

Abstract
Pancreatic cancer is a fatal disease. The five-year survival for patients with all stages of this tumor type is less than 10%, with a majority of patients dying from drug resistant, metastatic disease. Gemcitabine has been a standard of care for the treatment of pancreatic cancer for over 20 years, but as a single agent gemcitabine is not curative. Since the only therapeutic option for the over 80 percent of pancreatic cancer patients ineligible for surgical resection is chemotherapy with or without radiation, the last few decades have seen a significant effort to develop effective therapy for this disease. This review addresses preclinical and clinical efforts to identify agents that target molecular characteristics common to pancreatic tumors and to develop mechanism-based combination approaches to therapy. Some of the most promising combinations include agents that inhibit transcription dependent on BET proteins (BET bromodomain inhibitors) or that inhibit DNA repair mediated by PARP (PARP inhibitors).
Keywords: Pancreatic cancer, BET bromodomain inhibitors, PARP inhibitors, gemcitabine
Graphical abstract
1. Introduction
Pancreatic cancer is a highly aggressive lethal neoplasm, with a 5-year survival of <10% (1). While cancer associated deaths declined ~ 27% between 1991 and 2016, the incidence of and deaths associated with pancreatic cancer have continued to increase (2). Pancreatic cancer is expected to become second only to lung cancer as the leading cause of cancer related deaths in the United States by the year 2030 (3). The greatest non-modifiable risk factor associated with pancreatic cancer is age, with a median age at diagnosis in the United States of 70 years (4). Other well recognized risk factors include smoking, obesity, and type II diabetes (4-7). Pancreatic ductal adenocarcinoma (PDAC) is the most common histological type of pancreatic cancer. Although our overall understanding of the biology of this disease continues to expand, pancreatic cancer is frequently diagnosed at late stage due to non-specific symptoms and lack of early detection methods.
In addition to environmental and lifestyle risk factors, several genetic conditions also increase risk for PDAC. These include hereditary pancreatitis and Peutz-Jeghers syndrome (8-10). PDAC tumors also commonly harbor germline or sporadic mutations in KRAS (95%), CDKN2A (90%), TP53 (75%), and SMAD4 (50%) (11-13). Additional pathways or processes frequently altered in these tumors include proteins associated with Notch, WNT, and Hedgehog signaling, as well as proteins that contribute to DNA damage checkpoints and DNA repair (14). Histologically, these tumors are characterized by a unique desmoplastic stroma tumor microenvironment which is thought to minimize access of systemically administered anti-tumor agents to the tumor, support tumor cell proliferation, and contribute to chemoresistance (15).
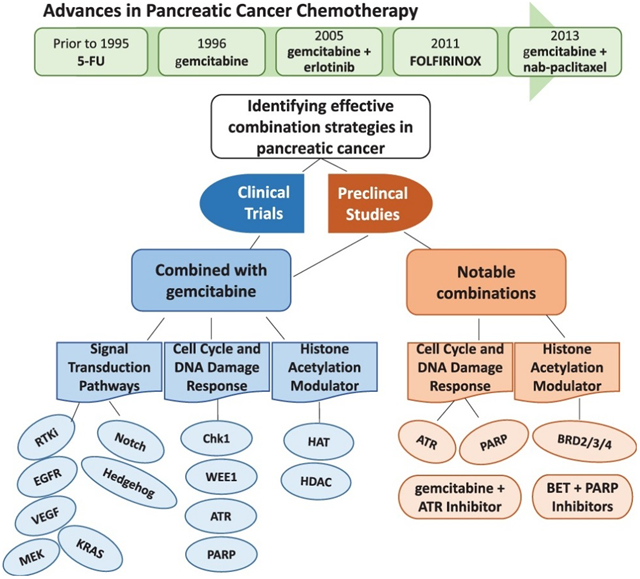
This review includes a brief history of approaches to the treatment of pancreatic cancer, a summary of the current standards of care, and a discussion of new drugs and drug combinations being evaluated preclinically and clinically. We propose that the more effective agents and combinations are those that target specific characteristics common to pancreatic tumors.
2. Standard of Care for Patients with Pancreatic Cancer
Surgical resection remains the only potentially curative treatment for patients with PDAC, but only 20% of these patients have surgically resectable disease at diagnosis (16). The remaining 80% of patients are diagnosed with locally advanced or metastatic disease and are considered ineligible for surgery. It wasn’t until the 1980s that conventional cytotoxic chemotherapeutics were introduced as standards of care for PDAC patients, when it was reported that the addition of a fluoropyrimidine such as 5-fluorouracil (5-FU) to radiation improved 1-year survival (17). Following this advance, the standard of care for these patients did not change for nearly two decades, until the nucleoside analog gemcitabine was shown to improve 1-year survival from 2% with 5-FU to 18% with gemcitabine (18). Subsequent combination therapies relied on combining other well-characterized conventional cytotoxic agents with the new standard of care, gemcitabine. Clinical trials were initiated to evaluate the efficacy and safety of gemcitabine in combination with cytotoxic agents such as cisplatin or the 5-FU prodrug capecitabine (19, 20). Although the combination of gemcitabine + cisplatin improved 6 month survival, this combination also increased the incidence of grade 3 and 4 toxicities (20). Similarly, a phase III trial in which gemcitabine + capecitabine was compared with gemcitabine alone as adjuvant therapy, the addition of capecitabine increased median overall survival from 25.5 to 28 months (P=0.032), but again with increased grade 3 and 4 toxicities (19). Recent phase I-II clinical trials demonstrate the utility of an albumin-bound form of paclitaxel (nab-paclitaxel, Abraxane) + gemcitabine in patients with advanced disease, and phase I-III trials with this combination are ongoing for patients with metastatic disease or as adjuvant therapy for patients with resectable disease (21-23). Another combination regimen that was introduced in 2010 and determined to be more effective than gemcitabine alone for the treatment of metastatic disease was FOLFIRINOX (leucovorin, fluorouracil, Irinotecan, oxaliplatin; PubChem CID: 136171075). Overall survival for patients treated with FOLFIRINOX was 11.1 months compared to 6.8 months with gemcitabine as a single agent (24). However, while the efficacy of FOLFIRINOX is greater than gemcitabine alone, the toxicity of this regimen limits its use to patients with an Eastern Cooperative Oncology Group (ECOG) performance status score of 0, 1 or 2, due to increased toxicity (24, 25).
3. Targeted Agents that Augment Gemcitabine Efficacy
In addition to identifying combinations of conventional agents that can be combined with gemcitabine to improve survival for patients with pancreatic cancer, recent studies attempt to identify targeted agents that can be effectively combined with this standard of care. This strategy has been facilitated by rapid advances in genetic, molecular, proteomic, and data analysis techniques that can be used to identify new therapeutic targets for pancreatic cancer. The following sections focus on agents that target dysfunction of pathways associated with overexpression or mutations in EGFR, VEGF, Notch, Hedgehog, MEK and KRAS proteins that occur commonly in pancreatic tumors. Table 1 summarizes combinations discussed in section 3.
Table 1.
Summary of preclinical and clinical studies of gemcitabine in combination with targeted agents to the most commonly dysregulated cell signal transduction pathways in pancreatic cancer.
| Combination | Type of Study | NCT # | Status | Reference |
|---|---|---|---|---|
| EGFR Inhibitors | ||||
| gemcitabine + erlotinib | Clinical (Phase III) | NCT00026338 | Completed | 29, 30 |
| gemcitabine + cetuximab | Clinical (Phase III) | NCT00075686 | Completed | 32,33,34 |
| VEGF Inhibitor | ||||
| gemcitabine + bevazucimab | Clinical (Phase II) | NCT00028834 | Completed | 39 |
| Notch Pathway (γ-Secretase Inhibitors) | ||||
| gemcitabine + PF-0384014 (Nirogacestat) | Preclinical | 45 | ||
| gemcitabine + MK-0752 | Clinical (Phase I) | NCT01098344 | Completed | 46 |
| Hedgehog Pathway (Smoothened inhibitors) | ||||
| gemcitabine + vismodegib | Clinical (Phase 1 b/lI) | NCT01064622 | Completed | 53 |
| gemcitabine + sonidegib | Clinical (Phase 1b) | NCT01487785 | Completed | 55 |
| gemcitabine + nab-paclitaxel + visdemogib | Clinical (Phase II) | NCT01088815 | Completed | 56 |
| sonidegib + pembrolizumab | Clinical (Phase I) | NCT04007744 | Not yet recruiting (expected to begin in 02/15/2020) | |
| MEK Inhibitors | ||||
| gemcitabine + pimasertib | Preclinical | 58 | ||
| Clinical (Phase II) | NCT01016483 | Completed | 61 | |
| gemcitabine + trametinib | Preclinical | 59, 60 | ||
| Clinical (Phase II) | NCT01231581 | Completed | 62 | |
| Prenylation Inhibitors | ||||
| gemcitabine + tipifarnib | Clinical (Phase II) | NCT00005832 | Completed | 69 |
3.1. Epidermal growth factor receptor (EGFR) inhibitors
EGFR belongs to the tyrosine kinase family of growth factor receptors, and is overexpressed in up to 95% of pancreatic tumors (26). This overexpression correlates with advanced and metastatic disease (27); and, therefore, has been evaluated as a potential therapeutic target for this tumor type. The small molecule EGFR inhibitor erlotinib (OSI-774, Tarceva) binds to the ATP binding domain of EGFR and inhibits EGFR-associated tyrosine kinase activity (28). Erlotinib (PubChem CID: 176870) was demonstrated in phase III clinical trials to increase 1-year survival of patients with pancreatic cancer from 17% with gemcitabine alone to 23% with gemcitabine + erlotinib (29). The FDA approved the use of this combination for locally advanced, unresectable or metastatic pancreatic cancer in 2005 (30).
