

Abstract
Recreational consumption of synthetic cannabinoid receptor agonists (SCRAs) is a growing crisis in public health in many parts of the world. AMB-FUBINACA is a member of this class of drugs and is responsible for a large proportion of SCRA-related toxicity both in New Zealand and internationally. Strikingly, little is currently known about the mechanisms by which SCRAs exert toxic effects or whether their activity through the CB1 cannabinoid receptor (the mediator of cannabinoid-related psychoactivity) is sufficient to explain clinical observations. The current study therefore set out to perform a basic molecular pharmacology characterization of AMB-FUBINACA (in comparison to traditional research cannabinoids CP55,940, WIN55,212–2, and Δ9-THC) in fundamental pathways of receptor activity, including cAMP inhibition, pERK activation, ability to drive CB1 internalization, and ability to induce translocation of β-arrestins-1 and −2. Activity pathways were then compared by operational analysis to indicate whether AMB-FUBINACA may be a biased ligand. Results revealed that AMB-FUBINACA is highly efficacious and potent in all pathways assayed. However, surprisingly, bias analysis suggested that Δ9-THC, not AMB-FUBINACA, may be a biased ligand, with it being less active in both arrestin pathways than predicted by the activity of the other ligands tested. These data may help predict molecular characteristics of SCRAs. However, more research is required to determine whether these molecular effects manifest in toxicity at tissue/system level.
Keywords: Synthetic cannabinoid receptor agonist, ligand bias, β-arrestin, AMB-FUBINACA, Δ9-THC, cannabinoid receptor
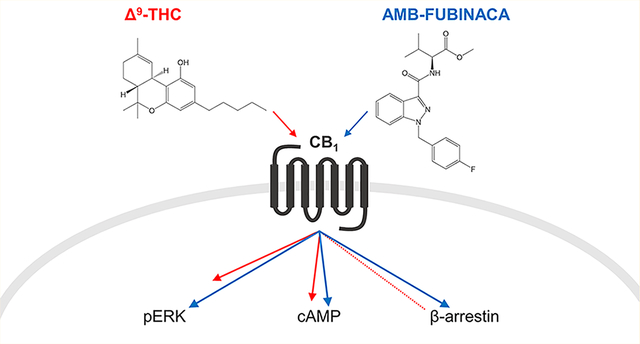
Graphical Abstract
INTRODUCTION
Substances that target CB1 and CB2 are a growing group of drugs of abuse. Indeed, in the decade 2009–2018, over 260 different synthetic cannabinoid receptor agonist (SCRAs) were identified by government agencies in surveyed United Nations Member States as “New Psychoactive Substances” (NPS), making them among the most rapidly proliferating classes of “designer drug”.1 Similarly, SCRAs were the most abundant (by tonnage) class of illicit synthetic NPS seized.1
SCRAs are increasingly being associated with severe toxicity and death.2–4 The Global Drug Survey found that SCRA use is more likely to lead to emergency medical treatment than any other drug, and that the overall risk of seeking emergency medical attention is at least 30 times greater after taking SCRAs than cannabis.5 Media reports also emphasize the increasing scale of this crisis: more than 25 people were killed and 700 hospitalized in a mass overdose involving the SCRA MDMB-FUBINACA in Russia in 2014.6 Outbreaks of 5F-ADB toxicity (with fatalities) have been reported in Minneapolis, MN7 and Japan8 (among other locations). A mass-intoxication in New York in 2016, reported at the time as a “zombie outbreak”, was attributed to AMB-FUBINACA,9 and a string of at least 45 deaths in New Zealand in 2017 was attributed to SCRAs, again predominantly AMB-FUBINACA,10,11 a drug now seemingly implicated in fatalities globally.12 This drug has therefore emerged as a globally prevalent case study for SCRA-associated toxicity.
AMB-FUBINACA (also known as MMB-FUBINACA, FUBAMB) is an indazole-3-carboxamide which was first reported in 2014, in Sweden to the European Monitoring Centre for Drugs and Drug Addiction,13 and is one of many emerging synthetic cannabinoids to show highly potent and efficacious agonism at CB1 and CB2.14,15 Previous molecular pharmacological characterization has shown that AMB-FUBINACA acts with high efficacy to promote CB1-mediated GTPγS accumulation and similarly acts at CB2 but with slightly lower efficacy than CP55,940.14 In the same study, AMB-FUBINACA was shown to act through CB1 to inhibit forskolin-induced cAMP with high potency and efficacy, but its effects on this pathway were not obviously different from those of CP55,940 or a number of other “traditional” synthetic cannabinoids. In vivo, AMB-FUBINACA behaved comparably to other ligands in a drug discrimination paradigm (which aligns with drug abuse liability), by potently substituting for Δ9-tetrahydrocannabinol (Δ9-THC) in mice trained to discriminate Δ9-THC from vehicle.14 This discriminative effect has since been confirmed in another study,16 which also noted that high concentrations of AMB-FUBINACA induced tremors. Interestingly, the authors’ interpretation of this finding (in the context of the toxicity experienced by recreational users of this drug) was that the concentration window between active and toxic doses is small; but they further noted that the subjective effects of cannabinoids sought by users may be independent of the toxic effects.16 The mechanism underlying these different pathways, however, remains unknown. Indeed, pharmacological properties of SCRAs described to date do not seem sufficient to explain the high levels of toxicity associated with these compounds.
In molecular pharmacology, the concept of ligand bias (functional selectivity) may help explain how different drugs act via the same target receptor but with greatly varying outcomes.17,18 Functional selectivity suggests that different ligands are capable of binding to their target in such a way that different receptor conformations are stabilized, which in turn modulates the extent to which different downstream pathways are activated.17 This idea has now been exploited in some receptor systems, where the consequences of different signaling pathways activities are comparatively well understood. The prototypical example of this is the mu-opioid receptor, which is thought to mediate a desirable analgesic effect through G protein-dependent pathways but adverse effects (including potentially fatal respiratory depression) through arrestin pathways (although characterization of this system continues and may not yet be fully understood19). Novel biased drugs with improved safety are now in clinical trial (reviewed in refs 20 and 21). In contrast to the mu-opioid receptor, the tissue/whole system-level roles of the different signaling pathways downstream of CB1 have not yet been dissected.
To investigate whether functional selectivity can help explain the toxicity associated with synthetic cannabinoids, we have undertaken a detailed molecular pharmacological study of AMB-FUBINACA. We compare the activity profiles of AMB-FUBINACA to those of several traditional, structurally and pharmacologically diverse cannabinoid agonists in a battery of standard in vitro assays for CB1 function. These reference drugs include Δ9-THC (the psychoactive constituent of plant cannabis), its “nonclassical” bicyclic analogue CP55,940, and the aminoalkylindole WIN55,212–2.22,23 Pathways investigated include inhibition of forskolin-induced cAMP production, production of pERK, and the canonical G protein-coupled receptor (GPCR) regulatory end points of ligand-mediated receptor internalization and recruitment of β-arrestin-1 and −2. Ligand pathway activities were compared for bias by operational analysis.24,25
RESULTS AND DISCUSSION
Affinity.
In order to confirm previous reports of AMB-FUBINACA affinity at CB1 and CB2,14 heterologous competition displacement radioligand binding assays were performed using [3H]-CP55,940 (all curves were consistent with one-site binding). In agreement with this previous report, AMB-FUBINACA exhibited high affinity binding at both CB1 and CB2 in the nanomolar (pKi 8.72 ± SEM 0.12, n = 5) and subnanomolar ranges (pKi 9.32 ± SEM 0.09, n = 5), respectively. Under matched conditions, AMB-FUBINACA affinity was similar to that of CP55,940 (pKd at CB1 of 8.79 and at CB2 of 9.3326). For comparative purposes, affinities were also obtained for WIN55,212–2 and Δ9-THC at CB1 and were found to be somewhat lower, with pKi 7.01 (± SEM 0.11, n = 5) and 7.28 (± SEM 0.14, n = 5), respectively.
Signaling.
Gαi/o is the canonical G protein effector of CB1 and acts to reduce cAMP levels by inhibiting adenylate cyclase. As shown in Figure 1 and Table 1, AMB-FUBINACA inhibited forskolin-induced cAMP with similar potency to that of CP55,940. Potencies of inhibition of cAMP by WIN55,212–2 and Δ9-THC were approximately 10-fold lower than those of AMB-FUBINACA and CP55,940, as expected from previously reported estimates.26 AMB-FUBINACA, CP55,940, and WIN55,212–2 were equi-efficacious in inhibiting cAMP production, and each of these drugs inhibited with significantly greater efficacy than Δ9-THC did (one-way ANOVA, F = 6.953; Holm-Šídák multiple comparisons post-test), consistent with the well-established partial agonist classification of this drug.27–29
Figure 1.
