

Abstract
Dynamics and conformational motions are important to the activity of enzymes, including protein tyrosine phosphatases. These motions often extend to regions outside the active site, so-called allosteric regions. In the tyrosine phosphatase, Vaccinia H1-Related (VHR) enzyme we demonstrate the importance of the allosteric interaction between the variable insert region and the active site loops in VHR. These studies include solution NMR, computation, steady-state, and rapid kinetics measurements. Overall, the data indicate concerted millisecond motions exist between the variable insert and catalytic acid loop in WT VHR. 150 ns computations studies show a flexible acid loop in WT VHR that opens during the simulation from its initial closed structure. Mutation of the variable insert residue, asparagine 74 to alanine results in a rigidification of the acid loop as observed by molecular dynamics simulation and a disruption of crucial active site hydrogen bonds. Moreover, enzyme kinetics analysis shows a weakening of substrate affinity in the N74A mutant and an over 2-fold decrease in substrate cleavage and hydrolysis rates. These data show that despite being nearly 20Å from the active site, the variable insert region is linked to the acid loop by coupled millisecond motions and that disruption of the communication between the Variable Insert and active site alters the normal catalytic function of VHR as well as perturbing the active site environment.
Keywords: Allostery, protein tyrosine phosphatase, NMR, computation, molecular dynamics
Graphical Abstract
Introduction
Protein tyrosine phosphatases (PTP) are critical regulators of important biological processes;1–4 Vaccinia H1-Related (VHR) phosphatase (Uniprot 541452) is a 20kDa PTP and its catalytic activity is crucial for proper cellular function. VHR was initially classified as a dual-specificity phosphatase (DUSP), but later studies demonstrated 103-fold preference for phospho-tyrosine (pY) containing substrates over phospho-Ser or phospho-Thr based substrates for VHR.5, 6 VHR is primarily a nuclear enzyme where it acts on a several different substrates including STAT5 (Signal Transducers and Activators of Transcription),7 the MAP Kinases ERK1 and ERK2,5 JNK1 (c-Jun N-terminal Kinase),8 EGFR (Epidermal Growth Factor Receptor) and ErbB2.9 In cervical cancer, VHR activity downregulates ERK1/2 and enhances cancer progression.10 In addition, it was shown that siRNA silencing of VHR results in cell cycle arrest at the G1/S and G2/M stage.11, 12 Furthermore, in certain cell models of breast cancer13 and non-small lung cell cancer cells,9 VHR expression is depressed and up-regulation of its expression slows cancer cell growth. These studies suggest that depending on the disease, inhibition or enhancement of VHR catalytic activity would be an avenue to therapeutic intervention.
Unfortunately, like all Class 1 PTPs, the high active site sequence conservation results in low specificity among competitive inhibitors making them poor drug candidates. In addition, the necessity of a negatively charged ligand for binding to the highly conserved, electropositive active-site P-loop prevents such small molecules from crossing the cellular membrane required of effective PTP inhibitors. These restrictive properties have resulted in PTP enzymes being dubbed undruggable.14, 15 However, allosteric ligands, which are not subject to the constraints of competitive inhibitors, represent a potential alternative.16–18 There are a significant number of allosteric ligands described in the literature. Specific to phosphatases, some notable examples are the drugs cyclosporin and FK506 that target the Ser/Thr phosphatase, calcineurin.19, 20 Allosteric ligands have also been discovered to inhibit the PTP enzyme PTP1B.21, 22 Moreover, allosteric sites have been described for the Ser/Thr phosphatase, PP2A that regulate its function23, 24 and it has been shown that PDZ domains, which among their many functions, can convey allosteric ligand binding information via changes in dynamics, which may be important for the catalytic activity of Protein Tyrosine phosphatase 1E.25, 26 Recent work in our lab using a combination of mutagenesis and solution NMR spectroscopy has identified two allosteric sites in VHR.27 However, an important next step is a mechanistic understanding of the linkage between the allosteric and active sites that results in alteration of catalytic activity.14, 28–30
VHR shares a similar catalytic mechanism with the more thoroughly characterized PTP enzymes YopH and PTP1B (Fig 1A).31, 32 The enzyme active site is flanked by several loop regions, which are important for its function. The phosphate group from the substrate binds to the highly conserved P-loop (residues 123–131 in VHR). Additional binding interactions occur via the recognition loop (residues 19–29). Subsequent to substrate binding, PTP enzymes remove the phosphate group from tyrosine residues in two steps, cleavage and hydrolysis. Cleavage of the phosphotyrosine protein substrate occurs by nucleophilic attack from the conserved cysteine (C124) residue in the P-loop (Fig. 1A & B). The phosphoenzyme intermediate is hydrolyzed in the second step to regenerate the active enzyme for the next catalytic cycle. Both steps involve the acid loop (88–99) that contains a conserved aspartic acid (D92) located in the active site. In the cleavage reaction, D92 donates a proton to the leaving group tyrosine. By analogy with PTP1B and YopH, D92 also helps coordinate a water molecule for the subsequent hydrolysis step (Fig. 1A).6, 31–36
Figure 1:
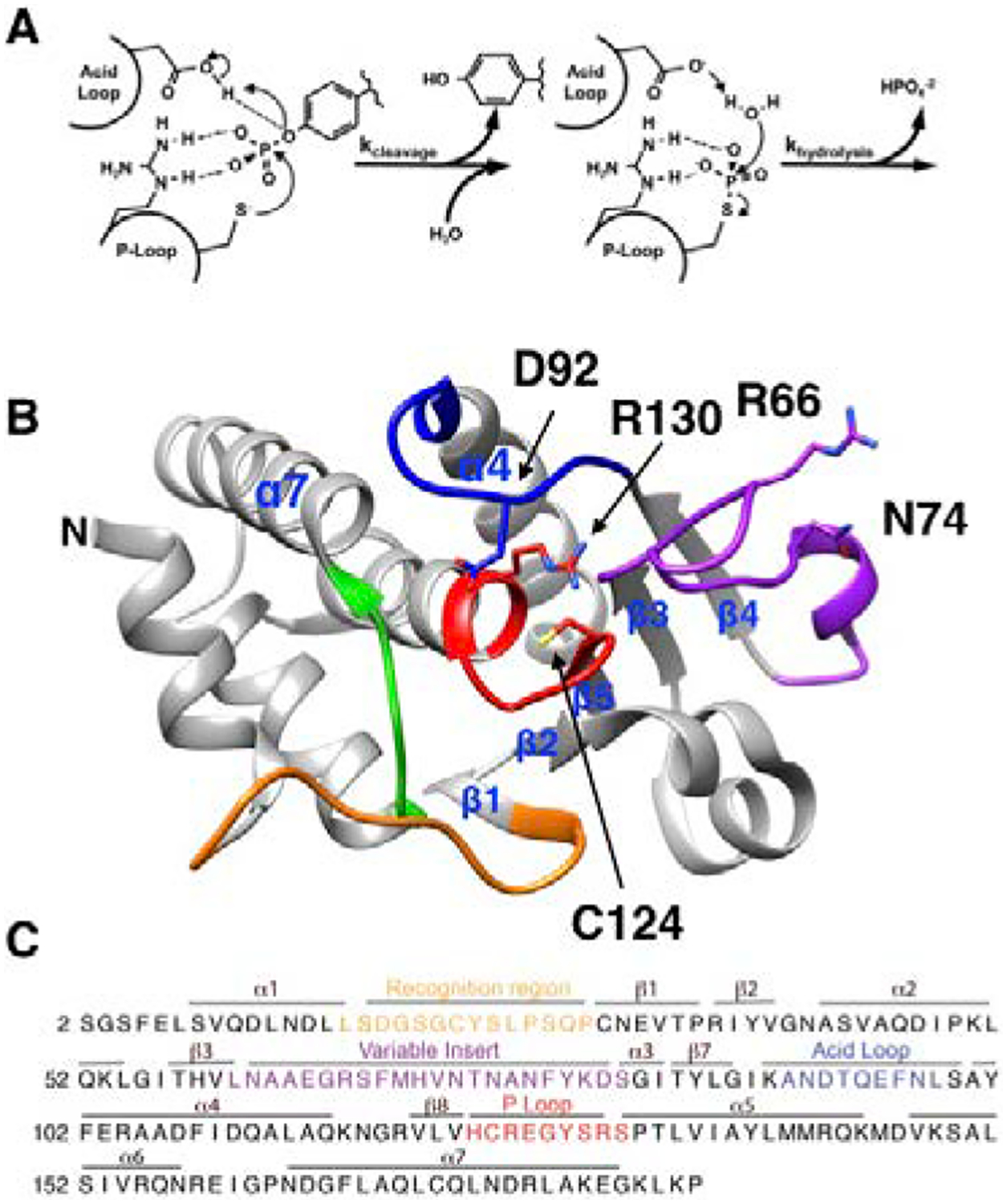
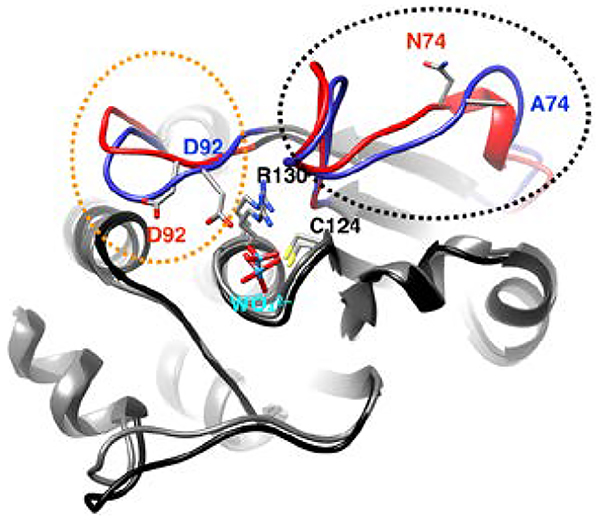
Mechanism and structure of VHR. A) General mechanism of Class I PTPs showing the cleavage step followed by the hydrolysis of the covalent intermediate. B) Structure of VHR (PDB ID: 1VHR) with the colored regions representing the recognition region (residues 19–29) in orange, variable insert (62–82) in purple, acid loop (89–99) in blue, P-loop (123–131) in red, and Q-loop (158–163) in green. The catalytic acid D92, active-site nucleophile C124, P-loop residue R130, and allosteric residues R66, and N74 are represented as sticks. Selected secondary structure elements are labeled. C) Primary sequence of VHR with relevant regions colored as in panel B.
There are a few notable differences between VHR, YopH, and PTP1B in regard to the active-site loops. Crystal structures of apo and ligand-bound forms of both PTP1B and YopH show distinct open and closed conformations of the acid loop, respectively. In the loop-open conformation of these enzymes, the catalytic acid is ~10 Å from where it needs to be to protonate the leaving group tyrosine.34, 37–41 Thus, loop closure is essential for catalysis and in the closed conformation the aspartic acid is brought in close proximity (~4 Å) to the substrate for efficient protonation. In contrast, the X-ray structures of VHR show the acid loop to be in the closed conformation,6, 42 although there is no available structure of VHR with an empty active site pocket. In the three published structures of VHR, the active site is occupied by peptide substrate (PDB 1J4X), the sulfonate moiety of HEPES buffer (PDB 1VHR), and a derivative of ethane sulfonic acid (PDB 3F81) are bound to the active site.6, 42, 43 It is therefore uncertain whether VHR utilizes a flexible acid loop in its catalytic cycle. An additional differentiating feature from PTP1B and YopH is the presence of the so-called variable insert (VI) loop (residues 62–82), which connects β3 and β4 and this connecting region is significantly shorter than in YopH or PTP1B (Fig 1B, C). The VI has been suggested to be a site that facilitates VHR dimerization,44 and is the location of an allosteric site that has been identified by solution NMR methods.27
Here, we provide initial NMR experiments on WT VHR and further examine the role of Variable Insert by mutation of a central residue in this region, N74, followed by characterization by solution NMR spectroscopy, computation, and enzyme kinetic assays. N74 is 12.4 Å (Fig. 1B) from the catalytic cysteine nucleophile yet alters aspects of VHR catalytic activity, ligand binding, and protein-wide NMR chemical shifts and molecular motions further bolstering the VI as an allosteric site in VHR.
