

Abstract
Background:
Cyclin-dependent kinase 12 (CDK12) loss occurs in 3–7% of metastatic prostate cancer patients and is characterized by a genomic instability signature, but the clinical implications of CDK12 loss are not well established.
Objective:
To determine the clinical course of patients with CDK12 mutant advanced prostate cancer compared with other genomic subtypes.
Design, setting, and participants:
A retrospective analysis of data from three academic medical centers, including 317 patients with advanced prostate cancer and prior next-generation sequencing from tumor tissue (n = 172) or circulating tumor DNA (n = 145), was performed. Forty-six patients had CDK12 mutations; 34 had biallelic CDK12 loss (79%).
Outcome measurements and statistical analysis:
Patients were stratified by mutation status (CDK12, homologous recombination deficiency [HRD; BRCA1/2 and ATM], TP53, and other cohort). The Kaplan-Meier method was used to evaluate time to event outcomes: time to development of metastatic disease, time to development of castration resistance, and time to prostate-specific antigen (PSA) progression after first-line androgen receptor pathway inhibitor (ARPI) therapy in a patient subset.
Results and limitations:
The median follow-up was 66.6 mo. Patients with CDK12 mutant prostate cancer exhibited shorter time to metastasis (median = 34.9 mo, p = 0.004) and development of castration-resistant disease (median = 32.7 mo, p < 0.001), compared with other genomic subtypes, with shorter time to PSA progression on first-line ARPI treatment of metastatic castration-resistant disease (median = 3.6 mo, p = 0.0219). CDK12 mutant patients did not have overall shorter time on treatment compared with other mutation subgroups, and CDK12 status did not demonstrate statistical significance in multivariate analysis. Limitations include variable center-dependent practice patterns and heterogeneity due to combining tumor and liquid biopsy data.
Conclusions:
Our data suggest that advanced prostate cancers harboring CDK12 mutations display aggressive clinical behavior, underscoring the need to fully delineate the molecular and clinical characteristics, and appropriate therapeutic approaches for distinct subtypes of advanced prostate cancers.
Patient summary:
In this report, we evaluate the clinical characteristics and outcomes of patients with prostate cancer and CDK12 mutation in their tumors. These patients seem to have more aggressive disease, with more high-grade Gleason ≥8 cancers and shorter time to developing metastatic cancer. Cases of advanced CDK12-mutated prostate cancer may warrant consideration of therapy intensification or combination approaches.
Keywords: Genomics, Next-generation sequencing, Prostate cancer, Cyclin-dependent kinase 12
1. Introduction
Metastatic castration-resistant prostate cancer (mCRPC) remains a lethal disease despite six Food and Drug Administration–approved therapies that prolong survival. While whole-exome and -genome sequencing studies have provided insights into the heterogeneous landscape of mutations and structural variants within advanced prostate cancer [1,2], the impact of these genomic alterations on clinical outcomes requires further investigation [3,4]. Recently, a novel molecular subtype of advanced prostate cancer harboring cyclin-dependent kinase 12 (CDK12) mutations has been reported in 3–7% of patients with mCRPC [1,2,5]. CDK12 functions in transcriptional regulation and RNA splicing [6], and regulates DNA damage repair genes involved in homologous recombination (HR [ie, BRCA1 and ATM]), suppresses intronic polyadenylation, and may increase susceptibility to poly ADP-ribose polymerase (PARP) inhibitors [7–9]. Recently, we and others showed that biallelic CDK12 loss in prostate cancer patients results in genomic instability and increased tandem duplications [2,5], which is also observed in CDK12-mutated ovarian cancer [10,11], but is distinct from tumors characterized by BRCA2 loss [2].
PTEN loss, RB loss, and TP53 mutations have been associated with poor outcomes in mCRPC [3,12–14]. The effects of alterations in the canonical HR gene BRCA2 remain an area of ongoing investigation [15]. BRCA2 mutations lead to decreased cancer-specific survival and poor clinical outcomes in the mCRPC setting, with a variable impact on response to therapy [16–18]. However, the prognostic relevance of CDK12 mutations remains unexplored. Therefore, we evaluated the clinical characteristics and outcomes of a multicenter cohort of patients with CDK12-mutated prostate cancer, comparing this genomic subtype with prostate cancers characterized by deficiency in canonical HR genes (ie, BRCA1, BRCA2, and ATM) and TP53.
