory washing-based chromatin immunoprecipitation followed by sequencing (MOWChIP-seq) for profiling histone modifications using as few as 100 cells per assay with a throughput as high as 8 assays in a run. MOWChIP-seq operation involves flowing of chromatin fragments through a packed bed of antibody-coated beads followed by vigorous microfluidic oscillatory washing. Our process is semi-automated for reduced labor and improved reproducibility. Using one 8-unit device, it takes 2 d to produce 8 sequencing libraries from chromatin samples. The technology is scalable. We used the protocol to study a number of histone modifications in various types of mouse and human tissues. The protocol can be conducted by a user who is familiar with molecular biology procedures and has basic engineering skills.
Introduction
The epigenome refers to the genome-wide molecular machinery that alters gene expression without changing DNA sequence. Epigenomic mechanisms form a critical layer in addition to genomic variations for regulating gene activities and phenotypes. Epigenetic variations such as histone modification, DNA methylation, and non-coding RNA regulation critically influence chromosome conformation and genome stability1. Aberrant epigenomic features are frequently involved in numerous human disorders ranging from cancer to neurodevelopmental defects1.
The comparison of disease and reference epigenomes provides the basis for functional annotation of epigenomic features and use of the knowledge for precision medicine2. The current efforts by groups such as the US NIH Roadmap Epigenomics Mapping Consortium are focused on establishing reference epigenomes for various tissues and cell types under normal development and in disease states. Once these references and the connections between epigenomic features and disease states are established, patients can be profiled and stratified for a personalized course of treatment. In current reference profiling efforts and future precision medicine tests on patients, only small quantities of tissue samples can be obtained. Furthermore, the cell-type specificity of epigenomes requires isolation of a homogeneous cell population of the same type. This practice further limits the quantity of the sample available for testing.
Genome-wide mapping of histone modifications is critical for understanding epigenetic dynamics during normal development and disease processes3,4. In eukaryotes, the nucleoprotein complex of chromatin is composed of nucleosomes that consist of a segment of DNA (147 bp) wrapped around a histone octamer. The histones H2A, H2B, H3, and H4 may be chemically modified or replaced by variants. Post-translational modifications such as methylation, acetylation, phosphorylation, and ubiquitination take place on histones and influence DNA-histone interactions in a profound way to form “histone code”3. Hundreds of histone modifications have been identified5 and at least tens of them are of known importance to epigenomic regulation6,7. Given the scarce amount of tissue samples typically available for examination and the large number of histone marks that are often of interest, it is critical to have low-input and high-throughput tools for mapping genome-wide histone modifications.
Chromatin immunoprecipitation coupled with next generation sequencing (ChIP-seq) has been the method of choice for mapping genome-wide histone modifications. In the ChIP-seq process, chromosomes are first released from cells and broken into fragments (200–500 bp) by either enzymatic digestion or sonication. Immunomagnetic beads coated with antibodies targeting a specific histone mark are mixed with the chromatin fragments and targeted chromatin fragments get pulled down to the bead surface during ChIP. ChIP DNA is then released from the bead surface and collected for library preparation and sequencing.
Applications of the method
MOWChIP-seq, or microfluidic oscillatory washing-based ChIP-seq, is a low-input method on a microfluidic platform developed in our lab8. The microfluidic platform offers special advantages for manipulating microscale immunomagnetic beads during the binding and washing steps that are critical for highly efficient ChIP. In MOWChIP-seq, we create a packed bed of immunomagnetic beads in a microfluidic chamber for flow-through adsorption of chromatin fragments. The highly efficient adsorption is followed by oscillatory washing that effectively removes nonspecific adsorption. The process typically yields ChIP DNA that is equivalent to 5–12% of the total genomic DNA with high enrichment. We produce ChIP-seq datasets with quality that is comparable to those generated by conventional methods in terms of peak number and correlation between replicates, using as few as 100 cells per assay.
We have applied MOWChIP-seq to map various genome-wide histone modifications with low input (100–10,000 cells per assay) and high throughput (8 assays in parallel). The MOWChIP operation is based on manipulation of chromatin fragments, thus in principle our method is not limited to a specific cell type or histone mark. The method has been practiced in our lab by various users (8 in total) to probe a number of histone marks (H3K4me3, H3K27ac, H3K27me3, H3K9me3, H3K36me3, and H3K79me2) in various cell types (GM12878 human lymphoblastoid cell line, U251 human glioblastoma cell line, THP-1 human monocytic cell line, mouse monocytes, mouse mammary, human breast, mouse and human brain tissues), starting with cells or nuclei. All the experiments follow the basic principles and procedures described in our original publication8, while we have updated and optimized the technology for user-friendliness and high-throughput operation in this protocol. In principle, MOWChIP-seq can also be used to profile binding of RNA polymerase II and transcription factors. The specific operational conditions for profiling a chromatin-binding factor may require additional optimization.
Comparison with other methods
Conventional ChIP-seq methods require millions of cells per assay. Nano-ChIP (10,000 cells per assay)9, LinDA (5000 cells per assay)10, iChIP (500–1000 cells per assay)11, MOWChIP-seq (100 cells per assay)8, and surfaceChIP-seq (30–100 cells per assay)12 have been developed in recent years to decrease the required input. Drop-ChIP was demonstrated to conduct ChIP-seq at the single cell level13. As an alternative to ChIP, CUT&RUN was developed to map histone modifications by cutting and releasing DNA fragments that interact with antibody-targeted histones into supernatant, requiring as few as 100 cells in the initial works14,15. Recently, CUT&RUN16 and its modified version (CUT&Tag)17 have been used to profile single cells. Another IP-free technology, chromatin integration labeling (ChIL-seq), produced data with 100–1000 cells and involves immunostaining, Tn5 transposition, in-situ transcription, and RNA-mediated linear amplification18. In addition to histone marks, LinDA10, iChIP11, CUT&RUN14, CUT&Tag17, ChIL-seq18 have also been used to profile chromatin-binding factors.
Unfortunately, Drop-ChIP only generates about 1000 unique reads on each cell and the coverage of the genome is very sparse13. Among the competing technologies requiring similar input as MOWChIP-seq, iChIP requires complex chemistry for indexing of various samples11 and surfaceChIP-seq (also developed by us) requires surface immobilization of the antibody inside a microfluidic channel12. In contrast, MOWChIP-seq demands setup of hardware for control of the system but requires no extra chemistry or molecular biology compared to conventional ChIP. Compared to low-input ChIP-seq technologies, histone modification data obtained by CUT&RUN appear to exhibit lower correlation with ChIP-seq data taken using conventional approaches (e.g. ENCODE data)14,15. The correlation also does not improve with increasing cell numbers (in the range of 100–6000 cells per assay)15. ChIL-seq involves complicated molecular process and many steps18. More importantly, the use of Tn5 transposition creates bias associated with chromatin openness and thus complicates the interpretation of the data. Nevertheless, these IP-free methods provide powerful insights into the epigenomic states.
Compared to various competing methods, MOWChIP-seq offers several distinct advantages. First, MOWChIP-seq generates high-quality data that rival those obtained with conventional methods using millions of cells per assay. For example, our MOWChIP-seq data sets on H3K4me3 produce 10,000–30,000 peaks and 2–5 million unique reads when 100–1000 cells per assay are used. For the hard-to-profile H3K27me3 mark, we produce 13,000 peaks and 12 million unique reads using 1000 nuclei. The data sets also present high reproducibility among replicates, with Pearson’s correlation coefficients larger than 0.90 for cell numbers larger than 1000 per assay. Thus, MOWChIP-seq facilitates production of data sets using scarce cell samples for use as reference epigenomes and patient profiles19,20. Second, in contrast to the manual procedures used in the vast majority of the competing technologies, chromatin immunoprecipitation and washing in MOWChIP-seq are semi-automated. Other than a couple of interventions to change input reagents, the operation of the microfluidic devices is largely programed and automated. This feature dramatically decreases the amount of labor required and improves data reproducibility. Third, MOWChIP-seq is a scalable technology and supports high-throughput processing of a large number of samples. We detail the operation of 8 MOWChIP units in parallel in this protocol. There is no barrier to running a large number of these devices in parallel with additional hardware. Finally, MOWChIP-seq implements simple and well-established immunoprecipitation chemistry to minimize burden associated with chemical and molecular manipulation.
Experimental Design
The entire process includes setup of the pneumatic control system, fabrication of MOWChIP devices, antibody coating of the IP beads, chromatin preparation, MOWChIP, ChIP DNA isolation, and library preparation (Fig. 1). In order to conduct MOWChIP-seq, a control system needs to be set up for running 8 microfluidic devices simultaneously (1 d). The control system is durable for running MOWChIP-seq experiments for a long period of time with very little maintenance required. For a MOWChIP-seq experiment that generates 8 ChIP-seq data sets, the microfluidic devices are first fabricated (0.5 d) and then the microfluidics-based ChIP process takes 1 d and the library preparation requires 1 d before the samples are ready for sequencing.
Figure 1 |.
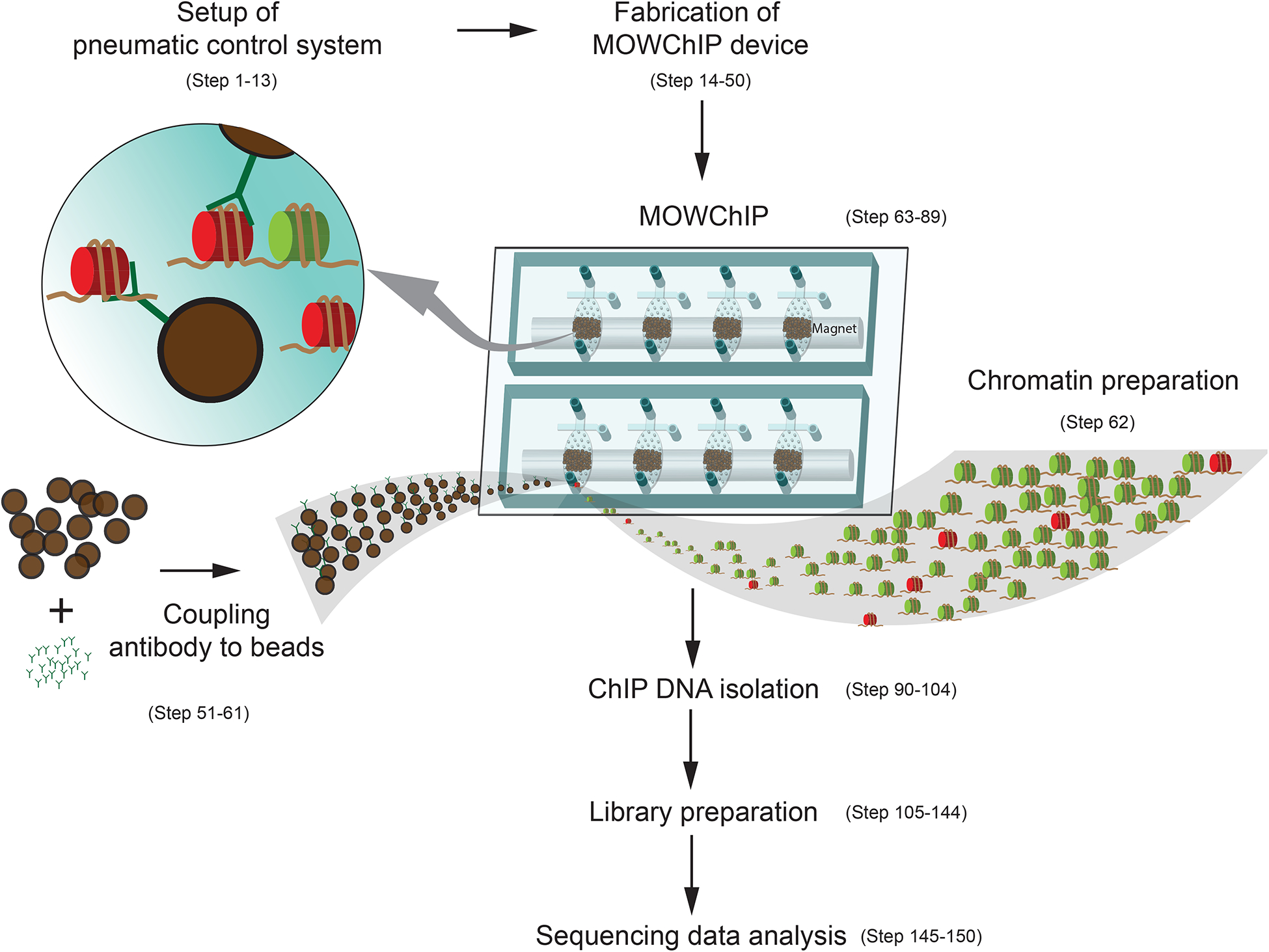
A schematic description of the MOWChIP-seq protocol.
Setup of the pneumatic control system.
A programmable pneumatic control system (Fig. 2, Fig. 3) was built for operation of a sieve valve (a monolithic pneumatic microvalve on the microfluidic device) and the microfluidic oscillatory washing. Our setup is similar to what is typically used for operating pneumatic microvalves21 (also known as Quake-style valves22,23). The mechanical and electrical connections are described in Fig. 2 and Fig. 3. The electrical part of the setup (Fig. 2b and 2d) consists of a computer, a data acquisition (DAQ) card, relays, and solenoid valves. The computer runs LabVIEW programs (Supplementary Data 1 and 2) during MOWChIP process and operates the solenoid valves via a DAQ card and relays. The mechanical connection schematics (Fig. 2c and 2e) show the connections among microfluidic devices, solenoid valves, pressure gauges and regulators, and pressure source via tubing and pipes. Operation of one single MOWChIP device (Fig. 2b and Fig. 2c) requires 3 solenoid valves, 3 relays, and 2 sets of pressure gauges/regulators. With 24 solenoid valves, 24 relays, 9 sets of pressure gauges/regulators, we are able to run 8 MOWChIP devices in parallel (Fig. 2d and 2e).
Figure 2 |.
