

Abstract
HBx is the smallest gene product of the Hepatitis B virus (HBV) and an oncogenic stimulus in chronic infections leading to liver disease. HBx interacts and interferes with numerous cellular processes, but its modes of action remain poorly understood. It has been invoked that HBx employs nucleotide hydrolysis to regulate molecular pathways or protein–protein interactions. In the present study, we reinvestigate the (d)NTP hydrolysis of recombinant HBx to explore its potential as a biochemical probe for antiviral studies. For our investigations, we employed existing soluble constructs (i.e., GST-HBx, MBP-HBx) and engineered new fusion proteins (i.e., DsbC-HBx, NusA-HBx), which are shown to serve as better systems for in vitro research. We performed mutational scanning of the computationally predicted NTP-binding domain, which includes residues associated with clinical cases. Steady-state and end-point activity assays, in tandem with mass-spectrometric analyses, reveal that the observed hydrolysis of all alleged HBx substrates, ATP, dATP, and GTP, is contingent on the presence of the GroEL chaperone, which preferentially copurifies as a contaminant with GST-HBx and MBP-HBx. Collectively, our findings provide new technical standards for recombinant HBx studies and reveal that nucleotide hydrolysis is not an operant mechanism by which HBx contributes to viral HBV carcinogenesis.
Introduction
Chronic Hepatitis B virus (HBV) infections affect approximately 350 million people worldwide and lead to the development of liver diseases such as cirrhosis and hepatocellular carcinoma (HCC).1−3 HBV infections account for more than 50% of the global HCC cases, making HBV the most dominant agent of hepatocellular malignancies.2 The viral oncogenic mechanisms are poorly understood, but significant evidence has identified a relationship between the 17 kDa HBV gene product, protein X (HBx), and disease pathogenesis.2,4−6 HBx acts as an oncoprotein by regulating viral replication and cellular functions via interactions with numerous pathways.1,7 However, key details about its modus operandi are missing, mainly because it lacks sequence homology to known proteins and is sparingly soluble, hindering biochemical studies. To manage the solubility challenges of HBx, the recombinant protein is commonly purified under denaturing conditions or under native conditions in the form of fusion to the glutathione S-transferase (GST) or the maltose-binding protein (MBP).8−10 Both GST-HBx and refolded, untagged HBx have been reported to hydrolyze ATP, dATP, and GTP.8,11,12 Such activities are particularly intriguing as they raise the possibility that HBx can modulate protein–protein interactions via nucleotide hydrolysis or phosphorylation of target proteins (including itself).13,14 Indeed, many of the proteins and pathways with which HBx interacts are highly regulated by phosphorylation, including p53-associated pathways, the Jak-STAT pathway, and the PI3K/Akt signaling pathway.15−17 We therefore reinvestigated the poorly understood NTP hydrolytic activity of HBx to allow for elucidation of its functional potential in cellular processes leading to disease. Moreover, nucleotide hydrolysis can be used for the development of generic methods for in vitro biochemical studies of HBx, which are currently lacking.
In the present study, we have reviewed the ability of four soluble fusion HBx proteins to hydrolyze nucleotides, with the aim to establish the kinetic parameters, resolve the protein regions involved in nucleotide interaction, and determine the range of possible substrates. Mutational mapping of the predicted ATP-binding region shows that activity is insensitive to the substitution of amino acids that are considered critical for NTP interactions and hydrolysis. Activity assays, combined with mass-spectrometric analyses, reveal that the GST-HBx and MBP-HBx constructs, which are often employed in recombinant HBx research,8,10,18 copurify with significant amounts of the Escherichia coli chaperone, GroEL. This chaperone is shown to be the sole source of the NTP hydrolytic activity previously attributed to HBx, and its presence poses a significant caveat for in vitro HBx studies.
Results and Discussion
The primary amino acid sequence of HBx does not have a canonical ATP-binding motif. However, the absence of such a motif does not prove the inability of protein polypeptide to bind and hydrolyze ATP.19 By employing prediction software,20 we identified a putative triphosphate-binding site in the highly conserved C-terminal region of HBx, spanning residues 130–141 (Figure 1A and Table S2). The predicted sequence region, [KVFVLGGCRHKL]130–141, weakly resembles a deviant Walker A nucleotide-binding motif (Figure 1B).19,21,22 Canonical and deviant Walker A motifs contain conserved glycine and lysine residues critical for nucleotide binding and hydrolysis.23,24 The role of glycines is to exclude water from the active site and allow for flexibility upon nucleotide binding, while lysines are required for stabilizing the negatively charged phosphate groups.25,26 Among such residues in the primary sequence of HBx, Gly-136 is predicted to have the highest probability for interaction with triphosphates and, together with Lys-130 and Lys-140, aligns well with the catalytic residues of both canonical and deviant Walker A motifs (Figure 1B and Table S2). We thus substituted Gly-136 and the two lysines with alanines, generating the following four HBx variants: G136A, K130A, K140A, and K130A/K140A.
Figure 1.