Materials and Methods:
Site-directed Mutagenesis
Mutants of VHR were prepared by PCR using oligonucleotide primers purchased from Keck Biotechnology Resource Laboratory (Yale University) and DNA sequences were confirmed by DNA sequencing performed by Keck Biotechnology Resource Laboratory. The sequences of the mutagenic primers used for N74A: 5’ – GTCCTTGTAGAAGTTGGCAGCGGTGTTGACGTGCATGAAG – 3’; 5’ – CTTCATGCACGTCAACACCGCTGCCAACTTCTACAAGGAC – 3’ and for K50A: 5’ – GCCTAGTTTCTGCAGCGCGGGGATGTCCTGAGCC – 3’ and 5’ – GGCTCAGGACATCCCCGCGCTGCAGAAACTAGGC – 3’.
Protein Expression and Purification
VHR was expressed and purified as previously described.27 The gene sequence for VHR contains a polyHis-tag preceding a TEV cleavage site at the N-terminus. The DNA for this protein construct in plasmid pet43.1b was transformed and expressed in BL21 (DE3) cells in either LB (for kinetic studies) or M9 minimal media (for NMR studies) in the presence of 100 mg/mL of carbenicillin. Expression started with a 10 mL culture of cells in LB placed in a 37°C incubator shaking at about 180 RPM overnight. Inoculation of 1 mL of this culture into either 500 mL of LB or 25 mL of 50% D2O M9 minimal media and placed back in the incubator. Minimal media cultures were incubated for 8 hours before inoculating 5 mL into 50 mL of 100% D2O M9 minimal media and shaken at 37°C overnight. This culture was then inoculated into 1 L of 100% D2O M9 minimal media supplemented with 15NH4Cl and 13C-glucose (when necessary) and placed back in the incubator. All E.coli cultures for protein expression were induced with 1 mM IPTG once the OD600 reached about 0.8, at which point the cultures were placed in a 25° C incubator rotating at 200 RPM for 16–18 hours. For selectively reverse-labeled samples, 500 mg/L of amino acids of natural isotope abundance were added to the minimal media cultures 15–45 minutes prior to induction, following previously published procedures.45 Eleven 2H, 13C, 15N labeled samples supplemented with an individual isotopically natural abundant amino acid (Ala, Arg, Asn, Gln, Gly, Lys, Met, Phe, Ser, Thr, Tyr) and two samples supplemented with nine (Ala, His, Leu, Lys, Ile, Met, Arg, Ser, Val) and three (Ile, Leu, Val) different amino acids were prepared using this method. Cells were harvested by centrifugation at 7,000 RPM for 45 minutes and stored at −80°C.
Purification of VHR started by resuspending cells in ~15–18 mL of lysis buffer [20 mM Tris Base, 500 mM NaCl, 20 mM imidazole, 5 mM 2-mercaptoethanol, and 5% glycerol (pH 7.4)] and lysed by sonication (3 seconds on/off for a total on-time of 2 minutes at 75% amplitude). The lysate was clarified by centrifugation at 13,000 RPM for 45 minutes and filtered through a 0.45 μm filter. The filtered lysate was then added to 5 mL of NTA-Ni resin and nutated for about 1 hour. The resin was washed with 75 mL of lysis buffer and VHR was subsequently eluted with 50 mL of elution buffer [20 mM Tris Base, 500 mM NaCl, 500 mM imidazole, 5 mM 2-mercaptoethanol, and 5% glycerol (pH 7.4)]. The purity of collected fractions was verified by PAGE before dialyzing into lysis buffer for > 4 hours at 4°C with 1:20 TEV:VHR. The fractions were added to another column with NTA-Ni resin using the identical protocol. The purity of collected fractions of VHR were again verified by PAGE and dialyzed into either the kinetics buffer [100 mM NaAc, 50 mM Bis-Tris, and 50 mM Tris (pH 5.5)] or NMR buffer [20 mM Bis-Tris propane, 100 mM NaCl, 1 mM TCEP, 1mM EDTA, 7% D2O, and 2% NaN3 (pH 6.5)]. The concentration of VHR was determined using the extinction coefficient at 280 nm of 11,500 M−1 cm−1.
Kinetic Assays
Steady-state kinetics were measured at 25° C for WT VHR, N74A, and K50A with para-NitroPhenylPhosphate (pNPP) at several concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1, 2, 4, 8, 10, 12, 20 mM) following a previously published protocol.46 Enzymes and pNPP were prepared in kinetics buffer and the reaction quenched with 1 M NaOH at 10, 20, 30, and 40 seconds after addition of enzyme to PNPP. The rate was determined by the slope of the absorbance at 405 nm using the extinction coefficient of PNP (18,000 M−1 cm−1) as a function of time. The [enzyme] adjusted rate as a function of the pNPP concentration was measured in triplicate and fit with the Michaelis-Menten equation in GraphPad Prism version 7 for MacOS X. Michaelis-Menten plots of WT, K50A, and N74A are provided in the supplemental information (Fig. S1A).
Transient kinetics assays were performed according to published methods on WT VHR, K50A, and N74A, at 25°C using a 100 mM sample of pNPP in kinetics buffer on a SX20 stopped-flow instrument (Applied Photophysics).46 The absorbance at 400 nm was recorded over time (≤ 1s) and the concentration of para-nitrophenolate (PNP) determined by the experimentally measured extinction coefficient (847 M−1 cm−1). Rapid kinetic data for five separate mixings were averaged. The change in concentration of PNP versus time was fit with the following equation:
where a is the burst size and proportional to the concentration of enzyme, k is the sum of the rate of cleavage (kcleavage) and the rate of hydrolysis (khydrolysis), and b is equal to kcleavage*khydrolysis/(kcleavage + khydrolysis). Measurements of the concentration of PNP as a function of time as a result of cleavage of pNPP by WT, K50A, and N74A are shown in the Supplemental Information (Fig. S1B).
Nuclear Magnetic Resonance Spectroscopy
All NMR experiments were performed on a 600 MHz, 700 MHz, or 800 MHz Varian spectrometers at 25°C. The standard triple-resonance experiments including the HNCA, HN(CA)CB, HN(CA)CO, HN(CO)CA, HN(COCA)CB, and HNCO were performed for resonance assignments. The assignment process was facilitated by the Computer-Aided Resonance Assignment (CARA, cara.nmr.ch) and SPARKY software packages.47, 48 To further aid the assignment process, HSQC and 2D HN(CO) spectra were collected for each sample of 13 different selectively reverse-labeled amino acid VHR samples. The spectra from these samples were then compared to the same spectra from a uniformly 2H, 13C, 15N labeled sample. The resonance assignments have been deposited in the BioMagResBank (BMRB) under accession number 27950.
The titration of phosphate to VHR was performed by preparing a 1.8 M solution of Na2HPO4/NaH2PO4 (pH 6.5) in NMR buffer and adding small aliquots (< 5 μL) to the enzyme sample until saturation ([PO4]/[VHR] = 84 for WT VHR). An HSQC spectrum was collected after each addition of the solution to the sample in order to track the chemical shift differences and to determine the saturation point. VHR saturation with phosphate was determined when additional titrations yielded no further change in the chemical shift. The tungstate titration was performed in an analogous manner with a 50 mM solution of Na2WO4 and larger aliquots (≤ 10 μL). Saturation for WT VHR was achieved when [WO4]/[VHR] = 5. The composite chemical shift differences (Δδ) were calculated as previously published:49
where δH and δN are the 1H and 15N chemical shifts and δapo and δbound are the chemical shifts of the apo and bound forms, respectively.
15N-CPMG relaxation dispersion experiments were performed on a 700 μM 2H-,15N-labeled VHR sample using 32 scans per 128 t1 increments. The 1H carrier frequency was set to the water resonance with the 15N carrier set to 120 ppm. Spectral widths in the 1H and 15N dimensions were 15 and 44 ppm, respectively. The 15N-CPMG relaxation dispersion experiments were performed at 600, 700, and 800 MHz with the spin-echo pulse train delay (tcp) set to 0.5, 0.625, 0.7143, 1.0, 1.25, 1.667, 2.0, 2.5, 3.333, 5.0, 10.0, and 20.0 ms in the relaxation-compensated experiment.50 A constant relaxation time of 30ms was used for each.51 The apparent transverse relaxation rate constant (R2,app) was determined using the in-house software. The R2,app values were then fit to the Carver-Richards52 using Mathematica version 12.1 Student Edition for MacOS X.
The TROSY Hahn-Echo experiment53 was performed on either 700 or 965 μM VHR with identical parameters spectral parameters except the number of acquisitions was 72, 72, and 576 for the α, zz, and β experiments, respectively. The Hahn Echo experiments were performed on apo WT, WO4-bound WT, and WO4-bound N74A with a relaxation delay time of 48.6 ms to obtain information on millisecond timescale motions.53 The experiment was collected in three separate spectra corresponding to the experiments measuring the transverse relaxation rate constants for the narrow (R2α) and broad (R2β) 15NH doublet components as well as the relaxation rate constant for the longitudinal two-spin order (R12HzNz ≈ R1H + R1N), which are referred to the α, β, and zz experiments. The β experiment is inherently weak in signal-to-noise therefore the number of acquisition scans was significantly increased relative to the other two experiments. The ratios of the signal intensities and the calculated Rex values were determined for each well-resolved resonance with sufficient signal to noise as previously described.53
Computational Studies
Molecular dynamics simulations were performed on WT VHR (PDB: 1VHR6) and two mutants (N74A and K50A). Tungstate was placed in the P-loop active site, in line with experimental conditions and reported crystallographic structures of other small molecules (e.g. phosphate) bound to VHR. Simulations were conducted in a periodic water box for 150 ns using the CHARMM3654 force field and NAMD package.55 The water box, including 150 mM NaCl was created by adding water for 15 Å in the positive and negative x, y, and z directions around the protein, yielding a rectangular box of size ~72 Å × 65 Å × 82 Å. Long range interactions were handled using the default Particle Mesh Ewald electrostatics algorithm. Default X-PLOR-type van der Waals switching and electrostatic shifting functions were used, with a cutoff of 12 Å and a switching distance of 10 Å.
Before performing the production run, the systems were minimized for 100 steps using a conjugate gradient algorithm and then gradually heated using a Langevin thermostat to 310 K three times. First, only the water and ions were minimized and heated. Next, the protein side chains were released to be minimized and heated with the solvent in an identical manner. To heat, the temperature was increased by 10 K at a time, equilibrated for one hundred, 2-fs time steps (0.2 ps), and increased again until 310 K was reached. The system was then equilibrated for 50 ps. Finally, the entire system was minimized and heated together. When the entire system was heated, the temperature step size was decreased to 1 K and the system was equilibrated at 310 K for 100 ps before initializing the production run. Post simulation analyses were performed using VMD56 (hydrogen bond and RMSD analyses). Hydrogen bonds were defined by a heavy atom distance less than 3 Å and an acceptor-donor-hydrogen angle less than 30°. Mutants were created in silico using standard procedures and energy minimized using the three-phase method described above prior to production MD runs.
Results:
NMR Backbone Assignments
VHR is a 185 amino acid residue (20.5 kDa) enzyme. Despite its modest size, by solution NMR standards, the resonance assignment process proved exceptionally difficult. This difficulty was due to resonance overlap and missing connectivities between a number of amino acids in the triple-resonance experiments due to low peak intensity. Therefore, in addition to acquisition of the standard backbone assignment experiments -TROSY-based HNCA, HNCACB, HNCACO, HNCOCA, HNCOCACB, and HNCO.57, 58 We also performed a number of amino acid-selective reverse-labeling experiments according to published methods45 to overcome these assignment problems. Eleven different samples of 2H, 13C, 15N – labeled VHR were prepared, each with a different naturally abundant 1H, 12C, 14N amino acid (alanine, arginine, asparagine, glutamine, glycine, lysine, methionine, phenylalanine, serine, threonine, or tyrosine). The HSQC and 2D HN(CO) spectra of these samples were compared to the spectra of a uniformly labeled 2H, 13C, 15N VHR sample. Two combinatorial reverse-labeled samples were also prepared in which one sample was reverse-labeled with natural abundant isotopic versions of alanine, histidine, isoleucine, leucine, valine, lysine, methionine, arginine, serine, glycine, and cysteine and a separate sample with added isoleucine, leucine, and valine. The former sample further reduced the spectral overlap and isolated the resonances in the acid loop. The latter sample was prepared because isoleucine, leucine, and valine metabolically scramble in E. coli and could not be prepared separately.47 The reverse-labeling protocol resulted in 14 additional assignments as well as confirmation of assignments obtained by standard triple-resonance experiments. This combination of methods results in a total of 158 out of 178 non-proline assignments.