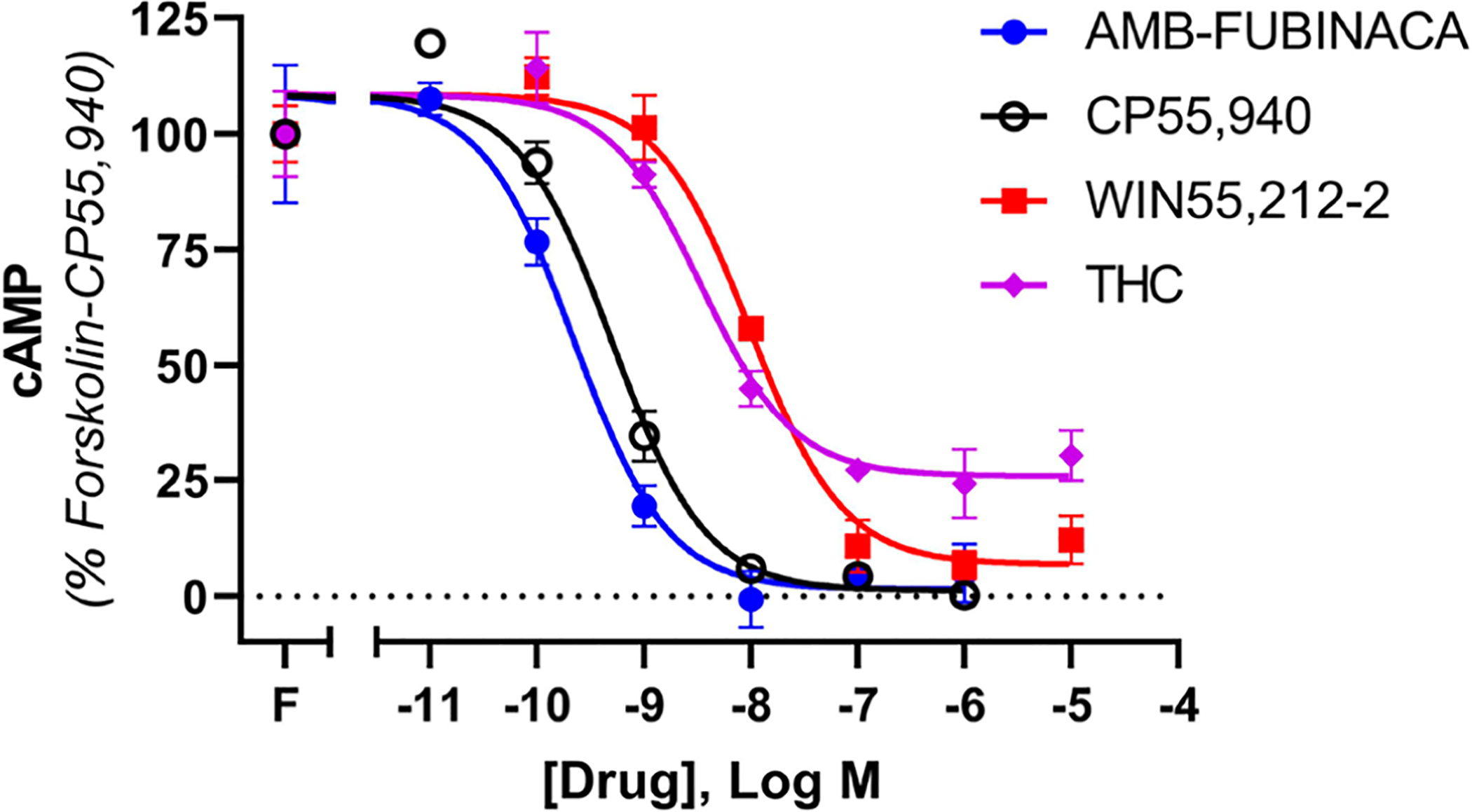
Concentration-response curves for cAMP signaling, showing 3HA-hCB1 HEK signaling on stimulation with 5 μM forskolin (F) and CP55,940, AMB-FUBINACA, WIN55,212–2, or Δ9-THC. Representative data are shown, demonstrating mean ± SEM of technical duplicates in the same assay.
Table 1.
Agonist Potencies and Efficacies in cAMP, pERK, Internalization, β-Arrestin-1 and −2 Pathways (pEC50 and EMAX ± SEM), n (Independent Determinations) = 3–5
| CP55,940 | AMB-FUBINACA | WIN55,212–2 | Δ9-THC | ||
|---|---|---|---|---|---|
| cAMP (n = 4) | pEC50 | 9.20 ± 0.10 | 9.18 ± 0.18 | 8.12 ± 0.14 | 7.78 ± 0.26 |
| Emax (span, %) | 99.4 ± 3.8 | 101.1 ± 5.8 | 92.1 ± 3.3 | 67.8 ± 8.8 | |
| pERK (n = 4, WIN n = 3) | pEC50 | 8.35 ± 0.18 | 8.80 ± 0.16 | 6.91 ± 0.11 | 7.23 ± 0.03 |
| Emax (span, %) | 100 ± 1.1 | 118.2 ± 3.9 | 113.8 ± 1.5 | 50.9 ± 5.9 | |
| internalization (n = 3) | pEC50 | 8.28 ± 0.13 | 8.14 ± 0.03 | 6.60 ± 0.17 | 7.14 ± 0.12 |
| Emax (1/MRT) | 0.0753 ± 0.0040 | 0.1197 ± 0.0082 | 0.1465 ± 0.0081 | 0.023 ± 0.0035 | |
| β-arrestin-1 (n = 4) | pEC50 | 7.29 ± 0.33 | 7.05 ± 0.09 | ~5.48 ± 0.17 | ND |
| Emax (span, %) | 19.1 ± 9.6 | 101.7 ± 2.5 | ND | ND | |
| β-arrestin-2 (n = 4) | pEC50 | 6.75 ± 0.14 | 7.26 ± 0.04 | ~5.89 ± 0.05 | ND |
| Emax (span, %) | 57.2 ± 2.6 | 102.4 ± 1.3 | ND | ND |
Canonically, activation (phosphorylation) of the mitogen-activated protein kinase, pERK, occurs downstream of activity of both Gα and Gβγ subunits of the G protein.18 In this signaling pathway, both AMB-FUBINACA and CP55,940 elicited peak pERK responses at 4 min (Figure 2A), so this time point was selected for subsequent concentration-response assays. These assays (Figure 2B, Table 1) revealed very similar activity profiles for all agonists, compared to their responses in cAMP: rank order potencies and efficacies were largely preserved. As not all compounds could be analyzed within a single assay, all assays included a CP55,940 concentration-response curve to enable normalization between experiments. This signaling pathway is activated with less efficiency than cAMP, so agonist potencies are right-shifted relative to cAMP inhibition. AMB-FUBINACA showed higher efficacy than the matched CP55,940 response but was not significantly more efficacious than WIN55,212–2 (which elicited a pERK effect mean slightly greater than the matched CP55,940 response). Δ9-THC stimulated ERK phosphorylation with significantly lower efficacy than the matched CP55,940 response (one-way ANOVA, F = 64.43; Holm-Šídák multiple comparisons post-test; note that as CP55,940 was used for normalization, it was not included in this analysis).
Figure 2.
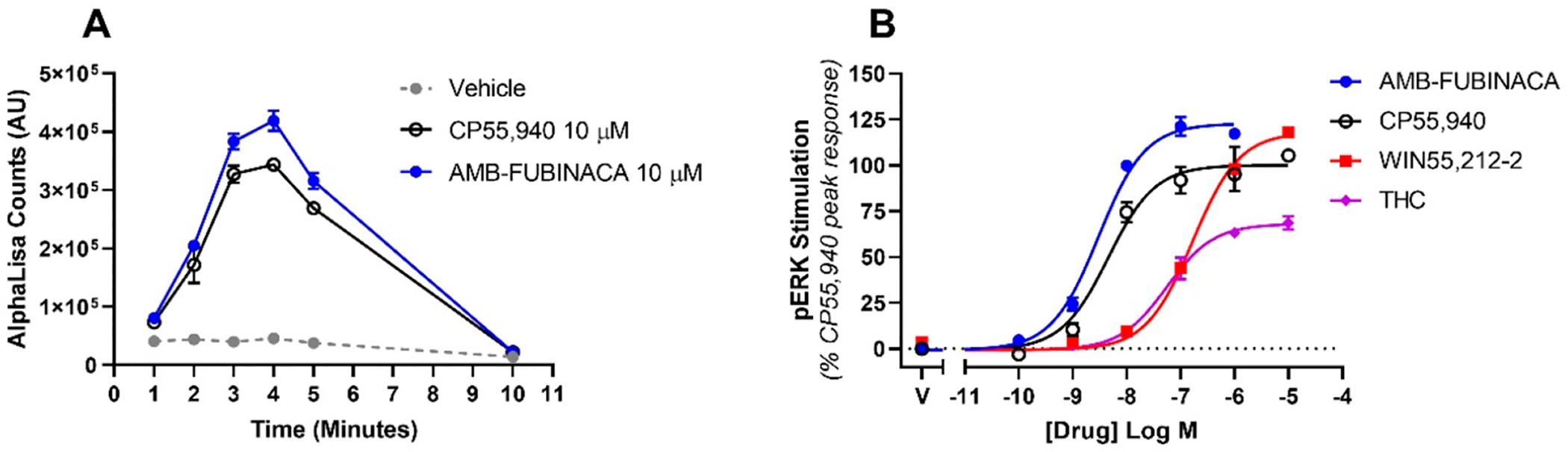
CB1-mediated phosphorylation of ERK in response to stimulation with cannabinoids, showing the time course of stimulation by high concentrations of two agonists of interest (A) and the agonist concentration responses at peak activity (4 min, B). Representative data are shown, demonstrating mean ± SEM of technical replicates in the same assay. AlphaLISA units (A) or normalized to the peak CP55,940 signal at 4 min (B).
Regulatory Responses to CB1 Agonism: Internalization and Arrestin Activation.
