

Abstract
Interferons are commonly utilized in the treatment of chronic hepatitis B virus (HBV) infection but are not effective for all patients. A deep understanding of the limitations of interferon treatment requires delineation of its activity at multiple “omic” levels. While myriad studies have characterized the transcriptomic effects of interferon treatment, surprisingly, few have examined interferon-induced effects at the proteomic level. To remedy this paucity, we stimulated HepG2 cells with both IFN-α and IFN-λ and performed proteomic analysis versus unstimulated cells. Alongside, we examined the effects of HBV transfection in the same cell line, reasoning that parallel IFN and HBV analysis might allow determination of cases where HBV transfection counters the effects of interferons. More than 6000 proteins were identified, with multiple replicates allowing for differential expression analysis at high confidence. Drawing on a compendium of transcriptomic data, as well as proteomic half-life data, we suggest means by which transcriptomic results diverge from our proteomic results. We also invoke a recent multiomic study of HBV-related hepatocarcinoma (HCC), showing that despite HBV’s role in initiating HCC, the regulated proteomic landscapes of HBV transfection and HCC do not strongly align. Special focus is applied to the proteasome, with numerous components divergently altered under IFN and HBV-transfection conditions. We also examine alterations of other protein groups relevant to HLA complex peptide display, unveiling intriguing alterations in a number of ubiquitin ligases. Finally, we invoke genome-scale metabolic modeling to predict relevant alterations to the metabolic landscape under experimental conditions. Our data should be useful as a resource for interferon and HBV researchers.
Introduction
Type-I interferon (IFN-I) stimulus is known to generate a stereotyped response.1 The response, as defined by regulation of key proteins or transcripts, does not seem limited to cell type2 and can be initiated by the presence of viruses, bacteria, parasites, and any number of purified components thereof [dsRNA, lipopolysaccharide (LPS), etc.].3,4 The use of type-I IFNs as a treatment against a wide range of infectious conditions has been shown efficacious.5 Additionally, IFN-I treatment has shown benefits in conditions ranging from multiple sclerosis6 to cancer7 because of immunomodulatory and/or proapoptotic effects.
An extraordinary number of transcriptomic studies have measured differential effects of IFN-I against controls in a wide variety of cell types and organisms; we estimate that the GEO database lists approximately 100 such studies in human cells alone.8 Given well-known disparities between mRNA and protein levels, it is then surprising to find that high-resolution proteomic studies of the IFN response are lacking. Searching the literature, a 2017 work in which IFN-α (a type-I interferon) and IFN-γ (type II) were applied to MRC-5 lung fibroblast cells would be the deepest study to date, emphasizing time course effects and differential IFN-α/IFN-γ results.9 Another deep proteomic analysis of IFN-γ effects on mesenchymal stromal/stem cells10 has been conducted, but such results would not be expected to be good proxies for IFN-I effects, given the differences in pathways that are triggered upon type I or II stimulation.11 We previously conducted a high-depth analysis of IFN type I and III effects on a human hepatoblastoma cell line transfected with HBV (HepG2.2.15), with the aim of testing potential therapeutic effects;12 however, this setting would not be ideal for the study of authentic IFN effects, as viral versus IFN stimulation of cells might well overlap upon analysis of altered protein expression.
In this study, we use modern technology to resolve the IFN-I-stimulated proteome in the HepG2 human hepatoblastoma cell line. We also examine the effects of IFN-III stimulation, which is thought to stimulate a response similar to that of IFN-I but may offer reduced side-effects by virtue of its specificity for epithelial cells.13 A comparison of HBV-transfected versus native HepG2 cells is also conducted, possibly allowing discrimination between a state induced by chronic viral infection and a state induced by IFN treatment. With the aim of providing a comprehensive analysis, we also incorporate data from our previous HBV-focused proteomic work.12 In all cases, multiple biological replicates are performed which, combined with modern methods, allow discernment of considerably more protein identifications than previous IFN-related studies. Our data are offered as a resource for further investigations at http://sysbio.chula.ac.th/IFN_HepG2_Proteomics/.
Results and Discussion
Data Overview
Prior to dissecting our data, we should point out a limitation of a portion of our work. In the case where HBV-transfected (HepG2.2.15) cells were compared against HepG2, a control vector was not transfected into HepG2. To retain the HBV-expression plasmid, cells were treated with the antibiotic Geneticin. This approach mirrors other studies in which HepG2.2.15 has been compared with HepG2.14−21 Nevertheless, the lack of a control vector and Geneticin within HepG2 cannot be excluded as a cause of differential results when comparing HepG2.2.15 cells against HepG2. Readers should also note that all differential proteomic results, unless otherwise noted, were acquired at 24 h post-treatment; longer or shorter treatment periods could result in significantly different profiles. Finally, these results were established in vitro; systemic delivery of IFN or infection with HBV in an appropriate in vivo model may also generate different results.
In order to dissect the IFN-response in HepG2 cells and to compare the IFN-response to an HBV-transfection induced state, we performed two experiments, with five replicates in each: HepG2 cells with/without IFN-I or IFN-III stimulation (IFN-α2 or IFN-λ3) and a comparison of HepG2 cells versus HepG2.2.15 cells; in addition, we also included our previously published work:12 HepG2.2.15 cells with/without IFN-I or IFN-III stimulation, into our bioinformatic analysis (Figure 1).
Figure 1.

Schematic overview of the experimental design and workflow.
Figure 2a,b attests to consistency between replicates in our two new proteomic studies, with proteins found in all five replicates being the dominant intersection. In all, we confidently identified a total of 6003 proteins in this study, of which 2830 were differentially regulated under at least one condition and 5708 proteins appeared under at least two conditions (P < 0.05, Student’s t-test, Figure 2c). Including results from our previous study of the effects of interferon treatment on HBV-transfected cells, we conducted PCA analysis on the differential protein expression data from the resulting 25 mass spectrometry (MS) samples (Figure 2d) from three dimethyl labeling experiments (HepG2.2.15 vs HepG2, HepG2 with IFN-α/λ vs control, and HepG2.2.15 with IFN-α/λ vs control). As expected, these three experiments are apparent in the distinct locations of ellipses in the figure. Clusters include the HBV-transfection group more tightly, possibly due to the aforementioned broad effects of transfection.
Figure 2.

Two dimethyl-labeling experiments consisting of 5 reps (samples) each were performed. Five-way Venn diagrams depict proteins identified in each sample. Only proteins found in at least 3 reps are counted. (a) Counts of proteins identified in the HepG2 control, IFN-α2-treated HepG2, and IFN-λ3-treated HepG2 comparisons are shown. (b) Counts of proteins in the HepG2.2.15 vs HepG2 control comparison are shown. Note that the predominant grouping is the case where a protein can be found in all five samples. (c) Venn diagram shows conditions under which proteins could be identified in both treatment and control conditions (note that the IFN-λ IDs are subsumed by the IFN-α IDs). (d) PCA diagram shows clustering of 25 samples (five experimental conditions and five repetitions).
The HepG2-IFN/HepG2 and HepG2.2.15/HepG2 comparisons are depicted as volcano plots (Figure 3). A total of 401 proteins were differentially expressed on IFN-I stimulation (260 up and 141 down), 711 on IFN-III stimulation (467 up and 244 down), and 2434 (954 up and 1480 down) in the HepG2.2.15 cells versus HepG2 comparison; 1689 proteins were not differentially expressed under any of the three conditions. Thus, in the IFN-I/III conditions, 12 and 7% of all proteins showed altered expression. Note that the MTT assays in our previous work12 were used to establish IFN treatment concentrations that were not detrimental to cell survival, so the above-mentioned differential results cannot be interpreted as alterations that would be expected because of cell death. Not surprisingly, proteins identified as upregulated/downregulated under IFN-I treatment strongly overlapped with those upregulated/downregulated under IFN-III treatment (P = 10–107/P = 10–46). As expected, the HBV-C protein was expressed only in the HBV-transfection condition. Several features of these plots should be pointed out. First, while IFN-treatment clearly elicits a powerful and stereotyped effect (see below), the extent to which HBV-transfection alters protein expression was surprising; 59% of all detected proteins were differentially expressed in the HepG2.2.15/HepG2 comparison, a figure not approached in either IFN treatment. Second, while it has been claimed, based on transcriptomics, that no interferon response is detectable upon HBV infection,22,23 this effect is obvious at the proteomic level in the comparison of HepG2.2.15 to HepG2, with innate immune components such as IFIT1 and IFIT3 upregulated with high significance.
Figure 3.
