

Patients with chronic phase CML (CP-CML) are effectively managed with BCR-ABL1 tyrosine kinase inhibitors (TKIs) and their survival approaches that of age-matched controls (1). However, only a minority of patients achieve treatment-free remission, while most require long-term TKI therapy to avoid recurrence of active CML, at considerable cost and potential toxicity (2). Current thinking holds that quiescent, fully leukemogenic CML stem cells (LSCs) are BCR-ABL1-independent, providing a reservoir of residual leukemia in TKI responders (3). Multiple mechanisms have been implicated in LSC survival in this setting, but thus far, this has not led to clinical solutions (4). BCR-ABL1 independence is not limited to residual CML, as many patients with clinical TKI resistance do not exhibit explanatory BCR-ABL1 kinase mutations, suggesting that alternative mechanisms support leukemia cell survival. There is overlap in the pathways enabling CML cell survival in these two scenarios. For instance, Wnt/β-catenin and PP2A have been implicated as mediators both of LSC survival and BCR-ABL1-independent TKI resistance (4).
Many malignant cells upregulate CRM1 (chromosome region maintenance 1/exportin 1)-dependent nuclear-cytoplasmic transport (NCT) to inactivate tumor suppressor proteins, and CRM1 inhibitors such as selinexor (KPT-330) are in clinical development for several cancer types, including myeloid malignancies (5, 6). We have previously shown that CD34+ cells from patients with BCR-ABL1-independent resistance depend on RAN, a small GTPase that provides the concentration gradient driving CRM1-dependent NCT. These cells are sensitive to selinexor and even more to selinexor/imatinib combinations (7). Based on this finding, we hypothesized that NCT may have a similar, critical function in LSCs, and that selinexor may synergize with TKIs to eliminate primitive CML cells.
We initially tested selinexor, imatinib, and the combination on CP-CML vs. cord blood (CB) CD34+38+ cells (representing mostly progenitor cells) and CD34+38− cells (enriched for primitive cells, including LSCs). Details on the patients are provided in Supplemental Table S1. We used drug concentrations previously identified as minimally toxic to normal CD34+ cells (7). Single-agent imatinib reduced colony formation of CML CD34+38+ cells by 87% compared to 23% for CB (Two-way ANOVA, p<0.0001), with a similar difference for CD34+38− cells, 91% vs. 36% (p<0.001). Remarkably, single-agent selinexor was also selectively toxic to CML cells, reducing colony formation of CML CD34+38+ cells by 58% vs. 22% for CB (p<0.01), with a more dramatic differential effect in CD34+38− cells [(reduction by 44% for CML vs. 4% for CB (p=0.001)]. KPT-330, combined with imatinib, decreased colony formation of CML CD34+38+ and CD34+38− cells by 92% and 97%, compared to 33% and 57% for CB (p<0.0001 and p<0.001, respectively) (Figure 1A, Supplemental Table S2). Apoptosis was assessed by annexin V staining after 72 hours of culture (Supplemental Figure 1). For single agents, the only significant increase in apoptosis was seen with imatinib in CML CD34+38− cells. The greater imatinib sensitivity of CD34+38− vs. CD34+38+ CML cells may reflect the relatively short culture period in a full complement of cytokines (3). Combined treatment increased apoptosis in CML CD34+38+ cells by 3-fold, but only by 2-fold in CB CD34+38+ cells (p<0.01 CML vs. CB). Differences were striking in CD34+38− cells, with a 9.2-fold increase of apoptotic cells over controls for CML vs. only 3.2-fold increase for CB (p<0.0001) (Figure 1B). These data are consistent with a large differential effect of the selinexor/imatinib combination on CML vs. normal hematopoietic cells that is most pronounced in primitive CD34+CD38− cells.
Figure 1. Inhibition of nuclear export selectively targets primitive CML cells.
