

Abstract
Genetically diverse inbred strains are frequently used in quantitative trait mapping to identify sequence variants underlying trait variation. Poor locus resolution and high genetic complexity impede variant discovery. As a solution, we explore reduced complexity crosses (RCCs) between phenotypically divergent, yet genetically similar, rodent substrains. RCCs accelerate functional variant discovery via decreasing the number of segregating variants by orders of magnitude. The simplified genetic architecture of RCCs often permit immediate identification of causal variants or rapid fine-mapping of broad loci to smaller intervals. Whole genome sequences of substrains make RCCs possible by supporting the development of array- and targeted sequencing-based genotyping platforms, coupled with rapid genome editing for variant validation. In summary, RCCs enhance discovery-based genetics of complex traits.
Keywords: GWAS, functional variant, rat genetics, positional cloning, substrain, QTL
Reduced Genetic Complexity Accelerates Variant Discovery
Genome-wide association studies (GWAS) and quantitative trait locus (QTL) mapping are discovery-based approaches to identify novel DNA variants, genes, and biological mechanisms underlying complex traits. While discovery-based genetics has been highly successful at identifying regions associated with trait variation, identification of the causal genes and variants remains a challenge[1]. There are two major impediments. First, several thousand genetically diverse individuals are often required to achieve sufficient power to detect loci responsible for heritable trait variation in a typical GWAS. Causal variants are rarely obvious. Second, classic linkage studies typically yield large loci containing hundreds of genes and thousands of potential variants. A surprising solution to these roadblocks has emerged as a byproduct of the cumulative fixation of residually heterozygous variants and spontaneous mutations (genetic drift, Box 1) in closely related, yet phenotypically divergent, inbred substrains. Advances in whole genome sequencing permit 1) identification of causal DNA variants underlying phenodeviation and 2) the development of high throughput genotyping platforms (targeted sequencing and custom DNA microarrays). The purpose of this review is to introduce reduced complexity crosses (RCCs) as a simple and powerful solution for rapid, high-confidence gene discovery for complex traits (Figure 1, Panel A).
TEXT BOX 1. Origin of substrains.
Two sources of genetic variation underlie genetic drift, enabling RCCs for variant mapping. First, residual heterozygosity (RH) describes unfixed, heterozygous genetic loci in a progenitor strain. If founders are separated prior to complete inbreeding (< 20 generations), RH makes a larger contribution to substrain variation. RH can linger for up to 40 generations of inbreeding[87]. The number of RH variants that become fixed depends on the filial (F) generation at which the populations diverged, the number of founders (more breeder pairs equals more RH loci), the number of heterozygous variants within segregating RH loci, and the founder breeding scheme. Strict sib-sib mating within founder lineages will rapidly generate new substrains; deliberate outcrossing of offspring derived from a large number of breeder pairs will delay, although not prevent, establishment of a substrain. Mutation rates in rats (per single site base pair substitution per generation) have been estimated at 4.2 × 10−9 from wild brown rats [88] and 2.96 × 10−9 based on evolutionary divergence from mice[89].
A second source of variation is de novo mutations. The rate at which mutations are acquired and fixed contributes to genetic drift. Early calculations of mutation rates in rodents were based on reporter constructs at loci in single mouse lines. Meta-analysis estimated the per generation mutation rate as 38 × 10−9 per single site base pair substitution per generation (200 mutations per diploid genome per generation, assuming a 2.6 billion nucleotide genome[90]). Next-generation sequencing facilitates unbiased identification of de novo mutations and estimation of mutation rates in rodents is an ongoing endeavor. The mutation rate for C57BL/6J following 20 generations of sibling mating was estimated as 5.4 × 10−9 or 28 per diploid genome and generation for single nucleotide variants using DNA sequencing [91]. Notably, the mutation rate estimated by sequencing was an order of magnitude less than estimated from reporter constructs. Leveraging sequences permitted estimation of the mutation rate for homozygous (line average of 4.25 × 10−9) and heterozygous (line average 5.45 × 10-9) single nucleotide variants and the mutation rate of insertions and deletions (indels; 3.1 × 10−9)[91]. Mutation rates for homozygous and heterozygous variants were similar and the strategy of sibling breeding at each generation possibly contributed to greater genetic fixation of de novo mutations. Additional DNA sequencing studies are needed to obtain an unbiased, mechanistic understanding of rodent mutation rates, drift, and impact on trait variation.
Figure 1Key Figure. Reduced Complexity Crosses (RCCs) for Complex Trait Analysis.
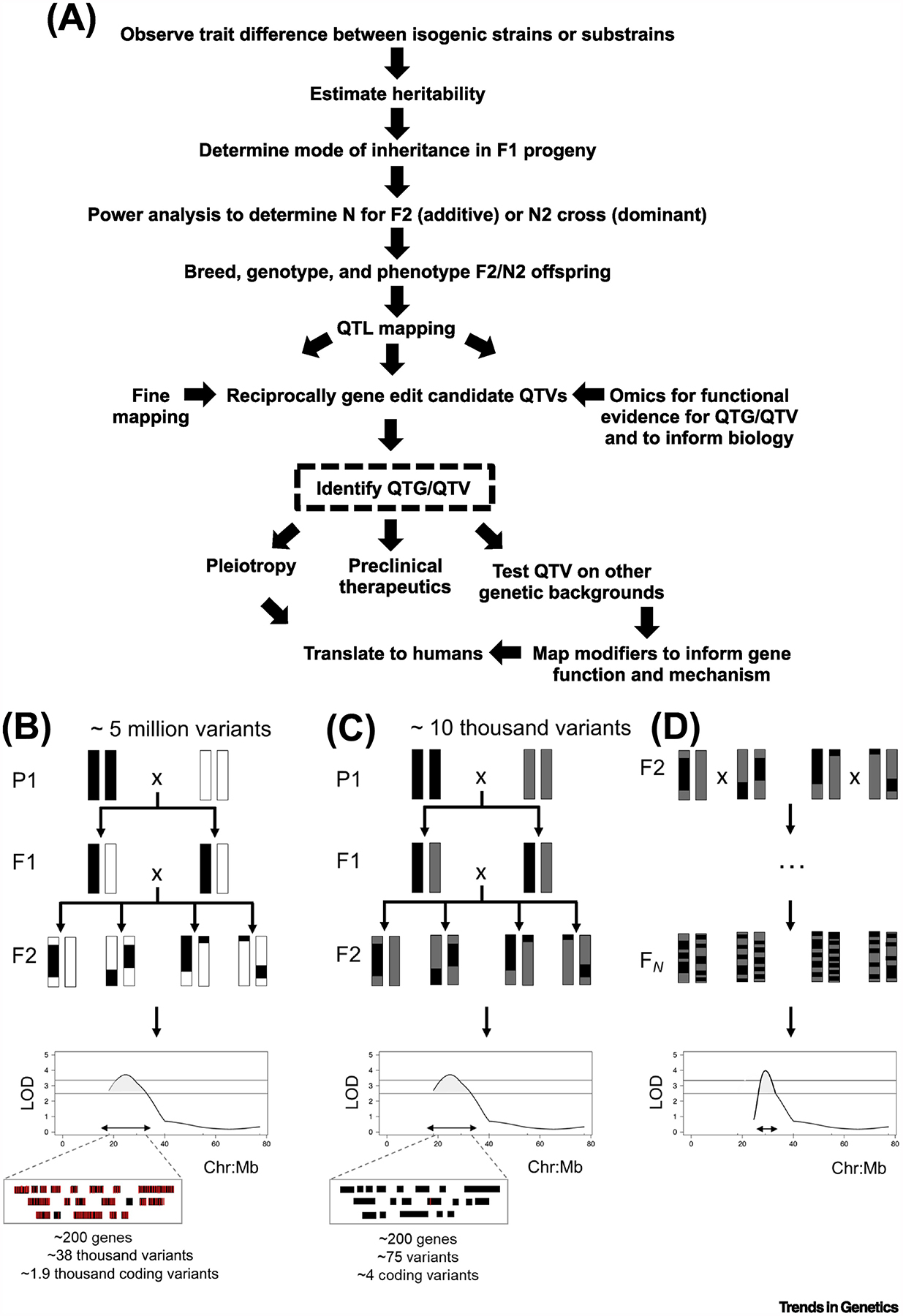
(A) RCC flow chart. (B) A classical F2 intercross between two inbred strains. Isogenic F1 offspring are generated by crossing two inbred strains. Every F1 individual has one chromatid from each parental strain for each chromosome. F1 offspring are intercrossed to generate recombinant F2 individuals. Historically, progenitor inbred strains segregate hundreds of thousands to millions of variants (5 million variants is used as an example here). (C) A reduced complexity F2 intercross. RCCs are generated by intercrossing strains the genomes of which are similar and, thus, segregate orders of magnitude fewer variants compared with more divergent inbred strains. As an example, a RCC between C57BL/6 substrains segregates between 10 000 and 20 000 variants (SNPs plus indels), or in other words, 250 000–500 000-fold fewer variants than a classical F2 cross. Even though the number of historical recombination events and the QTL resolution in an F2 cross are low (~20–40 Mb), the number of candidate causal variants underlying each locus is much smaller. For (B) and (C), the number of genes and variants within each QTL interval was determined based on a genome size of 2.64 B bases, a gene model that included 26 000 genes, and a random distribution model of genes and variants across the genome. (D) A reduced complexity advanced intercross (RCAI). The reduced complexity outcross strain (RCO) addresses the low resolution of an F2 cross by increasing the number of recombination events, which yields a narrower quantitative trait locus (QTL) interval and eliminates the need for fine mapping. The required number of generations for intercrossing to provide sufficient quantitative trait gene (QTG)/quantitative trait variant (QTV) resolution increases as genetic complexity increases.