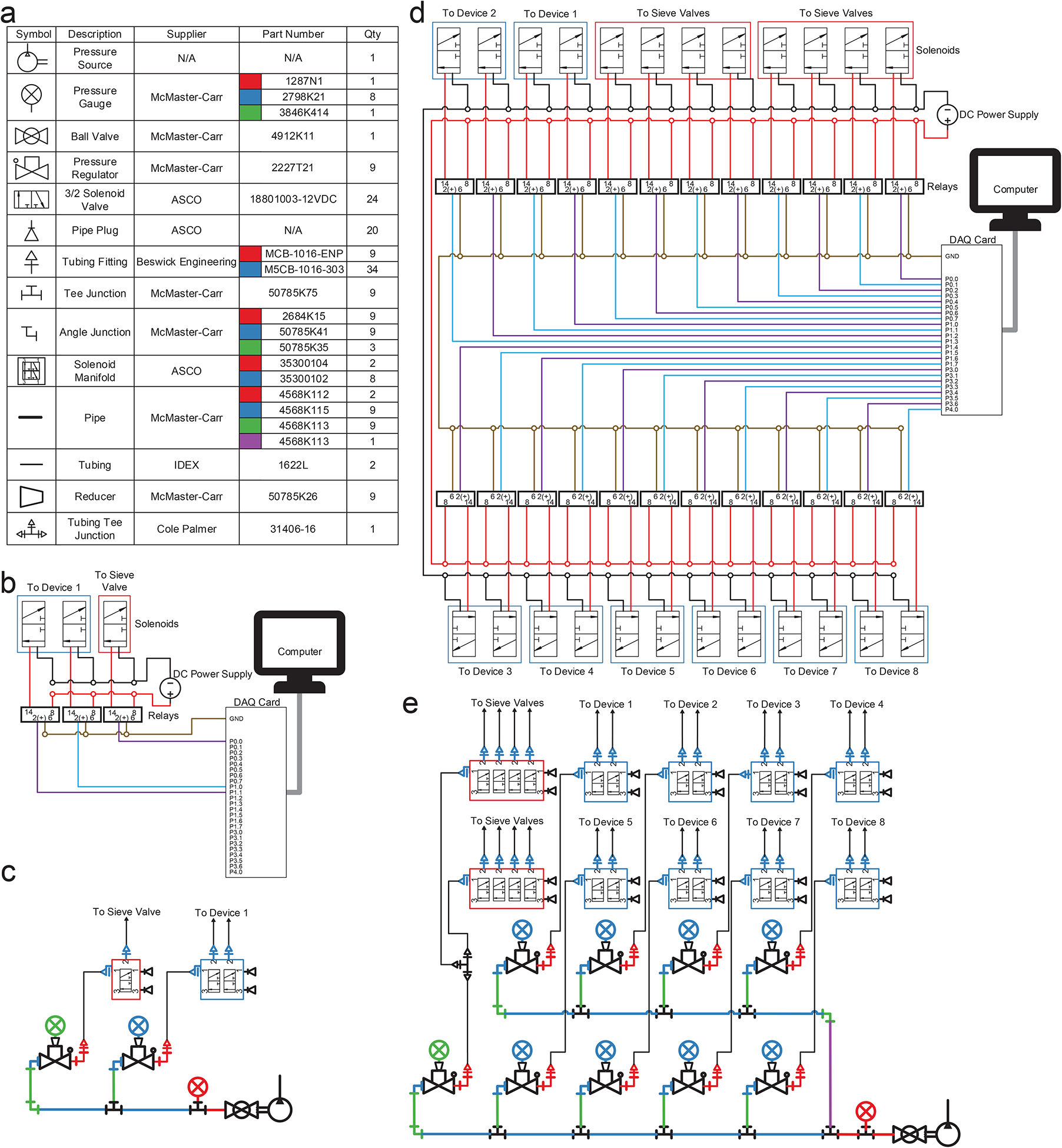
Schematics for electrical and pressure control systems associated with single-unit and 8-unit MOWChIP devices. (a) List of mechanical parts. The quantity listed is what is needed for operating 8 units simultaneously. (b) Electrical connections for automating 3 solenoid valves associated with a single-unit device. (c) Connection of various parts that constitute the pneumatic control system for a single-unit device. (d) Electrical connections for automating 24 solenoid valves associated with an 8-unit device. (e) Connection of various parts that constitute the pneumatic control system for an 8-unit device.
Figure 3 |.

The assembly of pneumatic control system for the 8-unit MOWChIP device shown in step-by-step fashion. All the individual parts are numbered and their information is listed in the “Equipment” list. (a-b) The assembling of a pressure gauge (#1), its associated pressure regulator (#3), and pipe (#6). (c-d) The assembling of a digital pressure gauge (#8), its associated pressure regulator, and pipes. (e-f) The assembling of all the pressure gauges and regulators. Gauge #1 and its associated regulator adjust pressure used in closing the sieve valves (at ~30 p.s.i.). 8 digital gauges (#8) and their associated regulators create pressure for oscillatory washing (at ~0.65 p.s.i.) in 8 units. The #12 gauge monitors the overall input pressure (90–100 p.s.i., from a pressured air source of the building in our case) which can be turned on or off by a ball valve (#15). (g-h) The assembling of parts associated with a 2-set solenoid valve. (i) Pressure gauges/regulators and solenoid valves are fixed to a pegboard. (j-m) Setting up reed relays on a breadboard for enabling control of solenoid valves by the DAQ card. (n-o) The assembled pneumatic control system. The inlets at the left of the solenoid valve manifolds (#19) are connected to the pressure regulators (#3) via PFA tubing.
Fabrication of the microfluidic devices.
The two-layer microfluidic MOWChIP devices are made of polydimethylsiloxane (PDMS). We fabricate the devices using multilayer soft lithography22–24 with minor modification. We first create masters (serving as molds) with photoresist features on 3-inch silicon wafers that can be repeatedly used. The device is then fabricated by molding PDMS layers and bonding of the fluidic and control layers. Each device has one inlet, one outlet and one microfluidic sieve valve8. The cost of the microfluidic device is low compared to that of the rest of the assay. We recommend using new devices each time to avoid cross contamination among samples.
Functionalization of magnetic beads with an antibody.
As in conventional ChIP-seq assays, we use magnetic beads functionalized with an antibody that specifically targets the modified histone of interest. The quality of ChIP-grade antibody and the coating conditions such as the antibody concentration and reaction time are of paramount importance for the final data quality.
Cell/nuclei harvesting.
ChIP-seq assays operate on chromatin fragments and thus are applicable to essentially all cell types. We have applied MOWChIP-seq protocols to examine cells from cell cultures or primary tissues and nuclei isolated from brain tissues. The studies of brain epigenomics often require sorting of neuronal and glial nuclei using fluorescence-activated cell sorter (FACS)12,25. Cells or nuclei are processed to yield chromatin fragments before ChIP. Prolonged storage of isolated cells or nuclei may lead to changes in the epigenomic state and should be avoided. We typically store isolated cells or nuclei on ice for less than 1 h before processing to yield chromatin fragments.
Chromatin fragmentation.
We have used two types of procedures for generating chromatin fragments from cells or nuclei. In one set of procedures, we conduct cell cross-linking for 5 min with 1% formaldehyde followed by sonication. A Covaris focused-ultrasonicator is used for sonication. Optimal sonication conditions yield chromatin fragments in the range of 200–500 bp. We recommend using the shortest sonication time that generates this size profile. Although we have demonstrated using cross-linking/sonication protocol on as few as 100 cells8, this protocol tends to lead to chromatin loss when the sample size is less than 100 cells. Alternatively, we also use cell lysis followed by micrococcal nuclease (MNase) digestion for chromatin fragmentation without crosslinking (that is, native ChIP-seq). Compared to sonication-based procedure, this approach offers better compatibility with analysis of scarce samples (30–100 cells per assay). Although previous studies have suggested that MNase digestion creates sequence-dependent bias during fragmentation26,27, our results showed that ChIP-seq data sets generated by crosslinking/sonication and lysis/MNase digestion presented very high correlations (Pearson correlation coefficients of 0.89–0.97 when 1000 cells per assay was used) that were similar to those between the technical replicates.
MOWChIP.
The most unique feature of our technology is the use of a microfluidic device for ChIP followed by oscillatory washing. Our MOWChIP process takes advantage of a packed bed in a microfluidic chamber for high-efficiency ChIP and an automated oscillatory washing step to effectively remove nonspecific binding. The MOWChIP process takes 2 h at room temperature, compared to overnight in conventional protocols. In our initial report, we operated a single MOWChIP microfluidic device to generate the data8. In this protocol, we illustrate the setup and operation of 8 units in parallel for high-throughput processing. All the units are operated independently and failure in one unit does not compromise the operation in other units.
Library preparation and sequencing.
The state-of-the-art library preparation kits require a minimum of 10–20 pg DNA as starting material. In our recent work, we have been using Swift Bioscience Accel-NGS 2S Plus DNA Library kit for library preparation. We use an Illumina HiSeq 4000 to sequence the samples with single-end 50 bp reads. For histone modification profiles with narrow peaks (for example, H3K4me3 or H3K27ac), 15–20 million reads per sample is used. We use a sequencing depth of 20–40 million reads per sample when histone marks with broad peaks (for example, H3K27me3) are examined, as suggested by previous literature28.
Limitations
MOWChIP-seq, in its current form without barcoding, requires generation of sufficient amount of ChIP DNA (10–20 pg) from a cell sample for preparation of a sequencing library. Thus it does not support single-cell analysis. MOWChIP-seq is the method of choice when a small cell population (100–10,000 cells) is available and cell-to-cell variability is not of interest. MOWChIP-seq produces roughly two orders of magnitude more unique reads than pooling single-cell Drop-ChIP data sets13 because our technology avoids loss in genomic DNA due to barcoding. Furthermore, the fabrication, setup and operation of the microfluidic device require engineering knowledge and efforts. These may be difficult for molecular biologists and clinicians.
Materials
Biological materials:
Human lymphocytes (Coriell Institute, ID no. GM12878) cultured in GM cell culture media (described below, in Reagent Setup section) under 37 °C in a 5% CO2 humidified cell growth incubator. Cells were sub-cultured every two days to maintain them in exponential growth phase. The cell line was authenticated by PCR targeting glucose-6-phosphate dehydrogenase gene8 and tested for mycoplasma contamination using Universal Mycoplasma Detection Kit every 6 months. CAUTION: The cell lines used in your research should be regularly checked to ensure they are authentic and not infected with mycoplasma.
Mouse prefrontal cortex (obtained from Dr. Javier González-Maeso of Virginia Commonwealth University). Mouse prefrontal cortex was dissected from 10-week old male CD-1 mice (Charles River Laboratories). The frozen samples were delivered to Virginia Tech by overnight shipment. We have complied with all relevant ethical regulations associated with animal works. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University. CAUTION: Any experiments involving live mice must conform to relevant Institutional and National regulations.
Reagents:
▲CRITICAL Prepare all solutions with deionized water. Store all solutions and regents in refrigerator (4 °C), unless otherwise indicated.
Photoresist SU-8 2025 (Microchem) !CAUTION Wear safety glasses, gloves and protective clothing when you are handling this material. Adequate ventilation is highly recommended in order to avoid breathing the vapors or mist.
Photoresist developer (Microchem) !CAUTION Wear safety glasses, gloves and protective clothing when you are handling this material. Adequate ventilation is highly recommended in order to avoid breathing the vapors or mist.
Acetone (Sigma-Aldrich, cat. no. 179973–4L). Store at room temperature.
Poly(dimethylsiloxane) (PDMS; Momentive, cas. no. RTV615). Store at room temperature.
Universal Mycoplasma Detection Kit (ATCC, cat. no. 30–1012K)
Dynabeads Protein A for immunoprecipitation (Life Technologies, cat. no. 10001D)
UltraPure Tris-HCl, 1M, pH 8.0 (Life Technologies, cat. no. 15568–025)
UltraPure EDTA, 0.5 M, pH 8.0 (Life Technologies, cat. mo. 15575–038)
UltraPure 10% SDS solution (w/v; Life Technologies, cat. no. 153–035). Store at room temperature.
Sodium chloride solution, 5 M, molecular biology grade (NaCl; Sigma-Aldrich, cat. no. 7647-14-5). Store at room temperature.
EGTA, 0.5 M, sterile solution, pH 8.0 (bioWorld, cat. no. 40520008-2). Store at room temperature.
Sodium deoxycholate, >97%, (Sigma-Aldrich, cat. no. 302-95-4). Store at room temperature.
Triton X-100, molecular biology grade (Sigma-Aldrich, cat. no. T8787-100ml)
RPMI-1640, Sterile-filtered (ATCC, cat. no. 30-2001)
Fetal Bovine Serum, 15%, qualified (w/v; FBS; Gibco, cat. no. 26140-079). Store at −20 °C.
Penicillin-streptomycin, 10,000 U/ml (PS; Gibco, cat. no. 15140122). Store at −20 °C.
Protease inhibitor cocktail (PIC; Sigma-Aldrich, cat. no. P8340). Prepare five 200 μl aliquots and store at −20 °C.
Phosphate Buffered Saline, pH 7.4 (PBS; Gibco, cat. no. 10010-031)
Phenylmethanesulfonyl fluoride, >98.5% (PMSF; Sigma-Aldrich, cat. no. P7626–1G). Store at −20 °C.
DL-Dithiothreitol solution, ~1M, molecular biology grade (DTT, Sigma-Aldrich, cat. no. 43816–10ml)
Sucrose, >99.5%, molecular biology grade (Sigma-Aldrich, cat. no. S0389–500G). Store at room temperature.
Calcium chloride dehydrate, molecular biology grade (CaCl2; Sigma-Aldrich, cat. no. 21097–50G). Store at room temperature.
Magnesium chloride hexahydrate, >99.0%, molecular biology grade (MgCl2; Sigma-Aldrich, cat. no. M2670–100G). Store at room temperature.
OptiPrep Density Gradient Medium (60% iodixanol solution; Sigma-Aldrich, cat. no. D1556–250ml)
Magnesium acetate tetrahydrate, >99%, molecular biology grade (Sigma-Aldrich, cat. no. M5661–50G). Store at room temperature.
10% Normal Goat Serum (10% NGS, Life Technologies, cat. no. 50062Z)
Dulbecco’s Phosphate Buffered Saline (dPBS, Life Technologies, cat. no. 14190–144)
Sodium bicarbonate, molecular biology grade (Sigma-Aldrich, cat. no. S7277–250G). Store at room temperature.
Ethyl alcohol, Pure, 200 proof, molecular biology grade (Sigma-Aldrich, cat. no. E7023–500ml). Store at room temperature.
Isopropanol, 99.5%, for molecular biology (Thermo Fisher Scientific, cat. no. AC327272500). Store at room temperature.
