

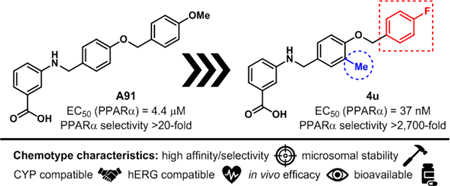
Abstract
Peroxisome proliferator-activated receptor alpha (PPARα) is expressed in retinal Müller cells, endothelial cells, and in retinal pigment epithelium, and agonism of PPARα with genetic or pharmacological tools ameliorates inflammation, vascular leakage, neurodegeneration, and neovascularization associated with retinal diseases in animal models. As such, PPARα is a promising drug target for diabetic retinopathy and age-related macular degeneration. Herein, we report proof-of-concept in vivo efficacy in an STZ-induced vascular leakage model (rat) and preliminary pharmacokinetic assessment of a first-generation lead 4a (A91). Additionally, we present the design, synthesis, and evaluation of second-generation analogs, which led to the discovery of 4u and related compounds that reach cellular potencies <50 nM and exhibit >2,700-fold selectivity for PPARα over other PPAR isoforms. These studies identify a pipeline of candidates positioned for detailed PK/PD and pre-clinical evaluation.
Keywords: PPARα, age-related macular degeneration, diabetic retinopathy, PPAR selectivity, structure-based design
Graphical Abstract
INTRODUCTION
Diabetic retinopathy (DR) is a common complication of diabetes (~30% of diabetics) and is the leading cause of blindness in the working population.1 Currently, >40% of the patient population fails to respond to the gold-standard treatment, direct intraocular injection of vascular endothelial growth factor (VEGF) antibodies.2 New therapies that are superior or complementary to current approaches would be of great value to patients, especially for those that remain refractory to current options. Ideally, unlike current treatment options, new approaches should be non-invasive (to the eye), affordable, and not reliant on specialized facilities.
The promise of peroxisome proliferator-activated receptor alpha (PPARα) agonism as a novel strategy for treating DR has been confirmed in two independent human clinical trials (FIELD and ACCORD), wherein orally administered Fenofibrate (Feno), a clinically used drug for hyperlipidemia, demonstrated robust protective effects against DR and retinal neovascularization (NV) in type 2 diabetic patients.3–4 This protection greatly reduced the need for patients to undergo disease modifying laser photocoagulation therapy. Since the report of these results, mechanistic and genetic studies have determined that the protective effects of Feno are unrelated to its lipid-lowering activity, but rather result from agonism of PPARα by the Feno metabolite fenofibric acid (FenoFA).5–6 Feno/FenoFA however, suffer from low ocular distribution, low affinity and selectivity for PPARα, and dose-limiting toxicities, all of which will likely limit its use as a DR therapy. Proof-of-concept, however, for targeting retinal PPARα with systemically available small molecules has been established with Feno and revealed that small molecule agonists with improved PK/PD profiles have high promise to become first-in-class non-invasive treatment options for DR and related ocular conditions (e.g., age-related macular degeneration and retinopathy of prematurity).7–8
As important members of the nuclear receptor superfamily, PPARs play essential roles in regulating cellular differentiation, development, metabolism, and tumorigenesis of higher organisms. Thus, these receptors have been considered attractive targets for treating metabolic disorders and inflammatory conditions.9–10 PPARs consist of 3 subtypes: PPARα, PPARγ and PPARδ/β. Although the ligand-binding domains share high sequence similarity (60–70%), the isoforms exhibit distinct tissue distribution, biological functionality, and can be targeted selectively.11 Within this family, PPARα has been implicated in hyperlipidemia, diabetes and inflammatory disorders; and agonism of this isoform provides pharmacological benefits for these conditions.12–13 Only recently, however, have the roles of PPARα in regulating VEGF, inflammation, mitochondrial function, apoptosis, and NV in diabetic retinae been revealed, thus elucidating a new avenue for PPARα agonists as therapeutics for oculovascular diseases.14–15 Specifically, previous studies demonstrate that PPARα knockout alone results in retinal mitochondrial dysfunction, a declined electroretinogram (ERG) response, and retinal cell apoptosis.16 Furthermore, PPARα levels in the retina are decreased in diabetic animal models and in diabetic humans and that agonism of PPARα restores balance.14–15 Although PPARα agonism was intensely investigated in the 1990s as a therapeutic approach for dyslipidemia, and numerous potent agonists exist in literature, none of the known chemotypes have been evaluated in animal models for efficacy against retinal conditions. Rather than attempting to repurpose known compounds, often lacking full disclosure of physicochemical and PK/PD profiles, we decided to embark on a discovery process to identify and develop novel PPARα agonistic chemotypes as new compositions for DR and AMD leads.
Recently, we published first-generation structure activity relationship (SAR) studies on a new PPARα agonistic chemotype, 4-carboxy-2-phenylquinolines, represented by Y-0452 (Figure 1).17 Guided by in-silico studies, we proposed that deconstruction of the rigid quinoline core would provide a more synthetically tractable and flexible scaffold, expected to exhibit improved complementarity to the U-shaped ligand binding pocket. Based on this design strategy, we developed the 4-benzyloxy-benzylamino chemotype to assess our hypothesis. In general, this small molecule family exhibits improved potency and selectivity over both Y-0452 (initial hit) and FenoFA, thus validating our rationale for further development of this chemotype and pursuing proof of concept studies. Compound A91 (Figure 1), a representative analogue from this series was advanced through rigorous biochemical evaluation to demonstrate typical downstream responses of PPARα agonism including PPARα upregulation, induction of target genes (e.g., Acadm, Cpt1a, Fabp3, and Slc25a20) and attenuation of cell migration.17 Having identified a new class of PPARα agonists and demonstrating useful SAR, the purpose of the work presented herein was to assess the preliminary PK and in vivo protection against vascular leakage of A91 to establish proof-of-concept and develop second-generation derivatives aimed at improving potency and selectivity of this chemotype.
Figure 1.
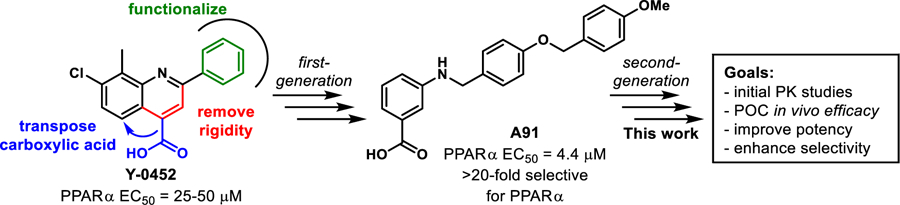
Overall strategy for hit optimization and the objective of the studies presented herein.
RESULTS AND DISCUSSION
PK/PD Profile of First-Generation Lead.
To gain insight about the metabolic stability and rate of clearance, both of which influence bioavailability, A91 was assessed for intrinsic clearance in human and rat (Sprague-Dawley) liver microsomes. As shown in Figure 2A, compound A91 proved to be rather stable, with the mean half-life (t1/2) exceeding 60 minutes in both human and rat microsomes. These results indicate that this chemotype is likely to exhibit low clearance and that phase-I hepatic metabolism is unlikely to limit bioavailability.
Figure 2. Pharmacokinetic profile of A91.
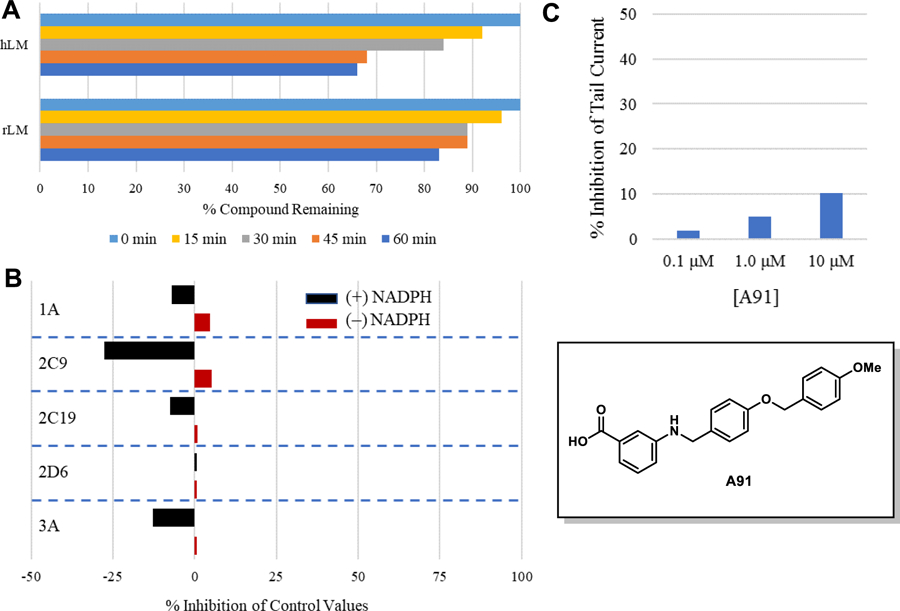
(A) Stability in liver microsomes. A91 was pre-incubated with pooled liver microsomes in phosphate buffer (pH 7.4) for 5 min at 37 °C. The system was activated by the addition of an NADPH-regenerating system and incubated for 0, 15, 30, 45, and 60 min before being quenched with an acetonitrile/methanol mixture. Samples were processed and analyzed by HPLC-MS/MS to determine the peak area remaining. Experiments completed in duplicate and mean values are depicted. (B) Assessment of A91 (10 μM) for irreversible inhibition of the major CYP450 drug metabolizing isoforms. Peak areas corresponding to the metabolite of known substrates for each isoform are recorded. The percent of control activity was calculated by comparing the peak area obtained in the presence of A91 to that obtained in the absence of A91. Percent inhibition was calculated by subtracting the percent control activity from 100. Time-dependent CYP inhibition is demonstrated if the percent inhibition from samples pre-incubated with the NADPH-regenerating system is larger than those without NADPH-regeneration. Negative values are a reflection of the variability in the background noise of the experiment. Experiments completed in duplicate and mean values are depicted. (C) Assessment of A91 for inhibition of the hERG channel. The degree of inhibition (%) was obtained by measuring the tail current amplitude, which is induced by a one-second test pulse to −40 mV after a two-second pulse to +20 mV, before and after drug incubation. Experiments completed in duplicate and mean values are depicted.
In parallel, A91 was assessed for evidence of time-dependent inhibition against the major drug metabolizing cytochrome P450 isoforms (i.e., 1A, 2C9, 2C19, 2D6, and 3A). In this assay, peak areas corresponding to the metabolite of control substrates were recorded in the presence or absence of A91. This analysis was completed in the presence and absence of NADPH, to reveal time-dependence of inhibition. Test compounds which exhibit higher inhibition of metabolite formation in the absence of NADPH are deemed time-dependent inhibitors. As shown in Figure 2B, A91 exhibits no evidence of time-dependent inhibition for any of the CYP450s evaluated. This observation is especially important for CYP3A, as this family is responsible for the metabolism of statins, a drug class required for a majority of diabetics. These results indicate that CYP drug-drug interactions due to irreversible or tight binding are unlikely with this chemotype.
Lastly, A91 was evaluated for inhibition of the human ether-á-go-go (hERG) potassium channel. Inhibition of this channel is a flagship sign of cardiac toxicity. For this experiment, the automated whole cell patch-clamp (Qpatch 48) technique was used to record outward potassium currents from a single cell in the presence and absence of the compound of interest. As shown in Figure 2C, compound A91 exhibits low potency for the hERG channel (compounds that exhibit ≥50% inhibition at 10 μM are flagged as toxic), indicating that this chemotype is unlikely to exhibit hERG-related cardiotoxicity, especially if potency for PPARα is improved. It is also worth noting that A91 exhibits no measurable cytotoxicity in the retinal pigment epithelium photoreceptor cell-line 661W at concentrations as high as 200 μM as measured in a colorimetric enzyme coupled LDH detection cytotoxicity assay (supplementary).
In vivo Efficacy of First-Generation Lead.
With a promising initial PK profile, and cellular potency and selectivity surpassing FenoFA, we advanced A91 to proof-of-concept in vivo efficacy studies in a well-established streptozotocin (STZ)-induced rat model of DR, which assesses the ability of a compound of interest to attentuate retinal vascular leakage—a major culprit behind diabetic macular edema and consequential vision loss. As shown in Figure 3A, systemic administration (i.p.) of compound A91 reduces retinal vascular leakage in diabetic rats to the range of the non-diabetic cohort at relatively the same dose as FenoFA. Of interesting note, A91 seems to lack signs of hepatomegaly (Figure 3B), a common side-effect of Feno/FenoFA observed in rodent models but not in humans. These in vivo results paired with the initial PK assessment, demonstrate that A91 1) exhibits in vivo efficacy in a relevant DR model following systemic administration, 2) crosses the blood-retinal barrier and reaches the retina, 3) is bioavailable, 4) survives first-pass metabolism and clearance mechanisms well enough to maintain efficacy, and 5) demonstrates a relatively safe profile (no observable toxicity) after daily i.p. injection for one-month.
Figure 3.

(A) Effects on retinal permeability and (B) liver phenotype for compound A91 in comparison to FenoFA. 7–8 week-old male Brown Norway rats were injected with streptozotocin (STZ, 55 mg/kg) to induce diabetes. Two weeks after STZ injections, daily treatment (i.p. injection) with compound A91 or FenoFA commenced and lasted for 28 days. Student’s t-test was used to compare groups for statistical significance and values are mean ± SD, # P<0.05 (vs. FenoFA), Retinal permeability was measured using Evans blue as tracer and normalized by total retinal protein. *P<0.05 (vs. STZ-DMSO).
Inspired by promising PK and in vivo results for A91, we commenced second-generation SAR studies with an aim of enhancing potency and isoform selectivity of this chemotype for PPARα. Since no co-crystal structure of A91 existed at the onset of this work, we developed an in-silico model using the Schrodinger Drug Discovery Suite to gain insight into the putative binding mode and guide our evolution of this chemotype. For these studies we selected PDB 2P54, a co-crystal structure of a known PPARα selective agonist GW590735 bound to human PPARα (hPPARα). To validate our docking approach, constraints, and parameters, GW590735 was extracted, exposed to MM2 energy-minimization, and re-docked into the hPPARα ligand binding domain to ensure that the results reproduced the bound conformation of the ligand. The overlay of co-crystallized and docked GW590735 showed excellent congruence (RMSD = 0.155 Å). Maintaining the same constraints and parameters, A91 was docked into hPPARα and the results were visually inspected for strategies to improve or introduce key interactions. Since our gatekeeper assay is a cell-based assessment and thus not based on direct binding affinity, scores were not used to prioritize ligands at this stage. Instead, visual inspection and structural modification hypotheses dictated the prioritization of derivatives selected for synthesis and assessment.