Surface receptor number is an essential component of efficacy. Indeed, receptor number has appeared in receptor theory as a contributor to effect size since the 1960s,30,31 and its modulation by receptor internalization helps prevent toxic system overstimulation.32,33 The profile of AMB-FUBINACA-mediated internalization of CB1 is therefore highly pertinent in attempts to understand molecular contributions to SCRA toxicity. Canonically, GPCR internalization occurs following agonist stimulation and homologous desensitization.34 Receptors are phosphorylated by G protein-coupled receptor kinases, which are thought to facilitate a decrease in affinity of G protein to the intracellular face of the receptor, resulting in signal desensitization.35
Phosphorylation is also understood to promote recruitment of β-arrestin-1 and β-arrestin-2 (arrestin-2 and arrestin-3) to receptors.36,37 β-Arrestins are scaffolding proteins, which are considered to be predominantly involved in receptor internalization via clathrin-coated vesicle formation and dynamin-facilitated endocytosis. However, a more recent concept is that arrestins may also be responsible for a second complement of receptor-mediated signaling, a G protein-independent set of responses.38,39 As previously noted, this idea was also an important foundation for the current study, because of the importance of G protein versus arrestin bias in adverse effects for other receptor systems, such as the opioid receptors20,21,40 (though it should be noted that the question of whether arrestins are sufficient to drive signaling responses alone is not fully settled41).
In the current study, the different agonists screened produced highly variable profiles of agonist-induced receptor internalization (Figure 3). Indeed, high concentrations of each agonist resulted in maximum half-lives (T1/2, minutes for 50% of surface receptors to internalize) ranging from (mean, ±SEM, n = 3) 1.92 min ± 0.60 (WIN55,212–2, Figure 3C; note that these data were first reported in Zhu et al.42), 2.46 min ± 0.29 (AMB-FUBINACA, Figure 3A), 5.16 min ± 0.25 (CP55,940, Figure 3B), to 19.05 min ± 3.29 min (Δ9-THC, Figure 3D). WIN55,212–2, AMB-FUBINACA and CP55,940-induced internalization rates were not significantly different, but all three induced significantly faster receptor internalization than Δ9-THC (one-way ANOVA, F = 23.68; Holm-Šídák multiple comparisons post-test). Data were analyzed for inverse mean residence time (MRT; the ratio of the concentration versus time area-under-the-curve and the area under the first moment curve), as previously described,42 in order to convert kinetic data into a form analyzable with the EMAX and operational models (Figure 3E, Table 1). Inverse MRT therefore reflects the average time a labeled receptor remains on the cell surface. In this form, WIN55,212–2 was slightly but significantly more efficacious than AMB-FUBINACA; both were significantly more efficacious than CP55,940; and all three were significantly more efficacious than Δ9-THC (one-way ANOVA, F = 72.78; Holm-Šídák multiple comparisons post-test). However, compared to cAMP and pERK signaling, inverse MRT analysis showed largely similar relative potencies, with AMB-FUBINACA and CP55,940 exerting effects with similar and high potencies, and WIN55,212–2 and Δ9-THC stimulating their effects at potencies approximately 2 orders of magnitude lower.
Figure 3.
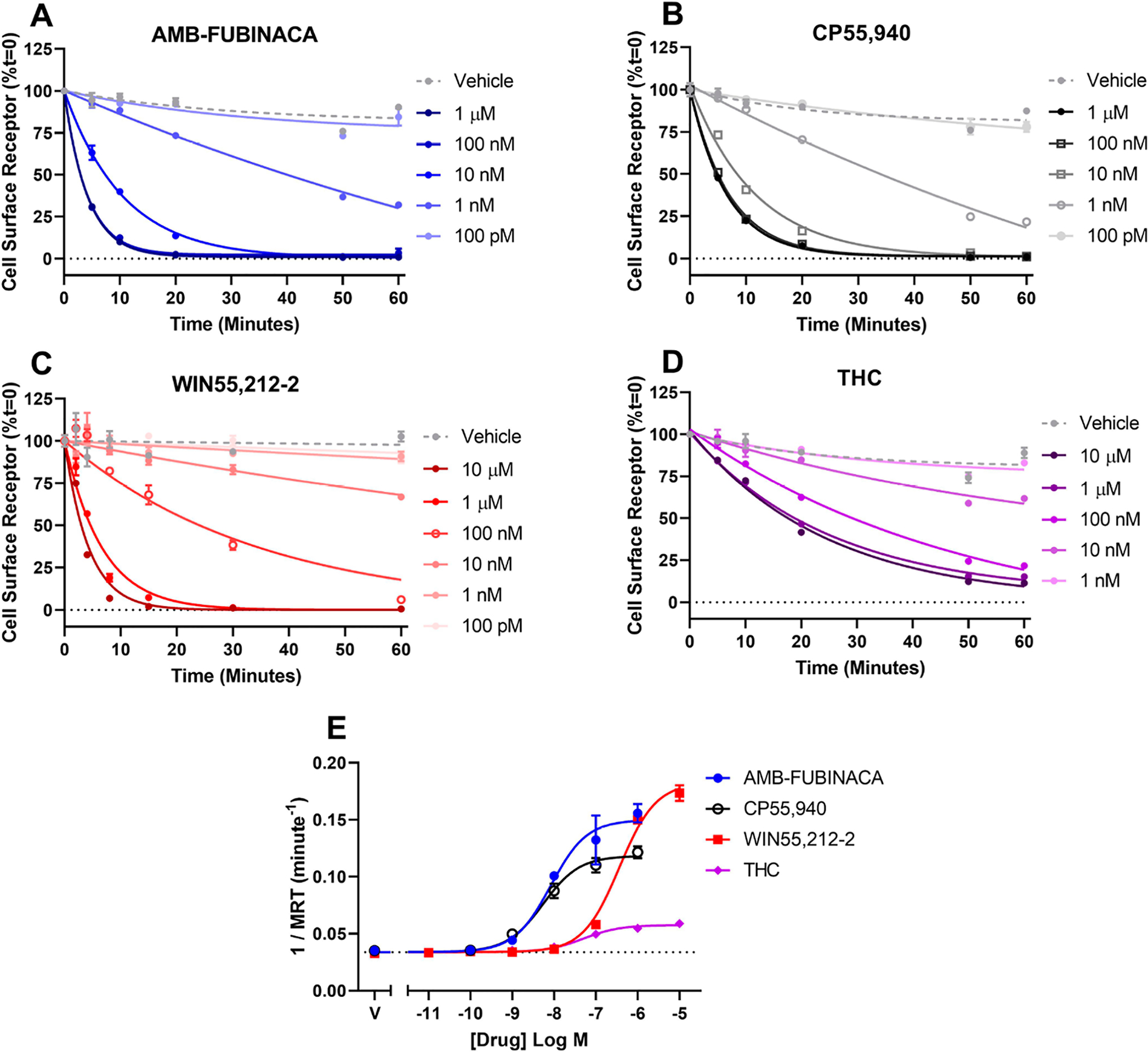
CB1 internalization following stimulation with AMB-FUBINACA, CP55,940, WIN55,212–2, or Δ9-THC. Data show time courses of internalization for each agonist (A–D), which have been analyzed for inverse mean residence time (1/MRT, E). Representative data are shown, demonstrating mean ± SEM of technical duplicates.
In general, the activity profiles of AMB-FUBINACA are largely consistent in the signaling and internalization pathways characterized thus far-its primary attribute is demonstrable high efficacy and potency in all pathways. Assays to quantify CB1-dependent stimulation of arrestin translocation revealed that AMB-FUBINACA recruits both β-arrestin-1 (Figure 4A, C) and −2 (Figure 4B, D) with generally equivalent maximal rates and efficacies compared to reference ligands; a finding that is consistent with recent reports.43,44 However, as observed in other pathways (above), these AMB-FUBINACA effects were elicited with substantially higher potency (Table 1) than WIN55,212–2 both for β-arrestin-1 and −2. In both cases, the WIN55,212–2 responses were such low potency that the curves were poorly determined, making the reported pEC50 values for WIN55,212–2 approximations only.
Figure 4.