Formaldehyde, 16% (wt/vol), electron microscopy grade (Pierce; Thermo Fisher Scientific, cat. no. 28906). !CAUTION Formaldehyde is toxic if inhaled, ingested or absorbed through the skin. Always wear a lab coat and gloves, and work in a chemical hood. All Formaldehyde waste must be kept inside a chemical hood or in sealed containers, and collected for disposal by the proper authorities.
-
Anti-H3K4me3 antibody (Abcam, cat. no. ab8580)
▲CRITICAL Store at −20°C
-
Anti-H3K27ac antibody (Abcam, cat. no. ab4729)
▲CRITICAL Store at −20°C.
-
Anti-H3K27me3 antibody (EMD Millipore, cat. no. 07–449, lot no. 2717675; Active Motif cat. no. 39155)
▲CRITICAL Store at −20°C
-
Anti-H3K9me3 antibody (Abcam, cat. no. ab8898, lot no. GR3217826–1)
▲CRITICAL Store at −20°C.
-
Anti-H3K36me3 antibody (Active Motif, cat. no. 61101, lot no. 10218004)
▲CRITICAL Store at −20°C.
-
Anti-H3K79me2 antibody (Active Motif, cat. no. 39143, lot no. 27710001)
▲CRITICAL Store at −20°C.
Proteinase K (Sigma-Aldrich, cat. no. P2308–100mg). Store at −20 °C.
Glycerol, for molecular biology, >99% (Sigma-Aldrich, cat. no. 56-81-5)
Glycine, UltraPure (Thermo Fisher Scientific, cat. no. 15527013)
KCl (Sigma-Aldrich, cat. no. P9333). Store at room temperature.
Anti-NeuN Antibody, clone A60, Alexa Fluor 488 conjugated (Millipore, cat. no. MAB377X)
Micrococcal Nuclease Solution (MNase, Thermo Fisher Scientific, cat. no. 88216). Store at −20 °C.
Glycogen (5 mg/ml, Thermo Fisher Scientific, cat. no. AM9510). Store at −20 °C.
Ammonium acetate, BioXtra, >98% (Sigma-Aldrich, cat. no. A7330–100g).
EvaGreen dye, 20X in water (Biotium, cat. no. 31000)
Low EDTA TE buffer (Swift Biosciences, cat. no. 90296)
PEG NaCl solution (Swift Biosciences, cat. no. 90196)
SPRIselect beads (Beckman Coulter, cat. no. B23317). Store at room temperature.
SYBR Green PCR Master Mix (Thermo Fisher Scientific, cat. no. 4312704)
Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences, cat. no. 21024). Store at −20 °C.
2S Indexing Kit (Swift Biosciences, cat. no. 21024). Store at −20 °C.
KAPA Library Quantification Kit (KAPA Biosystems, cat. no. KK4824). Store at −20 °C.
High Sensitivity D1000 ScreenTape (Agilent Technologies, cat. no. 5067–5584)
Primers (Table 1; Integrated DNA Technologies)
Mouse Negative Control Primer Set 1 (Active Motif, cat. no. 71011). Store at −20 °C.
Mouse Negative Control Primer Set 3 (Active Motif, cat. no. 71013). Store at −20 °C.
Mouse Positive Control Primer Set Actb-1 (Active Motif, cat. no. 71015). Store at −20 °C.
Mouse Positive Control Primer Set Gapdh-2 (Active Motif, cat. no. 71018). Store at −20 °C.
Mouse Positive Control Primer Set Hoxc10 (Active Motif, cat. no. 71019). Store at −20 °C.
Table 1.
Primers used in ChIP-qPCR for quality control. The designed primers were ordered from Integrated DNA Technologies, Coralville, IA, USA.
| Species | Histone mark | Positive (P) or negative (N) locus | Locus name | Primer |
|---|---|---|---|---|
| Human | H3K4me3 (designed for GM12878 cells) | P | C9orf3 (F) | CCT CCT CAG TTC TCC CAG ACT |
| C9orf3 (R) | AGC TGA GGT GGT AAG ATG TGA C | |||
| N | N1 (F) | TCA TCT GCA AAT GGG GAC AA | ||
| N1 (R) | AGG ACA CCC CCT CTC AAC AC | |||
| Mouse | H3K4me3 | P | Gapdh-2 | Active motif cat. no. 71018 |
| N | Negative set 1 | Active motif cat. no. 71011 | ||
| H3K27me3/H3K9me3 | P | Hoxc10 | Active motif cat. no. 71019 | |
| N | Negative set 3 | Active motif cat. no. 71013 | ||
| H3K36me3/H3K79me2 | P | Actb-1 | Active Motif cat. no. 71015 | |
| N | Negative set 1 | Active motif cat. no. 71011 | ||
| H3K27ac (designed for mouse brain) | P | Zranb3 (F) | GAA TGT ACC AGC GTC TCT TCT C | |
| Zranb3 (R) | TTT CTC TTT GGC AGC CTC TC | |||
| N | N3 (F) | GCC CAT AAA GAA GGA GCT AGA G | ||
| N3 (R) | GCA GCT AGA GAC AAG AGT TCT G |
Equipment:
Silicon wafer (3 inch in diameter; University Wafer, cat. no. P(100)1–100ohm-cm SSP, test grade).
10-cm Petri dish (Fisher Scientific, cat. no. 08-757-100D)
1.5 ml Eppendorf microcentrifuge tube (VWR, cat. no. 20170-038)
Spin coater (Laurell, model: WS-400BZ-6NPP/LITE)
Hot plate (Thermo Fisher Scientific, model: HP8885794)
Vacuum pump (Welch Vacuum, model: WOB-L 2522B-01)
Precleaned glass slide (VWR, cat. no. 48300–026)
UV-lamp (OmniCure, series no. 1000)
Sonicator (Covaris, model: M220 Foused-ultrasonicator)
PDMS hole puncher (Harris, cat. no. 69036–20)
Plasma cleaner (Harrick Plasma, cat. no. PDC-32G)
Vacuum pump (Dekker vacuum technologies, model: RVR003H-01)
Thread seal tape (Teflon tape, 1/2” x 520”, NDA Distributors LLC, item #: M3)
Pressure gauge (McMaster-Carr, cat. no.:3846K414; 2798K211; 1287N1), #1, #8, #12 in Fig. 3
Pressure regulator (McMaster-Carr, cat. no.: 2227T21), #3, in Fig. 3
Angle junction (McMaster-Carr, cat. no: 2684K15; 50785K41; 50785K35), #2, #4, #7 in Fig. 3
Tubing fitting (Beswick Engineering, part no.: MCB-1016-ENP; M5CB-1016–303), #5 (including #21), #19 (including #21) in Fig. 3
Pipe (McMaster-Carr, cat. no.: 4568K112; 4568K115; 4568K113; 4568K125), #14, #10, #6, #13 in Fig. 3
Reducer (McMaster-Carr, cat. no.: 50785K26), #9 in Fig. 3
Tee junction (McMaster-Carr, cat. no.: 50785K75), #11 in Fig. 3
Ball Valve (McMaster-Carr, cat. no.: 4912K11), #15 in Fig. 3
3/2 Solenoid Valve (ASCO, cat. no.: 18801003–12VDC), #17 in Fig. 3
Solenoid subbase (ASCO, cat. no.: 35300104; 35300102), #18 in Fig. 3
Front and rear ferrules (Beswick Engineering, part no.: MCB-16–303-FR; MCB-16–303-FF), #20 in Fig. 3
Tubing Tee junction (Cole Palmer, item no.: SK-31406–16)
Solenoid wires (ASCO, cat. no.: 88118803)
10 color stranded wire kit assortment (Jameco Electronics, part no.: 2187876), #22 in Fig. 3
Reed Relays Mini Reed PCB Relay SPST, 0.5A (Schneider Electric, part no.: 107DIP-5), #23 in Fig. 3
830-Point Solderless Breadboard 6.5” x 2.125” (Jameco Valuepro, part no.: WBU-202-R), #24 in Fig. 3
Multifunction Data Acquisition (DAQ) card (National Instruments, cat.no.: SCB-100), #25 in Fig. 3
Rotator mixer (Labnet, cat. no. H5500)
7 ml Dounce tissue grinder set, glass (Sigma-Aldrich, cat. no. D9063-1SET)
40 μm Sterile Cell Strainers (Fisher Scientific, cat. no. 22-363-547)
Centrifuge (Eppendorf, model: 5415D)
DynaMag-Spin Magnet (Thermo Fisher Scientific, cat. no. 12320D)
Perfluoroalkoxyalkane (PFA) high-purity tubing (IDEX Health & Science, cat. no. 1622L), #16 in Fig. 3
C-Flex clear tubing (Cole-Parmer Instrument, cat. no. ZX-06422–01)
Long tubing assembly (10 cm C-Flex clear tubing connected with 3 cm PFA tubing). It is reusable after washing with 70% ethanol solution followed by deionized water and exposing to UV light for 4 h. (Supplementary Figure 1)
Short tubing assembly (6 cm C-Flex clear tubing connected with 3 cm PFA tubing). It is reusable after washing with 70% ethanol solution followed by deionized water and exposing to UV light for 4 h. (Supplementary Figure 1)
1 ml BD Luer-lok Syringe (BD, cat. no. 309628)
Female to Female Luer Lock (Dolomite-microfludics, cat. no. 3000311)
Flangeless fitting for 1/16” OD tubing (Chrom Tech, cat. no. XP-202 & P200)
Syringe-tubing assembly (1 ml syringe connected with 25 cm PFA tubing via luer lock and fitting). It is reusable after washing with 70% ethanol solution followed by deionized water and exposing to UV light for 4 h. (Supplementary Figure 1)
PFA tubing elbow (11 cm PFA tubing). It is reusable after washing with 70% ethanol solution followed by deionized water and exposing to UV light for 4 h (Supplementary Figure 1)
Syringe pump (Chemyx, model: Fusion 400)
Pipettes package (max. volume 1 ml, 200 μl, 20 μl, 10 μl, and 2.5 μl) (Eppendorf, cat. no. 2231000224)
Pipette tips (VWR, cat. no. 37001–162(10 μl), 53503–294(200 μl), 83007–372(1 ml))
15 ml centrifuge tubes (Corning, cat. no. 430790)
Vortex mixer (VWR, cat. no.)
Magnetic rack (DiaMag0.2 ml; Diagenode, cat. no. B04000001)
Magnet bar (1/2 in x 1/2 in x 2 in; Applied Magnets, model: NB023–1)
Multi-therm heat/cool shaker incubator (Benchmark, cat. No. 3539234)
TapeStation (Agilent, model: G2962/A)
T100 Thermal Cycler (Bio-Rad, cat. no. 186–1096)
CFX Connect™ Real-Time PCR Detection System (Bio-Rad, cat. no. 1855200)
PCR 8-tube strips & caps (Bio-Rad, cat. no. TBC0802)
Cell Growth Incubator (Symphony, VWR, model: 3074)
Flask for cell culture (Corning, cat. no. 430639)
Hard-shell PCR Plates, 96-well, thin-wall (Bio-Rad, cat. no. HSP9601)
LayoutEditor: http://www.layouteditor.net/download.php5
Trim Galore: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore
fetchChromsizes: http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/
samtools30: https://gist.github.com/adefelicibus/f6fd06df1b4bb104ceeaccdd7325b856
bedtools31: https://bedtools.readthedocs.io/en/latest/content/installation.html
UCSC-wigtobigwig: https://genome.ucsc.edu/goldenpath/help/bigWig.html
IGV34,35: https://software.broadinstitute.org/software/igv/download
Reagent setup
GM cell culture media Mix 500 ml of RPMI-1640, 90 ml of FBS, and 6 ml of PS together. The media can be stored for at least one year at 4 °C.
Cross-linking solution Mix 9.375 ml of PBS with 0.625 mL of 16% (wt/vol) formaldehyde to generate 1%(wt/vol) formaldehyde in PBS. The solution should be prepared freshly every time.
2 M glycine Dissolve 15.04 g of glycine in 100 ml of Milli-Q water. The solution can be stored at least two years at 4 °C.
100 mM PMSF Dissolve 0.174 g of PMSF in 10 ml of isopropanol. The solution can be stored at least two years at −20 °C.
5 M Ammonia Acetate Dissolve 3.854 g of ammonia acetate in Milli-Q water and make the volume 10 ml. The solution can be stored at least two years at 4°C.
100 mM CaCl2 Dissolve 0.111 g of calcium chloride dehydrate in 10 ml of Milli-Q water. The solution can be stored at least three years at 4 °C.
Sonication buffer 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, and 0.1% (w/v) SDS, 1% PIC in Milli-Q water. Add PIC immediately before use. The buffer can be stored at 4 °C for at least one year.
NE buffer 0.32 M sucrose, 5 mM CaCl2, 3 mM Mg(Ac)2, 0.1 mM EDTA, 10 mM Tris-HCl, and 0.1% Triton X-100 in Milli-Q water. The buffer can be stored at 4 °C for at least one year.
50% iodixanol solution Mix 4 ml of 60% iodixanol with 0.8 ml of Milli-Q water containing 150 mM KCL, 30 mM MgCl2, and 120 mM Tris-HCl (pH 8.0). The solution can be stored at 4 °C for at least one year.
2% NGS/dPBS solution Mix 800 μl of dPBS with 200 μl of 10% NGS solution. The solution should be prepared freshly before use.
2x lysis buffer 4% (v/v) Trition X-100, 100 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 30 mM MgCl2 in Milli-Q water. The buffer can be stored at 4 °C for at least one year.
1x lysis buffer 2% (v/v) Trition X-100, 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, and 15 mM MgCl2 in Milli-Q water. The buffer can be stored at 4 °C for at least one year.
IP buffer 20 mM Tris-HCl (pH8.0), 140 mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1%(w/v) sodium doxycholate, 0.1% SDS, 1%(v/v) Triton-100X in Milli-Q water. The buffer can be stored at 4 °C for at least one year.
Low salt washing buffer 20 mM Tris-HCl (pH 8.0),150 mM NaCl, 2 mM EDTA, 0.1%SDS, 1%(v/v) Triton-100X in Milli-Q water. The buffer can be stored at 4 °C for at least three years.
High salt washing buffer 20 mM Tris-HCl (pH 8.0),500 mM NaCl, 2 mM EDTA, 0.1%SDS, 1%(v/v) Triton-100X in Milli-Q water. The buffer can be stored at 4 °C for at least three years.
