

Abstract
Protein complementation assays (PCA) have been incorporated as pharmacological tools, enabling a wide array of applications, ranging from studies of protein-protein interactions to second messenger effects. Methods to detect activities of G protein-coupled receptors (GPCRs) have particular relevance for drug screening. Recent development of an engineered luciferase NanoLuc has presented a possibility to generate a novel PCA, which in turn could open a new avenue for developing drug screening assays. Here we identified a novel split position for NanoLuc and demonstrated its use in a series of fusion constructs to detect the activity of GPCRs. The split construct can be applied to a variety of pharmacological screening systems.
Introduction
The PCA approach utilizing various split protein fragments such as fluorescent proteins (1), luciferases (2), and other enzymes (3,4) has been introduced to investigate protein-protein interactions in vitro in intact cells. Many of these have been incorporated into pharmacology settings because of their versatility when fused to proteins of interest (5). Among them, luciferase complementation has been intensively studied in luciferases of various species origin (6). These proteins show a utility in reliable kinetic measurement, and some split luciferases exhibit reversibility, making them very useful as fusion fragments (7–9).
Currently, PCA-based pharmacological assays rely mostly on a fusion of bulky protein fragments. In addition, the signal brightness of luciferase complementation approach should be improved particularly to make it amenable for higher throughput applications. Consequently, smaller split fragments with brighter signals in fewer number of cells would make the assay more adaptable. NanoLuc, which was engineered from Oplophorus luciferase (10), is superior to other luciferases because of its small size and luminescence intensity (11). Compared to Renilla luciferase (Rluc), it is about half a size (19 kDa vs. 36 kDa) and produces luminescence ~150 fold greater (11). To improve on the shortcomings of previously reported split luciferases of other species (e.g., bulkiness and signal level), we developed a novel bimolecular NanoLuc complementation (BiNC) pair of fragments and investigated its functional properties.
Besides the novelty in PCA tool-making, the BiNC approach holds a key to advance the development of robust and compact pharmacology assays. In GPCR drug screening, receptor-G protein coupling as well as G protein subunit rearrangement can be used effectively as ways to measure agonist and antagonist behaviors (12). While this manuscript was in preparation, a thorough analysis of split positions for NanoLuc was reported (13). The split position we selected in this current work happens to be near the several residues tested in Dixon et al. 2016. In the current study, the BiNC fusion is integrated to the GPCR activation scheme mentioned above to develop a variety of novel pharmacology assays beyond protein recruitment to a receptor. The establishment of reliable pharmacological assays in various GPCRs demonstrate the validity and utility of BiNC approach reported herein.
Material and Methods
DNA constructs and transfection. FRB or FKBP was C-terminally fused to a short linker (SGGGGS) followed by either an Rluc8 N-terminal fragment (L1; residues 1–229), an Rluc8 C-terminal fragment (L2; residues 230–311) (14), a NanoLuc N-terminal fragment (Nluc1; residues 1–97; VFTLEDFVGDWRQTAGYNLDQVLEQGGVSSLFQNLGVSVTPIQRIVLSGENGLKIDIHVIIPYEGLSGDQMGQIEKIFKVVYPVDDHHFKVILHYG), or a NanoLuc C-terminal fragment (Nluc2; residues 98–171; TLVIDGVTPNMIDYFGRPYEGIAVFDGKKITVTGTLWNGNKIIDERLINPDGSLLFRVTINGVTGWRLCERILA). Human receptor constructs, dopamine D1, D2, adrenoceptor β2A, adenosine A2A, and muscarinic M4, were N-terminally fused with a signal peptide followed by a Flag epitope tag for enhanced cell surface expression (15) and their detection (16). For the split fusions, either L1, L2, Nluc1, or Nlu2 was fused C-terminally. A linker was inserted between the receptor and the split fusion for D1 (made also without the linker), D2, and M4. The following effector constructs were made: Gαs-Nluc1 (inserted at residue 67), Gαs-Nluc2 (inserted at residue 67), Gαi1-Nluc1 (inserted at residue 91), Gαi1-Nluc2 (inserted at residue 91), Nluc1-Gγ2, Nluc2-Gγ2, and Nluc2-β-arrestin2. Untagged Gβ1, Gγ2, and G protein coupled receptor kinase 2 (GRK2) were also used for co-transfection. All the constructs were confirmed by sequencing analysis. A constant amount of plasmid cDNA (15 μg) was transfected into human embryonic kidney cells 293T (HEK-293T) using polyethylenimine (PEI; Sigma-Aldrich, St. Louis, MO) in a 1 to 2 ratio in 10 cm plates. Cells were maintained in culture with Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and kept in an incubator at 37°C and 5% CO2. The transfected amount and ratio among the receptor and heterotrimeric G proteins were optimized for the greatest dynamic range in drug-induced luminescence. Experiments were performed approximately 48 hr post-transfection.
Luciferase complementation assay. The time course of luciferase complementation was measured by rapamycin (Tocris)- and FK506 (Tocris)-induced complex formation and dissociation between FRB-L1 and FKBP-L2 or FRB-Nluc1 and FKBP-Nluc2. Four modes of luciferase complementation assays were performed to detect receptor ligand-induced events for i) Receptor-Gα engagement, ii) Receptor-Gγ interaction, iii) Gα-γ protein activation, and iv) Receptor-βarrestin2 recruitment. For each configuration, both complementation pairs were tested (i.e. Nluc1 fusion-Nluc2 fusion pair and Nluc2 fusion-Nluc1 fusion pair). Cells were harvested, washed, and resuspended in phosphate-buffered saline (PBS). Approximately 200,000 cells/well were distributed in 96-well plates, and 5 μM coelenterazine H (Nanolight) or 0.03% furimazine (Promega) was added to each well. One minute after addition of coelenterazine H or furimazine, ligands [dopamine (Sigma), L-(–)-norepinephrine (Sigma), 5’-N-Ethylcarboxamidoadenosine (NECA, Tocris), isoproterenol (Tocris), or carbachol (Tocris)] were added to each well. The complemented luminescence was quantified with an open filter for 1 sec using a Mithras LB940 (Berthold Technologies, Bad Wildbad, Germany) after 10 min of substrate incubation. Drug-induced luminescence values are expressed as the basal subtracted luminescence change in the dose-response graphs. Data and statistical analysis were performed with Prism 5 (GraphPad Software).