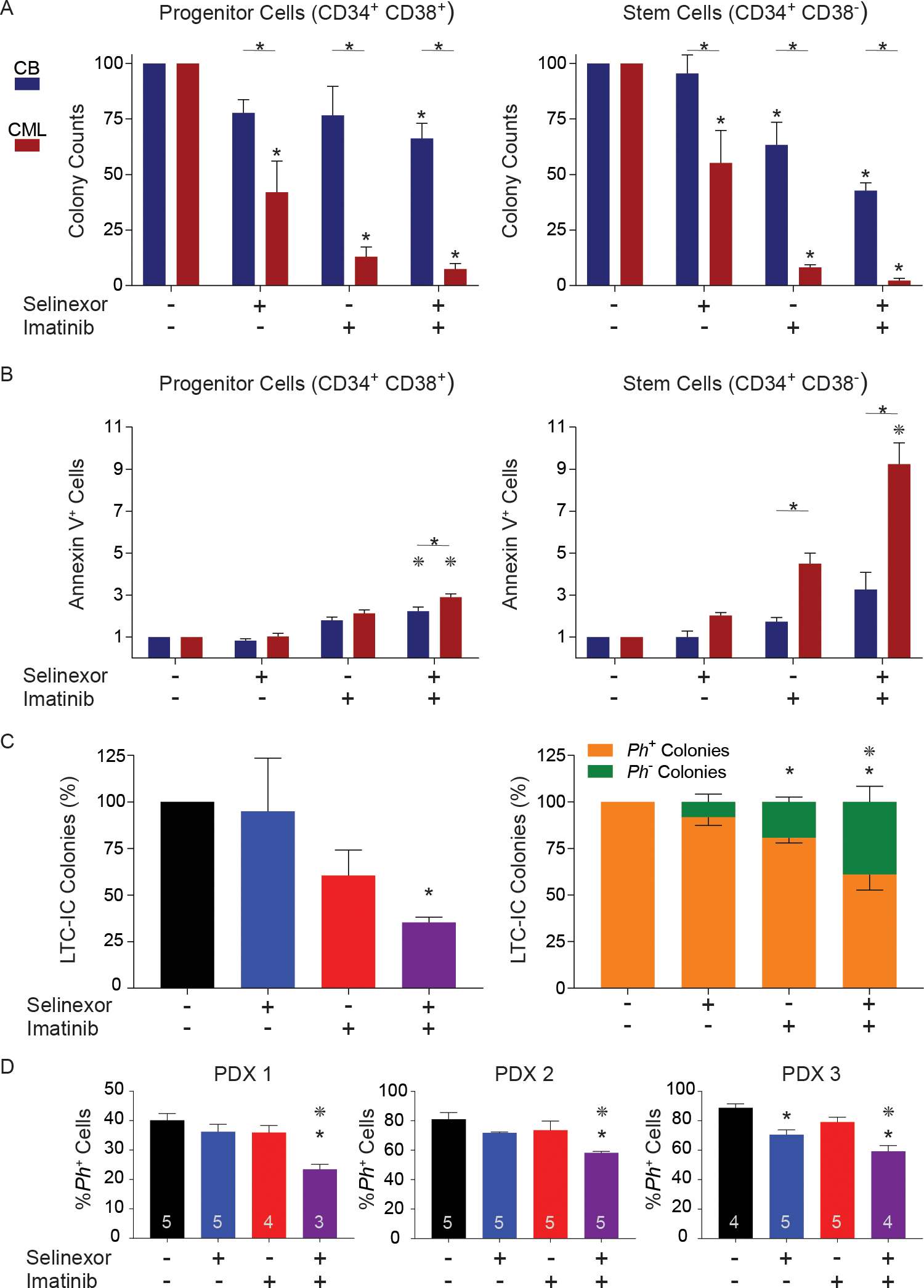
(A) CD34+38+ and CD34+38− cells from CML patients (n=4) and CB samples (n=3) were plated in cytokine-supplemented Methocult H4230, in the absence or presence of selinexor (50 nM), imatinib (2.5 μM) or the combination. CFU-GM colonies were counted after 14 days. A * above a bar represents p<0.05 vs untreated and a * above a line spanning two bars represents p<0.05 for CML vs CB. (B) CD34+38+ and CD34+38− cells from CML patients (n=3) and healthy CB samples (n=3) were cultured with or without selinexor (50 nM) and/or imatinib (2.5 μM) for 72 hours and apoptosis was measured by annexin V-APC and 7-AAD. A * above a line spanning two bars represents p<0.05 for CML vs CB. A * above a bar represents p<0.05 vs. imatinib. (C) CD34+ CML cells (n=6) were cultured ± selinexor (50 nM), imatinib (2.5 μM) or the combination for 72 hours, plated in LTC-IC assays, and CFU-GM colonies were counted and genotyped for BCR-ABL1 by FISH. Total LTC-IC colonies and Ph+ colonies were significantly reduced by the combination. (D) CD34+ CML cells (n=4 patients), treated with or without selinexor (50 nM), imatinib (2.5 μM) or the combination for 72 hours, were injected into sub-lethally irradiated female NSG mice (5 mice per treatment arm). At 12 weeks, engraftment of human cells was determined by immunophenotyping for hCD45+. Sorted hCD45+ cells were genotyped for BCR-ABL1 by FISH. The number of mice analyzed is shown inside each bar. * shows p<0.05 vs untreated and * shows p<0.05 vs imatinib single agent. Error bars represent standard error of the mean. CML, chronic myeloid leukemia; CB, cord blood; CFU-GM, colony forming unit granulocyte-macrophage; APC, allophycocyanin; 7-AAD, 7-aminoactinomycin D; LSCs, leukemic stem cells; LTC-IC, long-term culture-initiating cells; FISH, fluorescence in situ hybridization; NSG, non-obese diabetic severe combined immunodeficiency interleukin-2Rgamma null; PDX, patient-derived xenografts