Cetuximab (IMC-225, ABX-EGFR, PubChem SID: 46507042) is a monoclonal antibody that binds to the extracellular domain of EGFR and inhibits EGFR downstream signaling (31). Phase I, II and III clinical trials with this antibody in combination with gemcitabine did not increase overall survival (OS) (32, 33). For example, in phase III trials, a combination of cetuximab (400 mg/m2 followed by 250 mg/m2 weekly) + gemcitabine (1,000 mg/m2 weekly, for 7 weeks of an 8-week cycle and 3 weeks of each 4-week cycle) was associated with a 6.3 month median survival compared to a 5.9 month median survival with gemcitabine as a single agent (33). A recent review of similar published studies also concluded that cetuximab adds no benefit to standards of care for patients with pancreatic cancer (34).
3.2. Vascular endothelial growth factor (VEGF) inhibitor
Among VEGF members, VEGF-A is the most well characterized. VEGF-A is a secreted protein that binds to VEGF receptors (VEGFRs), primarily VEGFR-2, on endothelial cells, to facilitate downstream signaling and increase vascular permeability (35, 36). Early immunohistochemical studies show that VEGF-A is expressed in up to 65% pancreatic tumors, and that this expression correlates with local disease progression (36, 37). A second generation VEGF-A inhibitor, the recombinant humanized monoclonal antibody bevacizumab (Avastatin, PubChem SID: 46504473), binds to VEGF-A to limit the interaction of VEGF-A with VEGFR (38). Unfortunately, randomized and controlled phase III trials that included 602 patients with pancreatic cancer demonstrated that bevacizumab did not meet the designated primary end point of the study and the trial was discontinued (39). Although clinical trials have not demonstrated the utility of this monoclonal antibody for the treatment of pancreatic cancer, preclinical studies are ongoing to evaluate its potential utility in treating other tumor types (40, 41).
3.3. Notch inhibitors
In normal cells, Notch signaling plays a key role in embryonic development, cell proliferation and differentiation (42). In tumor cells, the role of Notch signaling in tumor development and progression is somewhat controversial. The literature indicates that this pathway may contribute to both oncogenesis and tumor suppression in tumor types that include pancreatic cancer (42). Notch signaling is thought to be required for Ras-dependent oncogenesis (43), and proteins involved in this pathway are expressed at higher levels in pancreatic tumors compared to normal pancreas (42). Gamma secretases (γ-secretases), a family of enzymes responsible for proteolytic processing of Notch receptors, have been investigated as potential therapeutic targets (44). Preclinical studies of one such inhibitor, PF-03084014 (nirogacestat, PubChem CID: 46224413), showed that PF-03084014 + gemcitabine suppressed the growth of subcutaneous pancreatic tumors and also suppressed metastatic tumor growth in orthotopic models of pancreatic cancer (45). PF-0384014 is in phase III clinical trials for desmoid tumors and aggressive fibromatosis; but that trial does not include patients with pancreatic cancer (NCT03785964). Data from a phase I trial in 2018 with a second γ-secretase inhibitor, MK-0752 (PubChem CID: 9803433), demonstrated that MK-0752 + gemcitabine in PDAC concluded these two agents are well tolerated by these patients at doses of MK-0752 1,800 mg/m2 weekly + gemcitabine 1,000 mg/m2 on days 1, 8,15 of a 28-day cycle (46).
3.4. Hedgehog inhibitors
The hedgehog (Hh) pathway contributes to embryonic development as well as to the development of tumors such as basal cell carcinoma (47). Three ligands activate Hedgehog signaling: Sonic hedgehog (Shh), Indian hedgehog (Ihh) and Desert hedgehog (Dhh), each of which binds to a plasma membrane receptor called patched (Ptch) to activate signaling (48). In several tumor types, Hh pathway activation contributes to tumor development (49). In pancreatic cancer, Shh a downstream target of oncogenic KRASG12D, is overexpressed and is activated by mutant KRAS (50, 51). Shh regulates the binding of the G protein-coupled receptor protein Smoothened (Smo) to Ptch1. Inhibitors of Smo would be anticipated to inactivate downstream events that facilitate cell proliferation (49). Inhibitors of Smo include vismodegib (GDC-0449, Erivedge; PubChem CID: 24776445) and sonidegib (LDE225, Odomzo; PubChem CID: 24775005). Vismodegib was approved for the treatment of advanced basal cell carcinoma in 2012 (52). However, vismodegib + gemcitabine was not superior to gemcitabine as a single agent in a phase Ib/II trial in patients with metastatic pancreatic cancer, with an overall survival of 6.3 months compared to 5.4 months for gemcitabine alone (53). Sonidegib was approved for the treatment of locally advanced basal cell carcinoma in 2015 (54). A phase Ib trial of sonidegib (400 mg daily) + gemcitabine (1,000 mg/m2 on days 1, 8, 15) on a 28-day cycle showed that although this combination was well-tolerated, median progression free survival (PFS) for this combination did not differ from that with gemcitabine alone (NCT01487785) (55). Similarly, a phase II clinical trial combining gemcitabine + nab-paclitaxel ± visdemogib in newly diagnosed patients with metastatic pancreatic cancer did not support further investigation of Hh inhibitors in this disease setting (NCT01088815) (56). Although the literature does not provide strong support for combining Hh inhibitors with gemcitabine for the treatment of pancreatic cancer, there are ongoing trials to evaluate the toxicity and efficacy of the Smo inhibitor sonidegib + the anti PD-1 monoclonal antibody pembrolizumab for patients with advanced solid tumors including metastatic and refractory PDAC (NCT04007744). The results of these trials have not yet been reported.
3.5. Mitogen-activated protein kinase (MAPK)/ extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitors
Over ninety percent of pancreatic tumors harbor KRAS mutations that constitutively activate this oncogene and contribute to pancreatic tumorigenesis and progression (57). Based on the hypothesis that targeting proteins downstream of KRAS such as RAF, MEK or ERK would inhibit tumor development and progression, inhibitors of these effectors have been developed. Preclinical studies with dual MEK1/2 kinase inhibitors demonstrate that, for example, when PDAC cells were exposed sequentially to the MEK1/2 inhibitor pimasertib (AS-703026, MSC1936369B, PubChem CID: 44187362) followed by gemcitabine, this combination was synergistic in vitro and inhibited ribonucleotide reductase subunit 1 (RRM1) (58). Further, in vivo data demonstrated that pimasertib + gemcitabine delayed the growth of orthotopic pancreatic tumors, compared to either drug as a single agent (P<0.05). Another preclinical study with the MEK1/2 inhibitor trametinib (PubChem CID: 11707110) showed that a 2-week regimen of trametinib + gemcitabine was more effective than trametinib alone in patient-derived orthotopic pancreatic cancer (59, 60). However, while preclinical data appeared encouraging, clinical trials of MEK1/2 inhibitors + gemcitabine have not supported the use of this combination. For example, a randomized phase II trial of pimasertib (AZD6244, PubChem CID: 10127622) + gemcitabine as front line treatment for metastatic pancreatic cancer patients did not meet its primary end point for progression free survival (PFS) (NCT01016483) (61). Another trial comparing trametinib + gemcitabine with gemcitabine alone showed a similar result: trametinib + gemcitabine did not improve overall survival (OS), PFS or duration of response (DOR), compared to gemcitabine alone (NCT01231581) (62). The literature indicates that MEK inhibitors + gemcitabine does not warrant further clinical evaluation for treatment of patients with pancreatic cancer.