Results and Discussion
Rapid kinetic complementation of novel NanoLuc splits
Based on the previously predicted beta strand repeats (10), which were later confirmed in the crystal structure that emerged during the preparation of this manuscript (17), a split position was chosen at residue 96. In order to assess the suitability of the split position, the amino acid sequences encoding the N-terminal (Nluc1) and C-termminal (Nluc2) fragments were fused C-terminally to FRB and FKBP for testing rapamycin-induced (18,19) bimolecular NanoLuc complementation (BiNC) as the macrolide rapamycin induces complex formation between the FRB and FKBP domains (Figure 1A–C). Using the same scheme, Rluc split constructs (Renilla luciferase bimolecular complementation (BiRC)) were fused to FRB and FKBP and compared for complementation kinetics (Figure 1E–G). When incubated in 100 nM rapamycin for 3 hours, at the end of which the substrate coelenterazine H (5 μM, a non-saturating but effective concentration; Supplementary Figure 1A–B) was added, cells expressing the FRB-NanoLuc N-terminal fragment (FRB-Nluc1) and the FKBP-NanoLuc C-terminal fragment (FKBP-Nluc2) showed an increase of 1.74 × 106 counts (arbitrary units), a 5.5 fold increase from the vehicle treated background (Figure 1A orange). The same fold increase was maintained for over 40 min of detection after the substrate was added while the absolute counts decayed. This increase in BiNC is in contrast to the 1.44 × 104 counts and 24% increase for rapamycin treated cells expressing FRB-Rluc N-terminal fragment (FRB-Rluc1) and FKBP-Rluc C-terminal fragment (FKBP-Rluc2) in the BiRC configuration (Figure 1E orange). FK506 is a competitive inhibitor for rapamycin and therefore disrupts complex formation between the FRB and FKBP domains. Three-hour incubation of 10 μM FK506 caused only 8% drop in the average level of luminescence in BiNC (Figure 1A blue vs. black) at 5 min after the substrate addition indicating that basal level of complex formation can only be reversed very weakly by FK506. A smaller change (2%) by FK506 was observed in BiRC (Figure 1E blue). Next the competitive inhibition of rapamycin by FK506 was assessed. Addition of 10 μM FK506 together with 100 nM rapamycin for 3 hours decreased the BiNC luminescence by 28% (Figure 1A red), whereas the co-administration of the drugs only decreased the BIRC luminescence by 3% (Figure 1E red) showing a partial but substantial degree of competition by FK506 in the BiNc configuration. Stability of the BiNC was tested for the same conditions in the presence of 30 μg/ml cycloheximide, an inhibitor of protein biosynthesis (20), as newly synthesized luciferase fragments can replenish the complemented pool. Cycloheximide did not cause a significant luminescence change in the response to rapamycin or FK506 treatment (Figure 1A orange open and blue open) confirming that no new protein synthesis contributed to BiNC formation and BiNC was stable for at least 3 hours. Because of its relatively low cost, we carried out the bulk of our assays with coelenterazine H. We also tested the more stable but much more expensive substrate furimazine (11) for its kinetics (Supplementary Figure 1C–D). In this case, BiNC showed a steady level of maintained or slightly increased luminescence over the time period measured. Notably, the rapamycin-induced fold increase in luminescence was similar to that with coelenterazine H, and the remainder of our studies were carried out with colenterezine H. As expected, with furimazine, BiRC did not yield luminescence as high as BiNC indicating the preference of substrate-enzyme selectivity.
Figure 1:
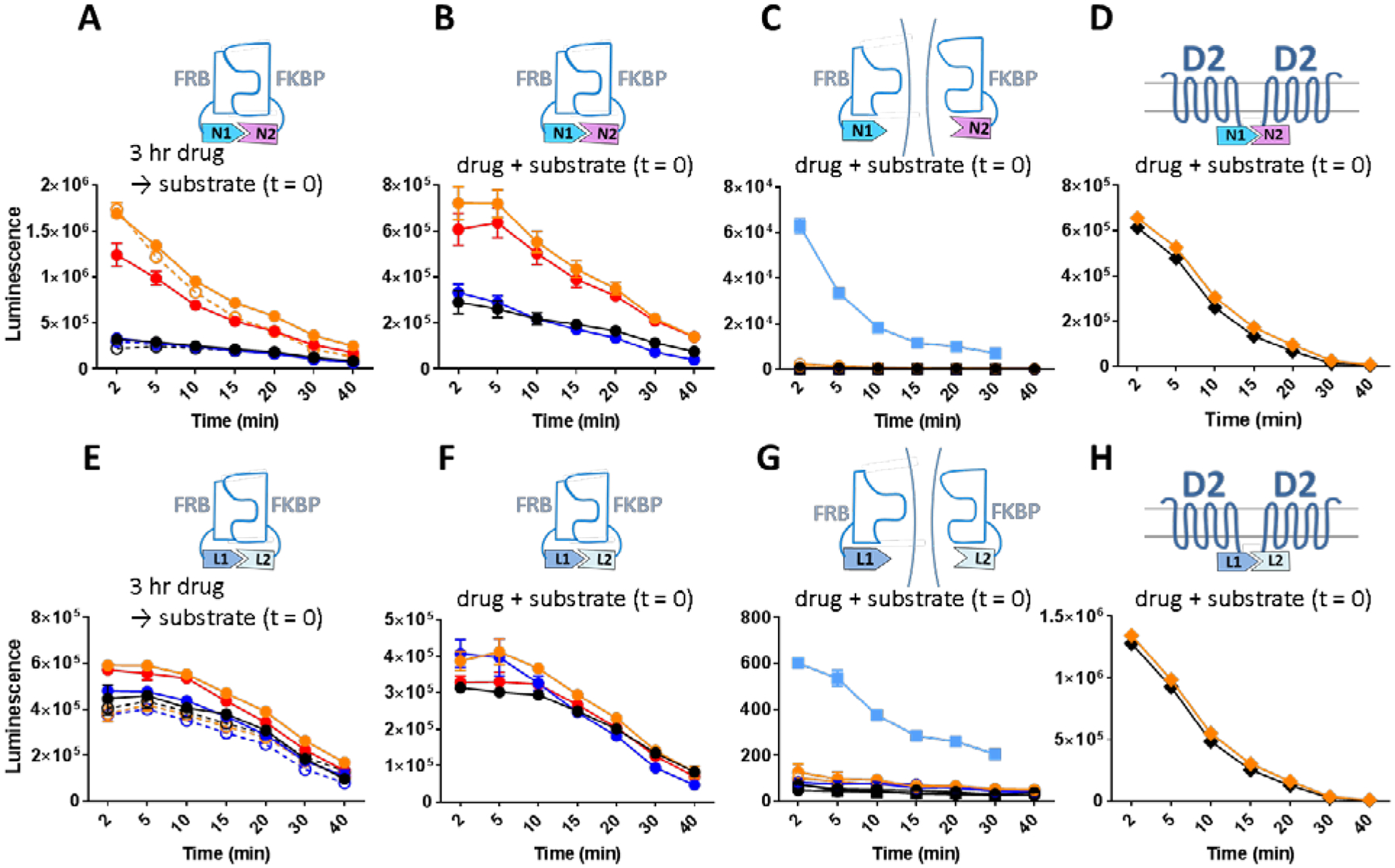