2. Patients and methods
We conducted a retrospective chart review of patients with advanced prostate cancer at the University of Michigan (MI), University of California San Francisco (UCSF), and University of British Columbia (UBC). University of Washington patients sequenced at MI via Stand up to Cancer (SU2C) were included in the MI cohort. CDK12 mutant and HRD patients were identified from each institution’s available next-generation sequencing (NGS) and clinical data. The remaining patients had previously undergone NGS analysis and were selected consecutively.
All participating sites received Institutional Review Board approval for this study. Deidentified clinical patient data and somatic mutation status of select genes were shared between institutions in a Health Insurance Portability and Accountability Act-compliant fashion.
NGS was performed on patients’ metastatic prostate cancer biopsy samples, primary prostatectomy samples, or plasma cell-free DNA (cfDNA). At MI, metastatic biopsies were analyzed (n = 94) via the CLIA/CAP–approved MIONCOSEQ NGS program (both tumor and germline genomic aberrations were assessed). At UCSF, NGS was performed with the UCSF500 Cancer Gene Panel or Foundation Medicine platform on metastatic biopsy samples (n = 69) and primary prostatectomy samples (n = 9). At UBC, cfDNA from patients (n = 145) was subjected to deep targeted sequencing with a 72-gene panel, as previously described (see Fig. 1A) [18].
Fig. 1 –

(A) CONSORT diagram depicting the patients included in this cohort based on NGS source and frequency of each mutation group. Patients were stratified by genomic mutation types. Graphs depict the proportion of patients with (B) Gleason score ≥8 or <8 at diagnosis and (C) localized (M0) or metastatic disease (M1) at diagnosis for each subgroup.
CDK12 = cyclin-dependent kinase 12; ctDNA = circulating tumor DNA; HRD = homologous recombination deficiency; mCRPC = metastatic castration-resistant prostate cancer; MI = University of Michigan; NGS = next-generation sequencing; n.s. = nonsignificant; pts = patients; UCSF = University of California San Francisco, UBC = University of British Columbia; UCSF = University of California San Francisco.
**p = 0.009 by chi-square testing.
Given the relatively low frequency of CDK12 biallelic loss in this patient population, we combined the metastatic tumor biopsy and liquid biopsy datasets. Importantly, same-patient cfDNA and prostate cancer tumor biopsies have demonstrated high concordance for detected somatic alterations [19,20]. However, differing outcomes have been reported regarding patients with mCRPC and DNA damage repair alterations [16,18,21]. To reduce heterogeneity between the tumor and liquid biopsy cohorts, UBC patients with germline HR deficiency (HRD) mutations were included in this analysis. Patients with somatic HRD mutations were excluded given that: (1) a higher circulating tumor DNA (ctDNA) burden is required to confidently call somatic genomic alterations and (2) a high ctDNA fraction already represents patients with a differential prognosis. However, patients for whom a germline HRD mutation was identified in the leukocyte DNA were still included even with a ctDNA fraction of <2%.
Molecular subgroups were as follows: the CDK12 cohort included any CDK12 alteration (Fig. 2 and Supplementary Table 1). The HRD cohort (n = 60) included BRCA1 (n = 4, 6.7%), BRCA2 (n = 46, 77%), or ATM (n = 10, 17%) mutations (Supplementary Table 2). BRCA1/2 and ATM alterations were heterogeneous, with deletions, point mutations, and frameshift mutations; 36 of 60 (60%) patients harbored germline mutations. TP53 alterations were assigned to the TP53 cohort; those with both CDK12 and TP53 mutations were included in the CDK12 cohort. All other patients were assigned to the other cohort. We excluded patients with mismatch repair mutations [22] and patients whose molecular testing failed internal quality control measures. UBC patient samples with ctDNA fraction ≥2% and no more than monoallelic CDK12, HRD, or TP53 alteration were classified into the other cohort.