3.6. Prenylation inhibitors
Prenylation is a posttranslational modification of proteins such as GTPases by farnesyl or geranylgeranyl transferases (63). Farnesyltransferase belongs to the family of prenylation enzymes (64). Farnesylation of Ras proteins, including KRAS, is required for protein function, and oncogenic KRAS and its effector signaling contribute to pancreatic cancer initiation, development and progression (65, 66). Therefore, farnesylation was one of the first proteins proposed as a potential therapeutic target for pancreatic cancer, and inhibitors of farnesylation have been in development since the early 1990s (67). Over the last two decades, farnesylation inhibitors have been evaluated for their ability to inhibit the oncogenic function of KRAS and to induce apoptosis in pancreatic cancer cells (68). One of these inhibitors, tipifarnib (Zarnestra, R115777; PubChem CID: 159324), was evaluated in a phase II trial for pancreatic cancer patients with advanced ‘systemic therapy-naïve’ disease (69). This randomized, double-blind, placebo-controlled phase II study compared tipifarnib + gemcitabine with placebo + gemcitabine. Tipifarnib (200mg BID) was given on an oral daily dosing schedule and gemcitabine (1,000 mg/m2 weekly dosing) was administered for 7 consecutive weeks on an 8 week cycle, followed by 3 consecutive weeks on a 4 week cycle (69). This combination did not prolong overall survival, compared to placebo + gemcitabine. Evaluation of potential uses for this agent is ongoing in a phase II trial for patients with HRAS-mutant head and neck cancer (NCT02383927) (70).
Interestingly, when farnesyltransferase is inhibited KRAS is geranylgeranylated (66). Therefore, it was suggested that simultaneous inhibition of both prenylation processes might have anti-proliferative effects. To address this hypothesis, Lobell et al. (2001), for example, compared the cytotoxicity of farnesnyltransferase inhibitors FTY-I or -II and the geranylgeranyl transferase inhibitors GGTI-I or II as single agents or in combination in multiple in vitro models that included the PSN-1 pancreatic cancer cell line (65). These investigators observed potent inhibition of pancreatic cancer cell proliferation by the combination. However, subsequent preclinical in vivo studies by this group demonstrated that the combination was toxic, and anti-tumor efficacy could not be evaluated (65). More recently, Sebti et al. (2018) reported that the ‘dual farnesyl and geranylgeranyl transferase inhibitor’ FGTI-2734 (PubChem CID: 49783195) was effective in inhibiting tumor growth in KRAS mutant patient-derived xenograft models of pancreatic cancer (71). In depth clinical evaluation of this dual inhibitor in KRAS mutated pancreatic cancer is anticipated.
4. Agents targeting DNA Damage Response Pathways
Gemcitabine remains the standard of care for patients with pancreatic cancer. While gemcitabine may have multiple mechanisms of action, it is well documented to be a DNA damaging agent (72). Intuitively, effective combination therapies might include agents that augment or complement the efficacy of this front-line agent by inhibiting repair of the DNA damage induced by gemcitabine. The predominant approaches toward this end have focused on either inhibiting DNA repair itself or on inhibiting the function of cell cycle checkpoint regulators to allow progression of cells through the cell cycle even though DNA damage is present. Either approach would be envisioned to induce apoptosis, when combined with a DNA damaging agent such as gemcitabine. This section provides a rationale and summary for preclinical and clinical efforts to develop inhibitors of cell cycle regulatory proteins Chk1, WEE1, or ATR or indirect inhibition of DNA repair using PARP inhibitors that can be effectively combined with gemcitabine. Table 2 summarizes combinations discussed in section 4.
Table 2.
Summary of preclinical and clinical studies of agents targeting cell cycle checkpoint regulatory proteins and DNA damage and repair proteins.
| Combination | Type of Study | NCT # | Status | Reference |
|---|---|---|---|---|
| Chk1 Inhibitor | ||||
| gemcitabine + PD321852 | Preclinical | 74 | ||
| gemcitabine + MK-8776 + radiation | Preclinical | 75 | ||
| Clinical (Phase I) | NCT00779584 | Completed | 76 | |
| gemcitabine + LY2603618 | Clinical (Phase I/II) | NCT00839332 | Completed | 77 |
| WEE1 Inhibitor | ||||
| gemcitabine + MK-1775 (Adavosertib) + radiation | Preclinical | 80, 81 | ||
| Clinical (Phase I) | NCT02037230 | Completed | 82 | |
| ATR Inhibitors | ||||
| gemcitabine + VE-822 + radiation | Preclinical | 86 | ||
| gemcitabine + AZD6738 (Ceralasertib) | Preclinical | 87 | ||
| gemcitabine + VX-970 | Clinical (Phase I) | NCT02157792 | Active, Not recruiting | 88 |
| AZD6738 (Ceralasertib) + olaparib | Clinical (Phase II) | NCT03682289 | Recruiting | |
| PARP Inhibitors | ||||
| gemcitabine + olaparib | Clinical (Phase I) | NCT00515866 | Completed | 98 |
4.1. Checkpoint kinase 1 (Chk1) inhibitors
Phosphorylation of Chk1 at Ser317 and/or Ser345 by protein kinases activates Chk1. Activation of Chk1, in turn, deactivates CDC25A and arrests cells in the G2 phase of the cell cycle (73). DNA damaging agents, such as gemcitabine, induce phosphorylation of Chk1 and arrest cell cycle progression, to allow repair of DNA damage before progression through the cell cycle. Inhibition of Chk1 would be predicted to minimize cell cycle arrest and allow cells to proceed through the cell cycle in the presence of DNA damage, resulting in accumulation of damage and induction of apoptosis. Parsels et al. (2009) evaluated this strategy using pancreatic cancer cell line models and the small molecule Chk1 inhibitor PD-321852 in combination with gemcitabine (73). This study demonstrated that PD-321852 1) increased gemcitabine-induced apoptosis up to 17-fold at IC50 concentrations, 2) increased gemcitabine-induced levels of the DNA damage marker γH2AX (P<0.05), and 3) inhibited the formation of gemcitabine-induced RAD51 foci >10-fold (74). Further, Engelke, et al. (2013) reported that a second Chk1 inhibitor, MK-8776, sensitized homologous recombination repair proficient pancreatic cancer cell lines to gemcitabine + radiation in vitro and in vivo (75). A phase I dose escalation trial of the Chk1 inhibitor MK-8776 with or without gemcitabine for patients with advanced solid tumors recommended doses of MK-8776 200 mg/m2 + gemcitabine 1,000 mg/m2 for phase II trials (NCT00779584) (76). A subsequent phase II trial was conducted to assess the impact on overall survival of the Chk1 inhibitor LY2603618 with or without gemcitabine in 99 patients with stages II-IV pancreatic cancer, but LY2603618 + gemcitabine failed to demonstrate better outcome than LY2603618 (NCT00839332) (77). The study concluded that the data did not support further investigation of this combination for the treatment of pancreatic cancer patients.
4.2. Wee1-like protein kinase (WEE1) inhibitors
WEE1 belongs to the Ser/Thr protein kinase family that inhibits cyclin dependent kinase 1 (CDK1) activity, to prevent cells from proceeding through the G2 phase of the cell cycle (78). WEE1 is upregulated or activated by genotoxic stress as would be mediated by cytotoxic agents, and this upregulation or activation results in G2/M arrest (79). The WEE1 inhibitor MK-1775 (AZD1775, Adavosertib) sensitized DNA damage and repair (DDR)-proficient pancreatic cancer cells to gemcitabine + radiation (80).
Interestingly, Rajeshkumar et al. (2011) showed that MK-1775 + gemcitabine had greater antitumor efficacy in p53 mutated patient-derived xenograft models of pancreatic cancer than in p53 wild type models (81). The combination of MK-1775 + gemcitabine + radiation in patients with locally advanced pancreatic cancer was well tolerated in a completed phase I trial (NCT02037230) (82).
4.3. Ataxia-telangiectasia and RAD3-related (ATR) kinase inhibitors
ATR kinase contributes to the DNA damage response (DDR) pathway by detecting single- and double-strand DNA breaks and other types of genotoxic stress, and transducing signals to effector proteins to initiate DNA repair and halt cell cycle progression (83, 84). Chk1 is thought to be a direct downstream effector of ATR; therefore, an ATR inhibitor would be predicted to allow cell cycle progression in the presence of DNA damage. Prevo et al. reported that the ATR inhibitor VE-821 sensitized pancreatic cancer cells to gemcitabine (P<0.05) and to radiation (P<0.01) in vitro (85). Further, Fokas et al. showed that the ATR inhibitor VE-822 + gemcitabine + radiation delayed the progression of pancreatic tumor xenografts, compared to the gemcitabine + radiation (P<0.001) (85, 86). Wallez et al. (2018) used murine and human pancreatic cancer cell lines to demonstrate that a second ATR inhibitor, AZD6738 (Ceralasertib, PubChem CID: 54761306), inhibited Chk1 activation and augmented the effect of gemcitabine (87). Further, combined administration of gemcitabine + AZD6738 induced regression of allografts derived from KRAS- and TP53-mutant KPC mouse pancreatic cancer cell lines (87). The authors suggested further clinical investigation of this combination is warranted. AZD6738 is in a phase II trial as a single agent and also in combination with the PARP inhibitor olaparib in patients with solid tumors including all stages of pancreatic cancer (NCT03682289). Plummer et al. (2017) reported a phase I trial of ATR inhibitor VX-970 (M6620) in combination with gemcitabine in patients with advanced solid tumors including 2 pancreatic cancer patients (NCT02157792) (88). This trial is currently active.