Drug induced complementation of split luciferase fragments. Throughout, time scale corresponds to substrate (5 μM coelenterazine H) incubation time. A. Kinetic measurement of NanoLuc complementation between FRB-Nluc1 and FKBP-Nluc2 after three hours of incubation with vehicle (black), 100 nM rapamycin (orange), 10 μM FK506 (blue), 100 nM rapamycin + 10 μM FK506 (red), 30 μg/ml cycloheximide + vehicle (black open), 30 μg/ml cycloheximide + 100 nM rapamycin (orange open), 30 μg/ml cycloheximide + 10 μM FK506 (blue open). B. Kinetic measurement of NanoLuc complementation between FRB-Nluc1 and FKBP-Nluc2 with simultaneous addition of vehicle (black), 100 nM rapamycin (orange), 10 μM FK506 (blue), 100 nM rapamycin +10 μM FK506 added after 5 min (red). C. Time dependent drug induced change of NanoLuc activity in cells separately expressing split fragments immediately after addition of vehicle with FRB-Nluc1 fragment (black top hemi circle), 100 nM rapamycin with FRB-Nluc1 fragment (orange top hemi circle), 10 μM FK506 with FRB-Nluc1 fragment (blue top hemi circle), vehicle with FKBP-Nluc2 fragment (black bottom hemi circle), 100 nM rapamycin with FKBP-Nluc2 fragment (orange bottom hemi circle), 10 μM FK506 with FKBP-Nluc2 fragment (blue bottom hemi circle), vehicle with FRB-Nluc1 fragment cells and FKBP-Nluc2 fragment cells combined (black square), and 100 nM rapamycin with FRB-Nluc1 fragment cells and FKBP-Nluc2 fragment cells combined (light blue square). D. NanoLuc complementation between D2R-Nluc1 and D2R-Nluc2 immediately after three hours of incubation with vehicle (black), and 100 nM rapamycin (orange). E-H. Rluc complementation (same experimental scheme and legend for A-D applied).
Next the kinetics of drug-induced BiNC was measured by adding the drugs and substrate at the same time without prior incubation. When added at the same time as colenterezine H, rapamycin increased the luminescence 2.3 fold (Figure 1B orange) and FK506 alone decreased 14% (Figure 1B blue) compared to the vehicle on average between 2–40 min. Interestingly, when FK506 was added 5 min after rapamycin (Figure 1B red), the degree of luminescence drop caused by FK506 (8%) was similar to FK506 alone (Figure 1B blue) indicating that FK506 does not reverse rapamycin’s effect. To confirm the lack of luciferase enzyme activity in the individual fragments of Nluc1 and Nluc2, FRB-Nluc1 or FKBP-Nluc2 was expressed separately. As expected, when FRB-Nluc1 or FKBP-Nluc2 was expressed alone, there was no luminescence nor with vehicle, rapamycin, or FK506 treatment (Figure 1C). The same results were observed for FRB-Rluc1 and FKBP-Rluc2 (Figure 1G). Somewhat surprisingly, when FRB-Nluc1 expressing and FKBP-Nluc2 expressing cells were combined in the same well, rapamycin induced a dramatic increase in luminescence compared to vehicle treatment (Figure 1C black square and light blue square). In comparison, combination of FRB-Rluc1 and FKBP-Rluc2 expressing cells did not yield much luminescence (Figure 1G black square and light blue). The complementation happened likely due to the cytosolic protein leaking out to the extracellular space via compromised plasma membrane or potentially from fragments that were secreted Finally, the specificity of the rapamycin effect was tested by examining the effects of rapamycin on BiNC between two proteins that lack the FKBP and FRB domains. Dopamine receptor D2 (D2R) fused with either Nluc1 or Nluc2 C-terminally was both co-transfected and rapamycin was added. Substantial complementation took place akin to the level of rapamycin-induced FRB-Nluc1 and FKBP-Nluc2 (Figure 1B), but as expected, rapamycin did not cause a significant change in the luminescence. Simiar results were observed for the D2R-Rluc fusion complementation counterparts (Figure 1D and H).
