

Abstract
Histone post-translational modifications (PTMs) are crucial for many cellular processes including mitosis, transcription, and DNA repair. The cellular readout of histone PTMs is dependent on both the chemical modification and histone site, and the array of histone PTMs on chromatin is dynamic throughout the eukaryotic life cycle. Accordingly, methods that report on the presence of PTMs are essential tools for resolving open questions about epigenetic processes and for developing therapeutic diagnostics. Reader domains that recognize histone PTMs have shown potential as advantageous substitutes for anti-PTM antibodies and engineering efforts aimed at enhancing reader domain affinities would advance their efficacy as antibody alternatives. Here we describe engineered chromodomains from D. melanogaster and humans that bind more tightly to H3K9 methylation (H3K9me) marks and result in the tightest reported reader:H3K9me interaction to date. Point mutations near the binding interface of the HP1 chromodomain were screened in a combinatorial fashion, and a triple mutant was found that binds 20-fold tighter than the native scaffold without any loss in PTM-site selectivity. The beneficial mutations were then translated to a human homolog, CBX1, resulting in an even tighter interaction with H3K9me3. Furthermore, we show that these engineered readers (eReaders) increase detection of H3K9me marks in several biochemical assays and outperform a commercial anti-H3K9me antibody in detecting H3K9me-containing nucleosomes in vitro, demonstrating the utility of eReaders to complement antibodies in epigenetics research.
Graphical Abstract
The organization of the eukaryotic genome into chromatin permits multiple levels of regulation of DNA-dependent processes, ranging from DNA replication to gene expression.1, 2 A major element governing chromatin architecture and function lies in the pattern of post-translational modifications (PTMs) primarily found on unstructured N-terminal regions of histone proteins, commonly referred to as the histone code.3, 4 These covalent modifications to amino acid side chains - including methylation, phosphorylation, and acylation - are recognized by histone “reader” domains, and reader-histone association directs the activity of enzymatic complexes that effect downstream biological processes.5–8 The significance of the histone code is highlighted by the numerous diseases and disorders that have been linked to aberrant PTM levels that perturb the homeostasis of epigenetic regulation.9–11 Thus, it has become increasingly clear that the ability to readily and reliably detect these modifications is crucial in order to better understand the complex functions of histone PTMs throughout the eukaryotic life cycle and in human disease.
To date, antibodies have been the primary tool used to map patterns of histone PTMs.5 Antibody-epitope interactions are commonly in the low nanomolar range, which provides robust detection of these marks. However, anti-histone PTM antibodies have several limitations that have been identified and have been documented recently.12–18 For example, the histone sequences surrounding key lysine methylation sites, such as K9 and K27 on histone 3 (H3), are highly similar, and off target recognition by anti-PTM antibodies due to low sequence selectivity is a significant issue when attempting to track and study the epigenetic outcome of a specific PTM site.12 Polyclonal anti-PTM antibodies also suffer from batch-to-batch variability, which can complicate data reproducibility.17, 19 Although this problem has been addressed with the development of monoclonal and recombinant antibodies, these alternatives can be prohibitively expensive and require routinely purchasing probes from the same company to maintain consistent PTM detection.
To overcome some of these limitations, researchers have explored using reader protein scaffolds as recognition tools for PTM detection.20–26 Reader protein families have evolved to be site-specific binders of different histone PTMs, and the mechanisms by which reader domains recognize these marks are generally well characterized.27 The recognition capability of reader scaffolds has been exploited to enrich chromatin samples for mass spectrometry analysis26, 28, to generate artificial histone PTM-dependent transcriptional activators29, 30, to visualize multiple PTMs simultaneously in stem cells31, and to employ histone interacting domains in chromatin immunoprecipitation assays.21, 22, 32, 33 While the utilization of reader domains as tools has promising potential given their small size and evolved specificity, a major limitation is the generally weak dissociation constants for their respective PTM ligands, often in the midmicromolar range.27 Efforts have been made to link multiple domains together to improve binding via avidity effects28–31, 34, however few mutagenesis studies have focused on improving the affinity of a single reader domain, thereby maintaining an advantageously small scaffold size.35
We report the development of engineered reader domains that recognize H3K9me3 to increase binding affinity, while maintaining selectivity, and thus better detection tools in histone interaction assays. Using site-directed mutagenesis focused on optimizing the noncovalent interactions at the ligand binding interface of the HP1α chromodomain from D. melanogaster, we identified a triple mutant with a 20-fold increase in affinity for H3K9me3. We utilized both histone peptide microarrays and peptide pulldowns to demonstrate that the engineered HP1α chromodomain had improved detection of H3K9me3 relative to wild-type and additionally confirmed that the activating mutations had not compromised epitope specificity. Furthermore, grafting beneficial mutations into the human ortholog CBX1 resulted in similar enhancements in ligand binding and detection in histone interaction assays. These engineered scaffolds, which we call eReaders, resulted in the tightest reader:H3K9me binding interaction reported to date and were shown to outperform an anti-H3K9me3 antibody. In sum, eReaders circumvent known issues with anti-PTM antibodies, are easily tunable to the desired application, and are readily accessible as detection reagents for epigenetics research.
RESULTS AND DISCUSSION
Rational mutagenesis of HP1α chromodomain.
To evaluate the possibility of developing an engineered reader protein as a detection reagent, we initiated our studies with the HP1α chromodomain from D. melanogaster as there have been significant mutagenesis studies probing its binding mechanism and selectivity.36–38 The HP1α chromodomain recognizes H3K9me3 with a binding affinity of 14 μM. The molecular determinants of binding consist of sequestration of the Kme3 sidechain in an aromatic cage as well as recognition of the surrounding sequence within a 3-stranded β-sheet.39, 40 Specific cross strand interactions within the β-sheet region impart selectivity for the histone sequence favoring H3K9me3 over neighboring H3K27me3, which differ by a single amino acid at the i-3 position relative to Kme3. We previously identified single point mutations in proximity to the aromatic cage and the β-sheet region that result in improved binding affinity to H3K9me3, ranging from 2-fold to 10-fold.37, 38 These mutations were selected to increase affinity through improved cross-strand interactions on the β-sheet and through optimized electrostatic interactions near the aromatic cage. Combining several of these mutations, if additive, would result in an improved affinity in the nanomolar range, suitable for many biochemical assays as a detection reagent. Accordingly, we investigated the effect of combining four different mutations in the HP1α chromodomain scaffold (Fig. 1a–b): A25 and D62 both flank the T6 position in the histone tail, making a 3-stranded β-sheet, but neither Ala nor Asp have high beta-sheet propensities41 or are known to form favorable cross-strand interactions with Thr.42, 43 Mutation to A25T and D62F had previously been shown to improve binding by 2-fold and 3-fold, respectively, through improvement of both beta-sheet propensity and cross-strand interactions (Fig. 1a).38 Additionally, mutation of two residues near the aromatic cage, K46A and E52D, also improved binding by 10-fold and 2-fold, respectively, due to optimized electrostatic interactions (Fig. 1b).37
Figure 1.