We next assessed selinexor effects on LSCs functionally defined as long-term culture-initiating cells (LTC-ICs). CD34+ cells from untreated CP-CML patients (n=6) were first cultured with selinexor, imatinib or the combination for 72 hours, then grown in LTC-IC assays for 6 weeks without inhibitors, followed by colony assays. Single-agent selinexor had no effect. Compared to untreated controls, imatinib reduced colony formation by 39% (p=0.06), whereas the combination resulted in 64% reduction (p<0.01) (Figure 1C, left panel). LTC-IC-derived colonies were genotyped by fluorescence in situ hybridization (FISH). Compared to untreated controls, single-agent selinexor had no effect on BCR-ABL1+ colonies (p=0.25, NS), single-agent imatinib reduced BCR-ABL1+ colonies by 19% (p<0.05), and the selinexor/imatinib combination by 39% (p<0.0001). Compared to single-agent imatinib, combined selinexor/imatinib was significantly more effective in depleting BCR-ABL1+ over BCR-ABL1− LTC-ICs (p<0.05) (Figure 1C, right panel).
We next injected CD34+ cells from four different CML patients, treated ex vivo with each single inhibitor or the combination for 72 hours, into sub-lethally irradiated female NOD SCID gamma (NSG) mice (5 mice per treatment arm) (Supplemental Figure 2). At 12 weeks, BM hCD45 cells were quantified by FACS and genotyped for BCR-ABL1 by FISH (Figure 1D). Human engraftment ranged from 78.1–81.7% across all treatment groups in the first patient-derived xenograft (PDX1), 0.9–3.4% in PDX2, and 3.2–7.7% in PDX3. In PDX4, mice transplanted with imatinib-treated cells died between days 28 and 30 from an unknown cause. In the remaining mice, engraftment at 12 weeks was 0.5–2.4% (Supplemental Figure 2). FISH revealed 40%, 81%, 88%, and 90% BCR-ABL1+ cells in the untreated groups of PDX1–4, respectively. Single-agent imatinib had no effect, and single-agent selinexor decreased BCR-ABL1+ cells only in PDX3 (89 vs. 71%; p=0.002, one-way ANOVA). The combination of selinexor/imatinib consistently reduced BCR-ABL1+ cells compared to the untreated groups (by 42, 28, 33, 31%; p<0.006 for PDX1–4, respectively), and compared to imatinib in PDX1–3 (p<0.02 for each) (Figure 1D).