RCCs between substrains consist of drastically reduced genetic diversity (complexity) in a segregating cross, yielding QTLs with far fewer candidate variants using a smaller number of individuals. Separation and maintenance of substrains that trace their descent from inbred strains has created a rich resource of nearly isogenic sets of substrains that vary genetically from less than 100 to up to ~ 100,000 sequence variants (Table 1). A rapidly growing number of genetically similar sets of substrains have now been fully sequenced, creating a database of variants that can be used both as genetic markers for mapping and positional candidate variants linked to trait variation. Several of these sets of substrains demonstrate robust and heritable differences in phenotypes, despite very low genetic diversity. For these traits, the RCC strategy is ideal for rapid identification of large-effect quantitative trait genes (QTGs) and variants (QTVs)[2,3]. QTLs in RCCs contain orders of magnitude fewer variants compared to crosses between divergent parental inbred strains, thus facilitating QTV identification and exemplifying that physical precision is not always necessary (Figure 1, Panels B and C). Validation of QTVs from a RCC is now easily and efficiently achieved via CRISPR-Cas9 gene editing to demonstrate that a variant is both necessary (i.e. repair of the QTV in the mutant line) and sufficient (i.e. introduction of the QTV on the wild-type, ancestral line) for the predicted phenotype in both backgrounds.
Table 1.
Exemplary mouse and rat substrains and phenotypes that can be subjected to RCC analysis.
| Mouse progenitor strains | Sequenced Mouse Substrains (on miniMUGA array) | Behavioral differences | Physiological, disease model differences | Oellular differences | Molecular differences |
|---|---|---|---|---|---|
| A | A/J, A/JOlaHsd | Muscle dysfunction [T1] | |||
| BALB/c | BALB/cJ, BALB/cByJ | Aggressio n[T2], alcohol preference[T3], anxiety-like behavior[T4], cognitive flexibility[T5], inhibitory control[T6], epilepsy and neuroanatomical abnormalities[28],[T7 ] |
Allergic orchitis and encephalomyelitis [T8,T9], immune response to infection[T10], Grave’s hyperthyroidism[T11], experimental arthritis and spondylitis[T12], GABA transmission and anterior cingulate volume[T13,T14], cardiac calcinosis[T15], dexamethasone-induced osteonecrosis[T16], diet-induced fatty liver[T17], streptozotocin-induced diabetes[T18] | Sperm abnormalities[T19], antibody-mediated immunity[T20], hepatocyte invasion following infection[T21, virus-induced demyelination[T22] | Copy number variants[T23], amino acid and monoamine neurotransmitter content in caudate[T24] |
| C3H | C3H/HeJ, C3H/HeNCrl, C3H/HeNRj, C3H/HeH, C3H/HeNHsd, C3H/HeNTac | Nest building[T25], paw preferenc e[T26] | Skeletal[T25], immune reactivity[T27], LPS responsiveness[T28], experimental leprosy[T29], spontaneous colitis[T30], experimental arthritis and spondylitis[T31], absence seizures[T32] | Cytotoxic activity of lymphocytes in cancer model[T33], | Toll-like receptor 4[T34], Gpr179[T35] |
| C57BL/6 | C57BL/6NJ, C57BL/6NCrl, C57BL/6JBom Tac, C57BL/6ByJ, C57BL/6JOlaH sd, C57BL/6NTyr<c>/BrdCrC rl, C57BL/6NJRj |
Several. Reviewed by [5]. See also [6], [T36], and main text, corticoster one-induced depressive-like behaviors[T37], | Several. Stroke[25], metabolic traits[T38], immune response[T39]. See also[6], kidney stones[T40], severity of Dravet syndrome model with Scn1a +/− [T41], circadian disruptive effects on behavior[T42], ocular lesions[T43], liver production of reactive oxygen species[T44], pain[5,24], viral-induced inflammation[T45], hindlimb unloading-induced bone loss[T46], inflammation-induced neutrophil recruitment[T47], high fat diet-induced obesity[T48], and metabolic and skeletal dysfunction[T49], impaired glucose secretion[T50], auditory physiology and pathology[T51], blood pressure[T52] | Several[6], cardiac fibrogenic response to angiotensin[T53], acetaminophen-induced hepatotoxicity[T54], hypoxic-ischemic brain injury[T55] | Gabra2[67], Cyfip2[10,11], Crb1[T56], Nlrp12[T57] |
| DBA/1 | DBA/1LacJ, DBA/1OlaHsd | Giant lysosomes in the kidney proximal tubules[T58], collagen-induced arthritis[T59] | C5[T60] | ||
| DBA/2 | Methamph etamine intake[21], acoustic startle [T61, T62] | Viral infection susceptibility[T63] | Klrd1 (CD94)[T64], Taar1[21] | ||
| FVB | FVB/NJ, FVB/NCrl, FVB/NRj, FVB/NHsd, FVB/NTac | Breast cancer[T65], pituitary abnormalities[T66], T-cell dysfunction and cutaneous pathology[T67] | |||
| NOD | NOD/ShiLtJ, NOD/MrkTac | Cataracts[T68], diabetes resistance[T69] | Structural variants in chromosome3 and Icam2 (chromosome 11)[T70] | ||
| 129 | 129Ps/OlaHsd, 129S1/SvImJ, 129S2/SvHsd, 129S2/SvPas OrlRj, 129S5/SvEvBrd, 129T2/SvEmJ, 129X1/SvJ | Opioid addiction-related behaviors[T71,T72], fear conditioning[T73,T74], anxiety-like behavior[T74,T75,T76], habituation[T74], reversal learning[T77], spatial learning/memory[T77], forced swim test[T76], m, k,.cocaine motivation [T78] | Inhibition of NaCl response in chorda tympani[T79] | Disc1 deletion[T80] | |
| Rat progenitor strains | Sequenced Rat Substrains | Behavioral differences | Physiological differences | Cellular differences | Molecular differences |
| WKY | WKY/NCrl vs WKY/NHsd | Attention[T81,T82], anxiety-like behavior[T83, social interaction [T83] | Tyrosine hydroxylase, dopamine transporter[T84], hippocampal gene expression[T85] | ||
| SHR | SHRSP vs SHRSP5/Dmcr | progression of fibrosis induced by high fat diet[T86] | |||
| SHR | SHR vs SHRSP | cerebral stroke[T87] | |||
| WKY | WLI/Eer | Open field[T88], FST[T89], stress-enhanced fear conditioning and alcohol consumption[60], response to chronic stress[59], Premature memory decline[T90] | Resting state functional connectivity[T91], baseline and stress corticosterone levels[T92] | Blood transcriptome[T92], amygdala transcriptome[T89], hippocampal transcriptome[T89, T93] | |
| WKY SS |
WMI/Eer | Open field[T88], FST[T89], stress-enhanced fear conditioning[60], alcohol consumption[60], response to chronic stress [59], premature memory decline[T90] | Baseline and stress corticosterone levels[60], blood pressure[T94] | Blood transcriptome[104], hippocampal transcriptome [T89,T93], and amygdala transcriptome[T89] | |
| SS/Jr vs SS/JrHsdMcwi vs SR/Jr | |||||
| F344 | F344/NHsd vs F344/NCrHsd | Body weight gain, neuropeptide Y and agouti-related protein[T95] | |||
Theoretical Limits on Genetic Complexity in a RCC
How much should the segregating variant pool be “reduced” in order to be considered a RCC? Conversely, how “complex” can a cross be before it no longer affords the advantages of a RCC? The answers depend partly on the trait and its genetic architecture in a particular RCC. At the extremely low end of genetic diversity, there may not be a sufficient number of genome-wide variants to conduct genome-wide mapping and thus, identifying the causal variant(s) relies on deep, reliable whole genome sequencing, positional cloning, and gene editing. There is also an upper variant limit after which the RCC approach becomes equivalent to a standard F2 cross between divergent inbred strains and QTVs are no longer easily resolved. To address these questions, we consider a case of low versus moderate genomic complexity. A discussion regarding sources of genetic variation and mutation rates can be found in Box 1.