Our approach focused on three key principles. First, maintain a suitably positioned carboxylate (or isostere) to capture key hydrogen bonding interactions. Previous studies demonstrate the significance of hydrogen bond interactions between hPPARα Ser280, Tyr314, His440, and Tyr464 on the AF-2 helix and the carboxylate of known PPARα ligands.18–25 The ability to interact with all four of these residues is believed to be responsible for triggering full agonism of hPPARα.19 Poorer agonists tend to only interact with some of these hydrogen-bonding partners. It is worth noting that while historical PPARα literature suggests a carboxylic acid is required for activity (a pharmacophore), examples of PPARα agonists lacking this functionality have been reported,13, 26 demonstrating the -COOH motif can be altered. Secondly, we wanted to maintain the N-benzyl linkage to provide a complementary geometry for the U-shaped architecture of the ligand-binding domain. Third, our in-silico studies consistently predicted the aryl tail (C-ring) to bind a hydrophobic pocket comprised of helices 6 and 7 and defined by Ile241, Leu247, Ala250, Leu254, and Ala333. Literature precedence demonstrates that improvement of this interaction with other PPARα chemotypes has proven beneficial to enhancing ligand potency and selectivity.23–25
Stage I SAR: Linker Extension, C-Ring Modification, Carboxylate Location.
The first stage of our SAR (Figure 4) focused on assessing various hydrophobic substituents on the C-ring, probing the effects of linker extension, and determining optimal location for the carboxylate motif (or isostere).
Figure 4.

Multi-stage approach to structural improvement of A91. Model depicts 4a (A91) docked to PDBID 2P54.
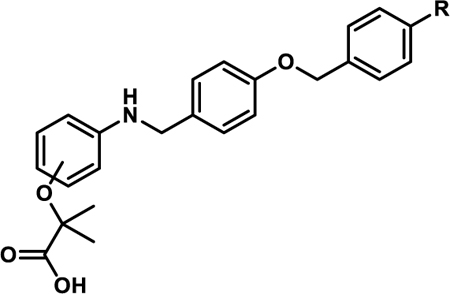
To generate stage I analogues, 4-hydroxybenzaldehyde (1) was coupled with various benzyl bromides (2a-l) to yield the corresponding 4-benzyloxy-benzaldehydes 3a-l (Scheme 1A). These precursors were then coupled with 3-aminobenzoic acid (or 4-aminobenzoic acid in the case of 4b*) via reductive amination to obtain 4a-4l and 4b*. Following a similar synthetic approach, 5a, 5b, 5b*, and 5f that include the typical fibrate head-group were synthesized (Scheme 1A). The only difference in the generation of 5a, 5b, 5b* and 5f was that the carboxylate remained protected as the ethyl ester until being revealed in the final step following saponification. Our previous studies indicated that the 4-benzyloxy-benzylamino chemotype deviates from the fibrates in that incorporation of the classical α-gem-dimethyl carboxylate “head-group” leads to pan-agonism and decreases potency. While these affects are obviously opposite to our goals, these analogues were generated to determine if this trend continued to propagate through this series.
Scheme 1.

Synthesis of stage I analoguesa
aReagents and conditions: A) (a) K2CO3, DMF; (b) i) 3-aminobenzoic acid or 4-amino benzoic acid, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 12 h, 32–97%; (c) i) ethyl 2-(3-aminophenoxy)-2-methylpropanoate or ethyl 2-(4-aminophenoxy)-2-methylpropanoate, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 12 h; (d) LiOH·H2O, THF/CH3OH/H2O, 12 h, 14–60%. B) (a) PBr3, diethyl ether; (b) 4-hydroxybenzaldehyde, K2CO3, DMF, 40–65%; (c) 3-aminobenzoic acid, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 12 h, 96%; (d) tyrosol, K2CO3, DMF, 26%; (e) 3-aminobenzoic acid, Et3N, DMF, 25%. All yields reported are unoptimized. Asterisks indicate the -COOH motif is attached para instead of meta.
To generate derivatives with extended linkers the synthesis began with the transformation of primary alcohols 6a-b into the corresponding alkyl bromides 7a-b (Scheme 1B). These precursors were then utilized to O-alkylate 4-hydroxybenzaldehyde to generate 8a-b, which were coupled with 3-aminobenzoic acid through reductive amination to provide 4m and 4n that included an extra –CH2– between the B and C-rings (Scheme 1B). Analogue 4o with an additional –CH2– between the A and B rings and the B and C rings was also generated. Chemoselective alkylation of tyrosol with 7b afforded 9, which was converted to the corresponding alkyl bromide 10 that was used to N-alkylate 3-aminobenzoic acid (Scheme 1B).
Stage I derivatives were evaluated for in vitro human PPARα (hPPARα) agonism in a commercially available hPPARα luciferase cell reporter assay (Indigo Biosciences). This assay utilizes non-human mammalian cells engineered to express hPPARα with the luciferase reporter gene functionally linked to a PPARα-response promoter. Thus, quantifying changes in luciferase expression in compound treated reporter cells provides a sensitive method to quantify changes in PPARα activity. As an initial first-pass assessment, all compounds were evaluated at two concentrations, 5 μM and 50 μM. This two-concentration approach is a cost-effective means to provide insight into 1) rough estimates of potency, 2) therapeutic window, and 3) relative levels of agonism. A 5:50 ratio <<1.0 indicates the compound is much more active at 50 μM than 5 μM, meaning the compound has poor potency. A 5:50 ratio ~1.0 indicates equal activity at both concentrations, meaning the EC50 is <5 μM and is non-cytotoxic to the cells at a 10-fold higher concentration, as cell death results in a decrease in luminescence production. As one would presume, a 5:50 ratio of >1.0 indicates a potency <5 μM, but also demonstrates toxicity at 50 μM. Therefore, 5:50 ratios of ~1.0 are desirable. GW590735, a highly selective (≥500-fold over PPARγ and PPARδ) PPARα partial agonist was employed as a positive control.24
In line with our hypothesis, the incorporation of lipophilic or electron withdrawing substituents on the C-ring of the benzoic acid chemotype (4b-l) provides an improvement in cellular activity in comparison to A91 (4a). Most analogues in the 4b-l series exhibit 5:50 ratios >0.5, indicating EC50 values <5 μM (Table 1). Comparison of 4b and 4b* demonstrates that the 3-carboxylate is favored over the 4-carboxylate. Within this class, we advanced 8 derivatives to multi-point dose-response analysis in the same luciferase assay. Compounds were chosen based on a combination of interest, 5:50 ratio, and level of agonism. As shown in Table 1, the 4-F (4b), 4-Br (4d), 2,4-difluoro (4j), and 3,4-difluoro (4k) analogues all exhibit submicromolar activity and are ~2–3 fold more active than the 4-I (4e) and 4-CF3 (4f). This result was somewhat forecasted by our in-silico results, which predicted that substituents larger than a bromine at the 4-position would be problematic. Even though the 3-F (4i) is less active than the 4-F (4b), it was still more active than A91. Thus, it is not surprising that inclusion of fluorine at both the 3- and 4-positions (4k) of this scaffold results in additive properties and is the most potent compound of this series. Inclusion of the typical fibrate head-group (5a, 5b, 5b*, and 5f) once again leads to decreased potency (Table 2) but often higher levels of agonism than the benzoic acid relatives (5a vs A91, 5b vs 4b, and 5f vs 4f). As shown in Table 3, analogs containing extended linkers 4m–4o exhibited poor potency (4m and 4n) or poor activation (4o) in comparison to related non-homologated derivatives.
Table 1.
In vitro Activity of 4a-la
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | Ratio (5/50)b | Fold-Signalc | EC50 hPPARα (μM)d | EC50 hPPARγ (μM) | EC50 hPPARδ (μM) | SIe |
| 4a (A91) | 4-OMe | 0.5 | 1.4 | 4.43±0.01 | >100 | >100 | >20 |
| 4b | 4-F | 0.8 | 1.3 | 0.77±0.03 | >100 | >100 | >125 |
| 4b* | 4-F | 0.3 | 0.4 | ||||
| 4c | 4-Cl | 0.6 | 1.5 | 0.83±0.04 | |||
| 4d | 4-Br | 0.8 | 1.3 | 0.97±0.14 | |||
| 4e | 4-I | 0.7 | 1.1 | 1.45±0.02 | |||
| 4f | 4-CF3 | 0.9 | 0.9 | 0.96±0.11 | |||
| 4g | 4-CN | 0.6 | 1.0 | ||||
| 4h | 2-F | 0.8 | 1.0 | ||||
| 4i | 3-F | 0.9 | 1.2 | 1.18±0.05 | |||
| 4j | 2,4-difluoro | 1.2 | 1.2 | 0.54±0.09 | >100 | >100 | >175 |
| 4k | 3,4-difluoro | 1.1 | 1.2 | 0.36±0.01 | |||
| 4l | 3,5-difluoro | 1.1 | 1.1 | ||||
| GW590735 | 1.0 | 1.0 | 0.015±0.002 | ||||
| Rosiglitazone | 0.28±0.3 | ||||||
| GW0742 | 0.0019±0.002 | ||||||
All analogues contain the carboxylate head-group at the meta-position of the A-ring unless otherwise indicated.
indicates a para-carboxylate.
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of maximal signal strength observed for the compound of interest to that obtained with GW590735.
EC50 values represent the mean ± SEM of atleast two separate experiments performed in triplicate.
SI = EC50 (PPARγ or PPARδ) / EC50 (PPARα). Blank cells indicate compound was not selected for testing in the corresponding assay.
Table 2.
All analogues contain the carboxylate head-group at the meta-position of the A-ring unless otherwise indicated.
indicates a para-carboxylate.
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of maximal signal strength observed for the compound of interest to that obtained with GW590735.
Table 3.
In vitro Activity of 4m-o
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of maximal signal (RLU) strength observed for the compound of interest to that obtained with GW590735.
From the stage I cohort, analogues 4b and 4j were selected to move forward to selectivity assessment, wherein equivalent luciferase assays were conducted on isogenic cell-lines engineered to overexpress either hPPARγ or hPPARδ. Neither compound produced significant levels of hPPARγ or hPPARδ agonism up to 100 μM compound concentration, indicating a selectivity of >125-fold (4b) and >175-fold (4j) for hPPARα. Rosiglitazone, a known selective PPARγ agonist, and GW0742, a known selective PPARδ agonist, were employed as positive controls in these isogenic cell-lines.
Stage II SAR: Strategic Methylation, Metabolism Blocking, Carboxylate Isosteric Replacement.
From the stage I results, compound 4b was selected as the base structure from this family for advancement. Three strategies were leveraged to advance the SAR in stage II 1) methylation at strategic positions within the scaffold that were expected to enhance complementarity with the shape and hydrophobicity of the PPARα ligand binding pocket; 2) blockage of predicted sites of metabolism; and 3) isosteric replacement of the carboxylate.
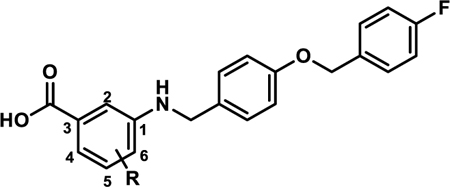
In silico prediction of major sites of metabolism for A91 with Schrodinger’s P450 Site of Metabolism software identified the A-ring as a potential liability for CYP450 oxidation (supplementary). As such, we synthesized 4p and 4q (Scheme 2) to interrogate the effects of fluorine blockage on activity. These two analogues were generated by coupling 3b to 5-amino-2-fluorobenzoic acid (11a) or 5-amino-3-fluorobenzoic acid (11b) to produce 4p and 4q, respectively. The 5-fluoro variant (4q) exhibits a ~3-fold improvement in activity over the 4-fluoro derivative (4p) and is nearly equipotent to 4b (Table 4). Interestingly, incorporation of a chlorine at the 5-position of the A-ring (4r) greatly decreases potency (5:50 ratio = 0.11), while incorporation of a bromine at the 5-position (4s) maintains potency in comparison to 4b but introduces apparent cytotoxicity at elevated concentrations (5:50 ratio = 1.3). These results suggest that fluorine incorporation should be a viable strategy to block sites on the A-ring if proven to be problematic as this series progresses through development.
Scheme 2.
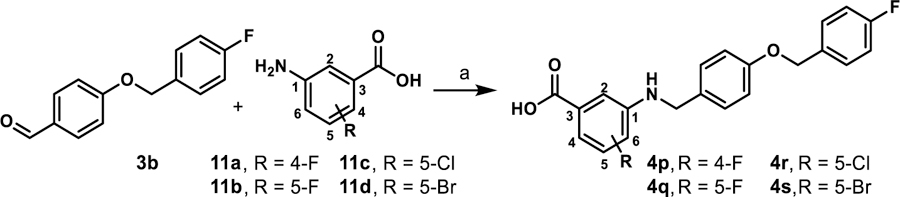
Synthesis of 4p-4sa
aReagents and conditions: (a) i) 11, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 25 °C, 12 h, 78–86%.
Table 4.
In vitro Activity of 4p-s
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | Ratio (5/50)a | Fold-Signalb | EC50 hPPARα (μM)c | EC50 hPPARγ (μM) | EC50 hPPARδ (μM) | SId |
| 4p | 4-F | 0.8 | 1.2 | 1.61±0.06 | |||
| 4q | 5-F | 1.1 | 1.1 | 0.58±0.04 | >100 | >100 | >170 |
| 4r | 5-Cl | 0.1 | 1.1 | ||||
| 4s | 5-Br | 1.3 | 1.4 | 1.12±0.08 | >100 | >100 | >100 |
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of signal maximal signal (RLU) strength observed for the compound of interest to that obtained with GW590735.
EC50 values represent the mean ± SEM of atleast two separate experiments performed in triplicate.
EC50 (PPARγ or PPARδ) / EC50 (PPARα). Blank cells indicate compound was not selected for testing in the corresponding assay.
Inspired by frequently reported methyl effects in drug discovery,27 analogues containing one or two methyl groups at different positions were synthesized (Scheme 3) using variations of chemistry discussed in Schemes 1 and 2 and evaluated in the luciferase assay (4t-z, Table 5). Specifically, we interrogated the effects of methylation on the 2- and 3-position of the B-ring, the 4-position of the A-ring, the nitrogen linker, and all possible variations on the benzylic position of the linker between the B- and C-rings (another predicted site of metabolic liability). Interestingly, installation of a methyl group at the 3-position of B-ring (4u) resulted in a drastic improvement in cellular activity, dropping the EC50 to ~40 nM, a ~21-fold improvement from the des-methyl relative, 4b, and improving the selectivity for the PPARα isoform to >2,700-fold (Figure 5). However, methylation at alternate sites (4t and 4v-z) failed to induce similar effects on the activity, indicating that the improvement observed with 4u is not likely to arise from an increase in general hydrophobicity but rather from improving aspects of target engagement.
Scheme 3.

