

Abstract
A second-generation kilogram-scale synthesis of the psychedelic tryptamine psilocybin has been developed. The synthesis was designed to address several challenges first encountered with the scale-up of previously described literature procedures, which were not optimized for providing consistent yield and purity of products, atom economy, or being run in pilot plant-scale reactors. These challenges were addressed and circumvented with the design of the second-generation route, which featured an optimized cGMP large-scale Speeter–Anthony tryptamine synthesis to the intermediate psilocin with improved in-process control and impurity removal over the three steps. Psilocin was subsequently phosphorylated directly with phosphorous oxychloride for the first time, avoiding a tedious and poor atom economy benzyl-protecting group strategy common to all previously described methods for producing psilocybin. In this report, the challenges encountered in a 100 g scale first-generation literature-based synthesis are highlighted, followed by a detailed description of the newly developed second-generation synthesis to provide over one kilogram of high-purity psilocybin under cGMP.
Introduction
The last two decades have witnessed a renewed interest in the clinical study of psychedelics for the potential treatment of a range of mental illnesses,1 including major depressive disorder,2 treatment-resistant depression,3 end-of-life anxiety,4−6 obsessive-compulsive disorder,7 and addiction disorders.8,9 In contrast to currently available pharmacotherapies for the aforementioned conditions, a remarkable feature of psychedelic-mediated approaches is the apparent lasting effect produced by a single dose of a drug.10 Though more than 100 different chemicals built upon phenethylamine, tryptamine, or lysergamide scaffolds have been shown to produce characteristic psychedelic effects in humans mediated in part by agonist activity at the 5-HT2A receptor in the central nervous system,11−14 the fungal natural product psilocybin was selected as the most suitable candidate to usher in the modern era of clinical research into psychedelics for several reasons. Psilocybin has been likely consumed by humans for thousands of years,15 has been shown to be well tolerated across escalating doses (18–72 mg) with a clinically acceptable 4–6 h duration of action,16 and is generally free from the manufactured stigma associated with other known psychedelic molecules, such as lysergic acid diethylamide (LSD). Additionally, it was previously produced and tableted by the Swiss pharmaceutical company Sandoz in the 1960s under the tradename Indocybin and supplied to researchers for clinical study.17−19 As with most psychedelic substances, scientific research with psilocybin was stunted considerably in the early 1970s, regardless of apparent therapeutic efficacy, with the passing of the Controlled Substances Act in the United States. Now that modern clinical activities have gained momentum and research demand for psilocybin is rapidly growing, a reliable kilo-scale synthetic approach was required to provide active pharmaceutical ingredient (API) under current Good Manufacturing Practice (cGMP) guidelines. To address the criteria needed for advanced clinical trials and the potential for commercial manufacture, a series of processes were developed and evaluated against attributes including amenability to quality by design (QBD) principles, reproducibility at scale in a cGMP environment, yield, purity, and cost. The second-generation synthesis of psilocybin described below has proven advantageous to these criteria.
Results and Discussion
Overview of First-Generation Literature-Based Synthesis
Given the availability of the literature precedent on psilocybin synthesis, the first-generation multigram-scale approach was adapted from several known methods.20 The route depicted in Scheme 1 was based primarily on the published methods of Shirota et al.21 and Nichols et al.22 and ultimately provided 100 g of high-purity psilocybin, which enabled the initiation of clinical trials. Nevertheless, the first-generation route presented several challenges (Scheme 1) that were not well predicted by bench-scale development reactions that would limit the utility of this approach in large-scale campaigns.
Scheme 1. First-Generation Multigram-Scale Synthesis of Psilocybin.
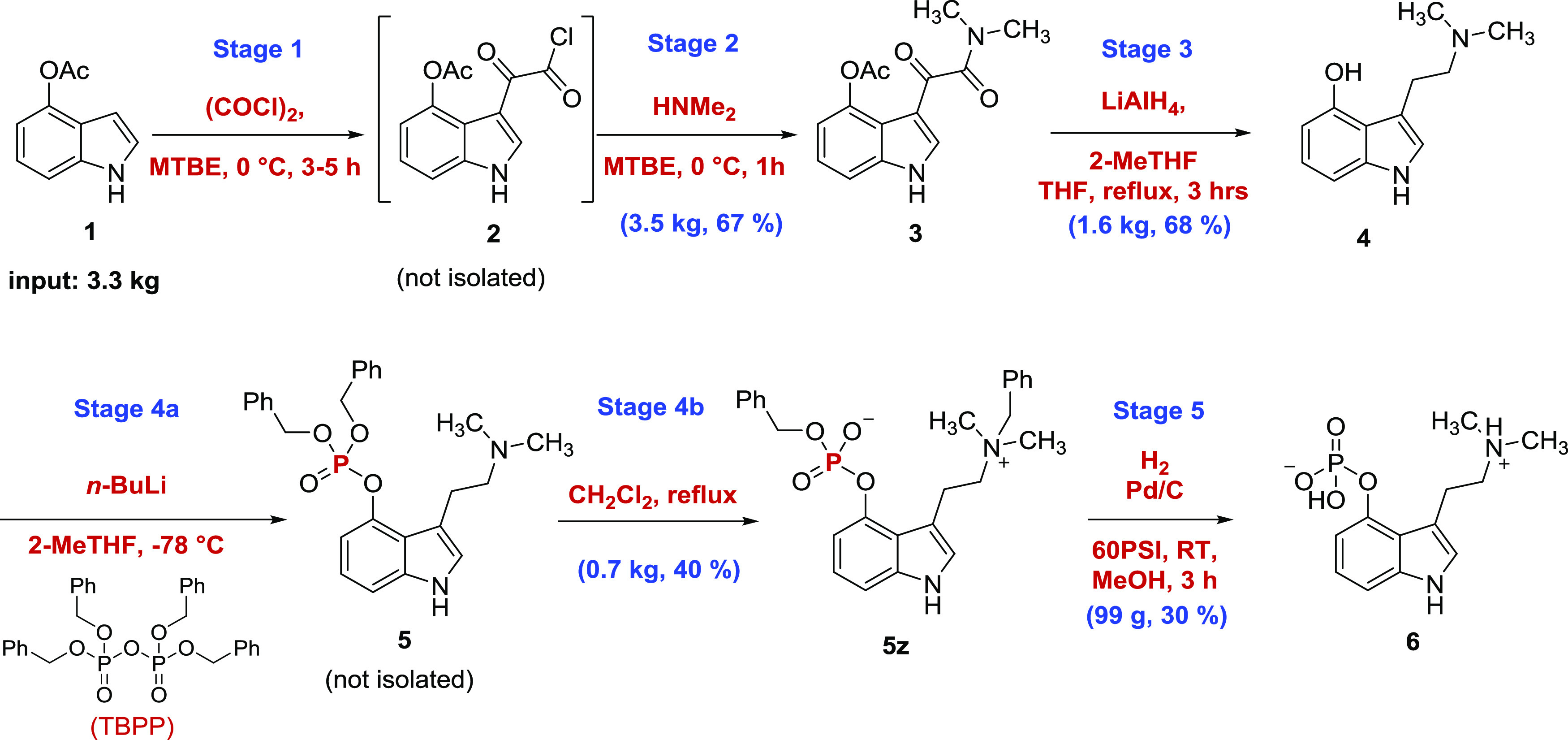
The problems encountered in the first-generation synthesis are delineated in the following:
-
1.