NanoLuc complementation-based tools for evaluating G protein activation
As the chemically-induced interaction between the NanoLuc splits was confirmed, next, BiNC induced by protein-protein interaction was next investigated in the context of GPCR-signaling (Figure 2A). The specificity of protein-protein interaction between dopamine D1 receptor (D1R) and its cognate Gα subunit, Gs, was used to explore NanoLuc complementation. Transfection with a fixed amount of D1R-Nluc1 and an increasing amount of co-transfected Gs-Nluc2 led to a dramatic saturable increase in luminescence, whereas FRB-Nluc2, which is not expected to interact with D1R, did not show increased luminescence (Figure 2A). In addition, non-cognate G protein coupling (i.e., D1R-Gi and D2R-Gs) was effectively absent without any ligand added (Supplementary Figure 2A). The results indicate that the BiNC can be enhanced by specific protein-protein interactions of fusion proteins.
Figure 2:
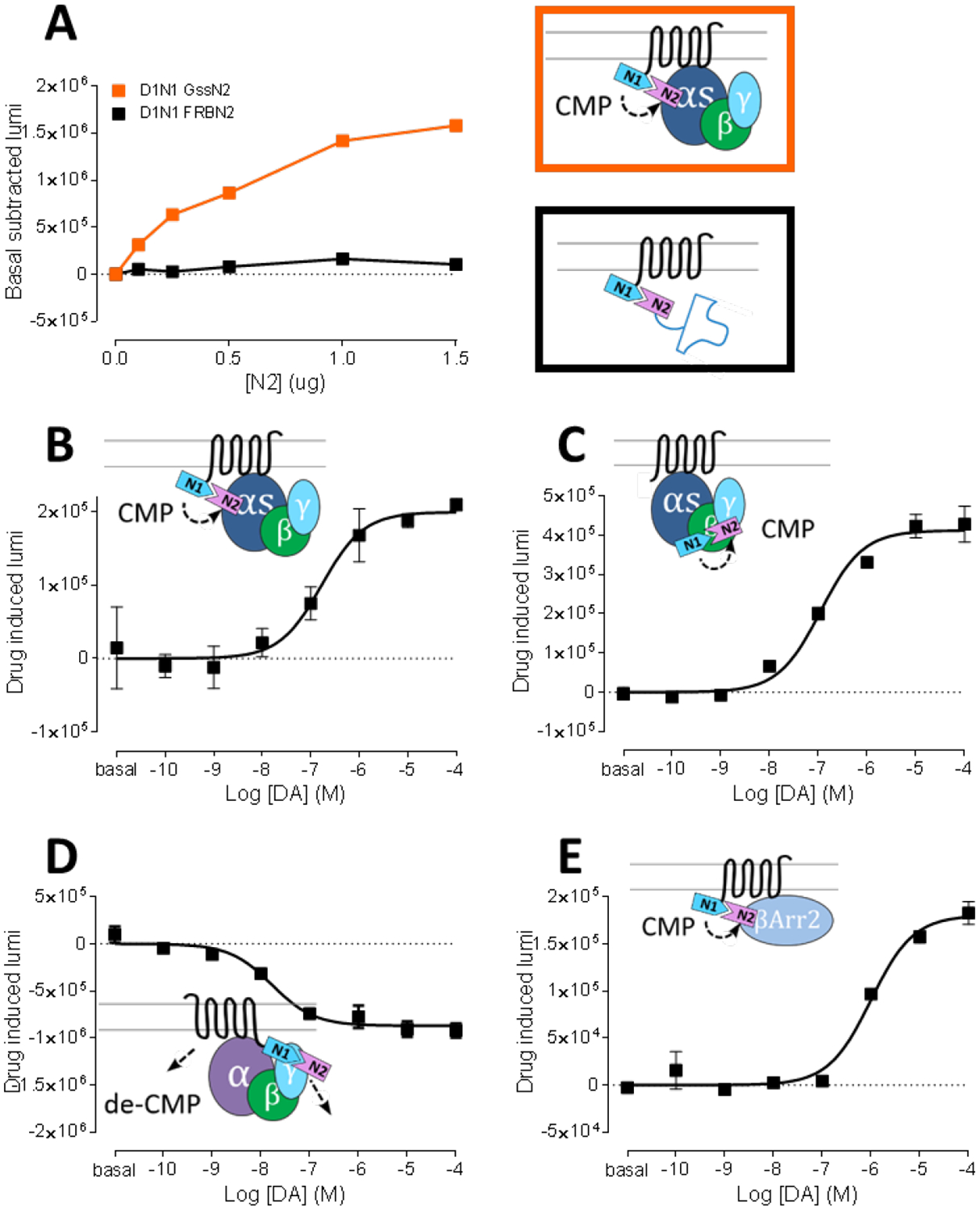
Dopamine (DA) induced G protein engagement, activation, and β-arrestin2 recruitment measured by NanoLuc complementation. A. Specific NanoLuc complementation between D1-Nluc1 (0.5 μg transfected) with non-interacting FRB-Nluc2 (black; transfected amount = y axis) or with cognate G protein Gαs-Nluc2 (orange; transfected amount = y axis). B-E. DA induced complementation change between D1-Nluc1 and Gαs-Nluc2 (B), Gαs-Nluc1 and γ2-Nluc2 (C), D1-Nluc1 and γ2-Nluc2 (D), and D1-Nluc1 and β-arrestin2-Nluc2 (E).
pEC50: D1N1 GsN2 – 6.78±0.25, D1 Gαs-Nluc1 γ2-Nluc2 – 6.96±0.11, D1-Nluc1 γ2-Nluc2 – 7.78±0.16, D1-Nluc1 β-arrestin2-Nluc2 – 6.00±0.10.