Synthesis of methylated derivatives 4t-za
aReagents and conditions: (a) i) 3-aminobenzoic acid or 3-amino-4-methylbenzoic acid, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 25 °C, 12 h, 70–92%; (b) paraformaldehyde, NaBH(OAc)3, EtOH, reflux 2 h, 25 °C, 12 h, 43%; (c) (R)-1-(4-fluorophenyl)ethan-1-ol, di-tert-butyl azodicarboxylate, PPh3, toluene; (d) LiOH·H2O, THF/CH3OH/H2O, 25 °C, 12 h, 65–75%.; (e) (S)-1-(4-fluorophenyl)ethan-1-ol, di-tert-butyl azodicarboxylate, PPh3, toluene; f) 4-hydroxybenzaldehyde, TFAA, DBU, CuCl2, MeCN, 8%; (g) i) 3-aminobenzoic acid, toluene, 130 °C, ii) NaBH(OAc)3, AcOH, THF, 12 h, 25%. All yields reported are unoptimized.
Table 5.
In vitro Activity of 4t-z
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | Ratio (5/50)a | Fold-Signalb | EC50 hPPARα (μM)c | SId |
| 4t | 2-Me | H | H | H | H | 0.8 | 1.2 | 1.45±0.06 | |
| 4u | 3-Me | H | H | H | H | 1.4 | 1.2 | 0.037±0.01 | >2700 |
| 4v | H | Me | H | H | H | 0.2 | 0.3 | ||
| 4w | H | H | Me | H | H | 0.8 | 1.7 | 0.90±0.08 | |
| 4x | H | H | H | Me | H | 0.1 | 0.6 | 25.6±3.5 | |
| 4y | H | H | H | H | Me | 0.2 | 1.6 | ||
| 4z | H | H | H | Me | Me | 0.2 | 0.8 | 8.52±1.83 | |
| 4a (A91) | 0.5 | 1.4 | 4.43 ± 0.01 | >20 | |||||
| 4b | 0.8 | 1.3 | 0.77 ± 0.03 | >125 | |||||
| GW590735 | 1.0 | 1.0 | 0.015±0.002 | ||||||
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of signal maximal signal (RLU) strength observed for the compound of interest to that obtained with GW590735.
EC50 values represent the mean ± SEM of atleast two separate experiments performed in triplicate.
EC50 (PPARγ or PPARδ) / EC50 (PPARα). Blank cells indicate compound was not selected for testing in the corresponding assay.
Figure 5.

Dose-response curves for 4u (blue squares, solid line) against PPARα, PPARδ, and PPARγ. Respective controls are provided as black circles/dashed line.
While the reason for the drastic improvement in activity arising from incorporation of the methyl group at the 3-position of the B-ring is not obvious, we have interrogated this phenomenon briefly by in silico methods. Results from these studies suggest that methylation at the 3-position (4u) results in a binding shift that allows for improved hydrogen bonding with the Ser280, Tyr314, His440, and Tyr464 tetrad (Figure 6A). Additionally, 4u is predicted to bind with the aromatic ring of the benzoic acid perpendicular to His440 thus allowing for T-shaped edge-to-face pi-interactions (Figure 6A). Methylation at the 2-position (4t), however, produces no such shift and 4t overlays very well with the des-methyl analogue 4b (Figure 6B). As such, it is not surprising that the activity of 4t and 4b are relatively similar.
Figure 6.
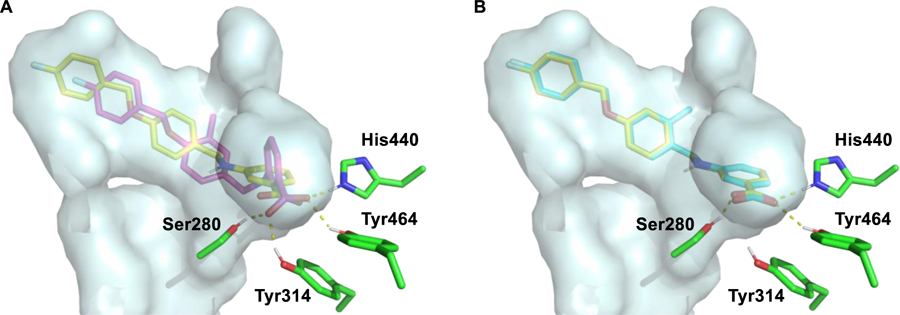
Predicted binding modes of 4b, 4t, and 4u, demonstrating the possible effects of methyl installation on the 2- or 3-position of the B ring. (A) Overlay of the predicted binding poses for 4b (yellow) and 4u (magenta). (B) Overlay of the predicted binding poses for 4b (yellow) and 4t (cyan). PDBID: 2P54. The pdb file of the docking model is included in the Supporting Information.
Coupling 3b to a variety of anilines containing carboxylic acid surrogates (18a-e) allowed us to assess the effect of isosteric replacement on PPARα agonistic activity (19a-e, Scheme 4). As seen in Table 6, replacement of the carboxylate with any of the isosteres resulted in dramatic decreases in activity, as noted by either a low 5:50 ratio or low overall signal. The exception being the tetrazole (19d), which exhibits decreased potency but comparable activation levels to the original carboxylate. This result confirms that the carboxylic acid is a key contributor to triggering PPARα agonism but can indeed be modified. As part of this series, the thio-variant of the fibrate head-group was also synthesized and assessed (19f). Interestingly, this derivative exhibits an attenuated level of agonism compared to the parent analogue, 5b. Differences in agonism levels now observed between the benzoic acid, fibrate head-group, and the thio-fibrate head-group strongly suggest that manipulation of this motif may provide a useful means to tune the level of agonism induced by a chemotype. This could be important as it is still unknown what level of stimulation (partial or full-agonism) is optimal for PPARα to have a therapeutic effect in the retina.
Scheme 4.
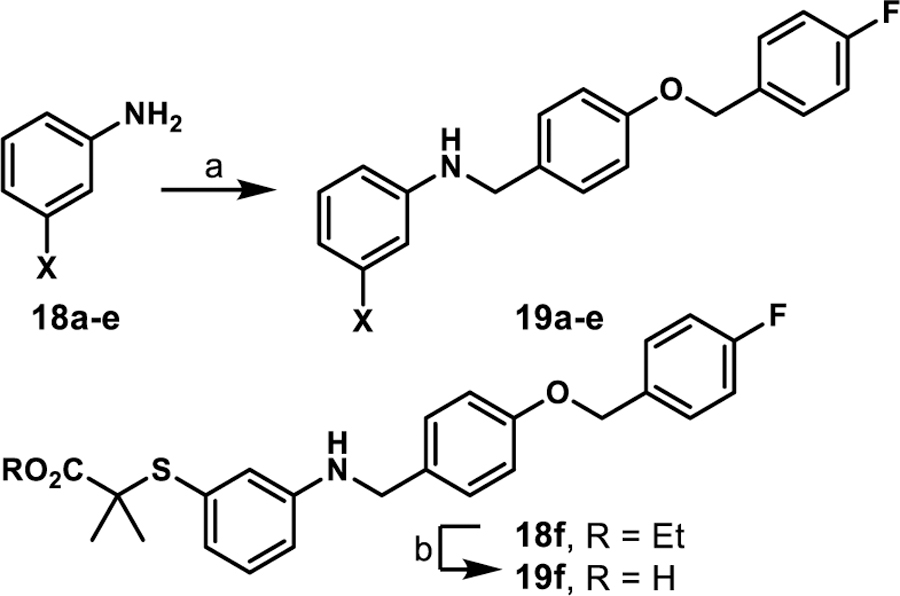
Synthesis of isostere containing derivatives 19a-fa
aReagents and conditions: (a) 3b, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 25 °C, 12 h, 8–80%; (b) LiOH·H2O, THF/CH3OH/H2O, 12 h, 13%.
Table 6.
In vitro Activity of Carboxylate Isosteres
| Compound | X | Ratio (5/50)a | Fold-Signalb |
|---|---|---|---|
| 19a |  |
1.0 | 0.1 |
| 19b | 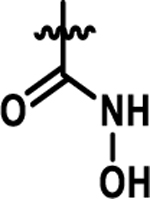 |
0.5 | 0.6 |
| 19c | 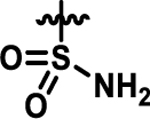 |
1.2 | 0.1 |
| 19d | 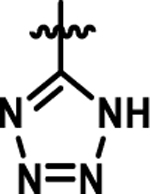 |
0.4 | 1.6 |
| 19e | 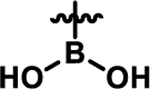 |
0.9 | 0.1 |
| 19f | 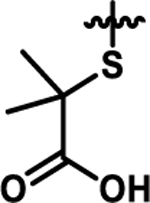 |
0.2 | 1.1 |
| 4b | 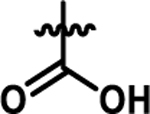 |
0.8 | 1.3 |
| GW590735 | 1.0 | 1.0 | |
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of signal maximal signal (RLU) strength observed for the compound of interest to that obtained with GW590735.
Additional Assessment of 4u.
As the most potent analogue identified in stages I and II, 4u was selected for additional assessment. To confirm target engagement, evaluate binding potency, and provide some insight into selectivity in a relevant human retinal cell line (MIO-M1, Müller cells) we subjected 4u to a CETSA (cellular thermal shift assay).28–29 Unlike the luciferase assay, MIO-M1 cells are not engineered to specifically report engagement with PPARα and thus provide a more realistic picture of compound behavior in a disease related cell setting. In an initial heat gradient, non-ligand associated PPARα displayed a temperature-dependent decay, with Tagg(50) and Tagg(75) at 40.8 °C and 45.5 °C, respectively (Figure 7A). In a subsequent test, dose-dependent potency of the PPARα agonists, A91 and 4u were tested at 45.5 °C, the temperature point at which 75% of free PPARα protein melted. As shown in Figure 7B, A91 improved PPARα stability incrementally with an EC50 of 0.3 μM. On the other hand, 4u rescued PPARα stability by 3-fold with an EC50 of 93 nM. The results for 4u correlate well with the EC50 value obtained from the luciferase assay (EC50 = 37 nM), demonstrating consistency of this compound within different contexts and eluding to excellent intracellular selectivity.
Figure 7.
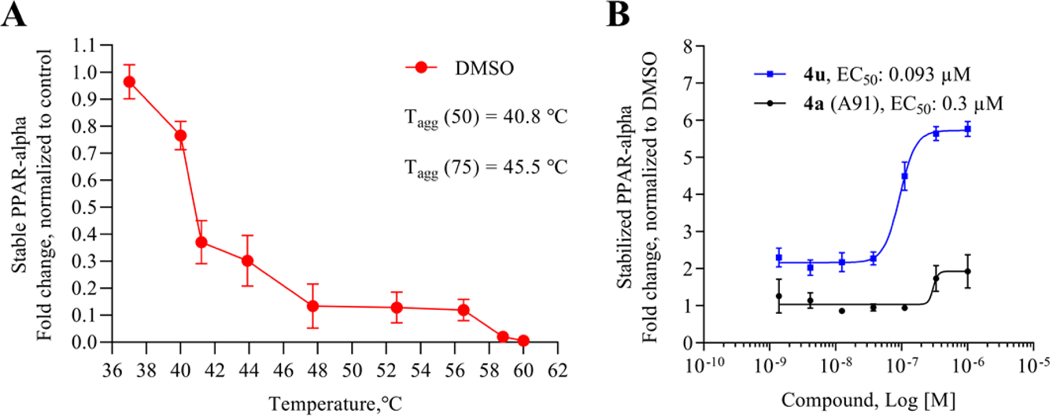
Cellular thermal shift assay results for A91 and 4u. (A) Thermal melting profile of PPARα. (B) Dose-dependent stabilization of PPARα by A91 and 4u. Experiment was run at 45.5 °C, the Tagg(75).
Compound 4u was assessed in the same PK studies as 4a (A91). As seen in Figure 8, this analogue exhibits significantly improved stability in human and rat liver microsomes (Figure 8A), shows no signs of irreversible inhibition of the major drug metabolizing CYP450s (Figure 8B) and is not considered a hERG liability (Figure 8C). It is important to note that while this compound exhibits 10% inhibition of hERG at 10 μM (the same as A91), this analogue is ~20-fold more potent than A91.
Figure 8. Pharmacokinetic profile of 4u.

(A) 4u was pre-incubated with pooled liver microsomes in phosphate buffer (pH 7.4) for 5 min at 37 °C. The system was activated by the addition of an NADPH-regenerating system and incubated for 0, 15, 30, 45, and 60 min before being quenched with an acetonitrile/methanol mixture. Samples were processed and analyzed by HPLC-MS/MS to determine the peak area remaining. Experiments completed in duplicate and mean values are depicted. (B) Assessment of 4u (10 μM) for irreversible inhibition of the major CYP450 drug metabolizing isoforms. Peak areas corresponding to the metabolite of known substrates for each isoform are recorded. The percent of control activity was calculated by comparing the peak area obtained in the presence of 4u to that obtained in the absence of 4u. Percent inhibition was calculated by subtracting the percent control activity from 100. Time-dependent CYP inhibition is demonstrated if the percent inhibition from samples pre-incubated with the NADPH-regenerating system is larger than those without NADPH-regeneration. Negative values are a reflection of the variability in the background noise of the experiment. Experiments completed in duplicate and mean values are depicted. (C) Assessment of 4u for inhibition of the hERG channel. The degree of inhibition (%) was obtained by measuring the tail current amplitude, which is induced by a one-second test pulse to −40 mV after a two-second pulse to +20 mV, before and after drug incubation. Experiments completed in duplicate and mean values are depicted.
Stage III SAR: Combining Lessons Learned.
Due to information gained from the stage I and II cohorts we designed and synthesized a series that combined beneficial features from each stage. Utilizing established synthetic routes, compounds 20-33 were synthesized and evaluated in similar fashion to stage I and II derivatives (Scheme 5). As shown in Table 7, all stage III analogues exhibit submicromolar activity in the cell luciferase assay, with many derivatives revealing EC50 values under 50 nM. Comparison of 24–29 to the corresponding des-methyl relatives demonstrates that the methyl effect is ~10–20-fold more potent for each derivative and thus exhibits exquisite consistency. Most notably, 22, which contains an iodine at the 4-position on the C-ring, exhibits a 50-fold improvement in activity in comparison to the des-methyl B-ring relative. Based on the predicted shift in the binding pocket (Figure 6) we suspect that room in the C-ring pocket is subsequently freed, allowing space for the larger iodine substituent that is not as easily accommodated with the des-methyl compound, 4e. Consistent with the effect observed between 4w and 4b, N-methylation of the linker results in ~2-fold decrease in potency (4u vs. 30). Furthermore, comparison of 4u and 31 reveals that incorporation of the 3-F on the A-ring results in a maintenance in potency, a phenomenon first observed between 4b and 4q. Overall, the results from stage III analogues confirm trends seen in stages I and II, corroborate in silico docking predictions, and provide compelling evidence that the SAR for this chemotype is consistent, a desirable yet often elusive property in lead optimization.