4.4. Poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP) inhibitors
PARP proteins are involved in many essential cell functions, including cell proliferation and death (89, 90). Of particular relevance to this review is the role of PARP enzymes in DNA repair. In 2005, the Ashworth group described the utility of PARP inhibitors in BRCA mutant cancer cells, based on the concept of ‘synthetic lethality’ (91-94). Synthetic lethality refers to conditions in which cells remain viable when each of two (or more) genes is dysfunctional, but cell death occurs when these genes are simultaneously dysfunctional. BRCA1/2 and PARP proteins both have DNA repair functions: BRCA1 and BRCA2 participate in homologous recombination repair of DNA double-strand breaks and PARP proteins function as DNA repair enzymes when mutations render BRCA1/2 nonfunctional (92, 95). Therefore, inhibiting PARP would be predicted to sensitize cancer cells with BRCA mutations, and selectively target cells with this genotype.
Preclinical data from multiple laboratories provide compelling data to support this concept (92, 96). Accordingly, the PARP inhibitor olaparib (PubChem CID: 23725625) was approved for the treatment of BRCA mutated ovarian cancer in 2018 (NCT02000622) and the PARP inhibitor talazoparib was approved for metastatic breast cancer in 2019 (NCT01945775) (97). The first phase I trial with olaparib + gemcitabine in patients with advanced pancreatic cancer was reported in 2015 (98). This randomized, dose escalation study showed that olaparib (100 mg BID, days 1-14) plus gemcitabine (600 mg/m2, days 1, 8, and 15) every 4 weeks was tolerated and without unexpected toxicities. These data support further clinical evaluation of olaparib + gemcitabine for patients with pancreatic tumors. A phase III trial to evaluate olaparib monotherapy in BRCA mutant metastatic pancreatic cancer demonstrated a median progression free survival of 7.4 months, compared to 3.8 months for placebo (P=0.004) (NCT02184195) (99). Additional studies using PARP inhibitors in combination with other targeted agents are discussed below.
5. Histone Acetylation Modulators
In addition to genetic lesions such as mutations in KRAS and TP53, other factors that impact therapeutic efficacy include the family of enzymes that regulate acetylation and deacetylation of lysine residues on chromatin associated histones (100, 101). Transcription of genes is regulated in part by acetylation and deacetylation of histones, a primary determinant of chromatin structure (102). In general, acetylated histones are thought to confer a more ‘open’ structure that supports transcription (103). Recent evidence suggests that the rate of acetylation/deacetylation also contributes to efficiency of transcription (104, 105). While it seems counterintuitive that both acetylation inhibitors and deacetylation inhibitors would have antiproliferative effects, preclinical data clearly support this conclusion. This section will discuss studies that focus on agents that inhibit the function of enzymes that acetylate or deacetylate chromatin-associated histones, and will also discuss the potential utility of each type of inhibitor.
Table 3 summarizes combinations discussed in sections 5 and 6.
Table 3.
Summary of preclinical and clinical studies evaluating the therapeutic efficacy of histone acetylation modulators in pancreatic cancer.
| Combination | Type of Study | NCT # | Status | Reference |
|---|---|---|---|---|
| HAT inhibitors | ||||
| gemcitabine + C646 | Preclinical | 108 | ||
| gemcitabine + curcumin | Clinical (Phase II) | NCT00192842 | Completed, results not reported | 109 |
| HDAC Inhibitors | ||||
| vorinostat + sorafenib | Preclinical | 114 | ||
| gemcitabine + vorinostat + sorafenib + radiation | Clinical (Phase I) | NCT02349867 | Recruiting | |
| vorinostat + capecitabine + radiation | Clinical (Phase I) | NCT00983268 | Completed | 115 |
| gemcitabine + romidepsin | Clinical (Phase I) | NCT00379639 | Completed | 117 |
| gemcitabine + CI-994 (Tacedinaline) | Clinical (Phase II) | NCT00004861 | Completed | 118 |
| gemcitabine + MGCD0103 (Mocetinostat) | Clinical (Phase I/II) | NCT00372437 | Completed, results not posted | |
| BET Inhibitors | ||||
| JQ1 + olaparib | Preclinical | 127 | ||
| JQ1 + vorinostat | Preclinical | 128 | ||
5.1. Histone Acetyltransferase (HAT) inhibitors
HATs are enzymes that add acetyl groups to lysine residues on histones or other proteins (106, 107). One of the synthetic small molecule HAT inhibitors that has been evaluated in vitro using pancreatic cancer cell lines is the p300 inhibitor C646 (108). Ono et al. (2016) reported that siRNA-mediated down regulation of p300 inhibited pancreatic cancer cell proliferation and increased levels of the apoptosis markers cleaved caspase 3,8,9 and cleaved PARP; therefore, inhibition of p300 activity would be anticipated to increase apoptosis in pancreatic cancer cells. Notably, C646 enhanced gemcitabine cytotoxicity in in vitro models (108); but preclinical studies or clinical trials with C646 have not yet been reported. The natural product curcumin is also thought to inhibit p300 HAT activity (109). The results of a phase II study with pancreatic cancer patients who received curcumin + gemcitabine have also not yet been reported (NCT00192842).
5.2. Histone Deacetylase (HDAC) inhibitors
The FDA has approved four HDAC inhibitors: vorinostat, romidepsin, belinostat and panobinostat. More than fifteen additional inhibitors are in phase I-III clinical trials (110).
5.2.1. Vorinostat (suberoylanilide hydroxamic acid [SAHA], Zolinza, PubChem CID: 5311)
Vorinostat is a pan HDAC inhibitor initially approved for the treatment of cutaneous T cell lymphoma (111). Two of the first studies to evaluate vorinostat in pancreatic cancer cell lines were those of Garcia-Morales et al. in 2005 and of Arnold et al. in 2007 (112, 113). These studies demonstrated that vorinostat inhibited the proliferation of pancreatic cancer cells and induced apoptosis. In 2008, Zhang et al. demonstrated that the tyrosine kinase inhibitor sorafenib + vorinostat was synergistic in several cell line models including pancreatic cell lines (114). Information regarding vorinostat in the clinical setting is relatively limited. A phase I trial with the combination of sorafenib + vorinostat with gemcitabine and radiation in pancreatic cancer patients is ongoing (NCT02349867). Data from a second phase I trial with vorinostat (100-400 mg daily) + capecitabine (100 mg QID on the days of radiation) + radiation (total of 30 Gy) determined an maximum tolerated dose (MTD) for vorinostat of 400 mg daily, and a median overall survival of 1.1 years (95% confidence interval 0.78-1.35) for patients with advanced pancreatic cancer (115)
5.2.2. Romidepsin (Istodax, PubChem CID: 5352062)
The HDAC inhibitor romidepsin was approved for the treatment of cutaneous T-cell lymphoma in 2006 (116). A phase I dose escalation trial of romidepsin + gemcitabine with advanced solid tumors including pancreatic cancer recommended doses of romidepsin of 12 mg/m2 and of gemcitabine of 800 mg/m2 (NCT00379639) (117). The authors suggest that this combination will require additional trials to evaluate safety and efficacy.
5.2.3. CI-994 (Tacedinaline, PubChem CID: 2746)
A phase II randomized clinical trial performed for the combination of gemcitabine + CI-994 for patients with advanced pancreatic cancer showed that the combination had no advantage over gemcitabine as a single agent in this cohort of patients with advanced disease (NCT00004861) (118).
5.2.4. MGCD0103 (Mocetinostat, PubChem CID: 9865515)
Data are not yet available for the phase I/II clinical trial of MGCD0103 + gemcitabine (NCT00372437) in patients with solid tumors.
The paucity of published preclinical and clinical data precludes conclusions regarding the utility of HAT inhibitors and HDAC inhibitors as single agents or in combination with gemcitabine, for treating patients with pancreatic cancer.