At this stage, many of the unassigned resonances were located in the loop regions (VI, P, and Q loops), which suggested that they were exchange broadened due to molecular motion. To overcome this problem, we produced two isotopically (2H,13C,15N) labeled VHR samples in which we titrated either inorganic phosphate or sodium tungstate, reaction product mimics, into the active site. Upon binding to these ligands, several additional resonances became resolved in the 2D 1H-15N-HSQC spectrum. The signal-to-noise of these additional peaks was sufficient such that they could be assigned using HNCA and HNCACB experiments. The residues assigned in this fashion were G65, V71, N74, I83, A90, D92, T93, N97, Y128, and R130. Using these complementary approaches, we assigned a total of 93% (166 out of 178 non-proline residues) of the VHR backbone. These assignments have been deposited in the BMRB with entry number 27950.59
Ligand titrations suggest allosteric sites in Wild-type VHR
In the process of the PO43– and WO42– titrations, we noticed chemical shift changes distant from the VHR active site. Even though the ligand titrations were intended to aid in the assignment process, the global response of VHR to binding of these anions was informative of the allosteric network throughout the enzyme.60 The composite chemical shift differences (Δδ) relative to the apo enzyme as a function of the residue number show which residues are significantly perturbed to values greater than 2σ above the 10% trimmed mean (Fig. 2 and Fig. S2). As expected, the largest perturbations were detected in the acid loop (89–99), P-loop (123–131), and Q-Loop (158–163), where direct interactions with these oxyanions occur. In these regions, A90, T93, E95, F96, L98, C124, R158, E159, and G161 have elevated Δδ values in the phosphate titration and K89, D92, E95, F96, L98, H123, C124, R158, E159, and G161 have elevated values in the tungstate titration. However, significantly elevated Δδ values were also observed in the previously identified allosteric sites27 in the variable insert (A62, A63, and V71 for both ligands and N74 only with tungstate). Residues in the α4-α7 region (A100, D164, G165, F166, L167, and A168 with phosphate and G165, F166, L167 with tungstate) also had elevated Δδ values. Other residues with significantly large Δδ values include residues in the recognition region (G19 and Q28), β1 (C30 and N31), β2 (G40), α2 (S43 and V44), β5 (V122), α5 (T133, V135, and Y138), and Q46 and I88 with phosphate and D18, G19, N31, (β2) N41, V44, Q46, I88, and V135 with tungstate. There are also subtle differences in the CSP patterns between phosphate and tungstate bound WT complexes that are suggestive of ligand-dependent allosteric differences in VHR. Overall these titration data indicate that the anion binding to the P-loop of VHR propagates to the distal regions of the enzyme including the VI and α7 and is consistent with the previous studies that identified these regions as allosteric in VHR.
Figure 2.
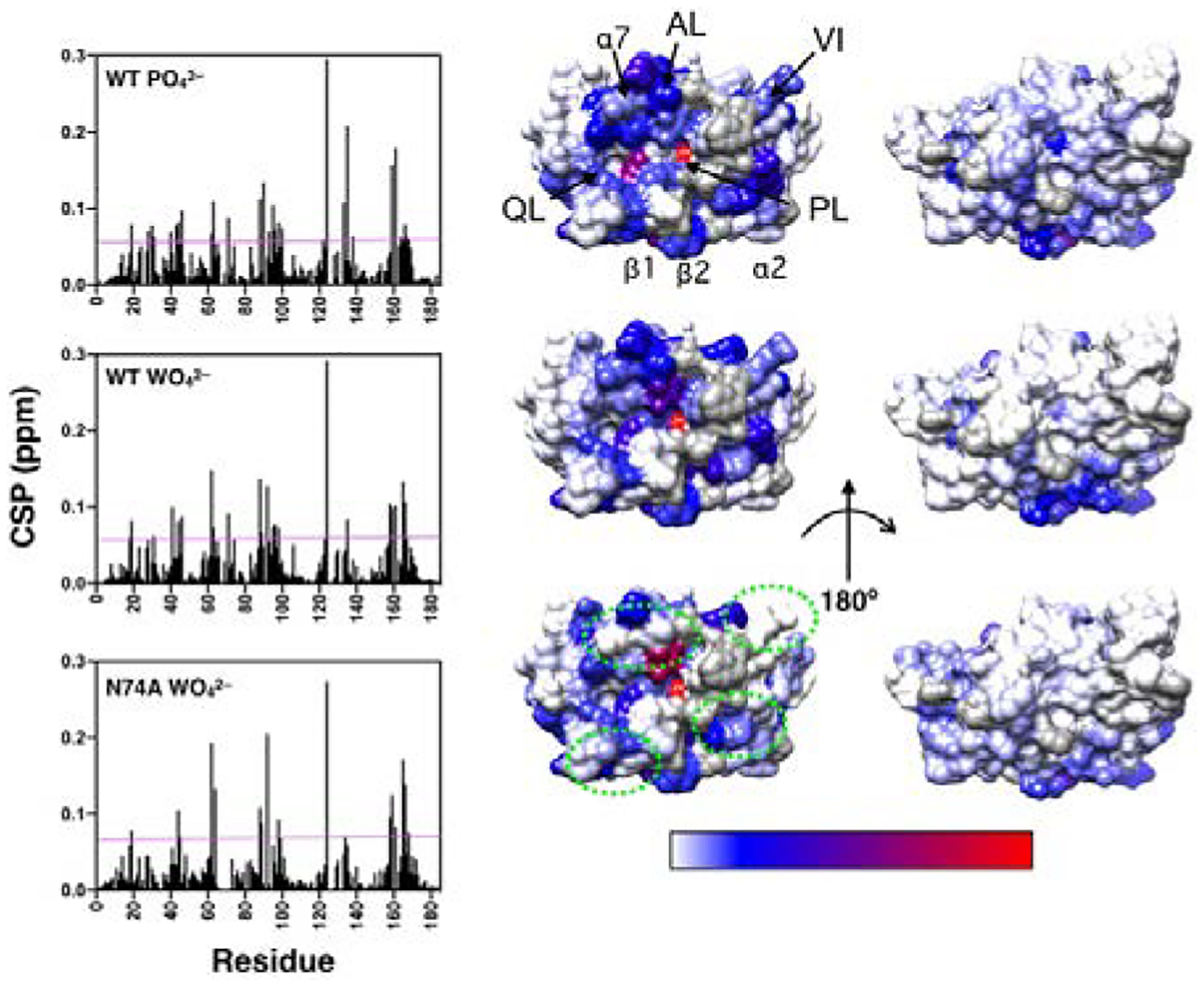
Ligand-induced chemical shift perturbations (CSP). The left panel shows CSP values versus amino acid residue for WT, and N74A with the respective complexes indicated in each graph. The magenta line represents 2σ above the 10% trimmed mean value for all three data sets. The right panels show CSP values mapped onto a surface rendering of VHR shown in the same orientation as in Figure 1B. CSP values above the magenta line are mapped according to magnitude (shown in the bottom bar) ranging from blue to red. Residues with CSP below this threshold are indicated in white. Residues that are unassigned, are prolines, or have poor signal-to-noise that precludes measurement are shown in gray. Regions that show significant differences from WT in the tungstate bound complex are enclosed in green dashes. The acid loop (AL), P-loop (PL), Q-loop (QL), Variable Insert (VI), and select 2° structure elements are denoted in the top structure.
Millisecond (ms) motions link the VI and acid loop in VHR
Some PTP family members possess a mobile acid loop61–63 whereas in other family members, such as VHZ the acid loop appears to be immobile.64 To investigate whether the acid loop or other regions of apo VHR are flexible, we performed 15N-Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments in order to assess the dynamics in the ms timescale. The apparent transverse relaxation rates (R2) as a function of the pulsing frequency (1/tcp) in the CPMG spin-echo pulse train were measured and the two-site Carver-Richards equation52 was fit to the relaxation data obtained at three different static magnetic fields (600, 700, and 800 MHz). A global fit65 of the relaxation data for the acid loop including residues K89, D92, E95, F96, L98, and S99 yielded rates of exchange (kex) values of 1400 ± 200 s–1, a population of the major conformation (pA) of 98.9 ± 0.1%, and residue-specific 15N chemical shift differences between the major (conformation A) and minor conformation (conformation B) (ΔωN = |ωN(A) – ωN(B)|) of 1040 ± 96, 840 ± 175, 1800 ± 400, 1170 ± 147, 1790 ± 321, and 700 ± 97 s–1, respectively. The Akaike information criterion (AIC) for this fit is 1014 and an F-test comparison yielded a p-value favoring the global model = 1.08 × 10−9 in which all residues share kex and population values.
These NMR relaxation experiments also revealed that the allosteric VI region exhibited considerable ms motions. A global fit for residues A63, R66, and N72 in the VI yielded similar fit values as obtained for the acid loop with kex = 2100 ± 460 s–1, pA = 98.5 ± 0.4%, |ΔωN| values of 920 ± 150, 1240 ± 230, and 1580 ± 360 s–1, and a global AIC of 457, and a p-value = 1.38 × 10−5. Considering the similarities in the kinetics and populations for both the acid loop and variable insert, we attempted to fit both regions to a single global conformational exchange process (Figure 3). This global fit yielded a kex of 1530 ± 200 s–1 and a pA of 98.8 ± 0.1% and statistical parameters of p-value = 8.2 × 10–14 and AIC = 1478 for the global model versus fitting the two regions separately. The global |ΔωN| values for A63, R66, N72, K89, D92, E95, F96, L98, and S99 for this fit are 930 ± 100, 1340 ± 170, 1810 ± 290, 1030 ± 80, 840 ± 160, 1740 ± 320, 1150 ± 120, 1610 ± 240, and 700 ± 90 s–1, respectively and are also shown in the bottom of each panel of Figure 3. The global fit for all residues is shown in Figure 3. Improvement of the error of the fit and the model-test parameters suggests that these two regions are likely involved in a concerted motion on the millisecond timescale.