Scheme 5.

Synthesis of Stage III Analogues 20–33a
aReagents and conditions: (a) K2CO3, DMF; (b) i) 3-aminobenzoic acid or 4-amino benzoic acid, toluene, 130 °C, 2–3 h; ii) NaBH(OAc)3, acetic acid, THF, 12 h, 36–84%; (c) paraformaldehyde, NaBH(OAc)3, EtOH, reflux 2 h, 25 °C, 12 h, 44%; (d) LiOH·H2O, THF/CH3OH/H2O, 12 h, 32%.
Table 7.
In Vitro Activity of Stage III Analogues
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | X | R1 | R2 | R3 | R4 | Ratio (5/50)a | Fold-Signalb | EC50 hPPARα (μM)c |
| 20 | COOH | 4-Cl | 3-Me | H | H | 1.2 | 1.2 | 0.029±0.001 |
| 21 | COOH | 4-Br | 3-Me | H | H | 1.2 | 1.3 | 0.031±0.008 |
| 22 | COOH | 4-I | 3-Me | H | H | 1.8 | 1.5 | 0.027±0.003 |
| 23 | COOH | 2,4-difluoro | 3-Me | H | H | 1.2 | 1.4 | 0.056±0.007 |
| 24 | COOH | 3,4-difluoro | 3-Me | H | H | 1.4 | 1.6 | 0.018±0.001 |
| 25 | COOH | 3,5-difluoro | 3-Me | H | H | 1.1 | 1.6 | |
| 26 | COOH | 4-F | 3-F | H | H | 0.7 | 1.7 | |
| 27 | COOH | 4-F | 3-Cl | H | H | 1.1 | 1.5 | |
| 28 | COOH | 4-OPFB | 3-OPFB | H | H | 1.0 | 1.4 | 0.81±0.17 |
| 29 | COOH | 4-OPFB | 2-OPFB | H | H | 0.9 | 1.2 | 0.93±0.02 |
| 30 | COOH | 4-F | 3-Me | H | Me | 0.8 | 2.1 | 0.067±0.01 |
| 31 | COOH | 4-F | 3-Me | F | H | 0.9 | 1.5 | 0.040±0.001 |
| 32 | COOH | 4-F | 3-Cl | F | H | 1.1 | 1.8 | 0.052±0.011 |
| 33 | OC(CH3)2COOH | 4-F | 3-Me | H | H | 1.0 | 1.6 | 0.74±0.04 |
| 4b | 0.8 | 1.3 | 0.77±0.03 | |||||
| 4a (A91) | 0.5 | 1.4 | 4.43±0.01 | |||||
| GW590735 | 1.0 | 1.0 | 0.015±0.002 | |||||
Ratio of relative light unit (RLU) signal at 5 μM and 50 μM compound concentrations.
Ratio of signal maximal signal (RLU) strength observed for the compound of interest to that obtained with GW590735.
EC50 values represent the mean ± SEM of atleast two separate experiments performed in triplicate. OPFB = para-fluorobenzyl. Numbering shown is based on the name of the resulting products. Blank cells indicate compound was not selected for testing in the corresponding assay.
Thermodynamic Binding Profiles.
To determine thermodynamic parameters relating to the formation of PPARα/ligand complexes, we subjected structurally related 4a, 4b, and 4u along with GW590735 (as a known agonist) to isothermal titration calorimetry (ITC). These analogues were selected, as they only differ in single functionalities that produce significant changes in cellular activity and thus allowed for the correlation of binding thermodynamics to specific molecular features. Replacement of the C-ring 4-OMe (4a) with a 4-F (4b) produces a ~3-fold improvement in binding affinity, an observation consistent with cellular activity. Based on the thermodynamic profile (Figure 9, Table 5), the observed improvement in binding affinity is likely due to a 4.73 kcal/mol lower entropic penalty of binding for 4b. Introduction of a methyl group on the B-ring (4u) dramatically improves (~38-fold) the binding affinity, also consistent with cellular results. As depicted in Figure 9, the incorporation of the methyl group at the 3-position of the B-ring produces a favorable –TΔS term while also maintaining a favorable ethalpic contribution. The full thermodynamic binding profile of 4u reveals enthalpically driven binding to PPARα, helping to explain the significant jump in affinity.
Figure 9.
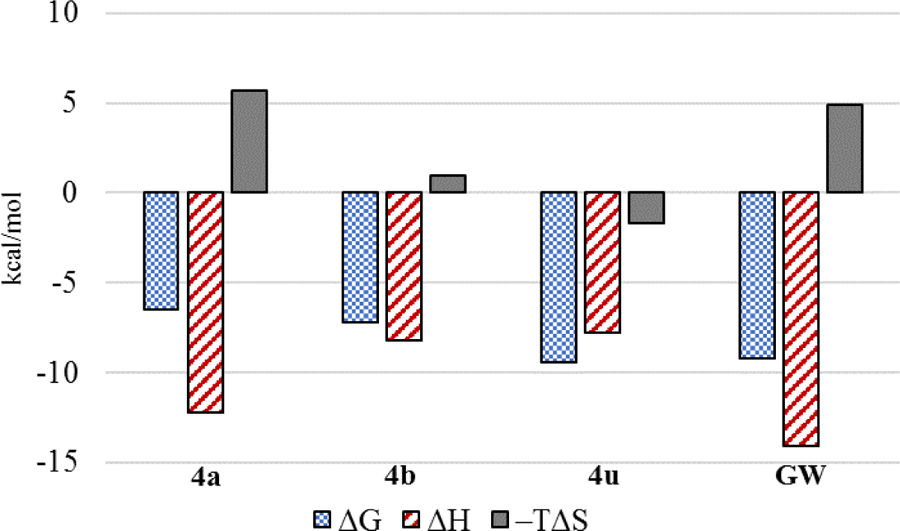
Thermodynamic binding profiles of select ligands.
Combined, these results suggest that the observed improvements in the cellular luciferase assay are likely due to improved binding profiles and not alterations in cellular permeability, solubility, or other physicochemical properties that may affect activity. Likewise, the proximities of Kd and EC50 values provides compelling evidence that this chemotype exhibits high specificity for PPARα. Attempts to obtain a Kd and thermodynamic binding profile for the fibrate headgroup containing member of this series (5b) failed to produce reproducable data due to solubility issues under assay conditions.
2.9. Conclusion
In conclusion, we have advanced our initial studies on the 4-benzyloxy-benzylamino chemotype as a novel PPARα agonistic scaffold by providing proof-of-concept in vivo efficacy, preliminary pharmacokinetic assessment and the design, synthesis, and evaluation of second-generation analogues that provide highly potent and isoform selective leads. The data demonstrate that systemic A91 (4a) treatment (i.p. injection) reduces retinal vascular leakage in diabetic rats to the level of non-diabetic controls at relatively the same dose as FenoFA, without signs of hepatomegaly. A full PK profile of this chemotype has yet to be determined (due to resource limitations), limiting PK/PD correlations for the in vivo data in comparison to FenoFA. However, embedded in these results is confirmation that A91 1) exhibits in vivo efficacy in a relevant DR model following systemic administration, 2) crosses blood-retinal barrier and reaches the retina, 3) is bioavailable, 4) survives first-pass metabolism and clearance mechanisms well enough to reach the site of disease and maintain efficacy, and 5) demonstrates a relatively safe profile (no observable toxicity) after daily intraperitoneal injection for one-month.
Utilizing A91 as a template we leveraged structure-guided design to develop second generation derivatives focused on improving the potency, affinity, and selectivity for PPARα (Figure 10). These efforts led to the discovery of 4u and related compounds that reach cellular potencies <50 nM and exhibit >2,700-fold selectivity for PPARα over other PPAR isoforms. Importantly, we discovered that incorporation of a methyl at the 3-position of the B-ring leads to dramatic improvements in target engagement, selectivity, and cellular activity. Cellular thermal shift analysis confirms tight binding of the top candidates to hPPARα and isothermal titration calorimetry further confirms a much-improved binding affinity and thermodynamic profile for 4u in comparison to A91 (4a). Initial pharmacokinetic assessment of 4u reveals microsomal stability in both human and rat microsomes, no evidence of irreversible inhibition of major drug metabolizing CYP450 enzymes, and a low risk for hERG interactions at relevant concentrations.
Figure 10.

Summary of current SAR on the 4-benxyloxy-benzylamino chemotype for PPARα agonism.
Detailed determination of a full PK/PD profile of this chemotype and advanced in vivo studies are ongoing and will be communicated in due time. Priority studies moving forward will focus on determining drug exposure in the retina after systemic administration and determining a more comprehensive metabolic profile, including phase-II conjugations. The data reported within, however, emphasize the promise of the 4-benzyloxy-benzylamino chemotype as a PPARα agonistic family with efficacy in the retina after systemic administration and identify a pipeline of candidates positioned for detailed PK/PD and pre-clinical evaluation.
EXPERIMENTAL SECTION
General Synthetic Information.
Starting materials, ACS grade DCM, methanol, ethyl acetate, toluene, anhydrous DMF, THF were purchased from TCI Chemicals, Oakwood, Alfa Aesar, Boron Molecular, Fisher Scientific or Sigma-Aldrich. All reactions requiring anhydrous conditions were run under a nitrogen atmosphere. NMR were collected on a 400, 500, and 600 MHz (specified below) Varian VNMRS Direct Drive spectrometer equipped with an indirect detection probe. NMR data were collected at 25 °C unless otherwise indicated. Pulse sequences were used as supplied by Varian VNMRJ 4.2 software. All NMR data was processed in MestReNova v11.0. High resolution mass spectrometry was obtained from and analyzed by the Mass Spectrometry Facility at the University of Oklahoma. All analogues evaluated in biological assays were >95% purity based on HPLC and NMR analyses.
Purity Analysis of Final Analogues.
Ultra-high-pressure liquid chromatography (UPLC) separation of samples (25 uM) was performed using an Agilent 1290 UPLC system equipped with an Agilent Zorbax Eclipse Plus reverse-phase column (ODS-18; 50 mm x 2.1 mm, 1.8− μm particle size). Mobile phase A consisted of 100% Optima® LC/MS submicron filtered water (Fisher Scientific). Mobile phase B consisted of 100% Optima® LC/MS acetonitrile. A binary gradient at 0.65 mL/min flow rate was applied as follows: 100% solvent A from 0 to 1 min linear gradient to 20% solvent B, linear gradient to 80% solvent B from 1 to 8 min, linear gradient to 100% solvent B from 8 to 9.1 min, 100% solvent B from 9.1 to 10.3 min, and linear gradient to return the mobile phase to 100% solvent A from 10.3 to 10.4 min, which was maintained for an additional 3 min before the next sample was injected. The UPLC column eluent was introduced into an Agilent 1290 Infinity Diode Array Detector. Absorbance was scanned from 190 to 400 nm. Absorbance intensity at 154 nm was quantitated by area integration. For accurate mass confirmation the eluent traveled in series from the DAD to an Agilent 6545 UHD Accurate Mass QTOF device equipped with an electrospray ionization source operated in negative ion mode. Nitrogen was used as a nebulizing gas (40 pounds/inch2) and a drying gas (325 °C and 10 L/min flow rate). Fragmentor, skimmer, and capillary voltages were 180, 45, and 2500 V, respectively. Data was collected with Mass Hunter Acquisition (B.08.00) and analyzed with Mass Hunter Qualitative (B.07.00).
General Procedure A (Reductive amination).
Requisite aniline (1.0 eq.) was dissolved in toluene (30 mL), and the requisite aldehyde (1.02 eq.) was added. The reaction mixture was heated for 2 h under reflux using a Dean–Stark trap. Subsequently, the solvent was evaporated under reduced pressure. The crude product was dissolved in THF (0.1 M) and cooled to 0 °C prior to the addition of neat sodium triacetoxyborohydride (2.0 eq.). The resulting reaction mixture was stirred for 15 min, after which acetic acid (0.01 eq.) was added to the reaction. The mixture was then allowed to stir for 12 h at 25 °C while monitoring by TLC. Upon complete consumption of the aniline, the reaction was quenched through slow addition of water. The mixture was extracted three times with EtOAc, the organic layers were combined and washed with brine. The organic layer was dried over anhydrous sodium sulfate, filtered, and evaporated under vacuum. The product was purified by column chromatography (SiO2) using dichloromethane/methanol (99.5:0.5) as the eluent.
General Procedure B (Ester saponification).
To a stirred solution of the requisite ethyl ester (1.0 eq.) in THF/MeOH/H2O mixture (0.1 M, 3:1:1) was added lithium hydroxide monohydrate (5.0 eq.) and the reaction mixture was stirred for 12 h at 25 °C. The reaction mixture was concentrated to remove organic solvents. The aqueous layer was acidified to pH 2.0 with 1 N aq. HCl and then extracted with EtOAc. The organic layer was washed with water and brine. The resulting organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The product was purified by column chromatography (SiO2) using dichloromethane/methanol (99:1) as the eluent.
3-((4-((4-methoxybenzyl)oxy)benzyl)amino)benzoic acid (4a):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 4a was prepared following General Procedure A with aldehyde 3a (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (210 mg, 80% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.46 (d, J = 7.7 Hz, 1H), 7.36 (d, J = 9.2 Hz, 3H), 7.27 (q, J = 6.3, 4.1 Hz, 4H), 6.94 (dd, J = 14.6, 8.1 Hz, 4H), 6.84 (d, J = 8.3 Hz, 1H), 4.99 (s, 2H), 4.30 (s, 2H), 3.82 (s, 3H) ppm; 13C NMR (101 MHz, Chloroform-d) δ 171.8, 159.6, 158.4, 148.3, 131.1, 130.2, 129.4, 129.3 (2C), 129.1, 128.9 (2C), 119.4, 118.2, 115.2 (2C), 114.2 (2C), 114.0, 70.0, 55.4, 47.8 ppm. HRESI m/z: 364.1449 (C22H21NO4 + H+ requires 364.1543).
3-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4b):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4b was prepared following General Procedure A with aldehyde 3b 0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (40 mg, 32% yield). 1H NMR (600 MHz, DMSO-d6) δ 7.48 (dd, J = 8.3, 5.5 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 7.21 (dd, J = 9.3, 8.9 Hz, 2H), 7.16 (dd, J = 1.9 Hz, 1H), 7.13 (dd, J = 7.7 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 8.7 Hz, 2H), 6.82 – 6.60 (m, 1H), 6.44 (t, J = 6.0 Hz, 1H), 5.04 (s, 2H), 4.21 (d, J = 5.8 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 161.7 (d, J = 243.5 Hz), 157.1, 148.7, 133.4 (d, J = 3.1 Hz), 131.9, 131.4, 129.8 (d, J = 8.4 Hz, 2C), 128.8, 128.3 (2C), 116.6, 116.3, 115.2 (d, J = 21.3 Hz, 2C), 114.6 (2C), 112.9, 68.4, 45.7. TOFMSESI m/z: 352.1357 (C21H18FNO3 + H+ requires 352.1304).
