

Abstract
Background
Overexpression of ATP‐binding cassette (ABC) transporter is a major contributor to multidrug resistance (MDR), in which cancer cells acquire resistance to a wide spectrum of chemotherapeutic drugs. In this work, we evaluated the sensitizing effect of sitravatinib, a broad‐spectrum tyrosine kinase inhibitor (TKI), on ATP‐binding cassette subfamily B member 1 (ABCB1)‐ and ATP‐binding cassette subfamily C member 10 (ABCC10)‐mediated MDR.
Methods
MTT assay was conducted to examine cytotoxicity and evaluate the sensitizing effect of sitravatinib at non‐toxic concentrations. Tritium‐labeled paclitaxel transportation, Western blotting, immunofluorescence analysis, and ATPase assay were carried out to elucidate the mechanism of sitravatinib‐induced chemosensitization. The in vitro findings were translated into preclinical evaluation with the establishment of xenograft models.
Results
Sitravatinib considerably reversed MDR mediated by ABCB1 and partially antagonized ABCC10‐mediated MDR. Our in silico docking simulation analysis indicated that sitravatinib strongly and stably bound to the transmembrane domain of ABCB1 human‐mouse chimeric model. Furthermore, sitravatinib inhibited hydrolysis of ATP and synchronously decreased the efflux function of ABCB1. Thus, sitravatinib could considerably enhance the intracellular concentration of anticancer drugs. Interestingly, no significant alterations of both expression level and localization of ABCB1 were observed. More importantly, sitravatinib could remarkably restore the antitumor activity of vincristine in ABCB1‐mediated xenograft model without observable toxic effect.
Conclusions
The findings in this study suggest that the combination of sitrvatinib and substrate antineoplastic drugs of ABCB1 could attenuate the MDR mediated by the overexpression of ABCB1.
Keywords: Sitravatinib
Abbreviations
- ABC
ATP‐binding cassette
- ABCB1
ATP‐binding cassette subfamily B member 1
- ABCC10
ATP‐binding cassette subfamily C member 10
- IF
immunofluorescence
- MDR
multidrug resistance
- MRP7
multidrug resistance‐associated protein 7
- NBD
nucleotide‐binding domain
- PBS
phosphate‐buffered saline
- PDGFR
platelet‐derived growth factor receptor
- PVDF
polyvinylidene difluoride
- SD
standard deviation
- TAM
TYR01, AXL, and MerTK
- TKI
tyrosine kinase inhibitor
- TMD
transmembrane domain
- VEGFR
vascular endothelial growth factor receptor
1. BACKGROUND
Chemotherapy is an important cancer treatment. One of the major causes of treatment failure is believed to be the development of intrinsic or acquired drug resistance to a variety of antineoplastic agents [1, 2]. In other words, the poor prognosis and short survival could sometimes result from multidrug resistance (MDR) [3]. To date, many researchers had figured out several mechanisms of action accounting for MDR including, but not limited to, reduced uptake and/or increased efflux of drugs [4, 5], cell cycle arrest [5], apoptosis regulation [5], re‐programmed drug metabolism [6, 7], DNA repair [8], oncogene mutations [9], and pathological transition [10]. Among them, the most common and typical factor is efflux function mediated by the ATP‐binding cassette (ABC) transporters found on the plasma membrane of certain cell types [11, 12]. The ABC transporter superfamily is composed of seven subfamilies from ABCA to ABCG [13]. Many of the transporters are responsible for the decreased intracellular concentration of drugs, such as by ATP‐binding cassette subfamily B member 1 (ABCB1) gene encoded ABCB1 (also known as P‐glycoprotein or MDR1) and ATP‐binding cassette subfamily C member 10 (ABCC10) gene encoded multidrug resistance‐associated protein 7 (MRP7) [7]. All subfamilies share some common structural features such as transmembrane domains (TMDs) and nucleotide‐binding domains (NBDs). TMDs mostly serve as ligand recognition and transportation sites, while NBDs mainly serve as ATP‐binding sites for hydrolyzing ATP [11].
Currently, there are some workable strategies to improve response and to overcome MDR, including chemosensitizers [14], gene therapy [15, 16], immune‐oncology [17], nanotechnology [18], or traditional Chinese medicine [19]. The chemosensitizers verapamil and cyclosporine A are known inhibitors of ABCB1 [20]. However, they were reported to have significant toxicity and pharmacokinetic issues [20]. Therefore, more improved and less toxic inhibitors of ABC transporters are urgently needed in both preclinical and clinical evaluation.
Tyrosine kinase inhibitors (TKIs) have been widely used in cancer targeted therapy in the past decades [21, 22, 23]. Mechanistically, they can down‐regulate the phosphorylation level of tyrosine residue of proteins that play important roles in cancer signaling pathways [24, 25]. It has been documented that several TKIs have the capacity to inhibit the function of ABC transporters [7, 26, 53]. Herein, we screened 40 TKIs via molecular docking analysis and cell viability assay, and found that sitravatinib has the potential to antagonize MDR mediated by ABCB1.
Sitravatinib, also known as MGCD516 or MG‐516, is a broad‐spectrum TKI that targets TAM (TYR01, AXL, and MerTK) receptors. Sitravatinib is one of the family members of vascular endothelial growth factor receptor (VEGFR) and platelet‐derived growth factor receptor (PDGFR) [27, 28]. In this study, we focused on the antagonizing activity of sitravatinib towards ABCB1‐mediated MDR.
2. MATERIALS AND METHODS
2.1. Cell lines
Human epidermoid carcinoma cell line KB‐3‐1 and its corresponding colchicine‐resistant cell line KB‐C2 were kindly provided by Dr. Shin‐Ichi Akiyama (Kagoshima University, Kagoshima, Japan). Human colon cancer cell line SW620, its corresponding doxorubicin‐resistant cell line SW620/Ad300, human embryonic kidney cell line HEK293, and the ABCB1‐/ABCC10‐transfected HEK293 cell lines were obtained from Drs. Susan E. Bates, Robert W. Robey and Suresh V. Ambudkar (National Cancer Institute [NCI], National Institutes of Health [NIH], Bethesda, MD, USA).
All cell lines mentioned above were cultured in Dulbecco's Modified Eagle Medium (DMEM; Corning, Steuben County, NY, USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Atlanta, GA, USA) and 1% penicillin‐streptomycin (Corning) at 37°C in a humidified incubator supplied with 5% CO2. KB‐C2 cells were cultured in complete medium with 2 µg/mL colchicine (Enzo Life Sciences, Farmingdale, NY, USA) [29]. SW620/Ad300 cells were maintained in complete medium with 300 ng/mL doxorubicin (Medkoo Sciences, Chapel Hill, NC, USA) [30]. All MDR cells were cultured in drug‐free medium for at least 3 weeks and passaged for at least 3 generations before use in experiments. All transfected cell lines, such as HEK293/pcDNA3.1, HEK293/ABCB1 and HEK293/ABCC10, were selected and maintained in complete medium with 2 mg/mL G418 (Enzo Life Sciences) [31].
2.2. Nude mice
Athymic NCR (nu/nu) nude mice (male, 4‐5 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All mice were housed and cared under standard conditions in the animal facility at the Chinese Academy of Medical Sciences and Peking Union Medical College. All animal procedures were performed under control and with approval according to the guidelines under the Animal Ethics Committee of Chinese Academy of Medical Sciences and Peking Union Medical College. At the end of the treatment period, the mice were euthanized using 100% carbon dioxide [32].
2.3. Human‐mouse chimeric ABCB1 model
The latest human‐mouse chimeric ABCB1 model [33] with near‐atomic resolution cryo‐EM structure is reported by Locher's group. The ABCB1 protein was bound to the human‐specific inhibitory antibody UIC2 and the ABCB1 inhibitor zosuquidar. We downloaded the ABCB1 model from the protein data bank (PDB: 6FN1) and used it for docking analysis.
2.4. Docking analysis of sitravatinib in human‐mouse chimeric ABCB1 model
The ligand‐receptor interaction was performed using Maestro v11.1 (Schrödinger, LLC, Cambridge, MA, USA) as previous reported [34]. ABCB1 protein model (PDB: 6FN1) [33] was prepared using Protein Preparation Wizard module, and the active site was defined using Receptor Grid Generation module in which the docking grid was generated at the TMD of ABCB1 model [35]. The structure of TKIs was drawn using 2D Sketcher module and then prepared with Ligprep module using default protocols. Glide XP docking was conducted with the pre‐processed ligands and protein model.