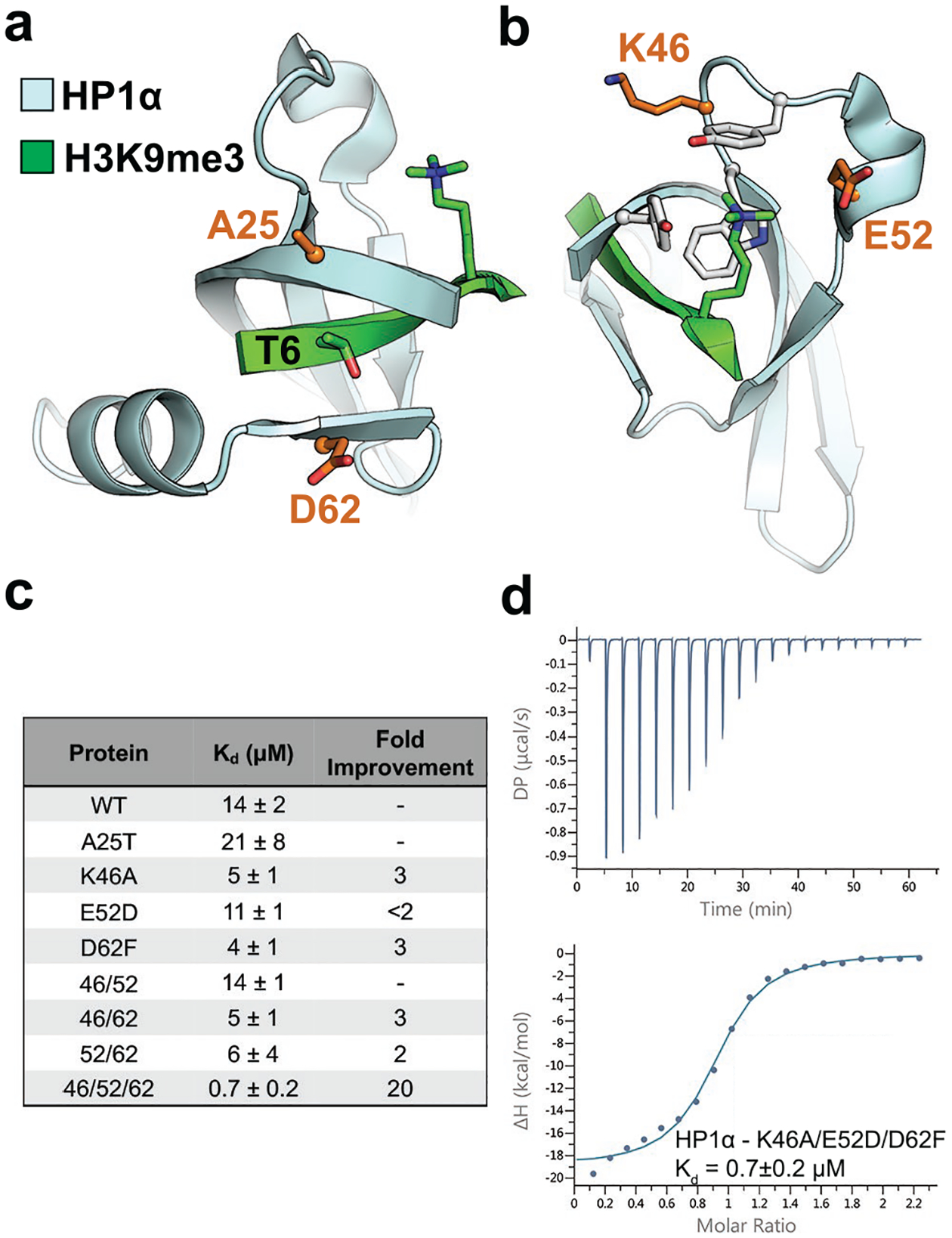
Rational mutation of the HP1α chromodomain increases binding affinity for H3K9me3. a) and b) HP1α chromodomain (cyan) recognizes H3K9me3 (green) through β-sheet formation with the histone tail and cation-π interactions with trimethylated lysine, and the mutated sites are highlighted in orange (PDB ID: 1KNE). c) Summary of results for ITC experiments for HP1α chromodomain variants showing the fold improvement in binding over wild-type. Data shown are representative of n ≥ 2 ITC binding experiments. d) ITC binding curve for the HP1α K46A/E52D/D62F engineered chromodomain.
The binding affinity of each of these single mutations had previously been measured by fluorescence anisotropy38, so we began with confirming their effects on binding using isothermal titration calorimetry (ITC, Fig. 1c). By ITC, the A25T mutation did not improve binding, and so we investigated the triple mutant, K46A/E52D/D62F, which is predicted to have a Kd of approximately 1.2 μM based on additivity of the single mutant binding affinities from ITC. While combining multiple mutations are often not synergistic, the observed Kd of the triple mutant was actually slightly tighter than expected at a value of 700 nM (Fig. 1d), which amounts to a 20-fold improvement in binding, indicating that the mutations are at least additive and perhaps weakly cooperative. Evaluation of each of the double mutants demonstrated that the triple mutant was necessary to achieve the desired sub-micromolar dissociation constant (Table S1).
A common result of introducing activating mutations in protein engineering efforts is a loss of overall protein stability. Therefore we investigated if the three beneficial mutations had destabilized the HP1α chromodomain. By circular dichroism (CD), the combinatorial mutations did not negatively affect the global fold or thermal stability of the chromodomain (Fig. S3). Having identified a variant that possessed a dissociation constant in the high nanomolar range, we investigated if the magnitude of this improvement in binding would impart greater potential for detection in histone interaction assays.
H3K9 methylation is recognized more efficiently by the engineered HP1α chromodomain.
Given that the engineered HP1α chromodomain had a tighter Kd for H3K9me3 as compared to wild-type, we next sought to evaluate if the increased affinity also affected binding to other histone modifications. To evaluate this possibility, we employed histone peptide microarrays consisting of >250 different types of histone tail modification states.44 Examination of the arrays suggested that peptides containing H3K9 methylation were bound by both wild-type and engineered domains, with the eReader binding to additional H3K9 methylated peptides and providing increased signal for shared hits (Fig. 2a and 2b). A scatter plot of the wild-type versus engineered chromodomain array data further revealed this trend, with newly visible hits bearing H3K9 methylation (Fig. 2c), and comparison of the magnitude of intensity showed that the HP1α chromodomain eReader yielded specific enhancement of binding to H3K9 methylation states over wild-type with a general trend of H3K9me3 > H3K9me2 > K3K9me1 (Fig. 2c and S9a). Importantly, mutation of the chromodomain did not incur spurious binding to H3S10p peptides nor to other types of modifications (Fig. 2c).
Figure 2.