4-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4b*): Reaction scale: 50 mg (0.36 mmol) of 4-amino benzoic acid. Compound 4b* was prepared following General Procedure A with aldehyde 3b (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (60 mg, 52% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.75 (d, J = 8.8 Hz, 2H), 7.45 (dd, J = 8.5, 5.5 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 7.13 – 7.04 (m, 2H), 6.95 (d, J = 8.6 Hz, 2H), 6.59 (d, J = 8.8 Hz, 2H), 5.04 (s, 2H), 4.30 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 170.8, 168.3 (d, J = 244.5 Hz), 168.1, 166.5, 162.6, 159.3, 154.4, 133.0 (d, J = 3.2 Hz), 132.6 (2C), 130.7, 130.6 (2C), 129.6 (d, J = 8.6 Hz, 2C), 118.5, 116.3 (d, J = 21.6 Hz, 2C), 116.0 (2C), 112.5 (2C), 70.3, 47.3. TOFMSESI m/z: 352.1357 (C21H18FNO3 + H+ requires 352.1304).
3-((4-((4-chlorobenzyl)oxy)benzyl)amino)benzoic acid (4c):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 4c was prepared following General Procedure A with aldehyde 3c (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (203.3 mg, 76% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.41 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 6.3 Hz, 2H), 7.28 (s, 1H), 7.25 (dt, J = 7.6, 1.3 Hz, 1H), 7.15 (t, J = 7.8 Hz, 2H), 6.93 (d, J = 8.6 Hz, 1H), 6.82 (ddd, J = 8.0, 2.5, 1.1 Hz, 1H), 5.03 (s, 2H), 4.26 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 169.3, 157.6, 148.8, 136.3, 133.1, 132.0, 131.0, 128.7 (2C), 128.5, 128.2 (2C), 128.1 (2C), 117.6, 117.0, 114.5 (2C), 113.2, 68.7, 46.5. TOFMSESI m/z: 366.0897 (C21H18ClNO3 - H+ requires 366.0902).
3-((4-((4-bromobenzyl)oxy)benzyl)amino)benzoic acid (4d):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4d was prepared following General Procedure A with aldehyde 3d (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (96 mg, 65% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.68 (s, 1H), 7.57 (d, J = 8.5 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 7.18 (dd, J = 2.0 Hz, 1H), 7.13 (dd, J = 7.6 Hz, 1H), 7.10 (ddd, J = 7.5, 1.5 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.78 (ddd, J = 8.0, 1.8 Hz, 1H), 6.44 (s, 1H), 5.05 (s, 2H), 4.21 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.3, 157.4, 149.1, 137.1, 132.5, 131.9, 131.8 (2C), 130.2 (2C), 129.3, 128.8 (2C), 121.3, 117.1, 116.7, 115.1 (2C), 113.4, 68.8, 46.2. TOFMSESI m/z: 412.0512 (C21H18BrNO3 + H+ requires 412.0543).
3-((4-((4-iodobenzyl)oxy)benzyl)amino)benzoic acid (4e):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 4e was prepared following General Procedure A with aldehyde 3e (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (210 mg, 80% yield).1H NMR (600 MHz, DMSO-d6) δ 7.74 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.17 (dd, J = 1.9 Hz, 1H), 7.14 (dd, J = 7.7 Hz, 1H), 7.13 – 7.07 (m, 1H), 6.94 (d, J = 8.6 Hz, 2H), 6.78 (ddd, J = 8.3, 1.7 Hz, 1H), 5.03 (s, 2H), 4.20 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.7, 157.0, 148.6, 137.2 (2C), 137.0, 132.0, 131.2, 129.8 (2C), 128.8, 128.3 (2C), 116.7, 116.4, 114.6 (2C), 112.9, 93.7, 68.4, 45.7. TOFMSESI m/z: 460.0424 (C21H18INO3 + H+ requires 460.0404).
3-((4-((4-(trifluoromethyl)benzyl)oxy)benzyl)amino)benzoic acid (4f):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 4f was prepared following General Procedure A with aldehyde 3f (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (170 mg, 58% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.60 (s, 1H), 7.75 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 8.7 Hz, 2H), 7.16 (dd, J = 2.6, 1.9 Hz, 1H), 7.13 (dd, J = 8.5, 7.7 Hz, 1H), 7.09 (ddd, J = 7.6, 1.6 Hz, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.78 (ddd, J = 8.1, 2.6, 1.2 Hz, 1H), 6.45 (t, J = 6.0 Hz, 1H), 5.20 (s, 2H), 4.21 (d, J = 5.8 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 156.9, 148.7, 142.1, 132.1, 131.3, 128.8, 128.4 (2C), 128.2 (q, J = 29.2, 30.8, 33.0 Hz), 127.9 (2C), 125.3 (q, J = 3.6 Hz, 2C), 124.2 (q, J = 271.2, 271.8, 272.3 Hz), 116.6, 116.3, 114.6 (2C), 112.8, 68.2, 45.7. TOFMSESI m/z: 400.1139 (C22H18F3NO3 - H+ requires 400.1166).
3-((4-((4-cyanobenzyl)oxy)benzyl)amino)benzoic acid (4g):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 4g was prepared following General Procedure A with aldehyde 3g (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (110 mg, 42% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.63 (s, 1H), 7.84 (d, J = 8.2 Hz, 2H), 7.62 (d, J = 7.9 Hz, 2H), 7.28 (d, J = 8.3 Hz, 2H), 7.19 (dd, J = 2.0 Hz, 1H), 7.14 (dd, J = 7.7 Hz, 1H), 7.12 (d, J = 8.7 Hz, 1H), 7.00 – 6.90 (m, 2H), 6.79 (dt, J = 7.8, 2.0 Hz, 1H), 6.45 (s, 1H), 5.18 (s, 2H), 4.22 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.9, 156.8, 148.7, 143.0, 132.4 (2C), 132.2, 131.3, 128.9, 128.4 (2C), 128.0 (2C), 118.8, 116.7, 116.4, 114.7 (2C), 112.9, 110.4, 68.2, 45.8. TOFMSESI m/z: 359.1406 (C22H18N2O3 + H+ requires 359.1390).
3-((4-((2-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4h):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4h was prepared following General Procedure A with aldehyde 3h (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (122 mg, 97% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.60 (s, 1H), 7.54 (t, J = 7.4 Hz, 1H), 7.41 (q, J = 6.4 Hz, 1H), 7.29 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 10.1 Hz, 1H), 7.22 (d, J = 7.4 Hz, 1H), 7.18 (s, 1H), 7.14 (t, J = 7.7 Hz, 1H), 7.10 (d, J = 7.6 Hz, 1H), 6.98 (d, J = 8.5 Hz, 2H), 6.79 (d, J = 7.8 Hz, 1H), 6.44 (s, 1H), 5.10 (s, 2H), 4.22 (d, J = 4.4 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 160.4 (d, J = 245.9 Hz), 157.1, 148.7, 132.1, 131.3, 130.6 (d, J = 4.1 Hz), 130.3 (d, J = 8.3 Hz), 128.9, 128.4 (2C), 124.5 (d, J = 3.5 Hz), 123.9 (d, J = 14.7 Hz), 116.7, 116.3, 115.4 (d, J = 21.1 Hz), 114.6 (2C), 112.9, 63.5 (d, J = 3.4 Hz), 45.8. TOFMSESI m/z: 374.1169 (C21H18FNO3 + Na+ requires 374.1163).
3-((4-((3-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4i):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4i was prepared following General Procedure A with aldehyde 3i (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (96 mg, 65% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 7.46 – 7.38 (m, 1H), 7.29 – 7.28 (m, 2H), 7.28 – 7.27 (m, 1H), 7.26 – 7.24 (m, 1H), 7.20 – 7.17 (m, 1H), 7.16 – 7.14 (m, 1H), 7.14 – 7.12 (m, 1H), 7.13 – 7.08 (m, 1H), 6.99 – 6.95 (m, 2H), 6.81 – 6.75 (m, 1H), 6.44 (t, J = 5.8 Hz, 1H), 5.10 (s, 2H), 4.21 (d, J = 4.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.8, 162.2 (d, J = 243.5 Hz), 157.0, 148.7, 140.2 (d, J = 7.3 Hz), 132.0, 131.3, 130.4 (d, J = 8.3 Hz), 128.9, 128.4 (2C), 123.4 (d, J = 2.8 Hz), 116.7, 116.4, 114.7 (2C), 114.5 (d, J = 20.9 Hz), 114.1 (d, J = 21.9 Hz), 112.9, 68.3, 45.8. TOFMSESI m/z: 352.1360 (C21H18FNO3 + H+ requires 352.1343).
3-((4-((2,4-difluorobenzyl)oxy)benzyl)amino)benzoic acid (4j):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4j was prepared following General Procedure A with aldehyde 3j (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (100 mg, 70% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.62 (s, 1H), 7.60 (ddd, J = 8.6, 6.7 Hz, 1H), 7.32 – 7.27 (m, 1H), 7.29 – 7.26 (m, 2H), 7.17 (dd, J = 1.9 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 7.13 – 7.11 (m, 1H), 7.10 – 7.09 (m, 1H), 6.98 (d, J = 8.6 Hz, 2H), 6.83 – 6.73 (m, 1H), 6.46 (t, J = 6.3 Hz, 1H), 5.07 (s, 2H), 4.22 (d, J = 4.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 161.4 (dd, J = 4.0, 12.6 Hz), 161.4 (dd, J = 12.1, 499.9 Hz), 157.0, 148.7, 132.2, 132.1 (dd, J = 5.7, 10.1 Hz), 131.3, 128.8, 128.4 (2C), 120.4 (dd, J = 3.4, 14.4 Hz), 116.7, 116.3, 114.6 (2C), 112.9, 111.5 (dd, J = 3.8, 21.3 Hz), 104.0 (t, J = 25.8 Hz), 63.1 (d, J = 2.9 Hz), 45.7. TOFMSESI m/z: 392.1085 (C21H17F2NO3 + Na+ requires 392.1069).
3-((4-((3,4-difluorobenzyl)oxy)benzyl)amino)benzoic acid (4k):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4k was prepared following General Procedure A with aldehyde 3k (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (106 mg, 79% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 7.51 (ddd, J = 11.6, 7.8, 2.0 Hz, 1H), 7.44 (ddd, J = 10.8, 8.4 Hz, 1H), 7.30 (d, J = 4.0 Hz, 1H), 7.27 (d, J = 8.4 Hz, 2H), 7.16 (dd, J = 1.9 Hz, 1H), 7.13 (dd, J = 7.7 Hz, 1H), 7.09 (ddd, J = 7.6, 1.3 Hz, 1H), 6.96 (d, J = 8.7 Hz, 2H), 6.77 (ddd, J = 8.0, 2.6, 1.2 Hz, 1H), 6.45 (t, J = 6.1 Hz, 1H), 5.06 (s, 2H), 4.21 (d, J = 5.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 156.9, 149.3 (dd, J = 12.7, 245.6 Hz), 148.9 (dd, J = 12.4, 245.3 Hz), 148.7, 135.0 (dd, J = 3.6, 5.8 Hz), 132.1, 131.3, 128.8, 128.3 (2C), 124.4 (dd, J = 3.5, 6.5 Hz), 117.5 (d, J = 17.1 Hz), 116.7 (d, J = 17.5 Hz), 116.6, 116.3, 114.6 (2C), 112.9, 67.8, 45.7. TOFMSESI m/z: 370.1238 (C21H17F2NO3 + H+ requires 370.1249).
3-((4-((3,5-difluorobenzyl)oxy)benzyl)amino)benzoic acid (4l):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 4l was prepared following General Procedure A with aldehyde 3l (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (196 mg, 73% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 7.28 (d, J = 8.3 Hz, 2H), 7.19 (dd, J = 2.4, 9.1 Hz, 1H), 7.17 (dd, J = 1.8 Hz, 2H), 7.15 (d, J = 3.7 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 7.10 (ddd, J = 1.4, 7.6 Hz, 1H), 6.97 (d, J = 8.6 Hz, 2H), 6.78 (ddd, J = 1.2, 2.5, 8.0 Hz, 1H), 6.45 (t, J = 5.8 Hz, 1H), 5.11 (s, 2H), 4.22 (d, J = 5.1 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 162.4 (dd, J = 13.0, 246.5 Hz, 2C), 156.7, 148.7, 141.9 (t, J = 9.2 Hz), 132.2, 131.3, 128.8, 128.4 (2C), 116.7, 116.3, 114.7 (2C), 112.9, 110.3 (dd, J = 5.2, 20.3 Hz, 2C), 103.0 (t, J = 25.8 Hz), 67.8, 45.7. TOFMSESI m/z: 368.1120 (C21H17F2NO3 - H+ requires 368.1104).
3-((4-phenethoxybenzyl)amino)benzoic acid (4m):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4m was prepared following General Procedure A with aldehyde 8a (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (121 mg, 96% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.39 (s, 1H), 7.28 (t, J = 2.0 Hz, 1H), 7.27 (s, 2H), 7.26 (d, J = 3.5 Hz, 2H), 7.24 (s, 2H), 7.19 (h, J = 4.1 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 8.6 Hz, 2H), 6.81 (dd, J = 1.9, 8.4 Hz, 1H), 4.23 (s, 2H), 4.13 (t, J = 6.9 Hz, 2H), 3.02 (t, J = 6.9 Hz, 2H). 13C NMR (151 MHz, Methanol-d4) δ 169.3, 157.9, 148.8, 138.5, 131.6, 131.1, 128.6 (2C), 128.5, 128.2 (2C), 128.0 (2C), 125.9 (2C), 117.6, 117.0, 114.2, 113.3, 68.5, 46.7, 35.3. TOFMSESI m/z: 346.1443 (C22H21NO3 - H+ requires 346.1449).
3-((4-(4-methoxyphenethoxy)benzyl)amino)benzoic acid (4n):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4n was prepared following General Procedure A with aldehyde 8b (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (130 mg, 96% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.28 (d, J = 2.2 Hz, 1H), 7.25 (d, J = 8.1 Hz, 2H), 7.25 (s, 1H), 7.18 (d, J = 8.2 Hz, 2H), 7.14 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 6.83 – 6.78 (m, 1H), 4.24 (s, 2H), 4.09 (t, J = 6.9 Hz, 2H), 3.75 (s, 3H), 2.96 (t, J = 6.9 Hz, 2H). 13C NMR (151 MHz, Methanol-d4) δ 170.7, 159.8, 159.4, 150.3, 133.0, 132.5, 131.8, 130.9 (2C), 129.9, 129.6 (2C), 119.0, 118.4, 115.7 (2C), 114.9 (2C), 114.8, 70.1, 55.7, 48.1, 35.9. TOFMSESI m/z: 378.1709 (C23H23NO4 + H+ requires 378.1700).