Figure 3:
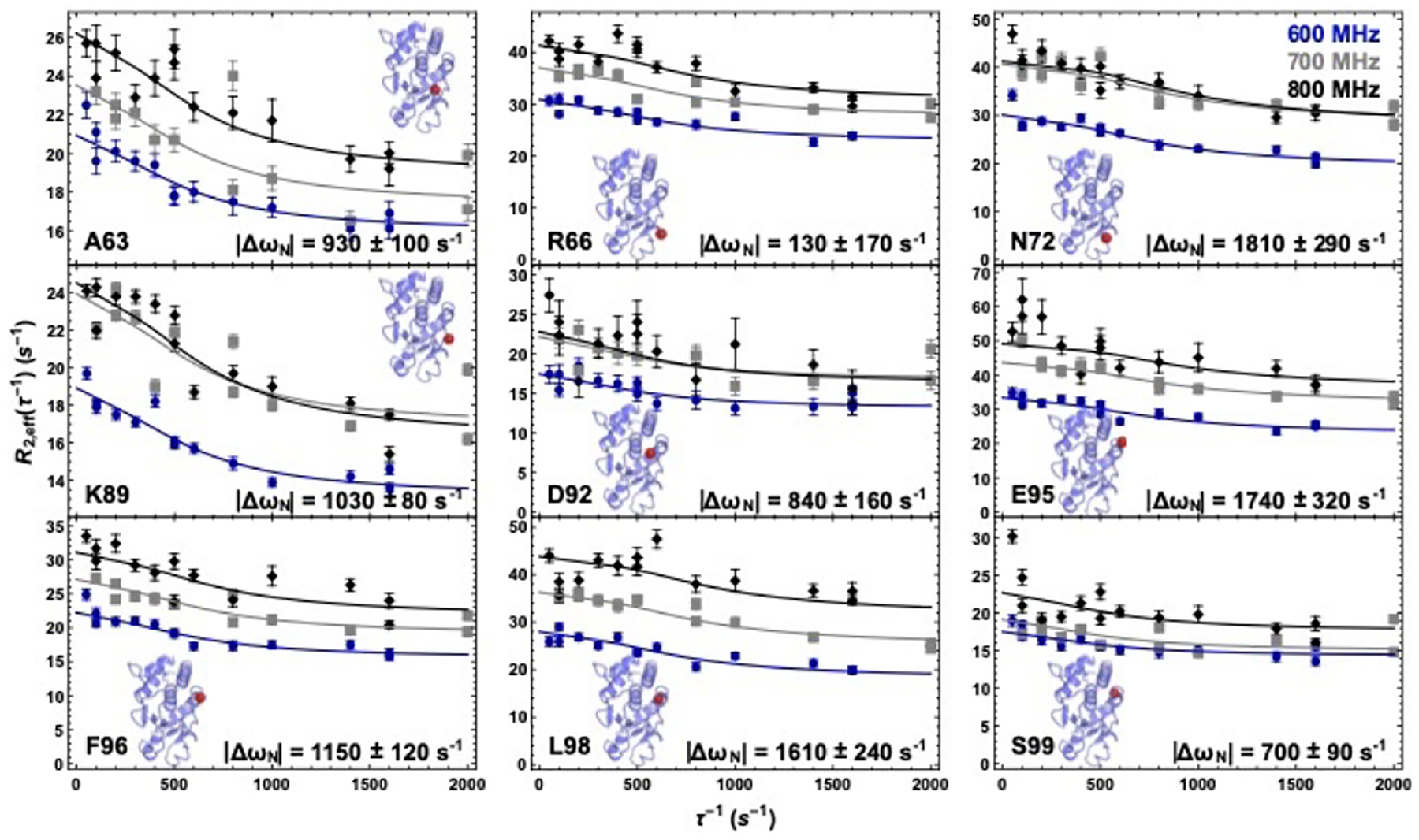
Millisecond motions in VHR. 15N-CPMG relaxation dispersion curves for residues A63, R66, N72, K89, D92, E95, F96, L98, and S99. The blue circles, gray squares, and black diamonds correspond to data collected at 600 MHz, 700 MHz, and 800 MHz. Inset figures show the crystal structure of VHR with the location of the corresponding residue represented as a red sphere. The curves represent a global fit to all nine data sets at three static magnetic field strengths. Δω values are given at the bottom of each graph.
Mutation in the Variable Insert allosteric site
Based on the chemical shift perturbation data with tungstate and phosphate and the NMR relaxation experiments that together suggest an allosteric interaction and concerted motions between the VI and active site acid loop, we examined the effect of mutation of N74 to alanine. N74A is located in the VI loop and was previously identified as allosterically linked to the active site acid loop and was therefore a logical first choice to address this issue. At its closest, N74 is 16 Å from C124, the active site nucleophile and the sidechain of N74 is solvent exposed. This alanine point mutation does not significantly perturb the overall fold of VHR as evidenced by the similarities in the HSQC spectra between WT and N74A that resulted in the small magnitude of the Δδ relative to WT (< 0.2 ppm) (Fig. S2). However, it is worth noting that several residues, mostly in the active site and VI, in the N74A mutant were broadened beyond detection in the apo enzyme of N74A, suggesting an enhancement of ms motions caused by mutation of this residue. Specifically, R66, V71, N72, I88, A90, D92, A100, R130, and G161 were broadened in the apo spectrum of N74A. Of these residues, those not in the VI such as I88, A90, D92, A100, R130, and G161, all are >10 Å from N74. This indicates that the effect of mutation is propagated beyond the local region of N74, reinforcing the allosteric impact of this residue and the close linkage between the VI and acid loop and active site (Figure 2).
As with the WT enzyme, a tungstate titration was performed with N74A to assess the response of this variant to binding of the product analog. The tungstate titration with N74A reveals 19 residues with elevated Δδ values. Seven of these residues are located in the active site (K89, E95, L98, S99, C124, R158, and E159) and five in the allosteric sites (A62, E64, G165, F166, and A168). Other residues with elevated Δδ values include D18, G19, N41, V44, A45, and L134. In addition, V71, I88, A90, D92, Q94, A100, R130, and G161 could not be analyzed due to exchange broadening as mentioned above. Overall, the tungstate perturbation of chemical shifts in N74A is similar to WT with some notable exceptions (green circles in Figure 2) in the AL, VI, and the Q-loop.
Millisecond motions in WT and N74A
A comparison of the dynamic behavior among WT VHR and N74A was investigated by solution NMR measurement of the chemical exchange rate constant (Rex) using the TROSY-based Hahn Echo experiment. The experiment was conducted on both the apo and tungstate-bound forms of the WT enzyme, and for the tungstate bound complex of N74A, which was necessary because of excessive exchange broadening in the apo form of this mutant (Figure 4). The 10% trimmed mean of the collective Rex values for all the enzymes is 0.33 ± 2.12 s–1. Rex values greater than two standard deviations (2σ = 4.57 s–1) above this mean are considered indicative of enhanced flexibility relative to the residues with measured Rex values below this threshold (Figure 4). In apo WT, the following residues have Rex > 4.57 s–1 R36 (6.7 ± 2.7 s–1), A63 (9.8 ± 1.0), R66 (12.9 ± 2.6), N72 (24.6 ± 3.6), Y78 (4.7 ± 0.6), K89 (9.2 ± 0.6), D92 (10.7 ± 1.6), E95 (24.5 ± 2.2), F96 (10.0 ± 2.5), L98 (15.1 ± 1.5), R119 (4.9 ± 1.4), C124 (4.6 ± 1.4), G161 (5.7 ± 1.1), and D164 (5.5 ± 0.6). These residues comprise the active site acid loop, the VI loop, and a second previously identified27 allosteric site α7, containing residues G161 and D164. Eight of these 14 residues were also identified as flexible in the relaxation-compensated CPMG experiment of the apo WT enzyme described above. The Rex values for these eight residues (A63, R66, N72, K89, D92, E95, F96, and L98) are within error of the Rex values determined from the CPMG relaxation dispersion experiment (with the exception of D92) and further verify the consistency between the two measurements. A table of the measured Rex values for each enzyme in the apo and tungstate-bound forms is provided in the Supplemental Information section (Table S1).
Figure 4.
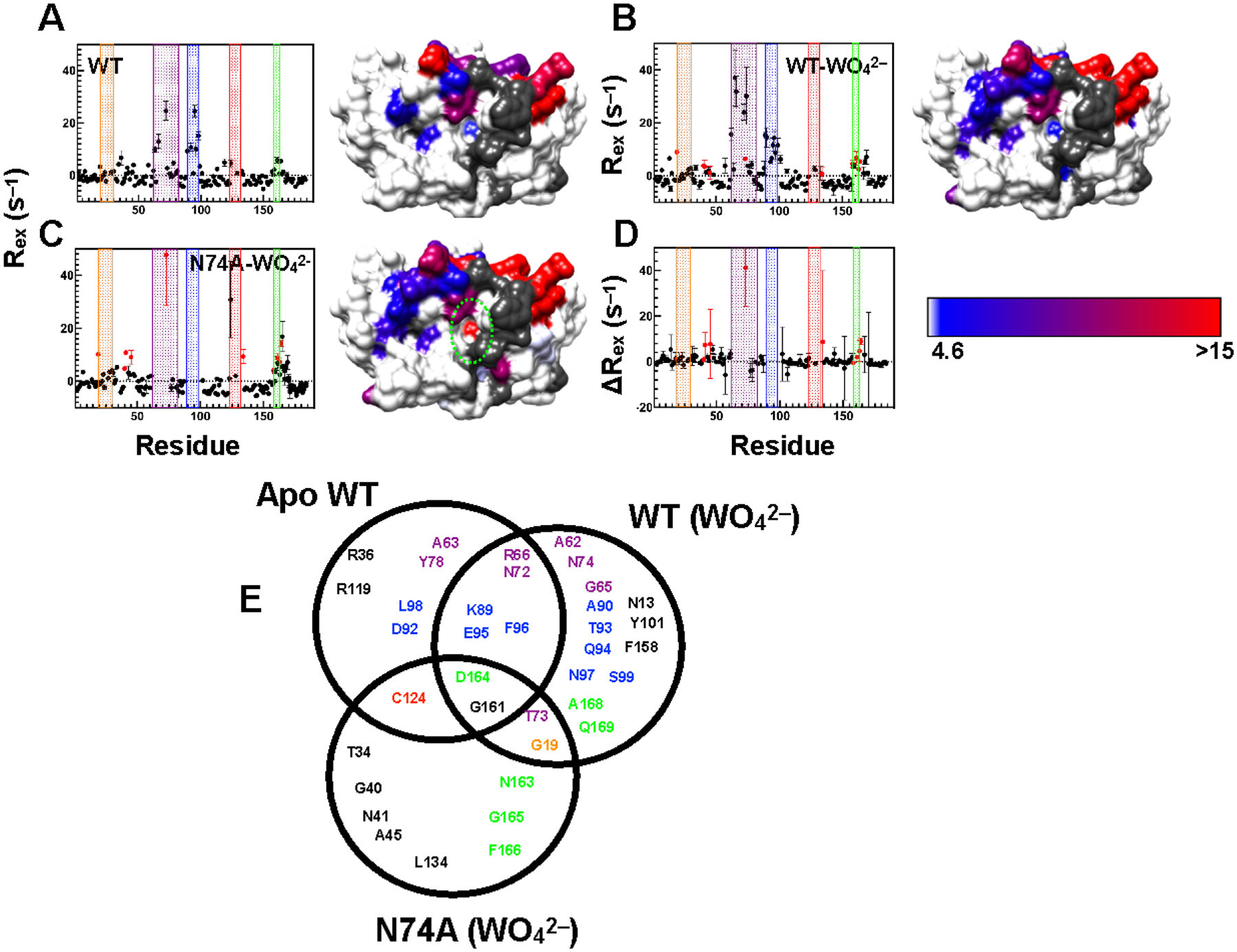
Millisecond motions in VHR. Residues with elevated Rex as determined by Hahn-Echo measurements are graphed versus amino acid sequence and mapped onto the surface rendering of WT VHR in the apo (A) and tungstate-bound complexes (B). In (C), residues with elevated Rex values in the tungstate bound form of N74A are shown. In (D) the difference in Rex between tungstate bound WT and tungstate bound N74A (N74A – WT) is shown versus residue number. Residues with Rex values below the 10% trimmed mean + 2σ (i.e. 4.6 s–1) are shown in white. Residues with Rex values greater than this threshold are colored from blue to red according to the color key located at the bottom left. Regions in gray represent unassigned, proline, or overlapped residues, as well as residues with insufficient signal-to-noise. In each graph, as in Figure 1, the recognition region, VI, acid loop, P-loop, and Q-loop are shaded orange, purple, blue, red, and green, respectively. Regions with different ms motions from the respective WT complex is enclosed within green dashes. (E) Venn diagram showing residues with elevated Rex values from the NMR Hahn-Echo experiment for apo WT and tungstate bound WT and N74A. Residues are color-coded according to the color scheme in Figures 1 and 4. The VHR orientation of the VHR structure in panels (A-C) is identical to that in Figures 1 and 2.
The profile of flexible residues of the tungstate-bound WT VHR is very similar to the apo form, yet with slightly elevated Rex values for some residues. Nearly half of residues with high Rex values in the bound form are in the active site, the recognition loop, and α7. Residues with elevated Rex values in the tungstate complexed form include N13, G19, A62, G65, R66, N72, T73, N74, K89, A90, T93, Q94, E95, F96, N97, S99, Y101, R158, G161, D164, A168, and Q169. The underlined residues are only spectroscopically observable in the presence of saturating tungstate. Overall, the increase in the number of flexible residues in tungstate bound WT versus the apo form is likely the result of additional residues becoming spectroscopically observable in the presence of tungstate. These results highlight the allostery present between the VI, the α4-α7 region and, the active site and indicate how the effects of ligand binding are propagated to distal regions of the enzyme.