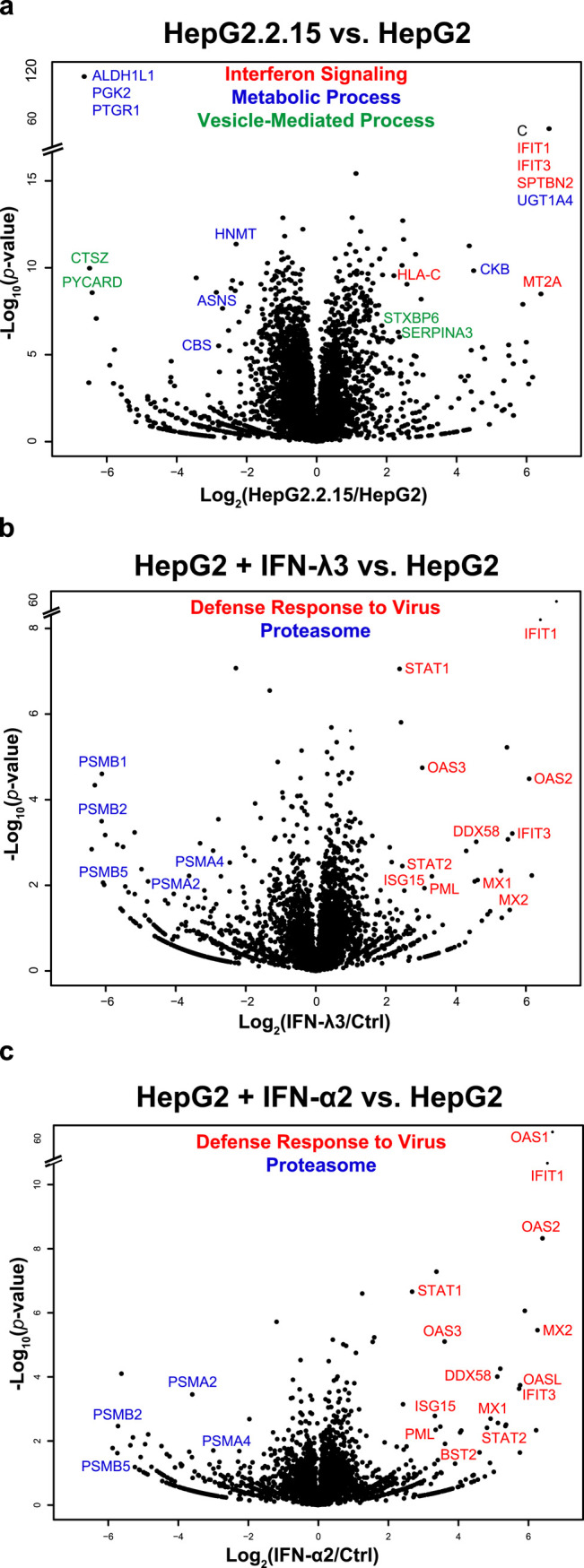
Volcanos. Volcano plots display the distribution of identified proteins according to significance and fold-change in (a) HBV-transfection condition, (b) IFN-III stimulation condition, and (c) IFN-I stimulation condition. Individual proteins exceeding minimum significance and fold-change thresholds are noted, with colors indicating membership in three prominent gene ontology groups. These groups were chosen such that genes in one group were not found in the other two groups. Proteins that exceeded fold-change and significance thresholds are labeled, with colors indicating proteins that fall into broad GO enrichment groups of interest. In the case of (a), the presence of the HBV core protein is noted, represented by “C”.
Proteomics Versus Transcriptomics
The correlations between differential transcriptomic and proteomic results are the subject of intense, ongoing debate.24,25 We constructed lists of the most commonly up- and downregulated transcripts upon IFN-I or III stimulation using 18 studies (Table S1). For inclusion in the list of upregulated transcripts, the transcript was required to appear at least four times and for downregulated transcripts, three times. No more than 200 transcripts were entered in these lists. The stereotypical nature of the IFN response is seen in the observation that three known interferon-inducible transcripts (IFIT1, USP18, and RSAD2)26−28 were shown to be upregulated in 16 of these studies; at the same time, only one transcript was downregulated more than 6 times in these studies (CERK). Comparing our HepG2 proteomic results against these transcriptomic lists, a strong overlap is seen in the case of upregulation (P = 10–17 on IFN-I treatment, 10–9 on IFN-III treatment, Fisher’s exact test) (Figure 4b). Similar results were seen in HepG2.2.15 (P = 10–15 on IFN-I treatment, 10–13 on IFN-III treatment). Interestingly, the overlaps were insignificant when downregulated transcripts were compared against downregulated HepG2 or HepG2.2.15 proteins. To further dissect transcript/protein relations, we conducted Pearson correlation tests using differential data from our own proteomic studies and 15 of the above-mentioned transcriptomic studies (in three cases, complete transcriptomic datasets were not available) (Figure 4a). In 14 of these studies, positive proteomic/transcriptomic correlations are seen, with P < 10–15 in 5 cases. Despite these correlations, the near-flat slope of these plots illustrates that under IFN-stimulation, proteins have a considerably wider dynamic range than transcripts. Figure S1 reinforces the aforementioned observation that proteome/transcriptome correlations are strongest in the case of upregulation; here, when downregulated proteins were removed from the underlying dataset, R values and slopes clearly increased, while P-values uniformly decreased.
Figure 4.

Proteomics vs transcriptomics. (a) Pearson correlation analysis of 15 transcriptomic studies in which IFN-λ3 and IFN-α2 were applied to various cell types vs our own IFN-treatment-based proteomic results. Axes depict log 2 fold alterations. Violin plot (lower right hand corner) depicts the distribution of R-values generated in these 15 tests. See Figure S1 for tests in which downregulated or upregulated proteins were excluded from the analysis; here, it becomes apparent that protein/transcript correlations are much stronger when examining upregulation vs downregulation. (b) Venn diagram displays intersection of transcripts canonically upregulated on IFN treatment against proteins upregulated on either IFN-α2 or IFN-λ3 treatment.
A hint as to the cause of the disparity between downregulated transcripts and proteins may be seen in a comparison of our IFN-I/III treatment results against two proteomic studies that sought to determine protein half-lives.24,29 We averaged IFN-I and IFN-III fold changes, selected only proteins that showed a significant change in at least one of these two IFN treatments, and compared these results against proteins shown to have long half-lives in mouse fibroblasts24 using Fisher’s exact test, defining 24 h (the duration of IFN exposure in our work) as the cutoff between “short-lived” and “long-lived” proteins. Here, only the overlap of proteins downregulated upon IFN treatment and proteins with long half-lives reached significance (P = 0.01). Changing the threshold of “long-lived” to be 100 h or more (approximately 25% of all proteins) resulted in P = 1.3 × 10–6. Performing the same exercise with Mathieson’s mouse hepatocyte data,29 we derive P = 0.001 when examining IFN-downregulated proteins against long-lived proteins. Again, significance increased when proteins with half-lives greater than 100 h (approximately 33% of all proteins) were intersected with proteins downregulated on IFN treatment: P = 8 × 10–6. It is possible, then, that IFN treatment has a general effect of decreasing proteins with long half-lives, suggesting increased degradation versus decreased production. A relative lack of RNA/protein correlation upon downregulation versus upregulation has been previously noted, suggesting a prominent role for protein degradation processes in widening the RNA/protein gap.30−32
Another potentially insightful exercise is to search for cases where the proteomic and transcriptomic data are in opposition. We identified only two cases, both involving proteins we found to be upregulated upon IFN-III treatment but typically downregulated at the transcriptomic level: RRM2 and BUB1B (no cases of protein downregulation/transcript upregulation were found). Interestingly, both are involved in the cell cycle, are highly expressed in numerous cancers, and are predictive of poor cancer outcomes.33−36 Such results point out how, in some cases, transcriptomics fails to predict the direction of protein differential regulation. To seek out overlooked proteins that may play prominent roles in the interferon response without being upregulated at the transcript level, we scanned our data for proteins that were highly significantly upregulated (P < 0.001) in both IFN-α2 and IFN-λ3 treatment conditions yet did not appear on our list of canonical IFN-upregulated transcripts in IFN treatments ranging from 6 h to 8 days. Four such proteins were found: CHDH, SRBD1, SLC25A31, and SFXN4. While the small size of this group makes enrichment analysis unprofitable, it is interesting that three of the four proteins are primarily mitochondrial (SRBD1 is not), hinting that a subset of mitochondrial proteins are stabilized upon IFN-α/λ treatment, or their transcripts are more efficiently translated, possibly hinting at an overlooked response to these IFNs.