3-((4-(4-methoxyphenethoxy)phenethyl)amino)benzoic acid (4o):
3-amino benzoic acid (50 mg, 0.36 mmol) and compound 10 (113 mg, 0.37 mmol) were dissolved in DMF. Triethylamine (102 uL, 0.72 mmol) was added at 25 °C and the reaction was stirred for 12 h upon which the reaction was deemed complete by TLC. The reaction was quenched with aq. 1N HCl (3 mL). The solution was diluted with EtOAc (30 mL). The organic layer was washed with water (3 × 20 mL) and brine (3 × 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under vacuum. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (36 mg, 25% yield). 1H NMR (500 MHz, Methanol-d4) δ 7.36 – 7.29 (m, 1H), 7.27 (ddd, J = 7.7, 1.6, 1.1 Hz, 1H), 7.19 (d, J = 6.6 Hz, 2H), 7.17 (d, J = 6.7 Hz, 2H), 7.14 (d, J = 7.7 Hz, 1H), 6.90 (ddd, J = 8.0, 2.4, 1.1 Hz, 1H), 6.84 (t, J = 2.0 Hz, 2H), 6.83 (t, J = 2.1 Hz, 2H), 4.41 (t, J = 6.9 Hz, 2H), 4.09 (t, J = 6.9 Hz, 2H), 3.75 (s, 3H), 2.97 (t, J = 7.0 Hz, 2H), 2.96 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, Methanol-d4) δ 168.5, 159.8, 159.1, 149.3, 132.2, 131.8, 131.4, 131.0 (2C), 130.9 (2C), 130.1, 120.8, 119.8, 116.7, 115.7 (2C), 114.8 (2C), 70.1, 66.8, 55.6, 35.9, 35.3. TOFMSESI m/z: 392.1870 (C24H25NO4 + H+ requires 392.1856).
2-fluoro-5-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4p):
Reaction scale: 50 mg (0.32 mmol) of 5-amino-2-fluorobenzoic acid (11a). Compound 4p was prepared following the General Procedure A with aldehyde 3b (0.33 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (98 mg, 82% yield). 1H NMR (600 MHz, DMSO-d6) δ 7.48 (dd, J = 8.5, 5.7 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.21 (t, J = 8.9 Hz, 1H), 7.01 – 6.97 (m, 1H), 7.00 – 6.93 (m, 3H), 6.75 (dt, J = 8.8, 3.5 Hz, 1H), 6.31 (s, 1H), 5.05 (s, 2H), 4.17 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 165.6, 161.7 (d, J = 243.5 Hz), 157.1, 152.9 (d, J = 244.7 Hz), 144.9, 133.4, 131.8, 129.9 (d, J = 8.2 Hz, 2C), 128.4 (2C), 117.3 (d, J = 7.4 Hz), 116.9 (d, J = 23.3 Hz, 2C), 115.3, 115.1, 114.7 (2C), 113.7, 68.4, 46.2. TOFMSESI m/z: 368.1093 (C21H17F2NO3 - H+ requires 368.1104).
3-fluoro-5-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4q):
Reaction scale: 50 mg (0.32 mmol) of 3-amino-5-fluorobenzoic acid (11b). Compound 4q was prepared following the General Procedure A with aldehyde 3b (0.33 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (102 mg, 86% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.45 – 7.37 (m, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.11 (t, J = 1.8 Hz, 1H), 7.10 – 7.00 (m, 2H), 6.96 – 6.89 (m, 2H), 6.87 (dt, J = 9.1, 1.9 Hz, 1H), 6.47 (dt, J = 11.4, 2.3 Hz, 1H), 4.98 (s, 2H), 4.22 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 169.4 (d, J = 3.3 Hz), 165.1 (d, J = 241.1 Hz), 163.8 (d, J = 244.7 Hz), 159.2, 152.0 (d, J = 11.1 Hz), 134.8 (d, J = 3.2 Hz), 134.1 (d, J = 9.9 Hz), 132.8, 130.6 (d, J = 8.1 Hz, 2C), 129.6 (2C), 116.2, 116.0 (2C), 111.1 (d, J = 2.0 Hz), 104.5 (d, J = 24.0 Hz, 2C), 103.9 (d, J = 26.0 Hz), 70.3, 47.8. TOFMSESI m/z: 368.1096 (C21H17F2NO3 - H+ requires 368.1104).
3-chloro-5-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4r):
Reaction scale: 50 mg (0.29 mmol) of 3-amino-5-chlorobenzoic acid (11c). Compound 4r was prepared following the General Procedure A with aldehyde 3b (0.30 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (88 mg, 78% yield). 1H NMR (500 MHz, Methanol-d4) δ 7.42 (dd, J = 8.4, 5.4 Hz, 2H), 7.26 (d, J = 8.3 Hz, 2H), 7.17 (dt, J = 16.7, 1.7 Hz, 2H), 7.07 (t, J = 8.8 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 6.75 (t, J = 2.1 Hz, 1H), 5.00 (s, 2H), 4.23 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.6, 161.7 (d, J = 243.5 Hz), 157.2, 150.1, 133.7, 133.3 (d, J = 3.0 Hz), 133.0, 131.3, 129.9, 129.8, 128.4 (2C), 115.5, 115.3, 115.1, 114.8, 114.7 (2C), 112.0, 68.5, 45.6. TOFMSESI m/z: 384.0812 (C21H17ClFNO3 - H+ requires 384.0808).
3-bromo-5-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (4s):
Reaction scale: 50 mg (0.23 mmol) of 3-amino-5-bromobenzoic acid (11d). Compound 4s was prepared following the General Procedure A with aldehyde 3b (0.23 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a pale amorphous solid (85 mg, 86% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.42 (dd, J = 8.5, 5.4 Hz, 2H), 7.29 (d, J = 1.7 Hz, 1H), 7.26 (d, J = 8.3 Hz, 2H), 7.21 (t, J = 1.8 Hz, 1H), 7.07 (t, J = 8.8 Hz, 2H), 6.93 (d, J = 8.4 Hz, 2H), 6.91 (t, J = 2.1 Hz, 1H), 5.00 (s, 2H), 4.23 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 169.1, 165.0, 161.9 (d, J = 146.3 Hz), 159.3, 151.5, 134.8, 134.2, 132.7, 130.6 (d, J = 8.2 Hz), 129.6 (2C), 123.7, 120.9, 119.9, 116.2, 116.04, 116.01 (2C), 113.6, 70.29, 47.67. TOFMSESI m/z: 428.0295 (C21H17BrFNO3 - H+ requires 428.0303).
3-((4-((4-fluorobenzyl)oxy)-2-methylbenzyl)amino)benzoic acid (4t):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 4t was prepared following General Procedure A with aldehyde 12a (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (100 mg, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.53 (s, 1H), 7.48 (dd, J = 8.5, 5.7 Hz, 2H), 7.25 – 7.16 (m, 2H), 7.18 (s, 1H), 7.17 (d, J = 4.0 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.11 (dt, J = 7.6, 1.5 Hz, 1H), 6.86 (d, J = 2.6 Hz, 1H), 6.79 (td, J = 8.6, 2.3 Hz, 2H), 6.31 – 5.95 (m, 1H), 5.05 (s, 2H), 4.15 (d, J = 4.5 Hz, 2H), 2.30 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.7, 157.1, 148.8, 148.1 (d, J = 330.2 Hz), 137.3, 133.4 (d, J = 2.9 Hz), 131.3, 129.7, 129.6, 129.4, 128.7 (d, J = 7.3 Hz, 2C), 116.7, 116.5, 116.1, 115.1 (d, J = 21.4 Hz, 2C), 112.5, 111.6, 68.3, 44.2, 18.7. TOFMSESI m/z: 364.1360 (C22H20FNO3 - H+ requires 364.1354).
3-((4-((4-fluorobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (4u):
Reaction scale: 100 mg (0.72 mmol) of 3-amino benzoic acid. Compound 4u was prepared following General Procedure A with aldehyde 12b (0.73 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (200 mg, 75% yield). 1H NMR (600 MHz, DMSO-d6) δ 7.49 (dd, J = 8.5, 5.7 Hz, 2H), 7.21 (dd, J = 8.9 Hz, 2H), 7.17 (dd, J = 2.0 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 7.14 (s, 1H), 7.11 (s, 1H), 7.10 (ddd, J = 7.5, 1.4 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 6.82 – 6.67 (m, 1H), 5.06 (s, 2H), 4.17 (s, 2H), 2.16 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.7, 161.6 (d, J = 243.3 Hz), 155.1, 148.7, 133.7 (d, J = 2.9 Hz), 131.5, 131.2, 129.5, 129.4 (d, J = 8.2 Hz, 2C), 128.8, 125.8, 125.6, 116.6, 116.3, 115.2 (d, J = 21.3 Hz, 2C), 112.8, 111.6, 68.5, 45.8, 16.2. TOFMSESI m/z: 388.1328 (C22H20FNO3 + Na+ requires 388.1319).
3-((4-((4-fluorobenzyl)oxy)benzyl)amino)-4-methylbenzoic acid (4v):
Reaction scale: 50 mg (0.33 mmol) of 3-amino-4-methylbenzoic acid. Compound 4v was prepared following the General Procedure A with aldehyde 3b (0.34 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (111 mg, 92% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.44 (t, J = 6.7 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 7.26 (s, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.08 (t, J = 8.6 Hz, 2H), 7.01 (d, J = 7.7 Hz, 1H), 6.95 – 6.91 (m, 2H), 5.02 (s, 2H), 4.36 (s, 2H), 2.18 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 161.7 (d, J = 243.6 Hz), 157.0, 145.9, 133.4 (d, J = 3.0 Hz), 132.2, 129.9, 129.8, 129.4, 128.0 (2C), 127.9, 126.2, 117.1, 115.3, 115.0, 114.6 (2C), 114.4, 110.0, 68.4, 45.7, 17.8. TOFMSESI m/z: 364.1342 (C22H20FNO3 - H+ requires 364.1354).
3-((4-((4-fluorobenzyl)oxy)benzyl)(methyl)amino)benzoic acid (4w):
Paraformaldehyde (5 mg, 0.16 mmol), compound 4b (30 mg, 0.08 mmol), sodium triacetoxyborohydride (6.2 mg, 0.096 mmol) and 2 mL of ethanol were combined in a 10 mL of round bottom flask with a reflux condenser. The mixture was heated for 2 h at 100 °C and subsequently cooled to 25 ° for 12 h. The solvent was removed under vacuum and the residue was dissolved in EtOAc (60 mL). The organic layer was washed with aq. 1N HCl (20 mL), water (20 mL), and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under vacuum. The compound was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (13 mg, 43% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.4 (d, J = 5.5 Hz, 1H), 7.4 – 7.4 (m, 2H), 7.3 (dt, J = 1.3, 7.6 Hz, 1H), 7.2 (t, J = 7.9 Hz, 1H), 7.1 (d, J = 8.6 Hz, 2H), 7.1 (t, J = 8.8 Hz, 2H), 7.0 (ddd, J = 1.1, 2.8, 8.3 Hz, 1H), 6.9 (d, J = 8.7 Hz, 2H), 5.0 (s, 2H), 4.5 (s, 2H), 3.0 (s, 3H). 13C NMR (101 MHz, Methanol-d4) δ 170.7, 163.8 (d, J = 244.4 Hz), 159.2, 151.1, 134.8 (d, J = 3.3 Hz), 132.4, 132.3, 130.6 (d, J = 8.3 Hz, 2C), 130.0, 129.2 (2C), 118.8, 118.2, 116.1 (d, J = 21.7 Hz, 2C), 116.0 (2C), 114.5, 70.3, 56.8, 38.9. TOFMSESI m/z: 388.1328 (C25H26FNO4 + H requires 388.1319). TOFMSESI m/z: 366.1509 (C22H20FNO3 + H+ requires 366.1500).
(R)-3-((4-(1-(4-fluorophenyl)ethoxy)benzyl)amino)benzoic acid (4x):
Compound 13 (53 mg, 0.15 mmol), (S)-1-(4-fluorophenyl)ethan-1-ol (21 mg, 0.15 mmol), and PPh3 (39 mg, 0.15 mmol) were dissolved in toluene (2.5 mL) and cooled to 0 °C. A solution of DtBAD (35 mg, 0.15 mmol) in toluene (5 mL) was added dropwise to the mixture. The reaction was warmed to 25 °C and stirred at for 12 h, at which time the reaction was deemed complete by TLC. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (SiO2, hexanes/ethyl acetate = 2: 1) to provide the desired product 14 as colorless oil. The resulting product 14 (50 mg, 0.114 mmol) was dissolved in a THF/MeOH/H2O (3:1:1) mixture and LiOH·added was (mmol 0.57, mg 24) H2O. The reaction stirred at 25 °C for 12 h upon which the reaction was deemed complete by TLC. The reaction was quenched with aq. 1N HCl (3 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were combined dried over Na2SO4, filtered and concentrated by vacuum. The product 4x was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (25 mg, 65% yield). [α]20D = +22.0 (c = 9.1 mg/mL, l = 0.5 dm, MeOH). 1H NMR (400 MHz, Methanol-d4) δ 7.35 (dd, J = 8.6, 5.4 Hz, 2H), 7.29 – 7.24 (m, 1H), 7.26 – 7.23 (m, 1H), 7.19 – 7.15 (m, 2H), 7.15 – 7.09 (m, 1H), 7.01 (t, J = 8.8 Hz, 2H), 6.80 (d, J = 6.6 Hz, 2H), 6.79 – 6.75 (m, 1H), 5.33 (q, J = 6.4 Hz, 1H), 4.17 (s, 2H), 1.54 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 170.7, 163.5 (d, J = 243.8 Hz), 158.2, 150.2, 140.7 (d, J = 3.1 Hz), 133.1, 132.4, 129.9, 129.5 (2C), 128.7 (d, J = 8.3 Hz, 2C), 119.0, 118.4, 117.1 (2C), 116.1 (d, J = 21.7 Hz, 2C), 114.7, 76.4, 48.0, 24.7. TOFMSESI m/z: 364.1363 (C22H20FNO3 - H+ requires 364.1354).