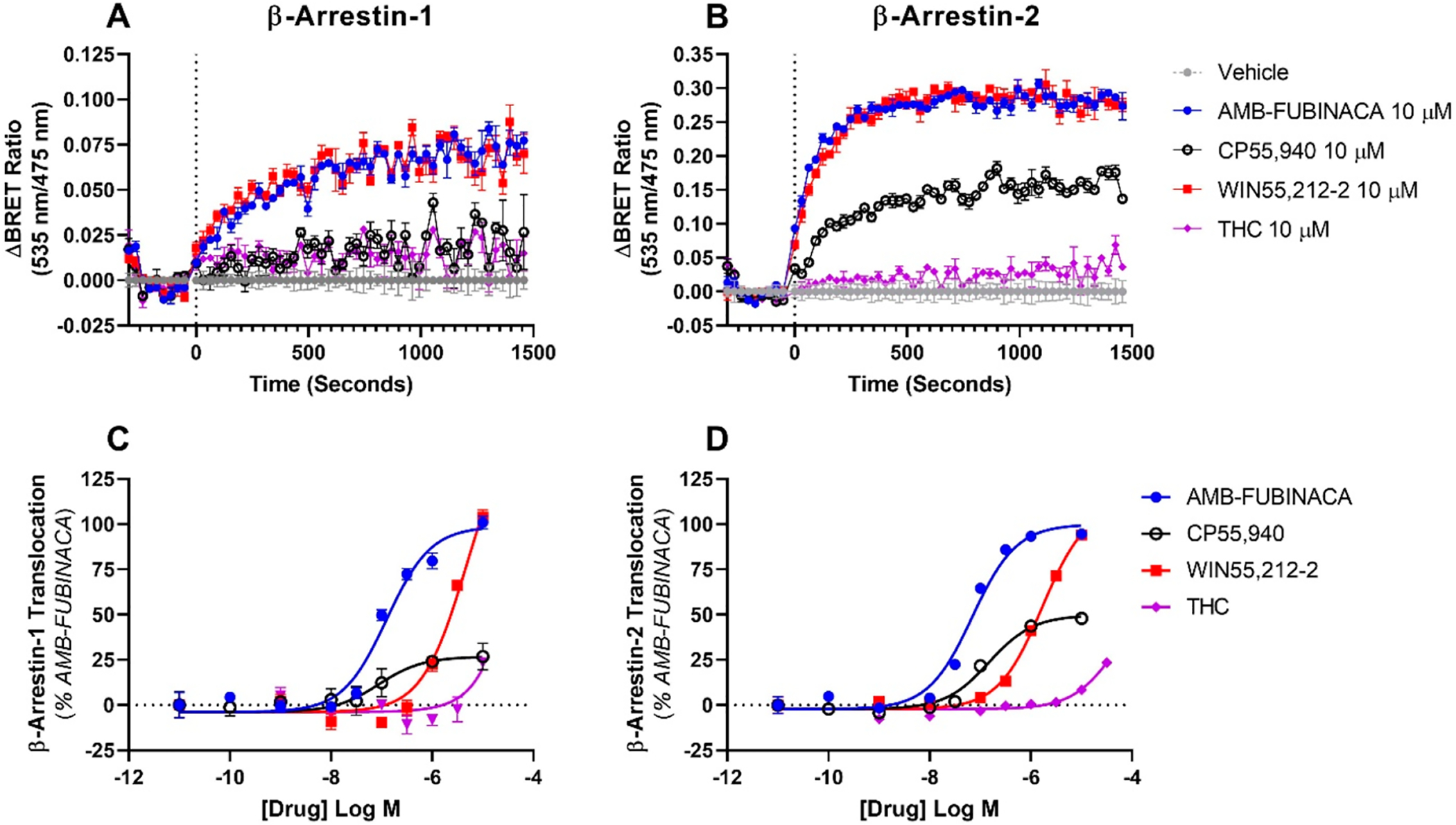
CB1-mediated translocation of β-arrestin-1 (A, C) and −2 (B, D) in response to stimulation with AMB-FUBINACA, CP55,940, WIN55,212–2, or Δ9-THC. Data presented are technical triplicates (mean ± SEM), shown as kinetic traces at high agonist concentrations (A, B), and concentration–response relationships (C, D).
More interestingly, activity profiles for AMB-FUBINACA were clearly distinct from CP55,940, with AMB-FUBINACA driving significantly higher efficacy responses for translocation of both β-arrestins-1 and −2 (two-tailed paired t tests). This was especially pronounced for β-arrestin-1, for which AMB-FUBINACA elicited a maximum efficacy approximately 5 times larger than that of CP55,940 (Table 1). Furthermore, comparison of AMB-FUBINACA with Δ9-THC was even more stark: Δ9-THC efficacy and potency were too small for EMAX model parameters to be accurately obtained in either arrestin pathway. The activity profiles of AMB-FUBINACA in both arrestin pathways therefore appears unique in comparison to the other compounds included in this study. Perhaps most notably, the arrestin responses elicited by AMB-FUBINACA may be the only arrestin responses of sufficient potency that they may occur in vivo; it would be challenging for micromolar concentrations to be achieved inside the human blood-brain barrier for exogenous substances, as would be necessary for WIN55,212–2-mediated β-arrestin activation, for example. The differing natures of AMB-FUBINACA and Δ9-THC effects in the activation of arrestin pathways may suggest that these pathways are involved in on-target toxic effects associated with AMB-FUBINACA.
One of the signaling responses suggested to follow arrestin activation is a kinetically novel pERK signal. Across the GPCR field, G protein-mediated pERK signaling appears as reported here (Figure 2) − a transient signal of several minutes’ duration, which occurs with rapid onset, and is G protein-mediated. However, the idea of a second “wave” of signaling, mediated by β-arrestin-1, has been proposed for CB1.18,38 We have not observed any delayed signal in experiments to date, but exploring the fundamental determinants of this response would be an interesting future avenue of research to pursue. CB1 is well-known to signal through noncanonical signaling pathways under specific conditions.26,45–47 This gives precedence to the idea that system factors may be important to unmask drug-specific differences in outcome, and may help explain on-target SCRA toxicity.
Operational Analysis for Functional Selectivity.
In light of these data, we hypothesized that the relatively high potency and much higher efficacy of AMB-FUBINACA may reflect functional selectivity toward arrestin pathways. Quantitative analysis for functional selectivity (agonist bias) was therefore performed, using the operational analysis for bias method (first described by Kenakin et al. in 2012,24 with later modifications25,48). Different cell types (including both native cells in vivo, but also the heterologous models employed in this study) are naturally heterogeneous in their signaling responses. Factors contributing to this will include the array of signaling effectors expressed by a particular cell, the abundance and stoichiometry of receptors, and many others. The approach taken in the current study therefore sets out to compare ligands under matched conditions. This means that the cell type used to produce data (in this case, a heterologous system) should not alter the conclusions of the ligand comparison analysis, even though the profiles of activity may well differ to those elicited in vivo.
The convention in operational analysis is to utilize a ligand that is fully efficacious in all pathways as the reference ligand. This ligand is then normalized to 1.0, and serves as a point of comparison for all the other ligands in the study. We have initially utilized WIN55, 212–2 as the reference ligand. In this analysis, notably different profiles emerged for AMB-FUBINACA compared to THC. Indeed, in most pathway comparisons involving either β-arrestins-1 or −2, Δ9-THC was consistently biased to the nonarrestin pathway by more than an order of magnitude of R (10ΔΔlogR). A possible exception to this trend is in the cAMP/β-arrestin-1 pathway comparison (Figure 5A, Table 2). By comparison, AMB-FUBINACA manifested as seemingly balanced in all pathways comparisons, meaning that its activity was effectively predicted by the relative activity of WIN55,212–2 in each pairwise pathway comparison.
Figure 5.
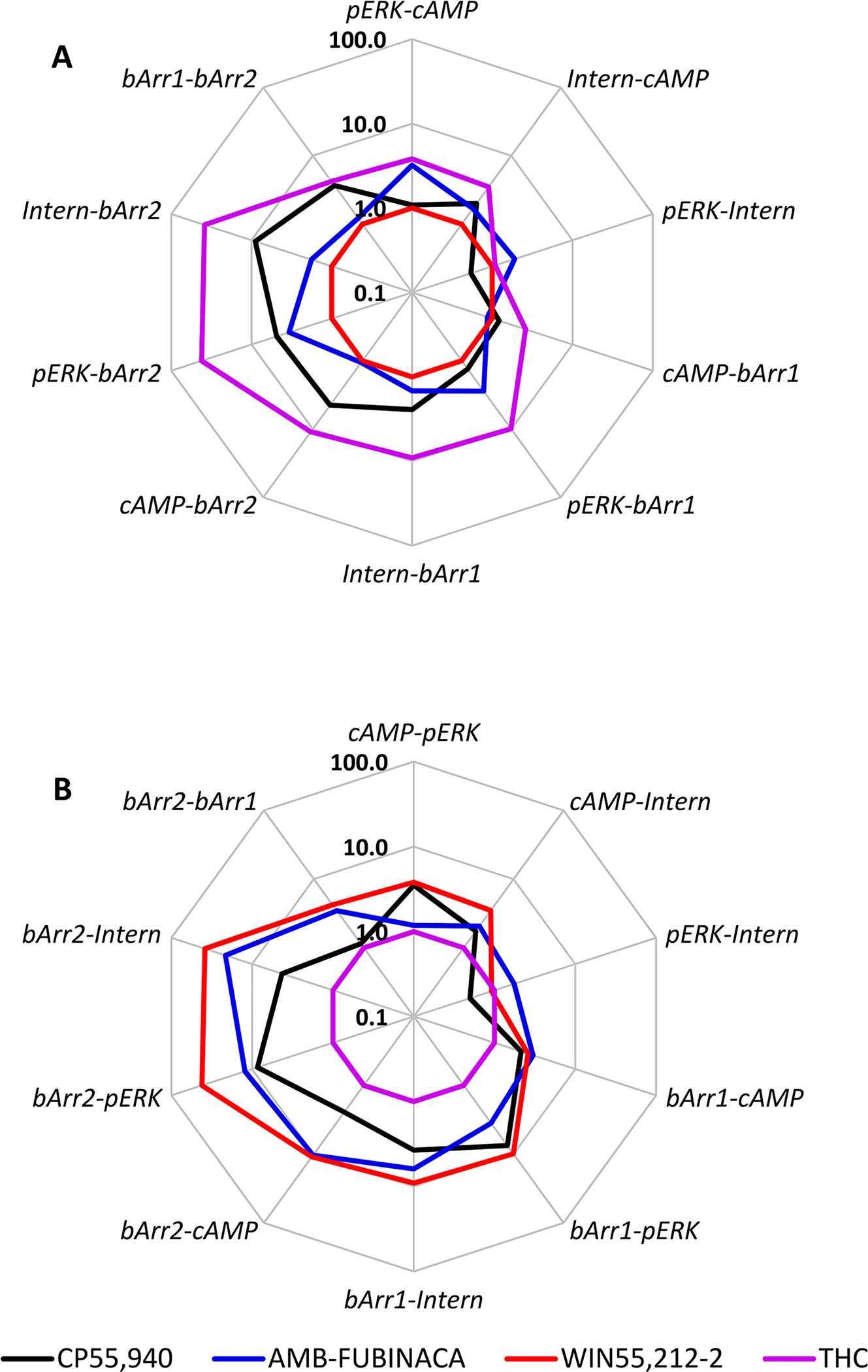
Radar plot showing operational analysis bias factors (10ΔΔlogR) for CP55,940, AMB-FUBINACA, WIN55,212–2, and Δ9-THC in all pathway comparisons assayed. The reference ligand is WIN55,212–2 (A) or Δ9-THC (B). Data (log R values) were collated from all available independent determinations (n = 3–4). Bias is indicated for the first pathway named in each comparison (each vertex) as bias factors > 1.0 (or vice versa). Note that the order of pathway comparisons is not always consistent between (A) and (B) in order that as many comparisons as possible will return values > 1.0.