In the first example, several BXD Recombinant Inbred (RI) strains were separated at different breeding colonies, which led to genetic drift and new substrains. The BXD29/TyJ-Tlr4Ips−2J (BXD29 mutant) substrain exhibits spontaneous bilateral subcortical heterotopias with partial callosal agenesis and deficits in auditory processing compared to the progenitor BXD29 strain[4]. The causal variants are unknown, but the heterotopia exhibits a two-locus autosomal recessive mode of inheritance. This is an extreme case of reduced complexity whereby sequencing identified ~1000 distinguishing variants. Although low, the number of variants is too high to identify causal QTVs by sequencing alone and mapping in a RCC is required.
The second example comes from the high demand for B6 mice that spawned multiple breeding locations and the creation of several B6 substrains currently separated by hundreds of generations at different institutions. In the 1950s, the C57BL/6N (N) lineage from NIH was separated from the C57BL/6J (J) breeding colony at The Jackson Laboratory at F32 which split the B6 lineage into two branches. Trait differences have been described, mainly between these lineages[5–7]. (Table 1). The number of variants distinguishing J versus N substrains is estimated between 10K and 20K (SNPs and indels)[6,8,9]. Causal gene variants for most phenotypic differences have yet to be identified and will require RCCs. Functional variants in Nnt and Crb1 with a large impact on phenotypes were identified[6] and a Cyfip2 variant demonstrated a pleiotropic impact on psychostimulant behaviors and binge eating[10,11]. Discovery of Cyfip2 required only 100 to 200 RCC individuals. However, some traits in B6 RCCs could exhibit polygenic inheritance underlying trait variation. A reasonable strategy would be to assume a one-locus model and power the study based on the parental effect size using R/qtlDesign[12]. If no QTLs or suggestive QTLs [e.g., a Logarithm of the Odds (LOD) score greater than 3 [13] or the permissive, yet conventional “suggestive” genome-wide p-value of less than 0.63 based on permutation analysis[14]] are identified, then multiple, smaller effect loci could underlie parental substrain differences and a large sample size will be required.
Ultimately, the lower limit of genetic complexity in a RCC with regard to variant load depends on genomic sequencing and bioinformatics to resolve the causal variant(s). Approaches such as ENU mutagenesis employed a similar mapping strategy for decades and genomic sequencing now provides the ability to map causal variants[15]. If parental substrains do not phenotypically deviate, then ENU mutagenesis followed by a RCC can be used to map the phenodeviant followed by sequencing to identify the causal QTV[16]. Validation of predicted high impact variants (e.g., variants within/near genes possessing cis-eQTLs) is then conducted using gene editing (Figure 1, Panel A). If a candidate causal variant is not revealed by sequencing, then generation and targeted mapping with a cohort of RCC individuals using candidate variants and locus-specific markers is required to resolve candidate QTGs and QTVs[17].
RCC with tens of thousands of variants have yielded clear causal variants, suggesting that the upper limit of genetic complexity providing RCC advantages has not been reached, However, at some point, increasing genetic complexity defeats the advantages of a RCC. As a case in point, consider the number of variants per 20 Mb interval (indicated in parentheses; assuming a genome size of 2.6 billion bases and random placement of variants) when the variant load is 1,000 (8), 10,000 (76), 100,000 (756), or 1 million (7,564). Once the number of segregating variants distinguishing progenitor strains swells into hundreds of thousands or millions, the number of variants within a candidate interval becomes intractable. As the number of segregating variants increases, so does the genetic complexity of a locus and the overall genetic architecture of a trait, such that additional additive or non-additive loci contribute to trait variation and obscure single-gene/variant identification. Thus, theoretical consideration of the total number of variants is worthwhile. For lower variant limits in a RCC, investigators should assess the likelihood that sequencing alone can identify causal variants with high confidence among nearly isogenic strains. If there are one or two obvious candidate variants, it is worth considering skipping the mapping step and moving straight to gene validation. For upper limits, investigators should consider trait and QTL effect size, possible underlying genetic architecture, QTL precision (the size of candidate loci) and the number of genes and variants likely to be harbored by a QTL.
RCC Design and Exemplary Behavioral Differences among Mouse and Rat Substrains
A successful RCC experiment requires heritable trait differences between substrains, a compendium of sequence variants, a marker panel and genotyping strategy, and QTL mapping software to link marker genotypes with trait variation to identify high confidence genomic intervals containing QTGs and QTVs (Figure 1, Panel A).
The first critical step is to identify phenotypic differences between substrains. Judicious selection of parental strains with desired trait differences can be difficult. The most efficient strategy is to screen related sets of substrains for reported or suspected phenotypic differences and then focus on the most closely related pair exhibiting robust trait divergence. Identification of the most genetically similar phenodeviant pair is useful because the resulting RCC will segregate fewer genetic variants, making causal variant identification easier.
Mouse substrains displaying robust phenotypic differences such as sequenced C57BL/6 substrains (Table 1), are ripe for constructing RCCs and span multiple facets of biology and diseases, including alcohol [18], opioid[19], cocaine[10], nicotine[20], and methamphetamine behaviors[21–23], eating disorders[11], pain[5,24], stroke[25,26], circadian activity[27], and brain morphology, including corpus callosum[28].
Inbred rat strains have also been housed for hundreds of generations at over 20 breeding facilities worldwide which spawned several rat substrains (Table 1). Recent progress with rat genomic sequencing (Box 2) has made rat RCCs feasible and more efficient. Several rat substrains (genomic sequencing in progress) exhibit robust, heritable differences in addiction-, stress- and depression related traits. Two examples, SHR substrains and Wistar Kyoto (WKY)-derived substrains, are discussed below.
TEXT BOX 2. Genomic Tool Development in Rats.