The tungstate-bound N74A complex has similar Rex values as WT, such as residues in and near the VI, acid loop, and α7 (T73, G161, N163, D164, G165, and F166) with a few exceptions. Residues G19 (recognition region), T34 (β1), G40 (β2), N41 (α2), A45 (α2), C124 (P-loop), L134 (α5) show significant flexibility. In WT, Rex values for C124, G165, and F166 could not be quantitated. It is also worth noting that most of the residues in the VI and acid loop in N74A remain exchange broadened and thus the Rex values could not be quantitated. Therefore, while these residues clearly retain or exceed WT-like ms motions, a direct comparison is not possible.
For the residues in both WT and N74A enzymes in which Rex values could be determined, the Rex values for those in N74A are within error of the WT value or slightly greater (Figure 4D). Notably, N41, A45, T73, N163, and D164 all have higher Rex values than WT. These enhancements in Rex are an indicator of increased conformational motion in N74A. Under the experimental conditions, considering the measured Kd for tungstate, and estimations of its dissociation rate constant, these elevated Rex values are not due to exchange of WO42– on and off of the enzyme, but rather real conformational exchange motions. A summary of residues with millisecond motions in each complex is shown in a Venn diagram in Figure 4E.
VHR catalytic activity
To determine the effects of these allosteric mutations on VHR catalytic activity, we measured steady-state and pre-steady state kinetics for WT, N74A, and K50A using the pseudo-substrate pNPP. K50A was used as a control because it is located near the VI yet its chemical shift is unperturbed in all of the acid loop mutants examined to date27 suggesting the absence of allosteric linkage to the active site. Moreover, the chemical shift of K50 is unperturbed in the presence of either phosphate or tungstate relative to the apo enzyme. Thus, we anticipate that the catalytic activity of K50A will be WT-like and that any differences from WT will be the result of experimental variability and provide an estimate of our measurement precision. Results of the steady-state kinetics assay reveal that K50A has the same kinetic parameters (kcat = 4.3 ± 0.3 s–1, Km = 2.1 ± 0.4 mM) as WT VHR (kcat = 4.6 ± 0.1 s–1, Km = 2.0 ± 0.1 mM) as seen in Table 1 and Figure S1A. Based on these data, we estimate the experimental variability in steady-state kinetics measurement is between 5–7% for Km and kcat. The N74A enzyme has both a slight (21%) decrease in kcat (3.4 ± 0.3 s–1) and an over 2-fold increase in Km (4.7 ± 1.1 mM) compared to WT and is outside the experimental ‘noise’ region defined above using K50A as a negative control. Thus, this allosteric mutant affects both the catalytic and substrate binding steps. To ascertain the effects of mutation on the individual reaction steps, we measured the cleavage and hydrolysis rates using stopped-flow kinetics experiments.
Table 1:
Results of the steady state (kcat and Km) and rapid kinetics of WT VHR, K50A, and N74A.
| Enzyme | kcat(s−1) | Km(mM) | kcleav (s–1) | khydr (s–1) |
|---|---|---|---|---|
| WT | 4.6±0.1 | 2.0± 0.1 | 19 ± 3 | 3.3 ± 0.1 |
| K50A | 4.3±0.3 | 2.1± 0.4 | 22 ± 2 | 3.1 ± 0.2 |
| N74A | 3.4±0.3 | 4.7±1.1 | 7.9 ± 0.8 | 1.17 ± 0.03 |
The reported rate constant for cleavage of pNPP by VHR (kcleavage = 34 s–1) is about 4 times faster than the rate of the hydrolysis step (7.6 s–1) at 30° C.46 Our experiments were performed on WT VHR, K50A, and N74A at 25°C on the same day in an effort to minimize experimental variation. The kinetic traces and fits with Equation 1 are shown in Figure S1B and the results are reported in Table 1. At 25°C for WT VHR, the measured cleavage rate constant, kcleavage = 19 ± 3 s–1 and khydrolysis = 3.3 ± 0.1 s–1. These values are slightly lower than the reported literature values most likely due to the lower temperature at which our experiments were conducted.46 The control enzyme, K50A, has kcleavage = 22 ± 2 s–1 and khydrolysis = 3.2 ± 0.2 s–1, values very similar to those for WT. The N74A mutant has a significant impact on both the kcleavage and khydrolysis compared to WT, where these values are 7.9 s–1 and 1.2 s–1 respectively, a roughly two-fold decrease for both kinetic parameters.
Computational analysis of VHR
Molecular dynamics simulations for the tungstate-bound forms of WT, N74A, and K50A VHR enzymes provide valuable mechanistic insights. First, for WT and K50A the simulations reveal that the acid loop moves from its starting closed position to an open conformation, further hinting at WT VHR possessing a flexible acid loop. A global comparison between WT, N74A, and K50A reveal very similar structures during the 150-ns simulations, with the root mean squared deviation (RMSD) of the Cα carbons between these enzymes being 0.642 Å (WT vs. N74A) and 0.796 Å (WT vs. K50A), indicating that neither mutation causes a large overall structural change. However, the N74A mutation produces significant changes in hydrogen bonding that affect the opening of the acid loop and the positioning of the tungstate ligand. Indeed, a close examination of select regions of VHR reveals critical changes caused by the mutation at N74. Figure 5 shows that the sidechain of R125 in the P-loop is displaced and becomes more rigid in the N74A mutant. Its displacement shifts the neighboring E126 away from the active site, where it would otherwise repel the sidechain of D92 and open the acid loop. The resulting changes in the acid loop motion correlate with a reduction of catalytic activity, as could result from suppression of substrate entry, or product exit, from the active site or a disruption in the timing of events necessary for optimal VHR catalysis.
Figure 5.
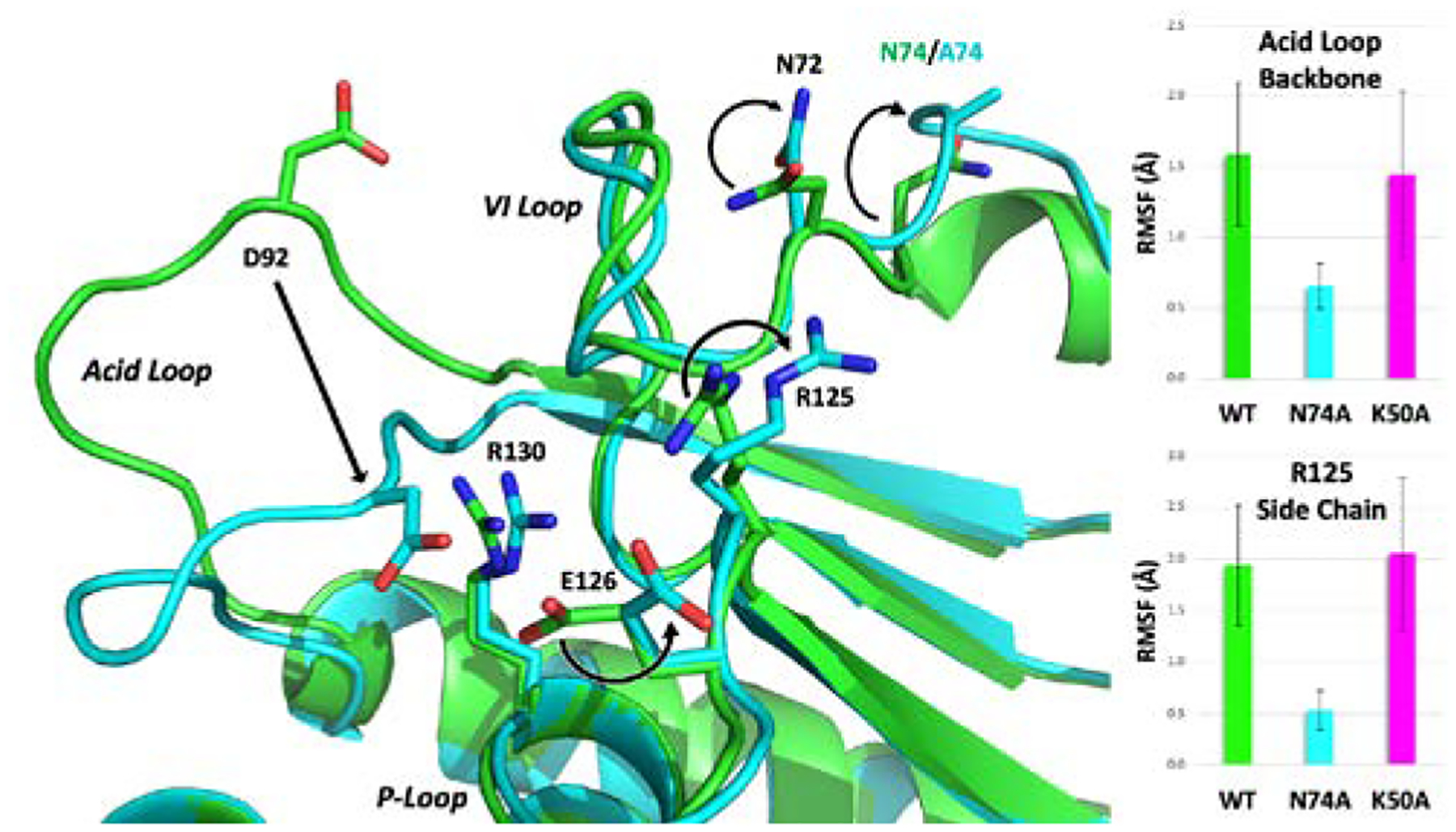
Structural differences revealed by the MD simulations of WT (green), K50A (magenta) and N74A (cyan) bound to tungstate (not shown). The right panel shows the root mean square fluctuations (RMSF) for the acid loop and sidechain of R125 for the latter 100 ns of the simulation, highlighting the flexibility differences between WT/K50A and N74A. The sidechain of R125 becomes more rigid in the N74A mutant (smaller RMSF). Its displacement shifts E126 away from the active site, consequently suppressing the electrostatic repulsion with D92, essential for opening of the acid loop (smaller RMSF). The resulting changes in the acid loop motion correlate with a reduction of catalytic activity, as could result from suppression of substrate entry or product exit from the active site.
Overall, the acid loop RMSF shown in Figure 5 reveals a large difference between WT and N74A, which is due to the acid loop remaining closed in the mutant. This is despite a distance of almost 20 Å between N74 and the active site. However, Figure 5 shows the path of perturbations that start at the mutation and lead to the locking of the acid loop in the closed position. The N74A mutation causes an unraveling of the short helix in the VI region comprising residues 74–78, which distorts the remainder of the VI loop. Figure 6 shows the hydrogen bonding changes that begin with N72 being pulled out of its hydrogen bonds with other residues in the VI loop to replace A74 as a solvent-exposed residue. This changes the orientation of the backbone of neighboring H70 just enough to form a stronger hydrogen bond with R125. With the R125 sidechain pulled to the side, E126 is also displaced. In WT and K50A, E126 and D92 jockey for space at the active site. Repulsion between them allows for the breakage of the hydrogen bond between R130 and the A90 backbone, which holds the acid loop closed. E126 also serves as a cap on the active site, keeping the opening small even when the loop is open (Figure 7). In N74A, E126 is less sterically hindered by R125 and is more willing to yield the active site to D92.
Figure 6.