2.5. Modified MTT colorimetric assay
A modified MTT colorimetric assay was performed to determine the cytotoxicity of chemotherapeutic agents with or without an inhibitor as described previously [34]. In short, each cell line was evenly seeded into 96‐well plates at 5,000 cells/well one day before treatment. The cells were pretreated with or without TKIs (sitravatinib was purchased from ChemieTek [Indianapolis, IN, USA], and other TKIs were kindly provided as free sample from Selleckchem (Houston, TX, USA)] at indicated concentrations or with a reference inhibitor to ABCB1 or ABCC10 (verapamil [Sigma‐Aldrich, St. Louis, MO, USA] or cepharanthine [Apexbio Technology, Houston, TX, USA] at 3 µmol/L, respectively). Following 2 h pretreatment, cells were treated with serial‐diluted chemotherapeutic agents (paclitaxel [Sigma‐Aldrich], vincristine [Sigma‐Aldrich], doxorubicin [Enzo Life Sciences], and cisplatin [Enzo Life Sciences]). After 68 h treatment, MTT (Fisher Sci., NJ, USA) was added at a final concentration of 4 mg/mL, and cells were incubated at 37°C for another 4 h protected from light. The formazan crystals were dissolved using dimethyl sulfoxide (DMSO; Sigma‐Aldrich) after the supernatant was discarded. The light absorbance at 570 nm was determined using accuSkan™ microplate spectrophotometer (Fisher Sci.). All experiments were performed independently at least 3 times.
2.6. [3H]‐Paclitaxel accumulation and efflux assay
Both sensitive and resistant cell lines were used for [3H]‐substrate accumulation assay as previously described [31]. Briefly, ABCB1‐mediated MDR cells and their parental cells were seeded into 24‐well plates at the final density of 1 × 106 cells/well and incubated one day prior to further assay. Following 2 h pretreatment with drug‐free medium, sitravatinib, or verapamil at indicated concentrations, cells were co‐incubated with [3H]‐paclitaxel (Moravek Biochemicals, Brea, CA, USA) and its corresponding treatment above at 37°C for another 2 h. After rinsed with ice‐cold phosphate‐buffered saline (PBS), cells were trypsinized, harvested, and placed in liquid scintillation cocktail (VWR Chemicals, Solon, OH, USA).
To further assess the efflux function mediated by ABC transporters, the [3H]‐substrate efflux assay was conducted based on established protocol [36]. In short, cells were prepared as described in above accumulation analysis. Then, cells were incubated in the absence or presence of inhibitors at indicated concentrations at 37°C for serial time frames (0, 0.5, 1, and 2 h). Subsequently, cells were washed with iced PBS twice, and then trypsinized, harvested, and transferred into liquid scintillation cocktail.
Radioactivity was quantified by Packard TRI‐CARB 1900CA liquid scintillation analyzer (Packard Instrument, Downers Grove, IL, USA). All the experiments were repeated at least 3 times independently.
2.7. Western blotting and immunofluorescence (IF) analysis
Protein expression level of ABCB1 after incubation with or without sitravatinib at 3 µmol/L was measured using Western blotting as previous mentioned [37]. Briefly, after treatment with the highest non‐toxic concentration of sitravatinib up to 72 h, cells were lysed with lysis buffer (10 mmol/L Tris, 1 mmol/L EDTA, 0.1% SDS, 150 mM NaCl, 1% Triton‐X, and protease inhibitor cocktail [VWR Chemicals, Solon, OH, USA]) on ice for 20 min, then centrifuged at 15,000 × g at 4°C for 20 min. The supernatant was harvested and then quantified by total protein concentration using the BCA Protein Kit (Thermo Scientific, Waltham, MA, USA). Protein was denatured using heat block at 70°C for 5 min. Total protein (20 µg) was loaded and separated by SDS‐PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% milk‐TBST for 2 h at room temperature, the membrane was incubated with primary antibodies against ABCB1 (Sigma‐Aldrich) at 1: 1000 dilution and GAPDH (Thermo Fisher Scientific, Rockford, IL, USA) at 1: 2000 dilution at 4°C overnight. Then, after washing with TBST, the membrane was incubated with HRP‐conjugated anti‐mouse IgG secondary antibody (Cell Signaling Technology, Dancers, MA, USA) at 1: 2000 dilution for 2 h at room temperature. The chemiluminescence signal was developed using ECL substrate (Thermo Fisher Scientific) per the manufacturer's instructions. Protein expression was quantified using Image J (NIH, Bethesda, MD, USA). All experiments were repeated at least 3 times independently.
IF assay was conducted to examine the subcellular localization of membrane protein ABCB1 as previously described [7]. In short, both parental and drug‐resistant cells were incubated with or without sitravatinib at its highest non‐toxic concentration for up to 72 h. After the treatment, cells were fixed in 4% formaldehyde (J.T. Baker Chemical, Phillipsburg, NJ, USA) and permeabilized in 0.1% Triton X‐100 (Sigma‐Aldrich) for 15 min. Cells were blocked with 6% BSA (Thermo Fisher Scientific) for 2 h. Then, primary antibody anti‐P‐gp at 1: 1000 dilution followed by Alexa Fluor 488 conjugated secondary antibody (Thermo Fisher Scientific) at 1:1000 dilution were added and incubated with cells. After that, cell nuclei were dyed with 1 µg/mL DAPI (Thermo Fisher Scientific). Images were captured with fluorescence microscope (Life Technologies, Gaithersburg, MD, USA).
2.8. ATPase assay
The vanadate‐sensitive ATPase activity of ABCB1 was quantified with similar protocol described previously [38]. In brief, we used ABCB1‐membrane to hydrolyze ATP and measure the inorganic phosphate (Pi) production. Reactions took place in assay buffer containing different types of ATPase inhibitors (1 mmol/L ouabain, 5 mmol/L sodium azide, 0.01 mol/L MgCl2, 2 mmol/L EGTA, 0.05 mol/L MES [pH 6.8], 0.05 mol/L KCl, and 2 mmol/L DTT). Then sitravatinib was incubated with membrane vesicles for 3 min. The ATP hydrolysis was initialized by adding 5 mmol/L of Mg‐ATP. After incubation at 37°C for 20 min, 5% SDS solution was added to terminate the reaction. Pi was measured at 880 nm in a spectrophotometer (Bio‐Rad, Hercules, CA, USA).
2.9. Xenograft model
The xenograft model was established as previously described [39]. SW620 and SW620/Ad300 cells were harvested and washed twice with PBS. Then, both cell lines (1 × 107 cells in 0.1 mL PBS) were injected subcutaneously under armpits of the nude mice. Once the tumor volume reached 500 mm3, the mice were euthanized, and the tumor tissues were dissected. Each tumor tissue, except the necrotic part, was cut into 2 × 2 mm in size. The small tumor pieces were implanted subcutaneously under armpits of the nude mice. When the tumor reached approximately 100 mm3, the mice were randomly assigned to the following four groups (n = 6 each treatment group). Sitravatinib was prepared in a final concentration of 0.5% hydroxypropyl methylcellulose (HPMC) and 0.1% Tween‐80 solution (pH 1.4) (vehicle A) [27], while vincristine was prepared in autoclaved water (vehicle B) [40]. Group 1 mice received vehicle A (administered orally) 1 h prior to vehicle B (administered intraperitoneally). Group 2 mice received sitravatinib (prepared in vehicle A, 2 mg/kg, administered orally) 1 h prior to vehicle B (administered intraperitoneally). Group 3 mice received vehicle A (administered orally) 1 h prior to vincristine (prepared in vehicle B, 0.4 mg/kg, administered intraperitoneally). Group 4 mice received sitravatinib 1 h prior to vincristine at indicated dosages. All treatments were given every other day for 14 days.
The dosage of vincristine was determined by Zhang et al. [40], and the dosage of sitravatinib was determined through preliminary experiments and previous pharmacokinetic studies [28]. During the treatment, the animal behavior was monitored daily. The body weights as well as the length and width of tumor tissues were recorded using vernier caliper every other day. At the end of treatment period, all mice were euthanized, and the tumor tissues were excised, weighed, and measured. The tumor volume (V) and the growth inhibition rates (IR) for tumor volume (IRV) and tumor weight (IRW) were calculated using followed formulae [26, 41‐43]:
2.10. Statistical analysis
All results are shown as mean ± standard deviation (SD). Comparisons were made between the control group and corresponding treatment group. The results were analyzed with one‐way or two‐way analysis of variance (ANOVA) following Tukey post hoc analysis. All statistical analyses were performed using GraphPad Prism version 6.02 for Windows (GraphPad Software, La Jolla, CA, USA). The value P < 0.05 indicates significant difference.