6. Bromodomain and extra-terminal (BET) bromodomain inhibitors
BET bromodomain proteins bind to acetylated lysine residues on specific chromatin-associated histones (103, 119). Proteins of this family (BRD2, BRD3, BRD4, BRDT) regulate transcription by controlling the binding of BET-dependent transcriptional complexes to promoter and enhancer regions of specific genes (103, 119). BET inhibitors are considered to function as acetylated lysine (K-Ac) mimetics that competitively inhibit the association of BET proteins with histones. This inhibition minimizes the recruitment of BET-dependent transcriptional complexes and inhibits transcription of a large subset of genes (103, 119, 120). The subset of genes affect by each family member may be cell type selective. BET bromodomain inhibitors such as JQ1 and I-BET762 were initially reported about ten years ago, and numerous published studies evaluate the potential use of this agent particularly in tumor types that overexpress the oncogene c-Myc (120-122). One of the first studies to evaluate the utility of BET inhibitors in pancreatic cancer cell lines in vitro showed that JQ1 or I-BET151 inhibited tumor cell proliferation in three-dimensional collagen (123). This study also determined that these BET inhibitors decreased expression of FOSL1 and high mobility group AT-Hook 2 (HMGA2) proteins in pancreatic cancer cells. Notably, HMGA2 is reported to confer resistance to gemcitabine in pancreatic cancer cells (124, 125).
Subsequently, our laboratory reported that the BET inhibitor JQ1 suppressed the growth of five patient-derived xenograft (PDX) models of PDAC (126). Using tumor models derived from independent primary human tumor specimens, we demonstrated that JQ1 suppressed tumor growth and decreased expression of the G2/M cycle regulator CDC25B. However, JQ1 as a single agent did not induce tumor regressions, and work is ongoing to evaluate the efficacy and toxicity of JQ1 in combination with other agents. Recently published results with JQ1 + olaparib are described below.
6. 1. JQ1 (BET inhibitor, PubChem CID: 46907787) + olaparib (PARP inhibitor, PubChem CID: 23725625): Preclinical study
In the preclinical studies mentioned above in which we observed that JQ1 as a single agent inhibited the growth of early passage tumors of human origin, we also observed that a well-tolerated regimen of JQ1 increased levels of the DNA damage marker γH2AX in vivo (127). Further, and consistent with the observed increase in DNA damage, this regimen of JQ1 inhibited DNA repair proteins Ku80 and RAD51. Based on these observations, we hypothesized that simultaneous inhibition of DNA repair by decreasing expression of Ku80 and RAD51 with JQ1 and inhibition of PARP activity with targeted small molecule PARP inhibitors would comprise effective ‘therapy’ (Figure 1). Sequential administration of the PARP inhibitor olaparib followed by the BET inhibitor JQ1 suppressed pancreatic tumor growth in two PDX models, and the efficacy of the combination was greater than either drug alone (127). No toxicity was observed. These preclinical data are encouraging, and suggest that combinations of BET inhibitors + PARP inhibitors warrant further investigation.
Figure 1. Model depicting complementary cytotoxicity of PARP inhibitors (PARPi) and JQ1 (BET bromodomain inhibitor).

(a) Schematic DNA double strand break (DSB) repair pathways: NHEJ and HR. (b) PARP recruits base excision repair (BER) enzymes to SSB loci. PARP inhibitors reduce SSB BER. Cells with reduced SSB repair result in DSB. (c) JQ1-mediated inhibition of BET protein (BRD2/3/4) activity inhibits expression of DSB repair proteins Ku80 and RAD51. Decreased DSB DNA repair sensitizes cells to PARP inhibitors. Multiple ongoing preclinical studies focus on the hypothesis that simultaneous inhibition of the expression or activity of enzymes that contribute to NHEJ, HR, and BER have complementary anti-tumor efficacy. Black font = normal pathway. Red font = effect of JQ1. Blue font = effect of PARPi. (Abbreviations: NHEJ, nonhomologous end joining; HR, homologous recombination; DSB, double-strand break; SSB, single-strand break; ac, acetylation).
6. 2. JQ1 (BET inhibitor) + vorinostat (HDAC inhibitor): Preclinical study
Mazur et al. reported that the BET inhibitor JQ1 25 mg/kg twice daily + the HDAC inhibitor vorinostat (SAHA) 25 mg/kg daily increased the median survival of Kras:p53 mutant genetically engineered mouse models of pancreatic cancer, compared to vehicle (P<0.001) or JQ1 alone (P<0.05) (128). This regimen also decreased tumor volume in two patient-derived PDX models of PDAC, compared to vehicle or JQ1 alone. No systemic toxicity or weight loss was observed with JQ1 + vorinostat.
7. Other Combinations Using Targeted Agents
7.1. MEK inhibitor + PI3K/mTOR (Phosphoinositide 3 kinase/Mammalian target of rapamycin) inhibitor
Alagesan et al. (2015) evaluated the combination of a MEK and a PI3K inhibition in a Kras:p53 mutant genetically engineered mouse model (GEMM) of pancreatic cancer (129). These investigators identified the MEK1/2 inhibitor AZD6244 (Selumetinib, PubChem CID: 10127622) as an effective inhibitor of pancreatic cancer cell viability using high-throughput screening (129). Subsequently, these investigators evaluated the efficacy of AZD6244 as a single agent or in combination with the PI3K inhibitor BKM120 (Buparlisib, PubChem CID: 16654980) or GDC-0941 (Pictilisib, PubChem CID: 17755052) in GEM models of PDAC (129). The combination of AZD6244 + BKM120 delayed development of detectable PDAC tumors from 31.5 days in the control group to 71 days in the group receiving the combination. Further, AZD6244 + BKM120 or AZD6244 + GDC-0941 reduced tumor size in 80% of mice compared to either drug alone. Notably these results were obtained when treatment was delayed until mice were 12 weeks of age, to mimic treatment of late stage PDAC. These investigators concluded that simultaneous inhibition of MEK and PI3K may have utility in pancreatic cancer patient populations.
A few clinical trials have been conducted with combinations of a MEK and a PI3K inhibitor for locally advanced or metastatic solid tumors, including pancreatic cancer. Trial NCT01347866 evaluated the PI3K/mTOR inhibitor PF-05212384 (Gedatolisib, PubChem CID: 44516953) + the MEK inhibitor PD-0325901 (PubChem CID: 9826528) or PF-05212384 + irinotecan. No results are available from that study, as it was terminated subsequent to internal prioritization review but not due to safety or efficacy concerns. Another phase I dose escalation trial NCT01390818 evaluated the combination of MEK inhibitor pimasertib (AS703026, MSC1936369B) with PI3K/mTOR inhibitor voxtalisib (SAR245409, PubChem CID: 16123056) in patients with locally advanced or metastatic solid tumors. Data from this study indicated that the combination was tolerated and a phase II trial was planned (130).
7.2. MEK inhibitor + EGFR inhibitor
Based on their earlier findings that MEK inhibition activated PI3K and EGFR pathways, Mirzoeva et al. (2013) evaluated the potential utility of simultaneous inhibition of MEK and EGFR pathways (131). These investigators demonstrated that MEK inhibitor (CI1040, PubChem CID: 6918454) + the EGFR inhibitor erlotinib were synergistic in vitro in PDAC cell lines. Further, PD0325901 7.5 mg/kg daily + erlotinib 35 mg/kg daily x 34 days inhibited the growth of HPAF-II cell line-derived xenograft tumors, compared to the control group or to groups that received a single agent. The effect of the combination was regarded as additive (131). Escalation of these doses to 12.5 mg/kg of PD-0325901 and 50 mg/kg for erlotinib was not tolerated, and the study was stopped at day 18. Based on these in vitro and preclinical findings, a phase II trial was conducted with the MEK1/2 inhibitor selumetinib + the EGFR inhibitor erlotinib for patients with chemorefractory advanced pancreatic ductal adenocarcinoma (132). Results from this trial indicated that 41% of patients had stable disease for over 6 weeks, with a median progression-free survival of 1.9 months and a median overall survival of 7.3 months. While the effect of the combination in this cohort of patients with advanced disease was modest, strong supporting mechanistic, in vitro and preclinical data suggest that it may be worthwhile to evaluate similar combinations in patients with lesser stage disease.