Next, agonist-induced G protein coupling was studied using an optimized 1:1 transfection ratio between D1R-Nluc1 and Gs-Nluc2. DA-induced luminescence followed a saturable dose response curve (Figure 2B; pEC50 = 6.78±0.25), while non-cognate pairs did not show DA-induced luminescence (Supplementary Figure 2B). In a configuration that detects a conformational change related to G protein activation (21) a DA-induced luminescence was measured between Gs-Nluc1 and Gγ2-Nluc2 (Figure 2C; pEC50 = 6.96±0.11). The result showed a positive BiNC with a similar EC50 for DA as the D1R-Nluc1 – Gs-Nluc2 configuration in Figure 2B. The agonist-induced signal increase in Gs- Gγ2 configuration is in agreement with other studies (12,21,22). To further characterize the conformational change within the D1R-G protein complex, an agonist-induced change between D1R-Nluc1 and Gγ2-Nluc2 was measured (Figure 2D; pEC50 = 7.78±0.16). Interestingly, in this configuration, DA caused a drop in luminescence indicating either dissociating movement or chromophore rearrangement between the receptor and γ subunit. Nonetheless, the pEC50 values of all the different configurations report in a relatively tight range between 6.78 and 7.78, indicating similar drug-induced conformational changes among different fusion constructs, albeit with somewhat different apparent potencies. The recruitment of the signaling protein, β arrestin, was also studied (Figure 2E; pEC50 = 6.00±0.10). As expected, DA caused a luminescence increase in D1R-Nluc1 and β arrestin 2-Nluc2 transfected cells, with slow kinetics consistent with the expected time frame of arrestin recruitment (data not shown).
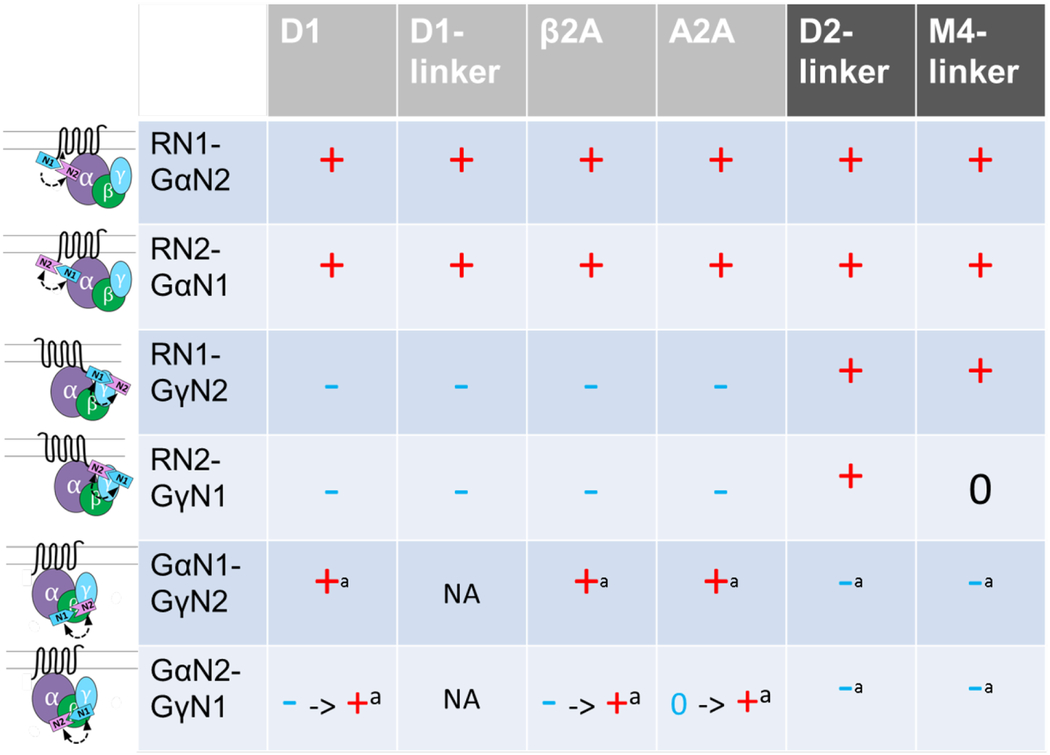
The BiNC configurations shown in Figure 2 were then tested for complementation using constructs in which Nluc1 and Nluc2 were reversed. As shown in the first column of Table 1, fusion replacement between Nluc1 and Nluc2 (e.g., D1N1-GsN2 vs. D1N2-GsN1, D1N1-Gγ2N2 vs. D1N2-Gγ2N1) results in the same directionality of agonist-induced response except for GsN1-Gγ2N2 vs. GsN2-Gγ2N1 in which GsN2- Gγ2N1 showed a time-dependent luminescence change in directionality of the agonist response from negative- to positive-induction.
Table 1:
Agonist induced complementation change between various combinations of Nluc1- and Nluc2-fusion constructs (top-bottom) for Gαs- and Gαi-coupled receptors (left-right). Signs indicate the drug induced luminescence change (-, 0, +) determined by 2, 10, 20, 30 min reading of dose effect curves (basal to 100 μM agonist). - → + or 0 → + = sign changing over 20 min. D1-, D2-, or M4-linker = linker (GGGGSGGGGSGGGGS) inserted between the receptor and NanoLuc fragment fusion. a = receptor with no NanoLuc fragment fusion is used. NA = not available. DA (D1 and D2), isoproterenol (β2A), NECA (A2A), or carbachol (M4) was used as a corresponding agonist.
pEC50: D1Nluc1 GsNluc2 – 6.78–7.77, D1-linkerNluc1 GsNluc2 – 6.17–7.43, β2ANluc1 GsNluc2 – 7.79–8.04, A2ANluc1 GsNluc2 – 6.68–6.87, D2-linkerNluc1 Gi1Nluc2 – 6.15–7.35, M4-linkerNluc1 Gi1Nluc2 – 5.89–6.95,
|
In order to validate the coupling findings and build pharmacological tools for different receptors, we made a series of constructs harboring Nluc1 and Nluc2 fusion in Gs- and Gi-coupled receptors (Table 1). Consistently, other Gs-coupled receptors, β2 adrenergic receptor (β2AR) and adenosine A2A receptor (A2AR), showed similar patterns of BiNC change as D1R. In contrast, Gi-coupled receptors (dopamine D2 receptor (D2R) and muscarinic M4 receptor (M4R)) showed quite different patterns of BiNC compared to the Gs-coupled receptors, consistent with their expected coupling to Gi. The receptor-Gα and receptor-Gγ configurations showed agonist-induced BiNC consistent with the conformational change involved in receptor engagement (Table 1 first four rows for D2R and M4R). In a G protein activation configuration, Gi-Gγ2 showed a negative BiNC indicative of distancing or dissociating movement between the two and in agreement with previous reports (12,21,22).