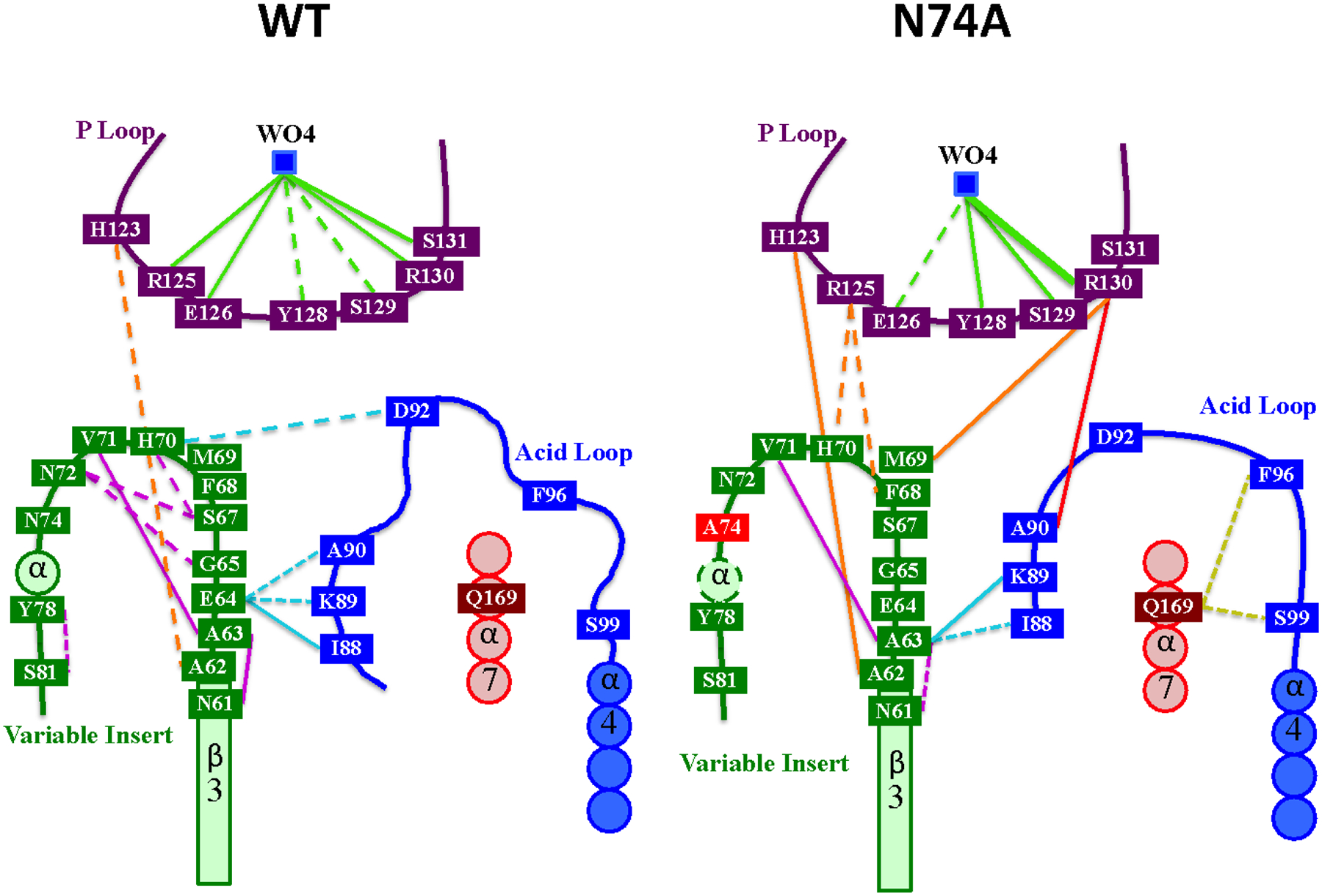
Summary of mutation effects of N74A. Lines indicated H-bonds observed during the molecular dynamics trajectory for 33–66% (dotted), > 66% (solid), and 100% (thick solid) of the entire trajectory.
Figure 7.
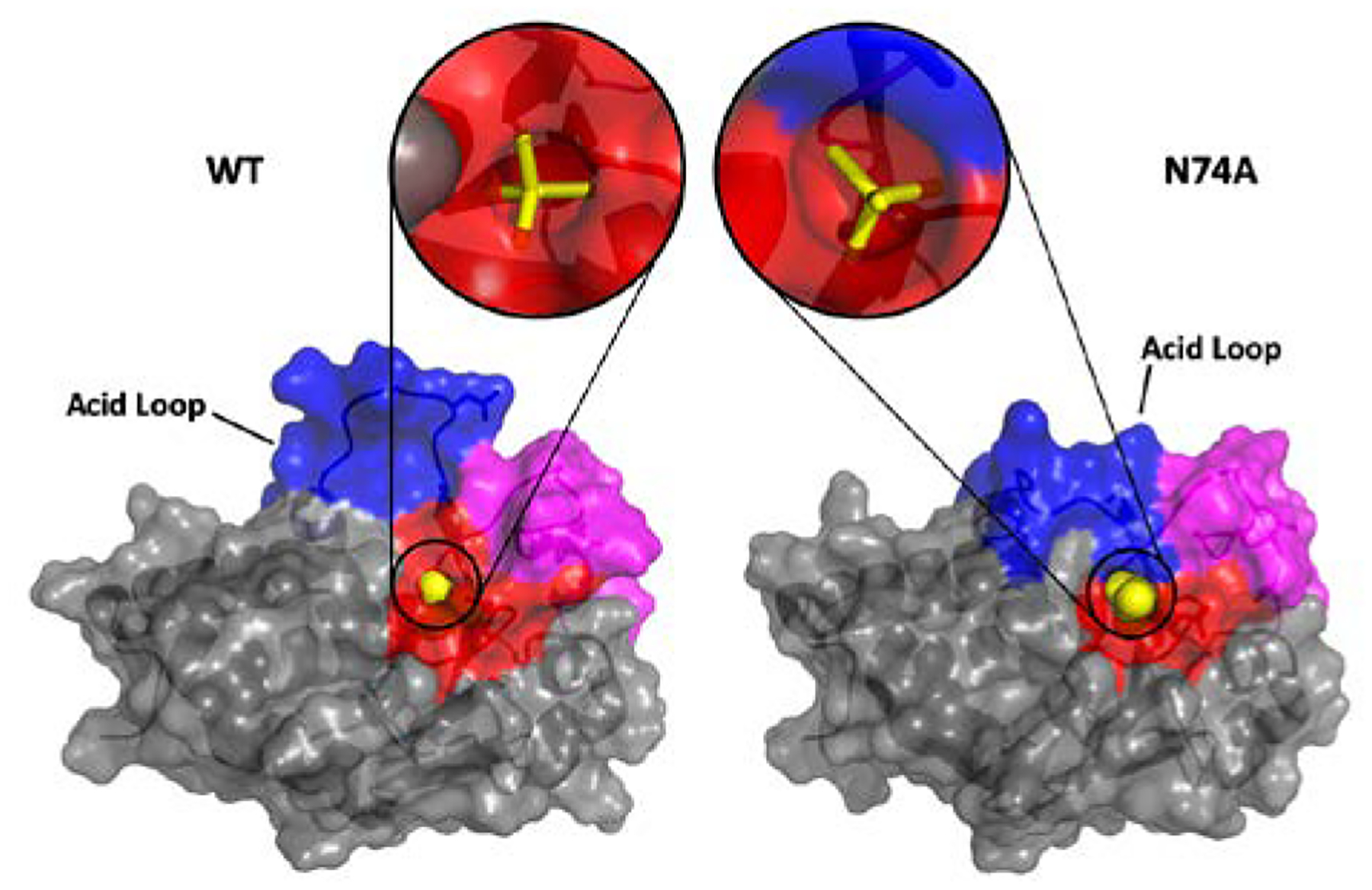
Surface rendering of WT (left) and the N74A mutant (right) for a representative configuration of the MD simulation. The acid loop is shown in blue, the VI loop in magenta, and the P-loop in red. The close-up view depicts WO42- (yellow) bound at the active site. The surfaces shown in the lower panel highlight that for the N74A mutant the acid loop is more open, although the WO42- molecule is more buried, due to changes in hydrogen bonds.
This structural change prevents N74A’s acid loop from opening and creates a distance of more than 5 Å between D92 in the mutant and D92 in the open configuration of WT. The acid loop in both WT and K50A is able to open fairly easily while the loop remains firmly closed in N74A (Figure 5). The flexibility of the acid loop in WT and K50A raises the loop’s RMSF over the final 100 ns of the simulation from 0.656 Å (N74A) to ~1.5 Å (WT and K50A) and the global backbone RMSF from 0.789 Å (N74A) to almost 1 Å in both WT and K50A and. Moreover, these differences in WT and K50A result in the tungstate molecule moving farther into the active site by ~1.4 Å, as compared to N74A. Predictably, this movement changes the hydrogen bonding partners of tungstate. Shown in Figure 6, the tungstate of WT shares strong hydrogen bonds with R125, E126, R130, and S131. The more raised location in N74A forms much stronger hydrogen bonds with R130 and favors Y128 and S129 over R125 and E126. The alteration of the position of the catalytic acid and the change in location of the product analog relative to its WT structure is likely responsible for the diminished catalytic activity of the N74A enzyme.
Given, the alterations in loop flexibility observed both experimentally and computationally, we compared the chemical shift differences, Δω determined by NMR relaxation dispersion studies of apo WT VHR, with Δδ chemical shift changes as a result of tungstate titration for WT VHR. Previously, correspondence between these values has been taken as evidence that the apo enzyme naturally samples the bound conformation in the absence of ligand.66–68 The comparison for VHR is shown in Figure 8. The data in Figure 8 show that for all residues in which measurement could be made that Δω values for loop residues are significantly larger than Δδ values for the same residues.
Figure 8.
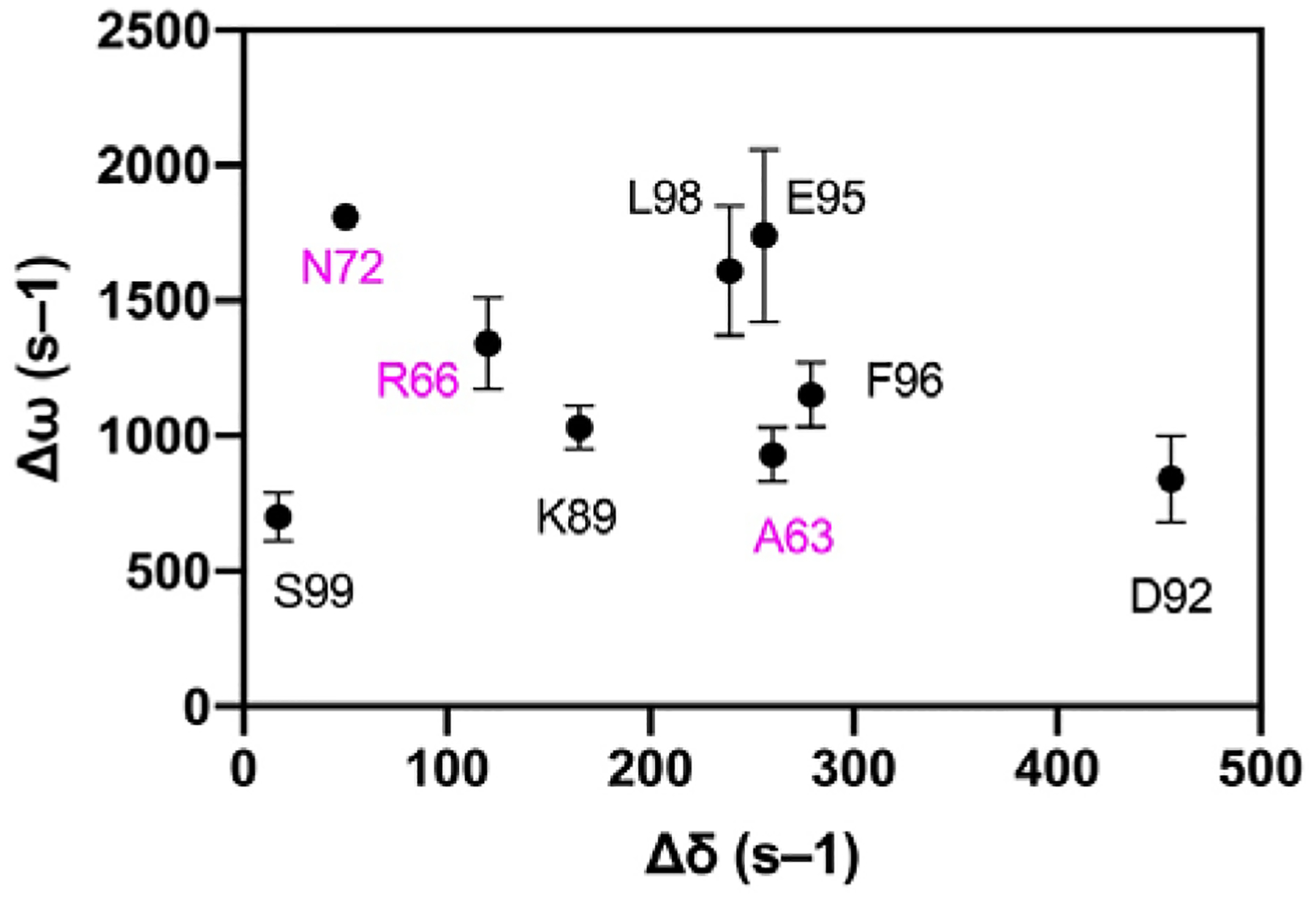
Chemical shift differences. Dynamic chemical shift changes (Δω) in apo VHR from CPMG relaxation dispersion experiments plotted against chemical shift changes (Δδ) from a tungstate titration into apo VHR. Residues are labeled and colored with VI residues in magenta and acid loop residues in black. The lack of correlation between the two values indicates that loop motions in the apo enzyme are distinct from those conformational changes that occur due to ligand binding.