(S)-3-((4-(1-(4-fluorophenyl)ethoxy)benzyl)amino)benzoic acid (4y):
Compound 13 (60 mg, 0.17 mmol), (R)-1-(4-fluorophenyl)ethan-1-ol (24 mg, 0.17 mmol), and PPh3 (45 mg, 0.17 mmol) were dissolved in toluene (2.5 mL) and cooled to 0 °C. A solution of DtBAD (39 mg, 0.17 mmol) in toluene (5 mL) was added dropwise to the mixture. The reaction was warmed to 25 °C and stirred at for 12 h, at which time the reaction was deemed complete by TLC. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography (SiO2, hexanes/ethyl acetate = 2: 1) to provide the desired product 15 as colorless oil. The resulting product 15 (24 mg, 0.05mmol) (23 mg, 0.049 mmol) was dissolved in a THF/MeOH/H2O (3:1:1) mixture and LiOH·added was (mmol 0.25, mg 10) H2O. The reaction stirred at 25 °C for 12 h upon which the reaction was deemed complete by TLC. The reaction was quenched with aq. 1N HCl (3 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated by vacuum. The product 4y was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (14 mg, 75% yield). [α]20D = −22.0 (c = 9.1 mg/mL, l = 0.5 dm, MeOH). 1H NMR (600 MHz, Methanol-d4) δ 7.40 – 7.34 (m, 2H), 7.28 – 7.24 (m, 1H), 7.27 – 7.22 (m, 1H), 7.21 – 7.16 (m, 2H), 7.13 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 8.8 Hz, 2H), 6.82 – 6.80 (m, 2H), 6.79 (ddd, J = 8.1, 2.5, 1.0 Hz, 1H), 5.36 (q, J = 6.4 Hz, 1H), 4.19 (s, 2H), 1.55 (d, J = 6.4 Hz, 3H). 13C NMR (151 MHz, Methanol-d4) δ 170.8, 163.7 (d, J = 243.9 Hz), 158.3, 150.4, 140.9 (d, J = 2.9 Hz), 133.3, 132.5, 130.0, 129.7 (2C), 128.8 (d, J = 8.4 Hz, 2C), 119.2, 118.6, 117.3 (2C), 116.3 (d, J = 21.6 Hz, 2C), 114.8, 76.5, 48.2, 24.8. TOFMSESI m/z: 364.1365 (C22H20FNO3 - H+ requires 364.1354).
4-((2-(4-fluorophenyl)propan-2-yl)oxy)benzaldehyde (17):
To a solution of compound 16 (1.5 g, 8.2 mmol) in MeCN (10 mL) at 0 °C was added DBU (3.7 g, 24.6 mmol). TFAA (1.14 mL, 8.2 mmol) was added dropwise and the solution was stirred for 30 min. Then the mixture was added dropwise to a solution of compound 1 (1 g, 8.2 mmol) and CuCl2·H2O (1.4 mg, 0.0082 mmol) in MeCN (10 mL) over a period of 5 min at 0 °C. The mixture was allowed to warm to 25 °C and stir for 12 h. The solvent was removed under reduced pressure and the residue was diluted in DCM (40 mL). The organic layer was washed sequentially with aq. 1N HCl (2 × 20 mL), aq. 1N NaOH (2 × 20 mL), sat. aq. NaHCO3 (2 × 20 mL), and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Compound 17 was purified by silica gel chromatography (SiO2, hexanes/ethyl acetate = 20:1) to provide the desired product as colorless oil. Yield: 8%. 1H NMR (500 MHz, Chloroform-d) δ 9.81 (s, 1H), 7.68 – 7.62 (m, 2H), 7.44 – 7.36 (m, 2H), 7.10 – 7.01 (m, 2H), 6.78 – 6.69 (m, 2H), 1.78 (s, 6H). 13C NMR (126 MHz, Chloroform-d) δ 190.9, 162.1 (d, J = 246.5 Hz), 161.6, 141.3 (d, J = 3.4 Hz), 131.4, 129.8, 126.9 (d, J = 8.0 Hz), 119.1, 115.7 (d, J = 21.4 Hz), 81.1, 29.8.
3-((4-((2-(4-fluorophenyl)propan-2-yl)oxy)benzyl)amino)benzoic acid (4z):
Compound 4z was prepared following the General Procedure A with aldehyde 17 (0.62 mmol). The desired product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (60 mg, 25% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.4 (s, 1H) 7.53 – 7.45 (m, 2H), 7.22 – 7.13 (m, 2H), 7.14 (d, J = 2.3 Hz, 1H), 7.16 – 7.08 (m, 2H), 7.12 – 7.05 (m, 1H), 7.07 (s, 1H), 6.70 (dt, J = 6.2, 2.9 Hz, 1H), 6.62 – 6.55 (m, 2H), 6.26 (t, J = 5.9 Hz, 1H), 4.13 (d, J = 5.4 Hz, 2H), 1.63 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 161.1 (d, J = 243.0 Hz), 154.1, 148.6, 142.3 (d, J = 3.1 Hz), 132.8, 128.5, 127.9 (2C), 127.3 (d, J = 8.0 Hz, 2C), 119.9 (2C), 116.8, 115.5, 115.0 (d, J = 21.0 Hz, 2C), 112.9, 102.2, 79.4, 45.8, 29.1 (2C). TOFMSESI m/z: 402.1482 (C23H22FNO3 + Na+ requires 402.1476).
2-(3-((4-((4-Methoxybenzyl)oxy)benzyl)amino)phenoxy)-2-methylpropanoic acid (5a):
Reaction scale: 99 mg (0.22 mmol) of ethyl 2-(3-((4-((4-methoxybenzyl)oxy)benzyl)amino)phenoxy)-2-methylpropanoate. Compound 5a was prepared following General Procedure B. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 100:1) to give a white amorphous solid (13 mg, 14% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.34 (d, J = 8.6 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 2.8 Hz, 1H), 6.92 (d, J = 3.1 Hz, 2H), 6.90 (d, J = 3.1 Hz, 2H), 6.34 – 6.26 (m, 1H), 6.19 (s, 1H), 6.16 (d, J = 8.6 Hz, 1H), 4.97 (s, 2H), 4.18 (s, 2H), 3.79 (s, 3H), 1.47 (s, 6H). 13C NMR (101 MHz, Methanol-d4) δ 160.9, 159.3, 157.8, 151.1, 133.5, 130.8, 130.3 (2C), 130.2, 130.0, 129.6 (2C), 116.0 (2C), 114.8 (2C), 111.4, 109.2, 109.0, 105.6, 70.9, 55.7, 48.2, 25.8 (2C). HRESI m/z: 444.1771 (C25H27NO5 + Na+ requires 444.1781).
2-(3-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenoxy)-2-methylpropanoic acid (5b):
Reaction scale: 96 mg (0.22 mmol) of ethyl 2-(3-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenoxy)-2-methylpropanoate. Compound 5b was prepared following General Procedure B. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 100:1) to give a white amorphous solid (19 mg, 21% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.58 – 7.39 (m, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.16 – 7.02 (m, 2H), 6.94 (d, J = 4.2 Hz, 1H), 6.92 (dd, J = 8.4, 1.5 Hz, 2H), 6.39 – 6.26 (m, 1H), 6.18 (t, J = 2.0 Hz, 1H), 6.16 (dd, J = 8.0, 2.0 Hz, 1H), 5.02 (s, 2H), 4.18 (s, 2H), 1.47 (s, 6H). 13C NMR (151 MHz, Methanol-d4) δ 168.9, 154.3 (d, J = 244.4 Hz), 149.6, 148.3, 141.6, 125.4 (d, J = 3.4 Hz), 124.2, 121.1 (d, J = 8.4 Hz, 2C), 120.7, 120.1 (2C), 106.6 (d, J = 21.8 Hz, 2C), 106.5 (2C), 99.8, 99.6, 96.2, 70.5, 60.8, 38.7, 16.3 (2C). TOFMSESI m/z: 410.1761 (C24H24FNO4 + H+ requires 410.1762).
2-(4-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenoxy)-2-methylpropanoic acid (5b*):
Reaction scale: 96 mg (0.22 mmol) of ethyl 2-(4-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenoxy)-2-methylpropanoate. Compound 5b* was prepared following General Procedure B. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 100:1) to give a white amorphous solid (62 mg, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.48 (dd, J = 8.3, 5.5 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 7.20 (t, J = 8.7 Hz, 2H), 6.95 (d, J = 8.1 Hz, 2H), 6.65 (d, J = 8.3 Hz, 2H), 6.46 (d, J = 8.3 Hz, 2H), 5.04 (s, 2H), 4.10 (s, 2H), 1.36 (s, 6H).13C NMR (101 MHz, DMSO-d6) δ 175.3, 161.7 (d, J = 243.5 Hz), 157.1, 145.4, 144.5, 133.5 (d, J = 3.1 Hz), 132.5, 129.9 (d, J = 8.3 Hz, 2C), 128.6, 121.5, 115.3 (d, J = 21.3 Hz, 2C), 114.6, 112.6, 78.8, 68.4, 46.5, 25.0 (2C). TOFMSESI m/z: 460.1732 (C24H24FNO4 + H+ requires 410.1762).
2-methyl-2-(3-((4-((4-(trifluoromethyl)benzyl)oxy)benzyl)amino)phenoxy)propanoic acid (5f):
Reaction scale: 107 mg (0.22 mmol) of ethyl 2-methyl-2-(3-((4-((4-(trifluoromethyl)benzyl)oxy)benzyl)amino)phenoxy)propanoate. Compound 5f was prepared following General Procedure B. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 100:1) to give a white amorphous solid (30 mg, 30% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.67 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.6 Hz, 2H), 6.98 – 6.90 (m, 2H), 6.92 (d, J = 8.0 Hz, 1H), 6.30 (ddd, J = 8.0, 2.2, 0.9 Hz, 1H), 6.17 (t, J = 2.2 Hz, 1H), 6.16 – 6.13 (m, 1H), 5.15 (s, 2H), 4.19 (s, 2H), 1.46 (s, 6H). 13C NMR (151 MHz, Methanol-d4) δ 168.8, 149.4, 148.2, 141.6, 134.0, 124.4 (d, J = 2.3 Hz), 121.3 (q, J = 32.1 Hz), 120.7, 120.2 (2C), 119.2 (2C), 116.8 (q, J = 3.9 Hz, 2C), 116.3 (q, J = 271.2 Hz), 106.4 (d, J = 2.2 Hz, 2C), 99.7 (d, J = 3.8 Hz), 99.6 (d, J = 1.8 Hz), 96.2 (d, J = 3.6 Hz), 70.5, 60.5 (d, J = 2.8 Hz), 38.7, 16.3 (2C). TOFMSESI m/z: 460.1732 (C25H24F3NO4 + H+ requires 460.1730).
N-(3-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenyl)methanesulfonamide (19a):
Reaction scale: 50 mg (0.27 mmol) of N-(3-aminophenyl)methanesulfonamide (18a). Compound 19a was prepared following General Procedure A with aldehyde 3b (0.28 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (27 mg, 25% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.39 (s, 1H), 7.48 (dd, J = 8.4, 5.7 Hz, 2H), 7.26 (d, J = 8.5 Hz, 2H), 7.20 (t, J = 8.8 Hz, 2H), 6.95 (d, J = 4.7 Hz, 2H), 6.94 (d, J = 4.1 Hz, 1H), 6.47 (d, J = 2.1 Hz, 1H), 6.38 – 6.32 (m, 1H), 6.31 (s, 1H), 6.33 – 6.26 (m, 1H), 5.05 (s, 2H), 4.13 (d, J = 5.9 Hz, 2H), 2.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.7 (d, J = 243.6 Hz), 157.0, 149.4, 139.0, 133.4 (d, J = 3.1 Hz), 132.1, 129.8 (d, J = 8.2 Hz, 2C), 129.4, 128.5 (2C), 115.2 (d, J = 21.4 Hz, 2C), 114.6 (2C), 108.0, 107.2, 103.5, 68.4, 45.8, 38.8. TOFMSESI m/z: 423.1160 (C21H21FN2O3S + H+ requires 423.1149).
3-((4-((4-fluorobenzyl)oxy)benzyl)amino)-N-hydroxybenzamide (19b):
Reaction scale: 50 mg (0.33 mmol) of 3-amino-N-hydroxybenzamide (18b). Compound 19b was prepared following General Procedure A with aldehyde 3b (0.34 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (96 mg, 80% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.44 (dd, J = 8.5, 5.5 Hz, 2H), 7.29 (d, J = 8.6 Hz, 2H), 7.15 (d, J = 7.8 Hz, 1H), 7.12 – 7.08 (m, 2H), 7.08 – 7.07 (m, 1H), 7.05 (d, J = 2.6 Hz, 1H), 6.93 (d, J = 8.6 Hz, 2H), 6.77 (dd, J = 7.8, 2.0 Hz, 1H), 5.02 (s, 2H), 4.27 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 201.5, 192.0 (d, J = 244.5 Hz), 187.3, 178.5, 163.8, 163.0 (d, J = 3.2 Hz), 161.6, 158.8 (d, J = 8.3 Hz, 2C), 158.2, 157.8 (2C), 145.5, 144.8, 144.3 (d, J = 21.8 Hz, 2C), 144.1 (2C), 141.0, 98.5, 76.1. TOFMSESI m/z: 351.1291 (C21H19FN2O3 OH- + 2H+ requires 351.1503).
3-((4-((4-fluorobenzyl)oxy)benzyl)amino)benzenesulfonamide (19c):
Reaction scale: 100 mg (0.58 mmol) of 3-aminobenzenesulfonamide (18c). Compound 19c was prepared following General Procedure A with aldehyde 3b (0.59 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (17 mg, 8% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.44 (dd, J = 8.6, 5.5 Hz, 2H), 7.31 – 7.26 (m, 2H), 7.20 (t, J = 7.9 Hz, 1H), 7.14 (t, J = 2.0 Hz, 1H), 7.12 – 7.09 (m, 2H), 7.07 (s, 1H), 6.97 – 6.91 (m, 2H), 6.78 (ddd, J = 8.2, 2.4, 1.0 Hz, 1H), 5.02 (s, 2H), 4.27 (s, 2H). 13C NMR (151 MHz, Methanol-d4) δ 163.8 (d, J = 244.4 Hz), 159.2, 150.5, 145.4, 134.8 (d, J = 2.9 Hz), 133.0, 130.6 (d, J = 8.1 Hz, 2C), 130.5, 129.7 (2C), 117.2, 116.1 (d, J = 21.8 Hz, 2C), 116.0 (2C), 114.6, 110.5, 70.3, 47.8. TOFMSESI m/z: 421.0645 (C20H19FN2O3S + Cl- requires 421.0794).