Fig. 2 –

Schematic of CDK12 mutations identified in the patient cohort. Mutations were primarily truncation mutations in the RS, PRM, or kinase domains, or alternatively missense mutations in the kinase domain. Additional details regarding the CDK12 mutations are outlined in Supplementary Table 1.
CDK12 = cyclin-dependent kinase 12; RS = arginine-serine domain; PRM = proline-rich motif.
Data were obtained from electronic medical records. The first date of availing clinical and genomic data was January 1, 1988; the data cutoff date was March 16, 2018. Patients were followed for each outcome until the date of death or last known follow-up. Median follow-up was 66.6 mo.
2.1. Statistical analysis
Demographic and clinical characteristics were summarized by genomic mutation types using descriptive statistics. Comparison of the continuous variables among mutation types was assessed by the analysis of variance. When normality assumption did not hold, the Kruskal-Wallis test was used. Chi-square test was used to evaluate the statistical association between each categorical variable and mutation types. Statistical significance was declared at p < 0.05. All statistical analyses were completed using R software (https://www.r-project.org/).
The Kaplan-Meier method and log-rank tests were used to characterize the relationship between each time to event outcome and genetic mutation types. Time to event outcomes included time from diagnosis to development of metastatic disease in patients presenting with localized disease, and time from diagnosis to development of castration-resistant prostate cancer (CRPC) in patients with localized and metastatic disease. Castration resistance was defined by two or more consecutively rising serum prostate-specific antigen (PSA) values and/or development of new radiographic metastases in the setting of continuous androgen deprivation therapy. In the UBC cohort, time to PSA progression (TTPP) on first-line androgen receptor (AR) pathway inhibitors (ARPIs: abiraterone acetate and enzalutamide) for mCRPC was examined. TTPP (a tertiary metric for disease progression as per the Prostate Cancer Clinical Trials Working Group 3 criteria) was defined as the number of months from drug initiation to a rise in PSA of ≥25% or from the documented nadir to an absolute PSA increase of ≥2 ng/ml [23]. PSA response (≥50% decline from baseline) rates were assessed and compared across these molecular subtype groups by chi-square test. To compare the proportion of patients who achieved a PSA ≥50% response for each therapy, the chi-square test was used. A univariate Cox-proportional hazard (cph) model was applied to evaluate the association between each continuous variable and the time to event outcome. Log rank test was used to evaluate the association between each categorical variable and the time to event outcome. Multivariate cph models were applied to evaluate the relationship between time to event outcomes and genetic mutation type with the variables detected in the univariate analysis (with p < 0.1).
To evaluate the use of various therapies and genomic mutation types across the institutions, chi-square test was used separately. To evaluate the duration of various therapies and genomic mutation types, the Kruskal-Wallis test was used.
Certain outcomes data were not available or estimable in all included patients. Imputation was not carried out for missingness.
3. Results
Baseline characteristics are shown in Table 1. A total of 317 patients were included in this analysis. At the time of censoring, 305 developed metastatic disease and 296 developed CRPC. Detailed mutation data were available for 43 of 46 patients with CDK12 mutations (Fig. 1A and 2, and Supplementary Table 1) with two distinct mutations in 34/43 patients (79%), consistent with biallelic loss (Supplementary Table 1). A significantly higher proportion (88%) of men with CDK12 mutations presented with Gleason score ≥8, compared with other genomic subtypes (p = 0.009; Fig. 1B and Table 1). Approximately half of the CDK12 patient cohort presented with localized disease at diagnosis (n = 24/43, 56%). The proportion of patients presenting with localized and de novo metastatic disease was comparable between all the genomic groups (p = 0.7; Fig. 1C and Table 1).