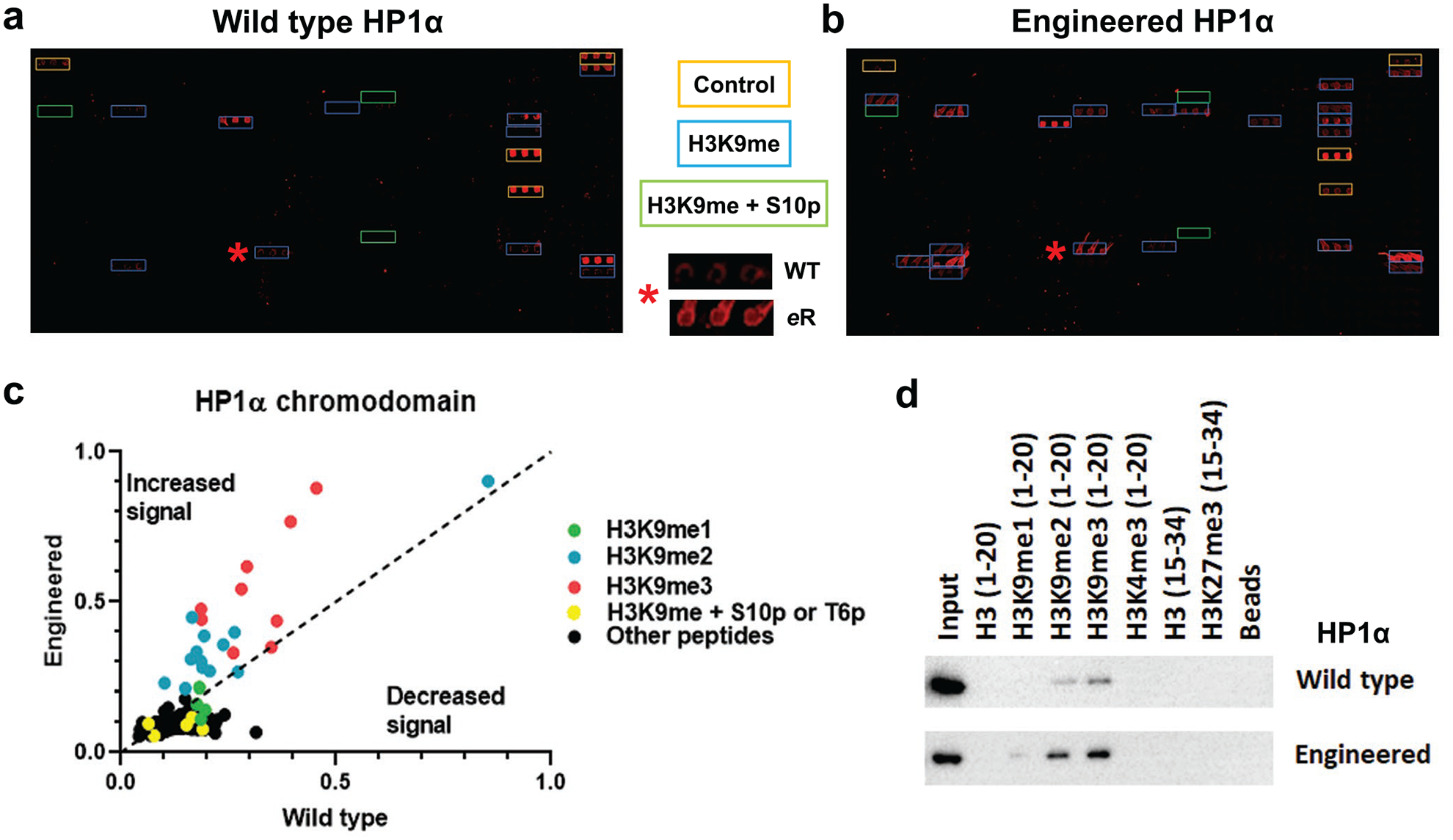
Mutation of the HP1α chromodomain increases binding to H3K9 methylation. a) and b) Images of histone peptide microarrays probed with either MBP-tagged wild-type (a) or engineered (b) HP1α chromodomain. Color coded boxes indicate peptide types and positive controls (IgG). The red star indicates a peptide (H3K9me3 containing) hit that has been zoomed in on (center panel), highlighting the change in binding between the two constructs. c) Scatter plot of the array data for HP1α chromodomains, scaled from 0–1 (further detail in Methods) and the indicated types of peptides color coded, where peptides with increased signal for the engineered protein fall above the dashed line and those with weaker signal below it. d) Western blot for peptide pulldowns of the wild-type and engineered HP1α proteins using peptides containing the known target sequences and potential off-targets for chromodomains. All arrays were performed using 500 nM of protein and all pulldowns with 5 pmol protein. All images and data are representative of n ≥ 3 experiments (subarrays or pulldowns).
To further validate these findings, we performed in solution peptide pulldowns, in which biotinylated histone tail peptides affinity-enrich binders from the sample mixture. Consistent with the arrays, we found that both proteins bind preferentially to higher methylation states of H3K9 but not to H3K4me3 or H3K27me3 (Fig. 2d). Moreover, the mutant protein bound more tightly to H3K9 methylation states compared to wild-type protein, consistent with the arrays (Fig. 2d). Taken together these data support the concept that the increased binding seen for the rationally designed HP1α chromodomain by ITC is specific to H3K9 methylation states, with a preference for H3K9me3, and that the mutations did not disrupt the specificity of the domain against other modification types.
Functional effects of HP1α mutations can be transferred to the human CBX1 chromodomain.
Although the sequence homology of chromodomain orthologs in humans that recognize H3K9me3 is about 60% of that of HP1α from D. melanogaster and the reported binding affinities and biological roles are distinct, the protein-protein binding interfaces have high sequence similarity (Fig. 3a).45 Moreover, sequence alignments indicate that the mutation sites are conserved among the CBX1, 3, and 5 chromodomains, so we investigated whether their positive effects on binding in the HP1α chromodomain would translate to the corresponding human scaffolds. Specifically, CBX1 (600 nM) and CBX3 (3 μM) are inherently tighter binders for H3K9me3, theoretically resulting in tighter affinities when compared to the engineered HP1α if the mutations are also synergistic in CBX chromodomains. Furthermore, the human orthologs are more thermally stable than HP1α, which is beneficial for both protein engineering and long-term stability for use as a biochemical tool (Fig. S5).
Figure 3.
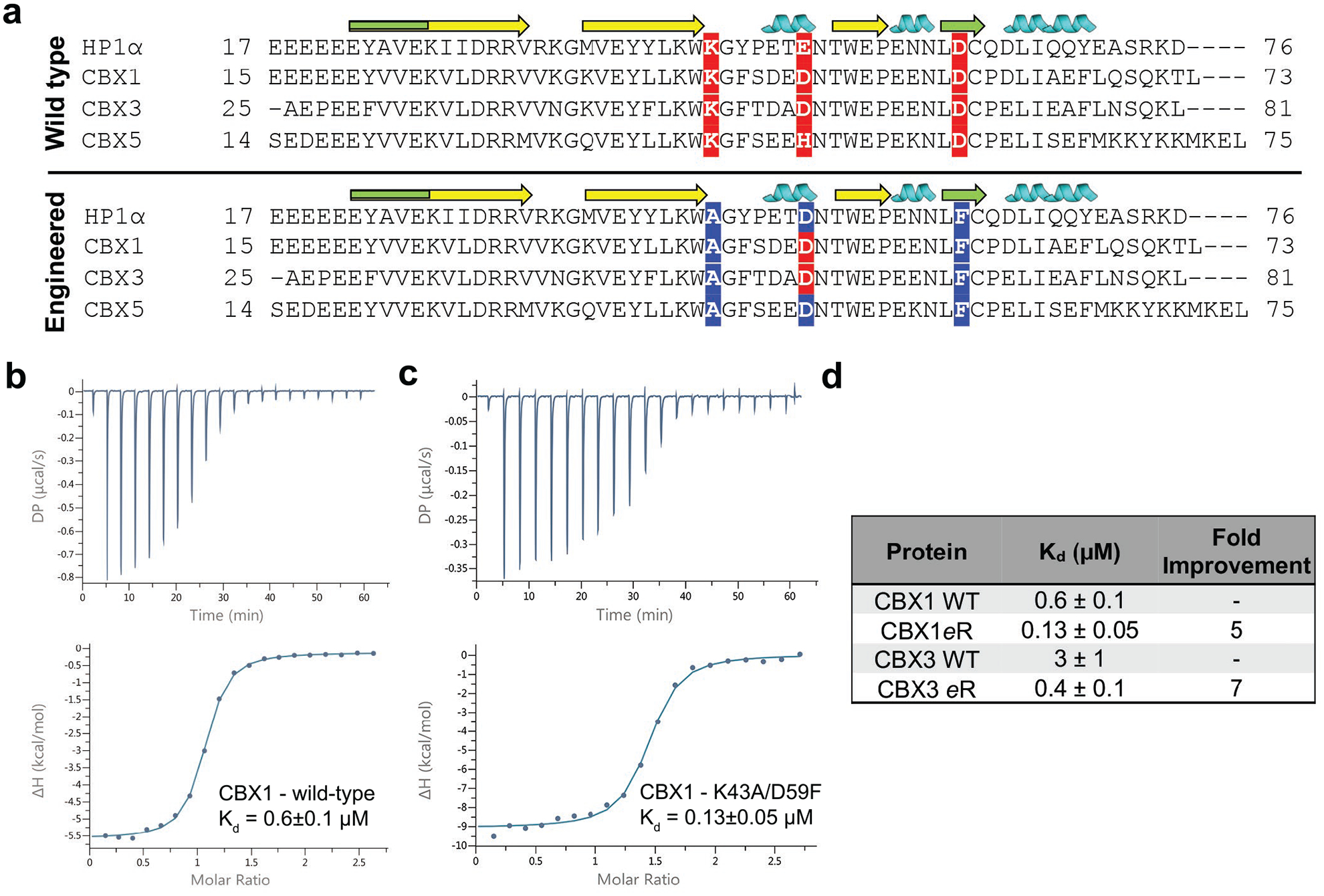
Mutations made in HP1α can be translated to the CBX1 and CBX3 chromodomains. a) Sequence alignment of wild-type Drosophila melanogaster HP1α with human homologs CBX 1,3, and 5. Green regions of the β-sheet (denoted as arrows) indicate binding interface with the histone tail. Beneficial mutations identified in HP1α along with their corresponding locations in CBX homologs are highlighted. Note that CBX1 and CBX3 are double mutants while CBX5 is a triple mutant. b) and c) ITC binding curves for the wild-type and mutant CBX1 chromodomains. d) Binding results for ITC experiments with the CBX1 and CBX3 wild-type and mutant chromodomains. Data shown are representative of ≥ 2 ITC binding experiments.