7.3. Anti-EGFR monoclonal antibody (Cetuximab) and anti-HER-2 monoclonal antibody (Trastuzumab)
Several reports in the literature suggest a potential anti-tumor effect for cetuximab + trastuzumab. Larbouret et al. (2007) documented that EGFR (HER-1) is expressed in 45-95% and HER-2 is expressed in 43-69% of pancreatic tumors (133). Worthylake et al. (1999) showed that HER-2 overexpression activates EGFR signaling by inhibiting EGFR internalization (134). Based on the possibility that simultaneous inhibition of both EGFR (HER-1) and HER-2 receptors would be synergistic and decrease the viability of pancreatic cancer cells, Larbouret et al. (2007) used cetuximab and trastuzumab to inhibit HER-1 and HER-2 activity, respectively, in a pancreatic cancer cell line-derived xenograft model. The combination had greater anti-tumor activity than either antibody alone (133). Based on this report, a phase I - II trial (THERAPY trial) in patients with metastatic pancreatic cancer was initiated. The phase I trial escalated the dose of trastuzumab with a fixed dose of cetuximab of 400 mg/m2 on day 1, weekly (135). The primary endpoint of the phase II trial was to achieve an objective response rate (ORR) of 5-20%, but results did not meet this criterion. Investigators who conducted these phase I-II trials suggested that definitions of dose limiting toxicities for ‘non-cytotoxic agents’ such as monoclonal antibodies may require different evaluation criteria than for cytotoxic agents, as adverse events such as the cutaneous lesions associated with these antibodies are not life-threatening, and traditional dose escalation studies may be inappropriate for therapeutic antibodies.
7.4. Immune checkpoint inhibitors
An emerging field of investigation for the treatment of solid tumors focuses on inhibiting the function of proteins that suppress the immune response, thereby allowing activation of endogenous immune response pathways. This type of inhibitor is termed an immune checkpoint inhibitor. Inhibition of immune checkpoint proteins such as PD-1 allows T cell activation, to facilitate the immune suppressive and anti-proliferative functions of these cells (136, 137). Thus far, most immune checkpoint inhibitors are monoclonal antibodies. Nivolumab, pembrolizumab and pidizumab, for example, bind to and inhibit the function of the immune checkpoint protein PD-1 (programmed cell death protein-1). Durvalumab inhibits PD-L1 (programmed death-ligand 1); ipilimumab and tremelimumab inhibit CTLA-4 (cytotoxic T lymphocyte-associated protein 4) (138, 139). More than ten phase I and II trials with immune checkpoint inhibitors (nivolumab, pembrolizumab or ipilimumab) are ongoing (140). A major advantage to this approach is the specificity of the therapeutic monoclonal antibody for its target protein.
However, unlike the clinical successes seen with these inhibitors for patients with, for example, lung adenocarcinoma and breast cancer, the strategy of inhibiting immune checkpoints to allow immunologic responses has been less successful in patients with pancreatic cancer (141). For example, O’Reilly et al. (2019) reported the first phase II trial of durvalumab ± tremelimumab in patients with metastatic PDAC (142). All 64 patients that participated in that trial tolerated the combination well, but no clinical benefit was observed. The authors of that study suggested that combining an immune checkpoint inhibitor with agents having a different mechanism of action might be superior to using two immune checkpoint inhibitors (142). Multiple ongoing trials evaluate combinations of immune checkpoint inhibitors + gemcitabine to other chemotherapeutic agents (143). It would be of interest to determine if, for example, sequential administration of gemcitabine followed by an immune checkpoint inhibitor(s) would prolong stable disease or tumor regressions produced by gemcitabine. Table 4 summarizes combinations discussed in section 7.
Table 4.
Summary of preclinical and clinical studies evaluating combination of targeted agents in pancreatic cancer.
| Combination | Type of Study | NCT # | Status | Reference |
|---|---|---|---|---|
| Combination of targeted agents | ||||
| selumetinib + BKM120 (Buparlisib) or GDC-0941 (Pictilisib) | Preclinical | 129 | ||
| PF-05212384 (Gedatolisib) + PD-0325901 or PF-05212384 + irinotecan | Clinical (Phase I) | NCT01347866 | Terminated | |
| pimasertib + voxtalisib | Clinical (Phase I) | NCT01390818 | Completed | 130 |
| erlotinib + CI1040 | Preclinical | 131 | ||
| selumetinib + erlotinib | Clinical (Phase II) | NCT01222689 | Completed | 132 |
| cetuximab + trastuzumab | Preclinical | 133 | ||
| Clinical (Phase I/II) | NCT00923299 | Completed | 135 | |
| durvalumab + tremelimumab | Clinical (Phase II) | NCT02558894 | Completed | 142 |
8. Conclusion
This review provides a brief overview of preclinical and clinical efforts in the past two decades to develop effective combination therapies for the treatment of pancreatic cancer. While numerous combinations have been evaluated, gemcitabine has remained the standard of care since 1996, with FOLFIRINOX or gemcitabine + nab-paclitaxel as alternative front line combinations for patients with unresectable PDAC (144). However, most of the trials or approaches to develop novel combination therapies have been marginally successful in the clinic, and overall survival for this tumor type has improved little.
Recent laboratory studies have identified many combinations that are not worth pursuing. However, we propose that two combinations in particular appear to deserve additional evaluation. These combinations are: 1) the BET inhibitor JQ1 + the PARP inhibitor olaparib, which was evaluated in two PDX models of PDAC (see section 6.1); and 2) the ATR inhibitor AZD6738 + gemcitabine (see section 4.3). With respect to the first of these combinations, our laboratory observed that a nontoxic regimen of JQ1 + olaparib induced stable disease and was more effective than either drug as a single agent (P<0.0001 to <0.05) in PDX models of PDAC (127). More than ten phase I-II trials are ongoing to evaluate BET inhibitors for the treatment of different types of solid tumors. Trials to evaluate BET inhibitors + with other classes of agents such as PARP inhibitors are anticipated. With respect to the second of these combinations, Wallez et al. observed that the ATR inhibitor AZD6738 + gemcitabine induced regressions in PDAC allograft models, and multiple phase I-II trials are underway using ATR inhibitors such as VX-970 or AZD6738 in combination with other classes of therapeutic agents in hematologic and solid tumor settings (87). A trial with AZD6738 + gemcitabine in patients with pancreatic cancer is also anticipated.
It is well accepted that combination therapies are usually more effective than monotherapies. We propose that effective combination therapies for pancreatic cancer will result from studies that focus on identifying agents that target genetic and molecular lesions common to pancreatic tumors and that synergize with gemcitabine, the current standard of care for this tumor type.
Acknowledgments
Funding
KJY is supported by National Institutes of Health grant R01 CA208272.
Abbreviations:
- PDAC
pancreatic ductal adenocarcinoma
- 5-FU
5-fluorouracil
- EGFR
epidermal growth factor receptor
- VEGF
vascular endothelial growth factor
- MEK
mitogen-activated protein extracellular signal-regulated kinase (MAP2K)
- ERK
extracellular receptor kinase (MAPK1)
- Chk1
Checkpoint kinase 1
- CDK1
cyclin dependent kinase 1
- DDR
DNA damage and repair
- ATR
ataxia-telangiectasia and RAD3-related
- PARP
poly (adenosine diphosphate [ADP]-ribose) polymerase
- HAT
histone Acetyltransferase
- HDAC
histone Deacetylase
- BET
bromodomain and extra terminal
- PDX
patient-derived xenograft
- PI3K
phosphoinositide 3 kinase
- mTOR
mammalian target of rapamycin
- GEMM
genetically engineered mouse model
- PD-1
programmed cell death protein-1
- PD-L1
programmed death-ligand 1
Footnotes
Declaration of Competing Interest
The authors declare that there is no competing interest related to this review manuscript.