We and others have shown overlap between the transcriptional profiles of BCR-ABL1 kinase-independent TKI resistance and blastic transformation (8). Similarly, several signaling pathways, including β-catenin, have been implicated both in the maintenance of quiescent LSCs and in overt TKI resistance in CML (9). Our data show that primitive CML cells [defined by CD34+38− immunophenotype, LTC-IC capacity or long-term engraftment in NSG mice] are sensitive to the selinexor/imatinib combination, promoting selection of residual BCR-ABL1-negative cells, suggesting that LSCs are more dependent on NCT than hematopoietic stem cells (HSCs). To identify potential CRM1 cargos involved in NCT, we performed a meta-analysis of five published CML microarray datasets from GEO to identify genes with differential expression between quiescent LSCs and HSCs (files downloaded from https://www.stemformatics.org/). A t-test (Satterthwaite approximation with no equivariance assumption) was performed for each probe and the probe with the lowest p-value for a gene was used to represent each gene. Fold changes ≥2 and a p-value of <0.05 in at least 3 of 5 studies was used to identify genes with evidence for consistent expression differences between LSCs vs. HSCs (Figure 2A). We identified 127 genes with increased expression and 48 genes with reduced expression in LSCs vs. HSCs (Supplemental Table S3). Kyoto Encyclopedia of Genes and Genomes (KEGG, http://amp.pharm.mssm.edu/Enrichr/) pathway analysis revealed enrichment for cell cycle genes (Figure 2B, C), consistent with the notion that primitive CML cells are poised to enter cell cycle (10). The top three upregulated genes (>10-fold higher expression in LSCs vs. HSCs), CD36, PIEZO2 and GAS2, have all been associated with TKI resistance in CML. CD36, also known as fatty acid translocase, is upregulated in primitive CML cells resistant to imatinib (11). Expression of PIEZO2, a mechanically activated ion channel, is increased in TKI-resistant vs. TKI-sensitive CML cells (12). GAS2 is a positive regulator of β-catenin, a key mediator of LSC self-renewal in CML (13, 14). No published CML association was identified for the top 3 genes downregulated in LSCs vs. HSCs.
Figure 2. Meta-analysis of CML stem cell microarray studies identifies potential targets of selinexor in primitive CML cells.
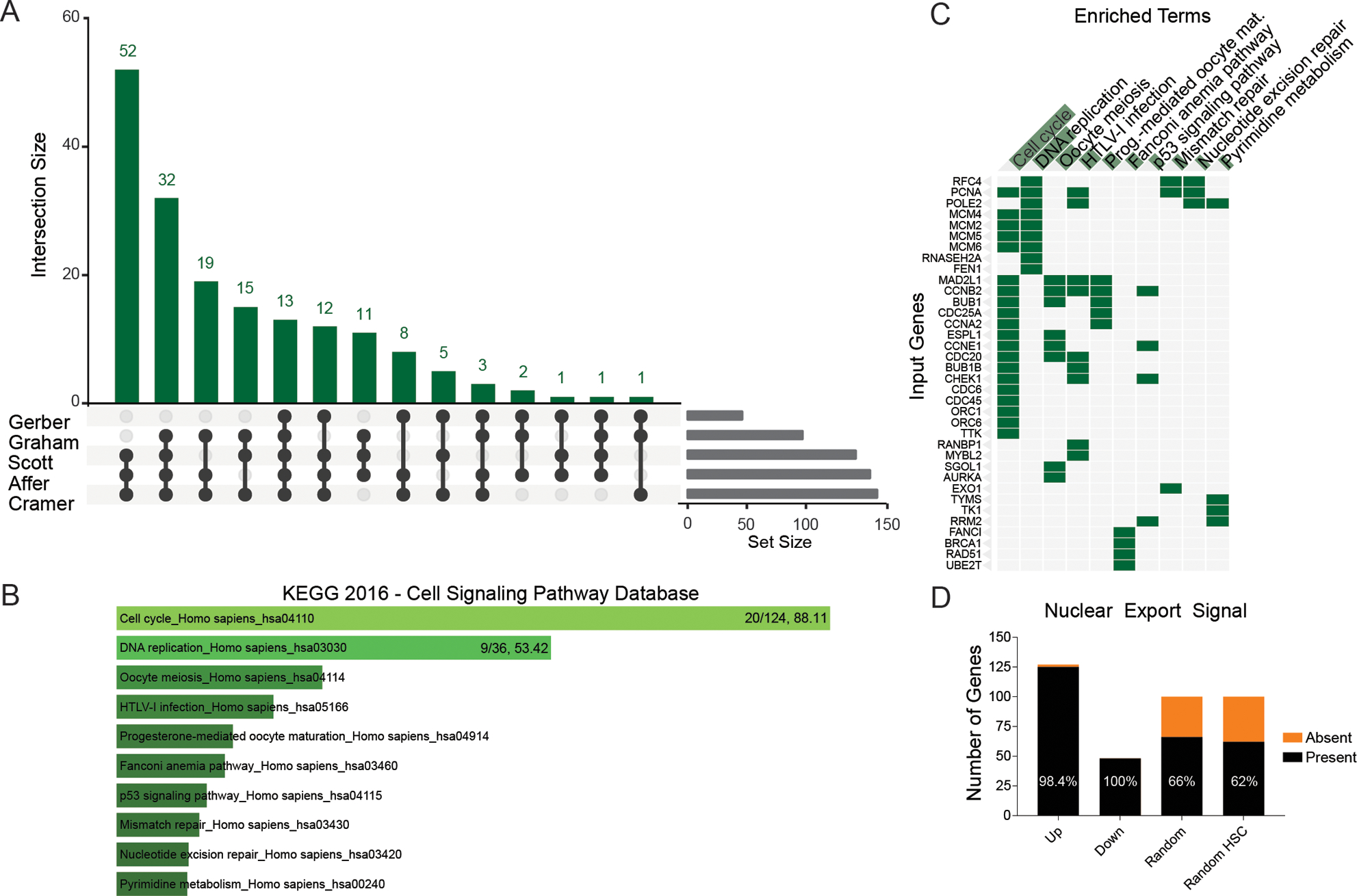