Reverse cross-linking buffer 200 mM NaCl, 50 mM Tris-HCl (pH 8.0), 10 mM EDTA, 1% SDS, 0.1 M NaHCO3 in Milli-Q water. The buffer can be stored at 4 °C for at least one year.
Elution buffer 10 mM Tris-HCl (pH 8.0), 50 mM NaCl, 10 mM EDTA, 0.03% SDS in Milli-Q water. The buffer can be stored at 4 °C for at least one year.
50% Glycerol Mix 10 ml glycerol with 10 ml Milli-Q water. The solution can be stored at 4 °C for at least one year.
Proteinase K Dissolve 100 mg of proteinase K in 5 ml of 50% glycerol. The solution can be stored at least two years at −20 °C.
Procedure:
Setup of the pneumatic control system •TIMING 1 d
▲CRITICAL During assembly of the system, thread seal tape should be used to seal every joint that does not have an O-ring seal. The integrity of the sealed system can be examined using soapy water applied to the joints with bubbles indicating the locations of leaking. All the tubing connected through compression fittings should be securely fitted. The connection schematics are provided in Fig. 2 and images in Fig. 3 guide the assembling. We demonstrate the setup of the pneumatic control system for 8-unit device in this section while a simpler system for running a single-unit device can also be built similarly.
-
1
Assemble a pressure gauge (#1), a pressure regulator (#3), three angle junctions (#2, #4, and #7), a tubing fitting (#5), and a pipe (#6) together as shown in Fig. 3(a–b).
-
2
Assemble a digital pressure gauge (#8), a reducer (#9), a pressure regulator (#3), two angle junctions (#2 and #4), a tubing fitting (#5), two pipes (#6 and #10), and a tee junction (#11) together as shown in Fig. 3(c–d).
-
3
Connect two parts from step 1 and step 2 together as shown in Fig. 3e.
-
4
Repeat step 2 to assemble 7 additional pressure gauge/regulator units.
-
5
Connect all the pieces from steps 3–4 together using a long pipe (#13), and attach two more pipes (#14), one tee junction (#11), a pressure gauge (#12) and a ball valve (#15) to the structure.
-
6
Immobilize two solenoid valves (#17) onto a 2-set solenoid manifold (#18) using screws, attach three tubing fittings (#19) and two pipe plugs to the manifold, and connect two pieces of PFA tubing to the manifold using two sets of ferrules (#20) and tubing fittings (#21), as shown in Fig. 3 (g–h).
-
7
Repeat step 6 to form seven additional 2-set solenoid valves
-
8
Similarly assemble two 4-set solenoid valves.
-
9
Attach the assembled pressure gauges and pressure regulators from step 5 and all the solenoid manifolds prepared from steps 6–8 to a pegboard by cable ties, as shown in Fig. 3i.
-
10
Connect each pressure regulator that is attached to a digital pressure gauge (#8) to a 2-set solenoid manifold via a piece of PFA tubing (with the tubing mostly running behind the pegboard).
-
11
Connect the pressure regulator shown in Fig. 3b to two 4-set solenoid manifolds via PFA tubing and a tubing tee junction.
-
12
Prepare a breadboard (#24) by attaching 8 relays (#23) and elbow angle wires (#22) as shown in Fig. 3(j–k). Repeat this step to prepare another two breadboards.
-
13
Connect all the 24 solenoid valves to a DAQ card (#25) via three breadboards as shown in Fig. 3(m–o).
Fabrication of SU-8 silicon masters •TIMING 3 h
▲CRITICAL Photomasks with microscale patterns are designed using LayoutEditor and printed on high-resolution (10,000 dots per inch) transparencies (produced by Fineline Imaging, Colorado Springs, CO, USA). The LayoutEditor files used for MOWChIP device fabrication are available as Supplementary Data 3. The SU-8 silicon masters are made in a fume hood because the solvent in the photoresist is toxic. The masters are used as molds for molding PDMS devices.
-
14
Pre-warm the photoresist SU-8 2025 to room temperature by placing the closed bottle in a fume hood for 1 h.
-
15
Fix a new pre-cleaned silicon wafer on the spinner chuck for the spin coater by vacuum.
-
16
Pour ~3g of the photoresist on the wafer.
-
17
Spin the wafer using a two-step program, 500 r.p.m. for 10 s followed by 2500 r.p.m. for 30 s.
▲CRITICAL STEP This step coats the wafer with SU-8 2025 photoresist of 36-μm thickness.
-
18
Release the wafer from the spinner chuck, and bake it on a hot plate at 95 °C for 8 min.
-
19
Place the wafer under the UV lamp (OFF, with power set as 160 mJ/cm2). Cover the wafer with the fluidic layer photomask. Turn on the UV light, and expose for 17 s.
-
20
Place the wafer on the hot plate and bake it at 95 °C for 8 min.
-
21
Immerse the wafer in a container with SU-8 Developer, develop the wafer for 5 min while shaking the container gently.
-
22
Wash the wafer with isopropanol and blow dry with pressurized air attached to the fume hood.
-
23
Bake the wafer on the hot plate at 150 °C for 15 min to strengthen SU-8 bonding to the wafer.
-
24
Repeat the steps 15–16.
-
25
Spin the wafer using a two-step program, 500 r.p.m. for 10 s followed by 1500 r.p.m. for 30 s.
▲CRITICAL STEP This step coats the wafer with SU-8 2025 photoresist of 60-μm thickness.
-
26
Repeat steps 18–23 while using the control layer photomask in step 19.
■PAUSE POINT The SU-8 silicon masters can be stored at room temperature in a closed container and repeatedly used for up to 1 year.
Fabrication of the microfluidic device •TIMING 9 h
-
27
Place the fluidic layer master (fabricated in Step 23) in a petri dish.
-
28
Mix 30 g of PDMS reagent A with 6 g of reagent B in a disposable plastic boat to create a 5:1 PDMS mixture. This amount of PDMS produces a ~5 mm thick fluidic layer. Mix 5 g of PDMS reagent A with 0.25 g of reagent B in another disposable plastic boat to create a 20:1 PDMS mixture.
-
29
Pour all of the 5:1 PDMS mixture on the fluidic layer master in the petri dish.
-
30
Place the petri dish (containing the fluidic layer master and 5:1 PDMS) and the plastic boat containing 20:1 PDMS in a desiccator connected to the vacuum pump at ~60 mTorr for 1 h to remove air bubbles.
-
31
Set the control layer master (fabricated in step 26) on the spinner chuck of the spin coater and apply vacuum.
-
32
After ensuring there are no bubbles in the mixture, carefully pour ~5 g of 20:1 PDMS mixture onto the control layer master.
▲CRITICAL STEP Perform this step carefully to prevent the formation of bubbles in PDMS.
-
33
Spin the master using a two-step program, 500 r.p.m. for 10 s followed by 1100 r.p.m. for 30 s.
-
34
Release the control layer master coated by ~108 μm PDMS from the spin coater, and carefully transfer it into a new petri dish.
-
35
Transfer both petri dishes containing fluidic layer and control layer masters to the oven set at 80 °C. Heat for 12 min to allow partial curing of PDMS.
▲CRITICAL STEP This PDMS curing step is finished when PDMS is not sticky upon touch.
-
36
Remove the petri dishes from the oven, and cool them to room temperature (22 °C).
-
37
Peel the fluidic layer slowly from the silicon wafer after cutting PDMS along the edge of the wafer in the petri dish.
-
38
Hold the fluidic layer and carefully assemble it onto the control layer, which is still on the silicon wafer.
▲CRITICAL STEP Having alignment marks on the wafer helps. Do the alignment slowly to avoid trapping an air pocket.
-
39
Place the wafer covered by both control and fluidic layers in the 80 °C oven for 2 h.
▲CRITICAL STEP In this step, PDMS is cured completely, and the control layer is bound to the fluidic layer permanently.
-
40
Take out the PDMS/wafer structure from the oven and cool it to room temperature.
-
41
Peel the PDMS slab from the control layer master, and cut the slab into several pieces with each containing 4 units.
-
42
Use a 2-mm hole puncher to punch holes in the PDMS slab. These holes serve as inlet, outlet and sieve valve port (Fig.4a).
-
43
Place a precleaned glass slide and 2 PDMS slabs of 4 units (to make an 8-unit device) into the chamber of the plasma cleaner, feature side up.
-
44
Close the vent valve and needle valve on the chamber lid by turning clock-wise all the way. Ensure RF LEVEL switch is OFF.
-
45
Place the lid over the chamber opening and turn on the PUMP ON/OFF switch to start vacuuming for 4 min.
-
46
Turn on the POWER ON/OFF switch, wait for 1 min before processing.
-
47
Set the RF LEVEL switch to HI level, open the needle valve on the chamber lid about 1/8 of a turn to let air flow into the chamber. Wait for 1 min.
▲CRITICAL STEP Look for a color change of the chamber from the top of the machine, from purple to pink, when you open the needle valve.
-
48
Switch the RF LEVEL to OFF, turn off the POWER switch and the PUMP ON/OFF switch, open the vent valve slowly and remove the chamber lid.
-
49
Immediately bring treated PDMS slab and the glass slide into contact, and then put the glass slide/PDMS device into the oven (80 °C) for 4 h.
-
50
Move the devices out of the oven, and cool down to room temperature.
■PAUSE POINT The microfluidic devices can be stored in a closed container at room temperature for up to 2 months.
Figure 4 |.
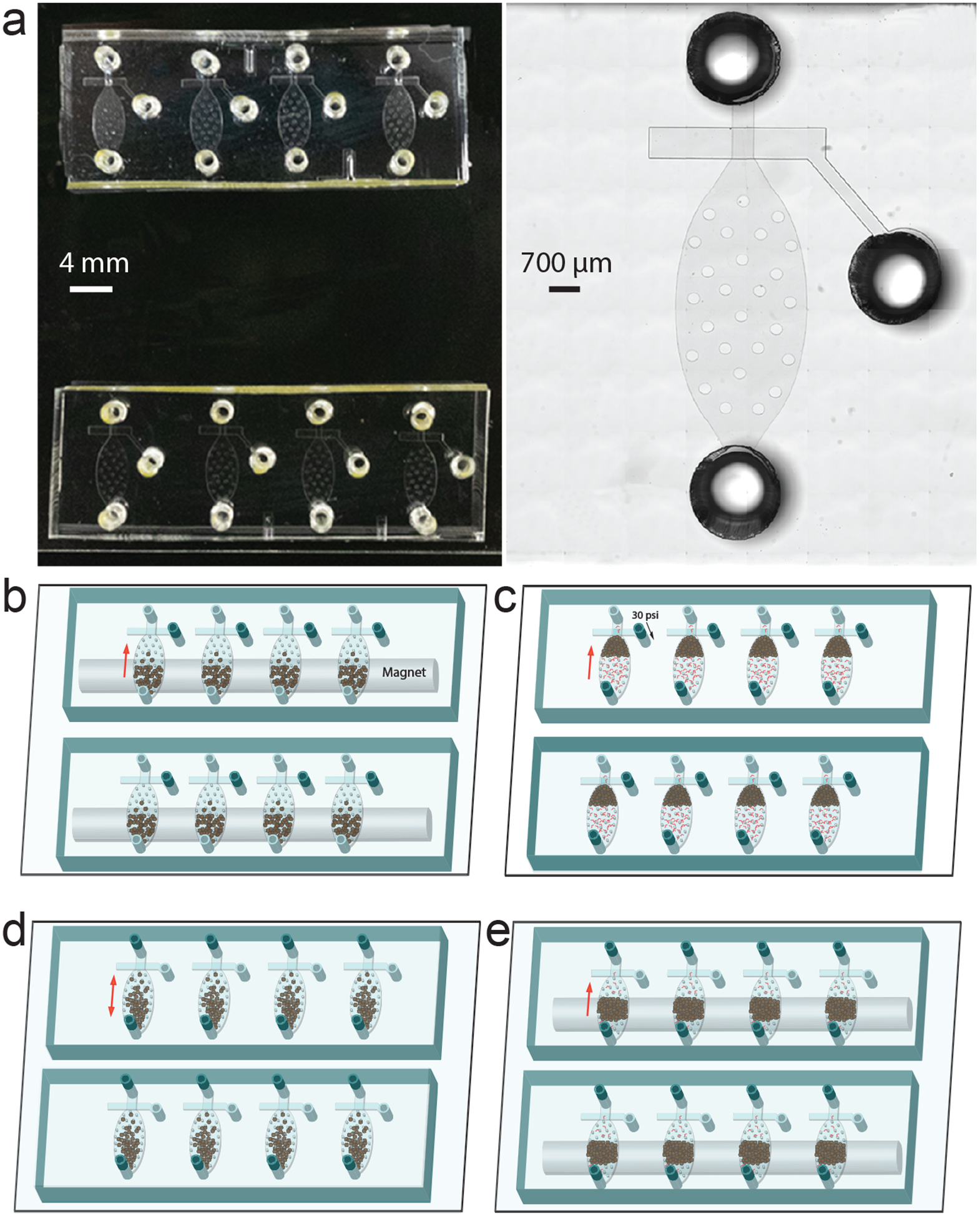
Design and operation of an 8-unit MOWChIP device. (a) Picture (left) and microscopic image (right) of an 8-unit MOWChIP device. (b) Loading immunomagnetic beads into the chamber with the assistance of a magnet. The sieve valves are open. (c) Flowing chromatin fragments through the packed bed of beads. The sieve valves are closed under 30 p.s.i. pressure. (d) Oscillatory washing under alternating pressure pulses applied via the inlet and outlet. Low salt and high salt washing buffers are used to remove nonspecifically bound chromatin. (e) Flushing out unbounded chromatin and debris while retaining the beads using the magnet.
Coupling an antibody to superparamagnetic beads •TIMING 2.5 h
▲CRITICAL We coat an antibody that targets a modified histone of interest on protein A-coated superparamagnetic beads one day before ChIP. The choice for the bead coating depends on the recommendation of the antibody manufacturer. In all histone marks discussed in this protocol, protein A-coated beads are recommended. The below procedure describes preparation of the beads for one assay starting with one tube of beads. Multiple tubes are used when beads are prepared for multiple ChIP assays (e.g. when running 8 assays on a device is desired).
-
51
Transfer 5 μl of protein A-coated Dynabeads into an Eppendorf microcentrifuge tube. Place the tube on the DynaMag-Spin Magnet for 5 s before removing supernatant by pipetting and discarding. Be careful not to disturb the pelleted beads.