Deconvoluting HBV-Transfection and IFN-Induced Effects
To further elucidate the proteome-level interferon response in HepG2 cells, we combined our previously generated data regarding the effect of IFN-treatment on HBV-transfected cells with our current data. The result is shown as a heatmap in Figure 5, with five clusters of expression patterns emerging from the five conditions. The first cluster can easily be described as “antiviral” in nature, with significant upregulation of interferon-response components under all conditions. The second cluster shows proteins that are strongly downregulated upon HBV-transfection but upregulated on IFN-treatment; we would term this cluster “IFN reverses the downregulation caused by HBV transfection.” Of the enrichment groups that appear in the figure, we would question the usefulness of terms such as “acetylation” given that PTMs occur at the peptide level, with modifications possibly being up- and downregulated on a single protein. Nevertheless, it is worth noting that proteins in this cluster have been described in individual studies as targets of PARP-ylation (log(P) = −3.3),37 sumoylation (−1.8),38 and phosphorylation (−1.7),39 in addition to acetylation. The third cluster is the converse of the second: “IFN diminishes the upregulation caused by HBV transfection.” The fourth cluster shows proteins that are strongly downregulated upon IFN stimulation but countered by HBV transfection; “IFN downregulation reversed by HBV transfection.” Here, enrichment is seen for proteasome core components, again suggesting that HBV actively alters the abundance of these components. The final group, cluster five, is rather perplexing. Here, proteins that are upregulated upon IFN treatment are downregulated upon HBV transfection, with IFN appearing to enhance, not reverse, this downregulation in HBV-transfected cells: “IFN enhances the downregulation caused by HBV transfection.” We note the presence of TBK1 in this group, a kinase that is targeted by numerous viruses in order to suppress the interferon response.40−42 One of the HBV proteins, HBx, has been shown to interact with TBK1, thus altering IFN signaling.43 One might speculate that HBV somehow redirects the activity of an interferon-stimulated kinase (such as TBK1) or ubiquitylase to target a subset of interferon-stimulated proteins, giving rise to the aforementioned perplexing behavior.
Figure 5.

Heatmap shows alterations in 193 proteins under five conditions: (1) IFN-α2 and (2) IFN-λ3 stimulation of HepG2 cells; (3) IFN-α2 and (4) IFN-λ3 stimulation of HepG2.2.15 cells; and (5) HepG2 vs HepG2.2.15 cells. For a protein’s entry into the heatmap, the following was required; of the five log 2 ratios associated with a given protein’s differential expression changes under different experimental conditions, the smallest ratio subtracted from the largest ratio must be greater than two. Note that expression under IFN-α2 and IFN-λ3 conditions is largely concordant. Proteins in five expression clusters are noted in boxes, and enrichment characteristics of these groups are shown in accompanying bar graphs.
Relevance to Immunoprocessing
Proteasome components were prominent in the volcano plots showing IFN-stimulated alterations, prompting further examination of this structure. Here, differences between the HepG2-IFN/HepG2 (IFN) conditions versus the HepG2.2.15/HepG2 (HBV-transfection) condition were clear. In the HBV-transfection condition, all proteasome components failed to exceed 2-fold up-/downregulation. Meanwhile, four proteasome components, all downregulated, exceeded this threshold upon IFN-stimulation (Figure 6, proteasome table). These large alterations all involved the core alpha and beta components of the constitutive proteasome. The downregulated PSMB5 protein is one of the three core components replaced in immunoproteasome formation, while PSME1 and PSME2, accessory components unique to the immunoproteasome, were upregulated upon IFN stimulation. We note that significant downregulation of the four aforementioned core components without concomitant alterations in “accessory” components has also been identified via MS in lymphocytes upon IL-2 and IL-15 stimulation.44 As with IFN-α/λ, these cytokines are expected to act on the JAK/STAT signaling pathway. Meanwhile, differential upregulation of immunoproteasome components (vs the constitutive proteasome) is seen at the transcript level on IFN-α treatment45 and extended (24 h vs 4) IFN-λ treatment46 in human liver culture.
Figure 6.

Immunoprocessing components under IFN treatment or HBV transfection. (a) Alteration of human 26S proteasome (PDB: 5L4G) components under IFN-treatment (left) and HBV-transfection (right). The mouse immunoproteasome cap (PDB: 5MX5) is shown once, as its two components were significantly altered under both the IFN and HBV-transfection conditions. Here, raspberry color serves only to distinguish PSME2 from PSME1. Strong red indicates significant upregulation, salmon indicates upregulation without significance, strong blue indicates significant downregulation, and light blue indicates downregulation without significance. If either of the IFN-α2 or IFN-λ3 treatments altered protein expression significantly, the protein in question was considered to be significantly altered. Tables showing alterations in (b) proteasome components, (c) immunoprocessing proteins downstream of the proteasome, (d) deubiquitylases, and (e) RING ligases and their accessory proteins. Bold lettering of fold-change data indicates significant alterations. Colors, from strong red (upregulation) to strong blue (downregulation), indicate the extent of fold change. Candidates for proviral modulation of the proteome.
Given the alterations of the proteasome upon IFN treatment, uncovered despite an absence of strong gene-enrichment statistics, we further examined our data against protein groups that are upstream and downstream of proteasome processing. These groups are expected to be relevant to HLA-peptide complex display and adaptive immune responses. A reasonable initial hypothesis would be that HBV transfection would tend to suppress components involved in positive regulation of this display, while IFN treatment would enhance them. Specifically, we examined upstream components including E3 ligases combined with their accessory factors, as well as deubiquitylases.47,48 Finally, our compilation would not be complete without a list of factors that are directly responsible for antigen processing and presentation, following proteasome-based peptide processing. We dissect these groups below.
Of the groups examined, the most significant alterations involved E3 ligases/accessories under the HBV-transfection condition. Here, while 28 such components were downregulated, only 11 were upregulated (Figure 6, E3 table). Adjusting for the fact that more proteins were significantly downregulated than upregulated in the HBV-transfection condition, we derive P = 0.03 via the binomial distribution. One might hypothesize that this pattern relates to the HBV-derived IFN response; however, the effect seems to be HBV-specific but not IFN-related, as only one ligase/accessory protein was found to be regulated in the same direction in both HBV-transfection and IFN conditions (FBXO22, downregulated). Upregulation of ligases and their accessories did not reach significance under IFN-treatment (P = 0.07). Deubiquitylases did not evince a significant trend of alteration (Figure 6, deubiquitylases table). The mechanism by which HBV would perform bulk downregulation of E3 ligases and their accessory factors is puzzling. Six factors directly involved in the HLA–peptide complex display were significantly upregulated in the HBV-transfection condition (P = 0.002) (Figure 6, antigen processing table). Here, however, IFN induction could be involved, as four of these factors were also upregulated in the IFN condition. None of the 11 factors in our HLA–peptide complex display list were downregulated upon IFN treatment, while six were upregulated (P = 0.09). In agreement with our initial hypothesis, IFN treatment upregulated factors directly involved in HLA–peptide complex display. Contrary to naive expectations, HBV transfection also tended to upregulate such factors, although a host-derived IFN response could be causative here. The expectation that HBV transfection would downregulate upstream components involved in positive regulation of HLA–peptide complex display, that is, E3 ligases and their accessories, was also met. The converse expectation, that IFN would upregulate these components, was not met.
Examining the literature, several downregulated E3 ligases/accessories under the HBV-transfection condition in this study stand out. TRIM25 has been described as “essential” for RIG-I-mediated antiviral activity49 and has previously been shown to exhibit anti-HBV effects.50 These anti-HBV effects were augmented by IFN-I treatment; TRIM25 was indeed upregulated on IFN treatment in our own study. Although not previously implicated in HBV inhibition, TRIM56 is also known to be repressed by a wide variety of viruses, undoubtedly due to its critical role in TLR3 antiviral signaling.51 CUL1 is also targeted by numerous viruses, and its interaction with c-Myc has been shown to be disrupted by the HBV HBx protein.52 Other downregulated E3 ligases and accessories implicated in antiviral processes (though not specifically HBV) include ATRX,53 HLTF,54 HUWE1,55 and RBBP6.56
While IFN-induced upregulation of E3 ligases and accessories as a group did not reach significance, it is worth noting the presence of several specific proteins that were significantly altered under IFN stimulation (Figure 6, E3 table). Under IFN-I and IFN-III conditions, DTX3L was strongly upregulated with high significance [10-fold (P = 10–7) and 5-fold (10–6), respectively]. The protein/transcript has shown to be upregulated under IFN-stimulation or viral infection.57 DTX3L ubiquitinates a subset of histones, altering the chromatin landscape to allow enhanced expression of interferon-stimulated genes; other functions and/or ubiquitylation targets have not, to our knowledge, been described. The effect on histones was not described as degradative, and our own work did not show significant downregulation of any histones. Several ligases/accessories were upregulated upon IFN stimulation without, to the best of our knowledge, previous mention in the interferon literature: SCAF11, ANKFY1, RNF149, and CUL4B. Of particular interest, both RNF149 and CUL4B ubiquitylate key components of the cell cycle (BRAF and various histones, respectively) relevant to cancer.58,59
We have suggested that HBV transfection triggers an IFN response. It is not surprising, then, that proteins upregulated in HepG2.2.15 versus HepG2 intersect significantly with those upregulated in HepG2 upon IFN-I (P = 10–7) or IFN-III (P = 10–8) treatment. What would be interesting, however, are cases where HBV-transfection apparently manipulates expression in a direction opposite to that seen on IFN treatment. This indeed occurred at significant levels; proteins significantly upregulated in the HepG2.2.15/HepG2 comparison intersected with proteins significantly downregulated on IFN-I treatment at P = 10–7 and on IFN-III treatment at P = 10–3 (case A), while proteins downregulated in the HepG2.2.15/HepG2 comparison intersected with those upregulated on IFN-I/III treatment at P = 10–3 and 10–5 (case B). Using the GSEA enrichment tool,60 case A proteins were enriched for involvement in oxidative phosphorylation (P = 10–4) and numerous metabolic groups (e.g. GO cofactor metabolic process, P = 10–3). We note two studies in which HBV infection was accompanied by broad upregulation of proteins and transcripts61 or metabolites62 relevant to glycolysis, the Krebs cycle, gluconeogenesis, lipid metabolism, and energy production. Case B proteins were enriched for poly-A RNA-binding (P = 10–5) and pyrimidine metabolism (P = 10–3).