The corresponding 52 position in CBX1 and CBX3 is natively an aspartate, therefore the double mutant (K46A/D62F, HP1α numbering) was generated for both chromodomains. All three mutations were made in CBX5, however the mutations resulted in protein aggregation, so this variant was not pursued further. The changes in binding affinity for the CBX1 and CBX3 double mutants were measured by ITC, and the double mutants yielded five-fold (130 nM) and seven-fold (400 nM) improvements in binding respective to the native domains (Fig. 3b, 3c and 3d). To the best of our knowledge, the CBX1 double mutant is the tightest H3K9me3 reader protein reported to date35. Analogous to HP1α, the mutations did not significantly perturb global folding or thermal stability, and the Tm measured for the engineered CBX readers was more than 15 °C greater than the engineered HP1α. (Fig. S5).
Since the binding affinity measured for the engineered CBX1 is roughly 5-fold tighter than the engineered HP1α chromodomain, we next probed its PTM selectivity and determined whether the stronger binding would lead to further improvement in detection. Evaluation of the engineered CBX1 on histone microarrays indicates that, as before, the intensity of hits for H3K9 methylated peptides was increased upon mutagenesis. Additionally, while the total number of new peptides detected by the engineered CBX1 protein was not as large as compared to engineered HP1α, the number of visible hits is greater for both wild-type and engineered CBX1 than for HP1α, which is consistent with the ITC binding data (Fig. 4a and 4b). A scatter plot of the array data also follows a similar trend to that observed for HP1α proteins: the greatest signal was that of H3K9 methylated peptides; moreover, comparison of the wild-type and eReader domains indicates that H3K9 methylated peptides have increased signal in general, again with a bias towards H3K9me3, but not to other modifications or H3S10p modified peptides (Fig. 4c and S9b). Peptide pulldowns also indicate that the engineered CBX1 protein has enhanced interactions with H3K9 methylated peptides with a preference for H3K9me3 relative to wild-type protein (Fig. 4d).
Figure 4.
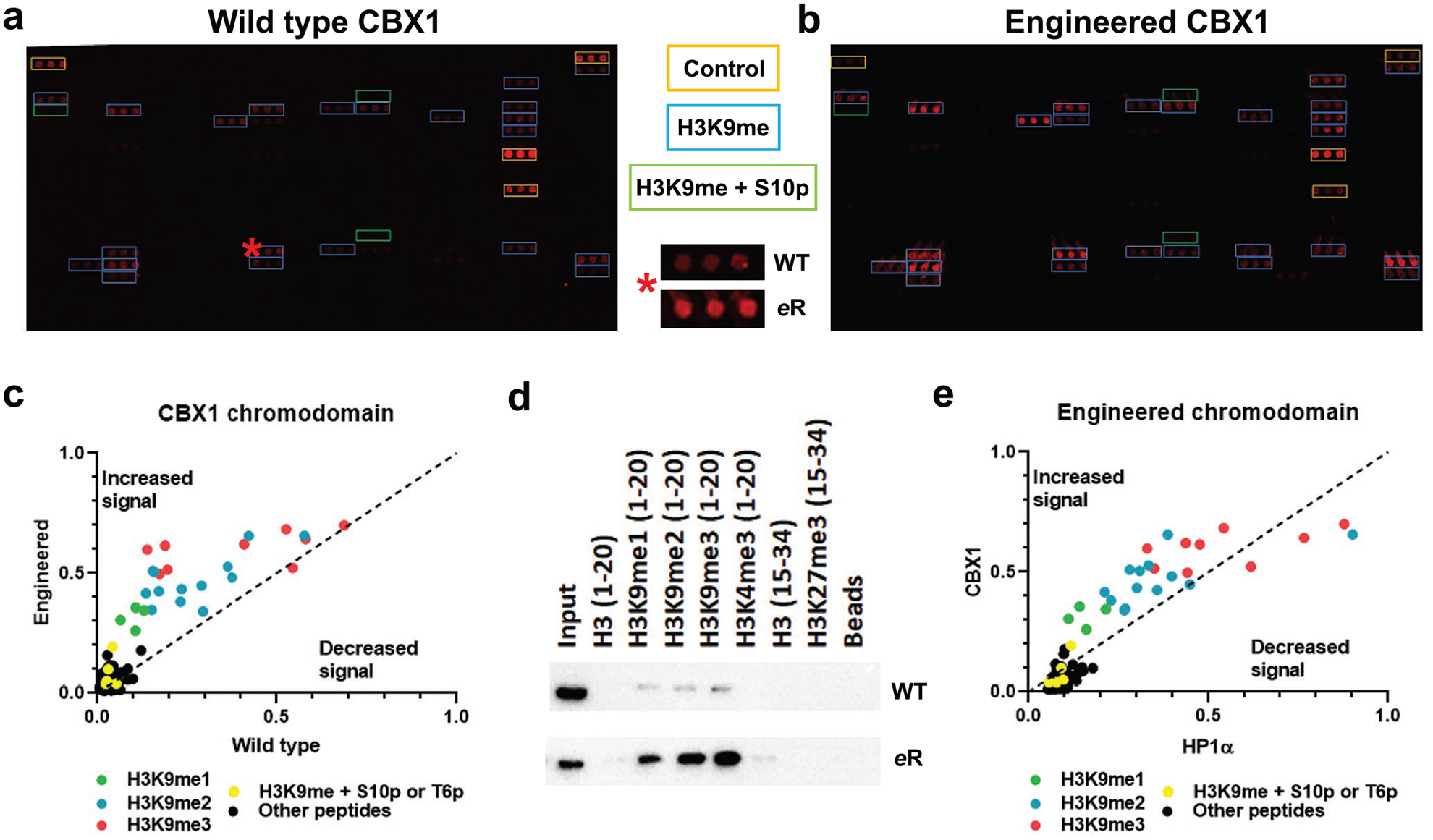
Equivalent mutation of the CBX1 chromodomain like HP1α also increases binding to H3K9 methylation. a) and b) Images of histone peptide microarrays probed with either MBP-tagged wild-type (a) or engineered (b) CBX1 chromodomain. Color coded boxes indicate peptide types and positive controls (IgG). The red star indicates a peptide (H3K9me3 containing) hit that has been zoomed in on in the center, highlighting the change in binding between the two constructs. c) Scatter plot of the array data of wild-type versus engineered CBX1 chromodomains, scaled from 0–1, with the indicated types of peptides color coded; enhanced signal of the mutant is above the dashed line and decreased signal below it. d) Images of peptide pulldowns of the wild-type and mutant CBX1 domains with the indicated peptides. e) Scatter plot of the array data of engineered HP1α versus engineered CBX1 chromodomains, scaled from 0–1, with the indicated types of peptides color coded; enhanced signal of the mutant is above the dashed line and decreased signal below it. All arrays were performed using 500 nM of protein and all pulldowns with 5 pmol protein. All images and data are representative of n ≥ 3 experiments.