3. RESULTS
3.1. Sitravatinib had a good affinity to human‐mouse chimeric ABCB1 model
To predict and compare the binding affinities of different TKIs with the human‐mouse chimeric ABCB1 model, docking simulation analysis was performed. Among all the 5 TKIs (Supplementary Figure S1A‐E), sitravatinib gave the highest docking score of –11.095 kcal/mol (Supplementary Figure S1F). We used verapamil, a reference ABCB1 inhibitor, as a control, whose docking score (–7.376 kcal/mol) was much lower than sitravatinib (Supplementary Figure S1F). Also, the 50% inhibition concentration (IC50) values of paclitaxel with or without TKIs in KB‐3‐1 and KB‐C2 cells are presented in Supplementary Table S1. According to the results, sitravatinib could significantly restore the sensitivity of KB‐C2 to paclitaxel. In contrast, other compounds did not reduce the IC50 values of paclitaxel to the similar level as verapamil in colchicine‐resistant cell line. For all TKIs, paclitaxel IC50 was not varied in control groups (parental cells). These preliminary experimental results indicated that sitravatinib has the best docking score and has significant inhibitory effect on ABCB1‐overexpressing cells compared with other TKIs. Hence, we conducted further mechanistic‐based assays using sitravatinib.
To further explore the interaction between the sitravatinib and human‐mouse chimeric ABCB1 model, the in silico docking simulation analysis was carried out. The overall interaction between ABCB1 human‐mouse chimeric model (PDB ID: 6FN1) and binding site of sitravatinib are shown in Figure 1A. The details on ligand‐protein binding are depicted in Figure 1B and C. Sitravatinib binded within the drug‐binding pocket of ABCB1 through hydrogen bonds and π‐π interaction. One amide group and the methoxy group of sitravatinib could form hydrogen bonds with Gln724 and Gln945 of ABCB1, respectively. Meantime, the pyridine ring binded with Tyr952 through the hydrogen bond and π‐π interaction. Sitravatinib could also be stabilized in the hydrophobic pocket of ABCB1 formed by the residues including Leu64, Ile305, Tyr306, Tyr309, Leu723, Phe727, Tyr952, Tyr949, Met948, Val990. These results indicated that sitravatinib has a good affinity to human‐mouse chimeric ABCB1 model.
FIGURE 1.
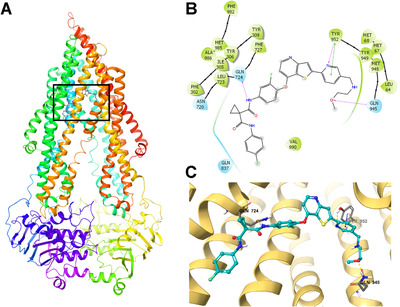
The in silico docking simulation analysis between sitravatinib and human‐mouse chimeric ABCB1 model (PDB ID: 6FN1) using Maestro. A. The overall structure of ABCB1 and the binding site of sitravatinib. B. The 2‐dimension diagram of the binding of sitravatinib with ABCB1. Blue bubbles indicate hydrophilic amino acids, and green bubbles indicate hydrophobic amino acids. Hydrogen bonds are shown with purple arrows, and green short line indicates π‐π interaction. C. The 3‐dimension diagram of the interaction of sitravatinib and ABCB1. Hydrogen bonds are shown with yellow dot lines, and π‐π interaction is indicated with blue dot line. The ABCB1 protein is depicted with yellow ribbon. Sitravatinib is shown with the following color codes: carbon (cyan), oxygen (red), nitrogen (blue), sulfur (yellow), fluoride (green), hydrogen (white). Abbreviations: ABCB1, ATP‐binding cassette subfamily B member 1
3.2. Sitravatinib potently enhanced the efficacy of chemotherapeutic drugs in ABCB1‐overexpressing cell lines, but moderately in ABCC10‐overexpressing cell lines
To further evaluate the biological function of sitravatinib in vitro, we examined the cytotoxic effect of sitravatinib on all cell lines used in this study. Based on the results, we selected the highest concentration that did not result in toxicity for further study, which was 3 µmol/L, at which concentration the cell survival rate was higher than 80% after incubation with sitravatinib for 72 h (Supplementary Figure S2).
Next, we tested the reversal effect of sitravatinib on ABCB1‐mediated MDR. Sitravatinib significantly decreased the IC50 values of various anticancer drugs which are substrates of ABCB1 (paclitaxel, doxorubicin, and vincristine) in drug‐selected cell lines (KB‐C2 and SW620/Ad300) in a concentration‐dependent manner (Figure 2). However, no significant change in IC50 value was observed in parental cell lines (KB‐3‐1 and SW620) after co‐incubation with sitravatinib at non‐toxic concentrations. Moreover, as shown in Figure 3A, sitravatinib also concentration‐dependently antagonized MDR in ABCB1‐transfected HEK293 cell line (HEK293/ABCB1). Nevertheless, sitravatinib could not significantly change the efficacy of chemotherapeutic agents mentioned above in corresponding sensitive cell line (HEK293/pcDNA3.1). More importantly, it is notable that the reversal effect of sitravatinib was not only comparable but also much better than the counterpart of positive control verapamil, a known inhibitor of ABCB1, at the same concentration level. In addition, in both sensitive and resistant cells, sitravatinib did not significantly change the antineoplastic effect of cisplatin, which is not a substrate of ABCB1 in ABCB1‐overexpressing cell lines (Figure 2, Figure 3A).
FIGURE 2.
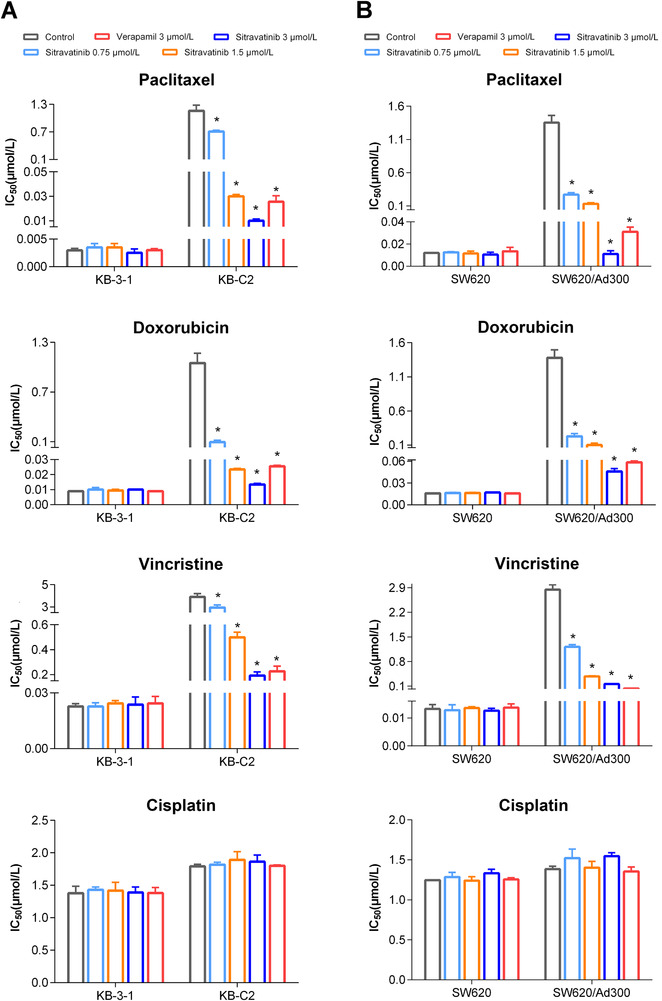
Effects of sitravatinib on IC50 values of chemotherapeutic drugs in drug‐selected resistant cell lines and their parental cell lines. A. IC50 values of paclitaxel, doxorubicin, vincristine, and cisplatin in ABCB1‐overexpressing KB‐C2 and KB‐3‐1 cells with or without inhibitor for 72 h. B. Cytotoxicity of ABCB1‐substrate drugs (paclitaxel, doxorubicin, vincristine) and cisplatin on SW620/Ad300 and parental SW620 cells with or without inhibitor for 72 h. Verapamil at 3 µmol/L was used as positive reversal agent. Data are shown as mean ± SD obtaining from at least 3 independent experiments. * P < 0.05 was considered statistically significant compared with control group.
FIGURE 3.