Analysis of Metabolic Protein Expression Changes
Given the aforementioned evidence of alterations in proteins associated with metabolism, we sought to identify metabolic functions or regions of metabolism associated with significant changes in protein expression. Our approach utilized a type of gene set analysis (GSA) whereby gene sets are defined based on the association of genes with other components of the metabolic network, namely metabolites and subsystems (metabolic pathways). This use of metabolites as gene sets is known as “reporter metabolite analysis.”63
The reporter metabolite analysis revealed a few metabolites associated with a significant enrichment in coordinated protein expression increases or decreases (Figure 7a). The results suggested increased activity in metabolic pathways involving long-chain acyl-CoAs (e.g., linoleoyl-CoA and palmitoyl-CoA) upon HBV transfection, attributed in part to an increased expression of ELOVL1, ACAD, and ACSL proteins involved in lipid biosynthesis and turnover (Figure S2A). These findings are consistent with the previous studies showing an effect of HBV infection on fatty acid metabolism.64 In particular, Yang et al.65 measured increased levels of palmitoleic acid in the livers of HBV transgenic mice compared to those of wild-type, in addition to observing a marked increase in the expression of lipid metabolic proteins (FABP5 and ACBP).
Figure 7.
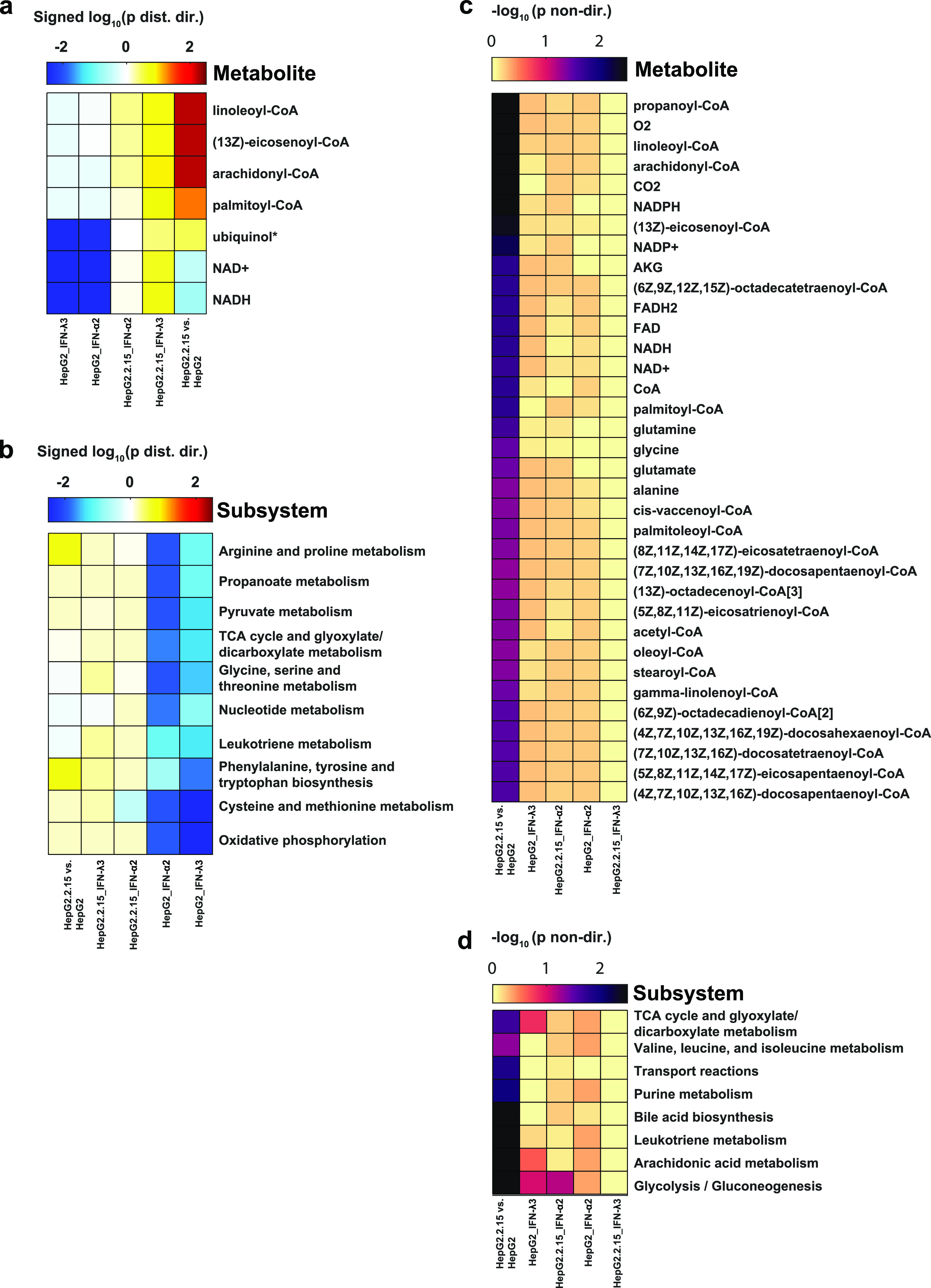
(a) Metabolite and (b) subsystem directional GSA results for the five comparisons. Shown are the log 10-transformed “distinct directional” P-values for metabolites and subsystems with P < 0.05 in at least one comparison. The log-transformed P-values are signed such that gene sets more significantly enriched in expression decreases are negative, whereas those enriched more in expression increases are positive. (c) Metabolite and (d) subsystem nondirectional GSA results for the five comparisons. Shown are the log 10-transformed “nondirectional” P-values for metabolites and subsystems with P < 0.05 in at least one comparison.
The enrichment of decreased protein expression associated with NADH and ubiquinol (and their oxidized counterparts) observed upon IFN treatment of HepG2 cells reflects the similar decrease observed in proteins associated with oxidative phosphorylation observed in the subsystem GSA (Figure 7b) and in the GSEA enrichment analysis described above. The subsystem GSA also revealed an enrichment of protein expression decreases upon IFN treatment in several other pathways, such as amino acid metabolism, nucleotide metabolism, and the TCA cycle (Figure 7b); however, no such decreases were enriched upon treatment of HBV-transfected HepG2 cells with IFN. The IFN-induced decrease of oxidative phosphorylation and TCA cycle enzyme expression have been reported previously; for example, Liu and colleagues observed a decreased expression of genes associated with the TCA cycle and glutamine catabolism upon treatment of macrophages with IFN-L and LPS,66 and Lewis et al. reported a decreased expression of mitochondrial gene expression and an electron transport chain function in IFN-treated murine fibroblasts and human lymphoblastoid cells.67 However, this effect appears to be a function of cell type, as others have measured an increase in oxygen consumption and oxidative phosphorylation activity in mouse dendritic cells, keratinocytes, and memory T cells upon IFN treatment.68
We next investigated metabolites and subsystems that were associated with significant protein expression changes but ignoring the direction of the change (increase or decrease). Many more metabolites were found to be significantly enriched in differential protein expression when the fold-change direction was excluded but only in the comparison of HBV-transfected versus nontransfected HepG2 cells (Figure 7c). The majority of the significant metabolites was acyl-CoAs, as observed in the directional GSA results, but some metabolites were associated with amino acid metabolism (glutamate, glutamine, glycine, and alanine) and the TCA cycle (α-ketoglutarate). These findings are supported by differences in metabolite exchange rates between HepG2.2.15 and HepG2 cells measured by Li et al.,61 where significant metabolites included glutamate, alanine, and α-ketoglutarate.