Discussion
The allosteric linkage between the variable insert region and the acid loop in VHR was originally verified when alanine scanning mutations of the acid loop resulted in chemical shift perturbations for residues located in the VI loop.27 The work here expands on those studies by showing that the enzymatic reaction product (inorganic phosphate) and product analog (tungstate) alter the chemical shifts not only at the active site but also for residues located in the VI including N74. Furthermore, NMR CPMG relaxation dispersion experiments indicate that the acid loop and VI are flexible on the ms timescale. And, the identical exchange rate constant and equilibrium populations that describe this motion suggest that the VI and acid loop motions are coupled. The implication of these data is that alterations to the VI should affect enzyme function, which we have observed in this work via steady- and pre-steady state kinetics for the N74A mutation showing that this mutation alters both chemistry steps and steady-state substrate binding. Interestingly, it has been previously shown using unnatural amino acid incorporation at the VI residue F68 that the VI can be a site for VHR dimerization and that this interaction inhibits enzyme activity.44 Wu et al also showed that VI residues F68 and M69 as well as residues in the substrate recognition loop, contribute to the binding of the inhibitor, SA3.43 Cumulatively, these data indicate that alterations to the VI sequence can disrupt VHR function and that residues in the VI could provide a binding platform for small molecule inhibitors. It further seems reasonable to speculate that in vivo, Nature may utilize the VI as a structural feature that is recognized by other protein regulators or substrates. For example, the pseudokinase, VRK3 binds to VHR and inhibits its catalytic activity.69 Although the binding interface between VRK3 and VHR is not known, these studies here suggest that the VI represents a potential starting point to identify such an interaction.
Molecular motions in WT VHR
To investigate the mechanism of the interaction between the VI and acid loop that results in altered enzyme function, we performed 150 ns molecular dynamics simulations for WT, K50A, and N74A all with tungstate bound to the active site P-loop. WT and K50A simulations show very similar behavior, again confirming the innocuous nature of mutation of residue 50. Therefore, the discussion that follows will focus on the WT enzyme and a comparison to N74A. As shown in Figure 6, the linkage between the VI and the acid loop is mediated by hydrogen bonds that connect E64 with the N-terminus of the acid loop (A90, K89, and I88). One additional interaction between these two regions in VHR is a hydrogen bond between H70 and the catalytic acid D92. Moreover, within the VI loop itself, there are hydrogen bonds that connect residues in the N-terminal portion of the VI with those in the center of the VI. These connections are likely responsible for the identical ms motions that are observed for residues in the VI and acid loop. On the faster timescale of the nanosecond simulations, the WT interactions between the VI and acid loop lead to an opening of the acid loop (Figure 7A).
Molecular motions in N74A VHR
In contrast, in N74A, the unraveling of the short helical region around residue 74 in the VI is propagated to the acid loop and active site in such a way that diminishes the hydrogen bonding between the VI and acid loop as well as the intra-VI hydrogen bonds (Figure 6). Moreover, new H-bonds are now formed between the VI and Q169 in α7 and between the acid loop and the P-loop of N74A (Figure 6). This rearrangement of hydrogen bonds results in a rigidification of the acid loop and a conformation that remains closed. In addition, new H-bonds are formed between the acid loop and the P-loop and between the VI and P-loop, which were not observed in the simulations of WT. These new interactions result in additional closing of the acid loop and a reduction of the WT-like interface around the active site, possibly limiting interaction with the substrate (Figure 7B). An assessment of the flexibility of the active site in the acid and P-loops (Figure 5) show that the fluctuations of the acid loop are decreased in N74A as well as in the P-loop sidechain R125. Thus, this allosteric mutation makes R125 and consequently the acid loop more rigid, likely weakening hydrogen bonds that stabilize the substrate at the active site. The allostery in VHR causes the VI loop to twist upon changes at residue 74, which gives R125 in the P-loop better access to the backbone of H70 in the VI. With R125 pulled to the side by a hydrogen bond, E126 is also able to move to one side, decreasing its interaction with D92 and the substrate. Without this interaction, tungstate is not pushed down into the pocket and the acid loop loses flexibility. The functional result of this mutation is a disruption in the ps – ns motion of the acid loop, a displacement of the catalytic acid D92, inefficient phosphoryl cleavage, and a change in the binding mode for the anionic portion of the substrate.
The results presented here are to be contrasted with the view of the VHR acid loop as being rigid in the WT enzyme. As noted in the Introduction, this current view is mainly due to the absence of an observed structure of VHR with an open acid loop conformation. In fairness, there is no true ‘apo’ structure of VHR as all three structures in the PDB have some variant of an anion bound to the P-loop and the acid loop is in the closed conformation in these structures. The CPMG dispersion and Hahn-echo NMR results here conclusively establish that the apo acid loop is moving on the millisecond timescale. In the tungstate bound form of VHR, these motions remain as shown by the Hahn-echo experiment. In addition, the computational studies show significant backbone motions during the faster timescale of the MD simulations. Thus, it would appear that VHR has a flexible acid loop like other members of the PTP family such as YopH and PTP1B.
However, in YopH and PTP1B,68 the NMR data suggested that the apo enzyme acid loop transiently sampled the closed conformation and conversely, when bound to substrate analog, the acid loop was predominantly closed yet sampled the open acid loop conformation. This conclusion was based on the similarity of chemical shift differences for loop residues between the open and closed conformation (|ΔδN|) with the dynamic chemical shift differences (|ΔωN|) determined by CPMG relaxation dispersion experiments. The agreement between these values seen in those enzymes is not observed for VHR (Figure 8). The |ΔωN| values of the acid loop residues from CPMG dispersion experiments are 1030, 840, 1740, 1150, 1610, and 700 s–1 for K89, D92, E95, F96, L98, and S99, respectively. These values are much greater than the |ΔδN| from the tungstate titrations, which are 165, 456, 256, 279, 239, and 17 s–1 for the same residues. The same trend is true for the VI residues in which |ΔωN| values from the CPMG dispersion experiments are 930, 1340, and 1810 compared to |ΔδN| values = 260, 120, and 50 s–1 for A63, R66, and N72 from the tungstate titrations suggesting that the innate conformational motions in the apo WT are different than the conformational changes as a result of ligand binding and the structures that are interconverting in VHR on the ms timescale remain unclear and represents an obvious next step in the studies of this enzyme.
Conclusion
Although all the details of the molecular motions of the acid loop and VI in VHR remain to be determined, the changes in motions, chemical shifts, and enzyme activity between WT and N74A suggest their importance in its dephosphorylation activity. There is an intimate connection between the acid loop and VI in VHR that is important to the binding and cleavage of substrate. Disruptions at residue 74 change the hydrogen bonding pattern and are propagated to the acid loop and P-loop. These changes affect the motions critical to the mechanism and cause inefficient binding and catalytic activity. The link between the VI and the acid loop in VHR may provide illumination on mechanisms of in vivo regulation and may also represent an avenue toward drugging VHR with small-molecule ligands. Furthermore, in a recent study of 30 dual specificity PTP enzymes, the majority of acid loops in these enzymes have been demonstrated to be closed, with a few showing open loops.70 However, like VHR all contained a negatively charged molecule at the acid site. Given the similarities between those enzymes and VHR, it seems possible that these enzymes also possess mobile acid loops, which could lend themselves to allosteric targeting via their variable insert regions.
Supplementary Material
ACKNOWLEDGMENT
VSB and KR acknowledge support from NIH Biophysics training grant T32 GM008283. JPL and VB acknowledge GM121781. JPL acknowledges MCB 1615415. VB acknowledges GM106121 and supercomputer resources from NSERSC.
Funding Sources
No competing financial interests have been declared.
Footnotes
Supporting Information. A pdf file containing a table listing Rex values for VHR WT and mutant enzymes along with figures containing enzyme kinetic data and 2D NMR 1H-15N HSQC spectra.
The Supporting Information is available free of charge on the ACS Publications website.
Accession Codes
The UniProt accession code for human Vaccinia H1-related phosphatase is 541452.