Table 1 –
Baseline patient characteristics across all institutions
| Variable | CDK12 | HRD | TP53 | Other | p value a | |||
|---|---|---|---|---|---|---|---|---|
| Mutation frequency, n (%) | 46/317 (15) | 60/317 (19) | 109/317 (34) | 102/317 (32) | ||||
| Age at diagnosis (yr), median (IQR) | 65 (58, 74) | 60 (57, 68) | 62 (57, 70) | 64 (57, 73) | 0.046 | |||
| Ethnicity, n (%) | ||||||||
| Caucasian | 34/39 (87) | 40/51 (78) | 60/66 (91) | 32/37 (87) | 0.3 | |||
| Non-Caucasian | 5/39 | 11/51 | 6/66 | 5/37 | ||||
| Missing | 7/46 (15) | 9/60 (15) | 43/109 (39) | 65/102 (64) | ||||
| PSA at diagnosis (ng/ml), median (IQR) | 20.9 (12.0, 138.0) | 18.0 (6.9, 75.0) | 11.3 (6.3, 32.7) | 13.0 (7.4, 57.2) | 0.064 | |||
| Time to continuous androgen deprivation therapy initiation (mo), median (IQR) | 1.8 (0.1, 13.6) | 3.3 (0.5, 36.5) | 1.8 (1.0, 50.6) | 4.0 (2.0, 51.1) | 0.06 | |||
| Gleason score, n (%) | ||||||||
| ≥8 | 36/41 (88) | 38/52 (73) | 74/103 (72) | 56/94 (60) | 0.009 | |||
| <8 | 5/41 | 14/52 | 29/103 | 38/94 | ||||
| Missing | 5/46 (11) | 8/60 (13) | 6/109 (5.5) | 8/102 (7.8) | ||||
| Stage at diagnosis, n (%) | ||||||||
| 0 (localized) | 24/43 | 38/60 | 62/104 | 64/99 | 0.7 | |||
| 1 (metastatic) |
19/43 (44) | 22/60 (37) | 42/104 (40) | 35/99 (35) | ||||
| Missing | 3/46 (6.5) | 0/60 (0) | 5/109 (4.6) | 3/102 (2.9) | ||||
| Lymph node metastasis, n (%) | ||||||||
| 0 (no) | 11/41 | 23/52 | 26/69 | 14/39 | 0.4 | |||
| 1 (yes) | 30/41 (73) | 29/52 (56) | 43/69 (62) | 25/39 (64) | ||||
| Missing | 5/46 (11) | 8/60 (13) | 40/109 (37) | 63/102 (62) | ||||
| Bone metastasis, n (%) | ||||||||
| 0 (no) | 9/39 | 17/55 | 24/106 | 23/99 | 0.7 | |||
| 1 (yes) | 30/39 (77) | 38/55 (70) | 82/106 (77) | 76/99 (77) | ||||
| Missing | 7/46 (15) | 5/60 (8.3) | 3/109 (2.8) | 3/102 (2.9) | ||||
| Visceral metastasis, n (%) | ||||||||
| 0 (no) | 30/39 | 41/54 | 79/106 | 83/97 | 0.3 | |||
| 1 (yes) | 9/39 (23) | 13/54 (24) | 27/106 (26) | 14/97 (14) | ||||
| Missing | 7/46 (15) | 6/60 (10) | 3/109 (2.8) | 5/102 (4.9) | ||||
CDK12 = cyclin-dependent kinase 12; HRD = homologous recombination deficiency; IQR = interquartile range; PSA = prostate-specific antigen.
p values based on the Cox proportional hazard model.
3.1. Time to metastases and time to CRPC
Patients with CDK12 mutations who presented with localized disease had statistically significantly shorter time from diagnosis to development of metastases (median = 34.9 mo) than those in the other genomic cohorts (overall p = 0.014 [Fig. 3A]; hazard ratios [HRs] to CDK12 of: HRD 0.46 [95% confidence interval {CI} 0.27, 0.80], TP53 0.59 [95% CI 0.37, 0.96], and other 0.48 [95% CI 0.30, 0.78]). Among patients with localized disease at presentation, those with CDK12 mutations also had statistically significantly shorter time to CRPC (median = 32.7 mo) compared with patients with all other genomic classifications (overall p = 0.008; Fig. 3B). The greatest difference in time to CRPC was observed between the CDK12 cohort and the other classified patients (median = 32.7 mo [CDK12] vs median = 72.8 mo [other], p = 0.001; HRs to CDK12 of: HRD 0.45 [95% CI 0.27, 0.80], TP53 0.51 [95% CI 0.30, 0.85], and other 0.42 [95% CI 0.25, 0.70]). This difference remained statistically significant when patients with localized and de novo metastatic disease at presentation were combined, suggesting that the difference in the time to CRPC is less likely driven by the stage of disease at presentation (overall p = 0.0056; Fig. 4).