Although both IFN treatments and HBV transfection caused significant expression changes in proteins involved in the TCA cycle, the IFN treatment was associated with a coordinated expression decrease, whereas HBV transfection was associated with a mixture of expression decreases and increases (Figure S2). The increased expression of some TCA cycle enzymes (e.g., CS, SDHB, and MDH2) upon HBV transfection observed here is consistent with protein measurements reported by Li et al. in their comparison of HepG2.2.15 with HepG2 cells.61 In addition to the TCA cycle, proteins involved in processes such as bile acid synthesis, purine metabolism, and glycolysis/gluconeogenesis exhibited significant expression changes upon HBV transfection, but such enrichment was not observed among the IFN treatment conditions (Figure 7d).
Relevance to HBV-Associated Hepatocarcinoma Treatment
A recent multiomics study examined HBV-associated hepatocarcinoma (HCC) against adjacent tissue from 159 patients at high depth at the proteomic level.69 As both Gao’s work and our own involve high-depth proteomics, liver cancer, and HBV infection/transfection, we took this opportunity to cross datasets in search of a potential insight that could point to treatment strategies. Beginning with the 3749 proteins in common in Gao’s work, our HBV transfection work and our IFN work, we used Fisher’s exact test to examine all combinations of our own results with Gao’s (e.g. upregulated in HCC vs downregulated on IFN treatment). Interestingly, the only strongly significant effect occurred when proteins downregulated by HBV-transfection were intersected with proteins upregulated in cancer (P < 5 × 10–7) (Figure 8); proteins upregulated in cancer tended to be downregulated in HBV-transfected cells. The underlying data and intersecting sets are shown in Table S2. It would appear, then, that HBV-transfection induces a cellular state that is in opposition to that found in HBV-related HCC, a surprising result given HBV’s well-documented propensity for inducing HCC. Interpreting this result is difficult. Interestingly, in Gao’s work HBV, protein levels were significantly (P < 10–12 in the case of core protein) higher in nontumorous tissue than tumorous tissue. Viral RNA levels also tended to be higher in the nontumorous tissue (P < 10–3 for envelope and X transcripts). Thus, assuming that cancer effects dominate viral effects in HBV-related HCC, two hypotheses may be proffered: (1) cancer upregulates antiviral proteins which are downregulated by HBV (i.e. cells transformed by HBV are hostile to further HBV replication) and (2) HBV downregulates procancer proteins upregulated in cancer. Examining proteins that are both upregulated by HCC and downregulated by HBV showed strong enrichment for factors involved in the cell cycle and DNA replication (Figure 8). While these factors are not typically considered “antiviral”, it is plausible that an environment of rapid cell division is not optimal for HBV fitness. While this view is undoubtedly simplistic and fails to address the mechanisms by which HBV initiates cancer, it does complicate any picture in which the cell cycle deregulation seen in both chronic HBV and advanced HCC are considered to be well-aligned.70
Figure 8.
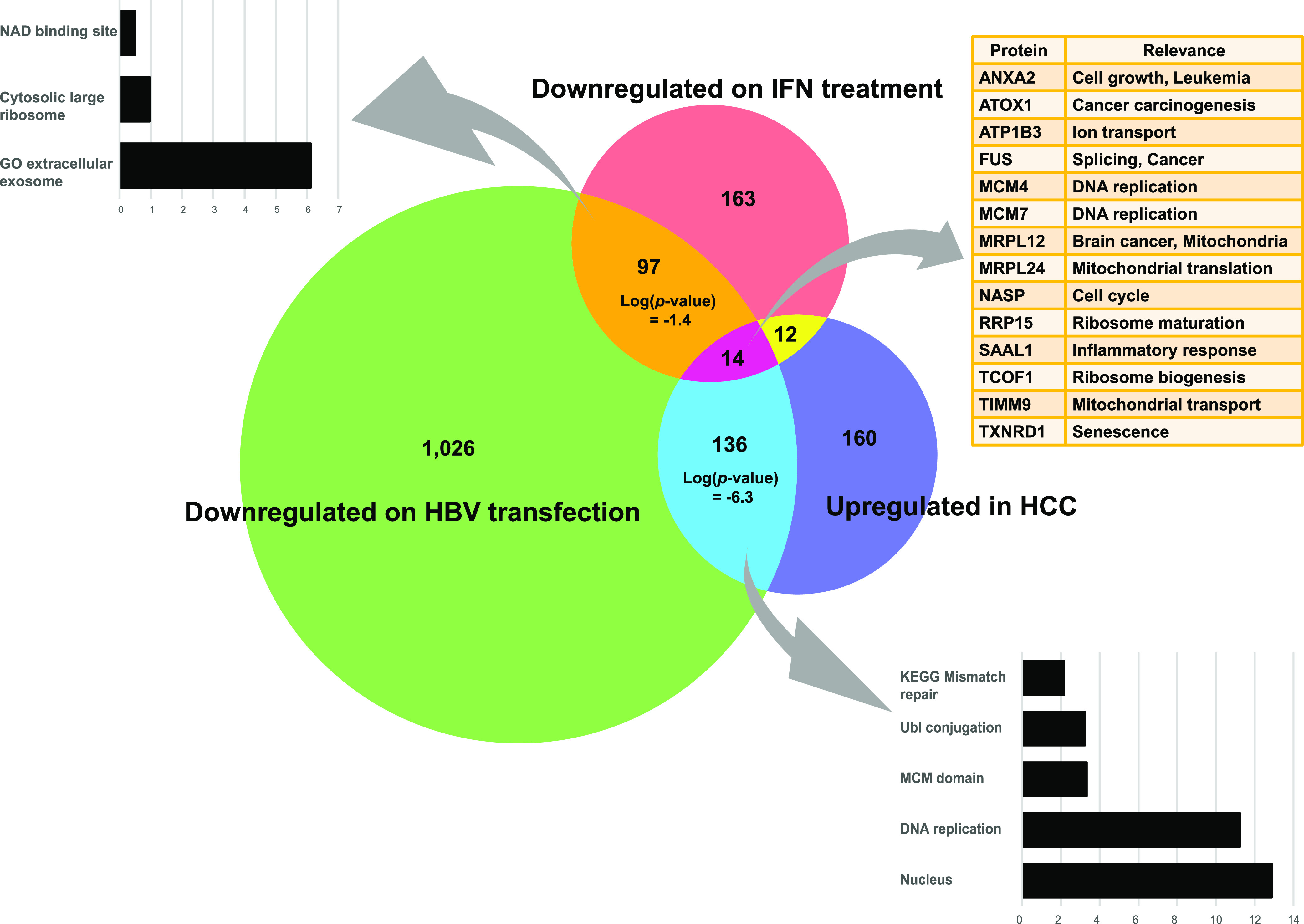
Relevance of IFN treatment and HBV transfection to HCC. Venn diagram showing the intersection of proteins significantly downregulated in our own IFN stimulation and HBV transfection work against Gao’s results showing proteins significantly upregulated in HBV-related HCC. Bar graphs show enriched gene sets found at the intersections of our two studies and the HCC study.
While HBV transfection appeared more potent than IFN in counteracting HCC’s proteomic effects, an HBV-based HCC therapy is rationally dismissed as ridiculous. However, 14 proteins upregulated in HCC were downregulated by both IFN and HBV-transfection (Figure 8), most of which play roles in the cell cycle. We note the presence of two minichromosome maintenance (MCM) proteins in this set, MCM4 and MCM7. Further, of the seven MCM complex proteins common in all three datasets (HCC, HBV-transfection, and IFN), six were downregulated in the HBV-transfection condition. Targeting of MCM complex proteins has indeed shown promise in combatting liver cancer,71 prostate cancer,72 and breast cancer.73 Other proteins within the 14 member set also figure prominently in cancer research. ANXA2 is repeatedly mentioned in the literature as promoting cancer invasiveness.74,75 ATOX1 has also been shown to play roles in cancer migration.76 Inhibitors of TXNRD1 have also been developed with the aim of sensitizing lung cancer cells to AKT suppression.77 Scanning GEO datasets,8 we note a number of studies in which drug treatments significantly reduced transcript levels of multiple proteins in the 14 member set (Table S3). These drugs vary widely in their mechanisms of action; erlotinib, for example, is a tyrosine kinase inhibitor, trichostatin A targets histone deacetylases, and pomalidomide has antiangiogenesis activity. Of note is the fact that two of the drugs are easily available at “health food” stores; octanoate (caprylic acid, derived from coconut oil) and melatonin. Further studies are required to examine the effects of these drugs at the protein level in liver cancer cells for development of potential treatments.