Rat genome annotation and genomic tools are rapidly improving[92,93]. Whole genome sequencing (WGS) of additional inbred rat strains and substrains[94] will increase translational relevance and expand the number of complex disease and neurobehavioral traits evaluated with the RCC approach[95]. There is a coordinated effort from several NIH-funded projects (PIs: Chen and Williams, University of Tennessee Health Science Center; Dwinell, Medical College of Wisconsin; Akil and Li, University of Michigan) to sequence 100 inbred rat strains, including two of the rat RI panels (HXB and LEXF). Sequencing substrains is also part of this effort and several substrains such as WLI/Eer, WMI/Eer, LEW/Crl, LEW/SSNHsd, F344/NCrl, F344/DuCrl, and F344/NHsd have already been sequenced (Table 1). The SHR substrains, including SHRSP (stroke-prone) will be sequenced in the near future using both Illumina short reads and high molecular weight linked-read libraries, which barcodes long DNA molecules (e.g. 50–100 kb) and thus has the advantage of more accurately identifying large *structural variants. This sequencing effort also revealed assembly errors in the current rat reference genome, rn6 (H.Chen et al., Complex Trait Consortium and Rat Genome, 2019, San Diego, CA USA). A coordinated effort with the Vertebrate Genomes Sequencing project (Sanger) is generating data from one male inbred Brown Norway rat (BN/NHsdMcwi) to construct a new reference genome for the rat.
Cocaine self-administration in the SHR/NCrl substrain was characterized on several indices of addiction liability[29–33]. SHR/NCrl acquired cocaine self-administration faster, exhibited escalated operant cocaine intake across doses; displayed greater reinforcement and motivation for cocaine, and greater reactivity to cocaine cues in reinstatement of cocaine seeking, compared to inbred WKY/NCrl and outbred Wistar (WIS/Crl) rats. SHR/NCrl also self-administered more heroin[34], d-amphetamine[35], and methylphenidate[36] compared to WKY/NCrl and/or WIS/Crl. Additionally, adolescent SHR/NCrl females consumed more ethanol than Sprague Dawley (SD)/Crl control rats[37]. These behaviors in SHR/NCrl capture distinct hallmarks of increased drug abuse liability. Critically, the SHR/NHsd substrain does not self-administer more drugs of abuse versus WKY/NHsd, WIS/Hsd, and/or SD/Hsd, including d-amphetamine or methylphenidate compared to WKY/NHsd, WIS/Hsd, and/or SD/Hsd controls[38–40]. With regard to nicotine, SHR/NHsd rats self-administered more nicotine via lever presses than WKY/NHsd at a dose of 30 but not 15 μg/kg[40,41]. Nicotine responses in SHR/NCrl have not been tested. High historical phenotypic diversity and low genetic complexity between SHR/NCrl and SHR/NHsd are ideal for a RCC to map the genetic basis of addiction vulnerability traits.
The WKY rat strain is also a well-established model for depression as its behavior mirrors symptoms of human major depression and anxiety including depressed mood, excessive anxiety, loss of interest or pleasure, disturbed sleep and appetite, and low energy[42–50]. Chronic treatments with antidepressants[51], electroshock administration (model for electroconvulsive therapy)[52] and deep-brain stimulation[53] can all reverse these depression-like behaviors.
The WKY strain was developed as the normotensive control for SHR strains. Louis and Howes[54] demonstrated that the WKY strain was distributed to different vendors and universities between F12 and F17 generations of inbreeding. RH segregating in WKY rats at these filial generations likely contributed to the genetic divergence within and between WKY colonies (see Box 1). Indeed, WKY demonstrated genetic and behavioral differences[55,56], including in the forced swim test (FST; a model for antidepressant efficacy in rodents). These results motivated bi-directional breeding in FST immobility[57]. Rats with the highest immobility and lowest climbing scores in the FST were bred, producing the WKY More Immobile (WMI) line. Rats with the lowest immobility and highest climbing scores were bred, producing the WKY Less Immobile (WLI) line. Rats showing the most extreme FST behavior within each line were selected for breeding, specifically avoiding sibling mating until F5, when sibling mating was initiated. These strains have surpassed 40 generations of inbreeding.
WMIs reliably show greater FST immobility than the WLIs[58]. With the exception of fluoxetine, antidepressant treatment (and an enriched environment in adulthood) alleviates depressive-like behavior of WMIs[57,59]. Behavioral and hormonal responsiveness to acute and chronic stress also differ between substrains[59]. In addition, stress-reactive WMIs showed greater stress-enhanced fear learning and alcohol consumption versus WLIs in a model for post-traumatic stress disorder[60]. Importantly, WMI and WLI differ by only ~4,500 SNPs and ~40 large structural variants based on combined ~100x coverage of WGS on three different sequencing platforms. Most variants are located in noncoding regions but there are some intriguing mutations in exons (e.g. Pclo) or splicing sites (e.g., Rab1a, Slc01a2, Ryr3, Lyg1, and Nap1l1). Taken together, WMI and WLI substrains can be used as RCCs to evaluate genetic factors mediating susceptibility to depression, stress reactivity, comorbidity between stress reactivity and motivation, learning, depression, anxiety and aging.
Sequenced genomes and pre-existing genotyping strategies accelerate QTG and QTV identification. The increasing availability of whole genome sequences for phenodeviant mouse and rat substrains necessitates efficient and affordable genotyping platforms for new RCCs. Only 200 or so genetic markers are required to conduct an F2 mapping study (spacing every 20 to 40 Mb or 10 to 20 cM). Optimal markers should be chosen based on pilot experiments and flanking DNA sequences. Two genotyping strategies include array-based technologies and targeted DNA sequencing. The miniMUGA DNA microarray was designed by Fernando P.M. de Villena and colleagues at UNC Chapel Hill and is sold commercially by NeoGen Genomics, Inc. (Lincoln, NE USA). This array contains complementary probes targeting between 200 and 400 SNPs for crosses between any two of over 40 inbred mouse substrains originating from 10 different parental strains[61] (Table 1). Microarrays can also be custom-designed (e.g., with rat substrains) to suit individual investigator needs.
Genotyping via multiplexed, targeted DNA re-sequencing is a high throughput and cost-effective genotyping strategy[62]. In this approach, specific genomic regions are selected and amplified using region-specific primers and multiplexed PCR followed by next-generation sequencing. Hundreds of markers can be profiled in hundreds of individuals simultaneously. Primers are designed to amplify each genomic interval containing the marker (SNP or indel). Each marker region from every RCC individual is amplified by PCR, barcoded, and sequenced at sufficient read coverage (at least 100 reads per targeted variant locus) using a next generation sequencing platform[62]. For every marker and each individual, the genotype is determined based on variant calling following read alignment to a reference genome. Commercial options are available (e.g. MonsterPlex from Floodlight Genomics LLC, Knoxville, TN USA).
Once heritable substrain differences in a trait are identified and a genotyping strategy is selected, the RCC is generated by a classic backcross or F2 intercross. Marker genotypes and trait values (e.g., behavior and gene expression) are measured for each RCC individual. Power analysis (e.g. the R package qtlDesign[12]) takes into account the estimated trait heritability and the additive effect of a locus in order to determine the sample size required to detect a single QTL of a given effect size and a set power level. The R package R/qtl2[63] and GeneNetwork[64] permit inclusion of additive and/or interactive covariates (e.g., Treatment, Sex, Cohort, etc.)[65] to statistically define genomic intervals containing variants that influence trait expression. The strength of linkage across the genome is represented by LOD scores, significance thresholds are generated via permutation analysis, and confidence intervals (e.g., 1.5-LOD or Bayesian credible) define the locus size[3,66]. In many cases the simple genetic architecture of RCCs enables efficient identification of large-effect QTGs and QTVs. For more complex RCCs and/or QTL intervals with no obvious candidate variant(s), rapid fine mapping can be applied to overcome low QTL resolution (Box 3).
TEXT BOX 3. Fine Mapping in RCCs.