Collectively, these results demonstrate that rational mutations made in the HP1α, chromodomain scaffold, are transferrable to another chromodomain, CBX1, despite low sequence homology, and these mutations yield similar trends of enhanced binding to H3K9me3 when compared to the wild-type chromodomain. Direct comparison of the array data for CBX1 and HP1α engineered domains reveals that CBX1 has greater potential to bind to H3K9 methylation, as expected based on ITC data (Fig. 4e). Because of this, we moved forward with the CBX1 eReader for further development.
Multiplexing of CBX1 wild-type or engineered chromodomains show similar enhanced H3K9me recognition.
Nature commonly utilizes multiple binding sites to interact with targets, including epitope recognition by antibodies and tandem reader domains in chromatin effectors. In previous work in which reader domains have been used as detection reagents, fusing multiple copies of the same domain was also used to increase affinity.29–31 Accordingly, we explored whether linking multiple copies of the CBX1 chromodomain would provide additional improvements beyond a single copy of the eReader.46 A (GlySer)10 linker was used to attach three wild-type or engineered CBX1 chromodomains together and the engineered 3x CBX1 eReader was compared to the 3x wild-type CBX1 on peptide arrays (Fig. 5a and b). In this platform, both show a similar degree of binding to H3K9 methylated peptides. This is not surprising, as both provide multivalency that likely dominates the 5-fold difference in Kd’s of the 1x readers. Importantly, the 3x readers do not introduce any off-target effects (Fig. 5c and S9c). These data demonstrate that multiplexing reader modules can enhance binding to the target H3K9me3 modification without inducing widespread off-target binding. eReader and multiple tandem CBX1 chromodomains have increased binding to H3K9 methylated substrates and to isolated nucleosomes as compared to an antibody.
Figure 5.
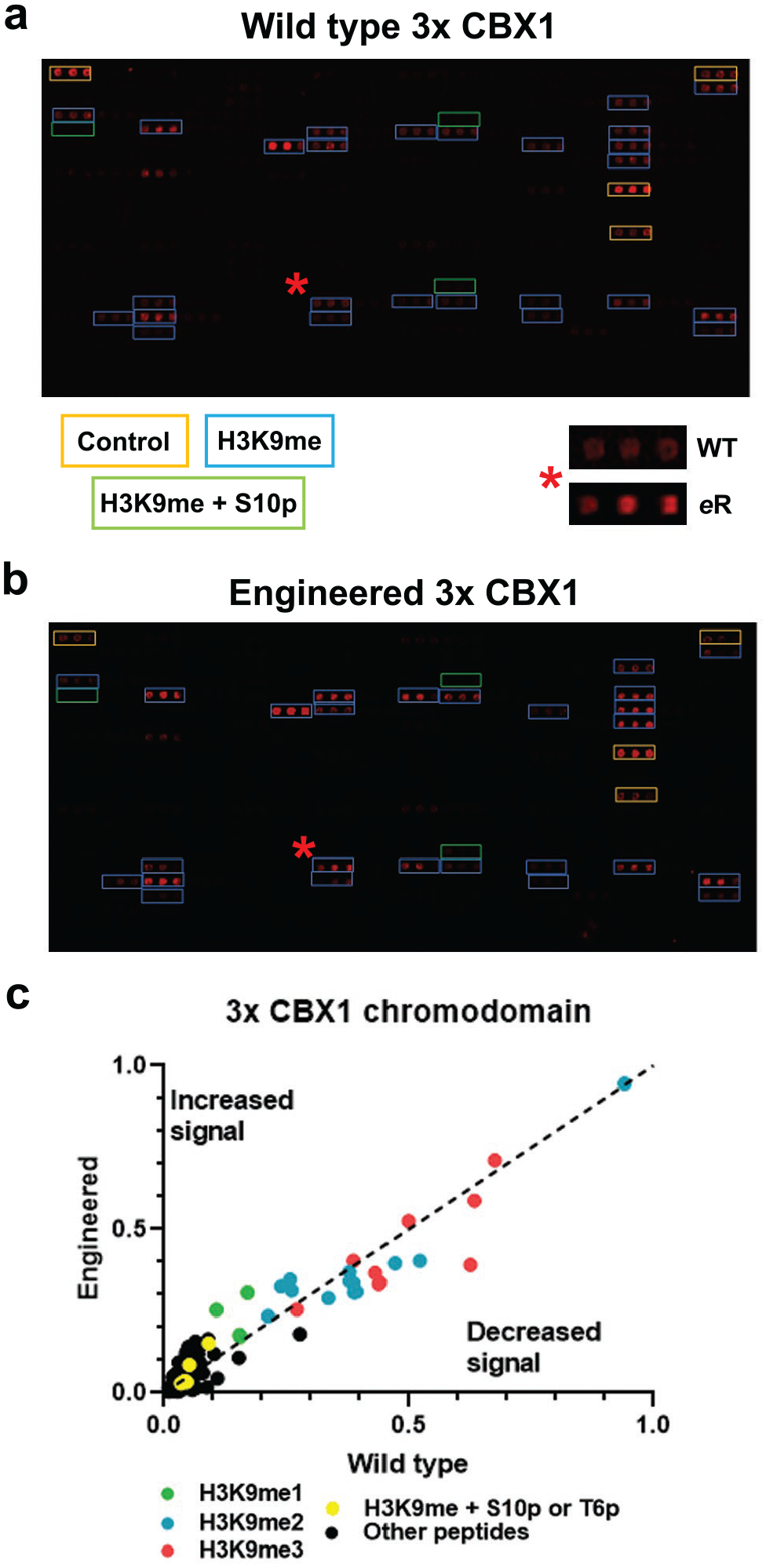
Multimerization of CBX1 wild-type and engineered chromodomains show similar binding to H3K9 methylated peptides. a) and b) Images of histone peptide microarrays probed with either MBP-tagged wild-type (a) or engineered (b) triple CBX1 chromodomains. The red star indicates a peptide (H3K9me3 containing) hit that has been zoomed in on to the right represents the change in binding between the two constructs. c) Graph of the triple reader domains with the engineered domain plotted against the wild-type construct so that enhanced signal for the mutant construct is above the dashed line and decreased signal is below the line; peptides with the specified modifications are color coded as indicated. All arrays were performed using 500 nM of protein. All images and data are representative of n ≥ 3 experiments.