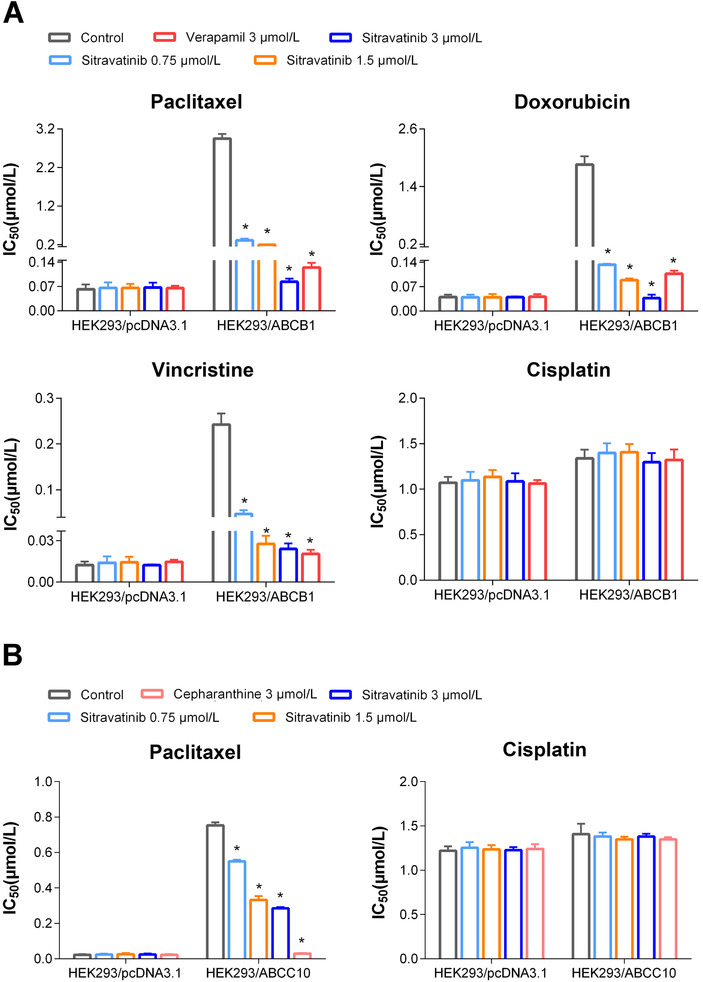
Effects of sitravatinib on IC50 values of antineoplastic drugs in ABCB1‐ or ABCC10‐transfected HEK293 cell line and its parental cell line HEK293 cell line transfected with pcDNA3.1. A. IC50 values of paclitaxel, doxorubicin, vincristine, and cisplatin incubated with or without inhibitor for 72 h on HEK293 cell line transfected with erither pcDNA3.1 vector or ABCB1 expression plasmid. Verapamil at 3 µmol/L acts as positive control. B. Cytotoxicity of paclitaxel and cisplatin in the absence or presence of inhibitor for 72 h on HEK293/pcDNA3.1 and ABCC10‐transfected HEK293 cell line. Cepharanthine at 3 µmol/L functions as positive control. Data are showed as mean ± SD, representive of 3 independent experiments. *, P < 0.05 indicates statistically significant. Abbreviations: ABCB1, ATP‐binding cassette subfamily B member 1; ABCC10, ATP‐binding cassette subfamily C member 10; SD, standard deviation
Because overexpression of ABCC10 confers resistance to paclitaxel and vincristine which are the substrates of ABCB1 as well [7], we also examined the reversal activity on ABCC10‐overexpressing MDR cell model. Sitravatinib partially sensitized HEK293/ABCC10 to paclitaxel (Figure 3B), but the effect was much weaker than that with equal concentration of cepharanthine as a positive control inhibitor of ABCC10. Thus, we did not further examine the mechanism of action on ABCC10‐overexpressing cells. Also, sitravatinib did not significantly change anticancer efficacy of cisplatin in both parental and resistant cell lines (Figure 3B).
These results indicated that sitravatinib can sensitize ABCB1‐mediated MDR in a concentration‐dependent manner. Further research is needed to understand the reversal mechanism of sitravatinib in ABCB1‐overexpressing cell lines.
3.3. Sitravatinib induced the intracellular accumulation of [3H]‐paclitaxel in ABCB1‐overexpressing cell lines
To obtain further information regarding reversal mechanism of sitravatinib in ABCB1‐mediated MDR, we quantified the intracellular accumulation of paclitaxel in parental and ABCB1‐overexpressing cells with or without sitravatinib. The intracellular [3H]‐paclitaxel levels were approximately 26‐fold and 30‐fold lower in KB‐C2 and SW620/Ad300 cell lines than those in KB‐3‐1 and SW620 cell lines, respectively (Figure 4A). Interestingly, after 2 h pretreatment with sitravatinib at 3 µmol/L, the intracellular radioactivity of [3H]‐paclitaxel was increased approximately from 3.8% to 77.6% in KB‐C2 cells or from 3.3% to 55.5% in SW620/Ad300 cells, compared with parental cells. In this assay, verapamil was used as a positive control inhibitor of ABCB1. Overall, the results indicated that sitravatinib increased the intracellular accumulation of [3H]‐paclitaxel in ABCB1‐mediated MDR cells.
FIGURE 4.
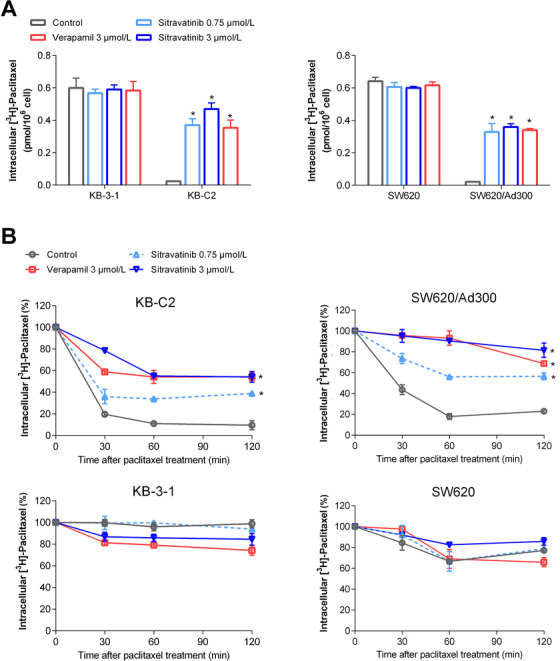
The effects of sitravatinib on intracellular concentration of tritium‐labeled paclitaxel. A. The effects of sitravatinib on the intracellular accumulation of tritium‐labeled paclitaxel in KB‐C2, KB‐3‐1, SW620/Ad300, and SW620 cells. B. The effects of sitravatinib on the efflux of tritium‐labeled paclitaxel in KB‐C2, KB‐3‐1, SW620/Ad300, and SW620 cells at 0, 30, 60, 120 min time point. Data are presented as mean ± SD obtained from 3 independent experiments. *, P < 0.05 was considered statistically significant compared with control group. Abbreviations: SD, standard deviation
3.4. Sitravatinib decreased the efflux of [3H]‐paclitaxel in ABCB1‐overexpressing cells in a time‐course study
To further determine the mechanism for enhanced intracellular accumulation of paclitaxel in MDR cells, an efflux assay was conducted to assess the pump function of ABCB1 through quantifying paclitaxel at a series of time points. As shown in Figure 4B, KB‐C2 or SW620/Ad300 cells in the control group pumped out 90% or 78% of [3H]‐paclitaxel respectively in 120 min incubation time. By contrast, sitravatinib at higher non‐toxic concentration could retain approximately 55% or 81% of [3H]‐paclitaxel in KB‐C2 or SW620/Ad300 cells, respectively. However, sitravatinib did not significantly change the efflux of [3H]‐paclitaxel in sensitive KB‐3‐1 or SW620 cells (Figure 4B). These results indicated that sitravatinib could interfere with the pump function of ABCB1 transporter, resulting in increased intracellular accumulation of tritium‐labeled paclitaxel by inhibiting the efflux pump.
3.5. Sitravatinib did not affect ABCB1 expression level or subcellular localization in ABCB1‐ overexpressing MDR cell lines
Western blotting was conducted to demonstrate the effect of sitravatinib on the protein expression level of ABCB1. Figure 5A and B showed that after exposure to sitravatinib at 3 µmol/L for up to 72 h, there was no downregulation of the ABCB1 expression in ABCB1‐overexpressing KB‐C2 or SW620/Ad300 cells.
FIGURE 5.