Because RCC traits exhibit a simple genetic inheritance on a nearly isogenic F2 background[10,11,67], fine mapping (if necessary) can be implemented immediately to overcome the coarse resolution of F2 QTLs. Fine mapping involves backcrossing and the introduction of new recombination events within a QTL interval to narrow down which subregions capture the phenotypic variance and thus, capture the causal QTGs and QTVs. Existing tools for fine-resolution QTL mapping such as advanced intercross lines, outcrosses (e.g., Diversity Outbred[96] but see Reduced Complexity Outcrosses section below), and interval-specific congenics are used to increase recombination events and improve QTL resolution, sometimes to near-single gene resolution[97–100]. Implementation of these fine mapping tools can take several years as they require several generations of breeding and unique genotyping assays. In contrast, fine mapping of a QTL in a RCC can begin with the very next generation of crossing, owing to a highly similar background and simplified genetic architecture of complex traits. Once a major QTL is identified in a RCC, F2 recombinant mice within the QTL interval are selected for immediate backcrossing and phenotypic screening at each consecutive generation containing new recombination events[3]. Because of the sparse density of informative markers, one limitation of fine mapping in the RCC is the potential to “run out” of markers before obtaining single-gene or, e.g.,1 Mb resolution. In this case, functional molecular analyses (e.g., transcriptome or proteome) can help further narrow the list of causal candidate genes before moving to validation. In other words, QTL mapping and DNA sequence are not always sufficient to pinpoint likely causal QTGs and QTVs. A systems genetic approach that employs cis- and trans-eQTL analysis (e.g., at the transcript or protein level) as well as transcript covariance analysis is always a welcome addition in establishing causality.
Candidate Variant Validation in a RCC: Necessity and Sufficiency
Validation of candidate QTGs/QTVs in a RCC is straightforward because the two highly similar genetic backgrounds provide the unique opportunity to demonstrate both necessity (via mutation correction[67] and sufficiency of the QTV (via mutation induction) with little risk that epistatic interactions will obscure the results. In contrast, nominating and validating QTVs is more difficult when using populations that emphasize increased genetic complexity[68]. When there are orders of magnitude more variants to sift through in nominating candidates, it is not always clear which genetic background is appropriate to demonstrate causality – this is especially true for the Diversity Outbred (DO) population that is composed of eight segregating genetic backgrounds[69]. The potential for epistasis and modifier loci to obfuscate predicted results is a genuine concern and is not reliably predicted based on typical sample sizes that are underpowered to detect pairwise epistasis. A seminal study exemplifying the power of epistasis showed that the detection, effect size and the directional effect of heterozygous knockout alleles for two GWAS genes (TCF7L2 and CACNA1C) differed across 30 different F1 backgrounds on which the heterozygous mutation was bred[70]. Even in a simpler scenario with a two-strain F2 cross, the potential is real for epistasis to obscure an allelic effect when placed on one of the parental strains (differing in millions of variants). The historical gold standard that approximates proof of QTG causality involves genetic complementation whereby mice heterozygous for a null mutation on one genetic background are crossed to the second background. If the effect of the knockout allele is rescued toward the wild-type ancestral level by the alternate strain allele, this demonstrates genetic complementation. A failure to complement as indicated by the retention of the mutant phenotype with the alternate allele is strong evidence that the causal QTG has been identified[71].
In the age of gene editing, one can provide stronger evidence for proof of the causal QTG[72] and/or QTV[67]. A major strength of RCCs is that they provide two similar genetic backgrounds with which to test for QTG/QTV causality. On the one hand, necessity can be demonstrated whereby the mutant allele (e.g., loss-of-function) is replaced (“corrected”) with the ancestral allele (e.g., regain of function) on the genetic background that normally harbors the mutation. For example, we identified a single intronic nucleotide deletion in Gabra2 (gene coding for the alpha 2 subunit of the GABA-A receptor) in the C57BL/6J substrain that caused a decrease in transcript and protein levels (gene expression is the quantitative trait exhibiting partial loss-of-function with the hypomorphic C57BL/6J allele) and was reversed by insertion of the corrected allele (regain of function) onto the same genetic background[67]. In a second example, a nonsynonymous missense SNP (proline->threonine) within the trace amine-associated receptor 1 (Taar1) of the DBA/2J strain was strongly implicated in higher methamphetamine intake in a subset of mice from the DBA/2J strain generated from 2001–2003 in oral intake of methamphetamine[21]. Replacement of the mutant allele in the high-selected line demonstrating high methamphetamine intake with the ancestral allele reduced intake toward wild-type (ancestral allele) levels, indicating that the mutant allele was necessary for increased methamphetamine intake[22]. On the other hand, sufficiency can be demonstrated whereby the mutant allele is edited onto the nearly identical genetic background that normally harbors the ancestral allele to show that the phenotype associated with the mutant allele can be induced on the genetic background that does not normally show it. To our knowledge, demonstration of both necessity and sufficiency of a QTV from a RCC has yet to be reported.
Reduced Complexity Advanced Intercrosses
Despite the advantages of RCCs, F2 intercrosses provide imprecise localization of QTLs and “rapid” fine mapping to resolve QTLs is still quite laborious (Box 3). A separate notion to improve QTL resolution in RCCs is the Reduced Complexity Advanced Intercross (RCAI) (Box 4) which involves continual intercrossing of unrelated individuals starting in F3 offspring and continuing for an infinite number of generations to introduce new recombination events and genetically unique individuals at each generation[73–76] (Figure 1, Panel D).
TEXT BOX 4. Advantages and Disadvantages of Reduced Complexity Advanced Intercrosses.
The RCAIs are advanced intercross lines produced from two closely related progenitor strains. We suggest that this approach will provide several advantages. First, cost-effective genotyping approaches are available, including exon capture and multiplexed PCR and sequencing[62]. Commercially available RCAIs would save the costs and time needed to generate, genotype, phenotype, and backcross F2 mice in order to fine map loci (if necessary). In addition, advanced intercrossing can improve mapping resolution to sub-Mb levels, depending on recombination frequency and the presence of markers that distinguish the progenitor strains. Random intercrossing of unrelated individuals in generating and maintaining RCAIs will yield a sufficient number of meiotic breaks within 10 to 15 generations (depending on the locus) to resolve the majority of individual QTVs [101]. Because of the large number of variants in genetically diverse crosses, only ~0.003% of variants are usually genotyped[96,102]. In contrast, the limited number of variants in an RCAI means that all variants can be evaluated cost-effectively. Genotyping ~100 animals will adequately interrogate the accumulated breakpoints in the RCAIs. Reduced complexity outcross strains (RCOs) are created by intercrossing animals from several closely related parental stocks. Compared to the RCAIs, the RCOs offer more opportunity for discovery but at the price of added genetic complexity. Two examples of genetically complex populations include Diversity Outbred (DO) mice originating from eight diverse founder strains and segregating for ~60 million variants[96] and the LG/J x SM/J Advanced Intercross Line (AIL) comprising two founder strains that have been continually outcrossed up to generation G56 at the time of the latest publication[99]. Each individual within these lines is genetically unique and thus, must be genotyped in order to conduct QTL mapping.
There are some limitations to the RCAI approach. First, sufficient power to detect phenotypic differences may require additional animals, depending on the trait variance and effect sizes. In addition, a lack of markers at narrowed loci can limit QTL resolution of RCAIs[3]. As an example, we mapped and narrowed a 30 Mb QTL to 3.7 Mb via fine mapping (Box 2) but we were unable to validate additional markers to detect additional recombination events. Some variants may also not be separable because of the presence of intervening recombination “cold spots”, strong selective advantage of linked variants, or transmission ratio distortion[103]. The final limitation (and perhaps the most challenging one) is that the resource has to be created for each pair or pairs of closely related strains or substrains.