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):10. [DOI] [PubMed] [Google Scholar]
- 2.Noone AM, Howlander N, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review. National Cancer Institute; 1975–2015;Bethesda, MD. [Google Scholar]
- 3.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. [DOI] [PubMed] [Google Scholar]
- 4.Rawla P, Sunkara T, Gaduputi V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol. 2019;10(1): 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, et al. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes. 2017;66(5):1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393(4):535–545. [DOI] [PubMed] [Google Scholar]
- 7.Rebours V, Gaujoux S, d'Assignies G, Sauvanet A, Ruszniewski P, Levy P, et al. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN). Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(15):3522–3528. [DOI] [PubMed] [Google Scholar]
- 8.Becker AE, Hernandez YG, Frucht H, Lucas AL. Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World journal of gastroenterology : WJG. 2014;20(32):11182–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korsse SE, Harinck F, van Lier MG, Biermann K, Offerhaus GJ, Krak N, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. Journal of medical genetics. 2013;50(1):59–64. [DOI] [PubMed] [Google Scholar]
- 10.Rustgi AK. Familial pancreatic cancer: genetic advances. Genes & development. 2014;28(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abramson MA, Jazag A, van der Zee JA, Whang EE. The molecular biology of pancreatic cancer. Gastrointestinal cancer research : GCR. 2007;1(4 Suppl 2):S7–S12. [PMC free article] [PubMed] [Google Scholar]
- 12.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–3130. [PubMed] [Google Scholar]
- 13.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India); 2012. [PubMed] [Google Scholar]
- 16.Riall TS, Lillemoe KD. Underutilization of surgical resection in patients with localized pancreatic cancer. Ann Surg. 2007;246(2):181–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moertel CG, Frytak S, Hahn RG, O'Connell MJ, Reitemeier RJ, Rubin J, et al. Therapy of locally unresectable pancreatic carcinoma: a randomized comparison of high dose (6000 rads) radiation alone, moderate dose radiation (4000 rads + 5-fluorouracil), and high dose radiation + 5-fluorouracil: The Gastrointestinal Tumor Study Group. Cancer. 1981;48(8):1705–1710. [DOI] [PubMed] [Google Scholar]
- 18.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1997;15(6):2403–2413. [DOI] [PubMed] [Google Scholar]
- 19.Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet. 2017;389(10073):1011–1024. [DOI] [PubMed] [Google Scholar]
- 20.Ouyang G, Liu Z, Huang S, Li Q, Xiong L, Miao X, et al. Gemcitabine plus cisplatin versus gemcitabine alone in the treatment of pancreatic cancer: a meta-analysis. World J Surg Oncol. 2016;14:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer discovery. 2012;2(3):260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine. 2013;369(18):1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(34):4548–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine. 2011;364(19):1817–1825. [DOI] [PubMed] [Google Scholar]
- 25.Peixoto RD, Ho M, Renouf DJ, Lim HJ, Gill S, Ruan JY, et al. Eligibility of Metastatic Pancreatic Cancer Patients for First-Line Palliative Intent nab-Paclitaxel Plus Gemcitabine Versus FOLFIRINOX. Am J Clin Oncol. 2017;40(5):507–511. [DOI] [PubMed] [Google Scholar]
- 26.Oliveira-Cunha M, Newman WG, Siriwardena AK. Epidermal growth factor receptor in pancreatic cancer. Cancers (Basel). 2011;3(2):1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y, Oida Y, et al. Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int J Mol Med. 2003;11(3):305–309. [PubMed] [Google Scholar]
- 28.Schettino C, Bareschino MA, Ricci V, Ciardiello F. Erlotinib: an EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev Respir Med. 2008;2(2):167–178. [DOI] [PubMed] [Google Scholar]
- 29.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(15):1960–1966. [DOI] [PubMed] [Google Scholar]
- 30.Adamska A, Domenichini A, Falasca M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. 2017;18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazzarella L, Guida A, Curigliano G. Cetuximab for treating non-small cell lung cancer. Expert Opin Biol Ther. 2018;18(4):483–493. [DOI] [PubMed] [Google Scholar]
- 32.Chakravarthy AB, Tsai CJ, O'Brien N, Lockhart AC, Chan E, Parikh A, et al. A phase I study of cetuximab in combination with gemcitabine and radiation for locally advanced pancreatic cancer. Gastrointestinal cancer research : GCR. 2012;5(4):112–118. [PMC free article] [PubMed] [Google Scholar]
- 33.Philip PA, Benedetti J, Corless CL, Wong R, O'Reilly EM, Flynn PJ, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(22):3605–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forster T, Huettner FJ, Springfeld C, Loehr M, Kalkum E, Hackbusch M, et al. Cetuximab in Pancreatic Cancer Therapy: A Systematic Review and Meta-Analysis. Oncology. 2020;98(1):53–60. [DOI] [PubMed] [Google Scholar]
- 35.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nature reviews Drug discovery. 2011;10(6):417–427. [DOI] [PubMed] [Google Scholar]
- 36.Craven KE, Gore J, Korc M. Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett. 2016;381(1):201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itakura J, Ishiwata T, Friess H, Fujii H, Matsumoto Y, Buchler MW, et al. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clinical cancer research : an official journal of the American Association for Cancer Research. 1997;3(8):1309–1316. [PubMed] [Google Scholar]
- 38.Pavlidis ET, Pavlidis TE. Role of bevacizumab in colorectal cancer growth and its adverse effects: a review. World journal of gastroenterology : WJG. 2013;19(31):5051–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(22):3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castro BA, Aghi MK. Bevacizumab for glioblastoma: current indications, surgical implications, and future directions. Neurosurg Focus. 2014;37(6):E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masuda C, Yanagisawa M, Yorozu K, Kurasawa M, Furugaki K, Ishikura N, et al. Bevacizumab counteracts VEGF-dependent resistance to erlotinib in an EGFR-mutated NSCLC xenograft model. International journal of oncology. 2017;51(2):425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nature medicine. 2002;8(9):979–986. [DOI] [PubMed] [Google Scholar]
- 44.Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67(5):1879–1882. [DOI] [PubMed] [Google Scholar]
- 45.Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, Korangath P, et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013;335(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook N, Basu B, Smith DM, Gopinathan A, Evans J, Steward WP, et al. A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. British journal of cancer. 2018;118(6):793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu D, Schlotman KE, Xie J. Deciphering the role of hedgehog signaling in pancreatic cancer. J Biomed Res. 2016;30(5):353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callejo A, Culi J, Guerrero I. Patched, the receptor of Hedgehog, is a lipoprotein receptor. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(3):912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes & development. 2008;22(18):2454–2472. [DOI] [PubMed] [Google Scholar]
- 50.Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. The Journal of biological chemistry. 2007;282(19):14048–14055. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Wang Z, Ma Q, Xu Q, Liu H, Duan W, et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(16):4326–4338. [DOI] [PubMed] [Google Scholar]
- 52.Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H, et al. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(9):2289–2293. [DOI] [PubMed] [Google Scholar]
- 53.Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(36):4284–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casey D, Demko S, Shord S, Zhao H, Chen H, He K, et al. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(10):2377–2381. [DOI] [PubMed] [Google Scholar]
- 55.Macarulla T, Tabernero J, Palmer DH, Sharma S, Yu KH, Sellami DB, et al. A phase Ib dose escalation, safety, and tolerability study of sonidegib in combination with gemcitabine in patients with locally advanced or metastatic pancreatic adenocarcinoma. Journal of Clinical Oncology. 2016;34(4_suppl):371–371. [Google Scholar]
- 56.Jesus-Acosta AD, O'Dwyer PJ, Ramanathan RK, Hoff DDV, Maitra A, Rasheed Z, et al. A phase II study of vismodegib, a hedgehog (Hh) pathway inhibitor, combined with gemcitabine and nab-paclitaxel (nab-P) in patients (pts) with untreated metastatic pancreatic ductal adenocarcinoma (PDA). Journal of Clinical Oncology. 2014;32(3_suppl):257–257.24297952 [Google Scholar]
- 57.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harbor perspectives in medicine. 2018;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vena F, Li Causi E, Rodriguez-Justo M, Goodstal S, Hagemann T, Hartley JA, et al. The MEK1/2 Inhibitor Pimasertib Enhances Gemcitabine Efficacy in Pancreatic Cancer Models by Altering Ribonucleotide Reductase Subunit-1 (RRM1). Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(24):5563–5577. [DOI] [PubMed] [Google Scholar]
- 59.Kawaguchi K, Igarashi K, Miyake K, Lwin TM, Miyake M, Kiyuna T, et al. MEK inhibitor trametinib in combination with gemcitabine regresses a patient-derived orthotopic xenograft (PDOX) pancreatic cancer nude mouse model. Tissue Cell. 2018;52:124–128. [DOI] [PubMed] [Google Scholar]
- 60.Kawaguchi K, Igarashi K, Murakami T, Kiyuna T, Lwin TM, Hwang HK, et al. MEK inhibitors cobimetinib and trametinib, regressed a gemcitabine-resistant pancreatic-cancer patient-derived orthotopic xenograft (PDOX). Oncotarget. 2017;8(29):47490–47496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Cutsem E, Hidalgo M, Canon JL, Macarulla T, Bazin I, Poddubskaya E, et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. International journal of cancer Journal international du cancer. 2018;143(8):2053–2064. [DOI] [PubMed] [Google Scholar]
- 62.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. European journal of cancer. 2014;50(12):2072–2081. [DOI] [PubMed] [Google Scholar]
- 63.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annual review of biochemistry. 1996;65:241–269. [DOI] [PubMed] [Google Scholar]
- 64.Shen M, Pan P, Li Y, Li D, Yu H, Hou T. Farnesyltransferase and geranylgeranyltransferase I: structures, mechanism, inhibitors and molecular modeling. Drug Discov Today. 2015;20(2):267–276. [DOI] [PubMed] [Google Scholar]
- 65.Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61(24):8758–8768. [PubMed] [Google Scholar]
- 66.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 2000;19(56):6584–6593. [DOI] [PubMed] [Google Scholar]