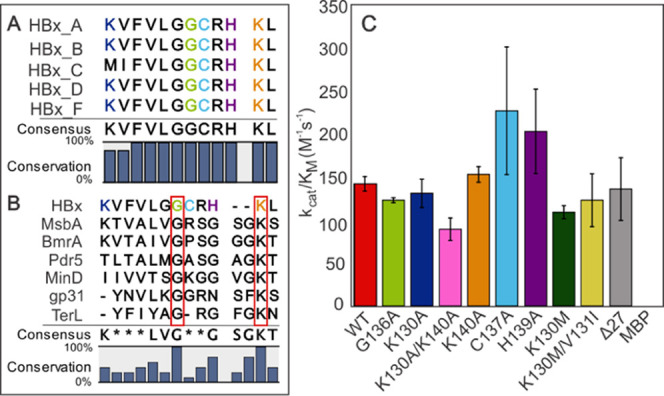
Predicted nucleotide-binding domain and ATP hydrolysis by WT and variant MBP-HBx. (A) Sequence alignment of the predicted HBx nucleotide-binding region in the well-studied HBx genotypes. Residues thought to be important for binding and/or are known to be essential for the HBx function are shown in colors. (B) Sequence alignment of the HBx (A2) putative ATP-binding domain and known Walker A motifs. MsbA, Pdr5, and BmrA contain canonical Walker A sequences, while MinD, gp31, and TerL utilize Deviant I, Deviant II, and Deviant III Walker A motifs, respectively. The red boxes highlight the residues conserved in all sequences. (C) Catalytic efficiency of MBP-HBx WT and variants of the putative ATP-binding domain. Error bars represent standard error. Error bars for MBP are not visible due to the y-axis scale.
The predicted C-terminal HBx region is associated with HCC development, and thus, this domain must contain residues important for the regulation of yet-to-be-described HBx functions, some of which may be ATP hydrolysis-based. We posited that any effect on the ATPase activity as a result of amino acid changes in this domain would provide details into the possible hydrolysis-based mechanisms in disease progression. In this respect, we generated a series of substitutions and deletions of residues within the predicted binding domain (Figure 1A) that are implicated in the transactivation function of HBx or in HCC. Cys-137 and His-139 were exchanged to alanines due to their importance in HBx-mediated HBV replication.27,28 The clinical double variant, K130M/V131I, occurs with a high frequency (∼66%) in patients with HBV-related HCC.29 We thus generated this variant and additionally introduced the single substitution, K130M, to address the effect of the lysine to methionine exchange. Finally, we generated the clinical C-terminal truncated variant, Δ27, which is known to promote cell proliferation and lacks the entirety of the predicted ATP-binding domain.30
HBx fused to MBP has been previously shown to be soluble, possess some tertiary structure through disulfide bonds, and exhibit transactivation activity.10,18 We therefore performed our biochemical investigations with MBP-HBx. The WT MBP-HBx hydrolyzes ATP with a catalytic constant of kcat = 0.017 ± 0.0004 s–1 and a catalytic efficiency of kcat/KM = 128 ± 4 M–1 s–1. This activity is significantly lower than that exhibited by most known ATPases, for which the kcat values range between approximately 0.1 and 90 s–1 and the catalytic efficiencies span a range of 103–104 M–1 s–1.31−33 All variants examined exhibit catalytic efficiencies similar to that of the WT MBP-HBx, and none of the substitutions, including a complete clinical truncation of the putative nucleotide-binding domain, result in complete abolishment of the activity (Figure 1C). In previous studies, extensive sequence deletions and truncations (Δ22–36, Δ33–68, Δ89–118) had only a negligible effect on ATP hydrolysis.8 The independence of HBx ATPase activity from all examined clinical mutations and truncations demonstrates that disease progression associated with these HBx variants does not proceed via ATP-related mechanisms. The poor ATP hydrolysis of WT MBP-HBx and the insensitivity of the catalytic efficiencies of MBP-HBx variants pose the obvious question of whether the observed ATP hydrolysis arises from a contaminating protein and not HBx itself.
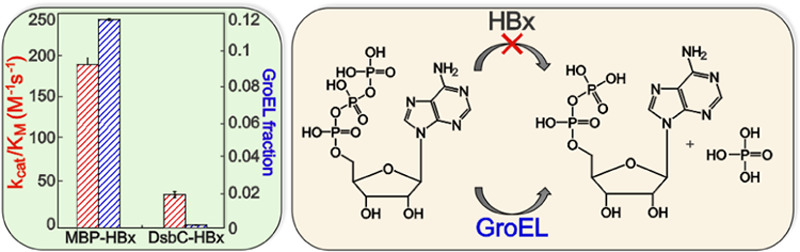
On the basis of SDS-PAGE analyses, MBP-HBx appears more than 95% pure, exhibiting a single band at the apparent molecular weight of the fusion protein (∼60 kDa) (Figure 2A) and thus the identity of a possible contaminating protein is difficult to infer. Therefore, we performed mass-spectrometric analyses on all our MBP-HBx samples to identify any otherwise undetectable proteins present that may contribute to the observed ATPase activity. Solution-based mass-spectrometric analyses show that all MBP-HBx samples contain significant amounts of the E. coli chaperone, GroEL (Table S3). GroEL has the same apparent molecular weight as MBP-HBx, which explains our inability to discern its presence by gel electrophoresis. GroEL is an ATP-dependent chaperone, for which the hydrolytic activity varies in the presence of client proteins and its co-chaperonin, GroES.34,35 While this variability prevents a direct correlation between the GroEL amount and MBP-HBx hydrolytic activity, the hydrolysis rates measured for MBP-HBx are comparable to those reported for GroEL (0.02–0.2 s–1).34,35 Moreover, the apparent Michaelis constant of WT MBP-HBx for ATP (135 ± 4 μM) agrees well with that measured for GroEL (255 ± 57 μM) (Figure S1), providing further support that the mass-spectrometrically identified GroEL is responsible for the observed ATP hydrolysis. Attempts at uncoupling the GroEL activity from the possible MBP-HBx activity via native, in-gel ATPase assays did not yield any interpretable results because MBP-HBx forms high-molecular-weight species in solution that cannot be separated from the native GroEL oligomer.36 Efforts to completely remove GroEL from the purified MBP-HBx were unsuccessful.
Figure 2.