To ascertain their efficacy in a more natural substrate, we used the four CBX1 scaffolds (1x and 3x, either wild-type or engineered) to probe a Western blot in which mononucleosomes derived from HeLa cells had been resolved, and compared detection to a commercially available antibody. The ability of each reader to detect histone methylation in the context of a nucleosome was evaluated at 100 nM and 10 nM of each reader protein. We found that, in agreement with other platforms using histone peptides, the CBX1 eReader was more robust than the wild-type, exhibiting detection even at 10 nM (Fig. 6a). Furthermore, the CBX1 triple reader cassettes showed the strongest binding toward nucleosomes, exhibiting an intense signal even at 10 nM. (Fig. 6a). Of note, the signal for the 3X readers at 10 nM (30 nM/reader) is more intense than 1X readers at 100 nM, strongly suggesting that avidity effects are playing a role in the enhanced binding. Significantly, the commercial ChIP-grade antibody only outperformed the single copy of wild-type CBX1 chromodomain and had weaker detection relative to all other engineered-CBX1 and multiple reader constructs (Fig. 6a). These data argue that reader domains can be engineered to provide tighter binding than is found in Nature and provide affinities that are competitive with, or even tighter than, antibodies.
Figure 6.
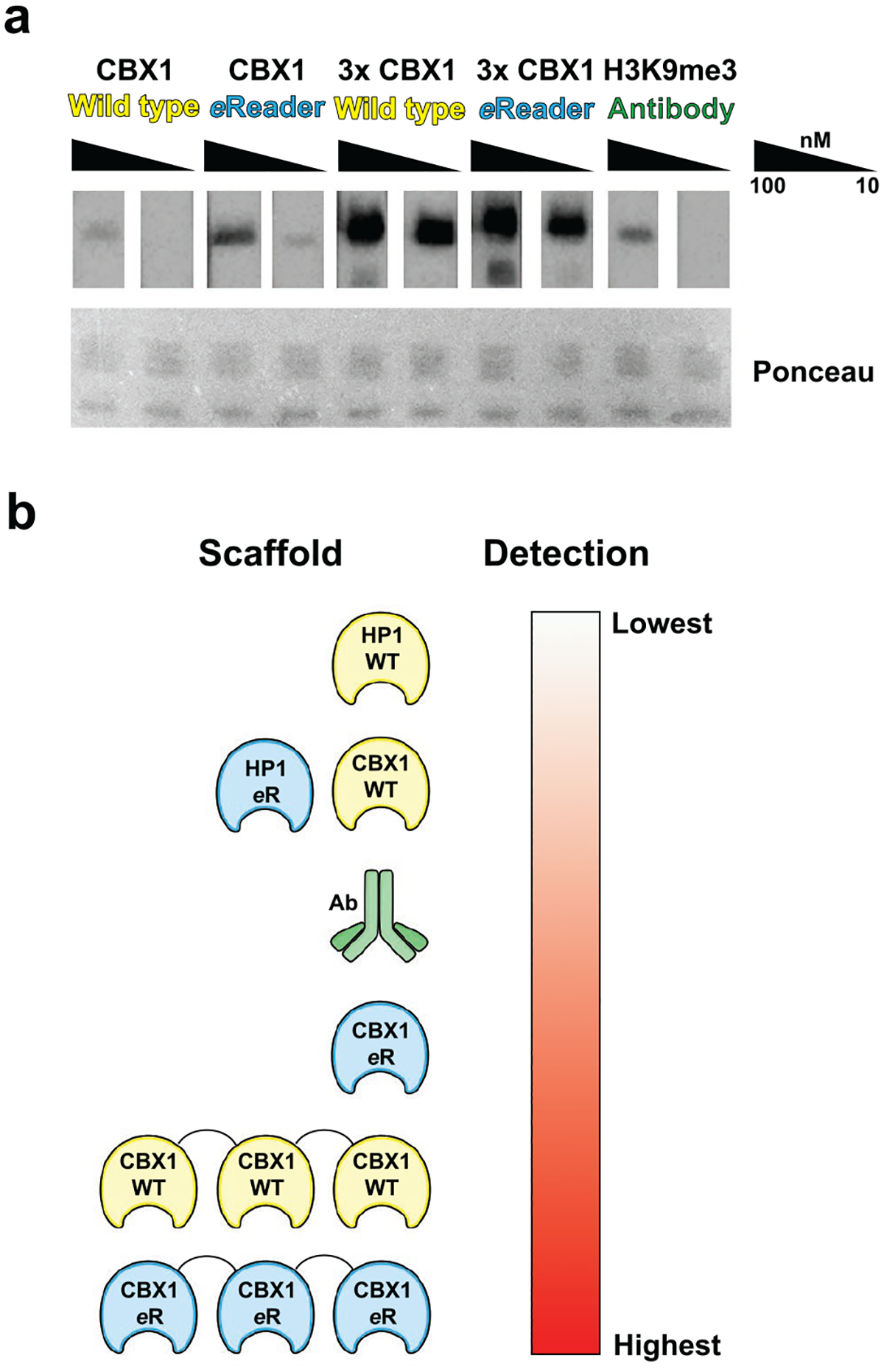
Engineered chromodomains bind to H3K9 methylated nucleosomes more effectively than wild-type or a H3K9me3 antibody. a) Nucleosome binding of the CBX1 constructs used previously as compared to a commercial anti-H3K9me3 antibody at 10 and 100 nM (upper) and Ponceau stain to show equivalent loading (lower). All images are representative of n ≥ 3 experiments. b) Summary of findings for the study, describing the increase in binding derived from mutagenesis and/or tandem arrangement of chromodomains.
CONCLUSIONS
While antibodies are the work-horse of biochemical and cellular assays, their limitations, particularly with respect to sensing histone PTMs, have come to the forefront in recent years. Several groups have attempted to use reader proteins directly as antibody replacements, but the readers generally suffer from weak binding affinities, making detection more challenging. Herein we have shown that the introduction of targeted mutations improved the binding affinity of the HP1α chromodomain towards H3K9me3 but did not incur significant changes in specificity for H3K9 methylation. These mutations were successfully translated to orthologous human CBX chromodomains, resulting in the tightest binding reader protein reported to date for H3K9me3. Additional engineering to give a 3x-concatenated construct led to further improvement in binding with no loss of selectivity. Investigation of binding to H3K9me3 in the context of whole nucleosomes indicated that the CBX1 eReader outperforms a commonly used antibody, and that the 3x constructs provide an even greater enhancement in signal. These results provide direct evidence that eReaders are a reliable and cost-effective tool that can supplement the use of commercial antibodies in a number of standard epigenetic assays. They eliminate batch-to-batch reproducibility issues and provide an inexpensive tool that can be expressed and purified using standard equipment in any biochemistry lab. We envision the single and triple domain eReaders have different advantages for various applications. For example, the single CBX1 eReaders may be advantageous in transfection-based experiments where smaller scaffold size will encourage nuclear localization. Alternatively, the 3x domains may be most beneficial for experiments where maximum sample enrichment is necessary. This work provides a starting point for developing a library of eReaders that meets the demands of epigenetic research by providing reliable, robust detection of histone PTMs and also offers more modular functionality than what is available with traditional antibodies and native reader domain scaffolds.
METHODS
Cloning of protein constructs.