(A) Meta-analysis was performed on five published CML microarray datasets to identify genes with differential expression between LCSs and HSCs (Supplemental Information). Primitive cells were defined by CD34+38− immunophenotype or G0 cell cycle status. Genes with concordant, ≥2-fold significant differences in expression between CML and normal controls in at least three of the datasets were further assessed for the presence of a predicted nuclear export signal based on the LocNES online tool. (B) KEGG pathway analysis results showing the top 10 pathways with upregulation in LSCs compared to HSCs. (C) The top 10 pathways shown in a clustergram, with hits per pathway and combined score shown for the top two pathways. (D) The 127 upregulated, 48 downregulated, 100 random, and 100 random HSC-expressed genes were analyzed with the LocNES platform and the percent that contain a nuclear export signal is shown.
Based on the higher sensitivity to selinexor of LSCs vs. HSCs we hypothesized that genes differentially expressed between LSCs and HSCs should be more likely to be subject to NCT than genes without differential expression. To test this, we used the LocNES algorithm to predict nuclear export signals (NESs) in the differentially expressed genes (Supplemental Table S3). Of 127 genes upregulated and 48 genes downregulated in LSCs vs. HSCs, 125 (98.4%) and 48 (100%), respectively, were predicted to contain an NES (Figure 2D). In contrast, only 66 of 100 randomly selected genes (p<0.00001, Fisher’s Exact test for comparison vs. differentially expressed genes) (Supplemental Table S4) and 62 of 100 genes expressed at high but comparable levels in LSCs and HSCs (p<0.00001 for comparison vs. differentially expressed genes) had a putative NES (Supplemental Table S5). These data suggest that genes differentially expressed in LSCs vs. HSCs are enriched for CRM1 substrates, consistent with the differential effects of selinexor on primitive cells. The three top genes with increased expression in LSCs vs. HSCs function in the cytoplasm (GAS2) or at the cell membrane (PIEZO2, CD36), and may accumulate in the nucleus upon selinexor treatment.
Altogether, our data suggest that selinexor affects multiple pathways in an LSC-specific manner by trapping select proteins in the nucleus. Selectivity may be afforded by targeting NES-containing proteins with differential expression between LSCs and HSCs. Functional validation will be required to identify CRM1 cargos critical for selinexor effects, the prediction being that multiple CRM1 clients are involved. Irrespective of this, we show that combining selinexor with imatinib selectively targets primitive LSCs over HSCs, potentially enabling elimination of residual CML. The recent approval of selinexor for multiple myeloma and the development of second-generation compounds such as eltanexor (KPT-8602), with improved tolerability, should facilitate the clinical translation of this concept (15).