Decreased diversity of RCAIs, combined with accumulation of numerous meiotic breakpoints for fine mapping of QTVs permits a nearly “comprehensive” genetic analysis of complex traits, in the sense that the contribution of all segregating variants to a phenotype within a population can be evaluated. The resulting insights will address several conundrums in mammalian genetics, including sources of missing heritability[77,78], percentage of variants that have an impact on complex traits[79,80], whether truly neutral variants exist[81], pleiotropy of QTVs among complex disease-relevant traits[82] and the omnigenic hypothesis[83]. Reduced complexity of the RCAIs also means reduced diversity, and so the resource may not answer these current issues in a generally applicable way. Nevertheless, deep phenotyping of the RCAIs, including molecular phenotypes and omics level analyses, combined with comprehensive QTV mapping of clinical traits, at the very least offers the prospect of a complete and satisfying evaluation of these issues within a simple, genetically defined segregating population[17], a prospect far beyond the reach of approaches with higher levels of genetic diversity. Another advantage of RCAIs relates to the observation that most QTVs are found in cis-regulatory regions and can be located at a considerable distance from the corresponding gene with many interposed “silent” variants[84,85], thus hindering QTG identification. QTLs derived from RCOs can quickly zero in on small regions with greater than two orders of magnitude reduced complexity which simplifies QTG/QTV identification. Epistasis contributes significantly to complex traits, but the large number of pair-wise combinations blunts statistical power to detect reliable interactions[86]. Decreased genetic complexity of RCOs makes epistatic analysis more tractable in the correspondingly rarer cases in which it clearly exists.
Concluding Remarks
RCCs offer key advantages over alternative mapping approaches that boost genetic complexity[69]. RCCs increase the speed and strength of evidence of QTG/QTV identification. However, RCCs are limited in the number of genetic factors that can be identified in a single cross as well as the number of phenotypes that can be measured. These disadvantages are offset by the rich array of existing rodent inbred substrains and the possibility of purposefully establishing new substrains. Low QTL yield and poor resolution in RCCs are mitigated by genetically simple loci, efficiency of fine mapping, and strength of evidence for causal identification. Ultimately, crosses comprising both decreased and increased genetic diversity offer unique, complementary advantages to complex trait analysis that should be fully pursued, given the emerging number of genome sequences and tools available for both approaches (see Outstanding Questions).
OUTSTANDING QUESTIONS.
Will the molecular mechanisms and biological pathways perturbed by gene variants in the RCC inform complex traits and diseases in humans?
How does genetic variation in the mutation spectrum and underlying mutational mechanisms differ among inbred rodent strains and how might this impact: germline mutation rate; generation of substrains; and the types of variants that drift to fixation?
What is the upper variant limit that defines the maximum utility of an RCC?
Can RCCs facilitate our understanding of the highly complex role that noncoding variants play in gene regulation?
As we discover new functional variants in B6 and other substrains, could “correcting” a portion or all of these mutations in a single parental inbred line serve as a useful resource?
What is the optimal number of outcrosses in a RCAI and how does this relate to the degree of genetic complexity across pairs of substrains?
Can we iteratively increase RCC complexity beyond a single major locus to improve our understanding of additive and epistatic interactions among genetic variants that influence biological pathways underlying complex traits and diseases?
Supplementary Material
HIGHLIGHTS.
Discovery of causal genes and variants underlying complex trait variation using traditional rodent crosses is limited by the combination of high genetic complexity and modest locus resolution.
Whole genome sequencing of nearly isogenic rodent substrains now enables a new type of cross— a reduced complexity cross (RCC) that can be highly efficient for variant discovery.
Residual heterozygosity and spontaneous mutations between rodent breeding colonies make RCCs an efficient approach for identifying quantitative trait genes and variants.
Compared to traditional crosses, RCCs segregate orders of magnitude fewer variants and accelerate causal quantitative trait gene and variant identification.
Gene editing strengthens causal gene discovery by providing an efficient means to demonstrate both necessity and sufficiency of variants on substrain backgrounds.
ACKNOWLEDGEMENTS
R01DA039168 (C.D.B.), R01CA22160 (M.I.D. & C.D.B.), U01DA041602 (D.J.S.), U01DA047638 (H.C. and R.W.W.), U01AA016662 (R.W.W.), and P30DA044223 (R.W.W. and M.K.M.)
GLOSSARY BOX
- Expression QTL
A chromosomal region associated with variance in gene expression. A cis-eQTL is located near or within the cognate gene. A trans-eQTL is located far away from the position of the cognate gene on the same chromosome or on a different chromosome.
- Genetic architecture
All DNA variants that influence trait variation, including the number, effect size, mode of inheritance, and their additive or epistatic interactions.
- Genetic drift
A change in allele frequency caused by the cumulative fixation of residually heterozygous variants (inbred strains) and spontaneous, de novo mutations following prolonged physical separation (vendors, institutes). Generations at earlier stages of inbreeding will have more residual heterozygosity and thus a larger number of variants that become fixed. The greater number of generations separating each population, the greater number of spontaneous mutations that will become fixed.
- Inbred strain
An isogenic, or genetically identical, strain with homozygous alleles at every DNA nucleotide. A strain is considered 98% inbred after 20 generations of strict brother/sister mating[87].
- Inbred substrain
Distinct, isogenic strains that arose via genetic drift following separation from a common lineage, typically at different institutions or breeding facilities. The level of substrain divergence depends on the number of generations of inbreeding that separates the last common ancestor and the number of generations separating each substrain. Substrains are differentiated by abbreviations that indicate their origin from different labs or commercial vendors. For example, the designations J, N, Crl and Tac represent sublines from The Jackson Laboratory, the NIH, Charles River Laboratories, and Taconic Biosciences, respectively.
- Logarithm of the Odds (LOD)
Statistical measure of the probability that a polymorphic genetic marker is physically linked to, and thus co-inherited with one or more causal variants contributing to overall phenotypic variance of a complex trait. A higher LOD score indicates a stronger statistical signal of linkage. A LOD score is akin to a negative logP value and thus, a LOD score of 3 indicates that the odds that the genetic marker and the causal variant(s) are linked are 1000 greater than the odds that they are not.
- Quantitative trait locus (QTL)
A statistically defined region of the genome for which there is a strong association between genotype and trait variation such that inheritance of one set of parental alleles in a genetic population is associated with higher trait expression.
- Quantitative trait gene (QTG)
The causal gene underlying a QTL.
- Quantitative trait variant (QTV)
The causal variant underlying a QTL.
- Reduced Complexity Cross (RCC)
A forward genetic cross (typically an F2 cross) between individuals from two substrains that contain very low genetic diversity that is used to generate a population of recombinant individuals for QTL mapping.