The D. melanogaster HP1α chromodomain gene (residues 17–76 and an N-terminal 6xHis-tag) was previously cloned into a pET11a vector using NdeI and BamHI restriction sites. The human CBX1 (residues 15 – 73), 3 (residues 25 – 81), and 5 (residues 14 – 75) chromodomain genes were cloned into a pET28a using NdeI and XhoI. To generate N-terminal maltose binding protein (MBP) fusions, the wild-type and mutant HP1α and CBX chromodomains were cloned into a pET28a-MBP-TEV vector using BamHI and XhoI. Mutations were generated using standard QuikChange and overlap extension PCR methods. The wild-type and mutant 3x-CBX1 chromodomain genes were also cloned into a pET28a-MBP-TEV vector using BamHI and XhoI. Successful mutagenesis was confirmed by Sanger sequencing (Genewiz). Sequence alignments and percent identity were determined using Clustal2.1. See the Supporting Information for additional details including all cloned protein sequences.
Protein Expression and Purification.
The HP1α and CBX constructs were expressed in BL21-DE3 gold cells (Agilent Technologies, 230132) by inducing cells with 0.1 mM IPTG at an OD600 of ~0.6. Expressions were allowed to incubate overnight at 18°C. Cell pellets were lysed in lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 30 mM imidazole, 0.25 mg mL−1 lysozyme) by incubating at 37°C with shaking for 30 min, and then sonicated on ice for 15 minutes. Lysate was clarified by centrifugation (19,000 RPM) for 45 minutes and filtered through a 0.45 μM filter.
The His-tagged chromodomains were first purified with Ni-affinity chromatography by loading clarified lysate onto an ÄKTA Purifier UPC 10 (GE Healthcare Life Sciences) equipped with a HisTrap- 5mL HP column (GE, 17524801). The MBP fusion proteins were first purified with MBP-affinity chromatography by loading clarified lysate onto an ÄKTAPurifier UPC 10 equipped with a MBPTrap- 5mL HP column (GE, 28918779).
Eluted fractions were pooled and concentrated with a 3 kDa Amicon Ultra-15 Centrifugal filter, then passed through a size exclusion column using a Superdex 200 10/300 GL equilibrated in buffer containing 50 mM sodium phosphate, pH 8.0, 25 mM NaCl, 2 mM DTT. Eluted fractions were pooled, concentrated and run on SDS-PAGE (Fig. S1) to confirm purity and size. Proper folding of variants was verified by circular dichroism spectroscopy using an Applied Photophysics Chirascan (Fig. S3A, S5A). Further information can be found in Supporting Information.
Isothermal titration calorimetry (ITC) binding measurements.
ITC experiments were performed by titrating H3K9me3 peptide (150 μM – 3.5 mM; Fig. S2) into HP1α and CBX1 and CBX3 wild-type and variants (10 μM – 230 μM) in 50 mM sodium phosphate, pH 7.4, 150 mM NaCl, 2 mM TCEP at 25°C using a Microcal AutoITC200. Peptide and protein concentrations were determined by measuring absorbance at 280 nm on a Cary 100 UV/Vis Spectrophotometer (Agilent Technologies). Heat of dilution was accounted for by subtracting the endpoint ΔH value from each prior injection. Data was analyzed using the One-Site binding model supplied in MicroCal Analysis software (Fig. S4, Fig. S6).
Histone peptide microarrays.
Histone peptide microarrays were performed as described previously.44 The arrays were imaged using a Typhoon imaging system and quantified with ImageQuant TL. The representative images were linearly adjusted for brightness and background were similar for visual comparison. Quantification was done as described previously.44 See Supporting Information for the average and standard deviations of individual peptides.
In solution peptide pulldowns.
The in solution peptide pulldowns were performed as described previously.44 See Supporting Information for details of the modified procedure.
Nucleosome binding blots.
Nucleosome binding tests were performed using purified HeLa mononucleosomes (Epicypher, 16–0002) resolved by 15% SDS-PAGE, transferred, and probed similar to a Western blot with modifications. For each lane, 1 μg of nucleosomes was run and after transfer, lanes were separated for individual incubation with the indicated reader domain or antibody. After blocking with 5% non-fat milk in 1 X TBST (10 mM Tris, 150 mM NaCl, 0.05% Tween-20, pH 7.6) for 15 minutes, a single lane was incubated with the indicated concentration of chromodomain in blocking buffer (or blocking buffer alone for the blots used for the commercial antibody) for one hour at room temperature with shaking. The strip blots were then washed three times for five minutes with 1 X TBST and subsequently incubated with 1:1000 anti-MBP antibody (ThermoFisher, PA1–989) or the indicated concentration of anti-H3K9me3 antibody (Active Motif, 39161) for one hour at room temperature with shaking. The blots were again washed three times for five minutes with 1 X TBST, after which they were incubated with anti-Rabbit-HRP at 1:20,000 (GE, NA934V) in blocking buffer for one hour, with shaking, at room temperature. Last, the blots were washed three times for five minutes and then imaged after incubation with a chemiluminescent substrate according to the manufacturer (GE, RPN2232).
Supplementary Material
ACKNOWLEDGMENT
We acknowledge funding through a Burroughs Wellcome fellowship to K. Albanese., an NIH T32 Training Fellowship for Integrated Training in Cancer Model Systems (T32CA009156) and an American Cancer Society Postdoctoral Fellowship (PF-19-027-01-DMC) to C. Petell., an NIH grant GM118499 to M. Waters., and NIH grant GM126900 to B. Strahl.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Protein sequences, protein and peptide characterization, binding data, microarray data, peptide pulldowns, and nucleosome blots (PDF)
Competing Interests
B.D.S. acknowledges that he is a co-founder of EpiCypher.
Data and materials availability
eReader plasmids will be made available on Addgene.