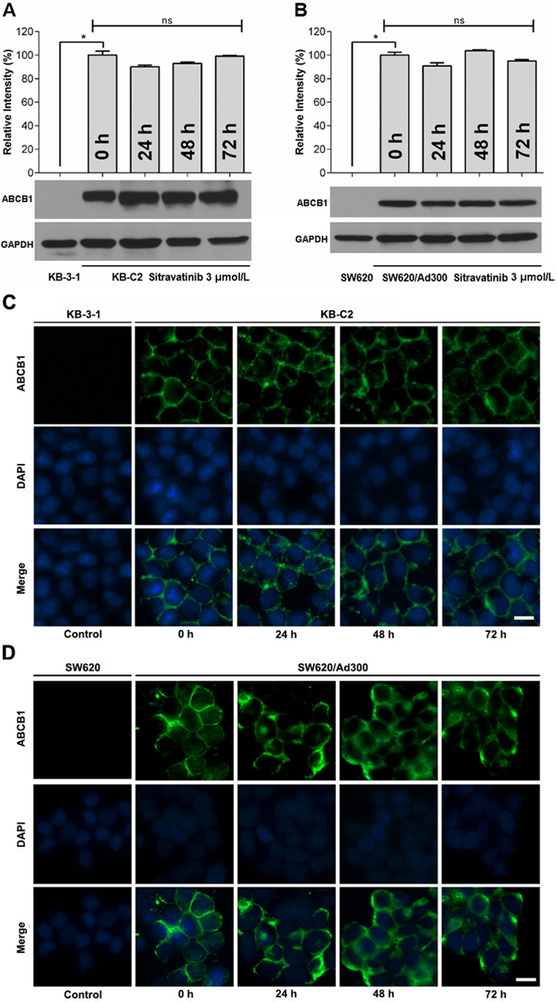
The expression level and subcellular localization of ABCB1. A‐B. The relative intensity of ABCB1 expression level in KB‐3‐1 and KB‐C2 (A), SW620 and SW620/Ad300 (B) in the presence of 3 µmol/L sitravatinib incubated for 0/24/48/72 h, respectively. Error bar represents SD value. *, P < 0.05 was considered statistically significant. C‐D. The subcellular localization of ABCB1 in KB‐3‐1 and KB‐C2 (C), SW620 and SW620/Ad300 (D) incubation for 0, 24, 48, 72 h with 3 µmol/L sitravatinib, separately. ABCB1 (green signals); DAPI (blue signals). Scale bar: 10 µm. Abbreviations: ABCB1, ATP‐binding cassette subfamily B member 1
The subcellular localization of transporters was detected by immunofluorescence in ABCB1‐mediated MDR cell lines. As shown in Figure 5C and D, the sensitive cell lines, KB‐3‐1 and SW620, showed no fluorescence signals, which was in consistent with the results of Western blotting. After treatment with 3 µmol/L sitravatinib for 0, 24, 48, 72 h separately, the membrane‐bound fluorescence signals were still enriched on KB‐C2 and SW620/Ad300 cell lines.
3.6. Sitravatinib inhibited the vanadate‐sensitive ABCB1 ATPase activity
The process of ATP hydrolysis provides ABC transporters with energy to transport a variety of endogenous ligands and exogenous drugs [44]. To further assess the effect of sitravatinib on ABCB1 function, the vanadate‐sensitive ATPase activity of ABCB1, in membrane protein obtained from the transporter‐overexpressing high‐five insect cells, was determined at different concentrations of sitravatinib. According to the result in Figure 6, sitravatinib showed a concentration‐dependent inhibition within 10 µmol/L on the ABCB1 transporter. ATPase activity reached a plateau of 10.7% of the basal activity for ABCB1. The inhibitory effect of sitravatinib reached 50% maximal at 4.4 µmol/L.
FIGURE 6.
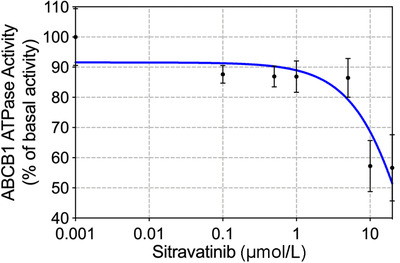
The effects of sitravatinib on ABCB1 ATPase activity. The vanadate‐sensitive ATPase activity of ABCB1, in membrane protein obtained from the transporter‐overexpressing high‐five insect cells, was determined at different concentrations of sitravatinib. ATP hydrolysis was monitored by measuring the amount of inorganic phosphate released using a colorimetric assay. Data are deminstrated as mean ± SD acquiring from 3 experiments independently. Abbreviations: ABCB1, ATP‐binding cassette subfamily B member 1; SD, standard deviation
3.7. Sitravatinib reversed ABCB1‐mediated MDR in SW620/Ad300 xenograft model
Following determining the reversal effect of sitravatinib on ABCB1‐mediated MDR in vitro, we further verified these findings on animal xenograft model. The images of excised tumors are shown in Figure 7A and B. Sitravatinib alone had 37% inhibitory effect on tumor volume (IRV) and 28% inhibitory effect on tumor weight (IRW) compared with the control group treated with vehicle only in the SW620 mice. The mice treated with vincristine alone or the combination of sitravatinib and vincristine had much smaller tumor size than the mice treated with vehicle or sitravatinib alone (approximately with 80% IRV and 70% IRW) (Figure 7C and E). In contrast, vincristine had limited inhibitory effect on SW620/Ad300 tumors due to ABCB1‐mediated MDR (approximately with 30% IRV and 20% IRW). Notably, the growth of xenograft tumors was significantly inhibited by combination of sitravatinib and vincristine with 76% IRV and 89% IRW (Figure 7D and F). Importantly, sitravatinib was well‐tolerated either applied alone or in combination with vincristine without obvious differences in body weight compared with counterpart before treatment at Day 0 in corresponding group (Figure 7G and H).
FIGURE 7.
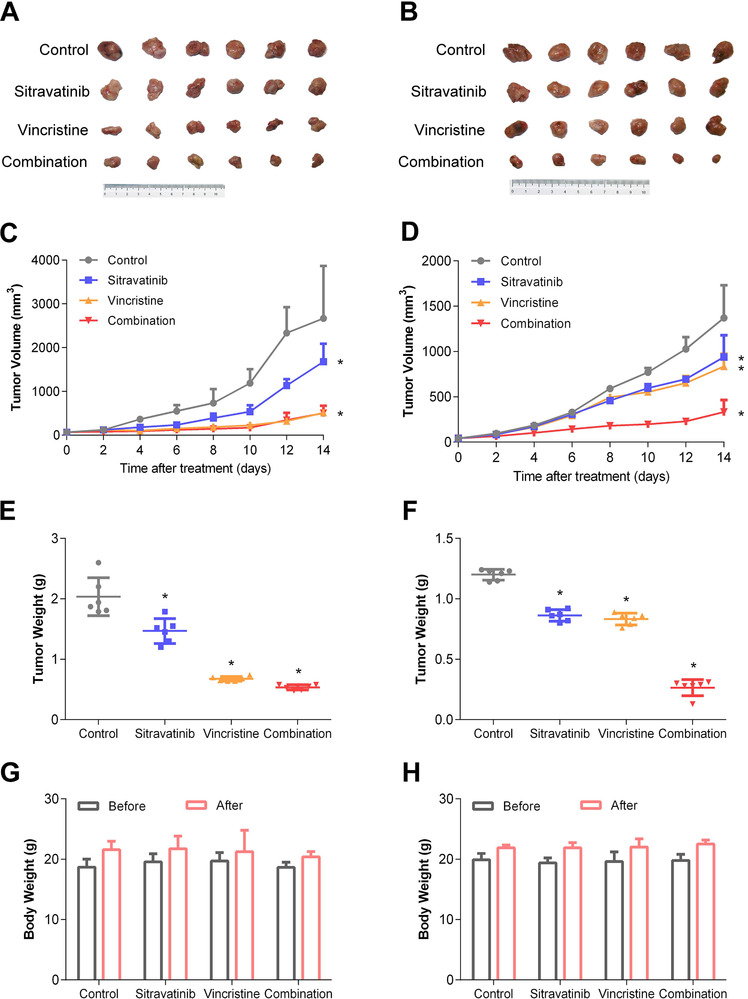
Effects of sitravatinib on the antitumor activity of vincristine in SW620 and SW620/Ad300 xenograft tumor models. A‐B. Images of excised SW620 and SW620/Ad300 tumor tissues from mice at the end of treatment period (n = 6). C‐D. The changes of tumor volume in SW620 and SW620/Ad300 xenograft model measured every other day after treatment. E‐F. The mean weight of excised SW620 and SW620/Ad300 tumor tissues from the mice treated with vehicle, sitravatinib, vincristine or the combination of sitravatinib and vincristine. G‐H. The changes of mean body weight before and after treatment for the SW620 and SW620/Ad300 xenograft model. Error bar represents SD value. *, P < 0.05, compared with control group, was considered statistically significant. Abbreviations: SD, standard deviation
These results indicated that sitravatinib enhanced the antitumor efficacy of vincristine in SW620/Ad300 ABCB1‐overexpressing xenograft tumor model without increasing in vivo toxicity in both SW620 and SW620/Ad300 models.
4. DISCUSSION
Several studies have shown that novel TKIs inhibit the ABCB1 transporter [7, 38, 45]. Thus, it is possible that TKIs have ability to work as chemosensitizers towards ABC transporters to conquer MDR by combination with anticancer drugs. Clinically, TKIs are used as first‐ or second‐line treatments for certain metastatic cancers [46]. However, TKIs have non‐specific and “off‐target” effects [47], thereby explaining why TKIs are used as alternative treatments in the clinical setting and restore the anticancer efficacy of chemotherapeutic drugs in the ABCB1‐mediated MDR model. Notably, it has been reported that sitravatinib has potent antitumor efficacy, that may be due, in part, to altering the tumor microenvironment and restoring the efficacy of immune checkpoint blockade (PD‐1) in diverse cancer models [28]. Dolan et al. [46] reported that sitravatinib could combat MDR caused by sunitinib and axitinib in metastatic human and mouse models. These studies provide a clue that sitravatinib might have the capability to antagonize MDR in cancer cells. It is notable that sitravatinib is currently in up to nine ongoing clinical trials for various indications (information from Clinicaltrials.gov, Supplementary Table S2), which suggests that sitravatinib does not produce intolerable adverse effects or toxic profiles.