From commercially available 4-acetoxyindole (1) and oxalyl chloride, acyl chloride 2 was not isolated and telescoped directly into the formation of ketoamide 3. Though it circumvented an isolation step, this process unavoidably introduced residual oxalyl chloride to stage 2, which reacted with excess dimethylamine to form the byproduct tetramethyloxamide, a persistent impurity in isolated ketoamide 3.
-
2.
To accommodate the variable levels of tetramethyloxamide impurity, which was reduced to tetramethylethylenediamine alongside 3, a large excess (10 equiv) of lithium aluminum hydride was found to be optimal at stage 3 to ensure complete reduction of all materials present and achieve a satisfactory conversion of ketoamide 3 to psilocin (4). Consequently, the resulting large volume of aluminum waste salts required extensive washing with tetrahydrofuran (THF) after quenching to maximize yield. The extended washing process was tedious given the reactive nature of psilocin, which is highly prone to degradation and forming blue oxidation products.23
-
3.
The phosphorylation was accomplished at stage 4a by the reaction of tetrabenzylpyrophosphate (TBPP) with the lithium phenolate of 4 using n-butyllithium under cryogenic conditions. The reaction also produced 1 equiv of lithium dibenzylphosphate as a byproduct. Following the solvent swap, the resulting dibenzyl-protected phosphate intermediate 5 underwent a spontaneous yet kinetically variable benzyl migration at stage 4b in refluxing methylene chloride to precipitate zwitterion 5z, as described in detail in Shirota et al.21
-
4.
The phosphorylation of psilocin in stages 4a and 4b is a complex reaction and the parameters that influence the benzyl migration from 5 to 5z remain little understood. As a result, the monitoring and in-process control (IPC) of stages 4a and 4b is complicated by the dynamics of all intermediates, products, and byproducts in flux. A key challenge on the multigram scale that was not predicted at the bench scale was the kinetics of the rearrangement of 5 to 5z, which was found to be sensitive to control of temperature as a function of reaction volume and the resulting impact on the particle size of the precipitated solid product. In one instance, the large-scale benzyl migration–precipitation stage 4b yielded an ultrafine particle that significantly impeded the final filtration, which was further exacerbated by volatile methylene chloride mother liquors evaporating from the bottom of the filter plate under vacuum leaving behind sticky dibenzylphosphate and reaction byproduct residues, resulting in a final product filtration that required more than 6 days to complete.
-
5.
The resulting zwitterion product (5z) was subjected to exhaustive catalytic hydrogenolysis at stage 5 to remove two benzyl groups and smoothly reveal zwitterionic psilocybin (6). A key observation in the development of an isolation process was the identification of several antisolvents, including acetone and isopropanol, with the capacity to precipitate psilocybin from aqueous solutions in purified form; this approach would also be used in the second-generation synthesis described below.20,24 A 0.65 kg input of 5z provided just 173 g (44% yield) of crude-isolated 6 with 99.6% high-performance liquid chromatography (HPLC) purity. The loss of two benzyl groups and corresponding forfeiture of molecular weight at stage 5 highlights the less-than-optimal atom economy of this process. Subsequent final API filtration and recrystallization from boiling water to provide a consistent crystalline form of psilocybin yielded 99 g (5% overall yield from 1) of psilocybin with 99.8% HPLC purity that met all other set specifications for API release.
Second-Generation Synthesis
Although the process described in the first-generation synthesis (Scheme 1) could provide multigram quantities of 6, further scale-up under cGMP conditions presented a process variability and control challenge. To overcome the challenges encountered in the five-step synthesis, it was imperative that the following questions were addressed: [1] Is there a way to minimize or eliminate the tetramethyloxamide byproduct at stage 2? The detrimental downstream effect of tetramethyloxamide required excess use of LAH in stage 3. The excess LAH byproducts at work-up led to degradation of the product and variability in meeting specifications. [2] Is there an approach that would quickly purify psilocin (4), while ideally avoiding chromatography? In the development work, psilocin was found to be unstable in the presence of excess lithium salts from the LAH reduction, especially when left over the filter cake for an extended period. The quick isolation of pure psilocin could help minimize a long hold time during step three and prevent degradation. [3] What are suitable conditions that could allow the direct phosphorylation of psilocin (4) to psilocybin (6)? This could potentially avoid phosphorylation using TBPP with all of the drawbacks enumerated above to its use and obliviate the need for catalytic hydrogenolysis, and [4] can boiling water be avoided in the purification and final isolation of psilocybin given that the degradation of psilocybin to psilocin was found to be enhanced at elevated temperatures in aqueous recrystallization conditions? Taken together, these inefficiencies contributed to reduced overall yield of 5% over five chemical steps, as shown in Scheme 1. Consequently, a second campaign was desired to design a more robust synthesis that would allow reworking of the first three steps and direct phosphorylation of psilocin to provide purified psilocybin. The improved, second-generation synthesis based on these areas of optimization is discussed below (Scheme 2).
Scheme 2. Improved Second-Generation Synthesis of Psilocybin Utilizing Direct Phosphorylation.