Table 2.
Operational Analysis Results (ΔΔlog R ± SEM), Showing All Possible Pathway Comparisons for CP55,940, AMB-FUBINACA, WIN55,212–2, and Δ9-THCa
| CP55,940 | AMB-FUBINACA | WIN55,212–2 | Δ9-THC | |||||
|---|---|---|---|---|---|---|---|---|
| ΔΔlog R | SEM | ΔΔlog R | SEM | ΔΔlog R | SEM | ΔΔlog R | SEM | |
| pERK-cAMP | 0.036 | 0.262 | 0.506 | 0.299 | 0.000 | 0.266 | 0.581 | 0.441 |
| Intern-cAMP | 0.302 | 0.262 | 0.228 | 0.285 | 0.000 | 0.320 | 0.544 | 0.460 |
| pERK-Intern | −0.266 | 0.300 | 0.279 | 0.274 | 0.000 | 0.312 | 0.037 | 0.225 |
| cAMP-βArr1 | 0.581 | 0.558 | −0.061 | 0.261 | 0.000 | 0.257 | 0.415 | 0.464 |
| pERK-βArr1 | 0.616 | 0.576 | 0.446 | 0.250 | 0.000 | 0.247 | 0.996 | 0.233 |
| Intern-βArr1 | 0.882 | 0.577 | 0.167 | 0.233 | 0.000 | 0.304 | 0.959 | 0.267 |
| cAMP-βArr2 | 0.651 | 0.228 | 0.025 | 0.232 | 0.000 | 0.219 | 1.041 | 0.436 |
| pERK-βArr2 | 0.687 | 0.271 | 0.532 | 0.219 | 0.000 | 0.207 | 1.621 | 0.168 |
| Intern-βArr2 | 0.953 | 0.271 | 0.253 | 0.199 | 0.000 | 0.272 | 1.585 | 0.213 |
| βArr1-βArr2 | 0.070 | 0.562 | 0.086 | 0.164 | 0.000 | 0.195 | 0.625 | 0.221 |
The reference ligand is WIN55,212–2 (n = 3−4).
One of the primary aims of the current study was to systematically compare the profiles of AMB-FUBINACA with Δ9-THC. Both of these drugs have been consumed by humans; however, AMB-FUBINACA is associated with severe acute toxicity, while Δ9-THC is recognized to have low acute toxicity. Thus, to more fully interrogate potential differences between these two compounds, we repeated ΔΔlog R calculations, this time using Δ9-THC as the reference ligand (Figure 5B; note that some pathway comparisons were calculated in the opposite order to Figure 5A in order to ensure that as many values as possible would remain >1.0). As expected from the Δ9-THC bias findings (see above), all three other agonists were more active in activating both β-arrestins-1 and −2 than would be predicted from the activity of Δ9-THC. No agonist appeared biased for any other pathway in this analysis.
Recently, a cryogenic electron microscopy structure of the MDMB-FUBINACA-bound CB1-Gαi complex was published.49 MDMB-FUBINACA is an analogue of AMB-FUBINACA, differing only by the addition of a single methyl group to the pendant valinamide side-chain of AMB-FUBINACA. MDMB- and AMB-FUBINACA share an indazole scaffold, which has been shown (for MDMB-FUBINACA) to interact with the F2003.36 residue in the CB1 binding pocket, allowing W3566.48 to rotate (“toggle twin switch”) and form a Gαi-interacting cavity on the cytoplasmic face of the receptor.49,50 This interaction has been shown to occur with high efficiency for MDMB-FUBINACA and is likely to be common for AMB-FUBINACA (based on structural similarity). Indeed, it has been postulated that this interaction is the crucial difference between high affinity agonists, and Δ9-THC.49 The structural rigidity of MDMB-FUBINACA (and likely AMB-FUBINACA) causes the toggle twin switch interaction, and therefore GTP cycling, and receptor activation much more readily than the comparatively flexible Δ9-THC.49 Naturally, the immediate downstream pathway of Gαi facilitates the canonical effects of the receptor, and we have shown here that AMB-FUBINACA is a high efficacy agonist with nanomolar potency matching that of CP55,940 (Figure 1), hitherto often considered a CB1 “full agonist” for inhibition of cAMP. Congruently, AMB-FUBINACA is more potent and efficacious than both WIN55,212–2 and Δ9-THC. This finding is supported by a previous study using a different cell line where the authors show similar cAMP potency to that defined in this study.14 Importantly, this suggests that the conformation of the receptor in the presence of Δ9-THC and AMB-FUBINACA is likely to be different; a key determinant of subsequent receptor bias.
Media reports have indicated that AMB-FUBINACA induces seizures (e.g., ref 51), consistent with observations for other synthetic cannabinoids in both rodents and humans,52–54 in contrast to typical responses to Δ9-THC in humans. It remains unclear whether these adverse effects are facilitated directly by the cannabinoid system or by other independent targets. Indeed, the seizurogenic effects of SCRAs are further confounded by a case report that has suggested that SR141716A (rimonabant, a widely used CB1-selective inverse agonist) may also promote seizures in susceptible patients.55 Evidence that other, noncannabinoid receptor systems may be involved in SCRA effects include the idea that naloxone (muopioid receptor antagonist) may have protective utility for sufferers of SCRA intoxication.56 In any case, such profound effects highlight the necessity of both investigating the seizurogenic potential of AMB-FUBINACA in comparison to “traditional” cannabinoids like Δ9-THC, but also systematically assessing polypharmacology (potential activity at noncannabinoid receptor targets) of AMB-FUBINACA and SCRAs in its class.
Pharmacokinetic considerations will also be highly important for understanding SCRA toxicity, encompassing attributes as diverse as route of administration and efficiencies of absorption, blood-brain barrier permeability (if toxic effects are indeed centrally mediated), and, importantly, pharmacological activity of metabolites (including the parent compound’s bioconversion rate). While substantial data are available for Δ9-THC (e.g., ref 57), little pharmacokinetics data are currently available for AMB-FUBINACA (particularly in humans), although forensic and in vitro data are now beginning to appear.43,58 Significantly, these new data suggest that AMB-FUBINACA metabolism occurs exceedingly quickly, with O-demethylation of the parent compound occurring in hepatocytes within minutes, leaving only 0.5% AMB-FUBINACA in the assay system after 60 min.43 It is therefore possible that the pharmacology of SCRA metabolites may be of greater importance for toxicity than the parent compound itself, a concept considered by other researchers also.59 Certainly, in the case of MDMB-FUBINACA, a recent study has suggested that its primary metabolite (ADB-FUBINACA-COOH) is active at CB1 (albeit with substantially reduced potency compared to the parent compound).60
SCRA-related cause of death has also not yet been resolved. Cannabis smoking has previously been associated with myocardial infarct,2 and indeed reports have appeared to suggest that this is a factor of SCRA toxicity.61,62 But acute respiratory failure has also been implicated,12 in addition to the seizure associations discussed above. It is therefore clear that many aspects of physiology will be profoundly affected by exposure to SCRAs. It is worth noting again that the mechanisms of toxicity may not be the same as the mechanism for the subjective effects of SCRAs.16 Researchers must therefore be incisive in assay design, lest potentially unique drug activities are associated with only one of these outcomes.
Many questions arise from the current study. Is the high potency and efficacy of AMB-FUBINACA in activating β-arrestin-1 and −2 a common feature for SCRAs, a “molecular fingerprint”, and if so is it an important mediator of toxic effects? What are the manifestations of toxicity at the tissue (forensic) level, and does this point to mechanisms of toxicity? Can central nervous system activity explain SCRA-related deaths? Do SCRAs such as AMB-FUBINACA have activity at targets other than CB1 and CB2? Nonetheless, this study points to clear differences between Δ9-THC and AMB-FUBINACA in CB1-mediated effects; future studies will aim to determine if these can be correlated with in vivo toxicity.
METHODS
Drugs. AMB-FUBINACA:
N-[[1-[(4-Fluorophenyl)methyl]-1H-indazol-3-yl]carbonyl]-l-valine, methyl ester. Stored at 10 mM in DMSO.
CP55,940:
(−)-cis-3-[2-Hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol, Cayman Chemical (AUS), cat#190–90084, lot# 0447745–19. Stored at 10 mM in absolute ethanol.
[3H]CP55,940:
(−)-cis-3-[2-Hydroxy-4-(1,1-dimethylheptyl)-phenyl]-trans-4-(3)hydroxypropyl) cyclohexanol [side chain-2, 3, 4–3H(N)] (PerkinElmer, Waltham MA), cat# NET1051001MC, lot#2248643. Stored at 4 μM in absolute ethanol.