In this study, we screened 40 TKIs via molecular docking analysis, and 5 compounds were selected with the highest docking scores to conduct MTT assay. We found that sitravatinib had the highest docking score and the strongest inhibitory effect on ABCB1‐mediated MDR. Our molecular modeling analysis indicated that the docking score of the binding of sitravatinib with human‐mouse chimeric ABCB1 model was –11.095 kcal/mol, and sitravatinib could interact with the drug‐binding pocket of ABCB1. Other TKIs with inhibitory activity to ABCB1, such as selonsertib, ulixertinib and glesatinib [7, 38, 45], received the docking scores of –11.094, –11.986, –12.639 kcal/mol, respectively. Also, the best‐scored pose of other well‐established ABCB1 inhibitors, such as verapamil and zosuquidar, received glide score of –7.376 and –8.646 kcal/mol. Hence, the affinity of sitravatinib and ABCB1 may be comparable with other known ABCB1 inhibitors.
The effect of sitravatinib was further evaluated by MTT assay, which confirmed that sitravatinib alone did not have different inhibitory effect on MDR cell lines and their corresponding sensitive cell lines. Then, a modified MTT assay was used to determine the cytotoxic effect of several chemotherapeutic agents with or without sitravatinib at non‐toxic concentrations, at which concentration the inhibition reached less than 20% (IC20). We found that the sensitizing effects of sitravatinib on different MDR cell lines were related to ABCB1. This is because sitravatinib did not significantly change the IC50 values of chemotherapeutic agents in parental cell lines; while sitravatinib would significantly restore the efficacy of antineoplastic drugs in resistant cell lines, in which ABCB1 protein level was significantly overexpressed as shown in Western blotting results. Furthermore, the antagonizing effects of sitravatinib were confirmed in ABCB1‐transfected HEK293 cell lines, in which ABCB1 overexpression is the only contributor to MDR. Moreover, sitravatinib did not have sensitizing effect towards cisplatin, which is not the substrate of ABCB1. Lastly, sitravatinib showed weak reversal effect on ABCC10‐transfected HEK293 cell line compared with cepharanthine, a reference inhibitor of ABCC10. Therefore, sitravatinib significantly antagonized MDR in ABCB1‐overexpressing cell line, but the reversal effect was not outstanding in ABCC10‐overexpressing cell line.
As previously reported, ABC transporters act as efflux pumps [48]. Therefore, we hypothesized that the sensitizing effects of sitravatinib was associated with improved intracellular accumulation profile in ABCB1‐mediated MDR cells. The results of the [3H]‐paclitaxel accumulation assay indicated that sitravatinib significantly enhanced drug accumulation in resistant cells without obvious influence on corresponding sensitive cells. Furthermore, since there are two explanations to interpret this phenomenon, decreased paclitaxel efflux process or increased paclitaxel uptake by the cells, a [3H]‐substrate efflux assay was conducted to further quantitatively evaluate the relationship between efflux and accumulation. Sitravatinib significantly decreased the drug efflux in ABCB1‐overexpressing cells. Thus, the mechanism of sitravatinib in antagonizing MDR is that sitravatinib decreases drug efflux and in turn increases the drug accumulation; as a result, it improves the intracellular concentration of anticancer drugs. Notably, in this assay, the short‐term treatment with a chemotherapeutic agent did not affect the expression level or subcellular localization on ABCB1 protein. However, the tritium‐labeled substrate accumulation and efflux assay we performed has limitations to overcome in the future. For example, there are other membrane transport methods, such as passive diffusion and active transport, depending on concentration gradient and energy. ABCB1 might not be the sole donor in determining the intracellular concentration of chemotherapeutic agents. Also, the increased intracellular accumulation is not the only factor leading to reduced efflux; there may be other factors, such as deactivation through an alternative metabolic pathway [49]. Further study is needed to exclude other underlying mechanisms resulting in increased intracellular concentration.
Mechanistically, deteriorated resistance to ABCB1 substrates may be related to multiple factors such as downregulating the expression of relative MDR proteins and/or altering the localization of ABC transporters [7, 31]. Normally, active ABC transporters with pump function are located on the lipid raft, which refers to the membrane domains enriched with receptors involved in many biological functions [50, 51]. Interestingly, our results showed that sitravatinib changed neither the expression level nor the subcellular localization of ABCB1 in MDR cells mediated by ABCB1‐overexpression.
It is known that the ATP hydrolysis of ABCB1 is the driving force of trapping and extrusion of substrates of ABCB1 [44]. This could be modulated by the presence of substrate or inhibitor [32, 48]. Hence, we performed an ATPase assay to examine the effect of sitravatinib towards ATPase. We found that sitravatinib inhibited ABCB1 ATPase in a concentration‐dependence manner, which suggested that sitravatinib could possibly work as an inhibitor of ABCB1 via impeding the function of ABCB1. Furthermore, these results may provide the clue that sitravatinib altered the efflux rate of tritium‐labeled paclitaxel over a time course. Also, we postulated that as more ATPase was inhibited, the active ATPase would become less; hence, sitravatinib reduced the drug efflux rate. However, this hypothesis needs further confirmation in the future.
The results from above showed that sitravatinib could bind to ABCB1, therefore inhibit hydrolysis of ATP and abolish the efflux function of ABCB1 without changing the expression level or the subcellular localization; as a result, it increases drug accumulation and improves the efficacy of anticancer drugs.
Recently, Wu et al. [52] showed that sitravatinib had similar inhibitory effect on ABCB1 and ABCG2 in vitro. Herein, we translated our in vitro findings into preclinical evaluation with the establishment of xenograft tumor models. The dosage and administration interval of vincristine were determined by previous studies [40]. Compared with the reported plasma concentration of sitravatinib [28], a remarkably lower dosage of sitravatinib was used to perform an in vivo study, because sitravatinib could induce microenvironment change or immunosuppression, besides directly targeting cancer cells [28]. Besides, this might also be the possible reason why the tumor volume of sitravatinib treatment group in SW620 model was much smaller than the counterpart of vesicle only group. However, this hypothesis needs to be validated in the future. Notably, sitravatinib is a broad‐spectrum TKI, and thus it would target several factors of the tumor microenvironment and reduce the possibility of developing resistance‐derived inhibition towards a single but vital biological pathway. In this work, we examined the sensitizing effect of sitravatinib on the antitumor activity of vincristine in SW620 and SW620/Ad300 tumor xenograft models. From our results, sitravatinib alone had little inhibitory effect towards tumor volume and tumor weight on both parental and resistant cell xenografts. Importantly, sitravatinib could significantly improve the antitumor activity of vincristine in ABCB1‐overexpressing xenograft model compared with the counterparts in sensitive tumor model. Also, each treatment regimen, including sitravatinib or vincristine alone and combination of sitravatinib and vincristine, did not considerably change the body weight before and after the treatment period, which demonstrated that these chemotherapeutic strategies can be well‐tolerated without observable toxic feature during the treatment period.
5. CONCLUSIONS
Collectively, this study demonstrated that sitravatinib at pharmacologically achievable doses could restore the efficacy of chemotherapeutic agents, particularly the substrate drugs of ABCB1 transporter. Sitravatinib could be used in combination with substrate antineoplastic drugs of ABCB1 for effective chemotherapy.
Supporting information
Supplementary Figure S1. Chemical structures and docking scores of TKIs screened in this study. A‐E. Chemical structure of sitravatinib (A), merestinib (B), gilteritinib (C), LOXO‐101 (D), and BMS‐777607 (E). F. The docking scores of the binding of TKIs to human‐mouse chimeric ABCB1 model (PDB ID: 6FN1). Verapamil serves as control.
Abbreviations: TKI, tyrosine kinase inhibitor; ABCB1, ATP‐binding cassette subfamily B member 1.
Supplementary Figure S2. The cytotoxity of sitravatinib on ABCB1‐ or ABCC10‐overexpressing MDR cell lines and their corresponding sensitive cell lines. The cell survival curves for KB‐C2 and parental KB‐3‐1 (A), SW620/Ad300 and parental SW620 (B), HEK293/ABCB1 and empty‐vector‐transfected HEK293 (HEK293/pcDNA3.1) (C), HEK293/ABCC10 and HEK293/pcDNA3.1 (D) cells after incubation with a serial concentration of sitravatinib for 72 h. The non‐toxic concentration, at which the cell survival rate was higher than 80% after incubation with sitravatinib for 72 h, is indicated by dash lines. Each point with error bar represents mean ± SD of 3 independent assays.