-
52
Immediately add 120 μl of IP buffer to the tube, lightly vortex for ~2 s to make the bead suspension uniform.
-
53
Centrifuge the mixture at 300 g for 5 s at room temperature to collect all the buffer and beads at the bottom of the tube.
-
54
Place the tube on the DynaMag-Spin Magnet and remove and discard supernatant.
-
55
Repeat steps 52–54 one more time to finish bead washing.
-
56
Resuspend the beads in 150 μl of IP buffer by pipetting up and down several times.
-
57
Add desired amount of antibody to the tube. We recommend using 0.5 μg of anti-H3K4me3, 0.4 μg of anti-H3K27me3, 0.6 μg of anti-H3K9me3, 0.25 μg of anti-H3K36me3, or 1 μg of anti-H3K79me2.
▲CRITICAL STEP The efficiency of antibody may varies from batch to batch. The researchers can start from the recommended amount of antibody and adjust a little based on the enrichment after ChIP.
-
58
Incubate the mixture at 4 °C on a rotator mixer at 24 r.p.m. for 2 h. Overnight incubation does not compromise the result.
-
59
Place the tube on the DynaMag-Spin Magnet and remove and discard supernatant.
-
60
Wash beads with 120 μl of IP buffer by gently pipetting up and down 6 times. Place the tube on the DynaMag-Spin Magnet and remove and discard supernatant. Repeat this step two more times.
-
61
Immediately suspend the beads in 5 μl of IP buffer, place on ice, and proceed with step 71 typically within 1 h.
Chromatin preparation
-
62
Prepare the chromatin solution. We prepare chromatin samples from cells and nuclei isolated from cell lines and primary tissues. The isolated cells or nuclei should be kept on ice for no longer than 1 h before processing. Here, we present two alternative approaches for preparing chromatin fragments of 200–500 bp: (A) Crosslinking and sonication; (B) Lysis and MNase digestion. We have used both approaches to treat cells and used (B) to treat nuclei isolated from mouse brains.
(A). Crosslinking and sonication •TIMING 1 h
▲CRITICAL we use the treatment of ~1200 GM12878 cells as an example to describe the process. The same process applies to 1000–100,000 cells. Crosslinking and sonication of samples containing as few as 100 cells are described in Box 1.
Box 1. Preparation of chromatin fragments from samples containing 100–500 cells •TIMING 1 h.
Transfer cells into a 1.5 ml Eppendorf microcentrifuge tube, remove and discard the supernatant after centrifugation at 300 g for 5 min at room temperature, resuspend the cell pellet in 10 μl of 10% (vol/vol) FBS in PBS.
Add 0.625 μl of 16% (wt/vol) formaldehyde to the cell suspension, mix well by pipetting.
Incubate the mixture at room temperature on a rotator mixer at 24 r.p.m. for 5 min.
Add 1.25 μl of 2.5 M glycine to the mixture to quench crosslinking, mix well by pipetting.
Incubate the tube on a rotator mixer at 24 r.p.m. for 5 min at room temperature.
Add 120 μl of ice-cold sonication buffer in order to sufficiently dilute formaldehyde in the mixture.
Sonicate the sample for 8 min with 5% duty factor, 105 W peak incident power and 200 cycles per burst.
Centrifuge the sonicated lysate at 14,000 g for 10 min at 4 °C.
-
Transfer the supernatant (~130 μl) which contains sonicated chromatin to a new 1.5 ml Eppendorf microcentrifuge tube.
■PAUSE POINT Sonicated chromatin suspension can be stored at −80°C for up to 12 months.
Harvest 1 ml of fresh GM12878 cell culture at room temperature into a 1.5 ml Eppendorf microcentrifuge tube, centrifuge at 300 g for 5 min at room temperature, remove and discard the supernatant.
Resuspend the cell pellet in 50 μl of PBS, count the cells with a hematocytometer, transfer ~1200 GM12878 cells resuspended in 18.75 μl PBS to a new 1.5 ml Eppendorf microcentrifuge tube.
Add 1.25 μl of 16% (wt/vol) formaldehyde to the cell suspension, mix well by pipetting.
-
Incubate the mixture at room temperature on a rotator mixer at 24 r.p.m. for 5 min.
▲CRITICAL STEP The cross-linking time used in this step is optimized for GM12878 cells. For other cell types, the conditions may be different. The degree of fixation affects chromatin fragmentation.
Centrifuge the tube at 200 g for 20 s at room temperature to collect all the solution at the bottom of the tube.
Add 1.33 μl of 2M glycine to the mixture, mix well by pipetting.
Incubate the tube on a rotator mixer at 24 r.p.m. for 5 min at room temperature.
Centrifuge the tube at 1,600 g for 5 min at room temperature, remove and discard the supernatant.
Rinse cell pellet with 20 μl of ice-cold PBS, taking care not to resuspend the cell pellet.
Centrifuge at 1,600 g for 5 min at room temperature, remove and discard the supernatant.
Carefully add 20 μl of ice-cold PBS, and repeat step 62Aix once more.
Resuspend the cell pellet in 60 μl of ice-cold sonication buffer.
-
Sonicate the sample for 8 min with 5% duty factor, 105 w peak incident power and 200 cycles per burst.
▲CRITICAL STEP The parameters for sonication should be optimized if a different amount or type of cells are used.
Centrifuge the sonicated lysate at 14,000 g for 10 min at 4 °C.
-
Transfer the supernatant (~55 μl), which contains sonicated chromatin, to a new 1.5 ml Eppendorf microcentrifuge tube.
■PAUSE POINT Sonicated chromatin suspension can be stored at −80°C for up to 12 months.
Transfer 50 μl of sonicated chromatin suspension to a new 1.5 ml Eppendorf microcentrifuge tube.
Add 0.5 μl of PIC and 0.5 μl of PMSF, mix well and place the tube on ice until use in step 72.
Transfer the remaining 5 μl of sonicated chromatin from step 62Axv to a new 1.5 ml Eppendorf microcentrifuge tube, and store on ice until use in step 90 as the input control which does not go through ChIP.
(B). Lysis and MNase digestion •TIMING 3.5 h
▲CRITICAL We use neuronal nuclei from prefrontal cortex of mouse brain as an example to describe the process here. We have also used the same procedure to treat GM12878 cells without the nuclei isolation and sorting steps. All the reagents used in this step are placed on ice to keep them ice-cold. Immunostaining and FACS sorting are used to isolate neuronal (NeuN+) and glial cells (NeuN-) from brain tissues12,25. The below process is for one ChIP assay using 1200 nuclei. Multiple batches of chromatin samples can be prepared if a number of ChIP assays are planned. We have also used the same process for samples containing 1000–10,000 nuclei or cells.
Put ~100 mg of frozen mouse prefrontal cortex (containing roughly 80,000 neuron nuclei) in a 7 ml glass dounce tissue grinder, add 5 ml of NE buffer, 50 μl of PIC, 5 μl of 100mM PMSF, and 5 μl of 1M DTT, and place the grinder on ice.
-
Once the tissue thaws, dounce the tissue with pestle A 15 times, followed by douncing with pestle B 25 times.
▲CRITICAL STEP The grinder should always be kept on ice during this step.
Place a new 15 ml centrifuge tube on ice. Filter the homogenized tissue through a 40-μm cell strainer into the tube.
Centrifuge the tube at 1000 g for 10 min at 4 °C.
Discard the supernatant and gently resuspend the crude nuclei pellet in 500 μl NE buffer by pipetting. Add 5 μl of PIC, 0.5 μl of 100 mM PMSF, and 0.5 μl of 1 M DTT to the nuclei suspension. Transfer the nuclei suspension into a 1.5 mL Eppendorf microcentrifuge tube. Place the tube on ice.
Add 750 μl of 50% (wt/vol)iodixanol solution, 7.5 μl of PIC, 0.75 μl of 100 mM PMSF and 0.75 μl of 1 M DTT to the tube. Mix the solution well before proceeding to the next step.
Centrifuge at 10,000 g for 20 min at 4 °C. Carefully remove and discard all the supernatant.
Add 500 μl 2% (wt/vol) NGS/dPBS, 5 μl of PIC, 0.5 μl of 100 mM PMSF, and 0.5 μl of 1 M DTT to the tube. Incubate on ice for 10 min. Resuspend the nuclei pellet by pipetting up and down gently.
Transfer 8 μl nuclei suspension to a new 1.5 ml Eppendorf microcentrifuge tube. Dilute to 50 μl with 2% NGS/dPBS solution and use the sample as unstained control.
Add 8 μl of 2 ng/μl anti-NeuN antibody conjugated with Alexa 488 into the tube containing 492 μl nuclei suspension. Mix at 4 °C on a rotator mixer at 24 r.p.m. for 1 h while protected from light.
-
After staining, transfer the nuclei suspension to a 15 ml centrifuge tube, and place the tube on ice during transportation to a FACS facility.
▲CRITICAL STEP Keep nuclei suspension on ice and in the dark during transportation.
-
Conduct FACS while using the unstained control to set the sorting parameters. Place the sorted nuclei samples (NeuN+ and NeuN-) on ice after FACS.
▲CRITICAL STEP We recommend proceeding to the next step quickly, because extended delay may compromise nuclei integrity.
Transfer 10 μl sorted nuclei suspension, containing ~1200 nuclei, to a 1.5 ml Eppendorf microcentrifuge tube. Add 0.1 μl PIC and 0.1 μl PMSF solution.
Add 10 μl of 2x Lysis buffer into the tube. Mix well, and incubate for 10 min at room temperature.
-
Add 1 μl of 100mM CaCl2 solution and 2 μl 10U/μl MNase (in PBS) into the tube, vortex for 5s. Incubate at room temperature for 10 min.
▲CRITICAL STEP Concentration of original MNase is 100 U/μl. Freshly dilute MNase with PBS to 10 U/μl before this step.
Add 2.2 μl 0.5M EDTA into the tube to stop MNase digestion. Rapidly mix. Incubate on ice for 10 min.
Centrifuge at 16,100 g for 10 min at 4 °C to precipitate cellular debris at the bottom of the tube.
-
Transfer supernatant (~22 μl) to a new 1.5 ml Eppendorf microcentrifuge tube (tube A). Take out 20 μl of the supernatant from tube A and dilute it to 50 μl with 1x lysis buffer in another 1.5 ml Eppendorf microcentrifuge tube (tube B); place both tubes on ice. The sample in tube B will be used in step 72 and the sample (~2 μl) in tube A will serve as the input control in step 90.
■PAUSE POINT The chromatin fragment samples can be stored on ice for up to 3–4 h.
MOWChIP •TIMING 1.5 h
▲CRITICAL The pneumatic control system allows us to conduct eight ChIP assays simultaneously. A smaller number of assays can be run when a subset of the solenoid valves are operated. In the supplementary files, we include LabVIEW programs for running either 1 or 8 units (Supplementary Data 1 and 2, respectively). The devices are taped down on a stage made by cutting a 3 x 3 inch square hole in a Styrofoam box (Fig. 5a). The stage allows placing the tubing on the PDMS side of the microfluidic devices while operating a magnet from the glass side of the devices. We use two types of tubing and the combinations of them at various locations: PFA tubing that is hard and C-Flex tubing that is flexible. The cost of the microfluidic device is trivial compared to the overall cost of the ChIP-seq assay. Thus we do not recommend reuse of the microfluidic device due to concern about potential cross contamination. The MOWChIP process is conducted at room temperature.
Figure 5 |.

The experimental setup for MOWChIP operation. (a) An 8-unit MOWChIP device connected to its ancillary fluid and pressure control systems. (b) Flow of chromatin samples through the packed beds. (c) Oscillatory washing of the beads. (d) Elution of the beads into tubes after MOWChIP.
-
63
Tape the MOWChIP devices on the stage made of a Styrofoam box (Fig 5a).
-
64
Draw 700 μl IP buffer into a syringe-tubing assembly (1 ml syringe connected with 25 cm PFA tubing via luer lock and fitting, Supplementary Fig. 1). Place the syringe-tubing assembly on a syringe pump. Adjust the pusher block so that it touches against the plunger flange of the syringe.
-
65
Fill a short tubing assembly (6 cm C-Flex clear tubing connected with 3 cm PFA tubing, Supplementary Fig. 1) with 30 μl water by pipetting from the free C-Flex tubing end. Plug the PFA-tubing-end of the tubing assembly into the port of the on-chip sieve valve and connect the free C-Flex tubing end with a PFA tubing that is attached to the solenoid manifold set.
▲CRITICAL STEP This step connects the on-chip sieve valve with a solenoid valve for actuation.
-
66
Set the pressure at 30 p.s.i. for the solenoid valve that is connected to the sieve valve. Wait till water slowly fills into the sieve valve under the pressure then stop the pressure.
-
67
Plug the PFA tubing of the syringe-tubing assembly into the inlet of the device.
-
68
Fill up the microchamber using IP buffer at a flow rate of 200 μl/min sustained by the syringe pump. The process takes 10–15 s.
? TROUBLESHOOTING
-
69
Decrease the flow rate to 1 μl/min, close the sieve valve, flow IP buffer for 2 min.
-
70
Stop the syringe pump. Unplug the PFA tubing of the syringe-tubing assembly from the inlet.
▲CRITICAL STEP During this step, there may be air bubbles in the inlet reservoir. Use a pipette to remove the bubbles and fill the reservoir with IP buffer.
-
71
Load the antibody-coated IP beads from Step 61(5 μl) into the inlet reservoir using a pipette while operating a permanent magnet (see ‘magnet bar’ in Reagents) from the glass side of the device to guide the beads into the microchamber.
-
72
Load the chromatin sample (50 μl) into a long tubing assembly (10 cm C-Flex clear tubing connected with 3 cm PFA tubing, Supplementary Fig. 1) from the free C-Flex tubing end using a pipette.
-
73
Connect the free C-Flex tubing end of the long tubing assembly with the PFA tubing of the syringe-tubing assembly. Leave an air bubble in the C-Flex tubing to separate the IP buffer and the chromatin sample.
-
74
Carefully plug the PFA tubing of the long tubing assembly into the inlet of the device, while using the permanent magnet to keep the beads in the chamber (Fig. 4b).
▲CRITICAL STEP Leave some space between the end of the PFA tubing and the bottom of the inlet reservoir to hold the air bubble at the end of the chromatin sample injection.
-
75
Set the flow rate at 1.5 μl/min to inject the chromatin sample into the chamber while using the permanent magnet to help move the IP beads to pack against the sieve valve to form a packed bed.