Fig. 3 –
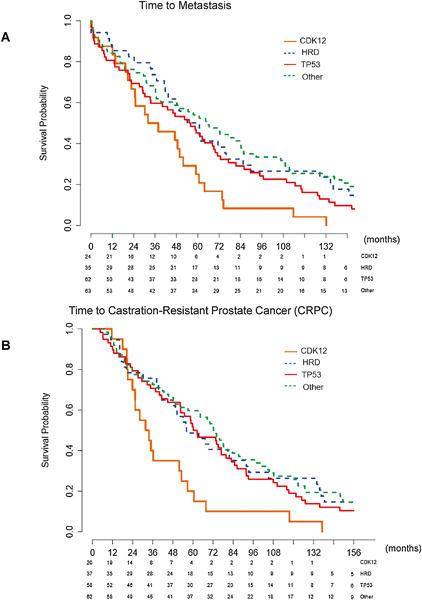
(A) Kaplan-Meier curve for time to development of metastasis, stratified by mutation type. Median time to metastasis: CDK12 34.9 mo, HRD 61.0 mo, TP53 55.6 mo, and other 64.7 mo. Overall p = 0.014. CDK12 versus HRD p = 0.0068, CDK12 versus TP53 p = 0.035, CDK12 versus other p = 0.0023, HRD versus TP53 p = 0.26, HRD versus other p = 0.94, and TP53 versus other p = 0.27. The greatest difference was in the CDK12 cohort compared with the other cohort (34.9 vs 64.7 mo, p = 0.0023). (B) Kaplan-Meier curve for time to development of CRPC in patients presenting with localized disease at presentation, stratified by mutation type. Median time to CRPC: CDK12 32.7 mo, HRD 56.2 mo, TP53 61.3 mo, and other 72.8 mo. Overall p = 0.008. CDK12 versus HRD p = 0.009, CDK12 versus TP53 p = 0.008, CDK12 versus other p = 0.001, HRD versus TP53 p = 0.62, HRD versus other p = 0.78, and TP53 versus other p = 0.28. The greatest difference was in the CDK12 compared with the other cohort (32.7 vs 72.8 mo, p = 0.001). The p values are based on log rank test.
CDK12 = cyclin-dependent kinase 12; CRPC = castration-resistant prostate cancer; HRD = homologous recombination deficiency.
Fig. 4 –
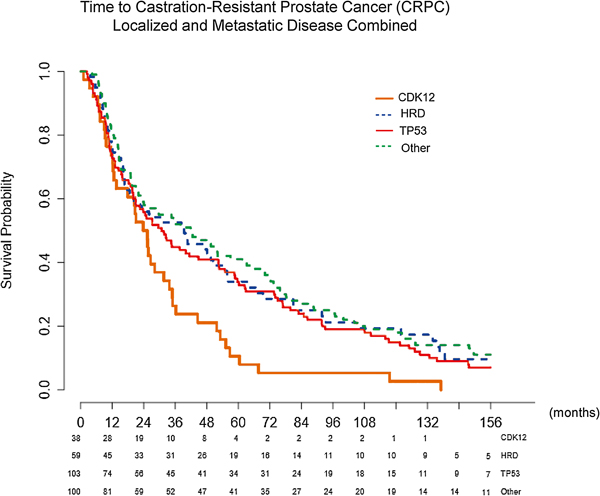
Time to development of CRPC with combined localized and metastatic disease at presentation, stratified by mutation type, using the Kaplan-Meier method. Median time to CRPC: CDK12 24.6 mo, HRD 39.4 mo, TP53 30.8 mo, and other 42.2 mo. Overall p = 0.0056. CDK12 versus HRD p = 0.0063, CDK12 versus TP53 p = 0.0097, CDK12 versus other p = 0.0004, HRD versus TP53 p = 0.58, HRD versus other p = 0.7, and TP53 versus other p = 0.21. The greatest difference in mutations was in the CDK12 compared with the other cohort (24.6 vs 42.2 mo, p = 0.0004). The p values are based on log rank test.