Pharmacological assessment of split receptor-G protein tools
Finally, the capability to parse out and characterize ligands with different pharmacological properties was tested in the receptor-Gα configuration for both Gs- and Gi-coupled receptors (Figure 3). For D1R, relative to DA (pEC50 = 6.78±0.25), the potency profile could be clearly differentiated between less potent norepinephrine (pEC50 = 5.90±0.21) and highly potent A77636 (pEC50 = 9.74±0.25). In terms of efficacy, a partial agonist SKF38393 showed the expected lower efficacy (48.5% of DA). A selective D1R antagonist SCH23390 counteracted 10 μM DA pre-activation and gave a negative BiNC change relative to the pre-activation level, consistent with inverse agonist activity. The antagonist also exhibited a high potency (pIC50 = 9.22±0.42). All of the characteristics of D1R ligands are in agreement with previous reports (23,24). D2R pharmacology was also in agreement with a previous study (25) in that norepinephrine showed less potency (pEC50 = 5.22±0.14) relative to DA (pEC50 = 6.61±0.10) and a selective D2R antagonist eticlopride showed complete blockade of 10 μM DA pre-activation and high potency (pIC50 = 9.47±0.46). Albeit narrower luminescence drug-induced ranges, furimazine showed similar potencies and efficacies (Supplementary Figure 2C–D). Taken together, for both Gs-coupled (D1R) and Gi-coupled (D2R) receptors, the receptor-Gα BiNC configuration can be used to assess pharmacological properties of ligands.
Figure 3:
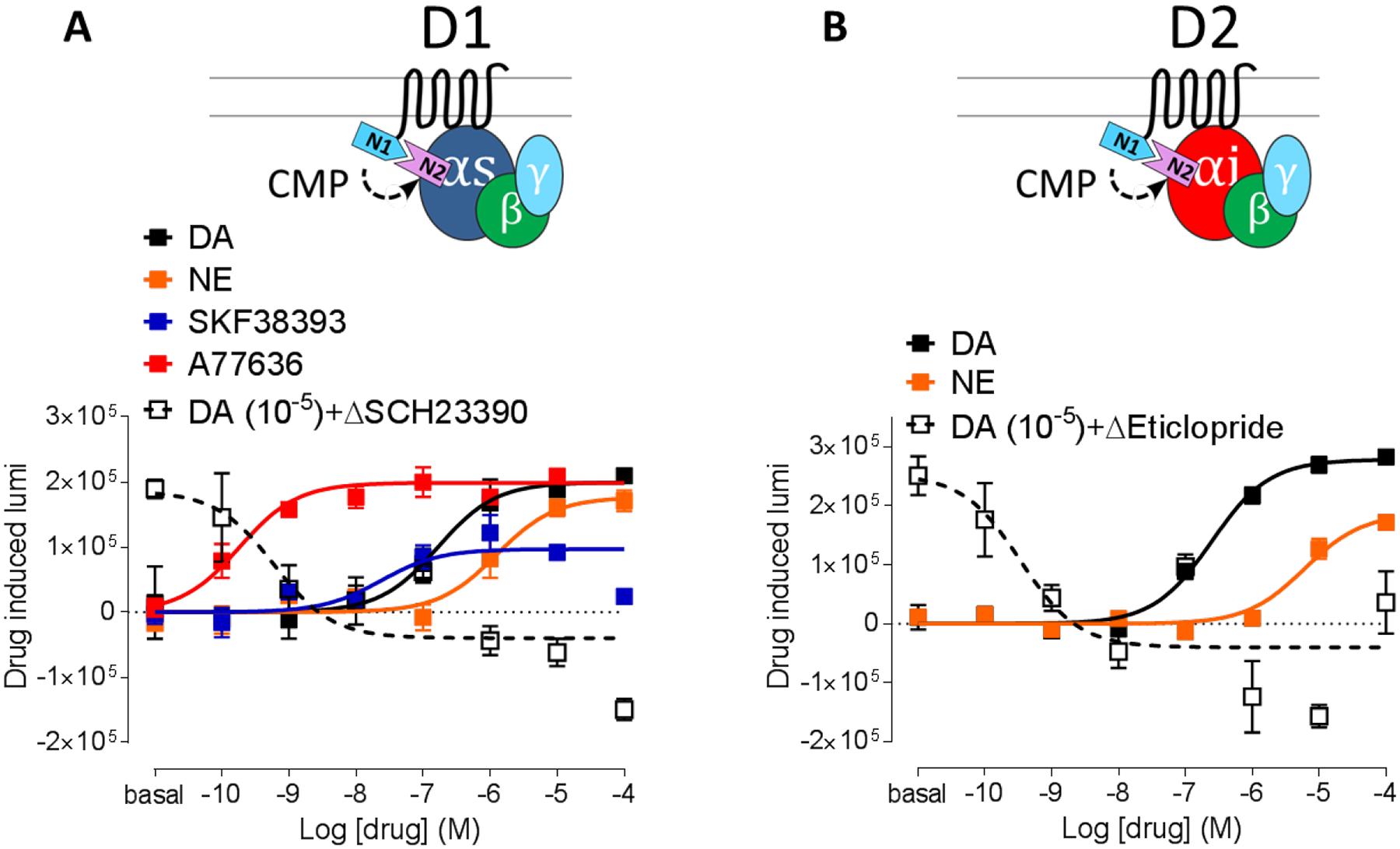
Confirmation of pharmacological characteristics in drug induced G protein engagement. A. D1-Nluc1 Gαs-Nluc2 engagement with dopamine (black), norepinephrine (orange), SKF38393 (blue), A77636 (red), 10 μM DA + SCH23390 (open black). B. D2-Nluc1 Gαi-Nluc2 engagement with dopamine (black), norepinephrine (orange), 10 μM DA + Eticlopride (open black).