▲CRITICAL STEP Do not pack the IP beads immediately because a sudden increase of pressure may break the sieve valve. Pack the beads around 5 min after the start of the sample injection.
-
76
Use Kimtech wipes to remove waste liquid from the outlets of the devices during the immunoprecipitation step.
-
77
The completion of the chromatin sample flow is indicated by the air bubble reaching the inlet reservoir and getting trapped there. Then flow IP buffer for 5 min at 1.5 μl/min.
-
78
Pull the long tubing assembly out of the device inlet. Separate the long tubing assembly from the PFA tubing of syringe-tubing system. Open the sieve valve by operating the solenoid valve.
▲CRITICAL STEP Pull the PFA tubing out first before opening the sieve valve to prevent bead loss.
? TROUBLESHOOTING
Oscillatory washing •TIMING 0.5 h
▲CRITICAL The oscillatory washing is conducted at room temperature.
-
79
Load 25 μl of Low Salt Washing Buffer into each of two short tubing assemblies from their free C-Flex tubing end.
▲CRITICAL STEP Use clean short tubing assemblies to prevent cross-contamination.
-
80
Connect each short tubing assembly to one end of the device (inlet or outlet) via its PFA tubing end and to one solenoid valve in a 2-set solenoid manifold via its C-Flex tubing end (Fig. 5c, Supplementary Fig. 1). Leave some distance (~3 mm) between the PFA tubing end and the bottom of the reservoir so that the beads are kept in the reservoirs during oscillatory washing (Supplementary Video 1).
▲CRITICAL STEP Try not to lose washing buffer in this step. Make sure that there is still buffer in C-Flex tubing when the setup is finished.
-
81
Set oscillatory washing parameters in the LabVIEW program. Set both duration and interval as 0.5 s for the alternating pressure pulses operated by the two solenoid valves attached to the same device (In the LabVIEW program, enter 0.5 for both “inlet duration” and “outlet duration”). Set the number of oscillations as 300 (generating a total washing time of 5 min). Click the ‘reset’ button in the program to reset time to be 0. Click the “active” button for the sets of solenoid manifolds that will be used in the run. Set ~0.65 p.s.i. as pressure by turning the knobs of the pressure regulators and the set pressure values will be shown in the connected pressure gauges.
▲CRITICAL STEP Each set of pressure regulator and gauge is connected with a 2-solenoid-valve manifold. Move the beads with the magnet to the center of the devices.
-
82
Click the “Oscillation” button in the LabVIEW program to start the oscillatory washing. After washing, pull the PFA tubing out of both ends of the device while using the magnet to retain the beads. Disconnect the short tubing assemblies from the long PFA tubing attached to the solenoid valves.
▲CRITICAL STEP During washing, use the magnet to pull beads back into the inlet/outlet reservoirs when the beads get into the PFA tubing.
-
83
Get another two clean short tubing assemblies; repeat the steps 79–82 with High Salt Washing Buffer. Carefully pull out the two short tubing assemblies from the device.
-
84
Close the sieve valve. Retain the beads with the permanent magnet. Plug the PFA tubing of the syringe-tubing assembly (that contains IP buffer) into the inlet of the device.
-
85
Set the flow rate at 2 μl/min on the syringe pump, and flow IP buffer for 2 min. Unbound chromatin fragments are flushed out of the device in this step.
-
86
Plug a 11 cm PFA tubing elbow (Supplementary Fig. 1) into the outlet of the device (Fig. 5d).
-
87
Place a 1.5 ml Eppendorf microcentrifuge tube at the other end of the PFA tubing elbow to collect eluted beads.
-
88
Set the flow rate at 200 μl/min on the syringe pump, and start the flow. Open and close the sieve valve several times to loosen the beads to facilitate elution.
-
89
Place the tubes (that contain IP beads) on the DynaMag-Spin Magnet for 1 min. Remove and discard all the supernatant.
ChIP DNA isolation •TIMING 2.5 h
▲CRITICAL We describe the extraction of DNA from one ChIP sample and the input below.
-
90
Release the tubes from the DynaMag-Spin Magnet. Add 100 μl of elution buffer into the tube, and then add 4 μl of 20 mg/ml proteinase K. Add 90 μl of elution buffer and 4 μl of proteinase K into the input tube from step 62Axviii (sonication) or 62Bxviii (MNase digestion).
▲CRITICAL STEP Use reverse cross-linking buffer instead of elution buffer if sonicated samples are treated here.
-
91
Put a caplock clip on each tube. Transfer tubes to a Multi-therm heat/cool shaker incubator. Incubate for 8 h at 65 °C (without shaking).
■PAUSE POINT After step 91, the samples can be incubated at 25 °C overnight in the Multi-therm shaker incubator.
-
92
Remove the caplock clip from the tubes. Vortex the mixture for 2 s. Centrifuge the tubes at 300 g for 30 s at room temperature.
-
93
Add 100 μl of phenol-chloroform into each tube. Vortex the mixture for 1 min.
-
94
Centrifuge at 16,000 g for 10 min at room temperature.
-
95
Add 480 μl of 100% ethanol, 60 μl of 5 M ammonium acetate and 6 μl of glycogen into a new 1.5 ml Eppendorf microcentrifuge tube.
-
96
Pipette the upper aqueous phase (eluted DNA) from the tubes of step 94 into the 1.5 ml Eppendorf microcentrifuge tubes from step 95.
-
97
Mix the samples by vortexing for 10 s.
-
98
Place the tubes in a freezer (-80 °C) for 2 h.
-
99
Centrifuge the tubes at 16,100 g at 4 °C for 10 min.
-
100
Carefully remove and discard the supernatant without aspirating the DNA pellet.
? TROUBLESHOOTING
-
101
Add 500 μl of ice-cold 70% ethanol into each tube. Centrifuge at 16,100g at 4 °C for 5 min.
-
102
Remove and discard the supernatant.
-
103
Centrifuge at 16,100g at 4 °C for 30s. Remove any residual supernatant from the tubes. Air-dry the DNA pellet in a biosafety cabinet by leaving the tubes open for 10 min.
▲CRITICAL STEP The pellet becomes crystal clear and hard-to-see when it is dry.
-
104
Resuspend the DNA pellets in 12 μl of low EDTA TE buffer.
■PAUSE POINT The samples can be stored at −20 °C for at least 2 months.
Enrichment check using qPCR •TIMING 3 h
-
105
Transfer 2 μl each of the sample and the input to two separate 1.5 ml Eppendorf microcentrifuge tubes.
-
106
Dilute each tube to 20 μl with low EDTA TE buffer. To assess the enrichment of ChIP DNA from human cells (e.g. GM 12878), follow option A; to assess the enrichment of ChIP DNA from mouse brain nuclei (e.g. nuclei from prefrontal cortex), follow option B.
(A). GM12878 cells
Thaw SYBR Green Master Mix, two primer pairs (e.g. C9orf3, N1 for human H3K4me3 with sequence information in Table 1) at room temperature.
Use SYBR Green Master Mix in a 25 μl reaction for each primer pair (12.5 μl SYBR Green, 2 μl of the primer pair, 4 μl DNA template, 6.5 μl low EDTA TE buffer) in a 96-well plate.
- Perform qPCR using following thermal cycling profile:.
Cycle Denature Anneal Extend 1 95°C, 10 min 2–46 95°C, 15 sec 58°C, 40 sec 72°C, 30 sec
(B). Nuclei from mouse prefrontal cortex
Thaw SYBR Green Master Mix, two primer pairs (e.g. GAPDH 2 and negative set 1 for H3K4me3, Hoxc10 and negative set 3 for H3K27me3, or Zranb3 and N3 for H3K27ac from Table 1) at room temperature.
Use SYBR Green Master Mix in a 20 μl reaction (10 μl SYBR Green, 1.4 μl of the primer pair, 8.6 μl DNA template) in a 96-well plate.
- Perform qPCR using following thermal cycling profile:
Cycle Denature Anneal Extend 1 95°C, 2 min 2–46 95°C, 15 sec 58°C, 20 sec 72°C, 20 sec
-
107Calculate the relative fold enrichment, which is the ratio of percent input between a positive locus and a negative locus. The percent input for a specific locus is calculated using the below equation
where Ctinput and CtChIP are the Ct value of input and ChIP DNA, respectively. DF is the dilution factor, which is defined as (sample volume of input + sample volume of ChIP DNA) / sample volume of input.(1) The enrichment can be calculated using the following equation(2) ? TROUBLESHOOTING
Library preparation •TIMING 6 h
▲CRITICAL Library preparation is performed using Accel-NGS 2S Plus DNA Library kit from Swift Biosciences. Minor modification is made to the manufacturer’s procedures as detailed below.
-
108
Thaw all the reagents in the library kit on ice. Freshly prepare 10 ml of 80% (vol/vol) ethanol solution.
-
109
Transfer 8 μl of the sample from step 104 to a 0.2 ml PCR tube. Dilute the sample to 40 μl with low EDTA TE buffer.
-
110
Add 13 μl of low EDTA TE buffer, 6 μl of Buffer W1, and 1 μl of Enzyme W2 into the sample. Mix on ice by pipetting.
▲CRITICAL STEP Vortex the reagents from the kit for 2 s each before use. Add all the reagents in the specified order.
-
111
Incubate the solution in a thermal cycler at 37 °C for 10 min.
-
112
Add 84 μl of SPRI beads to the solution. Pipette the mixture until it mixes well.
▲CRITICAL STEP Vortex the SPRIselect beads for 5 s before use.
-
113
Incubate for 5 min at room temperature. Place the tubes on a magnetic rack for 5 min. When necessary, use a magnetic bar to further collect the beads into a small pellet. Remove and discard the supernatant.
-
114
Add 180 μl of 80% (v/v) ethanol to the tube without disturbing the bead pellet. Wait for 30 s. Carefully remove and discard the supernatant.
-
115
Repeat step 114 once for a second wash.
-
116
Centrifuge the tube at 200 g for 15 s to further collect the liquid.
▲CRITICAL STEP Manipulate the bead pellet to the bottom of the tube using the magnet bar before the centrifugation.
-
117
Place the tube back to the magnetic rack, remove and discard any residual ethanol solution.
-
118
Air-dry the bead pellet by opening the tube cap in a biosafety cabinet. Keep watching the bead pellet to avoid over-drying. Stop drying upon appearance of a small crack in the pellet. This takes roughly 1–2 min.
-
119
Resuspend the pellet in 30 μl of low EDTA TE buffer.
-
120
Add 5 μl of Buffer G1, 13 μl of Reagent G2, 1 μl of Enzyme G3 and 1 μl of Enzyme G4 into the solution. Mix on ice by pipetting.
▲CRITICAL STEP Vortex all the reagents from the kit for 2 s before use. Add all the reagents in order.
-
121
Seal the tube with a new cap. Incubate the solution in a thermal cycler at 20°C for 20 min.
-
122
Add 60 μl of PEG solution to the tube. Mix by pipetting.
-
123
Repeat steps 113–118 for another bead clean-up.
-
124
Resuspend the pellet in 20 μl of low EDTA TE buffer.
-
125
Add 3 μl of Buffer Y1, 2 μl of Enzyme Y3, and 5 μl of Reagent Y2 into the solution. Mix on ice by pipetting.
▲CRITICAL STEP Vortex all the reagents from the kit for 2 s before use. Add all the reagents in order. Y2 contains the indexed adapter, which is provided separately in the Indexed Adapter kit. Use different adapters for each sample.
-
126
Seal the tube with a new cap. Incubate the solution in the thermal cycler at 25°C for 15 min.
-
127
Add 25.5 μl of PEG solution to the tube. Mix by pipetting.
-
128
Repeat the steps 113–118 for another bead clean-up.
-
129
Resuspend the pellet in 30 μl of low EDTA TE buffer.
-
130
Add 5 μl of Buffer B1, 2 μl of Reagent B2, 9 μl of Reagent B3, 1 μl of Enzyme B4, 2 μl of Enzyme B5, and 1 μl of Enzyme B6 into the solution. Mix on ice by pipetting.
▲CRITICAL STEP Vortex all the reagents from the kit for 2 s before use. Add all the reagents in order.
-
131
Seal the tube with a new cap. Incubate the solution in the thermal cycler at 40°C for 10 min.
-
132
Add 42.5 μl of PEG solution to the tube. Mix by pipetting.
-
133
Repeat steps 113–118 for another bead clean-up.
-
134
Resuspend the pellet in 22 μl of low EDTA TE buffer. Wait for 5 min at room temperature.
-
135
Place the tube on the magnetic rack to pellet the beads. Transfer the supernatant to a new PCR tube.
■PAUSE POINT The ChIP DNA samples can be stored at 4°C for 2–3 h, or at −20°C for up to 1 week.
-
136Create a 50 μl amplification mix in a 96-well plate as outlined below:
Component Amount (μl) low EDTA TE buffer 7.5 20X Evagreen dye 2.5 Reagent R1 5 Reagent R2 4 Buffer R3 10 Enzyme R4 1 ChIP DNA sample 20 Total 50 -
137Place the 96 well plate in the CFX Connect™ Real-Time PCR Detection System. Run the qPCR program:
Cycle Denature Anneal Extend 1 98°C, 30 sec 2–46 98°C, 10 sec 60°C, 30 sec 68°C, 60 sec ▲CRITICAL STEP The number of PCR cycles used for each sample is determined by monitoring its fluorescence intensity. Stop the amplification when the sample’s fluorescence intensity increases by 3000 relative fluorescence units (RFU). ChIP DNA from ~1000 cell/nuclei needs to be amplified 13–14 cycles.
?TROUBLESHOOTING
-
138
Carefully transfer the amplification product into a new PCR tube. Add 37.5 μl of SPRI beads to the tube. Mix by pipetting.
▲CRITICAL STEP Vortex the SPRIselect beads for 5 s before use.
-
139
Repeat steps 113–118 for library clean-up.
-
140
Resuspend the pellet in 10 μl of low EDTA TE buffer. Wait for 5 min at room temperature.
-
141
Place the tube on the magnetic rack to pellet the beads. Transfer the supernatant to a new 1.5 ml microcentrifuge tube. Label different samples properly.
■PAUSE POINT The prepared library can be stored at −80 °C for one year.
-
142
Repeat steps 105–107 to examine the enrichment of the library using qPCR. An enrichment (as defined by eqn 2) of >30 is desired for high-quality ChIP-seq data.