The first task was to develop a scalable process for the synthesis of acyl chloride 2 in good yield and purity. In the five-step campaign, it was telescoped into the next step and required excess use of oxalyl chloride in the acylation reaction. Excess oxalyl chloride needed to be effectively removed for the reasons outlined above in the first-generation synthesis. From the development work, the optimal conditions for the synthesis of 2 involved treatment of 4-acetoxyindole (1) with 1.2 equiv of oxalyl chloride at −10 °C in methyl tert-butyl ether (MTBE). Upon completion of the reaction, the mixture was diluted with heptane to precipitate 2 as a yellow solid. The collected solid was washed with 1:3 MTBE/heptane, ensuring that excess oxalyl chloride was completely removed. The end point of the washing was determined by checking the washes for oxalyl chloride via derivatization with aniline to permit detection by HPLC. The level was typically ≤1% after a few successive washes to provide 2 with an HPLC purity of >98% relative peak area and isolated in 92% yield. The long-term stability of 2 was not established beyond 24 h, and it was typically used for the next step within 1 day. Having obtained 2 in a relatively stable, pure, and high yielding form, the amidation step was straightforward. Addition of dimethylamine in THF to 2 followed by triethylamine to neutralize generated HCl gave crude 3. The use of triethylamine at this stage was an improvement over previously reported procedures that have relied on toxic pyridine for the same purpose.22 Optimal conditions for the amidation step necessitated the dilution of the reaction mixture with heptane, filtration, and recrystallization from isopropanol followed by water wash. During this step, the eluting aqueous filtrate was conveniently monitored for residual triethylamine-HCl by precipitation reaction with AgNO3 as a visual indicator. With the optimized process, ketoamide 3 was obtained in 83% yield and >98% area purity by HPLC.
Synthesis of Psilocin (4)
Given the challenges in providing the consistent product at stage 3 in the first-generation process, a considerable amount of time was spent developing a process that would provide satisfactory results at scale. The stability of 4 was found to greatly depend on the processing time in solution. Degradation in solution was rapid, and the solution transitioned from colorless to dark purple rapidly. The initial approach implemented the exothermic addition of LAH to a solution of 3 in THF at 0 °C followed by refluxing and required the use of excess LAH. Under these conditions, the reaction was typically still incomplete after refluxing for 5 h. An early-eluting intermediate peak on HPLC, which was later identified as the β-hydroxy psilocin intermediate,20 persisted and the reaction stalled with continued reflux. Alternatively, the reverse addition (i.e., a solution of 3 added to a solution of LAH) showed significant improvement. Furthermore, a solvent screen yielded 2-Me-THF as a promising candidate in terms of improving the reaction profile. 2-Me-THF permitted a higher reflux temperature, which both shortened the reaction time and assisted in reduction of the persistent early-eluting intermediate. Ketoamide 2 was not, however, readily soluble in 2-Me-THF so in the optimized process, 3 was added as a slurry in 2-Me-THF to a solution of LAH at >60 °C and then heated to reflux. With this approach, the reaction was less exothermic, had an improved purity profile, and only 8% of the β-hydroxy psilocin intermediate remained after 90 min; with continued reflux, the reaction was essentially complete after 3 h.
The approach to quenching the reaction mixture was also found to be important for a successful process. A quench with water and NaOH using well-known protocols25−27 gave a strong exotherm and off-gassing. The suspension was filtered and concentrated to obtain a white to off-white solid that turned dark green rapidly with a significant amount of the product still adhered to the filter cake. Subsequent reslurry of the filter cake with a 10% solution of 7 N NH3 in MeOH in THF led to isolation of the product in high purity. However, the conditions still did not suppress oxidation to a dark blue colored solid over time. Further reslurry of the solid with MTBE provided an off-white solid product with improved purity. In a separate experiment, a small amount of off-white solid 3 was treated with 1 N NaOH to give a greenish solution, which could indicate formation of the sodium-phenolate of the phenolic hydroxyl group that may be strongly retained on the waste alumina salts and impede product recovery. In light of the unsuitability of the Fieser work-up, an alternative quenching protocol was developed that involved a quench with THF/H2O (100:27), to which were added silica gel and anhydrous sodium sulfate followed by dilution with DCM/MeOH (9:1). The mixture was filtered through a small pad of silica gel and was followed by a DCM/MeOH rinse to give a colorless solution. Additionally, longer residence time on silica gel did not appear to adversely affect the product quality or yield. The crude product was isolated by solvent swap to heptanes, which resulted in a yellow precipitate that was further purified by reslurry in heptanes and diisopropyl ether to afford an off-white solid that remained colorless initially. Discoloration of isolated 4 was time-dependent on standing. With short-term storage under ambient conditions, discoloration was not significant, but over several weeks, the darkening of the product still appeared with oxygen exposure. On the kilogram scale, the yield of the product was >70%, with a purity of 94.4% peak area by HPLC.
Direct Phosphorylation of Psilocin to Psilocybin
Several strategies have utilized direct phosphorylation as part of both medicinal chemistry efforts and natural product synthesis.28−33 A recent example by Kempson and co-workers at Bristol-Myers Squibb utilized direct phosphorylation in their scale-up synthesis of a potent GluN2B inhibitor and its prodrug.34 The initial attempts at direct phosphorylation of 4 using various phosphorylating agents, including H3PO4/P2O5, PCl5, H3PO4/Cl3CCN, and Na4P2O7, failed to deliver psilocybin in isolable quantity. However, phosphorylation of 4 with POCl3 in THF showed promise, though it initially gave several process impurities in addition to 6 as seen by HPLC (Table 1). Additionally, the reaction tended to form a sticky precipitate that inhibited efficient stirring. To address these initial challenges, several optimizations were introduced.
Table 1. HPLC Relative Retention Times (RRT) of Selected Process Impurities and their Relative Percent Areas at Various Stages in the Development of the Direct Phosphorylation Reaction Using POCl3.

| entry | condition | pyrophosphate (10) RRT 0.58 | psilocybin (6) RRT 1.0 | RRT 1.9 | psilocin (4) RRT 2.21 | RRT 2.41 |
|---|---|---|---|---|---|---|
| 1 | 10 min post POCl3 addition | 0.15 | 69.46a | 9.27 | 1.9 | 2.08 |
| 2 | 1 h post POCl3 addition | 0.10 | 74.12a | 6.77 | 1.60 | 2.58 |
| 3 | 2.5 h post POCl3 addition | 0.94 | 77.31a | 0.99 | 0.45 | 2.23 |
| 4 | 24 h after quench | 1.18 | 80.27 | 0.78 | 2.69 | 11.99 |
| 5 | crude-isolated product | 0.24 | 97.61 | 0.33 | 1.38 | 0.24 |
| 6 | mother liquors from isolation | 6.43 | 46.01 | 1.32 | 25.32 | 15.99 |
| 7 | after MeOH reslurry | 0.11 | 98.93 | 0.11 | 0.13 | 0.56 |
| 8 | after 40 °C H2O reslurry (development scale) | 0.04 | 99.86 | ndb | 0.10 | nd |
| 9 | after 40 °C H2O reslurry dried product (kilo-scale) | 0.11 | 99.55 | nd | 0.11 | nd |
Indirect indication of formation of 7 by microquench of the aliquot prior to HPLC analysis.