The HBV HBx protein is widely thought to be largely responsible for HBV-associated cell cycle alterations and for HBV-associated carcinogenicity.78 On HBx overexpression, expression of two proteins, NASP and ANXA2, found in our 14 member group has been shown to actually increase.79 However, these proteins were sharply downregulated in HepG2.2.15 cells, with P < 10–7 in both cases. Such discrepancies may indicate the HBV stage of infection that is predominant in HepG2.2.15 cells, with HBV proteins found near the limits of MS detection,12 as well as the extreme levels of the HBx protein present in an overexpression context. In fact, contradictory pro- and antiapoptotic functions have been noted with respect to HBx,80 with speculation that such behavior relates to HBx abundance in infected cells.81
Conclusions
Given the depth of our underlying IFN-stimulation proteomic data, we hope that this data would be of use in interferon research in the years to come. The inclusion of equally deep HBV-transfection proteomic data allows for speculation into the means by which HBV, and possibly other viruses, may stymie the interferon response. We were legitimately surprised to note the relative absence of proteomic work on a perturbation that has been extensively characterized at the transcriptomic level. This observation, in turn, suggests that a number of proteomic studies on otherwise well-characterized perturbations (knockdowns/outs, overexpression, and drugs) await. Our own work points to some of the “low hanging fruit” that could be targeted; proteins that are directly involved in protein degradation or stabilization (e.g. proteasome components and E3 ligases) and, as a consequence, mediate the immunoprocessing effects of the interferon.
Materials and Methods
Cell Culture and IFN-Treatment
HepG2 cells were cultured in DMEM (Gibco, MA), FBS (Gibco), 1% MEM-NEAA (Gibco), and 1% penicillin/streptomycin (Gibco). HepG2.2.15 cells, a gift from Professor Antonio Bertoletti (Singapore Institute for Clinical Sciences, A*STAR), were cultured as mentioned above, with the addition of Geneticin at 150 μg/mL. Cells were maintained at 37 °C, 5% CO2, in a humidified incubator. In IFN-treatment versus control experiments, 5 × 106 cells were seeded into T-75 flasks well plates and grown for 24 h. Type I IFN in the form of IFN-α2a (11100-1, PBL Assay Science, NJ) and type III IFN in the form of IFN-λ3 (5259-IL-025, R&D Systems, MN) were added to final concentrations of 100 ng/mL, as previously optimized in ref (12).
Proteomics
In each experimental condition (IFN-stimulation or HBV-transfection), five biological replicates were performed. Cells were lysed using 5% sodium deoxycholate with the addition of 1X protease inhibitor (Thermo), incubation for 3 min, and sonication. Cell debris was eliminated via centrifugation. Protein concentrations of all samples were determined using the BCA protein assay (Thermo). All samples were reduced with dithiothreitol (DTT) at 37 °C for 30 min, followed by alkylation with iodoacetamide at room temperature for 30 min in the dark. A quenching step was conducted with further DTT treatment for 15 min, followed by overnight trypsin treatment at 37 °C. 5% trifluoroacetic acid treatment, and centrifugation was utilized to remove precipitates. Tryptic peptide amounts were determined using the Pierce Quantitative Fluorometric Peptide Assay (Thermo).
Dimethyl labeling of tryptic peptides followed, as previously described, with a 1 h incubation period. Ammonia followed by formic acid treatment was used to stop the reaction. All samples used for downstream experiments were labeled at greater than 99% efficiency. Samples were combined and dried (SpeedVac) at room temperature. To enhance identification of low-abundance peptides, the pooled peptides were fractionated into 10 fractions using the Pierce high pH reversed-phase peptide fractionation kit (Thermo). All fractions were again dried prior to LC–MS/MS analysis.
Samples were suspended in 0.1% FA to a volume of 15 μL. An EASY-nLC1000 system (Thermo) coupled to a Q-Exactive Orbitrap Plus mass spectrometer (Thermo) with a nanoelectrospray ion source was used for MS analysis.
Elution proceeded in 5–40% acetonitrile (ACN), 0.1% FA for 70 min, and 40–95% ACN, 0.1% FA, for 20 min with a 300 nL/min flow rate. A full MS scan at resolution 70,000 was followed by 10 data-dependent MS2 scans at resolution 17,500. The normalized collision energy of HCD fragmentation was 32%. The MS scan range was set to 350 to 1400 m/z. Precursors ions with unassigned charge states, states of +1 or greater than +8, were excluded. Dynamic exclusion of 30 s was set. Proteome Discoverer software 2.1 (Thermo) was used to generate peak lists, and the SEQUEST-HT engine was used for data processing. Databases used in search included a list of common contaminants (www.thegpm.org/crap), the human UniProt database (November 2018), and the hepatitis B virus UniProt database (November 2018). Search parameters were set for trypsin digestion, a maximum of 2 missed cleavages, up to 4 modifications, variable oxidation of methionine, and fixed modifications: carbamidomethylation of cysteine (+57.02146 Da), as well as light, medium, and heavy dimethylation of N termini and lysine (+28.031300, +32.056407, and +36.075670 Da). Mass tolerance for precursor ions was set to 10 ppm; and mass tolerance for fragment ions was set at 0.02 Da. Known contaminant ions were excluded. False discovery rate was based on Q-values, set to 1%. Differential ratios between various conditions were calculated by comparison of mean intensities; when fewer than three ratios could be calculated, the proteins in question were dropped from further analysis. An unpaired t-test was used to calculate the significance of differential results.
The precursor ions quantifier node in Proteome Discoverer software was employed to quantify the relative MS signal intensities of dimethyl labeled peptides. The vehicle channel was used as a denominator to generate abundance ratios of the IFN-λ3/vehicle and IFN-α2/vehicle. The HepG2 channel was used as a denominator to generate abundance ratios of HepG2.2.15/HepG2. Log 2 of the normalized ratio was used to calculate the mean and standard deviation of fold change across all five biological replicates. When these ratios were found in less than three experiments, the relevant proteins were excluded.
The MS data have been deposited to the ProteomeXchange Consortium via the PRIDE82 partner repository with the dataset identifier PXD017251.
Statistical Methods
Differential Analysis
In cases where we speak of protein “upregulation” or “downregulation”, significance at P < 0.05 (Student’s t-test) must be met. With respect to proteins identified via dimethyl labeling, the proteins were required to be identified in at least 3 of 5 reps in order to be considered for differential analysis. For volcano plots, the R package “plot” was used. In the heatmap, to whittle the list of proteins down to about 200 strongly altered proteins for the purpose of stringent clustering, we applied a filter to include only proteins that exhibited substantial differential changes (ratios) between experimental conditions. Specifically, we added a requirement for entry; of the five log 2 ratios associated with a given protein’s differential expression changes under different experimental conditions, the smallest ratio subtracted from the largest ratio must be greater than two. For heatmaps, the R package “heatmap.2” was used to construct the heatmap in Figure 5.
In the case of a list of canonical IFN-upregulated and downregulated transcripts that we constructed, a transcript must be upregulated in at least 6 of 18 studies or downregulated in 3 of 18, for inclusion (see Table S1 for the complete list of studies). When using DAVID for enrichment analysis, a background of 6003 confidently identified proteins was set. In cases where an option for input of “custom” backgrounds is not available (e.g. GSEA), we downloaded the underlying sets that showed apparent enrichment and then performed Fisher’s exact test using the R-programming function “fisher.test”.
When calculating binomial probabilities, probabilities of success were adjusted according to the total number of significantly up- and downregulated proteins in a study. For example, roughly 3 proteins were downregulated for every 2 upregulated under the HBV-transfection condition, meaning that a downregulated protein has an a priori 60% chance of success versus an upregulated protein.
Bioinformatics
PCA and Pearson Analysis
PCA was performed via the R function “prcomp” (tidyverse library) and plotted using the “fviz_pca_ind” function (factoextra). Any columns containing non-numerical data were removed. For Pearson analysis (R function “ggscatter” with the cor.method parameter set to “pearson”), removal of non-numerical data was not required, and the differential effects of IFN-I and IFN-III were combined.
Metabolite Analysis
Metabolite and subsystem gene set collections were extracted from the human genome-scale metabolic model, Human-GEM.83 GSAs were performed using the Piano package in R,84 which enables the incorporation of fold-change directionality information when evaluating the significance of gene set enrichment. A GSA was run for each of the five conditions: transfection of HepG2 with HBV, treatment of HepG2 with IFN-α2, treatment of HepG2 with IFN-λ3, treatment of HBV-transfected HepG2 with IFN-α2, and treatment of HBV-transfected HepG2 with IFN-λ3. All P-values reported for the metabolite and subsystem GSA were corrected for the false discovery rate using the Benjamini-Hochberg procedure.
Acknowledgments
K.H. is supported by the Rachadaphiseksompot Fund for Postdoctoral Fellowship, Chulalongkorn University, Thailand. J.R. is financially supported by the Knut and Alice Wallenberg Foundation as a part of the National Bioinformatics Infrastructure Sweden at SciLifeLab. S.K. is financially supported by the Thailand Research Fund (MRG6280206), Mahidol University, and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research, and Innovation Thailand. This project was supported by the Chulalongkorn Academic Advancement into its 2nd Century Project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01865.