DNA quantification and pooling for sequencing •TIMING 4 h
-
143
Transfer 1 μl of each library from step 141 into a PCR tube. Dilute it to 2 μl using the low EDTA TE buffer. Use the High Sensitivity DNA Analysis kit on the TapeStation to check library fragment size.
▲CRITICAL STEP Follow the instruction for High Sensitivity DNA Analysis kit. Mix solution well before loading them in the TapeStation. The desired fragment size is described in Figure 6.
? TROUBLESHOOTING
-
144
Transfer 1 μl of each library from Step 141 into a PCR tube. Make three serial 1:100 dilutions for each sample using low EDTA TE buffer. Quantify the diluted libraries using a KAPA Library Quantification kit, following the manufacturer’s instruction. Pool the libraries to generate at least 10 nM DNA concentration for sequencing by Illumina HiSeq 4000 with single-end 50-bp read.
▲CRITICAL STEP The number of reads for each sample can be estimated based on the overall number of reads produced per lane, the number of pooled libraries, and the concentrations of the libraries. For example, we pool 16 libraries with 8 of them on H3K4me3 at 8 nM each and the rest on H3K27me3 at 16 nM each. When all the libraries have the same volume (e.g. 2 μl each) and the sequencing lane yields a total of 340 million reads, each H3K4me3 library would have 14 million reads and each H3K27me3 library would have 28 million reads.
Figure 6 |.
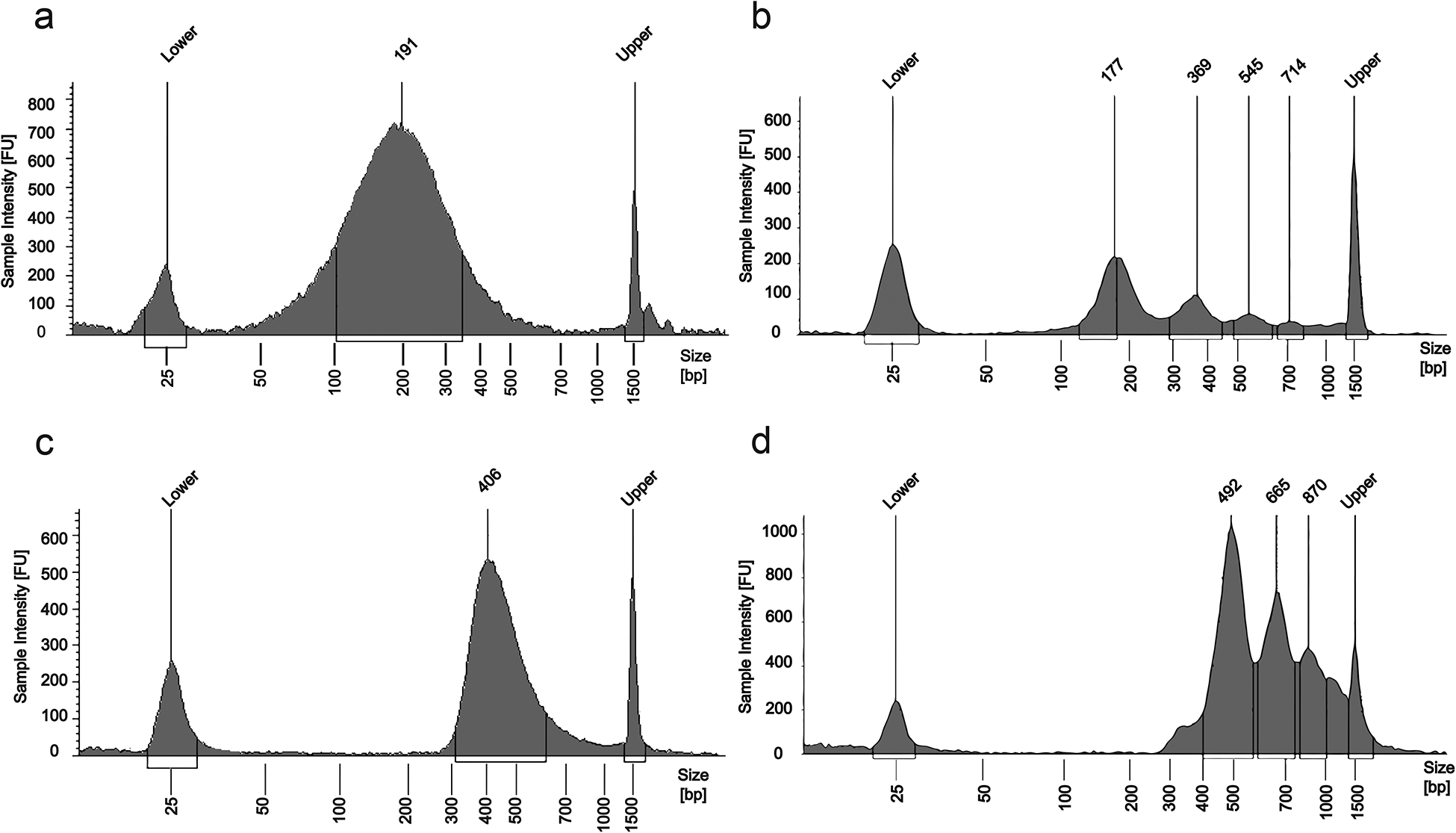
DNA fragment size distributions measured by a TapeStation. “FU” means fluorescence unit. “Lower” and “Upper” refer to 25 bp and 1500 bp peaks generated by the DNA ladder, respectively. (a) DNA from 10,000 GM12878 cells after crosslinking and sonication. (b) DNA from 2000 nuclei isolated from mouse prefrontal cortex after MNase digestion. (c) Sequencing library after H3K4me3 MOWChIP of 10,000 GM12878 cells and library preparation. (d) Sequencing library after H3K4me3 MOWChIP of 2000 mouse prefrontal cortex nuclei and library preparation.
Sequencing data analysis •TIMING 6 h
▲CRITICAL All the steps in data analysis can be combined in a single bash file. The script we use is included in this protocol (Supplementary Data 4).
-
145
Install software including Trim Galore!, Bowtie29, fetchChromSizes, samtools30, bedtools31, MACS232 and epic33 for trimming, alignment, and peak calling.
-
146
Trim the FASTQ files using Trim Galore! with default settings to remove low quality reads. Align the reads to a reference genome, such as hg38 (human) or mm10 (mouse), using Bowtie29.
-
147
Obtain uniquely mapped reads for the input and samples with the samtools view function. Next, generate a 100nt bin across the genome using fetchChromSizes to obtain genome sizes and bedtools makewindows to create the bins. Then, use bedtools coverage–counts to compute a signal for each 100nt bin across the genome with the mapped read files (SAM or BAM format) from both ChIP and input samples.
-
148Use the 100nt bin signal files from the sample and the input, along with the number of mapped reads obtained in Step 147, and normalize the ChIP signal in each 100nt bin using the following equation in a script:
Normalized signal in regions of interest or the entire epigenome for each sample can be used to calculate Pearson Correlation.(3) -
149
The mapped reads are used for narrow peak calling using MACS2 (q-value < 0.05)32 and broad peak calling using epic’s implementation of SICER (–windowSize 1000–gaps-allowed 3)33.
-
150
Extend the reads of both ChIP and input samples by 100 nt on either side then compute a normalized signal for each 100 nt bin as in steps 147–148. Generate a BigWig file and visualize the tracks in a genome browser such as Integrative Genomics Viewer34,35.
Troubleshooting
Troubleshooting advice can be found in Table 2.
TABLE 2│.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 68 | Trapped air bubbles | Sudden injection of IP buffer into the hydrophobic PDMS device | Set the flow rate at 50 μl/min. Close the sieve valve. Wait for 5 s, and then open the valve to flush the air bubble out with the help of pressure generated in the chamber. Alternatively, press the chamber with a pair of tweezers to push the bubble out. |
| 78 | Air bubbles in the chamber after chromatin solution loading | The air bubble that separates IP buffer and chromatin solution is too large. | C-Flex tubing in long-tubing assembly needs to be changed periodically (roughly after 20 experiments) because it may expand over time and hold large air bubble during solution loading. |
| 100 | No visible pellet at the bottom of the microtube | Low concentration of ammonium acetate or ethanol | Be sure to prepare 5 M ammonium acetate solution in ultrapure water accurately. Use high quality glycogen and ethanol. |
| 107 | Low enrichment (<10 when primers in Table 1 are used) for ChIP DNA detected by qPCR | Chromatin fragmentation is not optimal | Adjust conditions for cross-linking and sonication to achieve 200–500 bp fragment size. Keep the temperature of the sample around 4 °C during sonication. For MNase digestion-based fragmentation, try different concentrations of MNase to achieve 170–540 bp fragment size. |
| The quality of antibody is not good. | Test antibodies from other batches or suppliers. | ||
| Problem with the reagents used in the experiment. | Make sure to add protease inhibitors as instructed in the protocol. | ||
| Oscillatory washing conditions are not optimal. | Adjust the time and pressure associated with washing. Generally, increased washing improves enrichment but decreases DNA amount. Ideal percent input is lower than 1% for negative loci, and 10%-40% for positive loci. | ||
| Primer design is not optimal | Design alternative primer sets. | ||
| Selected loci are not optimal | Target other loci in the genome. Order commercial primer sets if they are available. | ||
| 137 | More than 16 cycles are needed in library amplification when the amount of ChIP DNA is > 10 pg | Too much DNA is lost during SPRI beads cleanup steps. | Perform the SPRI cleanup with extra attention and use correct buffer ratio. Be sure to dry the beads after 80% (vol/vol) ethanol washing and also avoid over-drying the beads. |
| 143 | A large amount of adapter dimer in the library (~120 bp) | Insufficient size-selection step | Do the size-selection cleanup with accurate SPRI-to-buffer ratio. |
TIMING
Steps 1–13, Setup of the pneumatic control system: 1 d
Steps 14–26, Fabrication of SU-8 silicon masters: 3 h
Steps 27–50, Fabrication of the microfluidic device: 9 h
Steps 51–61, Coupling an antibody to superparamagnetic beads: 2.5 h
Steps 62, Chromatin preparation: 1 h (option A) or 3.5 h (option B)
Steps 63–78, ChIP: 1.5 h
Steps 79–89, Oscillatory washing: 0.5 h
Steps 90–104, ChIP DNA isolation: 2.5 h
Steps 105–107, Enrichment quality check using RT-PCR: 3 h
Steps 108–142, Library preparation: 6 h
Steps 143–144, DNA quantification and pooling for sequencing: 4 h
Steps 145–150, Sequencing data analysis: 6 h
ANTICIPATED RESULTS:
Chromatin preparation
Optimal chromatin shearing after sonication results in fragments in the range of 200–500 bp (Fig. 6a). MNase digestion typically results in several peaks at ~180 bp, ~370 bp, and ~540 bp (Fig. 6b). About 120 bp increase in the DNA fragment size can be observed after library preparation (Fig. 6c and 6d). The cell type, conditions of cross-linking and sonication all affect the chromatin fragment size distribution observed by Tapestation. For MNase digestion, MNase concentration critically affects the result. The chromatin fragment size significantly affects ChIP DNA enrichment and ChIP-seq data quality. Thus careful tuning of the fragmentation conditions is important.
MOWChIP
There are several parameters associated with the MOWChIP process that need to be closely watched. First, the amount of ChIP DNA yielded by MOWChIP is typically in the range of 5–12% of the amount of DNA contained in the starting chromatin and this may vary depending on the mark being probed. A percentage that is significantly lower than this range possibly indicates excessive washing. On the other hand, a yield that is much higher than this range suggests insufficient washing and predicts poor enrichment. Second, the enrichment (calculated using Eqn 2) of ChIP DNA critically measures the success of the ChIP process. The enrichment depends on cell type, specific negative and positive loci selected, type of histone modification, and experimental conditions. Based on our experience, the enrichment of ChIP DNA needs to be >30 to generate ChIP-seq data of high-quality and >15 to generate data of acceptable quality, when primers in Table 1 are used. Measurement of ChIP DNA enrichment using qPCR allows optimization of experimental conditions without costly sequencing (Fig. 7).
Figure 7 |.
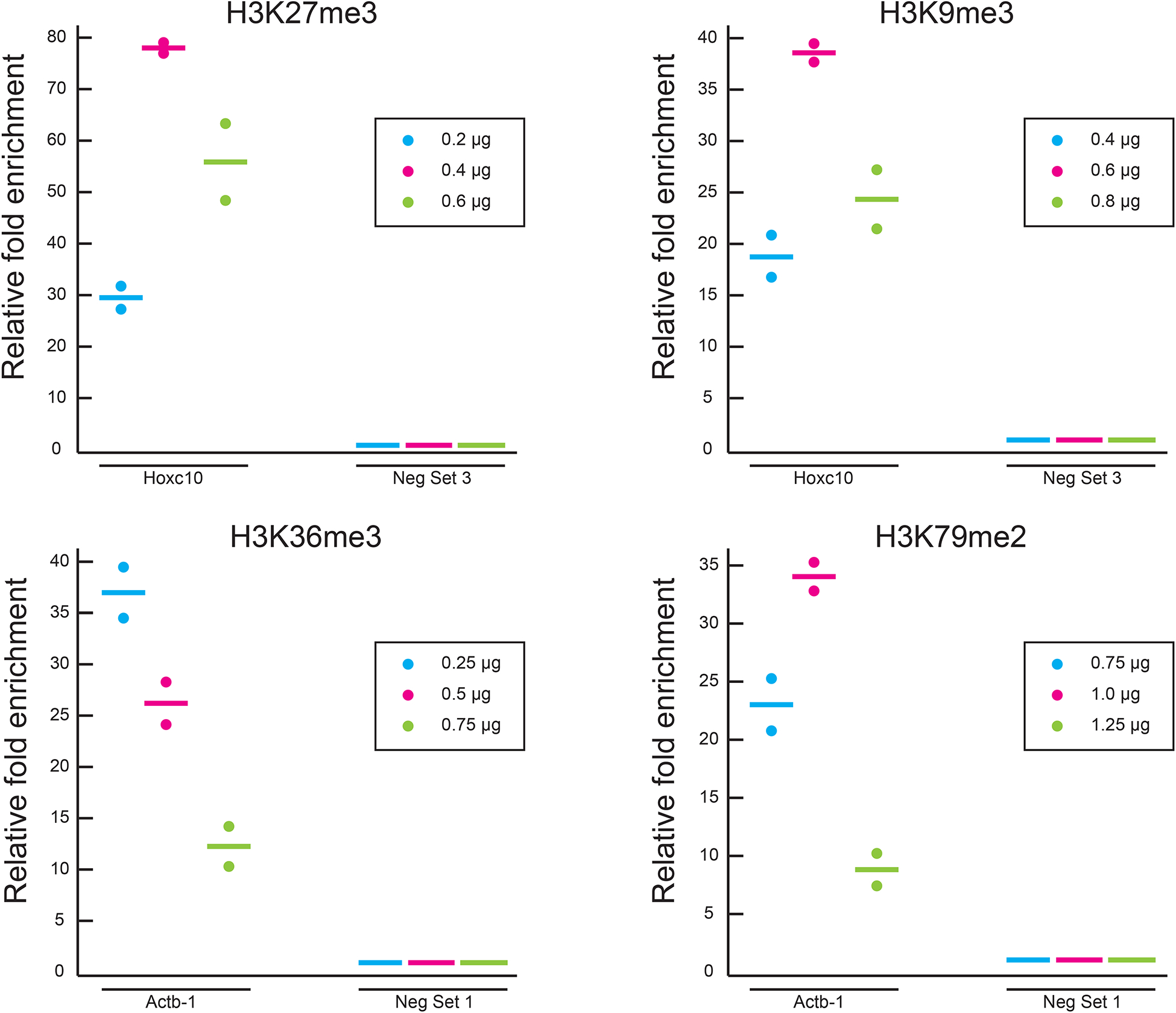
Optimization of antibody quantity using MOWChIP-qPCR for 4 histone marks. For each histone mark, 3 different quantities of antibody were used during IP bead coating and enrichment of ChIP DNA at positive (Hoxc10 for H3K27me3 and H3K9me3; Actb-1 for H3K36me3 and H3K79me2) and negative (Neg Set 3 for H3K27me3 and H3K9me3; Neg Set 1 for H3K36me3 and H3K79me2) loci. Enrichment was examined by qPCR under each condition. All experiments were conducted in duplicate and the horizontal lines represent the mean. The relative fold enrichment was normalized against that of Neg Set 3 or Neg Set 1. 1000 nuclei from mouse prefrontal cortex were used in each assay and MNase digestion was used for chromatin fragmentation. 10-week old CD-1 male mice were used in the study. The nuclei were not sorted and included neurons and glia. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University.