CDK12 = cyclin-dependent kinase 12; CRPC = castration-resistant prostate cancer; HRD = homologous recombination deficiency.
3.2. TTPP on ARPI therapy
We evaluated the TTPP on first-line ARPI therapy for mCRPC. Data were available only for the UBC cohort. A statistically significantly shorter TTPP was observed in the CDK12 (median = 3.6 mo) and HRD (median = 3.3 mo) cohorts, compared with the TP53 (median = 5.6 mo) and other (median = 7.4 mo) cohorts (overall p = 0.012 [Fig. 5]; HRs to CDK12 of: HRD 0.74 [95% CI 0.35, 1.57], TP53 0.40 [95% CI 0.19, 0.83], and other 0.44 [95% CI 0.23, 0.87]).
Fig. 5 –
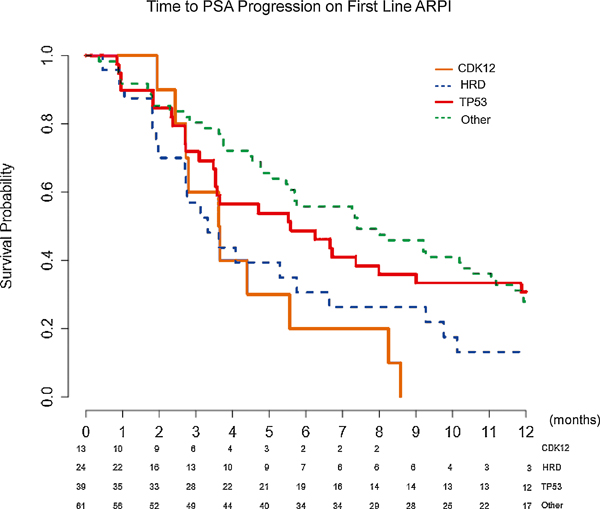
Kaplan-Meier curves for time to PSA progression on first-line AR pathway inhibitor (ARPI) therapy. Median time to PSA progression: CDK12 3.6 mo, HRD 3.3 mo, TP53 5.6 mo, and other 7.4 mo. Overall p = 0.012. CDK12 versus HRD p = 0.47, CDK12 versus TP53 p = 0.069, CDK12 versus other p = 0.0023, HRD versus TP53 p = 0.063, HRD versus other p = 0.022, and TP53 versus other p = 0.67. The greatest difference was between the CDK12 and the other cohort (3.6 vs 7.4 mo, p = 0.0023). The p values are based on log rank test.
AR = androgen receptor; CDK12 = cyclin-dependent kinase 12; HRD = homologous recombination deficiency; PSA = prostate-specific antigen.
3.3. Therapy use and duration of therapy
Evaluation of duration on various therapies revealed no statistically significant differences across all treatment types and genomic cohorts (Supplementary Table 3), but there was heterogeneity in therapy use among the treating centers (Supplementary Table 4). We also evaluated the proportion of patients who achieved a PSA ≥50% response for each therapy and found no significant differences between genomic classification cohorts (Supplementary Table 5).
3.4. Multivariable analysis
Finally, we conducted a multivariable analysis with prognostic factors, mutation classification, and therapeutic information (Supplementary Table 6). However, genomic subtype did not demonstrate statistical significance.