Purity and ATPase activity of HBx fusion proteins. (A) Coomassie-stained SDS-PAGE gel of HBx fusion proteins (expected molecular weight NusA-HBx: 76 kDa, MBP-HBx: 60 kDa, GST-HBx: 44 kDa, DsbC-HBx: 42 kDa). Due to proteolytic degradation, GST-HBx samples have high levels of free GST (27 kDa). This degradation is time-dependent and could not be alleviated with protease inhibitors, suggesting an intrinsic instability of the GST-HBx fusion, the molecular origin of which is outside the scope of our studies. The GST-HBx fusion protein band at a molecular weight of ∼44 kDa is boxed. GroEL, GST, and, to a lesser extent, HBx contribute to the ∼60 kDa band (indicated with a star), as determined by mass spectrometry. (B) Michaelis–Menten kinetics of HBx fusion proteins. Error bars are not shown for GST-HBx because the data exhibit a large degree of scattering due to the difficulty in consistently quantifying the amount and purity of the soluble fusion protein. (C) Correlation between the kcat/KM of HBx fusion proteins for ATP and the amount of GroEL present in the sample. Error bars indicate standard error.
To circumvent these limitations, we generated two new HBx fusion constructs in the anticipation that they might not copurify with the GroEL chaperone. HBx was thus fused to the N utilization substance A (NusA) and the disulfide bond isomerase C (DsbC). For these constructs, the yield of soluble HBx fusion is similar to or improved when compared to that of MBP-HBx (Table 1 and Figure 2A). The GST-HBx fusion was also generated, which, in the previous studies, showed (d)ATP hydrolysis with an apparent KM of 95 μM for dATP.8,11 However, GST-HBx exhibits significant proteolytic degradation leading to high levels of free GST (Figure 2A), and therefore, the total yield (and purity) of the fusion protein is much lower compared to that of the other constructs but similar to previous reports.8,9,11
Table 1. Yield and Purity of HBx Fusion Proteinsa.
| yield (mg/L) | % HBx fusion | % GroEL | |
|---|---|---|---|
| GST-HBx | 0.4 ± 0.3 | 3.5 ± 2.2 | 8.4 ± 2.2 |
| MBP-HBx | 8.9 ± 1.3 | 74.0 ± 0.4 | 11.7 ± 0.04 |
| DsbC-HBx | 6.6 ± 0.6 | 93.8 ± 0.5 | 0.1 ± 0.01 |
| NusA-HBx | 28.6 ± 4.7 | 82.6 ± 0.5 | 0.2 ± 0.01 |
The percentages of the HBx fusions and GroEL in the samples were quantified by mass spectrometry using the emPAI values.
Due to molecular mass differences, GroEL copurifying with these HBx fusions is evident via electrophoresis, unlike the case of MBP-HBx. In this respect, initial inspection of the SDS gels indicates a lack of GroEL in the NusA-HBx and DsbC-HBx samples, but not in the GST-HBx samples, for which a band at the same apparent molecular weight to that of GroEL is detected (Figure 2A). This band at ∼60 kDa was also observed in the previous studies of recombinant GST-HBx, but its chemical origin has been controversial depending on the method employed for its identification. Using an HBx antibody,8 the ∼60 kDa band was attributed to the GST-HBx dimer, while using mass spectrometry,9 the same apparent band was assigned to the GroEL chaperone (Hsp60 family). In our studies, the mass-spectrometric analysis of the ∼60 kDa SDS gel-excised band (Figures 2A and S2) shows its composition to be a mixture of GroEL, GST, and HBx (minor component). Our result is consistent with both previous reports and suggests that when employing an HBx antibody, GroEL was present but remained undetected, thus contributing to the ATPase activity falsely attributed to HBx.
Solution-based mass-spectrometric measurements on all four HBx fusions show that the amount of copurifying GroEL is strongly construct-dependent. GST-HBx and MBP-HBx copurify with the GroEL chaperonin, whereas DsbC-HBx and NusA-HBx samples contain minimal amounts of the chaperone (Table 1). It is unlikely that the preferential binding of GroEL is of biological relevance, as GroEL is known to assist in the folding of recombinantly expressed fusion proteins.37,38 While it is unclear if the GroEL interaction with MBP-HBx and GST-HBx is dependent on HBx or the solubility tag, the nature of this interaction is beyond the scope of our studies. The amount of HBx fusion and GroEL in each sample was quantified using the exponentially modified Protein Abundance Index (emPAI) values extracted from mass spectrometry.39 These emPAI values were used to estimate the total concentration of the fusion protein in each sample to afford a more genuine comparison of the catalytic efficiencies among the different constructs (Figure 2C). The amount of GroEL present in each of the HBx fusions positively correlates with the extent of observed ATP hydrolysis (Figures 2C and S3). DsbC-HBx and NusA-HBx, which copurify with very low amounts of the chaperone (Table 1), lack any significant hydrolytic activity. These results support that the hydrolytic activity stems from the chaperone rather than HBx itself.
HBx was also reported to act on other nucleotides, specifically dATP and GTP. We thus monitored hydrolysis of the three purported nucleotide HBx substrates with all fusion proteins, as well as purified GroEL. The extent of ATP, dATP, and GTP hydrolysis was determined via end-point reactions by reverse-phase high-performance liquid chromatography. MBP-HBx hydrolyzes ∼60% of ATP within 20 min, whereas DsbC-HBx and NusA-HBx exhibit ∼13 and ∼9% conversions, respectively (Figure 3A). Purified GroEL shows almost complete conversion under the same conditions (Figure 3B). For all HBx constructs, the extent of dATP hydrolysis is comparable to that of ATP, demonstrating an insensitivity of the activity to the oxy or deoxy form of ATP (Figure 3C). However, GTP hydrolysis is one- to three-fold lower, demonstrating a dependence on the chemical nature of the base. The small mismatches in the activities between the different HBx fusions and the exact GroEL amount are not surprising and can be explained considering that GroEL activity is influenced by the presence/absence of GroES and client proteins, which, in this case, may be the different HBx fusions.35 GroEL hydrolyzes dATP as efficiently as ATP and to a lesser extent GTP, showing that all nucleotides can also be processed by the chaperone. The dependence of the hydrolytic activity on the nature of the HBx construct and the consistent trend in nucleotide preference between GroEL and the different HBx samples (Figure 3C) provides convincing evidence that the chaperone, and not HBx, is responsible for the (d)NTP hydrolysis.