Δ9-Tetrahydrocannabinol (Δ9-THC):
(6aR,10aR)-6,6,9-Trimethyl-3-propyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromen-1-ol, THC Pharm GmbH (Germany). Stored at 31.6 mM in absolute ethanol.
WIN55-212-2:
(R)-(+)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate, Tocris Bioscience, Bristol, U.K., cat#1038, batch#32A/159481. Stored at 10 mM in absolute ethanol.
Membrane Preparation.
Methods utilized have been previously reported in Finlay et al.26 In brief, HEK cells stably expressing pplss-3HA-hCB1 (first reported in Finlay et al.26) were cultured in 175 cm2 polystyrene flasks until semiconfluency. Cells were then lifted with 5 mM EDTA in PBS, sedimented by centrifugation (200 x g), and snap frozen at −80 °C. Cell pellets were resuspended in sucrose buffer (200 mM sucrose, 50 mM Tris-HCl pH 7.4, 5 mM MgCl2, 2.5 mM EDTA) and homogenized on ice with a glass-glass dounce homogenizer. The homogenate was sedimented at low speed (200g, 10 min; P1 pellet). The supernatant was then sedimented at 26 916 x g for 30 min. The supernatant was discarded, and the remaining P2 pellet resuspended in sucrose buffer, aliquoted, and stored at −80 °C. Protein concentration was quantified using a protein assay kit (Bio-Rad, Hercules CA) according to the manufacturer’s instructions.
Radioligand Binding Assays.
Unlabeled drugs were diluted into ice-cold binding buffer (50 mM HEPES pH 7.4, 1 mM MgCl2, 1 mM CaCl2, 0.2% fatty-acid-free BSA; MP Biomedicals, Auckland, NZ) and incubated at 30 °C for 1 h with [3H]-CP55,940 (0.75–2.5 nM) and 3–5 μg of P2 membrane isolate in a V-bottom 96-well mixing plate (Hangzhou Gene Era Biotech Co., China). During incubation, a 96-well GF/C glass-filter harvest plate (PerkinElmer, Waltham, MA) was incubated for 1 h in branched PEI (polyethylimine, 0.1% w/v, Sigma-Aldrich, St. Louis, MO). Filter plates were washed with 200 μL of ice-cold wash buffer (50 mM HEPES pH 7.4, 500 mM NaCl, 0.2%) BSA on a vacuum manifold (Pall Corp., New York, NY) at 5–10 mmHg. The binding assay cocktail (radioligand, membrane, and displacer drug) was filtered through the harvest plate and washed three times with 200 μL of wash buffer and then dried overnight at room temperature. The plate bottom was sealed, and IRGASAFE Plus scintillation fluid (PerkinElmer, Waltham, MA) was added to each well. Plates were incubated in dark conditions for 30 min, and then radioactivity was detected in a Wallac TriLux MicroBeta2 scintillation counter (PerkinElmer, Waltham, MA, 2 min counts).
BRET-CAMYEL cAMP Measurement.
HEK/3HA-hCB1 were seeded at a density of 5 × 106 cells into a 10 cm culture dish (Corning, Corning, NY) and grown overnight. Medium was replaced, and cells were transfected with 5 μg of pcDNA-His-CAMYEL (ATCC, described by Jiang et al.,63 Cawston et al.64) with 1.5 μg of linear PEI (Sigma-Aldrich, St. Louis, MO) in 10 mM NaCl and incubated overnight. Transfected cells were lifted and plated at 60 000–80 000 cells/well into a PDL-coated (Sigma-Aldrich) white 96-well CulturPlate (PerkinElmer, Waltham MA) and grown overnight. On assay day, culture medium was aspirated and wells were washed with HBSS (Thermo Fisher Scientific, Waltham MA) and then preincubated in assay buffer (HBSS supplemented with 1 mg/mL BSA) for 30 min. Five minutes before stimulation, coelenterazine-h was dispensed (Nanolight, Pinetop, AZ; final concentration 5 μM) to assay wells and moved to a prewarmed, 37° LUMIstar (BMG Labtech GmbH, Ortenberg, BW, Germany). Drugs (cannabinoid serial dilutions with forskolin, 2.5 and 5 μM final concentrations were tested; all vehicle controlled) were prepared in assay buffer at 10× concentration and dispensed into the prepared assay wells, and luminescence was detected simultaneously for both 460 and 535 nm wavelengths for approximately 20–30 min. Inverse BRET ratios (460/535 nm) were calculated in Omega MARS software (V3.1 R5, BMG Labtech GmbH, Ortenberg, Germany) and exported for further analysis in GraphPad Prism. Real time BRET traces were converted to concentration-response format by performing area-under-the-curve analysis in GraphPad Prism, followed by normalizing to forskolin (100%) and maximum inhibition by CP55,940 (0%).
AlphaLISA SureFire pERK Detection.
HEK/3HA-hCB1 were seeded into the inner-60 wells of PDL-coated 96-well plates (Corning, NY) at 30 000 cells/well and grown overnight. Complete medium was aspirated, replaced with serum-free DMEM supplemented with 1 mg/mL BSA, and incubated overnight prior to stimulation. Drugs were diluted in DMEM with 1 mg/mL BSA and applied at appropriate time points over the course of the assay in a barely submerged 37 °C water bath. Detection of pERK was performed using an AlphaLISA SureFire Ultra kit (PerkinElmer, Waltham, MA) in accordance with the manufacturer’s instructions. Plates were read in a CLARIOstar (BMG Labtech GmbH, Ortenberg, BW, Germany) and analyzed in GraphPad Prism.
Receptor Internalization Assay.
Assays for receptor internalization were performed as described in Grimsey et al.65 with modifications as described in Zhu et al.42 In brief, agonist-mediated receptor internalization was measured using a “live-at-start” method. 3HA-hCB1 HEK cells were plated (40 000 cells/well) into PDL-coated, clear-plastic 96-well plates (Nunc) and cultured overnight. Culture medium was replaced with DMEM supplemented with 1 mg/mL BSA (assay medium) for 1 h. Primary mouse anti-HA 16B12 monoclonal antibody (BioLegend, San Diego, CA) was diluted in assay medium (1:500), warmed to 37 °C, and then applied to each well for 30 min prior to drug stimulation. Antibody-containing medium was aspirated, and wells were washed, followed by application of prewarmed drug dilutions. At the end of the time course, receptor trafficking was arrested with a 5 min incubation on ice. Well contents were aspirated, and secondary antibody was dispensed (AlexaFluor goat anti-mouse 488, diluted 1:300 in assay medium; Thermo Fisher Scientific, Waltham, MA). Secondary antibody was incubated at room temperature for 30 min, aspirated, and then wells were twice-washed with assay medium. Cells were fixed in 4% paraformaldehyde (Sigma-Aldrich) in phosphate buffer (0.1 M) for 10 min. After fixation, wells were aspirated and washed twice in PBS. Hoechst 33258 (4 mg/mL in Milli-Q water, diluted 1:500 in PBS supplemented with 0.2% Triton-X100/PBS+T) was then dispensed for 15 min. Wells were then washed twice in PBS+T and finally left for storage and imaging in 50 μL/well PBS+T supplemented with 0.4 mg/mL merthiolate (thiomersal).
Microplate imaging was performed using an ImageXpress Micro XLS High-Content System automatic microscope. Images were analyzed using the associated analysis platform MetaXpress v6.2.3.733 (Molecular Devices, San Jose, CA) as previously described.42,65,66 Inverse MRT was calculated for time-course fluorescence data as, as previously described.42
Measurement of β-Arrestin Translocation.
Arrestin translocation assays were performed as previously described in Ibsen et al.,67 with modifications. HEK wildtype cells were plated into 10 cm culture dishes (Corning, NY) and cultured overnight. Complete culture medium was replaced, and then a transfection cocktail was dispensed, containing (per dish): 2 μg of mem-linker-citrine-SH3 pcDNA3.1+, 50 ng of Rluc8-human-β-arrestin-1 or −2 pcDNA3.1+, 1.6 μg of pplss-3HA-hCB1 pEF4a, and 350 ng of empty pcDNA3.1+; combined with 36 μg of PEI-Max (polyethylimine 25K, Polysciences, Warrington, PA) in Opti-MEM reduced serum medium (Thermo Fisher Scientific, Waltham, MA) and incubated overnight. Transfected cells were lifted with trypsin and seeded at a density of 60 000 cells/well into PDL-coated white 96-well CulturPlates (PerkinElmer, Waltham, MA) and incubated overnight. Culture medium was aspirated, and wells were washed with PBS, medium was replaced with HBSS supplemented with 1 mg/mL BSA (assay buffer) for 30 min. Coelenterazine-h and drug addition was performed as described above (cAMP CAMYEL). Luminescence was detected at 37 °C for 25 min in a LUMIstar Omega plate reader. BRET ratios (535/460 nm) were exported from Omega MARS software and analyzed in Prism 8. Data were normalized as described by Ibsen et al.67 Raw BRET values for the predrug incubation were averaged, and both the predrug baseline and subsequent drug condition data were normalized to this average. “Vehicle” BRET ratios were subtracted from all matches drug responses, and net area-under-the-curve obtained for each assay point.
Operational Analysis.
Operational analysis was conducted as described in Zhu et al.,48 based on a previously developed model.24,25 Log R was estimated separately from each independent determination (biological replicate, n = 3), from which SEM was generated. For drugs with very low potency or efficacy (particularly CP55,940 and Δ9-THC in the arrestin pathways), operational analysis will still allow a fit, but the log R values obtained from these fits should be considered estimates only. However, we consider these estimates conservative, because partially defined operational analysis curves may result in log R values that assume greater efficacy than may be the case (i.e., the model may treat the top of the curve as being equal to the nominal full agonist). At the level of bias analysis (ΔΔlog R calculations), this would result in an under-reporting of bias (should the true efficacy be lower than estimated by the model).