4. Discussion
This study represents the largest assessment to date of clinical outcome measures in CDK12 mutant advanced prostate cancer. Of note, we report that 14.5% of patients in this cohort harbored CDK12 mutations. Given that we have purposefully included all available CDK12 patients at our institutions, we would expect our proportion of patients with CDK12 mutations to be artificially higher than the previously reported prevalence [1,5]. We identified several characteristics that suggest that CDK12 mutant prostate cancer may be a distinct clinical subtype with aggressive features, including higher Gleason scores at presentation, shorter time to metastasis, and CRPC. In a subset of patients, we found subsequent shorter TTPP on first-line ARPI therapy in the mCRPC setting. However, we did not find that patients with CDK12 mutations had shorter durations on therapy (Supplementary Table 5), which is likely related to several complex factors: patient preferences, comorbidities, and shared patient-physician decision making in the setting of rising PSA.
Despite its putative role in regulating homologous recombination, we found that the CDK12 and HRD cohorts had different clinical characteristics. These findings suggest that CDK12 may have functions distinct from those of BRCA2 and ATM. Prostate cancers with CDK12 mutations have a genomic instability signature distinct from the BRCA2 signature [2,11,24]. Whereas BRCA2 mutant tumors exhibit large chromosomal deletions with flanking microhomology, tumors with CDK12 inactivation have focal tandem duplications, leading to high copy-number gains of prostate cancer-relevant oncogenes (eg, MYC, AR, and CCND1) [2,5,11,25]. As AR amplification has been proposed as a mechanism leading to castration resistance [26–30], we hypothesize that these alterations downstream of CDK12 loss (ie, AR enhancer amplification) may allow tumor cells to adapt to antiandrogen therapies, leading to the comparatively shorter time to metastasis, CRPC, and progression on ARPIs. The genomic instability may also promote AR-independent tumor growth through amplification of other oncogenes. We previously reported that CDK12-mutated prostate cancers have novel gene fusions and increased T-cell infiltrates, suggesting a potentially higher likelihood of response to immunotherapy [5]. In this cohort, five of the 46 (10.9%) CDK12 patients received at least one dose of anti-PD-1 checkpoint inhibitor immunotherapy, and two of five attained a PSA50 response (Supplementary Table 5). None of the three CDK12 patients exposed to a PARP inhibitor attained a PSA50 response. These very small sample sizes preclude drawing any definitive conclusions from these data. However, CDK12 biallelic loss is currently being examined as a potential predictive biomarker for immunotherapy in an ongoing prospective clinical trial at our institutions (NCT03570619). A CDK12 loss prostate cancer response to PARP inhibitors is also under investigation.
As we continue to define the landscape of genomic alterations and structural variants within advanced prostate cancer, understanding the clinical implications and tailoring treatment approaches for these molecular subtypes will need to follow.
4.1. Limitations
Several factors contributed to the heterogeneity of our data. Measurable differences among the patient cohorts treated at the three institutions likely reflect differing patient demographics, referral and screening patterns, and center-dependent practice patterns. While high concordance between tumor biopsy testing and ctDNA analysis has been demonstrated previously, these differing methods likely contributed additional heterogeneity. Although liquid biopsies may potentially capture a broader sample of mutations, they may fail to detect ctDNA derived from malignant cells in patients with a low disease burden (ie, false negative). Furthermore, not all patients have metastatic disease amenable to biopsy and not all tissue biopsies yield enough tumor DNA for analysis. In addition, patients underwent NGS via various clinical-grade platforms, which may not accurately call loss of heterozygosity without a normal reference sample (ie, concurrent germline testing). Therefore, it was not always possible to definitively establish mono- or biallelic CDK12 loss; consequently, all cases were included in the CDK12 group. Nonetheless, our findings remained statistically significant with the biallelic CDK12 patients. Moreover, TTPP data on first-line ARPI treatment of mCRPC were available only from the UBC cohort. Finally, despite demonstrated shorter time to metastasis and CRPC, we did not identify mutation type to be a statistically significant independent variable in multivariable analysis. We speculate that this is due to the fact that CDK12 mutations may not be acting independently to impact clinical outcomes, but rather through inter-related clinical and biochemical variables that are not yet identified.
5. Conclusions
CDK12-mutated prostate cancers are a molecularly distinct subset of advanced prostate cancer, displaying clinical features suggestive of more aggressive disease and which may warrant intensification of therapy. Overall, our study illustrates the importance of molecularly subtyping advanced prostate cancers, given the apparent heterogeneity in clinical behavior, and serves as the foundation for future prospective investigations to further characterize the CDK12 subtype of prostate cancer.