- 67.Palsuledesai CC, Distefano MD. Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem Biol. 2015;10(1):51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. Oncologist. 2005;10(8):565–578. [DOI] [PubMed] [Google Scholar]
- 69.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22(8):1430–1438. [DOI] [PubMed] [Google Scholar]
- 70.Ho AL, Chau N, Bauman J, Bible K, Chintakuntlawar A, Cabanillas ME, et al. Preliminary results from a phase 2 trial of tipifarnib in squamous cell carcinomas (SCCS) with HRAS mutations. Annals of Oncology. 2018;29(Suppl_8). [Google Scholar]
- 71.Kazi A, Xiang S, Yang H, Chen L, Kennedy P, Ayaz M, et al. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019;25(19):5984–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. European journal of pharmacology. 2014;741:8–16. [DOI] [PubMed] [Google Scholar]
- 73.Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci. 2013;70(21):4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parsels LA, Morgan MA, Tanska DM, Parsels JD, Palmer BD, Booth RJ, et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Molecular cancer therapeutics. 2009;8(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Engelke CG, Parsels LA, Qian Y, Zhang Q, Karnak D, Robertson JR, et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(16):4412–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(9):1060–1066. [DOI] [PubMed] [Google Scholar]
- 77.Laquente B, Lopez-Martin J, Richards D, Illerhaus G, Chang DZ, Kim G, et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC cancer. 2017;17(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci. 2016;37(10):872–881. [DOI] [PubMed] [Google Scholar]
- 79.Lal S, Zarei M, Chand Sn, Dylgjeri E, Mambelli-Lisboa NC, Pishvaian MJ, et al. WEE1 inhibition in pancreatic cancer cells is dependent on DNA repair status in a context dependent manner. Sci Rep. 2016;6:33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kausar T, Schreiber JS, Karnak D, Parsels LA, Parsels JD, Davis MA, et al. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia. 2015;17(10):757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(9):2799–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2019;37(29):2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O'Connor MJ. Targeting the DNA Damage Response in Cancer. Molecular cell. 2015;60(4):547–560. [DOI] [PubMed] [Google Scholar]
- 85.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer biology & therapy. 2012;13(11):1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, et al. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Molecular cancer therapeutics. 2018;17(8):1670–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Plummer ER, Dean EJ, Evans TRJ, Greystoke A, Herbschleb K, Ranson M, et al. Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792). Journal of Clinical Oncology. 2016;34(15_suppl):2513–2513. [Google Scholar]
- 89.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342 ( Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 90.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nature reviews Cancer. 2010;10(4):293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(22):3785–3790. [DOI] [PubMed] [Google Scholar]
- 92.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. [DOI] [PubMed] [Google Scholar]
- 93.Kaelin wG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nature reviews Cancer. 2005;5(9):689–698. [DOI] [PubMed] [Google Scholar]
- 94.O'Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nature reviews Genetics. 2017;18(10):613–623. [DOI] [PubMed] [Google Scholar]
- 95.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature reviews Cancer. 2011;12(1):68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. [DOI] [PubMed] [Google Scholar]
- 97.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. The New England journal of medicine. 2018;379(8):753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bendell J, O'Reilly EM, Middleton MR, Chau I, Hochster H, Fielding A, et al. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol. 2015;26(4):804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Golan T, Locker GY, Kindler HL. Maintenance Olaparib for Metastatic Pancreatic Cancer. Reply. The New England journal of medicine. 2019;381(15):1492–1493. [DOI] [PubMed] [Google Scholar]
- 100.Lomberk GA, Iovanna J, Urrutia R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics. 2016;8(6):831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Omura N, Goggins M. Epigenetics and epigenetic alterations in pancreatic cancer. International journal of clinical and experimental pathology. 2009;2(4):310–326. [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO reports. 2015;16(11):1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews Drug discovery. 2014;13(5):337–356. [DOI] [PubMed] [Google Scholar]
- 104.Verdone L, Agricola E, Caserta M, Di Mauro E. Histone acetylation in gene regulation. Brief Funct Genomic Proteomic. 2006;5(3):209–221. [DOI] [PubMed] [Google Scholar]
- 105.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual review of biochemistry. 2007;76:75–100. [DOI] [PubMed] [Google Scholar]
- 106.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nature reviews Molecular cell biology. 2007;8(4):284–295. [DOI] [PubMed] [Google Scholar]
- 107.Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. 2016;8:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ono H, Basson MD, Ito H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget. 2016;7(32):51301–51310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116(20):4670–4676. [DOI] [PubMed] [Google Scholar]
- 110.Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harbor perspectives in medicine. 2016;6(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–1252. [DOI] [PubMed] [Google Scholar]
- 112.Arnold NB, Arkus N, Gunn J, Korc M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(1):18–26. [DOI] [PubMed] [Google Scholar]
- 113.Garcia-Morales P, Gomez-Martinez A, Carrato A, Martinez-Lacaci I, Barbera VM, Soto JL, et al. Histone deacetylase inhibitors induced caspase-independent apoptosis in human pancreatic adenocarcinoma cell lines. Molecular cancer therapeutics. 2005;4(8):1222–1230. [DOI] [PubMed] [Google Scholar]
- 114.Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(17):5385–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chan E, Arlinghaus LR, Cardin DB, Goff L, Berlin JD, Parikh A, et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother Oncol. 2016;119(2):312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, et al. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther. 2010;10(7):997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jones SF, Infante JR, Spigel DR, Peacock NW, Thompson DS, Greco FA, et al. Phase 1 results from a study of romidepsin in combination with gemcitabine in patients with advanced solid tumors. Cancer Invest. 2012;30(6):481–486. [DOI] [PubMed] [Google Scholar]
- 118.Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. 2006;17(7):1096–1102. [DOI] [PubMed] [Google Scholar]
- 119.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular cell. 2014;54(5):728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(4):912–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sahai V, Kumar K, Knab LM, Chow CR, Raza SS, Bentrem DJ, et al. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Molecular cancer therapeutics. 2014;13(7):1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ, et al. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011;71(3):1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dangi-Garimella S, Sahai V, Ebine K, Kumar K, Munshi HG. Three-dimensional collagen I promotes gemcitabine resistance in vitro in pancreatic cancer cells through HMGA2-dependent histone acetyltransferase expression. PloS one. 2013;8(5):e64566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016;35(7):833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miller AL, Fehling SC, Garcia PL, Gamblin TL, Council LN, van Waardenburg R, et al. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine. 2019;44:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nature medicine. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(2):396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heist RS, Gandhi L, Shapiro G, Rizvi NA, Burris HA, Bendell JC, et al. Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: Results of a phase Ib dose-escalation trial. Journal of Clinical Oncology. 2013;31(15_suppl):2530–2530.23752106 [Google Scholar]
- 131.Mirzoeva OK, Collisson EA, Schaefer PM, Hann B, Hom YK, Ko AH, et al. Subtype-specific MEK-PI3 kinase feedback as a therapeutic target in pancreatic adenocarcinoma. Molecular cancer therapeutics. 2013; 12(10):2213–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ko AH, Bekaii-Saab T, Van Ziffle J, Mirzoeva OM, Joseph NM, Talasaz A, et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Larbouret C, Robert B, Navarro-Teulon I, Thezenas S, Ladjemi MZ, Morisseau S, et al. In vivo therapeutic synergism of anti-epidermal growth factor receptor and anti-HER2 monoclonal antibodies against pancreatic carcinomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(11):3356–3362. [DOI] [PubMed] [Google Scholar]
- 134.Worthylake R, Opresko LK, Wiley HS. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. The Journal of biological chemistry. 1999;274(13):8865–8874. [DOI] [PubMed] [Google Scholar]
- 135.Assenat E, Azria D, Mollevi C, Guimbaud R, Tubiana-Mathieu N, Smith D, et al. Dual targeting of HER1/EGFR and HER2 with cetuximab and trastuzumab in patients with metastatic pancreatic cancer after gemcitabine failure: results of the "THERAPY"phase 1-2 trial. Oncotarget. 2015;6(14):12796–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nature reviews Immunology. 2018;18(3):153–167. [DOI] [PubMed] [Google Scholar]
- 138.Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.De Sousa Linhares A, Battin C, Jutz S, Leitner J, Hafner C, Tobias J, et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci Rep. 2019;9(1):11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pu N, Lou W, Yu J. PD-1 immunotherapy in pancreatic cancer: current status. Journal of Pancreatology. 2019;2(1):6–10. [Google Scholar]
- 141.Johansson H, Andersson R, Bauden M, Hammes S, Holdenrieder S, Ansari D. Immune checkpoint therapy for pancreatic cancer. World journal of gastroenterology : WJG. 2016;22(43):9457–9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.O'Reilly EM, Oh DY, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kabacaoglu D, Ciecielski KJ, Ruess DA, Algul H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front Immunol. 2018;9:1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vogl UM, Andalibi H, Klaus A, Vormittag L, Schima W, Heinrich B, et al. Nab-paclitaxel and gemcitabine or FOLFIRINOX as first-line treatment in patients with unresectable adenocarcinoma of the pancreas: does sequence matter? BMC cancer. 2019;19(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]