When compared to the BiRC, in addition to the smaller fragment sizes (i.e., 95 and 63 amino acids for Nluc1 and Nluc2 versus 229 and 83 amino acids for Rluc1 and Rluc2), the rapamycin-induced activity increase of BiNC-fusion constructs was more pronounced (5.5 fold compared to 1.2 fold for BiRC) and more kinetically responsive (135% increase after 2 min of rapamycin addition compared to 4% increase for BiRC). These characteristics show the clear advantages of BiNC over BiRC. Coincidentally, while preparing the manuscript, both a crystal structure (17) and new BiNC split positions (13) of NanoLuc were published. This split position was tested in the arrestin recruitment assay but not assessed for G protein activation.
Equally important to the enzymatic properties of BiNC is its applicability in functionally relevant fusion constructs. To this end, we evaluated five different Gs- and Gi-coupled receptors. As shown in Table 1 legend, the potencies were within a relatively tight range among the different configurations tested showing the utility of the diverse modes of detection of luminescence complementation. Also the EC50 values were in close agreement with reports using different methods for measuring G protein activation and receptor engagement (26–28). Overall, for the D1R and D2R, the potencies and efficacies of tested ligands were in agreement with previous reports (23,25,29,30) indicating the utility of the assay configuration for testing pharmacology of other receptors. Overall, the current study demonstrates that BiNC-fusion constructs of receptor-effector pairs can be used as an effective method to assay pharmacological properties of ligands that can be expanded to a higher throughput approach.
Supplementary Material
Supplementary Figure 1: Comaprison of coelenterazine H and fumirazine as Nanoluc substrates. A. Kinetic measurement of luminescence after three hour incubation of rapamycin for NanoLuc complementation between FRB-Nluc1 and FKBP-Nluc2. Curves are with (open symbol) or without (filled symbol) 100 nM rapamycin for 50 nM (black), 500 nM (orange), 5 μM (blue), and 50 μM (red) coelenterazine H. B. Kinetic measurement of luminescence after three hour incubation of rapamycin for Rluc complementation between FRB-Rluc1 and FKBP-Rluc2 with or without 100 nM rapamycin incubation for coelenterazine H (color /symbol scheme same as A). C. Kinetic measurement of luminescence after three hour incubation of rapamycin for NanoLuc complementation between FRB-Nluc1 and FKBP-Nluc2. Curves are with (open symbol) or without (filled symbol) 100 nM rapamycin for 0.001% (black), 0.01% (orange), 0.1% (blue), and 1% (red) furimazine. D. Kinetic measurement of luminescence after three hour incubation of rapamycin for Kinetic measurement of Rluc complementation between FRB-Rluc1 and FKBP-Rluc2 after three hour incubation with or without 100 nM rapamycin incubation for coelenterazine H (color /symbol scheme same as C).
Supplementary Figure 2: Specificity of cognate G protein coupling. A. NanoLuc complementation between D1-Nluc1 (0.5 μg transfected) with non-interacting Gαi1-Nluc2 (filled circle; transfected amount = y axis) or D2-Nluc1 (0.5 μg transfected) with non-interacting Gαs-Nluc2 (open circle; transfected amount = y axis). B. DA induced complementation change between D1-Nluc1 and Gαi1-Nluc2 (filled circle) and D2-Nluc1 and Gαis-Nluc2 (open circle). C. D1-Nluc1 Gαs-Nluc2 engagement with dopamine (filled square), norepinephrine (circle), 10 μM DA + SCH23390 (open square). D. D2-Nluc1 Gαi-Nluc2 engagement with dopamine (filled square), norepinephrine (circle), 10 μM DA + Eticlopride (open square).