Data and Statistical Analysis.
All raw data were processed and analyzed in Prism 8 (GraphPad Software Inc. San Diego, CA) unless otherwise specified.
Funding
D.B.F. is supported by a Lottery Health Research Postdoctoral Fellowship. J.J.M. and M.P. are recipients of University of Otago Doctoral Scholarships. Support for research expenses was provided by the University of Auckland School of Medical Sciences, the University of Otago School of Biomedical Sciences, and a grant from the Maurice and Phyllis Paykel Trust to M.G.
ABBREVIATIONS
- BRET
bioluminescence resonant energy transfer
- BSA
bovine serum albumin
- cAMP
3′,5′-cyclic adenosine monophosphate
- CB1
type 1 cannabinoid receptor
- CB2
type 2 cannabinoid receptor
- Δ9-THC
trans-Δ9-tetrahydrocannabinol
- DMEM
Dulbecco’s modified Eagle’s medium
- ERK
Extracellular signal-related kinase
- GPCR
G protein-coupled receptor
- HBSS
Hank’s buffered saline solution
- MRT
mean residence time
- pERK
phosphorylated extracellular signal-related kinase
- PDL
poly-d-lysine hydrobromide
- PBS
phosphate-buffered saline solution
- SCRA
synthetic cannabinoid receptor agonist
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.United Nations Office on Drugs and Crime (2019) World Drug Report (Booklet 2): Global Demand and Supply In World Drug Report 2019, pp 48–51, United Nations. [Google Scholar]
- 2.Drummer OH, Gerostamoulos D, and Woodford NW (2019) Cannabis as a Cause of Death: A Review. Forensic Sci. Int 298, 298–306. [DOI] [PubMed] [Google Scholar]
- 3.Labay LM, Caruso JL, Gilson TP, Phipps RJ, Knight LD, Lemos NP, McIntyre IM, Stoppacher R, Tormos LM, Wiens AL, et al. (2016) Synthetic Cannabinoid Drug Use as a Cause or Contributory Cause of Death. Forensic Sci. Int 260, 31–39. [DOI] [PubMed] [Google Scholar]
- 4.Trecki J, Gerona RR, and Schwartz MD (2015) Synthetic Cannabinoid-Related Illnesses and Deaths. N. Engl. J. Med 373 (2), 103–107. [DOI] [PubMed] [Google Scholar]
- 5.Winstock A, Lynskey M, Borschmann R, and Waldron J (2015) Risk of Emergency Medical Treatment Following Consumption of Cannabis or Synthetic Cannabinoids in a Large Global Sample. J. Psychopharmacol 29 (6), 698–703. [DOI] [PubMed] [Google Scholar]
- 6.25 killed, over 700 hospitalized: Cheap ‘Spice’ Designer drug causes severe poisoning across Russia. RT World News, https://www.rt.com/news/193700-russia-spice-deadly-drug/ (accessed Feb 27, 2019). [Google Scholar]
- 7.Arens AM, Olives TD, Simpson NS, Laes JR, Anderson DL, Bangh SA, Lee SC, Martin S, Banister SD, Gerona RR, et al. (2019) An Outbreak of Synthetic Cannabinoid Exposures Reported to a Regional Poison Center: “K2” Identified as 5F-ADB. Clin. Toxicol 57 (1), 69–71. [DOI] [PubMed] [Google Scholar]
- 8.Hasegawa K, Wurita A, Minakata K, Gonmori K, Nozawa H, Yamagishi I, Watanabe K, and Suzuki O (2015) Postmortem Distribution of MAB-CHMINACA in Body Fluids and Solid Tissues of a Human Cadaver. Forensic Toxicol 33, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, and Gerona R (2017) Zombie” Outbreak Caused by the Synthetic Cannabinoid AMB-FUBINACA in New York. N. Engl. J. Med 376 (3), 235–242. [DOI] [PubMed] [Google Scholar]
- 10.31 Christchurch synthetics users hospitalised in last 22 days. Newshub, https://www.newshub.co.nz/home/new-zealand/2018/10/31-christchurch-synthetic-cannabis-users-hospitalised-in-last-22-days.html (accessed Feb 27, 2019). [Google Scholar]
- 11.Synthetic drugs kill 45 people in one year - Coroner. Newshub, https://www.newshub.co.nz/home/politics/2018/07/synthetic-drugs-kills-45-people-in-one-year-coroner.html (accessed Feb 27, 2019). [Google Scholar]
- 12.Adamowicz P, Meissner E, and Maślanka M (2019) Fatal Intoxication with New Synthetic Cannabinoids AMB-FUBINACA and EMB-FUBINACA. Clin. Toxicol, 1–6. [DOI] [PubMed] [Google Scholar]
- 13.European Monitoring Centre for Drugs and Drug Addiction (2015) EMCDDA-Europol 2014 Annual Report on the Implementation of Council Decision 2005/387/JHA, Implementation Reports [Google Scholar]
- 14.Gamage TF, Farquhar CE, Lefever TW, Marusich JA, Kevin RC, McGregor IS, Wiley JL, and Thomas BF (2018) Molecular and Behavioral Pharmacological Characterization of Abused Synthetic Cannabinoids MMB- and MDMB-FUBINACA, MN-18, NNEI, CUMYL-PICA, and 5-Fluoro-CUMYL-PICA. J. Pharmacol. Exp. Ther 365 (2), 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banister SD, Longworth M, Kevin R, Sachdev S, Santiago M, Stuart J, Mack JBC, Glass M, Mcgregor IS, Connor M, et al. (2016) Pharmacology of Valinate and Tert-Leucinate Synthetic Cannabinoids 5F-AMBICA, 5F-AMB, 5F-ADB, AMB-FUBINACA, MDMB-FUBINACA, MDMB-CHMICA, and Their Analogues. ACS Chem. Neurosci 7 (9), 1241–1524. [DOI] [PubMed] [Google Scholar]
- 16.Gatch MB, and Forster MJ (2019) Cannabinoid-like Effects of Five Novel Carboxamide Synthetic Cannabinoids. Neuro-Toxicology 70, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional Selectivity and Classical Concepts of Quantitative Pharmacology. J. Pharmacol. Exp. Ther 320 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- 18.Ibsen MS, Connor M, and Glass M (2017) Cannabinoid CB 1 and CB 2 Receptor Signaling and Bias. Cannabis Cannabinoid Res 2 (1), 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, Williams JT, Christie MJ, and Schulz S (2019) Phosphorylation-Deficient G-Protein-Biased μ-Opioid Receptors Improve Analgesia and Diminish Tolerance but Worsen Opioid Side Effects. Nat. Commun 10 (1), 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siuda ER, Carr R, Rominger DH, and Violin JD (2017) Biased Mu-Opioid Receptor Ligands: A Promising New Generation of Pain Therapeutics. Curr. Opin. Pharmacol 32, 77–84. [DOI] [PubMed] [Google Scholar]
- 21.Madariaga-Mazón A, Marmolejo-Valencia AF, Li Y, Toll L, Houghten RA, and Martinez-Mayorga K (2017) Mu-Opioid Receptor Biased Ligands: A Safer and Painless Discovery of Analgesics? Drug Discovery Today 22 (11), 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huffman JW, Dai D, Martin BR, and Compton DR (1994) Design, Synthesis and Pharmacology of Cannabimimetic Indoles. Bioorg. Med. Chem. Lett 4 (4), 563–566. [Google Scholar]
- 23.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, et al. (1986) Structure-Activity Relationships in Cannabinoids. Pharmacol. Rev 38 (2), 75–149. [PubMed] [Google Scholar]
- 24.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, and Novick S (2012) A Simple Method for Quantifying Functional Selectivity and Agonist Bias. ACS Chem. Neurosci 3 (3), 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Der Westhuizen ET, Breton B, Christopoulos A, and Bouvier M (2014) Quantification of Ligand Bias for Clinically Relevant b 2 -Adrenergic Receptor Ligands: Implications for Drug Taxonomy S. Mol. Pharmacol 85 (3), 492–509. [DOI] [PubMed] [Google Scholar]
- 26.Finlay DB, Cawston EE, Grimsey NL, Hunter MR, Korde A, Vemuri VK, Makriyannis A, and Glass M (2017) Gα s Signalling of the CB 1 Receptor and the Influence of Receptor Number. Br. J. Pharmacol 174 (15), 2545–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pertwee RG (2008) The Diverse CB 1 and CB 2 Receptor Pharmacology of Three Plant Cannabinoids: Δ 9 -Tetrahydrocannabinol, Cannabidiol and Δ 9 -Tetrahydrocannabivarin. Br. J. Pharmacol 153 (2), 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pertwee RG (2008) Ligands That Target Cannabinoid Receptors in the Brain: From THC to Anandamide and Beyond. Addict. Biol 13 (2), 147–159. [DOI] [PubMed] [Google Scholar]
- 29.Pertwee RG, Howlett a C., Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, et al. (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB 1 and CB 2. Pharmacol. Rev 62 (4), 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furchgott RF (1966) The Use of β-Haloalkylamines in the Differentiation of Receptors and in the Determination of Dissociation Constants of Receptor-Agonist Complexes In Advances in Drug Research (Harper NJ, and Simmonds AB, Eds.), pp 22–55, Vol. 3, Academic Press, New York. [Google Scholar]
- 31.Rang HP (2006) The Receptor Concept: Pharmacology’s Big Idea. Br. J. Pharmacol 147 (Suppl 1), S9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanyaloglu AC, and von Zastrow M (2008) Regulation of GPCRs by Endocytic Membrane Trafficking and Its Potential Implications. Annu. Rev. Pharmacol. Toxicol 48 (1), 537–568. [DOI] [PubMed] [Google Scholar]