Supplementary Material
Acknowledgments
Funding/Support and role of the sponsor: This work is supported by the National Cancer Institute (1R01CA230516), Early Detection Research Network Grant (U01 CA214170), Prostate SPORE Grants (P50 CA186786 and P50 CA097186), and Prostate Cancer Foundation.
Financial disclosures: Felix Y. Feng certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Jonathan Chou was funded by the A.P. Giannini Foundation Postdoctoral Fellowship, the Rosenberg Fellowship in Genitourinary Oncology at UCSF, and a T32 Training Grant from the National Cancer Institute (CA108462–13S1). Arul M. Chinnaiyan is a recipient of the NCI Outstanding Investigator Award, and is a Howard Hughes Medical Institute Investigator, Taubman Scholar, and American Cancer Society Professor. Felix Y. Feng, Eric J. Small, Alexander W. Wyatt, and Arul M. Chinnaiyan are supported by grants from the Prostate Cancer Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018;175:889. [DOI] [PubMed] [Google Scholar]
- [3].Hamid AA, Gray KP, Shaw G, et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur Urol 2019;76:89–97. [DOI] [PubMed] [Google Scholar]
- [4].Chen WS, Aggarwal R, Zhang L, et al. Genomic drivers of poor prognosis and enzalutamide resistance in metastatic castration-resistant prostate cancer. Eur Urol. In press 10.1016/j.eururo.2019.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wu YM, Cieslik M, Lonigro RJ, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018;173:1770–82 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chila R, Guffanti F, Damia G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat Rev 2016;50:83–8. [DOI] [PubMed] [Google Scholar]
- [7].Dubbury SJ, Boutz PL, Sharp PA. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018;564:141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Blazek D, Kohoutek J, Bartholomeeusen K, et al. The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011;25:2158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res 2014;74:287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Popova T, Manie E, Boeva V, et al. Ovarian cancers harboring inactivating mutations in CDK12 display a distinct genomic instability pattern characterized by large tandem duplications. Cancer Res 2016;76:1882–91. [DOI] [PubMed] [Google Scholar]
- [11].Menghi F, Barthel FP, Yadav V, et al. The tandem duplicator phenotype is a prevalent genome-wide cancer configuration driven by distinct gene mutations. Cancer Cell 2018;34:197–210 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McNair C, Xu K, Mandigo AC, et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest 2018;128:341–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jamaspishvili T, Berman DM, Ross AE, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol 2018;15:222–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019;116:11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Warner EW, Yip SM, Chi KN, Wyatt AW. DNA repair defects in prostate cancer: impact for screening, prognostication and treatment. BJU Int 2019;123:769–76. [DOI] [PubMed] [Google Scholar]
- [16].Castro E, Romero-Laorden N, Del Pozo A, et al. PROREPAIR-B: a prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol 2019;37:490–503. [DOI] [PubMed] [Google Scholar]
- [17].Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov 2018;8:444–57. [DOI] [PubMed] [Google Scholar]
- [19].Wyatt AW, Annala M, Aggarwal R, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst 2017;109:djx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vandekerkhove G, Struss WJ, Annala M, et al. Circulating tumor DNA abundance and potential utility in de novo metastatic prostate cancer. Eur Urol 2019;75:667–75. [DOI] [PubMed] [Google Scholar]
- [21].Hussain M, Daignault-Newton S, Twardowski PW, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol 2018;36:991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abida W, Cheng ML, Armenia J, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol 2019;5:471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Menghi F, Inaki K, Woo X, et al. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc Natl Acad Sci U S A 2016;113:E2373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Viswanathan SR, Ha G, Hoff AM, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018;174:433–47 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer 2003;89:552–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33–9. [DOI] [PubMed] [Google Scholar]
- [28].Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 1995;9:401–6. [DOI] [PubMed] [Google Scholar]
- [29].Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol 2017;35:2149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.