Not detected.
Stoichiometry optimization indicated a smooth and complete conversion of 4 to the phosphorodichloridate intermediate 7 (Scheme 3) with 1.5 equiv POCl3. A key optimization was the observation that including celite in the reaction facilitated the formation of a stirrable mixture, which helped to circumvent the problematic insoluble sticky precipitate encountered in initial reactions. The reaction was monitored at various stages by performing a microquench on the aliquoted reaction mixture, then analyzing for 6 by HPLC, which indicated that consumption of 4 and the initial formation of the labile phosphorodichloridate intermediate 7 was very fast and typically near completion in the first 10 min, with undesirable side reactions also occurring relatively quickly thereafter (Table 1, entries 1–3). Stressing experiments indicated that the reaction should optimally be held for no more than 2 h, and that proceeding directly to the hydrolytic quenching step with minimal delay in hold time was critical to minimize unwanted side reactions.
Scheme 3. Direct Phosphorylation Showing intermediate 7 and Plausible Mechanism of Formation of Characterized Pyrophosphate Minor Impurity 10.
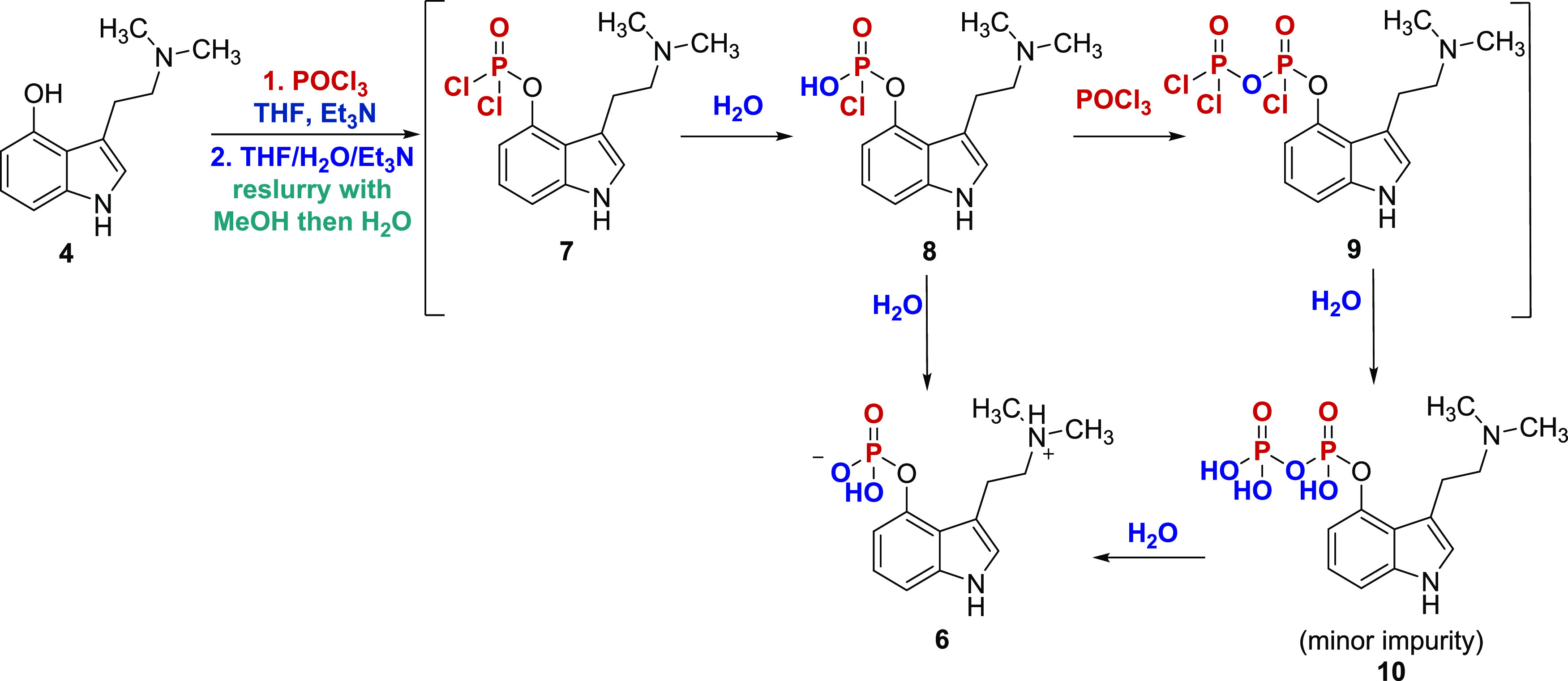
The hydrolysis step was completed by quenching the crude mixture containing primarily intermediate 7 into a premixed solution of 30% aqueous THF containing 6 equiv of triethylamine at ≤0 °C. Prolonged holds of the quenched reaction mixture (up to 72 h) caused slight degradation and hydrolysis of psilocybin back to psilocin. Typically, this was about 3% conversion after 24 h (Table 1, entry 4) and up to 15% after a 72 h hold, though high psilocin contamination was found to be tolerated and purged in downstream processing. Following the quench and hold time of at least 60 min, the resulting slurry was filtered, and the filter cake was washed with water to extract remaining 6 adhered to the celite to provide a biphasic filtrate. The upper organic phase typically contained residual 4 and was removed, with the majority of 6 in the lower aqueous phase. The aqueous phase was transferred back to the reactor, and the crude psilocybin was precipitated by addition of isopropanol followed by distillation of the solvent to a minimum volume. Crude psilocybin solid was subsequently collected by filtration from the aqueous slurry in typically 50–55% yield and 98% purity with psilocin as the highest-level impurity (Table 1, entry 5), where most of the psilocin and other process impurities were purged and remained in the filtrate (Table 1, entry 6).