- Reduced Complexity Advanced Intercross (RCAI)
Animals obtained by intercrossing progeny originating from two closely related strains over multiple generations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Baud A and Flint J (2017) Identifying genes for neurobehavioural traits in rodents: progress and pitfalls. Dis.Model.Mech 10, 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant CD (2011) The blessings and curses of C57BL/6 substrains in mouse genetic studies. Ann.N.Y.Acad.Sci 1245, 31–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant CD et al. (2018) Reduced complexity cross design for behavioral genetics In Molecular-Genetic and Statistical Techniques for Behavioral and Neural Research Book, Section vols. (Gerlai RT, ed), pp. 165–190 [Google Scholar]
- 4.Rosen GD et al. (2013) Bilateral subcortical heterotopia with partial callosal agenesis in a mouse mutant. Cereb. Cortex 23, 859–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant CD et al. (2008) Behavioral differences among C57BL/6 substrains: implications for transgenic and knockout studies. J.Neurogenet 22, 315–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon MM et al. (2013) A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 14, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashbrook DG et al. (2018) Post-genomic behavioral genetics: From revolution to routine. Genes Brain Behav. 17, e12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keane TM et al. (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yalcin B et al. (2011) Sequence-based characterization of structural variation in the mouse genome. Nature 477, 326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar V et al. (2013) C57BL/6N mutation in Cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science 342, 1508–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkpatrick SL et al. (2017) Cytoplasmic FMR1-Interacting Protein 2 Is a Major Genetic Factor Underlying Binge Eating. Biol.Psychiatry 81, 757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sen S et al. (2007) R/qtlDesign: inbred line cross experimental design. Mamm.Genome 18, 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lander E and Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature genetics 11, 241–7 [DOI] [PubMed] [Google Scholar]
- 14.Broman KW and Sen S (2009) A Guide to QTL Mapping with R/qtl, (1st edn) Springer-Verlag, Inc. [Google Scholar]
- 15.Bull KR et al. (2013) Unlocking the bottleneck in forward genetics using whole-genome sequencing and identity by descent to isolate causative mutations. PLoS Genet. 9, e1003219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyoshi C et al. (2019) Methodology and theoretical basis of forward genetic screening for sleep/wakefulness in mice. Proc. Natl. Acad. Sci. U.S.A 116, 16062–16067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant CD et al. (2018) Reduced complexity cross design for behavioral genetics, Molecular-Genetic and Statistical Techniques for Behavioral and Neural ResearchAcademic Press, London, United Kingdom. [Google Scholar]
- 18.Mulligan MK et al. (2008) Alcohol trait and transcriptional genomic analysis of C57BL/6 substrains. Genes Brain Behav. 7, 677–689 [DOI] [PubMed] [Google Scholar]
- 19.Kirkpatrick SL and Bryant CD (2015) Behavioral architecture of opioid reward and aversion in C57BL/6 substrains. Front.Behav.Neurosci 8, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akinola LS et al. (2019) C57BL/6 Substrain Differences in Pharmacological Effects after Acute and Repeated Nicotine Administration. Brain Sci 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed C et al. (2017) A Spontaneous Mutation in Taar1 Impacts Methamphetamine-Related Traits Exclusively in DBA/2 Mice from a Single Vendor. Front Pharmacol 8, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stafford AM et al. (2019) Taar1 gene variants have a causal role in methamphetamine intake and response and interact with Oprm1. Elife 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi X et al. (2016) Genetic Polymorphisms Affect Mouse and Human Trace Amine-Associated Receptor 1 Function. PLoS ONE 11, e0152581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryant CD et al. (2019) C57BL/6 substrain differences in inflammatory and neuropathic nociception and genetic mapping of a major quantitative trait locus underlying acute thermal nociception. Mol Pain DOI: 10.1177/1744806918825046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nowak TS and Mulligan MK (2019) Impact of C57BL/6 substrain on sex-dependent differences in mouse stroke models. Neurochem. Int 127, 12–21 [DOI] [PubMed] [Google Scholar]
- 26.Zhao L et al. (2019) Substrain- and sex-dependent differences in stroke vulnerability in C57BL/6 mice. J. Cereb. Blood Flow Metab 39, 426–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corty RW et al. (2018) Mean-Variance QTL Mapping Identifies Novel QTL for Circadian Activity and Exploratory Behavior in Mice. G3 (Bethesda) 8, 3783–3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wahlsten D (1989) Deficiency of the corpus callosum: incomplete penetrance and substrain differentiation in BALB/c mice. J. Neurogenet 5, 61–76 [DOI] [PubMed] [Google Scholar]
- 29.Harvey RC et al. (2011) Methylphenidate treatment in adolescent rats with an attention deficit/hyperactivity disorder phenotype: cocaine addiction vulnerability and dopamine transporter function. Neuropsychopharmacology 36, 837–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Somkuwar SS et al. (2013) Adolescent atomoxetine treatment in a rodent model of ADHD: effects on cocaine self-administration and dopamine transporters in frontostriatal regions. Neuropsychopharmacology 38, 2588–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan CJ et al. (2014) Cocaine-seeking behavior in a genetic model of attention-deficit/hyperactivity disorder following adolescent methylphenidate or atomoxetine treatments. Drug Alcohol Depend 140, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jordan CJ et al. (2016) Adolescent d-amphetamine treatment in a rodent model of attention deficit/hyperactivity disorder: impact on cocaine abuse vulnerability in adulthood. Psychopharmacology (Berl.) 233, 3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan CJ et al. (2016) Adolescent D-amphetamine treatment in a rodent model of ADHD: Pro-cognitive effects in adolescence without an impact on cocaine cue reactivity in adulthood. Behav. Brain Res 297, 165–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller ML et al. (2018) Ventral striatal regulation of CREM mediates impulsive action and drug addiction vulnerability. Mol. Psychiatry 23, 1328–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.dela Peña I et al. (2015) Gene expression profiling in the striatum of amphetamine-treated spontaneously hypertensive rats which showed amphetamine conditioned place preference and self-administration. Arch. Pharm. Res 38, 865–875 [DOI] [PubMed] [Google Scholar]
- 36.dela Peńa IC et al. (2011) Methylphenidate self-administration and conditioned place preference in an animal model of attention-deficit hyperactivity disorder: the spontaneously hypertensive rat. Behav Pharmacol 22, 31–39 [DOI] [PubMed] [Google Scholar]
- 37.Berger DF et al. (2010) The effects of strain and prenatal nicotine exposure on ethanol consumption by adolescent male and female rats. Behav. Brain Res 210, 147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer AC et al. (2010) Genetics of novelty seeking, amphetamine self-administration and reinstatement using inbred rats. Genes Brain Behav. 9, 790–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marusich JA et al. (2011) Strain differences in self-administration of methylphenidate and sucrose pellets in a rat model of attention-deficit hyperactivity disorder. Behav Pharmacol 22, 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han W et al. (2017) Social learning promotes nicotine self-administration by facilitating the extinction of conditioned aversion in isogenic strains of rats. Sci Rep 7, 8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H et al. (2012) Genetic factors control nicotine self-administration in isogenic adolescent rat strains. PLoS ONE 7, e44234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baum AE et al. (2006) Test- and behavior-specific genetic factors affect WKY hypoactivity in tests of emotionality. Behav. Brain Res 169, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dugovic C et al. (2000) Sleep in the Wistar-Kyoto rat, a putative genetic animal model for depression. Neuroreport 11, 627–631 [DOI] [PubMed] [Google Scholar]
- 44.Malkesman O et al. (2005) Reward and anxiety in genetic animal models of childhood depression. Behav. Brain Res 164, 1–10 [DOI] [PubMed] [Google Scholar]
- 45.Paré WP (1994) Open field, learned helplessness, conditioned defensive burying, and forced-swim tests in WKY rats. Physiol. Behav 55, 433–439 [DOI] [PubMed] [Google Scholar]
- 46.Paré WP (1994) Hyponeophagia in Wistar Kyoto (WKY) rats. Physiol. Behav 55, 975–978 [DOI] [PubMed] [Google Scholar]
- 47.Paré WP and Redei E (1993) Depressive behavior and stress ulcer in Wistar Kyoto rats. J. Physiol. Paris 87, 229–238 [DOI] [PubMed] [Google Scholar]
- 48.Redei EE et al. (2001) Paradoxical hormonal and behavioral responses to hypothyroid and hyperthyroid states in the Wistar-Kyoto rat. Neuropsychopharmacology 24, 632–639 [DOI] [PubMed] [Google Scholar]