Pearson correlation analysis in which either upregulated or downregulated proteins were excluded in protein/transcript comparisons and comparison of expression changes in proteins associated with various metabolites (PDF)
Data from the studies used to generate a list of canonical IFN-regulated transcripts; protein lists from all our studies and Gao’s from which P-values and Venn diagrams were generated; and drugs that, under various conditions and cell lines, downregulate transcripts in our “14 member set” (XLSX)
Author Contributions
# K.H. and J.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Waddell S. J.; Popper S. J.; Rubins K. H.; Griffiths M. J.; Brown P. O.; Levin M.; et al. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS One 2010, 5, e9753 10.1371/journal.pone.0009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry A. K.; Chen G.; Zheng D.; Tang H.; Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- de la Maza L. M.; Peterson E. M.; Goebel J. M.; Fennie C. W.; Czarniecki C. W. Interferon-induced inhibition of Chlamydia trachomatis: dissociation from antiviral and antiproliferative effects. Infect. Immun. 1985, 47, 719–722. 10.1128/iai.47.3.719-722.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiting D. P. Protozoan parasites and type I interferons: a cold case reopened. Trends Parasitol. 2014, 30, 491–498. 10.1016/j.pt.2014.07.007. [DOI] [PubMed] [Google Scholar]
- McNab F.; Mayer-Barber K.; Sher A.; Wack A.; O’Garra A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reder A. T.; Feng X. How type I interferons work in multiple sclerosis and other diseases: some unexpected mechanisms. J. Interferon Cytokine Res. 2014, 34, 589–599. 10.1089/jir.2013.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker B. S.; Rautela J.; Hertzog P. J. Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- Edgar R.; Domrachev M.; Lash A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megger D. A.; Philipp J.; Le-Trilling V. T. K.; Sitek B.; Trilling M. Deciphering of the Human Interferon-Regulated Proteome by Mass Spectrometry-Based Quantitative Analysis Reveals Extent and Dynamics of Protein Induction and Repression. Front. Immunol. 2017, 8, 1139. 10.3389/fimmu.2017.01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Q.; Ezzati P.; Spicer V.; Krokhin O.; Wall D.; Wilkins J. A. Interferon gamma induced compositional changes in human bone marrow derived mesenchymal stem/stromal cells. Clin. Proteomics 2017, 14, 26. 10.1186/s12014-017-9161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney S.; Delgado C.; Lenz L. L. Differential effects of type I and II interferons on myeloid cells and resistance to intracellular bacterial infections. Immunol. Res. 2013, 55, 187–200. 10.1007/s12026-012-8362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makjaroen J.; Somparn P.; Hodge K.; Poomipak W.; Hirankarn N.; Pisitkun T. Comprehensive Proteomics Identification of IFN-lambda3-regulated Antiviral Proteins in HBV-transfected Cells. Mol. Cell. Proteomics 2018, 17, 2197–2215. 10.1074/mcp.ra118.000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommereyns C.; Paul S.; Staeheli P.; Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.-H.; Tao Y.; Zhang Z.-Z.; Chen W.-X.; Cai X.-F.; Chen K.; et al. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor AP-1. J. Virol. 2014, 88, 2442–2451. 10.1128/jvi.02861-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Wang T. Z.; Kong D.; Zhang L.; Meng H. X.; Jiang Y.; et al. Hepatoma cell line HepG2.2.15 demonstrates distinct biological features compared with parental HepG2. World J. Gastroenterol. 2011, 17, 1152–1159. 10.3748/wjg.v17.i9.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Xie C.; Li Y.-m.; Huang Z.-l.; Zhao Q.-y.; Hu Z.-x.; et al. TMEM2 inhibits hepatitis B virus infection in HepG2 and HepG2.2.15 cells by activating the JAK-STAT signaling pathway. Cell Death Dis. 2016, 7, e2239 10.1038/cddis.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakpoor A.; Ni Y.; Chen A.; Ho Z. Z.; Oei V.; Yang N.; et al. Spatiotemporal Differences in Presentation of CD8 T Cell Epitopes during Hepatitis B Virus Infection. J Virol. 2019, 93 (4), e01457-18. 10.1128/jvi.01457-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Jiang J.; Ni C.; Xu Q.; Ye S.; Wu J.; et al. HBV Integration-mediated Cell Apoptosis in HepG2.2.15. J. Cancer 2019, 10, 4142–4150. 10.7150/jca.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Jiang D.; Zhang H.; Lv S.; Rao H.; Fei R.; et al. Proteome responses to stable hepatitis B virus transfection and following interferon alpha treatment in human liver cell line HepG2. Proteomics 2009, 9, 1672–1682. 10.1002/pmic.200800621. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhao R.; Wu Y.; Kong D.; Zhang L.; Wu D.; et al. Hepatitis B virus induces G1 phase arrest by regulating cell cycle genes in HepG2.2.15 cells. Virol. J. 2011, 8, 231. 10.1186/1743-422x-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Yang F.; Yang Y.; Hu Y.; Liu W.; Huang C.; et al. HBV Facilitated Hepatocellular Carcinoma Cells Proliferation by Up-Regulating Angiogenin Expression Through IL-6. Cell. Physiol. Biochem. 2018, 46, 461–470. 10.1159/000488614. [DOI] [PubMed] [Google Scholar]
- Wieland S.; Thimme R.; Purcell R. H.; Chisari F. V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 6669–6674. 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland S. F.; Chisari F. V. Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. 10.1128/jvi.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; et al. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Li J. J.; Bickel P. J.; Biggin M. D. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2014, 2, e270 10.7717/peerj.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A.; Lassnig C.; Eberle C.-A.; Górna M. W.; Baumann C. L.; Burkard T. R.; et al. IFIT1 is an antiviral protein that recognizes 5’-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- François-Newton V.; Magno de Freitas Almeida G.; Payelle-Brogard B.; Monneron D.; Pichard-Garcia L.; Piehler J.; et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One 2011, 6, e22200 10.1371/journal.pone.0022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Hinson E. R.; Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Mathieson T.; Franken H.; Kosinski J.; Kurzawa N.; Zinn N.; Sweetman G.; et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018, 9, 689. 10.1038/s41467-018-03106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D. B.; Gasch A. P. Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol. Biol. Cell 2008, 19, 4580–4587. 10.1091/mbc.e07-07-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. V.; Topper S. E.; Hubler S. L.; Hose J.; Wenger C. D.; Coon J. J.; et al. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 2011, 7, 514. 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic M.; Rooney M. S.; Mertins P.; Przybylski D.; Chevrier N.; Satija R.; et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 2015, 347, 1259038. 10.1126/science.1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putluri N.; Maity S.; Kommagani R.; Creighton C. J.; Putluri V.; Chen F.; et al. Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia 2014, 16, 390–402. 10.1016/j.neo.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grolmusz V. K.; Karászi K.; Micsik T.; Tóth E. A.; Mészáros K.; Karvaly G.; et al. Cell cycle dependent RRM2 may serve as proliferation marker and pharmaceutical target in adrenocortical cancer. Am. J. Cancer Res. 2016, 6, 2041–2053. [PMC free article] [PubMed] [Google Scholar]
- Davenport J. W.; Fernandes E. R.; Harris L. D.; Neale G. A. M.; Goorha R. The mouse mitotic checkpoint gene bub1b, a novel bub1 family member, is expressed in a cell cycle-dependent manner. Genomics 1999, 55, 113–117. 10.1006/geno.1998.5629. [DOI] [PubMed] [Google Scholar]
- Zhong W.-d.; Fu X.; Chen G.; Cai Z. D.; Wang C.; Liu Z. Z.; Lin Z. Y.; et al. Overexpression of BUB1B contributes to progression of prostate cancer and predicts poor outcome in patients with prostate cancer. OncoTargets Ther. 2016, 9, 2211–2220. 10.2147/ott.s101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson B. A.; Zhang Y.; Jiang H.; Hussey K. M.; Shrimp J. H.; Lin H.; et al. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016, 353, 45–50. 10.1126/science.aaf7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz K.; Wagner K.; Mendler L.; Hölper S.; Dehne N.; Müller S. SUMO Signaling by Hypoxic Inactivation of SUMO-Specific Isopeptidases. Cell Rep. 2016, 16, 3075–3086. 10.1016/j.celrep.2016.08.031. [DOI] [PubMed] [Google Scholar]