Early patent data on psilocybin had indicated boiling water or methanol as useful recrystallization solvents.35 With concern for the stability of psilocybin in boiling water for the extended hold times required on large-scale, thermal stability studies on psilocybin in water at 90 °C were performed, which demonstrated about 17% hydrolysis of psilocybin back to psilocin after 1 h; at a lower temperature of 70 °C, psilocybin in water was found to hydrolyze to psilocin at a rate of 3–4% per hour. The final purification was accomplished, circumventing the hydrolysis problem, by employing sequential reslurry in methanol followed by reslurry in warm (45–55 °C) H2O to minimize degradation. After the MeOH reslurry, the purity of the crude psilocybin was typically improved to about 98.9% (Table 1, entry 7) and after filtration provided the known crystalline methanol solvate36 indicated by X-ray powder diffraction (XRPD) (see the Supporting Information). Following the warm water reslurry, the typical purity observed in development batches (up to 90 g scale) was improved to 99.9% with all impurities below the desired specifications (Table 1, entry 8). From the water reslurry, XRPD demonstrated that the psilocybin was isolated as a crystalline trihydrate; the previously reported anhydrous polymorphic form37 was subsequently obtained by drying the trihydrate at 45–55 °C for 24 h. Higher temperature or longer drying time was found to induce a slow conversion to another polymorphic form.
Although development data had indicated that the level of one earlier eluting impurity observed at RRT 0.58 (Table 1) was typically reduced during the final water reslurry step and drying process (Table 1, entry 8), during kilo-scale manufacturing the level of this impurity remained above the ICH Q3A guidance for unidentified impurities at 0.11%, where the target was ≤0.10% for individual unidentified impurities (Table 1, entry 9).38 At this juncture, the identification and characterization of the impurity at RRT 0.58 were initiated. The unidentified impurity was isolated by preparative reversed-phase HPLC and subsequently characterized by high-resolution mass spectrometry and 1H-, 13C-, 31P-NMR, all of which ultimately supported the identification of pyrophosphate structure 10 (Scheme 3) with high confidence (full experimental details described in the Supporting Information). The requirement for the excess use of POCl3 in the optimized conditions generated the pyrophosphate impurity by the putative mechanism outlined in Scheme 3. In a separate experiment, the impurity was found to hydrolyze to psilocybin in acidic aqueous environments, suggesting that it could be eliminated in future batches by a slight modification of aqueous hold times prior to product isolation. With the 0.11% pyrophosphate impurity identified, the batch met the requirements of ICH Q3A.38
Conclusions
The second-generation synthetic approach to psilocybin ultimately enabled the manufacture of 1.21 kg of API in 17% overall yield from 4-acetoxyindole with 99.7% HPLC assay purity with 31% yield for the newly developed direct phosphorylation reaction. This first production run has provided sufficient cGMP API to meet current clinical demand and represents a viable approach toward future commercial manufacture. The newly developed second-generation synthesis was designed in response to variability in the performance of known literature procedures when adapted to the required scale. The key features of the improved synthesis were an optimized three-step process to psilocin, highlighted by increased targeted purities and yields for individual products and expanded in-process controls. The final step featured a novel direct phosphorylation, which completely avoided an unacceptable protecting group strategy required by previously known procedures. The scalability, controllability, and reproducibility inherent to the newly developed procedure make it amenable to meeting current clinical and future commercial needs.
Experimental Section
3-(2-Chloro-2-oxoacetyl)-1H-indol-4-yl Acetate (2)
A solution of oxalyl chloride (4.29 kg, 33.8 mol, 1.2 equiv) in MTBE (19.8 L, 4 vol) was prepared and cooled to 0–10 °C. Another solution of 4-acetoxyindole (4.94 kg, 28.2 mol, 1 equiv) in MTBE (29.6 L, 6 vol) was prepared by stirring at 20–25 °C for 30–45 min. This was added to the oxalyl chloride solution over 60–75 min, maintaining the internal temperature within the range 0–10 °C. A bright yellow precipitate was observed ca. one-fifth way through addition, which remained throughout. The funnel was rinsed with MTBE (1.98 L, 0.4 vol) at the end of the addition. The reaction was maintained at 0–10 °C for 2–4 h, and reaction completion was monitored by HPLC targeting ≤2% area remaining 4-acetoxyindole. Heptane (26.7 L, 5.4 vol) was charged at a steady rate over 45–60 min at 0–5 °C to the reaction mixture and stirred for 60–75 min at 0–5 °C. The precipitated yellow solid was filtered under a blanket of nitrogen, and the cake was washed at 20–25 °C with MTBE/heptane (1:3) (24.68 L, 5 vol). Remaining oxalyl chloride in the wet cake was analyzed by aniline derivatization of oxalyl chloride in the wash (targeted <1% in the wash). The product was dried for at least 30 min under N2 atmosphere on the filter, and the material (92% yield, 6.86 kg, 98.9% area HPLC) was used as is in the subsequent step.
3-(2-(Dimethylamino)-2-oxoacetyl)-1H-indol-4-yl Acetate (3)
Tetrahydrofuran (73 L, 10.7 vol) was charged to the vessel containing 3-(2-chloro-2-oxoacetyl)-1H-indol-4-yl acetate (6.81 kg, 25.6 mol, 1 equiv) under N2 atmosphere and stirred until complete dissolution. The mixture was cooled to 0–5 °C under a nitrogen atmosphere. A solution of 2 M dimethylamine in THF (13.1 kg, 30.7 mol, 1.2 equiv) was added over 1 h at 0–10 °C via a dip pipe (to prevent dimethylamine hydrochloride precipitating in the reactor headspace). The solution turned to a slurry during the addition. Triethylamine (3.19 kg, 31.53 mol, 1.23 equiv) diluted in tetrahydrofuran (12.3 L, 1.8 vol) was charged over at least 30 min at 0–10 °C followed by tetrahydrofuran (12.3 L, 1.8 vol) to rinse the addition line. The mixture was warmed to 15–20 °C and stirred for at least 3 h. Heptane (90.7 L, 13.3 vol) was charged over at least 30 min at 15–20 °C (a stirrable suspension is afforded). The mixture was cooled to 0–5 °C over at least 30 min and maintained within this temperature range for at least 1 h. The mixture was filtered under a N2 blanket (prevents tendency to darken at the surface) and washed with heptane at 15–20 °C (27.3 L, 2 vol). The solid was pulled dry under vacuum and nitrogen blanket for at least 15 min until no more filtration liquors were observed. The wet cake was discharged to the vessel and 2-propanol (81.8 L, 12 vol) was added. The mixture was heated to 80–85 °C and held for 15–60 min until a complete solution was formed. The vessel was cooled down to 18–22 °C over at least 2 h (typically precipitation occurs at 65–70 °C). The slurry was further cooled to 0–5 °C over at least 30 min and maintained within this temperature range for at least 30 min. The mixture was filtered, washed with 2-propanol (13.6 L, 2 vol) at 0–10 °C, and pulled dry under vacuum for at least 15 min. The solid was washed with water (3 × 17.1 L, 3 × 2.5 vol) at 5–10 °C and then with heptane (2 × 13.6 L, 2 × 2 vol) at 10–20 °C. The solid was dried for at least 16 h in vacuo at 35–40 °C to afford the desired compound (83% yield, 5.83 kg, 99.7% area HPLC). The solid was stored at 20 °C protected from light and moisture.