- 49.Solberg LC et al. (2004) Sex- and lineage-specific inheritance of depression-like behavior in the rat. Mamm. Genome 15, 648–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solberg LC et al. (2001) Altered hormone levels and circadian rhythm of activity in the WKY rat, a putative animal model of depression. Am. J. Physiol. Regul. Integr. Comp. Physiol 281, R786–794 [DOI] [PubMed] [Google Scholar]
- 51.Jeannotte AM et al. Desipramine Modulation of a-, g-Synuclein, and the Norepinephrine Transporter in an Animal Model of Depression. [DOI] [PubMed]
- 52.Kyeremanteng C et al. (2012) A Study of Brain and Serum Brain-Derived Neurotrophic Factor Protein in Wistar and Wistar-Kyoto Rat Strains after Electroconvulsive Stimulus. Pharmacopsychiatry 45, 244–249 [DOI] [PubMed] [Google Scholar]
- 53.Falowski SM et al. (2011) An evaluation of neuroplasticity and behavior after deep brain stimulation of the nucleus accumbens in an animal model of depression. Neurosurgery 69, 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Louis WJ and Howes LG (1990) Genealogy of the spontaneously hypertensive rat and Wistar-Kyoto rat strains: implications for studies of inherited hypertension. J. Cardiovasc. Pharmacol 16 Suppl 7, S1–5 [PubMed] [Google Scholar]
- 55.Kurtz TW et al. (1989) Molecular evidence of genetic heterogeneity in Wistar-Kyoto rats: implications for research with the spontaneously hypertensive rat. Hypertension 13, 188–192 [DOI] [PubMed] [Google Scholar]
- 56.Paré WP and Kluczynski J (1997) Differences in the stress response of Wistar-Kyoto (WKY) rats from different vendors. Physiol. Behav 62, 643–648 [DOI] [PubMed] [Google Scholar]
- 57.Will CC et al. (2003) Selectively bred Wistar-Kyoto rats: an animal model of depression and hyper-responsiveness to antidepressants. Mol. Psychiatry 8, 925–932 [DOI] [PubMed] [Google Scholar]
- 58.Andrus BM et al. (2012) Gene expression patterns in the hippocampus and amygdala of endogenous depression and chronic stress models. Mol. Psychiatry 17, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mehta-Raghavan NS et al. (2016) Nature and nurture: environmental influences on a genetic rat model of depression. Transl Psychiatry 6, e770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim PH et al. (2018) Genetic Model to Study the Co-Morbid Phenotypes of Increased Alcohol Intake and Prior Stress-Induced Enhanced Fear Memory. Front Genet 9, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shorter JR et al. (2019) Whole Genome Sequencing and Progress Toward Full Inbreeding of the Mouse Collaborative Cross Population. G3 (Bethesda) 9, 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wingo TS et al. (2017) MPD: multiplex primer design for next-generation targeted sequencing. BMC Bioinformatics 18, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broman KW et al. (2019) R/qtl2: Software for Mapping Quantitative Trait Loci with High-Dimensional Data and Multiparent Populations. Genetics 211, 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulligan MK et al. (2017) GeneNetwork: A Toolbox for Systems Genetics. Methods Mol.Biol 1488, 75–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broman KW et al. (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics (Oxford, England) 19, 889–90 [DOI] [PubMed] [Google Scholar]
- 66.Mott R and Flint J (2013) Dissecting quantitative traits in mice. Annu.Rev.Genomics Hum.Genet 14, 421–439 [DOI] [PubMed] [Google Scholar]
- 67.Mulligan MK et al. (2019) Identification of a functional non-coding variant in the GABAA receptor α2 subunit of the C57BL/6J mouse reference genome: Major implications for neuroscience research. Frontiers in Genetics DOI: 10.3389/fgene.2019.00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Czechanski A et al. (2014) Derivation and characterization of mouse embryonic stem cells from permissive and nonpermissive strains. Nat Protoc 9, 559–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saul MC et al. (2019) High-Diversity Mouse Populations for Complex Traits. Trends Genet. 35, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sittig LJ et al. (2016) Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 91, 1253–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yalcin B et al. (2004) Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat.Genet 36, 1197–1202 [DOI] [PubMed] [Google Scholar]
- 72.Turner TL (2014) Fine-mapping natural alleles: quantitative complementation to the rescue. Mol.Ecol 23, 2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gonzales NM and Palmer AA (2014) Fine-mapping QTLs in advanced intercross lines and other outbred populations. Mamm Genome 25, 271–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chesler EJ (2014) Out of the bottleneck: the Diversity Outcross and Collaborative Cross mouse populations in behavioral genetics research. Mamm Genome 25, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mott R and Flint J (2002) Simultaneous detection and fine mapping of quantitative trait loci in mice using heterogeneous stocks. Genetics 160, 1609–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Solberg Woods LC and Palmer AA (2019) Using Heterogeneous Stocks for Fine-Mapping Genetically Complex Traits In Rat Genomics 2018 (Hayman GT et al. , eds), pp. 233–247, Springer; New York: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Génin E (2019) Missing heritability of complex diseases: case solved? Hum Genet DOI: 10.1007/s00439-019-02034-4 [DOI] [PubMed] [Google Scholar]
- 78.Young AI (2019) Solving the missing heritability problem. PLoS Genet 15, e1008222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sohail M et al. (2019) Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yengo L et al. (2018) Meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Hum Mol Genet 27, 3641–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pouyet F et al. (2018) Background selection and biased gene conversion affect more than 95% of the human genome and bias demographic inferences. eLife 7, e36317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Watanabe K et al. (2019) A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet 51, 1339–1348 [DOI] [PubMed] [Google Scholar]
- 83.Boyle EA et al. (2017) An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gallagher MD and Chen-Plotkin AS (2018) The Post-GWAS Era: From Association to Function. Am J Hum Genet 102, 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Visscher PM et al. (2017) 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 101, 5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritchie MD and Van Steen K (2018) The search for gene-gene interactions in genome-wide association studies: challenges in abundance of methods, practical considerations, and biological interpretation. Ann Transl Med 6, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taft RA et al. (2006) Know thy mouse. Trends Genet. 22, 649–653 [DOI] [PubMed] [Google Scholar]
- 88.Ness RW et al. (2012) Nuclear gene variation in wild brown rats. G3 (Bethesda) 2, 1661–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deinum EE et al. (2015) Recent Evolution in Rattus norvegicus Is Shaped by Declining Effective Population Size. Mol. Biol. Evol 32, 2547–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lynch M (2010) Evolution of the mutation rate. Trends Genet. 26, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Uchimura A et al. (2015) Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res. 25, 1125–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tutaj M et al. (2019) Rat Genome Assemblies, Annotation, and Variant Repository. Methods Mol. Biol 2018, 43–70 [DOI] [PubMed] [Google Scholar]
- 93.Laulederkind SJF et al. (2019) Rat Genome Databases, Repositories, and Tools. Methods Mol. Biol 2018, 71–96 [DOI] [PubMed] [Google Scholar]
- 94.Hermsen R et al. (2015) Genomic landscape of rat strain and substrain variation. BMC Genomics 16, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parker CC et al. (2014) Rats are the smart choice: Rationale for a renewed focus on rats in behavioral genetics. Neuropharmacology 76 Pt B, 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saul MC et al. (2019) High-Diversity Mouse Populations for Complex Traits. Trends Genet 35, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yazdani N et al. (2015) Hnrnph1 Is A Quantitative Trait Gene for Methamphetamine Sensitivity. PLoS Genet. 11, e1005713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheng R et al. (2010) Genome-wide association studies and the problem of relatedness among advanced intercross lines and other highly recombinant populations. Genetics 185, 1033–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gonzales NM et al. (2018) Genome wide association analysis in a mouse advanced intercross line. Nat Commun 9, 5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shirley RL et al. (2004) Mpdz is a quantitative trait gene for drug withdrawal seizures. Nature neuroscience 7, 699–700 [DOI] [PubMed] [Google Scholar]
- 101.Darvasi A and Soller M (1995) Advanced intercross lines, an experimental population for fine genetic mapping. Genetics 141, 1199–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Svenson KL et al. (2012) High-resolution genetic mapping using the mouse diversity outbred population. Genetics 190, 437–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chesler EJ et al. (2016) Diversity Outbred Mice at 21: Maintaining Allelic Variation in the Face of Selection. G3 (Bethesda) 6, 3893–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pajer K et al. (2012) Discovery of blood transcriptomic markers for depression in animal models and pilot validation in subjects with early-onset major depression. Transl Psychiatry 2, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.