REFERENCES
- [1].Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, and Mustelin T (2004) Protein tyrosine phosphatases in the human genome, Cell 117, 699–711. [DOI] [PubMed] [Google Scholar]
- [2].Barr AJ (2010) Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development, Future medicinal chemistry 2, 1563–1576. [DOI] [PubMed] [Google Scholar]
- [3].Tautz L, Critton DA, and Grotegut S (2013) Protein tyrosine phosphatases: structure, function, and implication in human disease, Methods Mol Biol 1053, 179–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease, Nature reviews. Molecular cell biology 7, 833–846. [DOI] [PubMed] [Google Scholar]
- [5].Todd JL, Tanner KG, and Denu JM (1999) Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway, J Biol Chem 274, 13271–13280. [DOI] [PubMed] [Google Scholar]
- [6].Yuvaniyama J, Denu JM, Dixon JE, and Saper MA (1996) Crystal structure of the dual specificity protein phosphatase VHR, Science 272, 1328–1331. [DOI] [PubMed] [Google Scholar]
- [7].Hoyt R, Zhu W, Cerignoli F, Alonso A, Mustelin T, and David M (2007) Cutting edge: selective tyrosine dephosphorylation of interferon-activated nuclear STAT5 by the VHR phosphatase, J Immunol 179, 3402–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Todd JL, Rigas JD, Rafty LA, and Denu JM (2002) Dual-specificity protein tyrosine phosphatase VHR down-regulates c-Jun N-terminal kinase (JNK), Oncogene 21, 2573–2583. [DOI] [PubMed] [Google Scholar]
- [9].Wang JY, Yeh CL, Chou HC, Yang CH, Fu YN, Chen YT, Cheng HW, Huang CY, Liu HP, Huang SF, and Chen YR (2011) Vaccinia H1-related phosphatase is a phosphatase of ErbB receptors and is down-regulated in non-small cell lung cancer, J Biol Chem 286, 10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Henkens R, Delvenne P, Arafa M, Moutschen M, Zeddou M, Tautz L, Boniver J, Mustelin T, and Rahmouni S (2008) Cervix carcinoma is associated with an up-regulation and nuclear localization of the dual-specificity protein phosphatase VHR, BMC Cancer 8, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ishibashi T, Bottaro DP, Chan A, Miki T, and Aaronson SA (1992) Expression cloning of a human dual-specificity phosphatase, Proc Natl Acad Sci U S A 89, 12170–12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rahmouni S, Cerignoli F, Alonso A, Tsutji T, Henkens R, Zhu C, Louis-dit-Sully C, Moutschen M, Jiang W, and Mustelin T (2006) Loss of the VHR dual-specific phosphatase causes cell-cycle arrest and senescence, Nat Cell Biol 8, 524–531. [DOI] [PubMed] [Google Scholar]
- [13].Hao L, and ElShamy WM (2007) BRCA1-IRIS activates cyclin D1 expression in breast cancer cells by downregulating the JNK phosphatase DUSP3/VHR, International journal of cancer. Journal international du cancer 121, 39–46. [DOI] [PubMed] [Google Scholar]
- [14].Zhang ZY (2017) Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases, Acc Chem Res 50, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stanford SM, and Bottini N (2017) Targeting Tyrosine Phosphatases: Time to End the Stigma, Trends Pharmacol Sci 38, 524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].De Amici M, Dallanoce C, Holzgrabe U, Trankle C, and Mohr K (2010) Allosteric Ligands for G Protein-Coupled Receptors: A Novel Strategy with Attractive Therapeutic Opportunities, Medicinal Research Reviews 30, 463–549. [DOI] [PubMed] [Google Scholar]
- [17].Kanuma K, Aoki T, and Shimazaki Y (2010) Recent patents on positive allosteric modulators of the metabotropic glutamate 5 receptor as a potential treatment for schizophrenia, Recent Pat CNS Drug Discov 5, 23–34. [DOI] [PubMed] [Google Scholar]
- [18].Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, Garcia JA, Naidoo J, Longgood J, Frantz DE, Tambar UK, Gardner KH, and Bruick RK (2013) Allosteric inhibition of hypoxia inducible factor-2 with small molecules, Nat Chem Biol 9, 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jin L, and Harrison SC (2002) Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin, Proc Natl Acad Sci U S A 99, 13522–13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW (1995) Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex, Nature 378, 641–644. [DOI] [PubMed] [Google Scholar]
- [21].Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, Jensen MR, Gauss CM, Page R, Blackledge M, Muthuswamy SK, Peti W, and Tonks NK (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor, Nat Chem Biol 10, 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr BJ, Zhong M, Taylor L, Randal M, McDowell RS, and Hansen SK (2004) Allosteric inhibition of protein tyrosine phosphatase 1B, Nat Struct Mol Biol 11, 730–737. [DOI] [PubMed] [Google Scholar]
- [23].Peti W, and Page R (2015) Strategies to make protein serine/threonine (PP1, calcineurin) and tyrosine phosphatases (PTP1B) druggable: achieving specificity by targeting substrate and regulatory protein interaction sites, Bioorg Med Chem 23, 2781–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang X, Bajaj R, Bollen M, Peti W, and Page R (2016) Expanding the PP2A Interactome by Defining a B56-Specific SLiM, Structure 24, 2174–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, and Lee AL (2009) Hidden dynamic allostery in a PDZ domain, Proc Natl Acad Sci U S A 106, 18249–18254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang J, Sapienza PJ, Ke H, Chang A, Hengel SR, Wang H, Phillips GN, and Lee AL (2010) Crystallographic and nuclear magnetic resonance evaluation of the impact of peptide binding to the second PDZ domain of protein tyrosine phosphatase 1E, Biochemistry 49, 9280–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cui DS, Beaumont V, Ginther PS, Lipchock JM, and Loria JP (2017) Leveraging Reciprocity to Identify and Characterize Unknown Allosteric Sites in Protein Tyrosine Phosphatases, Journal Of Molecular Biology. 429, 2360–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Abdel-Magid AF (2015) Allosteric modulators: an emerging concept in drug discovery, ACS Med Chem Lett 6, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Abdel-Magid AF (2017) Therapeutic Advantage of the Positive Allosteric Modulators of the GABA-B Receptor, ACS Med Chem Lett 8, 474–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vagnarelli P, and Alessi DR (2018) PP1 Phosphatase Complexes: Undruggable No Longer, Cell 174, 1049–1051. [DOI] [PubMed] [Google Scholar]
- [31].Grzyska PK, Kim Y, Jackson MD, Hengge AC, and Denu JM (2004) Probing the transition-state structure of dual-specificity protein phosphatases using a physiological substrate mimic, Biochemistry 43, 8807–8814. [DOI] [PubMed] [Google Scholar]
- [32].Hengge AC, Denu JM, and Dixon JE (1996) Transition-state structures for the native dual-specific phosphatase VHR and D92N and S131A mutants. Contributions to the driving force for catalysis, Biochemistry 35, 7084–7092. [DOI] [PubMed] [Google Scholar]
- [33].Denu JM, and Dixon JE (1995) A catalytic mechanism for the dual-specific phosphatases, Proc Natl Acad Sci U S A 92, 5910–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brandao TA, Robinson H, Johnson SJ, and Hengge AC (2009) Impaired acid catalysis by mutation of a protein loop hinge residue in a YopH mutant revealed by crystal structures, J Am Chem Soc 131, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rigas JD, Hoff RH, Rice AE, Hengge AC, and Denu JM (2001) Transition state analysis and requirement of Asp-262 general acid/base catalyst for full activation of dual-specificity phosphatase MKP3 by extracellular regulated kinase, Biochemistry 40, 4398–4406. [DOI] [PubMed] [Google Scholar]
- [36].Pavic K, Duan G, and Kohn M (2015) VHR/DUSP3 phosphatase: structure, function and regulation, FEBS J 282, 1871–1890. [DOI] [PubMed] [Google Scholar]
- [37].Brandao TA, Hengge AC, and Johnson SJ (2010) Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps, J Biol Chem 285, 15874–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brandao TA, Johnson SJ, and Hengge AC (2012) The molecular details of WPD-loop movement differ in the protein-tyrosine phosphatases YopH and PTP1B, Arch Biochem Biophys 525, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barford D, Flint AJ, and Tonks NK (1994) Crystal structure of human protein tyrosine phosphatase 1B, Science 263, 1397–1404. [PubMed] [Google Scholar]
- [40].Denu JM, Lohse DL, Vijayalakshmi J, Saper MA, and Dixon JE (1996) Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis, Proc Natl Acad Sci U S A 93, 2493–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Phan J, Lee K, Cherry S, Tropea JE, Burke TR Jr., and Waugh DS (2003) High-resolution structure of the Yersinia pestis protein tyrosine phosphatase YopH in complex with a phosphotyrosyl mimetic-containing hexapeptide, Biochemistry 42, 13113–13121. [DOI] [PubMed] [Google Scholar]
- [42].Schumacher MA, Todd JL, Rice AE, Tanner KG, and Denu JM (2002) Structural basis for the recognition of a bisphosphorylated MAP kinase peptide by human VHR protein Phosphatase, Biochemistry 41, 3009–3017. [DOI] [PubMed] [Google Scholar]
- [43].Wu S, Vossius S, Rahmouni S, Miletic AV, Vang T, Vazquez-Rodriguez J, Cerignoli F, Arimura Y, Williams S, Hayes T, Moutschen M, Vasile S, Pellecchia M, Mustelin T, and Tautz L (2009) Multidentate small-molecule inhibitors of vaccinia H1-related (VHR) phosphatase decrease proliferation of cervix cancer cells, J Med Chem 52, 6716–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pavic K, Rios P, Dzeyk K, Koehler C, Lemke EA, and Kohn M (2014) Unnatural amino acid mutagenesis reveals dimerization as a negative regulatory mechanism of VHR’s phosphatase activity, ACS chemical biology 9, 1451–1459. [DOI] [PubMed] [Google Scholar]
- [45].Bellstedt P, Seiboth T, Hafner S, Kutscha H, Ramachandran R, and Gorlach M (2013) Resonance assignment for a particularly challenging protein based on systematic unlabeling of amino acids to complement incomplete NMR data sets, J Biomol NMR 57, 65–72. [DOI] [PubMed] [Google Scholar]
- [46].Zhang ZY, Wu L, and Chen L (1995) Transition state and rate-limiting step of the reaction catalyzed by the human dual-specificity phosphatase, VHR, Biochemistry 34, 16088–16096. [DOI] [PubMed] [Google Scholar]
- [47].Lee W, Tonelli M, and Markley JL (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy, Bioinformatics 31, 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Keller R (2004) The computer aided resonance assignment tutorial (CANTINA), Verlag, http://www.cara.nmr-software.org/.
- [49].Grzesiek S, Stahl SJ, Wingfield PT, and Bax A (1996) The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR, Biochemistry 35, 10256–10261. [DOI] [PubMed] [Google Scholar]
- [50].Loria JP, Rance M, and Palmer AG (1999) A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy, Journal of the American Chemical Society 121, 2331–2332. [Google Scholar]
- [51].Mulder FA, Skrynnikov NR, Hon B, Dahlquist FW, and Kay LE (2001) Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme, J. Am. Chem. Soc 123, 967–975. [DOI] [PubMed] [Google Scholar]
- [52].Carver JP, and Richards RE (1972) A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation, J. Magn. Res 6, 89–105. [Google Scholar]
- [53].Wang C, Rance M, and Palmer AG 3rd. (2003) Mapping chemical exchange in proteins with MW > 50 kD, J Am Chem Soc 125, 8968–8969. [DOI] [PubMed] [Google Scholar]
- [54].Huang J, and MacKerell AD Jr. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data, J Comput Chem 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005) Scalable molecular dynamics with NAMD, J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics, J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- [57].Loria JP, Rance M, and Palmer AG 3rd. (1999) Transverse-relaxation-optimized (TROSY) gradient-enhanced triple-resonance NMR spectroscopy, J Magn Reson 141, 180–184. [DOI] [PubMed] [Google Scholar]
- [58].Salzmann M, Wider G, Pervushin K, Senn H, and Wuthrich K (1999) TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins, J. Am. Chem. Soc 121, 844–848. [Google Scholar]
- [59].Lee W, Westler WM, Bahrami A, Eghbalnia HR, and Markley JL (2009) PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy, Bioinformatics 25, 2085–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Selvaratnam R, Chowdhury S, VanSchouwen B, and Melacini G (2011) Mapping allostery through the covariance analysis of NMR chemical shifts, Proc Natl Acad Sci U S A 108, 6133–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Khajehpour M, Wu L, Liu S, Zhadin N, Zhang ZY, and Callender R (2007) Loop dynamics and ligand binding kinetics in the reaction catalyzed by the Yersinia protein tyrosine phosphatase, Biochemistry 46, 4370–4378. [DOI] [PubMed] [Google Scholar]
- [62].Schubert HL, Fauman EB, Stuckey JA, Dixon JE, and Saper MA (1995) A ligand-induced conformational change in the Yersinia protein tyrosine phosphatase, Protein Sci 4, 1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang F, Li W, Emmett MR, Hendrickson CL, Marshall AG, Zhang YL, Wu L, and Zhang ZY (1998) Conformational and dynamic changes of Yersinia protein tyrosine phosphatase induced by ligand binding and active site mutation and revealed by H/D exchange and electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Biochemistry 37, 15289–15299. [DOI] [PubMed] [Google Scholar]
- [64].Kuznetsov VI, Hengge AC, and Johnson SJ (2012) New aspects of the phosphatase VHZ revealed by a high-resolution structure with vanadate and substrate screening, Biochemistry 51, 9869–9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kovrigin EL, Kempf JG, Grey M, and Loria JP (2006) Faithful estimation of dynamics parameters from CPMG relaxation dispersion measurements, J. Magn. Reson 180, 93–104. [DOI] [PubMed] [Google Scholar]
- [66].Beach H, Cole R, Gill ML, and Loria JP (2005) Conservation of μs-ms enzyme motions in the apo- and substrate-mimicked state, J. Am. Chem. Soc 127, 9167–9176. [DOI] [PubMed] [Google Scholar]
- [67].Boehr DD, McElheny D, Dyson HJ, and Wright PE (2006) The dynamic energy landscape of dihydrofolate reductase catalysis, Science 313, 1638–1642. [DOI] [PubMed] [Google Scholar]
- [68].Whittier SK, Hengge AC, and Loria JP (2013) Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases, Science 341, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kang TH, and Kim KT (2006) Negative regulation of ERK activity by VRK3-mediated activation of VHR phosphatase, Nat Cell Biol 8, 863–869. [DOI] [PubMed] [Google Scholar]
- [70].Jeong DG, Wei CH, Ku B, Jeon TJ, Chien PN, Kim JK, Park SY, Hwang HS, Ryu SY, Park H, Kim DS, Kim SJ, and Ryu SE (2014) The family-wide structure and function of human dual-specificity protein phosphatases, Acta Crystallogr D Biol Crystallogr 70, 421–435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.