(2-(Dimethylamino)ethyl)-1H-indol-4-ol (4)
To a clean, dry reactor under a nitrogen atmosphere was charged 3-(2-(dimethylamino)-2-oxoacetyl)-1H-indol-4-yl acetate (5.70 kg, 20.8 mol, 1 equiv) followed by 2-Me-THF (68.4 L, 12 vol) at 20–25 °C. The vessel was heated to 62–74 °C, and a yellow-colored slurry was observed. To the slurry, LiAlH4 (2.4 M in THF) (29.4 kg, 77.9 mol, 3.75 equiv) was carefully charged over at least 150 min, maintaining the internal temperature at 62–74 °C (target 67 °C). This charge was exothermic. The thick yellow mixture was heated to 74–80 °C and held at this temperature for at least 4 h. The vessel was cooled to 20–25 °C and THF (14.3 L, 2.5 vol) was slowly charged to dilute the reaction mixture. The reaction contents were cooled to 0–20 °C and the mixture was slowly quenched with a solution of THF/H2O (100:27) (28.5 L, 5 vol), maintaining the internal temperature at 0–20 °C. Note: the quench is very exothermic and gas evolution is observed. Typically, the reaction mixture turns to yellow/green in color. The quenched mixture was stirred for at least 15–30 min at 0–20 °C to ensure that any material stuck to the vessel walls is exposed to the quench solvent. Silica gel (5.70 kg, 1 wt) and anhydrous sodium sulfate (5.70 kg, 1 wt) were charged to the reaction mixture. The temperature of the reaction contents was adjusted to 20–25 °C. A solution of DCM/MeOH (90:10) (5.70 L, 10 vol) was charged to the reaction mixture. This charge is slightly exothermic, and the reaction mixture typically turns green in color. To a pad of celite (depth ca. 3–4 cm) and with a filter cloth on top was poured the reaction mixture and the filtrate was drummed up to avoid unnecessary air exposure. The reactor contents and celite pad were washed in turn with 2 × DCM/MeOH (90:10) (2 × 114 L, 20 vol) and the respective filtrates drummed up separately. The filtrates were poured in turn through a pad of silica (28.5 kg, 5 wt) and the pad was washed with 4 × DCM/MeOH (90:10) (4 × 114 L, 20 vol). The combined filtrates were concentrated to 5 vol, maintaining the internal temperature <50 °C. Heptane (57.0 L, 10 vol) was charged to the reactor and concentrated to 5 vol again, maintaining the internal temperature <50 °C whereupon the product precipitated. The reactor contents were heated to 35–45 °C, and diisopropyl ether (28.5 L, 5 vol) was charged to the reactor at 35–45 °C. The vessel was cooled to 20–25 °C over at least 2 h. The resultant yellow-colored slurry was stirred for at least 12 h. The mixture was filtered under the nitrogen atmosphere and washed twice with heptane/diisopropyl ether (4:1) (11.4 L, 2 vol). The wet cake was pulled dry under the nitrogen atmosphere for at least 30 min and the solid was dried under vacuum at 30–40 °C for at least 24 h affording an off-white solid (73% yield, 3.09 kg, 94.4% area HPLC).
3-(2-(Dimethylamino)ethyl)-1H-indol-4-yl Dihydrogen Phosphate (6)
To a clean, dry reactor under a nitrogen atmosphere were charged THF (28.0 L, 10 vol) and phosphorus oxychloride (3.15 kg, 20.6 mol, 1.5 equiv) at 20–25 °C. This charge is not exothermic. The vessel was cooled to −5 to −15 °C. Separately, to a second clean, dry reactor under the nitrogen atmosphere was charged psilocin (2.80 kg, 13.7 mol, 1 equiv) and celite (2.80 kg, 1 wt) followed by THF (42.0 L, 15 vol). The resultant slurry was held at 18–25 °C for at least 2 h. The reactor contents were then cooled to 0 to −15 °C. The psilocin/celite/THF slurry was slowly charged to the POCl3 solution via the pump while maintaining the internal temperature at −15 to 0 °C. This charge is exothermic. The mixture was stirred for ideally no more than 2 h at −15 to 0 °C. Longer holds are possible if the reaction mixture is maintained at the lower end of the temperature range. During this time, to a clean reactor was prepared a quench solution of THF/H2O (70:30) (28.0 L, 10 vol) and Et3N (8.32 kg, 82.2 mol, 6 equiv). The vessel containing the quench mixture was cooled to −20 to 0 °C, and the crude psilocin reaction mixture was slowly added into the THF/H2O/Et3N solution, maintaining the internal temperature at −20 to 0 °C. THF (2 × 5.60 L, 2 × 2 vol) was charged to the psilocin reactor, cooled to 0 to −5 °C, and used as a rinse into the quench medium, maintaining the internal temperature of the quenched mixture at −20 to 0 °C. Purified water (8.40 L, 3 vol) was charged to the psilocin reactor, cooled to 2–7 °C, and used as a rinse into the quench medium, maintaining the internal temperature at −20 to 0 °C. The mixture was stirred at −20 to 0 °C for at least 60 min. The mixture was filtered and the cake was washed with water at 5–10 °C (2 × 5.60 L, 2 × 2 vol). This was to dissolve any psilocybin stuck to the celite cake, and the filtration was typically fast. The biphasic filtrate was transferred back to the reactor. A rinse with water (1.40 L, 0.5 vol) can be used as part of the transfer. The temperature was adjusted to 18–25 °C, and the lower aqueous phase was separated. The organic phase was removed. The lower aqueous phase contains psilocybin and the upper organic phase typically contains residual psilocin. The aqueous phase was transferred back to the reactor. A rinse with water (1.40 L, 0.5 vol) can be used as part of the transfer. IPA (28.0 L, 10 vol) was charged to the aqueous phase. The mixture was concentrated at <45 °C internal temp to ca. 5 vol of the remaining water. Further additions of IPA (14.0 L, 5 vol) or purified water (5.60 L, 2 vol) can be added to aid azeotropic distillation of water. Upon reaching the aqueous distillation, volume purified water (14.0 L, 5 vol) was charged at 18–25 °C and the solution was stirred for at least 24 h. Psilocybin normally precipitates at this time. The reactor contents were filtered under the nitrogen atmosphere and the cake was washed with cold (2–6 °C) purified water (2 × 5.60 L, 2 × 2 vol) and pulled dry for at least 60 min under the nitrogen atmosphere. The solid was dried at 35–45 °C under vacuum for at least 24 h. The crude psilocybin was charged to a clean, dry reactor under the nitrogen atmosphere at 20–25 °C. Methanol (10 vol, based on crude psilocybin discharge weight) was charged to the reactor at 20–25 °C and the contents were stirred for at least 12 h at 20–25 °C. The mixture was filtered under nitrogen and the cake rinsed with methanol (2 × 1.5 vol, based on crude psilocybin discharge weight) at 20–25 °C. The solid was pulled dry for at least 2 h under nitrogen then charged to a clean, dry reactor under the nitrogen atmosphere. Purified water (10 vol, based on crude psilocybin discharge weight) was charged to the reactor at 20–25 °C, and the contents were heated to 45–55 °C for at least 24 h. The contents were cooled to 20–30 °C at a rate of 10 degrees per hour and held for at least 2 h. The mixture was filtered under the nitrogen atmosphere and washed in turn with 20–25 °C purified water (1 × 1 vol, 1 × 2 vol) (based on crude psilocybin discharge weight) and pulled dry under the nitrogen atmosphere for at least 2 h. The solid was dried at 35–45 °C under vacuum for at least 24 h and subsequently at 50–60 °C (target 55 °C) under vacuum for at least 24 h. This is to convert the trihydrate form initially isolated to the desired anhydrate form A by XRPD. A white solid was afforded (31% yield, 1.21 kg, 99.7% area ultraperformance liquid chromatography (UPLC)).