Figure 3.
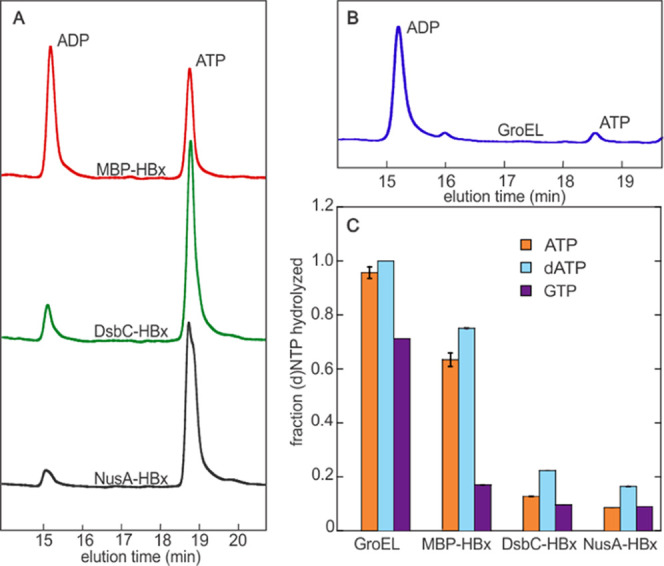
Hydrolysis of (d)ATP and GTP by HBx and GroEL. (A) HPLC traces comparing the hydrolysis of ATP by MBP-HBx, DsbC-HBx, and NusA-HBx in end-point assays (20 min). (B) HPLC trace of ATP hydrolysis by GroEL in an end-point assay (20 min). (C) Fraction of (d)NTPs hydrolyzed by GroEL and HBx fusions. Data are an average of two replicates. Error bars indicate standard error. Experimental conditions: [protein] = 5 μM, [(d)NTP] = 100 μM, reaction time: 20 min.
Conclusions
Recombinant HBx is biologically active and reported to exhibit NTP hydrolysis.40 In the present study, we carried out a detailed investigation on a series of variant and soluble WT HBx fusions to quantify and characterize their hydrolytic activities. We reveal that the molecular origin of this activity lies in the copurifying GroEL chaperone and not in HBx. Thus, clinical variants in the predicted NTP-interacting region must modulate HBx via molecular mechanisms different from ATP hydrolysis. We show that GST-HBx and MBP-HBx, two widely employed constructs for in vitro HBx studies, copurify with the chaperone, a fact that can interfere with antiviral studies employing recombinant HBx. We engineered two new HBx soluble fusions that are superior for in vitro biochemical assays of HBx in terms of both their improved solution behavior and purity. These fusions lack the contaminating chaperone and thus any detectable ATP hydrolysis, offering new standards for in vitro HBx studies and the development of new HBV therapeutic avenues. Collectively, our findings challenge the current notion that HBx hydrolyzes nucleotides and show that this activity is an artifact, allowing for disambiguation of one of its many attributed roles within the cell. The presumed role of HBx in oncogenesis via ATP hydrolysis-based mechanisms must be reconsidered, as HBx does not harbor any such activity.
Experimental Section
Overexpression of Fusion HBx
The HBx sequence genotype A2 (NCBI accession: P69713) was inserted into the pMtac vector (kindly gifted by Dr. Michael Marr, Brandeis University, MA) which allows for its expression as a fusion with the maltose-binding protein (MBP) with a cleavable tobacco etch virus (TEV) recognition site. Additionally, the HBx A2 sequence was inserted into the pET-40b(+) vector (kindly gifted by Mehmet Berkmen, NEB) for expression as a fusion with the disulfide bond isomerase (DsbC).41 The vector was modified such that the S-tag and thrombin sites were replaced with a His6 tag and a TEV recognition site. For the expression of HBx as a fusion with the N utilization substance A protein (NusA-HBx) and the glutathione S-transferase (GST-HBx), the HBx sequence was inserted into the pDB.His.NusA and pDB-HisGST vectors (Berkeley Structural Genomics Center, CA), respectively. Both vectors express the protein of interest as a fusion with an N-terminal His6 tag followed by the solubility enhancement tag (i.e., NusA or GST) and a TEV recognition site. All constructs confer kanamycin resistance. The HBx plasmid was transformed into E. coli T7 express cells (NEB, Ipswich, MA), and the cells were grown in Luria-Bertani (LB) media with 50 mg/L kanamycin at 37 °C with shaking (200 rpm) until they reached an OD600 of ∼0.6, at which time they were cold-shocked at 4 °C for 1 h. Protein expression was induced by the addition of 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), and the cultures were additionally supplemented with 0.25 mM (NH4)2Fe(SO4)2·6H2O. The cells were then incubated with shaking (200 rpm) at 18 °C for 18–20 h and harvested by centrifugation at 7000 rpm for 15 min at 4 °C. Cell pellets were flash-frozen in liquid N2 and stored at −80 °C prior to further usage.
Generation of MBP-HBx Variants
Single and multiple amino acid substitutions and sequence truncations of HBx (when attached to an N-terminal MBP-tag) were generated using the Q5 Hot Start Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA), with the primers listed in Table S1. All WT and variant sequences were confirmed by Sanger Sequencing (Genewiz Inc., NY).