Supplementary Material
Acknowledgements
This work was supported by a Leukemia & Lymphoma Society (LLS) Translational Research Program Award (6086-12) (M.W.D.) and an LLS Specialized Center of Research Program Award (GCNCR0314A-UTAH) (M.W.D.). This work was also supported by the National Institutes of Health (NIH) National Cancer Institute grant R01CA178397 (M.W.D. and T.O.). CCM was funded by the pediatric cancer program which is supported by the Intermountain Healthcare and Primary Children’s Hospital Foundations, Department of Pediatrics, Division of Pediatric Hematology/Oncology, and the University of Utah. H.T. was a visiting scholar from Singapore, and was supported by Research Training Fellowship from National Medical Research Council of Singapore. D.Y. is supported by the International Award from the Lady Tata Memorial Trust following the Special Fellow Award from the Leukemia & Lymphoma Society. We thank James Marvin, Director of the Flow Cytometry Facility at The University of Utah for assistance with experiments. The University of Utah Flow Cytometry Facility was supported by the National Cancer Institute through award 5P30CA042014-24 and the National Center for Research Resources of the National Institutes of Health under award 1S10RR026802-01.
Footnotes
Disclosure of Conflicts of Interest
M.W.D. is on the advisory board and is a consultant for Incyte, Novartis and Pfizer, and serves on the advisory board for Takeda, Blueprint and Galena BioPharma. His laboratory receives research funding from Novartis and Pfizer. S.S. is the President and Chief Scientific Officer of Karyopharm Therapeutics.
References
- 1.Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N Engl J Med. 2017;376(10):917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saussele S, Richter J, Guilhot J, Gruber FX, Hjorth-Hansen H, Almeida A, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;19(6):747–57. [DOI] [PubMed] [Google Scholar]
- 3.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. The Journal of Clinical Investigation. 2011;121(1):396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nature Reviews Cancer. 2012;12(8):513–26. [DOI] [PubMed] [Google Scholar]
- 5.Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122(17):3034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013;27(1):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khorashad JS, Eiring AM, Mason CC, Gantz KC, Bowler AD, Redwine HM, et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood. 2015;125(11):1772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McWeeney SK, Pemberton LC, Loriaux MM, Vartanian K, Willis SG, Yochum G, et al. A gene expression signature of CD34+ cells to predict major cytogenetic response in chronic-phase chronic myeloid leukemia patients treated with imatinib. Blood. 2010;115(2):315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, et al. Genetic and Pharmacologic Inhibition of β-Catenin Targets Imatinib Resistant Leukemia Stem Cells in CML. Cell Stem Cell. 2012;10(4):412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham SM, Vass JK, Holyoake TL, Graham GJ. Transcriptional analysis of quiescent and proliferating CD34+ human hemopoietic cells from normal and chronic myeloid leukemia sources. Stem Cells. 2007;25(12):3111–20. [DOI] [PubMed] [Google Scholar]
- 11.Landberg N, von Palffy S, Askmyr M, Lilljebjorn H, Sanden C, Rissler M, et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica. 2018;103(3):447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh N, Tripathi AK, Sahu DK, Mishra A, Linan M, Argente B, et al. Differential genomics and transcriptomics between tyrosine kinase inhibitor-sensitive and -resistant BCR-ABL-dependent chronic myeloid leukemia. Oncotarget. 2018;9(54):30385–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu D, Marquis K, Pei J, Fu SC, Cagatay T, Grishin NV, et al. LocNES: a computational tool for locating classical NESs in CRM1 cargo proteins. Bioinformatics. 2015;31(9):1357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Zhou W, Saberwal G, Konieczna I, Horvath E, Katsoulidis E, et al. Interferon Consensus Sequence Binding Protein (ICSBP) Decreases β-Catenin Activity in Myeloid Cells by Repressing GAS2 Transcription. Molecular and Cellular Biology. 2010;30(19):4575–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Etchin J, Berezovskaya A, Conway AS, Galinsky IA, Stone RM, Baloglu E, et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia. 2017;31(1):143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.