Acknowledgments
We would like to thank Dr. Paul Daley of The Alexander Shulgin Research Institute for performing GC/MS analyses for the initial phases of this work and for his recommendation to explore POCl3 as a direct phosphorylation reagent.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02387.
Certificate of analysis for GMP psilocybin; psilocybin solubility data; XRD diffractograms; UPLC methodology and chromatograms; isolation and characterization of the pyrophosphate impurity (PDF)
Author Contributions
∇ R.B.K. and A.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Sherwood A. M.; Prisinzano T. E. Novel Psychotherapeutics - A Cautiously Optimistic Focus on Hallucinogens. Expert Rev. Clin. Pharmacol. 2018, 11, 1–3. 10.1080/17512433.2018.1415755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. W.; Griffiths R. R. Potential therapeutic effects of psilocybin. Neurotherapeutics 2017, 14, 734–740. 10.1007/s13311-017-0542-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris R. L.; Bolstridge M.; Rucker J.; Day C. M.; Erritzoe D.; Kaelen M.; Bloomfield M.; Rickard J. A.; Forbes B.; Feilding A.; et al. Psilocybin With Psychological Support For Treatment-Resistant Depression: An Open-Label Feasibility Study. Lancet Psychiat. 2016, 3, 619–627. 10.1016/S2215-0366(16)30065-7. [DOI] [PubMed] [Google Scholar]
- Griffiths R. R.; Johnson M. W.; Carducci M. A.; Umbricht A.; Richards W. A.; Richards B. D.; Cosimano M. P.; Klinedinst M. A. Psilocybin produces substantial and sustained decreases in depression and anxiety in patients with life-threatening cancer: A randomized double-blind trial. J. Psychopharmacol. 2016, 30, 1181–1197. 10.1177/0269881116675513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S.; Bossis A.; Guss J.; Agin-Liebes G.; Malone T.; Cohen B.; Mennenga S. E.; Belser A.; Kalliontzi K.; Babb J.; et al. Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J. Psychopharmacol. 2016, 30, 1165–1180. 10.1177/0269881116675512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob C. S.; Danforth A. L.; Chopra G. S.; Hagerty M.; McKay C. R.; Halberstadt A. L.; Greer G. R. Pilot study of psilocybin treatment for anxiety in patients with advanced-stage cancer. Arch. Gen. Psychiatry 2011, 68, 71–78. 10.1001/archgenpsychiatry.2010.116. [DOI] [PubMed] [Google Scholar]
- Moreno F. A.; Wiegand C. B.; Taitano E. K.; Delgado P. L. Safety, tolerability, and efficacy of psilocybin in 9 patients with obsessive-compulsive disorder. J. Clin. Psychiatry 2006, 67, 1735–1740. 10.4088/JCP.v67n1110. [DOI] [PubMed] [Google Scholar]
- Johnson M. W.; Garcia-Romeu A.; Griffiths R. R. Long-term follow-up of psilocybin-facilitated smoking cessation. Am. J. Drug Alcohol Abuse 2017, 43, 55–60. 10.3109/00952990.2016.1170135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. W.; Garcia-Romeu A.; Cosimano M. P.; Griffiths R. R. Pilot study of the 5-HT2AR agonist psilocybin in the treatment of tobacco addiction. J. Psychopharmacol. 2014, 28, 983–992. 10.1177/0269881114548296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agin-Liebes G. I.; Malone T.; Yalch M. M.; Mennenga S. E.; Ponte K. L.; Guss J.; Bossis A. P.; Grigsby J.; Fischer S.; Ross S. Long-term follow-up of psilocybin-assisted psychotherapy for psychiatric and existential distress in patients with life-threatening cancer. J. Psychopharmacol. 2020, 34, 155–166. 10.1177/0269881119897615. [DOI] [PubMed] [Google Scholar]
- Shulgin A.; Shulgin A.. PIHKAL: A Chemical Love Story; Transform: Berkeley, CA, 1991. [Google Scholar]
- Shulgin A.; Shulgin A.. TIHKAL: The Continuation; Transform Press: Berkeley, CA, 1997. [Google Scholar]
- Nichols D. E. Hallucinogens. Pharmacol. Ther. 2004, 101, 131–181. 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Nichols D. E. Psychedelics. Pharmacol. Rev. 2016, 68, 264–355. 10.1124/pr.115.011478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samorini G. The oldest representations of hallucinogenic mushrooms in the world (Sahara Desert, 9000-7000 BP). Integration 1992, 2, 69–78. [Google Scholar]
- Brown R. T.; Nicholas C. R.; Cozzi N. V.; Gassman M. C.; Cooper K. M.; Muller D.; Thomas C. D.; Hetzel S. J.; Henriquez K. M.; Ribaudo A. S.; Hutson P. R. Pharmacokinetics of Escalating Doses of Oral Psilocybin in Healthy Adults. Clin. Pharmacokinet. 2017, 56, 1543–1554. 10.1007/s40262-017-0540-6. [DOI] [PubMed] [Google Scholar]
- Troxler F.; Seemann F.; Hofmann A. Abwandlungsprodukte von Psilocybin und Psilocin. 2. Mitteilung über synthetische Indolverbindungen. Helv. Chim. Acta 1959, 42, 2073–2103. 10.1002/hlca.19590420638. [DOI] [Google Scholar]
- Hofmann A.; Troxler F.. Esters of indoles. U.S. Patent US3,075,9921963.