Purification of WT and Variant MBP-HBx
MBP-HBx cell pellets were resuspended in 50 mM HEPES (pH 8.0) and 300 mM NaCl (Buffer 1). Phenylmethylsulfonyl fluoride (PMSF) was added to the cell suspension to a final concentration of 45 μg/mL. The suspension was then lysed via sonication for a total time of 30 min (15 s pulse, 59 s pause, 60% amplification) and centrifuged at 22 000g for 30 min. The clarified lysate was loaded onto an MBP-trap (2 × 5 mL, GE Healthcare) equilibrated in Buffer 1. The column was washed with Buffer 1 to remove nonspecifically bound proteins. The bound protein was eluted using 50 mM HEPES (pH 8.0), 300 mM NaCl, 10 mM maltose (Buffer 2). The fractions containing the protein of interest were concentrated at 4000g using a 30 K MWCO Amicon Centrifugal Filter (Millipore, Sigma). In all cases, the fractions eluted from the MBP-trap contained MBP-HBx and endogenous MBP. An additional size exclusion chromatography purification step was performed using a HiLoad 16/600 Superdex-200 column (120 mL, GE Healthcare) equilibrated with 50 mM HEPES (pH 8.0), 150 mM NaCl, 10% glycerol (Buffer 3). The fractions containing pure MBP-HBx were combined and concentrated at 4000g using a 30 K MWCO Amicon Centrifugal Filter (Millipore, Sigma-Aldrich). Protein purity was assessed via SDS-PAGE with Coomassie staining, and the total protein concentration was determined via the Bradford assay. The molar percentage of the HBx fusion protein was calculated from mass spectrometry (vide supra) by normalizing the HBx exponentially modified Protein Abundance Index (emPAI) value against the sum of all emPAI values in the data set used.
Purification of NusA-HBx and DsbC-HBx
NusA-HBx and DsbC-HBx cell pellets were resuspended in 50 mM HEPES (pH 8.0), 300 mM NaCl, and 10 mM imidazole (Buffer 4). PMSF was added to the suspension to a final concentration of 45 μg/mL. The suspension was then lysed via sonication for a total time of 30 min (15 s pulse, 59 s pause, 60% amplification) and centrifuged at 22 000g for 30 min. The supernatant was loaded onto a Ni-NTA immobilized affinity chromatography column (∼20 mL, McLab, CA) equilibrated in Buffer 4. The column was first washed with Buffer 4, followed by washing with 50 mM HEPES (pH 8.0), 300 mM NaCl, 30 mM imidazole (Buffer 5) to remove nonspecifically bound proteins. The bound protein was eluted with 50 mM HEPES (pH 8.0), 300 mM NaCl, 250 mM imidazole. The protein-containing fractions were combined and concentrated at 4000g using a 30 K MWCO Amicon Centrifugal Filter (Millipore, Sigma-Aldrich). Protein purity was assessed via SDS-PAGE with Coomassie staining, and the total protein concentration was determined via the Bradford assay. Mass spectrometry was employed to quantify the exact concentration of the HBx fusion protein. The molar percentage was calculated by normalizing the HBx emPAI value against the sum of all emPAI values in the data set used.
Purification of GST-HBx
Purification of GST-HBx was adapted from previous reports with slight modifications.8 Frozen cells were resuspended in 20 mM HEPES (pH 7.5), 0.5 mM EDTA, 10% glycerol, 0.1% NP-40, 12.5 mM KCl, 10 mM β-mercaptoethanol, and 1.05 M NaCl (Buffer 6). A Pierce Protease Inhibitor tablet (EDTA free) was added to the suspension, and the solution was lysed via sonication for a total time of 30 min (15 s pulse, 59 s pause, 60% amplification) and centrifuged at 22 000g for 30 min. The supernatant was loaded onto a GST-trap (5 mL, GSTrap HP, GE Healthcare) that was equilibrated in Buffer 6. The column was first washed with Buffer 6 followed by washing with 20 mM HEPES (pH 7.5), 0.5 mM EDTA, 10% glycerol, 0.1% NP-40, 12.5 mM KCl, 10 mM βME, and 50 mM NaCl (Buffer 7). The bound protein was eluted from the column with Buffer 7 containing an additional 5 mM reduced glutathione and 100 mM glutamate. The fractions containing the protein of interest were combined and dialyzed overnight against 20 mM HEPES (pH 7.9), 10% glycerol, 2 mM MgCl2, 1 mM DTT, 0.5 mM EDTA, and 50 mM KCl (Buffer 8). The dialyzed sample was then subjected to a cation-exchange column (5 mL, HiTrap SP HP column, GE Healthcare) followed by an anion-exchange column (5 mL, HiTrap Q FF column, GE Healthcare). In both cases, the columns were first washed with Buffer 8 and eluted by a gradient with Buffer 8 containing 1 M KCl. GST-HBx and free GST eluted off both columns during the initial wash. The fractions containing GST-HBx were concentrated at 4000g using a 30 K MWCO Amicon Centrifugal Filter (Millipore, Sigma-Aldrich). The total protein concentration was determined via the Bradford assay, and the exact amount of the fusion protein was calculated from the emPAI as described for the other constructs.