References
- 1.Kerppola TK 2008. Bimolecular Fluorescence Complementation (BiFC) Analysis as a Probe of Protein Interactions in Living Cells. Annual Review of Biophysics 37:465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azad T, Tashakor A, and Hosseinkhani S. 2014. Split-luciferase complementary assay: applications, recent developments, and future perspectives. Analytical and Bioanalytical Chemistry 406:5541–5560. [DOI] [PubMed] [Google Scholar]
- 3.Kerppola TK 2006. Complementary methods for studies of protein interactions in living cells. Nat Meth 3:969–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Remy I and Michnick SW. 2007. Application of protein-fragment complementation assays in cell biology. Biotechniques 42:137–145. [DOI] [PubMed] [Google Scholar]
- 5.Morell M, Ventura S, and Aviles FX. 2009. Protein complementation assays: approaches for the in vivo analysis of protein interactions. FEBS Lett 583:1684–1691. [DOI] [PubMed] [Google Scholar]
- 6.Thorne N, Inglese J, and Auld DS. 2010. Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology. Chemistry & Biology 17:646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, and Michnick SW. 2007. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proceedings of the National Academy of Sciences 104:16916–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villalobos V, Naik S, Bruinsma M, Dothager RS, Pan M-H, Samrakandi M, Moss B, Elhammali A, and Piwnica-Worms D. 2010. Dual-Color Click Beetle Luciferase Heteroprotein Fragment Complementation Assays. Chemistry & Biology 17:1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Remy I and Michnick SW. 2006. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Meth 3:977–979. [DOI] [PubMed] [Google Scholar]
- 10.Inouye S and Sasaki S. 2007. Overexpression, purification and characterization of the catalytic component of Oplophorus luciferase in the deep-sea shrimp, Oplophorus gracilirostris. Protein Expression and Purification 56:261–268. [DOI] [PubMed] [Google Scholar]
- 11.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, et al. 2012. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chemical Biology 7:1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE, and Bouvier M. 2005. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Meth 2:177–184. [DOI] [PubMed] [Google Scholar]
- 13.Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, et al. 2016. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chemical Biology 11:400–408. [DOI] [PubMed] [Google Scholar]
- 14.Paulmurugan R and Gambhir SS. 2003. Monitoring Protein–Protein Interactions Using Split Synthetic Renilla Luciferase Protein-Fragment-Assisted Complementation. Analytical Chemistry 75:1584–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan XM, Tong Sun K, and Kobilka BK. 1992. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. Journal of Biological Chemistry 267:21995–21998. [PubMed] [Google Scholar]
- 16.Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, and Javitch JA. 2008. Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomabechi Y, Hosoya T, Ehara H, Sekine S.-i., Shirouzu M, and Inouye S. 2016. Crystal structure of nanoKAZ: The mutated 19 kDa component of Oplophorus luciferase catalyzing the bioluminescent reaction with coelenterazine. Biochemical and Biophysical Research Communications 470:88–93. [DOI] [PubMed] [Google Scholar]
- 18.Remy I and Michnick SW. 1999. Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proceedings of the National Academy of Sciences 96:5394–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liberles SD, Diver ST, Austin DJ, and Schreiber SL. 1997. Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proceedings of the National Academy of Sciences 94:7825–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helinek TG, Devlin TM, and Ch’ih JJ. 1982. Initial inhibition and recovery of protein synthesis in cycloheximide-treated hepatocytes. Biochemical Pharmacology 31:1219–1225. [DOI] [PubMed] [Google Scholar]
- 21.Gales C, Van Durm JJJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, and Bouvier M. 2006. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol 13:778–786. [DOI] [PubMed] [Google Scholar]
- 22.Frederick AL, Yano H, Trifilieff P, Vishwasrao HD, Biezonski D, Meszaros J, Urizar E, Sibley DR, et al. 2015. Evidence against dopamine D1/D2 receptor heteromers. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Xiong B, Zhen X, and Zhang A. 2009. Dopamine D1 receptor ligands: Where are we now and where are we going. Medicinal Research Reviews 29:272–294. [DOI] [PubMed] [Google Scholar]
- 24.Conroy JL, Free RB, and Sibley DR. 2015. Identification of G Protein-Biased Agonists That Fail To Recruit β-Arrestin or Promote Internalization of the D1 Dopamine Receptor. ACS Chemical Neuroscience 6:681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sánchez-Soto M, Bonifazi A, Cai NS, Ellenberger MP, Newman AH, Ferré S, and Yano H. 2016. Evidence for Noncanonical Neurotransmitter Activation: Norepinephrine as a Dopamine D2-Like Receptor Agonist. Molecular Pharmacology 89:457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, and Klotz KN. 2004. Comparative pharmacology of human beta-adrenergic receptor subtypes--characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol 369:151–159. [DOI] [PubMed] [Google Scholar]
- 27.Dionisotti S, Ongini E, Zocchi C, Kull B, Arslan G, and Fredholm BB. 1997. Characterization of human A(2A) adenosine receptors with the antagonist radioligand [3H]-SCH 58261. British Journal of Pharmacology 121:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Figueroa KW, Griffin MT, and Ehlert FJ. 2009. Selectivity of Agonists for the Active State of M(1) to M(4) Muscarinic Receptor Subtypes. The Journal of Pharmacology and Experimental Therapeutics 328:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunahara RK, Guan H-C, O’Dowd BF, Seeman P, Laurier LG, Ng G, George SR, Torchia J, et al. 1991. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 350:614–619. [DOI] [PubMed] [Google Scholar]
- 30.Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, et al. 1998. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA research monograph 178:440–466. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Comaprison of coelenterazine H and fumirazine as Nanoluc substrates. A. Kinetic measurement of luminescence after three hour incubation of rapamycin for NanoLuc complementation between FRB-Nluc1 and FKBP-Nluc2. Curves are with (open symbol) or without (filled symbol) 100 nM rapamycin for 50 nM (black), 500 nM (orange), 5 μM (blue), and 50 μM (red) coelenterazine H. B. Kinetic measurement of luminescence after three hour incubation of rapamycin for Rluc complementation between FRB-Rluc1 and FKBP-Rluc2 with or without 100 nM rapamycin incubation for coelenterazine H (color /symbol scheme same as A). C. Kinetic measurement of luminescence after three hour incubation of rapamycin for NanoLuc complementation between FRB-Nluc1 and FKBP-Nluc2. Curves are with (open symbol) or without (filled symbol) 100 nM rapamycin for 0.001% (black), 0.01% (orange), 0.1% (blue), and 1% (red) furimazine. D. Kinetic measurement of luminescence after three hour incubation of rapamycin for Kinetic measurement of Rluc complementation between FRB-Rluc1 and FKBP-Rluc2 after three hour incubation with or without 100 nM rapamycin incubation for coelenterazine H (color /symbol scheme same as C).
Supplementary Figure 2: Specificity of cognate G protein coupling. A. NanoLuc complementation between D1-Nluc1 (0.5 μg transfected) with non-interacting Gαi1-Nluc2 (filled circle; transfected amount = y axis) or D2-Nluc1 (0.5 μg transfected) with non-interacting Gαs-Nluc2 (open circle; transfected amount = y axis). B. DA induced complementation change between D1-Nluc1 and Gαi1-Nluc2 (filled circle) and D2-Nluc1 and Gαis-Nluc2 (open circle). C. D1-Nluc1 Gαs-Nluc2 engagement with dopamine (filled square), norepinephrine (circle), 10 μM DA + SCH23390 (open square). D. D2-Nluc1 Gαi-Nluc2 engagement with dopamine (filled square), norepinephrine (circle), 10 μM DA + Eticlopride (open square).