REFERENCES
- 1.Ramakrishnan V (1997) Histone structure and the organization of the nucleosome, Annu. Rev. Biophys. Biomol. Struct 26, 83–112. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mader AW, Richmond RK, Sargent DF, and Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution, Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD, and Allis CD (2000) The language of covalent histone modifications, Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- 4.Rando OJ, and Chang HY (2009) Genome-wide views of chromatin structure, Annu. Rev. Biochem 78, 245–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su Z, and Denu JM (2016) Reading the Combinatorial Histone Language, ACS Chem. Biol 11, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothbart SB, and Strahl BD (2014) Interpreting the language of histone and DNA modifications, Biochim. Biophys. Acta 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres IO, and Fujimori DG (2015) Functional coupling between writers, erasers and readers of histone and DNA methylation, Curr. Opin. Struct. Biol 35, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang T, Cooper S, and Brockdorff N (2015) The interplay of histone modifications - writers that read, EMBO Rep. 16, 1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, Shurtleff SA, Mulder HL, Boggs K, Vadordaria B, Cheng J, Yergeau D, Song G, Becksfort J, Lemmon G, Weber C, Cai Z, Dang J, Walsh M, Gedman AL, Faber Z, Easton J, Gruber T, Kriwacki RW, Partridge JF, Ding L, Wilson RK, Mardis ER, Mullighan CG, Gilbertson RJ, Baker SJ, Zambetti G, Ellison DW, Zhang J, and Downing JR (2014) The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes, Nat. Commun 5, 3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson MA (2017) The cancer epigenome: Concepts, challenges, and therapeutic opportunities, Science 355, 1147–1152. [DOI] [PubMed] [Google Scholar]
- 11.Feinberg AP (2018) The Key Role of Epigenetics in Human Disease Prevention and Mitigation, N. Engl. J. Med 378, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, Krajewski K, Guo AH, Shanle EK, Josefowicz SZ, Fuchs SM, Allis CD, Magnuson TR, Ruthenburg AJ, and Strahl BD (2015) An Interactive Database for the Assessment of Histone Antibody Specificity, Mol. Cell 59, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blow N (2007) Antibodies: The generation game, Nature 447, 741–744. [DOI] [PubMed] [Google Scholar]
- 14.Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung M-S, Day DS, Gadel S, Gorchakov AA, Gu T, Kharchenko PV, Kuan S, Latorre I, Linder-Basso D, Luu Y, Ngo Q, Perry M, Rechtsteiner A, Riddle NC, Schwartz YB, Shanower GA, Vielle A, Ahringer J, Elgin SCR, Kuroda MI, Pirrotta V, Ren B, Strome S, Park PJ, Karpen GH, Hawkins RD, and Lieb JD (2011) An assessment of histone-modification antibody quality, Nat. Struct. Mol. Biol 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strahl BDF, Stephen M (2011) Antibody recognition of histone post-translational modifications: emerging issues and future prospects, Epigenomics 3, 247–249. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs SM, Krajewski K, Baker RW, Miller VL, and Strahl BD (2011) Influence of Combinatorial Histone Modifications on Antibody and Effector Protein Recognition, Curr. Biol 21, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bock I, Dhayalan A, Kudithipudi S, Brandt O, Rathert P, and Jeltsch A (2011) Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays, Epigenetics 6, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishikori S, Hattori T, Fuchs SM, Yasui N, Wojcik J, Koide A, Strahl BD, and Koide S (2012) Broad ranges of affinity and specificity of anti-histone antibodies revealed by a quantitative peptide immunoprecipitation assay, J. Mol. Biol 424, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busby M, Xue C, Li C, Farjoun Y, Gienger E, Yofe I, Gladden A, Epstein CB, Cornett EM, Rothbart SB, Nusbaum C, and Goren A (2016) Systematic comparison of monoclonal versus polyclonal antibodies for mapping histone modifications by ChIP-seq, Epigenet. Chromatin 9, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bock I, Kudithipudi S, Tamas R, Kungulovski G, Dhayalan A, and Jeltsch A (2011) Application of Celluspots peptide arrays for the analysis of the binding specificity of epigenetic reading domains to modified histone tails, BMC Biochem. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kungulovski G, Kycia I, Tamas R, Jurkowska RZ, Kudithipudi S, Henry C, Reinhardt R, Labhart P, and Jeltsch A (2014) Application of histone modification-specific interaction domains as an alternative to antibodies, Genome Res. 24, 1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kungulovski G, Mauser R, Reinhardt R, and Jeltsch A (2016) Application of recombinant TAF3 PHD domain instead of anti-H3K4me3 antibody Epigenetics & Chromatin Application of recombinant TAF3 PHD domain instead of anti-H3K4me3 antibody, Epigenet. Chromatin 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lungu C, Pinter S, Broche J, Rathert P, and Jeltsch A (2017) Modular fluorescence complementation sensors for live cell detection of epigenetic signals at endogenous genomic sites, Nat. Commun 8, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mauser R, Kungulovski G, Meral D, Maisch D, and Jeltsch A (2018) Application of mixed peptide arrays to study combinatorial readout of chromatin modifications, Biochimie 146, 14–19. [DOI] [PubMed] [Google Scholar]
- 25.Carlson SM, Moore KE, Green EM, Martín GM, and Gozani O (2014) Proteome-wide enrichment of proteins modified by lysine methylation, Nat. Protoc 9, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore KE, Carlson SM, Camp ND, Cheung P, James RG, Chua KF, Wolf-Yadlin A, and Gozani O (2013) A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation., Mol. Cell 50, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taverna SD, Li H, Ruthenburg AJ, Allis CD, and Patel DJ (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers, Nat. Struct. Mol. Biol 14, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryson BD, Del Rosario AM, Gootenberg JS, Yaffe MB, and White FM (2015) Engineered bromodomains to explore the acetylproteome, Proteomics 15, 1470–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tekel SJ, Vargas DA, Song L, Labaer J, Caplan MR, and Haynes KA (2018) Tandem Histone-Binding Domains Enhance the Activity of a Synthetic Chromatin Effector, ACS Synth. Biol 7, 842–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tekel SJ, Barrett C, Vargas D, and Haynes KA (2018) Design, Construction, and Validation of Histone-Binding Effectors in Vitro and in Cells, Biochemistry 57, 4707–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delachat AM-F, Guidotti N, Bachmann AL, Meireles-Filho ACA, Pick H, Lechner CC, Deluz C, Deplancke B, Suter DM, and Fierz B (2018) Engineered Multivalent Sensors to Detect Coexisting Histone Modifications in Living Stem Cells, Cell Chem. Biol 25, 51–56.e56. [DOI] [PubMed] [Google Scholar]
- 32.Su Z, Boersma MD, Lee JH, Oliver SS, Liu S, Garcia BA, and Denu JM (2014) ChIP-less analysis of chromatin states, Epigenet. Chromatin 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su Z, and Denu JM (2015) MARCC (Matrix-Assisted Reader Chromatin Capture): An Antibody-Free Method to Enrich and Analyze Combinatorial Nucleosome Modifications., Curr. Protoc. Mol. Biol 111, 21.32.21–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tekel SJ, and Haynes KA (2017) Molecular structures guide the engineering of chromatin, Nucleic Acids Res. 45, 7555–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hard R, Li N, He W, Ross B, Mo GCH, Peng Q, Stein RSL, Komives E, Wang Y, Zhang J, and Wang W (2018) Deciphering and engineering chromodomain-methyllysine peptide recognition, Sci. Adv 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baril SA, Koenig AL, Krone MW, Albanese KI, He CQ, Lee GY, Houk KN, Waters ML, and Brustad EM (2017) Investigation of Trimethyllysine Binding by the HP1 Chromodomain via Unnatural Amino Acid Mutagenesis, J. Am. Chem. Soc 139, 17253–17256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisert RJ, and Waters ML (2011) Tuning HP1 chromodomain selectivity for di- and trimethyllysine, ChemBioChem 12, 2786–2790. [DOI] [PubMed] [Google Scholar]
- 38.Eisert RJ, Kennedy SA, and Waters ML (2015) Investigation of the β-sheet interactions between d hp1 chromodomain and histone 3, Biochemistry 54, 2314–2322. [DOI] [PubMed] [Google Scholar]
- 39.Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis CD, and Khorasanizadeh S (2001) Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3., EMBO J. 20, 5232–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs SA, and Khorasanizadeh S (2002) Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail, Science 295, 2080–2083. [DOI] [PubMed] [Google Scholar]
- 41.Smith CK, Withka JM, and Regan L (2002) A Thermodynamic Scale for the .beta.-Sheet Forming Tendencies of the Amino Acids, Biochemistry 33, 5510–5517 [DOI] [PubMed] [Google Scholar]
- 42.Smith CK, and Regan L (1995) Guidelines for Protein Design: The Energetics of β Sheet Side Chain Interactions, Science 270, 980–982. [DOI] [PubMed] [Google Scholar]
- 43.Wouters MA, and Curmi PMG (1995) An Analysis of Side Chain Interactions and Pair Correlations Within Antiparallel P-Sheets: The Differences Between Backbone Hydrogen-Bonded and Non-Hydrogen-Bonded Residue Pairs, Proteins 22, 119–131. [DOI] [PubMed] [Google Scholar]
- 44.Petell CJ, Pham AT, Skela J, and Strahl BD (2019) Improved methods for the detection of histone interactions with peptide microarrays, Sci. Rep 9, 6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney GA, Li Z, Vedadi M, Schapira M, Min J, and Arrowsmith CH (2011) Recognition and Specificity Determinants of the Human Cbx Chromodomains, J. Biol. Chem 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma R, and Zhou MM (2015) Partners in crime: The role of tandem modules in gene transcription, Protein Sci. 24, 1347–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.