Preparation of E. coli GroEL
Purification of GroEL was adapted from the previous reports with slight modifications.42E. coli T7 express cells were transformed with the pT-GroE vector for the expression of GroEL and GroES. The transformed cells were grown in LB media with 34 μg/L chloramphenicol shaking at 37 °C until the OD600 reached ∼0.6. The cells were induced by the addition of 0.4 mM IPTG and incubated at 37 °C while shaking at 220 rpm for 4 h. The cultures were centrifuged at 7000 rpm for 15 min, and the cell pellets were flash-frozen in liquid nitrogen and stored at −80 °C. The cell pellets were resuspended in 20 mM HEPES (pH 8.0), 100 mM NaCl, 0.1% NP-40, and 10 mM EDTA (Buffer 9). PMSF was added to a final concentration of 45 μg/mL, together with 10 μg/mL of lysozyme and 1 μg/mL DNase. The cells were lysed via sonication (30 min total time), and the cellular debris was pelleted by centrifugation at 22 000g for 30 min. One percent streptomycin sulfate was added to the clarified lysate slowly over 30 min while stirring on ice. The lysate was then centrifuged for 15 min at 22 000g, and MgCl2·6H2O (10 mM final concentration) was added to the supernatant. The sample was further purified by the addition of 32% (w/v) ammonium sulfate added slowly over 1 h. The precipitate was isolated by centrifugation at 22 000g for 15 min, and the pellet was resuspended in 15 mL Buffer 9. The sample was then concentrated at 4000g using a 30 K MWCO Amicon Centrifugal Filter and further purified using a Superdex-200 column (120 mL resin) equilibrated with Buffer 3. The fractions containing GroEL were concentrated, and the protein concentration was determined by the Bradford assay.
ATPase Assays
ATP hydrolysis was monitored in a coupled enzymatic assay employing pyruvate kinase (PK) and lactate dehydrogenase (LDH), in which the regeneration of hydrolyzed ATP is coupled to the oxidation of NADH.43 The spectrophotometric assay was performed in 150 μL reactions in a 96-well plate in a Powerwave XS2 plate reader. The final concentrations of the reagents were 400 μM NADH, 800 μM phosphoenolpyruvate (PEP), 100 U/mL PK, 12 U/mL LDH, and varying ATP concentrations between 0 and 2 mM. The reaction mixture contained MgSO4·7H2O in a 10-fold excess with respect to ATP. After the addition of NADH, PEP, PK, LDH, and ATP, the plate was shaken for 30 s and allowed to incubate at 25 °C for 10 min prior to the addition of 1–5 μM fusion protein, with which the reactions were initiated and followed for 20 min. Consumption of NADH was monitored by following the absorbance at 340 nm. The traces were fit by linear regression using the Kaleidagraph software.
(d)NTP Hydrolysis Assays by High-Performance Liquid Chromatography (HPLC)
HBx fusion proteins at a final concentration of 10 μM were incubated with 100 μM ATP, dATP, or GTP and 100 μM MgSO4·7H2O for 20 min at room temperature in Buffer 3. The reactions were quenched by heat denaturation at 98 °C for 5 min. The samples were then centrifuged at 21 130g to remove any precipitated proteins, and the supernatant was then applied to a 0.22 μm nylon Spin-X Centrifuge Tube Filter (Corning Incorporated, Corning, NY) to ensure complete removal of any particulates. The reactions were analyzed on a 1260 Infinity Liquid Chromatography system equipped with a 1260 Infinity Photodiode Array Detector WR. The samples were injected on an Agilent reverse-phase C18-A Polaris column (particle size 5 μm, 150 × 4.6 mm2), and the products were separated by a gradient utilizing a water-based mobile phase (10 mM KH2PO4 and 10 mM tetrabutylammonium hydroxide (TBAH), pH 6, Solvent A) and an organic-based mobile phase (methanol with 10 mM TBAH, Solvent B). Chromatographic separation of the substrates and products was achieved with a gradient of 95% solvent A and 5% solvent B to 50% solvent A and 50% solvent B at a flow rate of 1.5 mL/min for 25 min. Comparison of the integrated peak intensities to that of internal standards (substrates and products) of known concentration enabled quantification of the analyses. Nucleotides were detected at a wavelength of 254 nm.
Mass Spectrometry of HBx Proteins
Gel bands were excised, reduced, alkylated, and digested with trypsin at 37 °C overnight, with the resulting peptides being extracted, concentrated to 20 μL, and placed in 300 μL autosampler vials. Solution-based samples were precipitated with trichloroacetic acid (TCA), resuspended in a tris(hydroxymethyl)aminomethane (Tris)/urea buffer, reduced, alkylated, and digested with trypsin at 37 °C overnight. The resulting peptides were concentrated to 20 μL and placed in 300 μL autosampler vials. Ten microliters of the digested samples was injected onto a Waters NanoAcquity HPLC equipped with a self-packed Aeris 3 μm C18 analytical column (0.075 mm by 20 cm, Phenomenex). The peptides were eluted using standard reverse-phase gradients. The effluent from the column was analyzed using an Orbitrap Elite (ThermoFisher) mass spectrometer (nanospray configuration) operated in a data-dependent manner. The resulting fragmentation spectra were correlated against custom databases using Mascot (Matrix Science), with the output files being uploaded to Scaffold Q+S (Proteome Software) for visualization. Custom databases included the RefSeq entries for E. coli BL21-Gold(DE3)pLysS AG (Taxonomy ID: 866768), HBx fusion sequences, and common contaminants.
Acknowledgments
The authors are grateful to Eric Spooner (Whitehead Institute, Boston, MA) for his assistance with the mass spectrometry measurements and data interpretation. The authors would also like to thank Xingchen Liu and Chansik Kim for their contribution in generating many of the mutations expressed in this paper. Finally, the authors thank Vincent Sutera for his assistance in the generation of a ΔgroEE. coli strain.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01762.
The supporting information includes the procedures for protein expression, purification, activity assays, and mass-spectrometric analyses; Table S1 lists the primers for mutagenesis; Table S2 lists the NTP prediction probabilities of the HBx amino acid residues; Table S3 reports the emPAI values of HBx fusions from mass spectrometry; and Figures S1 and S2 contain the GroEL ATP activity and Western blot for GST-HBx, respectively (PDF)
Accession Codes
HBx genotype A2 (NCBI ID: P69713). E. coli GroEL (UniProtKB: C4NV17).
Author Contributions
The manuscript was written through contributions of all authors, and all authors have approved the final version of the manuscript.