Abbreviations: ABCB1, ATP‐binding cassette subfamily B member 1; ABCC10; ATP‐binding cassette subfamily C member 10; SD, standard deviation.
Supporting information
Supporting information
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The animal study in this article was approved by Animal Ethics Committee of Chinese Academy of Medical Sciences and Peking Union Medical College.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
This work was partially supported by NIH (No. 1R15GM116043‐01) and National Natural Science Foundation of China (No. 81872901) as well as some supports from Department of Pharmaceutical Sciences, St. John's University.
AUTHORS' CONTRIBUTIONS
Y.Y. and Z.S.C. designed the study. Y.Y., N.J., J.Q.W. and C.Y.C. gave contribution to perform experiments. Z.N.L., Q.X.T., Z.X.W., Q.C. and Y.P. provided technical and material support. Y.Y. wrote the first draft. All authors discussed the results and implications and developed the manuscript at all stages.
ACKNOWLEDGEMENTS
We thank Dr. Shin‐Ichi Akiyama (Kagoshima University, Kagoshima, Japan), and Drs. Susan E. Bates, Robert W. Robey and Suresh V. Ambudkar (NCI, NIH, Bethesda, Maryland, USA) for kindly providing the cell lines. Thanks are also given to Dr. Tanaji T. Talele (St. John's University, New York, USA) for providing the computing resources for the docking analysis.
Yang Y, Ji N, Cai C‐Y, et al. Modulating the function of ABCB1: in vitro and in vivo characterization of sitravatinib, a tyrosine kinase inhibitor. Cancer Communications. 2020;40:285–300. 10.1002/cac2.12040
Contributor Information
Ning Ji, Email: jining@ihcams.ac.cn.
Zhe‐Sheng Chen, Email: chenz@stjohns.edu.
REFERENCES
- 1. Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10(2):147‐56. 10.1038/nrc2789. [DOI] [PubMed] [Google Scholar]
- 2. Cui Q, Wang J‐Q, Assaraf YG, Ren L, Gupta P, Wei L, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1‐25. 10.1016/j.drup.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 3. Gillet J‐P, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47‐76. 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 4. Kathawala RJ, Gupta P, Ashby CR, Jr. , Chen Z‐S. The modulation of ABC transporter‐mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1‐17. 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 5. Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, et al. Overcoming ABC transporter‐mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat. 2016;27:14‐29. 10.1016/j.drup.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 6. Rashmi R, Huang X, Floberg JM, Elhammali AE, McCormick ML, Patti GJ, et al. Radioresistant Cervical Cancers Are Sensitive to Inhibition of Glycolysis and Redox Metabolism. Cancer Res. 2018;78(6):1392‐403. 10.1158/0008-5472.CAN-17-2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. You X, Jiang W, Lu W, Zhang H, Yu T, Tian J, et al. Metabolic reprogramming and redox adaptation in sorafenib‐resistant leukemia cells: detected by untargeted metabolomics and stable isotope tracing analysis. Cancer Commun. 2019;39(1):17 10.1186/s40880-019-0362-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Centurione L, Aiello FB. DNA Repair and Cytokines: TGF‐β, IL‐6, and Thrombopoietin as Different Biomarkers of Radioresistance. Front Oncol. 2016;6:175 10.3389/fonc.2016.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xue M, Cheng J, Zhao J, Zhang S, Jian J, Qiao Y, et al. Outcomes of 219 chronic myeloid leukaemia patients with additional chromosomal abnormalities and/or tyrosine kinase domain mutations. International journal of Laboratory Hematology. 2019;41(1):94‐101. 10.1111/ijlh.12928. [DOI] [PubMed] [Google Scholar]
- 10. Chen Y, Tang WY, Tong X, Ji H. Pathological transition as the arising mechanism for drug resistance in lung cancer. Cancer Communications (London, England). 2019;39(1):53 10.1186/s40880-019-0402-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang YK, Wang YJ, Gupta P, Chen ZS. Multidrug Resistance Proteins (MRPs) and Cancer Therapy. The AAPS Journal. 2015;17(4):802‐12. 10.1208/s12248-015-9757-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446(7137):749‐57. 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 13. Dean M, Allikmets R. Complete characterization of the human ABC gene family. J Bioenerg Biomembr. 2001;33(6):475‐9. 10.1023/a:1012823120935. [DOI] [PubMed] [Google Scholar]
- 14. Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW. Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett. 2017;405:100‐10. 10.1016/j.canlet.2017.07.023. [DOI] [PubMed] [Google Scholar]
- 15. Lage H. Gene Therapeutic Approaches to Overcome ABCB1‐Mediated Drug Resistance. Recent Results Cancer Res. 2016;209:87‐94. 10.1007/978-3-319-42934-2_6. [DOI] [PubMed] [Google Scholar]
- 16. Shi R, Peng H, Yuan X, Zhang X, Zhang Y, Fan D, et al. Down‐regulation of c‐fos by shRNA sensitizes adriamycin‐resistant MCF‐7/ADR cells to chemotherapeutic agents via P‐glycoprotein inhibition and apoptosis augmentation. J Cell Biochem. 2013;114(8):1890‐900. 10.1002/jcb.24533. [DOI] [PubMed] [Google Scholar]
- 17. Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second‐ and third‐generation drugs for immuno‐oncology treatment‐The more the better? Eur J Cancer. 2017;74:55‐72. 10.1016/j.ejca.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 18. Yin J, Lang T, Cun D, Zheng Z, Huang Y, Yin Q, et al. pH‐Sensitive Nano‐Complexes Overcome Drug Resistance and Inhibit Metastasis of Breast Cancer by Silencing Akt Expression. Theranostics. 2017;7(17):4204‐16. 10.7150/thno.21516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu WL, Shen HL, Ao ZF, Chen BA, Xia W, Gao F, et al. Combination of tetrandrine as a potential‐reversing agent with daunorubicin, etoposide and cytarabine for the treatment of refractory and relapsed acute myelogenous leukemia. Leuk Res. 2006;30(4):407‐13. 10.1016/j.leukres.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 20. Zhang H, Jiang H, Wang X, Chen B. Reversion of multidrug resistance in tumor by biocompatible nanomaterials. Mini Rev Med Chem. 2010;10(8):737‐45. [DOI] [PubMed] [Google Scholar]
- 21. Cai W, Kong W, Dong B, Zhang J, Chen Y, Xue W, et al. Pretreatment Serum Prealbumin as an Independent Prognostic Indicator in Patients With Metastatic Renal Cell Carcinoma Using Tyrosine Kinase Inhibitors as First‐Line Target Therapy. Clin Genitourin Cancer. 2017;15(3):e437‐e46. 10.1016/j.clgc.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 22. Cai W, Zhong H, Kong W, Dong B, Chen Y, Zhou L, et al. Significance of preoperative prognostic nutrition index as prognostic predictors in patients with metastatic renal cell carcinoma with tyrosine kinase inhibitors as first‐line target therapy. Int Urol Nephrol. 2017;49(11):1955‐63. 10.1007/s11255-017-1693-9. [DOI] [PubMed] [Google Scholar]
- 23. Shibata M, Shizu M, Watanabe K, Takeda A. Uterine metastasis of lung adenocarcinoma under molecular target therapy with epidermal growth factor receptor tyrosine kinase inhibitors: A case report and review of the literature. J Obstet Gynaecol Res. 2018;44(2):352‐8. 10.1111/jog.13493. [DOI] [PubMed] [Google Scholar]