- Sharma K.; D’Souza R. C. J.; Tyanova S.; Schaab C.; Wiśniewski J. R.; Cox J.; et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Prins K. C.; Cárdenas W. B.; Basler C. F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. 10.1128/jvi.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati M.; Li Z.; Liu F.; Sousa A. M. M.; Nakagawa N.; Li M.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. 10.1016/j.celrep.2016.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q.; Cao X.; Lu J.; Huang B.; Liu Y.-J.; Kato N.; et al. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. 10.1016/j.jhep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Tang H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 2010, 1, 1106–1117. 10.1007/s13238-010-0141-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osinalde N.; Sanchez-Quiles V.; Akimov V.; Guerra B.; Blagoev B.; Kratchmarova I. Simultaneous dissection and comparison of IL-2 and IL-15 signaling pathways by global quantitative phosphoproteomics. Proteomics 2015, 15, 520–531. 10.1002/pmic.201400194. [DOI] [PubMed] [Google Scholar]
- Maiwald T.; Schneider A.; Busch H.; Sahle S.; Gretz N.; Weiss T. S.; et al. Combining theoretical analysis and experimental data generation reveals IRF9 as a crucial factor for accelerating interferon alpha-induced early antiviral signalling. FEBS J. 2010, 277, 4741–4754. 10.1111/j.1742-4658.2010.07880.x. [DOI] [PubMed] [Google Scholar]
- Bolen C. R.; Ding S.; Robek M. D.; Kleinstein S. H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. 10.1002/hep.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvar B.; Raghuram V.; Pisitkun T.; Sarkar A.; Knepper M. A. Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation. Physiol. Genomics 2016, 48, 502–512. 10.1152/physiolgenomics.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z.; Chen J.-X.; Zhao Y.; Medvar B.; Knepper M. A. Data integration in physiology using Bayes’ rule and minimum Bayes’ factors: deubiquitylating enzymes in the renal collecting duct. Physiol. Genomics 2017, 49, 151–159. 10.1152/physiolgenomics.00120.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack M. U.; Shin Y. C.; Joo C.-H.; Urano T.; Liang C.; Sun L.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Tan G.; Xiao Q.; Song H.; Ma F.; Xu F.; Peng D.; et al. Type I IFN augments IL-27-dependent TRIM25 expression to inhibit HBV replication. Cell. Mol. Immunol. 2018, 15, 272–281. 10.1038/cmi.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Li N. L.; Wang J.; Liu B.; Lester S.; Li K. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 2012, 287, 36404–36413. 10.1074/jbc.m112.397075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra N.; Kumar V. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc. FEBS Lett. 2006, 580, 431–436. 10.1016/j.febslet.2005.12.034. [DOI] [PubMed] [Google Scholar]
- Jurak I.; Silverstein L. B.; Sharma M.; Coen D. M. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J. Virol. 2012, 86, 10093–10102. 10.1128/jvi.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahouassa H.; Blondot M.-L.; Chauveau L.; Chougui G.; Morel M.; Leduc M.; et al. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 5311–5316. 10.1073/pnas.1600485113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S. P.; Okawa K.; Nakano T.; Sano K.; Ogawa K.; Masuda T.; et al. Huwe1, a novel cellular interactor of Gag-Pol through integrase binding, negatively influences HIV-1 infectivity. Microbes Infect. 2011, 13, 339–349. 10.1016/j.micinf.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Batra J.; Hultquist J. F.; Liu D.; Shtanko O.; Von Dollen J.; Satkamp L.; et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175, 1917–1930. 10.1016/j.cell.2018.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Mao D.; Roswit W. T.; Jin X.; Patel A. C.; Patel D. A.; et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 2015, 16, 1215–1227. 10.1038/ni.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.-W.; Jin D.-H.; Shin J.-S.; Moon J.-H.; Na Y.-S.; Jung K.-A.; et al. Ring finger protein 149 is an E3 ubiquitin ligase active on wild-type v-Raf murine sarcoma viral oncogene homolog B1 (BRAF). J. Biol. Chem. 2012, 287, 24017–24025. 10.1074/jbc.m111.319822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa L. A.; Wu M.; Ye T.; Kobayashi R.; Sun H.; Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- Subramanian A.; Tamayo P.; Mootha V. K.; Mukherjee S.; Ebert B. L.; Gillette M. A.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Zhu W.; Zhang L.; Lei H.; Wu X.; Guo L.; et al. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. 10.1038/srep08421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne R. J.; Casciano J. C.; Bouchard M. J. A broad investigation of the HBV-mediated changes to primary hepatocyte physiology reveals HBV significantly alters metabolic pathways. Metabolism 2018, 83, 50–59. 10.1016/j.metabol.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil K. R.; Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 2685–2689. 10.1073/pnas.0406811102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.-X.; Huang C.-J.; Yang Z.-G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J. Gastroenterol. 2016, 22, 8161–8167. 10.3748/wjg.v22.i36.8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Yan S.; He Y.; Wang F.; Song S.; Guo Y.; et al. Expression of hepatitis B virus proteins in transgenic mice alters lipid metabolism and induces oxidative stress in the liver. J. Hepatol. 2008, 48, 12–19. 10.1016/j.jhep.2007.06.021. [DOI] [PubMed] [Google Scholar]
- Liu L.; Lu Y.; Martinez J.; Bi Y.; Lian G.; Wang T.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1alpha-dependent. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 1564–1569. 10.1073/pnas.1518000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. A.; Huq A.; Najarro P. Inhibition of mitochondrial function by interferon. J. Biol. Chem. 1996, 271, 13184–13190. 10.1074/jbc.271.22.13184. [DOI] [PubMed] [Google Scholar]
- Wu D.; Sanin D. E.; Everts B.; Chen Q.; Qiu J.; Buck M. D.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. 10.1016/j.immuni.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q.; Zhu H.; Dong L.; Shi W.; Chen R.; Song Z.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 1240. 10.1016/j.cell.2019.10.038. [DOI] [PubMed] [Google Scholar]
- Benn J.; Schneider R. J. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 11215–11219. 10.1073/pnas.92.24.11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y.; Zhang B.; Zhou J. N.; Wang D. X.; Xu Y. C.; Zeng Q.; et al. Arsenic trioxide inhibits liver cancer stem cells and metastasis by targeting SRF/MCM7 complex. Cell Death Dis. 2019, 10, 453. 10.1038/s41419-019-1676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y.-b.; Yang D.-r.; Nong S.-j.; Ni J.; Hu C.-h.; Li J.; et al. Breviscapine (BVP) inhibits prostate cancer progression through damaging DNA by minichromosome maintenance protein-7 (MCM-7) modulation. Biomed. Pharmacother. 2017, 93, 103–116. 10.1016/j.biopha.2017.06.024. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Li W.; Liu J.; Li J.; He F.; Jiang Y.; et al. Simvastatin suppresses the DNA replication licensing factor MCM7 and inhibits the growth of tamoxifen-resistant breast cancer cells. Sci. Rep. 2017, 7, 41776. 10.1038/srep41776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Yuan J.; Zhang J.; Tian R.; Ji W.; Zhou Y.; et al. Anxa2 binds to STAT3 and promotes epithelial to mesenchymal transition in breast cancer cells. Oncotarget 2015, 6, 30975–30992. 10.18632/oncotarget.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Zhang F.; Yu M.; Zhao P.; Ji W.; Zhang H.; et al. Up-regulation of Anxa2 gene promotes proliferation and invasion of breast cancer MCF-7 cells. Cell Proliferation 2012, 45, 189–198. 10.1111/j.1365-2184.2012.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blockhuys S.; Wittung-Stafshede P. Copper chaperone Atox1 plays role in breast cancer cell migration. Biochem. Biophys. Res. Commun. 2017, 483, 301–304. 10.1016/j.bbrc.2016.12.148. [DOI] [PubMed] [Google Scholar]
- Dai B.; Yoo S.-Y.; Bartholomeusz G.; Graham R. A.; Majidi M.; Yan S.; et al. KEAP1-dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013, 73, 5532–5543. 10.1158/0008-5472.can-13-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slagle B. L.; Bouchard M. J. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. 10.1016/j.coviro.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H.; Li X.; Niu D.; Chen W. N. Protein profile in HBx transfected cells: a comparative iTRAQ-coupled 2D LC-MS/MS analysis. J. Proteomics 2010, 73, 1421–1432. 10.1016/j.jprot.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Ye L.; Dong N.; Wang Q.; Xu Z.; Cai N.; Wang H.; et al. Progressive changes in hepatoma cells stably transfected with hepatitis B virus X gene. Intervirology 2008, 51, 50–58. 10.1159/000120289. [DOI] [PubMed] [Google Scholar]
- Kew M. C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 144–152. 10.1111/j.1440-1746.2010.06546.x. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Ríos D.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J. L.; Kocabas P.; Wang H.; Cholley P. E.; Cook D.; Nilsson A.; et al. An atlas of human metabolism. Sci. Signaling 2020, 13, eaaz1482 10.1126/scisignal.aaz1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo L.; Nielsen J.; Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. 10.1093/nar/gkt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.