This research has been supported by the National Institutes of Health (Grant Nos. GM111978 and GM126303 to M.E.P.) and the Brandeis University Innovation SPROUT Program (to M.L. and M.E.P.).
The authors declare no competing financial interest.
Supplementary Material
References
- Bouchard M. J.; Schneider R. J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. 10.1128/JVI.78.23.12725-12734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamor P. J.; deLemos A. S.; Russo M. W. Viral hepatitis and hepatocellular carcinoma: etiology and management. J. Gastrointest. Oncol. 2017, 8, 229–242. 10.21037/jgo.2017.03.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen M.-F.; Chen D.-S.; Dusheiko G. M.; Janssen H. L. A.; Lau D. T. Y.; Locarnini S. A.; Peters M. G.; Lai C.-L.; Hepatitis B. Infection. Nat. Rev. Dis. Primers 2018, 4, 1–20. 10.1038/s41572-018-0001-z. [DOI] [PubMed] [Google Scholar]
- Kim C. M.; Koike K.; Saito I.; Miyamura T.; Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- Ma N.-F.; Lau S. H.; Hu L.; Xie D.; Wu J.; Yang J.; Wang Y.; Wu M. C.; Fung J.; Bai X.; Tzang C. H.; Fu L.; Yang M.; Su Y. A.; Guan X.-Y. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin. Cancer Res. 2008, 14, 5061–5068. 10.1158/1078-0432.CCR-07-5082. [DOI] [PubMed] [Google Scholar]
- Lucifora J.; Arzberger S.; Durantel D.; Belloni L.; Strubin M.; Levrero M.; Zoulim F.; Hantz O.; Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Murakami S. Hepatitis B virus X protein: a multifunctional viral regulator. J. Gastroenterol. 2001, 36, 651–660. 10.1007/s005350170027. [DOI] [PubMed] [Google Scholar]
- De-Medina T.; Haviv I.; Noiman S.; Shaul Y. The X Protein of Hepatitis B Virus Has a ribo/deoxy ATPase activity. Virology 1994, 202, 401–407. 10.1006/viro.1994.1356. [DOI] [PubMed] [Google Scholar]
- Zhang S. M.; Sun D. C.; Lou S.; Bo X. C.; Lu Z.; Qian X. H.; Wang S. Q. HBx protein of hepatitis B virus (HBV) can from complex with mitochondrial HSP60 and HSP70. Arch. Virol. 2005, 150, 1579–1590. 10.1007/s00705-005-0521-1. [DOI] [PubMed] [Google Scholar]
- Jiang T.; Liu M.; Wu J.; Shi Y. Structural and biochemical analysis of Bcl-2 interaction with the hepatitis B virus protein HBx. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 2074–2079. 10.1073/pnas.1525616113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De-Medina T.; Shaul Y. Functional and structural similarity between the X protein of hepatitis B virus and nuceloside diphosphate kinases. FEBS Lett. 1994, 351, 423–426. 10.1016/0014-5793(94)00900-7. [DOI] [PubMed] [Google Scholar]
- Dopheide T. A.; Azad A. A. The hepatitis B virus X protein is a potent AMP kinase. J. Gen. Virol. 1996, 77, 173–176. 10.1099/0022-1317-77-2-173. [DOI] [PubMed] [Google Scholar]
- Quadri I.; Maguire H. F.; Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Biochemistry 1995, 92, 1003–1007. 10.1073/pnas.92.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga S.; Rawat S.; Ajenjo M.; Bouchard M. J. Hepatits B virus (HBV) X protein-mediated regulation of hepatocyte metabolic pathways affects viral replication. Virology 2016, 498, 9–22. 10.1016/j.virol.2016.08.006. [DOI] [PubMed] [Google Scholar]
- Benn J.; Schneider R. J. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 10350–10354. 10.1073/pnas.91.22.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F.; Schneider R. J. Hepatitis B virus HBx protein activates transcription factor NF-kappaB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J. Virol. 1996, 70, 4558–4566. 10.1128/JVI.70.7.4558-4566.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-H.; Yun Y. HBx Protein of Hepatitis B Virus Activates Jak1-STAT Signaling. J. Biol. Chem. 1998, 273, 25510–25515. 10.1074/jbc.273.39.25510. [DOI] [PubMed] [Google Scholar]
- Sidhu K.; Kumar S.; Reddy V. S.; Kumar V. Mass Spectrometric Determination of Disulfide Bonds in the Biologically Active Recombinant HBx Protein of Hepatitis B Virus. Biochemistry 2014, 53, 4685–4695. 10.1021/bi500140t. [DOI] [PubMed] [Google Scholar]
- Mitchell M. S.; Rao V. B. Novel and deviant Walker A ATP-binding motifs in bacteriophage large terminase-DNA packaging proteins. Virology 2004, 321, 217–221. 10.1016/j.virol.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Chen K.; Mizianty M. J.; Kurgan L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics 2012, 28, 331–341. 10.1093/bioinformatics/btr657. [DOI] [PubMed] [Google Scholar]
- Hayashi I.; Oyama T.; Morikawa K. Structural and functional studies of MinD ATPase: implications for the molecular recognition of the bacterial cell division apparatus. EMBO J. 2001, 20, 1819–1828. 10.1093/emboj/20.8.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C.; Dalmas O.; Gros P.; Di Pietro A.; Jault J.-M. The Conserved Glutamate Residue Adjacent to the Walker-B Motif Is the Catalytic Base for ATP Hydrolysis in the ATP-binding Cassette Transporter BmrA. J. Biol. Chem. 2003, 278, 47002–47008. 10.1074/jbc.M308268200. [DOI] [PubMed] [Google Scholar]