- 24. Tsukita S, Oishi K, Akiyama T, Yamanashi Y, Yamamoto T, Tsukita SJTJoCB. Specific proto‐oncogenic tyrosine kinases of src family are enriched in cell‐to‐cell adherens junctions where the level of tyrosine phosphorylation is elevated. 1991;113(4):867‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paiment A, Hocking J, CJJob Whitfield. Impact of phosphorylation of specific residues in the tyrosine autokinase, Wzc, on its activity in assembly of group 1 capsules in Escherichia coli. 2002;184(23):6437‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang YK, Wang YJ, Lei ZN, Zhang GN, Zhang XY, Wang DS, et al. Regorafenib antagonizes BCRP‐mediated multidrug resistance in colon cancer. Cancer Lett. 2019;442:104‐12. 10.1016/j.canlet.2018.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patwardhan PP, Ivy KS, Musi E, de Stanchina E, Schwartz GK. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti‐tumor activity in preclinical models of sarcoma. Oncotarget. 2016;7(4):4093‐109. doi:10.18632/oncotarget.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI insight. 2018;3(21). 10.1172/jci.insight.124184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J‐Q, Wang B, Lei Z‐N, Teng Q‐X, Li JY, Zhang W, et al. Derivative of 5‐cyano‐6‐phenylpyrimidin antagonizes ABCB1‐ and ABCG2‐mediated multidrug resistance. Eur J Pharmacol. 2019;863:172611 10.1016/j.ejphar.2019.172611. [DOI] [PubMed] [Google Scholar]
- 30. Lai GM, Chen YN, Mickley LA, Fojo AT, Bates SE. P‐glycoprotein expression and schedule dependence of adriamycin cytotoxicity in human colon carcinoma cell lines. Int J Cancer. 1991;49(5):696‐703. 10.1002/ijc.2910490512. [DOI] [PubMed] [Google Scholar]
- 31. Wu Z‐X, Teng Q‐X, Cai C‐Y, Wang J‐Q, Lei Z‐N, Yang Y, et al. Tepotinib reverses ABCB1‐mediated multidrug resistance in cancer cells. Biochem Pharmacol. 2019;166:120‐7. 10.1016/j.bcp.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 32. Zhang GN, Zhang YK, Wang YJ, Gupta P, Ashby CR, Jr. , Alqahtani S, et al. Epidermal growth factor receptor (EGFR) inhibitor PD153035 reverses ABCG2‐mediated multidrug resistance in non‐small cell lung cancer: In vitro and in vivo. Cancer Lett. 2018;424:19‐29. 10.1016/j.canlet.2018.02.040. [DOI] [PubMed] [Google Scholar]
- 33. Alam A, Kung R, Kowal J, McLeod RA, Tremp N, Broude EV, et al. Structure of a zosuquidar and UIC2‐bound human‐mouse chimeric ABCB1. PNAS. 2018;115(9):E1973‐e82. 10.1073/pnas.1717044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ji N, Yang Y, Cai CY, Lei ZN, Wang JQ, Gupta P, et al. VS‐4718 Antagonizes Multidrug Resistance in ABCB1‐ and ABCG2‐Overexpressing Cancer Cells by Inhibiting the Efflux Function of ABC Transporters. Frontiers in Pharmacology. 2018;9:1236 10.3389/fphar.2018.01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cai C‐Y, Zhai H, Lei Z‐N, Tan C‐P, Chen B‐L, Du Z‐Y, et al. Benzoyl indoles with metabolic stability as reversal agents for ABCG2‐mediated multidrug resistance. Eur J Med Chem. 2019;179:849‐62. 10.1016/j.ejmech.2019.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang B, Ma L‐Y, Wang J‐Q, Lei Z‐N, Gupta P, Zhao Y‐D, et al. Discovery of 5‐Cyano‐6‐phenylpyrimidin Derivatives Containing an Acylurea Moiety as Orally Bioavailable Reversal Agents against P‐Glycoprotein‐Mediated Mutidrug Resistance. J Med Chem. 2018;61(14):5988‐6001. 10.1021/acs.jmedchem.8b00335. [DOI] [PubMed] [Google Scholar]
- 37. Ji N, Yang Y, Cai CY, Wang JQ, Lei ZN, Wu ZX, et al. Midostaurin Reverses ABCB1‐Mediated Multidrug Resistance, an in vitro Study. Front Oncol. 2019;9:514 10.3389/fonc.2019.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ji N, Yang Y, Lei ZN, Cai CY, Wang JQ, Gupta P, et al. Ulixertinib (BVD‐523) antagonizes ABCB1‐ and ABCG2‐mediated chemotherapeutic drug resistance. Biochem Pharmacol. 2018;158:274‐85. 10.1016/j.bcp.2018.10.028. [DOI] [PubMed] [Google Scholar]
- 39. Ding Y, Gao H, Zhang Y, Li Y, Vasdev N, Gao Y, et al. Alantolactone selectively ablates acute myeloid leukemia stem and progenitor cells. J Hematol Oncol. 2016;9(1):93 10.1186/s13045-016-0327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang H, Patel A, Ma SL, Li XJ, Zhang YK, Yang PQ, et al. In vitro, in vivo and ex vivo characterization of ibrutinib: a potent inhibitor of the efflux function of the transporter MRP1. Br J Pharmacol. 2014;171(24):5845‐57. 10.1111/bph.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Naito S, von Eschenbach AC, Giavazzi R, Fidler IJ. Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 1986;46(8):4109‐15. [PubMed] [Google Scholar]
- 42. Pearson AT, Finkel KA, Warner KA, Nor F, Tice D, Martins MD, et al. Patient‐derived xenograft (PDX) tumors increase growth rate with time. Oncotarget. 2016;7(7):7993‐8005. doi:10.18632/oncotarget.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24(3):148‐54. [DOI] [PubMed] [Google Scholar]
- 44. Dean M, Allikmets R. Complete characterization of the human ABC gene family. J Bioenerg Biomembr. 2001;33(6):475‐9. [DOI] [PubMed] [Google Scholar]
- 45. Cui Q, Cai CY, Gao HL, Ren L, Ji N, Gupta P, et al. Glesatinib, a c‐MET/SMO Dual Inhibitor, Antagonizes P‐glycoprotein Mediated Multidrug Resistance in Cancer Cells. Frontiers in Oncology. 2019;9:313 10.3389/fonc.2019.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dolan M, Mastri M, Tracz A, Christensen JG, Chatta G, Ebos JML. Enhanced efficacy of sitravatinib in metastatic models of antiangiogenic therapy resistance. PLoS One. 2019;14(8):e0220101 10.1371/journal.pone.0220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mastri M, Lee CR, Tracz A, Kerbel RS, Dolan M, Shi Y, et al. Tumor‐Independent Host Secretomes Induced By Angiogenesis and Immune‐Checkpoint Inhibitors. Mol Cancer Ther. 2018;17(7):1602‐12. 10.1158/1535-7163.Mct-17-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr Drug Targets. 2006;7(7):893‐909. [DOI] [PubMed] [Google Scholar]
- 49. Herling A, Konig M, Bulik S, Holzhutter HG. Enzymatic features of the glucose metabolism in tumor cells. FEBS J. 2011;278(14):2436‐59. 10.1111/j.1742-4658.2011.08174.x. [DOI] [PubMed] [Google Scholar]
- 50. Klappe K, Hummel I, Hoekstra D, Kok JW. Lipid dependence of ABC transporter localization and function. Chem Phys Lipids. 2009;161(2):57‐64. 10.1016/j.chemphyslip.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 51. Sonnino S, Prinetti A. Membrane domains and the “lipid raft” concept. Curr Med Chem. 2013;20(1):4‐21. [PubMed] [Google Scholar]
- 52. Wu CP, Hsiao SH, Huang YH, Hung LC, Yu YJ, Chang YT, et al. Sitravatinib Sensitizes ABCB1‐ and ABCG2‐Overexpressing Multidrug‐Resistant Cancer Cells to Chemotherapeutic Drugs. Cancers. 2020;12(1). 10.3390/cancers12010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang Y, Ji N, Teng Q‐X, Cai C‐Y, Wang J‐Q, Wu Z‐X, Lei Z‐N, Lusvarghi S, Ambudkar SV, Chen Z‐S. Sitravatinib, a Tyrosine Kinase Inhibitor, Inhibits the Transport Function of ABCG2 and Restores Sensitivity to Chemotherapy‐Resistant Cancer Cells in vitro. Frontiers in Oncology. 2020;10: 10.3389/fonc.2020.00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Chemical structures and docking scores of TKIs screened in this study. A‐E. Chemical structure of sitravatinib (A), merestinib (B), gilteritinib (C), LOXO‐101 (D), and BMS‐777607 (E). F. The docking scores of the binding of TKIs to human‐mouse chimeric ABCB1 model (PDB ID: 6FN1). Verapamil serves as control.
Abbreviations: TKI, tyrosine kinase inhibitor; ABCB1, ATP‐binding cassette subfamily B member 1.
Supplementary Figure S2. The cytotoxity of sitravatinib on ABCB1‐ or ABCC10‐overexpressing MDR cell lines and their corresponding sensitive cell lines. The cell survival curves for KB‐C2 and parental KB‐3‐1 (A), SW620/Ad300 and parental SW620 (B), HEK293/ABCB1 and empty‐vector‐transfected HEK293 (HEK293/pcDNA3.1) (C), HEK293/ABCC10 and HEK293/pcDNA3.1 (D) cells after incubation with a serial concentration of sitravatinib for 72 h. The non‐toxic concentration, at which the cell survival rate was higher than 80% after incubation with sitravatinib for 72 h, is indicated by dash lines. Each point with error bar represents mean ± SD of 3 independent assays.
Abbreviations: ABCB1, ATP‐binding cassette subfamily B member 1; ABCC10; ATP‐binding cassette subfamily C member 10; SD, standard deviation.
Supporting information
Supporting information