- 33.Calebiro D, and Godbole A (2018) Internalization of G-Protein-Coupled Receptors: Implication in Receptor Function, Physiology and Diseases. Best Pract. Res. Clin. Endocrinol. Metab 32 (2), 83–91. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh C, Brown S, Derleth C, and Mackie K (1999) Internalization and Recycling of the CB1 Cannabinoid Receptor. J. Neurochem 73 (2), 493–501. [DOI] [PubMed] [Google Scholar]
- 35.Luttrell LM, and Lefkowitz RJ (2002) The Role of Beta-Arrestins in the Termination and Transduction of G-Protein-Coupled Receptor Signals. J. Cell Sci 115 (Pt 3), 455–465. [DOI] [PubMed] [Google Scholar]
- 36.Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C, and Mackie K (1999) Distinct Domains of the CB1 Cannabinoid Receptor Mediate Desensitization and Internalization. J. Neurosci 19 (10), 3773–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daigle TL, Kearn CS, and Mackie K (2008) Rapid CB1 Cannabinoid Receptor Desensitization Defines the Time Course of ERK1/2 MAP Kinase Signaling. Neuropharmacology 54 (1), 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nogueras-Ortiz C, and Yudowski GA (2016) The Multiple Waves of Cannabinoid 1 Receptor Signaling. Mol. Pharmacol 90 (5), 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahn KH, Mahmoud MM, Shim J-Y, and Kendall DA (2013) Distinct Roles of β-Arrestin 1 and β-Arrestin 2 in ORG27569-Induced Biased Signaling and Internalization of the Cannabinoid Receptor 1 (CB1). J. Biol. Chem 288 (14), 9790–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen X-T, Pitis PM, Gotchev D, Yuan C, et al. (2013) A G Protein-Biased Ligand at the -Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. J. Pharmacol. Exp. Ther 344 (3), 708–717. [DOI] [PubMed] [Google Scholar]
- 41.O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, Kawakami K, Aoki J, Messer K, Sunahara R, et al. (2017) Genetic Evidence That β-Arrestins Are Dispensable for the Initiation of Β2-Adrenergic Receptor Signaling to ERK. Sci. Signaling 10 (484), No. eaal3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu X, Finlay DB, Glass M, and Duffull SB (2019) Model-free and Kinetic Modelling Approaches for Characterising Non-equilibrium Pharmacological Pathway Activity: Internalisation of Cannabinoid CB 1 Receptors. Br. J. Pharmacol 176, 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fabregat-Safont D, Mardal M, Noble C, Cannaert A, Stove CP, Sancho JV, Linnet K, Hernández F, and Ibáñez M (2019) Comprehensive Investigation on Synthetic Cannabinoids: Metabolic Behaviour and Potency Testing, Using 5F-APP-PICA and AMB-FUBINACA as Model Compounds. Drug Test. Anal, No. dta.2659. [DOI] [PubMed] [Google Scholar]
- 44.Antonides LH, Cannaert A, Norman C, Vives L, Harrison A, Costello A, Daeid NN, Stove CP, Sutcliffe OB, and McKenzie C (2019) Enantiospecific Synthesis, Chiral Separation, and Biological Activity of Four Indazole-3-Carboxamide-Type Synthetic Cannabinoid Receptor Agonists and Their Detection in Seized Drug Samples. Front. Chem 7, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glass M, and Felder CC (1997) Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Augments CAMP Accumulation in Striatal Neurons: Evidence for a Gs Linkage to the CB1 Receptor. J. Neurosci 17 (14), 5327–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kearn CS, Blake-Palmer K, Daniel E, Mackie K, and Glass M (2005) Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Enhances Heterodimer Formation: A Mechanism for Receptor Cross-Talk? Mol. Pharmacol 67 (5), 1697–1704. [DOI] [PubMed] [Google Scholar]
- 47.McIntosh BT, Hudson B, Yegorova S, Jollimore CAB, and Kelly MEM (2007) Agonist-Dependent Cannabinoid Receptor Signalling in Human Trabecular Meshwork Cells. Br. J. Pharmacol 152 (7), 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu X, Finlay DB, Glass M, and Duffull SB (2018) An Evaluation of the Operational Model When Applied to Quantify Functional Selectivity. Br. J. Pharmacol 175 (10), 1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krishna Kumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, Latorraca NR, Kato HE, Hilger D, and Maeda S (2019) Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 176, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, and Abood ME (2004) Structural Mimicry in Class A G Protein-Coupled Receptor Rotamer Toggle Switches: The Importance of the F3.36(201)/W6.48(357) Interaction in Cannabinoid CB1 Receptor Activation. J. Biol. Chem 279 (46), 48024–48037. [DOI] [PubMed] [Google Scholar]
- 51.Sherwood S Warning issued over synthetic cannabis use after 10 people hospitalised https://www.stuff.co.nz/national/107268892/warning-issued-over-synthetic-cannabis-use-after-10-people-hospitalised (accessed Jun 26, 2019).
- 52.van Amsterdam J, Brunt T, and van den Brink W (2015) The Adverse Health Effects of Synthetic Cannabinoids with Emphasis on Psychosis-like Effects. J. Psychopharmacol 29 (3), 254–263. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz MD, Trecki J, Edison LA, Steck AR, Arnold JK, and Gerona RR (2015) A Common Source Outbreak of Severe Delirium Associated with Exposure to the Novel Synthetic Cannabinoid ADB-PINACA. J. Emerg. Med 48 (5), 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kevin RC, Anderson L, McGregor IS, Boyd R, Manning JJ, Glass M, Connor M, and Banister SD (2019) CUMYL-4CNBINACA Is an Efficacious and Potent Pro-Convulsant Synthetic Cannabinoid Receptor Agonist. Front. Pharmacol 10, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braakman HMH, van Oostenbrugge RJ, van Kranen-Mastenbroek VHJM, and de Krom MCTFM (2009) Rimonabant Induces Partial Seizures in a Patient with a History of Generalized Epilepsy. Epilepsia 50 (9), 2171–2172. [DOI] [PubMed] [Google Scholar]
- 56.Jones JD, Nolan ML, Daver R, Comer SD, and Paone D (2017) Can Naloxone Be Used to Treat Synthetic Cannabinoid Overdose? Biol. Psychiatry 81 (7), No. e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huestis MA (2007) Human Cannabinoid Pharmacokinetics. Chem. Biodiversity 4 (8), 1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu D, Zhang W, Li J, Wang J, Qin S, and Lu J (2019) Analysis of AMB-FUBINACA Biotransformation Pathways in Human Liver Microsome and Zebrafish Systems by Liquid Chromatography-High Resolution Mass Spectrometry. Front. Chem 7, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cannaert A, Storme J, Franz F, Auwärter V, and Stove CP (2016) Detection and Activity Profiling of Synthetic Cannabinoids and Their Metabolites with a Newly Developed Bioassay. Anal. Chem 88 (23), 11476–11485. [DOI] [PubMed] [Google Scholar]
- 60.Wouters E, Mogler L, Cannaert A, Auwärter V, and Stove C (2019) Functional Evaluation of Carboxy Metabolites of Synthetic Cannabinoid Receptor Agonists Featuring Scaffolds Based on L-valine or L- Tert -leucine. Drug Test. Anal 11, 1183. [DOI] [PubMed] [Google Scholar]
- 61.Hamilton RJ, Keyfes V, and Banka SS (2017) Synthetic Cannabinoid Abuse Resulting in ST-Segment Elevation Myocardial Infarction Requiring Percutaneous Coronary Intervention. J. Emerg. Med 52 (4), 496–498. [DOI] [PubMed] [Google Scholar]
- 62.Ozturk HM, Yetkin E, and Ozturk S (2019) Synthetic Cannabinoids and Cardiac Arrhythmia Risk: Review of the Literature. Cardiovasc. Toxicol 19 (3), 191–197. [DOI] [PubMed] [Google Scholar]
- 63.Jiang LI, Collins J, Davis R, Lin K-M, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, et al. (2007) Use of a CAMP BRET Sensor to Characterize a Novel Regulation of CAMP by the Sphingosine 1-Phosphate/G 13 Pathway. J. Biol. Chem 282 (14), 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, and Glass M (2013) Real-Time Characterization of Cannabinoid Receptor 1 (CB 1) Allosteric Modulators Reveals Novel Mechanism of Action. Br. J. Pharmacol 170 (4), 893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grimsey NL, Narayan PJ, Dragunow M, and Glass M (2008) A Novel High-Throughput Assay for the Quantitative Assessment of Receptor Trafficking. Clin. Exp. Pharmacol. Physiol 35 (11), 1377–1382. [DOI] [PubMed] [Google Scholar]
- 66.Finlay DB, Joseph WR, Grimsey NL, and Glass M (2016) GPR18 Undergoes a High Degree of Constitutive Trafficking but Is Unresponsive to N-Arachidonoyl Glycine. PeerJ 4, No. e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ibsen MS, Finlay DB, Patel M, Javitch JA, Glass M, and Grimsey NL (2019) Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front. Pharmacol 10, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]