Library preparation
It is important to minimize the number of amplification cycles in order to keep the enrichment in ChIP DNA during library preparation. Only the minimal quantity of DNA required for sequencing should be produced from library preparation. The enrichment of the library is highly predictive of the ChIP-seq data quality. The enrichment of the library needs to be higher than 30 in order to generate high-quality ChIP-seq data in most cases.
Sequencing results
The number of called peaks varies with the cell type and the histone mark. The number of peaks yielded by MOWChIP-seq using 100–10,000 cells or nuclei is typically similar to that yielded by conventional assays (for example, the data sets by ENCODE). In addition to H3K4me3 and H3K27ac data on various cell types in our previous publications8,36, we show 4 data sets on H3K27me3, 2 data sets on H3K9me3, 2 data sets on H3K36me3, and 2 data sets on H3K79me2 generated using 1000 nuclei from mouse prefrontal cortex per assay (Fig. 8a). Our technology works well with hard-to-profile marks such as H3K27me3 and H3K9me3. We called 15K-20K peaks in these datasets. The Pearson correlations among replicates of these histone makers are in the range of 0.89–0.96 (0.94 for H3K27me3, 0.93 for H3K9me3, 0.96 for H3K36me3, and 0.89 for H3K79me2) (Fig. 8b).
Figure 8 |.
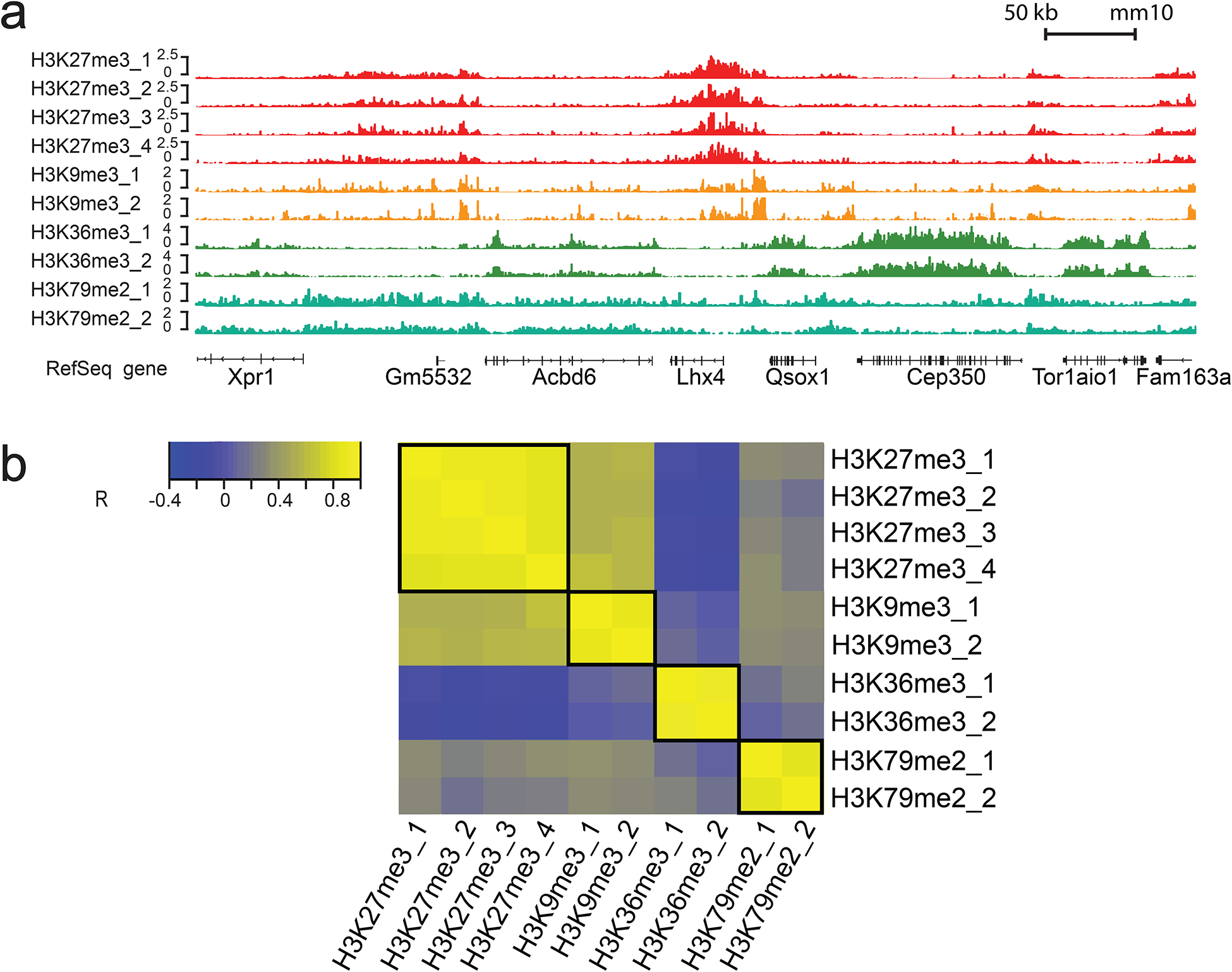
MOWChIP-seq data on H3K27me3, H3K9me3, H3K36me3, and H3K79me2 generated using 1000 nuclei from mouse prefrontal cortex per assay. H3K27me3 data (4 replicates) were produced by 4 units of an 8-unit MOWChIP device while H3K9me3, H3K36me3, and H3K79me2 data (2 replicates each) were produced by 6 units of another 8-unit MOWChIP device. 10-week old CD-1 male mice were used in the study. The nuclei were not sorted and included neurons and glia. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Virginia Commonwealth University. (a) Normalized H3K27me3, H3K9me3, H3K36me3, and H3K79me2 signal. (b) Cross correlations of H3K27me3, H3K9me3, H3K36me3, and H3K79me2 ChIP-seq signals in the promoter regions. R represents Pearson correlation coefficient.
Supplementary Material
Supplementary Video 1. A video on major steps involved in MOWChIP.
Supplementary Figure 1. Various tubing assemblies used in the protocol.
Supplementary Data 1. A LabVIEW program for operating the single-unit MOWChIP device.
Supplementary Data 2. A LabVIEW program for operating the 8-unit MOWChIP device.
Supplementary Data 3. A LayoutEditor file for the 8-unit MOWChIP device.
Supplementary Data 4. A custom script for ChIP-seq data analysis.
Acknowledgements
We thank Dr. Javier González-Maeso of Virginia Commonwealth University for providing mouse brain samples. This work was supported by US National Institutes of Health grants CA214176 (CL), EB017235 (CL), HG009256 (CL), HG008623 (CL), and seed grants from Center for Engineered Health of Virginia Tech Institute for Critical Technology and Applied Science.
Footnotes
Data availability. Gene Expression Omnibus: MOWChIP-seq data on H3K27me3, H3K9me3, H3K36me3, and H3K79me2 in mouse prefrontal cortex (Figure 8) are deposited under accession number GSE123606 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123606).
Code availability. A custom script is included as Supplementary Data 4.
Competing interests
C.L. declares that he is a co-inventor of a US patent (9,732,377) issued on MOWChIP-seq. The remaining authors declare no competing financial interests.
Related links
Key references using this protocol
Cao, Z. et al. Nat. Methods 12, 959–962 (2015) https://doi.org/10.1038/nmeth.3488
Cox, M. et al. ACS Biomaterials Science & Engineering 5, 1544–1552 (2019) https://doi.org/10.1021/acsbiomaterials.9b00161
References
- 1.Egger G, Liang GN, Aparicio A & Jones PA Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463, (2004). [DOI] [PubMed] [Google Scholar]
- 2.Campos EI & Reinberg D Histones: annotating chromatin. Annu. Rev. Genet 43, 559–599, (2009). [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45, (2000). [DOI] [PubMed] [Google Scholar]
- 4.Zhou VW, Goren A & Bernstein BE Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet 12, 7–18, (2011). [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y & Garcia BA Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb Perspect Biol 7, a025064, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I & Zhao K High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837, (2007). [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ & Zhao K Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet 40, 897–903, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Z, Chen C, He B, Tan K & Lu C A microfluidic device for epigenomic profiling using 100 cells. Nat. Methods 12, 959–962, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adli M, Zhu J & Bernstein BE Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat. Methods 7, 615–618, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shankaranarayanan P, Mendoza-Parra MA, Walia M, Wang L, Li N, Trindade LM & Gronemeyer H Single-tube linear DNA amplification (LinDA) for robust ChIP-seq. Nat. Methods 8, 565–U565, (2011). [DOI] [PubMed] [Google Scholar]
- 11.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, Friedman N & Amit I. Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma S, Hsieh Y-P, Ma J, Lu C Low-input and multiplexed microfluidic assay reveals epigenomic variation across cerebellum and prefrontal cortex. Sci. Adv 4, eaar8187, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA & Bernstein BE Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skene PJ, Henikoff JG & Henikoff S Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019, (2018). [DOI] [PubMed] [Google Scholar]
- 16.Hainer SJ, Boskovic A, McCannell KN, Rando OJ & Fazzio TG Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 177, 1319–1329, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K & Henikoff S CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harada A, Maehara K, Handa T, Arimura Y, Nogami J, Hayashi-Takanaka Y, Shirahige K, Kurumizaka H, Kimura H & Ohkawa Y A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol 21, 287–296, (2019). [DOI] [PubMed] [Google Scholar]
- 19.Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C & Snyder M An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T & Kellis M Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brower K, Puccinelli RR, Markin CJ, Shimko TC, Longwell SA, Cruz B, Gomez-Sjoberg R & Fordyce PM An open-source, programmable pneumatic setup for operation and automated control of single- and multi-layer microfluidic devices. HardwareX 3, 117–134, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thorsen T, Maerkl SJ & Quake SR Microfluidic large-scale integration. Science 298, 580–584, (2002). [DOI] [PubMed] [Google Scholar]
- 23.Unger MA, Chou H-P, Thorsen T, Scherer A & Quake SR Monolithic Microfabricated Valves and Pumps by Multilayer Soft Lithography. Science 288, 113–116, (2000). [DOI] [PubMed] [Google Scholar]
- 24.Brower K, White AK & Fordyce PM Multi-step Variable Height Photolithography for Valved Multilayer Microfluidic Devices. J. Vis. Exp, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma S, de la Fuente Revenga M, Sun Z, Sun C, Murphy TW, Xie H, González-Maeso J & Lu C Cell-type-specific brain methylomes profiled via ultralow-input microfluidics. Nat. Biomed. Eng 2, 183–194, (2018). [PMC free article] [PubMed] [Google Scholar]
- 26.Horz W & Altenburger W Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res. 9, 2643–2658, (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dingwall C, Lomonossoff GP & Laskey RA High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 9, 2659–2673, (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung YL, Luquette LJ, Ho JW, Ferrari F, Tolstorukov M, Minoda A, Issner R, Epstein CB, Karpen GH, Kuroda MI & Park PJ Impact of sequencing depth in ChIP-seq experiments. Nucleic Acids Res. 42, e74, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R & Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W & Liu XS Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137–R137, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu S, Grullon S, Ge K & Peng W Spatial Clustering for Identification of ChIP-Enriched Regions (SICER) to Map Regions of Histone Methylation Patterns in Embryonic Stem Cells. Methods in molecular biology (Clifton, N.J.) 1150, 97–111, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorvaldsdóttir H, Robinson JT & Mesirov JP Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics 14, 178–192, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G & Mesirov JP Integrative Genomics Viewer. Nat. Biotechnol 29, 24–26, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox MCD,C; Naler L, Lu C; Verbridge SS Effects of culture condition on epigenomic profiles of brain tumor cells. ACS Biomater. Sci. Eng 5, 1544–1552, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. A video on major steps involved in MOWChIP.
Supplementary Figure 1. Various tubing assemblies used in the protocol.
Supplementary Data 1. A LabVIEW program for operating the single-unit MOWChIP device.
Supplementary Data 2. A LabVIEW program for operating the 8-unit MOWChIP device.
Supplementary Data 3. A LayoutEditor file for the 8-unit MOWChIP device.
Supplementary Data 4. A custom script for ChIP-seq data analysis.