- Walker J. E.; Saraste M.; Runswick M. J.; Gay N. J. Distantly related sequences in the alpha- and beta- subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon G.; Albers S. V.; Dijkstra B. W.; Driessen A. J.; Thunnissen A. M. Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucelotide-bound conformations. J. Mol. Biol. 2003, 330, 343–358. 10.1016/S0022-2836(03)00575-8. [DOI] [PubMed] [Google Scholar]
- Hemmer W.; McGlone M.; Tsigelny I.; Taylor S. S. Role of the Glycine Triad in the ATP-binding Site of cAMP-dependent Protein Kinase. J. Biol. Chem. 1997, 272, 16946–16954. 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- Sharma D.; Say A. F.; Ledford L. L.; Hughes A. J.; Sehorn H. A.; Dwyer D.; Sehorn M. G. Role of the conserved lysine within the Walker A motif of human DMC1. DNA Repair 2013, 12, 53–62. 10.1016/j.dnarep.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S. A.; Lee T.-H.; Butel J. S.; Slagle B. L. Hepatitis B Virus X Protein Interferes with Cellular DNA Repair. J. Virol. 1998, 72, 266–272. 10.1128/JVI.72.1.266-272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan D.; Xing W.; Beran R. K.; Chemuru S.; Rohrs H.; Niedziela-Majka A.; Marchans B.; Mehra U.; Zabransky A.; Dolezal M.; Hubalek M.; Pichova I.; Gross M. L.; Kwon H. J.; Fletcher S. P. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93, 1–18. 10.1128/JVI.00250-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Zhuang L.; Lu Y.; Xu Q.; Tang B.; Chen X. Naturally occuring basal core promoter A1762T/G1764A dual mutations increase the risk of HBV-related hepatocellular carcinoma: a meta-analysis. Oncotarget 2016, 7, 12525–12536. 10.18632/oncotarget.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Shan C.; Li N.; Zhang X.; Zhang X.; Xu F.; Zhang S.; Qiu L.; Ye L.; Zhang X. Identification of a natural mutant of HBV X protein truncated 27 amino acids at the COOH terminal and its effect on liver cell proliferation. Acta Pharmacol. Sin. 2008, 29, 473–480. 10.1111/j.1745-7254.2008.00764.x. [DOI] [PubMed] [Google Scholar]
- van der Does C.; Presenti C.; Schulze K.; Dinkelaker S.; Tampe R. Kinetics of the ATP Hydrolysis Cycle of the Nucleotide-binding Domain of MdI1 Studeied by a Novel Site-specfic Labeling Technique. J. Biol. Chem. 2006, 281, 5694–5701. 10.1074/jbc.M511730200. [DOI] [PubMed] [Google Scholar]
- Hofacker M.; Gompf S.; Zutz A.; Presenti C.; Haase W.; van der Does C.; Model K.; Tampe R. Structural and Functional Fingerprint of the Mitochondrial ATP-binding Cassette Transporter Mdl1 from Saccharomyces cerevisiae. J. Biol. Chem. 2006, 282, 3951–3961. 10.1074/jbc.M609899200. [DOI] [PubMed] [Google Scholar]
- Noske R.; Cornelius F.; Clarke R. J. Investigation of the enzymatic activity of the Na+, K+-ATPase via isothermal titration microcalorimetry. Biochim. Biophys. Acta 2010, 1797, 1540–1545. 10.1016/j.bbabio.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Rye H. S.; Roseman A. M.; Chen S.; Furtak K.; Fenton W. A.; Saibil H. R.; Horwich A. L. GroEL-GroES Cycling: ATP and Nonnative Polypeptide Direct Alternation of Folding-Active Rings. Cell 1999, 97, 325–338. 10.1016/S0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zheng Z.; Ramsey A.; Chen L. Analysis of Peptides and Proteins in Their Binding to GroEL. J. Pept. Sci. 2010, 16, 693–700. 10.1002/psc.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhai T.; Heidrich N. G.; Dencher N. A.; Seelert H. Highly sensitive detection of ATPase activity in native gels. Electrophoresis 2009, 30, 3622–3625. 10.1002/elps.200900114. [DOI] [PubMed] [Google Scholar]
- Rohman M.; Harrison-Lavoie K. J. Separation of copurifying GroEL from glutathione-S-transferase fusion proteins. Protein Expression Purif. 2000, 20, 45–47. 10.1006/prep.2000.1271. [DOI] [PubMed] [Google Scholar]
- Douette P.; Navet R.; Gerkens P.; Galleni M.; Levy D.; Sluse F. E. Escherichia coli fusion carrier proteins act as solubilizing agents for recombinant uncoupling protein 1 through interactions with GroEL. Biochem. Biophys. Res. Commun. 2005, 333, 686–693. 10.1016/j.bbrc.2005.05.164. [DOI] [PubMed] [Google Scholar]
- Ishihama Y.; Oda Y.; Tabata T.; Sato T.; Nagasu T.; Rappsilber J.; Mann M. Exponentially Modified Protein Abuncance Index (emPAI) for Estimation of Absolute Protein Amount in Proteomics by the Number of Sequenced Peptides per Protein. Mol. Cell. Proteomics 2005, 4, 1265–1272. 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- Jameel S.; Siddiqui A.; Macguire H. F.; Rao K. V. S. Hepatitis B Virus X Protein Produced in Escherichia coli Is Biologically Functional. J. Virol. 1990, 64, 3963–3966. 10.1128/JVI.64.8.3963-3966.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkman M. Production of disulfide-bonded proteins in Escherichia coli. Protein Expression Purif. 2012, 82, 240–251. 10.1016/j.pep.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Molugu S. K.; Li J.; Bernal R. A. Separation of E. coli chaperonin groEL from β-galactosidase without denaturation. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015, 1007, 93–99. 10.1016/j.jchromb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Roskoski R. J.Assays of Protein Kinase. In Methods in Enzymology; Academic Press, 1983; Vol. 99, pp 3–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.