- Hofmann A.; Brack A.; Kobel H.; Heim R.; Cailleux R.. Method of Inducing Therapeutic Tranquilization with Psilocybin and Psilocin. U.S. Patent US3,192,1111965.
- Sherwood A. M.; Meisenheimer P.; Tarpley G.; Kargbo R. B. An Improved, Practical, and Scalable Five-Step Synthesis of Psilocybin. Synthesis 2020, 52, 688–694. 10.1055/s-0039-1691565. [DOI] [Google Scholar]
- Shirota O.; Hakamata W.; Goda Y. Concise Large-Scale Synthesis of Psilocin and Psilocybin, Principal Hallucinogenic Constituents of “Magic Mushroom”. J. Nat. Prod. 2003, 66, 885–887. 10.1021/np030059u. [DOI] [PubMed] [Google Scholar]
- Nichols D. E. Improvements to the Synthesis of Psilocybin and a Facile Method for Preparing the O-Acetyl Prodrug of Psilocin. Synthesis 1999, 1999, 935–938. 10.1055/s-1999-3490. [DOI] [Google Scholar]
- Lenz C.; Wick J.; Braga D.; Garcia-Altares M.; Lackner G.; Hertweck C.; Gressler M.; Hoffmeister D. Injury-Triggered Blueing Reactions of Psilocybe ″Magic″ Mushrooms. Angew. Chem., Int. Ed. 2020, 59, 1450–1454. 10.1002/anie.201910175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood A. M.; Halberstadt A. L.; Klein A. K.; McCorvy J. D.; Kaylo K. W.; Kargbo R. B.; Meisenheimer P. Synthesis and Biological Evaluation of Tryptamines Found in Hallucinogenic Mushrooms: Norbaeocystin, Baeocystin, Norpsilocin, and Aeruginascin. J. Nat. Prod. 2020, 83, 461–467. 10.1021/acs.jnatprod.9b01061. [DOI] [PubMed] [Google Scholar]
- Fieser L. F.; Fieser M.. Reagents for Organic Synthesis; J. Wiley & Sons: New York, NY, 1967; Vol. 1, pp 1472. [Google Scholar]
- Basolo F.; Murmann R. K.; Chen Y. T. Dissociation Constants of Substituted Ethylenediamines. J. Am. Chem. Soc. 1953, 75, 1478–1480. 10.1021/ja01102a507. [DOI] [Google Scholar]
- Micovic V.; Mihailovic M. The reduction of acid amides with lithium aluminum hydride. J. Org. Chem. 1953, 18, 1190–1200. 10.1021/jo50015a017. [DOI] [Google Scholar]
- Ao W.; Ma X.; Lin Y.; Wang X.; Song W.; Wang Q.; Zhang X.; Xu H.; Zhang Y. Synthesis And Biological Evaluation Of Deuterated Sofosbuvir Analogs As Hcv Ns5B Inhibitors With Enhanced Pharmacokinetic Properties. J. Labelled Compd. Radiopharm. 2019, 62, 215–229. 10.1002/jlcr.3715. [DOI] [PubMed] [Google Scholar]
- Ruda G. F.; Wong P. E.; Alibu V. P.; Norval S.; Read K. D.; Barrett M. P.; Gilbert I. H. Aryl phosphoramidates of 5-phospho erythronohydroxamic acid, a new class of potent trypanocidal compounds. J. Med. Chem. 2010, 53, 6071–6078. 10.1021/jm1004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsico F.; Turshatov A.; Weber K.; Wurm F. R. A metathesis route for BODIPY labeled polyolefins. Org. Lett. 2013, 15, 3844–3847. 10.1021/ol401461h. [DOI] [PubMed] [Google Scholar]
- Harusawa S.; Shioiri T. Diethyl phosphorocyanidate (DEPC): a versatile reagent for organic synthesis. Tetrahedron 2016, 72, 8125–8200. 10.1016/j.tet.2016.09.070. [DOI] [Google Scholar]
- Granger E.; Solomianko K.; Young C.; Erb J. Exploration Of Chiral Lewis Acid Mg2+ Catalysts In The Synthesis Of Aryl Organophosphate Triesters From Phosphorus Oxychloride Through A Three-Step, Two-Pot Substitution Sequence. Tetrahedron Lett. 2018, 59, 1404–1408. 10.1016/j.tetlet.2018.02.066. [DOI] [Google Scholar]
- Kozak W.; Rachon J.; Daśko M.; Demkowicz S. Selected Methods for the Chemical Phosphorylation and Thiophosphorylation of Phenols. Asian J. Org. Chem. 2018, 7, 314–323. 10.1002/ajoc.201700638. [DOI] [Google Scholar]
- Kempson J.; Zhang H.; Wong M. K.; Li J.; Li P.; Wu D.-R.; Rampulla R.; Galella M. A.; Dabros M.; Traeger S. C.; et al. Evolution of a Scale-Up Synthesis to a Potent GluN2B Inhibitor and Its Prodrug. Org. Process Res. Dev. 2018, 22, 846–855. 10.1021/acs.oprd.8b00120. [DOI] [Google Scholar]
- Heim R.; Hofmann A.; Brack A.; Kobel H.; Cailleux R.. Obtaining Psilocybin and Psilocin From Fungal Material. U.S. Patent US3,183,1721965.
- Weber H. P.; Petcher T. J. Crystal structures of the Teonanácatl hallucinogens. Part I. Psilocybin C12H17N2O4P. J. Chem. Soc., Perkin Trans. 2 1974, 942–946. 10.1039/P29740000942. [DOI] [Google Scholar]
- Folen V. A. X-Ray Powder Diffraction Data For Some Drugs, Excipients, And Adulterants In Illicit Samples. J. Forensic Sci. 1975, 20, 348–372. 10.1520/JFS10282J. [DOI] [PubMed] [Google Scholar]
- Impurities in New Drug Products Q3A; International Conference on Harmonization (ICH).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.