N-(4-((4-fluorobenzyl)oxy)benzyl)-3-(1H-tetrazol-5-yl)aniline (19d):
Reaction scale: 100 mg (0.62 mmol) of 3-(1H-tetrazol-5-yl)aniline (18d). Compound 19d was prepared following General Procedure A with compound 3b (0.63 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (100 mg, 43% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.45 (dd, J = 8.5, 5.6 Hz, 2H), 7.37 – 7.30 (m, 2H), 7.30 – 7.24 (m, 2H), 7.20 (dt, J = 7.5, 1.3 Hz, 1H), 7.14 – 7.04 (m, 2H), 7.00 – 6.92 (m, 2H), 6.83 (ddd, J = 8.1, 2.5, 1.1 Hz, 1H), 5.03 (s, 2H), 4.32 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 163.8 (d, J = 244.6 Hz), 159.2, 157.7, 151.0, 134.8 (d, J = 3.4 Hz), 133.2, 131.0, 130.6 (d, J = 8.1 Hz, 2C), 129.7 (2C), 125.6, 117.1, 116.1 (d, J = 18.6 Hz, 2C), 116.01, 115.99 (2C), 111.7, 70.3, 47.8. TOFMSESI m/z: 376.1575 (C21H18FN5O + H+ requires 376.1568).
(3-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenyl)boronic acid (19e):
Reaction scale: 100 mg (0.58 mmol) of (3-aminophenyl)boronic acid (18e). Compound 19e was prepared following General Procedure A with aldehyde 3b (0.59 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (109 mg, 42% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.43 (dd, J = 8.6, 5.5 Hz, 2H), 7.28 (d, J = 8.6 Hz, 2H), 7.11 – 7.08 (m, 1H), 7.07 (d, J = 8.8 Hz, 2H), 6.98 – 6.89 (m, 2H), 6.84 (s, 1H), 6.84 – 6.79 (m, 1H), 6.67 (ddd, J = 8.1, 2.5, 1.2 Hz, 1H), 5.01 (s, 2H), 4.23 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 165.0, 162.6, 159.1, 149.3, 134.9 (d, J = 3.3 Hz), 133.8 (2C), 130.6 (dd, J = 8.5, 23.6 Hz), 129.6 (d, J = 21.7 Hz), 129.3 (d, J = 6.9 Hz), 123.0 (2C), 119.1 (2C), 116.7 – 116.0 (m), 115.9 (2C), 115.6 (d, J = 11.9 Hz), 70.3, 48.3. TOFMSESI m/z: 352.1537 (C20H19BFNO3 + H+ requires 352.1515).
2-((3-((4-((4-fluorobenzyl)oxy)benzyl)amino)phenyl)thio)-2-methylpropanoic acid (19f):
Reaction scale: 190 mg (0.56 mmol) of ethyl 2-((3-aminophenyl)thio)-2-methylpropanoate (18f). Compound 19f was prepared following General Procedure B with compound 3b (0.57 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 100:1) to give a white amorphous solid (30 mg, 13% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.47 – 7.38 (m, 2H), 7.30 – 7.23 (m, 2H), 7.13 – 6.97 (m, 2H), 7.01 (t, J = 7.8 Hz, 1H), 6.96 – 6.88 (m, 2H), 6.76 (dd, J = 2.4, 1.6 Hz, 1H), 6.72 (ddd, J = 7.5, 1.7, 1.0 Hz, 1H), 6.63 (ddd, J = 8.2, 2.4, 1.0 Hz, 1H), 5.01 (s, 2H), 4.21 (s, 2H), 1.37 (s, 6H). 13C NMR (101 MHz, Methanol-d4) δ 178.3, 163.8 (d, J = 244.5 Hz), 159.1, 150.3, 134.8 (d, J = 3.1 Hz), 133.5, 133.3, 130.6 (d, J = 8.1 Hz, 2C), 130.0, 129.7 (2C), 125.7, 121.6, 116.1 (d, J = 21.7 Hz, 2C), 115.9 (2C), 115.1, 70.2, 52.0, 48.0, 26.6 (2C). TOFMSESI m/z: 448.1358 (C24H24FNO3S + Na+ requires 448.1359).
3-((4-((4-chlorobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (20):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 20 was prepared following General Procedure A with 4-((4-chlorobenzyl)oxy)-3-methylbenzaldehyde (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (50 mg, 36% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 7.45 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 7.14 (d, J = 1.9 Hz, 1H), 7.13 (d, J = 2.5 Hz, 1H), 7.13 – 7.10 (m, 1H), 7.09 (d, J = 6.7 Hz, 1H), 7.08 – 7.04 (m, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.75 (ddd, J = 8.0, 2.5, 1.2 Hz, 1H), 6.41 (t, J = 6.0 Hz, 1H), 5.06 (s, 2H), 4.15 (d, J = 5.6 Hz, 2H), 2.15 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.3, 164.5, 158.2, 146.0, 141.7, 141.0, 140.8, 139.0, 138.6 (2C), 138.3, 137.9 (2C), 135.3, 135.1, 126.1, 125.8, 122.3, 121.1, 77.8, 55.3, 25.7. TOFMSESI m/z: 404.1006 (C22H20ClNO3 + Na+ requires 404.1024).
3-((4-((4-bromobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (21):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 21 was prepared following General Procedure A with 4-((4-bromobenzyl)oxy)-3-methylbenzaldehyde (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (70 mg, 45% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.62 (s, 1H), 7.58 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 2.1 Hz, 1H), 7.17 – 7.14 (m, 1H), 7.16 – 7.12 (m, 1H), 7.13 – 7.09 (m, 1H), 7.12 – 7.07 (m, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.77 (dd, J = 8.0, 1.1 Hz, 1H), 6.42 (t, J = 6.0 Hz, 1H), 5.07 (s, 2H), 4.17 (d, J = 5.4 Hz, 2H), 2.17 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 155.0, 148.8, 137.0, 131.5, 131.3 (2C), 131.2, 129.5, 129.4 (2C), 128.8, 125.8, 125.6, 120.7, 116.6, 116.3, 112.9, 111.6, 68.3, 45.9, 16.2. TOFMSESI m/z: 426.0648 (C22H20BrNO3 + H+ requires 426.0699).
3-((4-((4-iodobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (22):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 22 was prepared following General Procedure A with 4-((4-bromobenzyl)oxy)-3-methylbenzaldehyde (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (90 mg, 52% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.62 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 7.9 Hz, 2H), 7.16 (s, 1H), 7.15 (s, 1H), 7.14 (d, J = 7.9 Hz, 1H), 7.11 (d, J = 8.1 Hz, 1H), 7.09 (d, J = 7.8 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H), 6.40 (t, J = 6.1 Hz, 1H), 5.05 (s, 2H), 4.17 (d, J = 5.5 Hz, 2H), 2.17 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 155.0, 148.7, 137.3, 137.2 (2C), 131.5, 131.3, 129.5 (2C), 129.4, 128.8, 125.8, 125.6, 116.6, 116.2, 112.9, 111.6, 93.6, 68.5, 45.9, 16.2. TOFMSESI m/z: 472.0399 (C22H20INO3 - H+ requires 472.0415).
3-((4-((2,4-difluorobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (23):
Reaction scale: 40 mg (0.29 mmol) of 3-amino benzoic acid. Compound 23 was prepared following General Procedure A with 4-((2,4-difluorobenzyl)oxy)-3-methylbenzaldehyde (0.30 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (87 mg, 78% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 7.61 (ddd, J = 8.5, 6.6 Hz, 1H), 7.30 (ddd, J = 10.5, 2.6 Hz, 1H), 7.16 (dd, J = 1.9 Hz, 1H), 7.15 (s, 1H), 7.14 (s, 1H), 7.14 – 7.13 (m, 1H), 7.11 (d, J = 8.1 Hz, 1H), 7.09 (d, J = 7.5 Hz, 1H), 7.01 (d, J = 8.2 Hz, 1H), 6.80 – 6.72 (m, 1H), 6.43 (t, J = 6.0 Hz, 1H), 5.08 (s, 2H), 4.18 (d, J = 5.5 Hz, 2H), 2.12 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 161.5 – 160.8 (m), 159.6, 155.0, 148.7, 131.7 (d, J = 13.3 Hz), 131.7 – 131.5 (m), 131.3, 129.5, 128.8, 125.9, 125.7, 120.7, 116.6, 116.3, 112.9, 111.8, 111.5, 104.0 (t, J = 25.8 Hz), 63.3, 45.8, 16.1. TOFMSESI m/z: 406.1243 (C22H19F2NO3 + Na+ requires 406.1225).
3-((4-((3,4-difluorobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (24):
Reaction scale: 100 mg (0.73 mmol) of 3-amino benzoic acid. Compound 24 was prepared following General Procedure A with 4-((3,4-difluorobenzyl)oxy)-3-methylbenzaldehyde (0.74 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (170 mg, 61% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.60 (s, 1H), 7.51 (tt, J = 8.0, 4.0 Hz, 1H), 7.45 (ddd, J = 10.7, 9.9, 8.4 Hz, 1H), 7.31 (dd, J = 6.4 Hz, 1H), 7.16 (d, J = 1.8 Hz, 1H), 7.16 (s, 1H), 7.15 – 7.13 (m, 1H), 7.13 – 7.10 (m, 1H), 7.09 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.80 – 6.75 (m, 1H), 6.42 (t, J = 6.0 Hz, 1H), 5.07 (s, 2H), 4.17 (d, J = 5.4 Hz, 2H), 2.18 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 154.9, 149.9 (dd, J = 12.5, 73.4 Hz), 148.7, 148.3 (dd, J = 12.6, 73.0 Hz), 135.3 (dd, J = 3.7, 5.8 Hz), 131.7, 131.3, 129.5, 128.8, 125.8, 125.6, 124.1 (dd, J = 3.4, 6.7 Hz), 117.5 (d, J = 17.2 Hz), 116.6, 116.34 (d, J = 10.7 Hz), 116.26, 112.9, 111.6, 67.9, 45.8, 16.2. TOFMSESI m/z: 406.1243 (C22H19F2NO3 + Na+ requires 406.1225).
3-((4-((3,5-difluorobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (25):
Reaction scale: 40 mg (0.29 mmol) of 3-amino benzoic acid. Compound 25 was prepared following General Procedure A with 4-((3,5-difluorobenzyl)oxy)-3-methylbenzaldehyde (0.30 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (83 mg, 74% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.28 (d, J = 2.3 Hz, 1H), 7.25 (d, J = 7.7 Hz, 1H), 7.19 (s, 1H), 7.15 (d, J = 7.9 Hz, 1H), 7.13 (s, 1H), 7.08 – 7.02 (m, 2H), 6.89 – 6.87 (m, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.81 (dd, J = 7.9, 2.3 Hz, 1H), 5.10 (s, 2H), 4.24 (s, 2H), 2.26 (s, 3H). 13C NMR (151 MHz, Methanol-d4) δ 164.6 (dd, J = 13.0, 247.4 Hz, 2C), 156.7, 150.3, 143.9 (t, J = 9.2 Hz), 133.4, 131.0 (2C), 129.8, 127.9, 127.0 (2C), 119.0, 118.2, 114.8, 112.6 (dd, J = 2.4, 4.2 Hz), 110.6 (dd, J = 5.3, 20.7 Hz, 2C), 103.6 (t, J = 25.8 Hz), 69.6, 48.1, 16.5. TOFMSESI m/z: 382.1268 (C22H19F2NO3 - H+ requires 382.1260).
3-((3-fluoro-4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (26):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 26 was prepared following General Procedure A with 3-fluoro-4-((4-fluorobenzyl)oxy)benzaldehyde (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (100 mg, 75% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.64 (s, 1H), 7.49 (dd, J = 8.5, 5.7 Hz, 2H), 7.23 – 7.20 (m, 2H), 7.20 – 7.19 (m, 1H), 7.18 (d, J = 1.9 Hz, 1H), 7.16 – 7.15 (m, 1H), 7.14 (d, J = 7.8 Hz, 1H), 7.11 (d, J = 1.4 Hz, 1H), 7.10 (q, J = 1.6 Hz, 1H), 6.77 (ddd, J = 7.9, 2.5, 1.2 Hz, 1H), 6.50 (t, J = 6.1 Hz, 1H), 5.12 (s, 2H), 4.23 (d, J = 5.9 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.7, 161.9 (d, J = 243.9 Hz), 151.7 (d, J = 243.9 Hz), 148.5, 144.6 (d, J = 10.7 Hz), 133.4 (d, J = 5.0 Hz), 132.8 (d, J = 2.9 Hz), 131.3, 130.0 (d, J = 8.5 Hz, 2C), 128.9, 123.0 (d, J = 3.0 Hz), 116.9, 116.4, 115.4, 115.3 (d, J = 21.3 Hz, 2C), 114.7 (d, J = 18.3 Hz), 112.9, 69.6, 45.3. TOFMSESI m/z: 368.1110 (C21H17F2NO3 - H+ requires 368.1104).
3-((3-chloro-4-((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (27):
Reaction scale: 100 mg (0.72 mmol) of 3-amino benzoic acid. Compound 27 was prepared following General Procedure A with 3-chloro-4-((4-fluorobenzyl)oxy)benzaldehyde (0.73 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (120 mg, 43% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.65 (s, 1H), 7.50 (dd, J = 8.3, 5.6 Hz, 2H), 7.42 (d, J = 2.0 Hz, 1H), 7.27 (d, J = 8.5 Hz, 1H), 7.23 (t, J = 8.7 Hz, 2H), 7.19 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 2.5 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 7.11 (d, J = 7.5 Hz, 1H), 6.79 – 6.69 (m, 1H), 6.51 (s, 1H), 5.15 (s, 2H), 4.23 (d, J = 5.9 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 161.8 (d, J = 243.5 Hz), 152.2, 148.4, 133.6, 132.8 (d, J = 3.1 Hz), 131.4, 129.7 (d, J = 8.4 Hz, 2C), 128.9, 128.5, 126.9, 121.4, 116.9, 116.3, 115.3 (d, J = 21.3 Hz, 2C), 114.3, 112.9, 69.3, 45.2. TOFMSESI m/z: 384.0820 (C21H17ClFNO3 - H requires 384.0808).
3-((3,4-bis((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (28):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 28 was prepared following General Procedure A with 3,4-bis((4-fluorobenzyl)oxy)benzaldehyde (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (95 mg, 58% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.62 (s, 1H), 7.45 (dd, J = 2.1, 6.2 Hz, 2H), 7.44 (dd, J = 2.4, 5.8 Hz, 2H), 7.24 – 7.16 (m, 2H), 7.17 (d, J = 2.5 Hz, 2H), 7.19 – 7.12 (m, 1H), 7.13 (d, J = 7.6 Hz, 1H), 7.11 (s, 1H), 7.10 (d, J = 5.9 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 6.88 (dd, J = 1.9, 8.2 Hz, 1H), 6.77 (dt, J = 2.0, 7.7 Hz, 1H), 6.42 (t, J = 6.0 Hz, 1H), 5.06 (s, 2H), 5.05 (s, 2H), 4.19 (d, J = 5.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.8, 161.7 (d, J = 243.3 Hz), 160.5 (d, J = 243.5 Hz), 148.7, 148.1, 147.0, 133.6 (d, J = 3.2 Hz), 133.4 (d, J = 3.1 Hz), 132.9, 131.3, 129.8 (d, J = 8.3 Hz, 2C), 129.7 (d, J = 8.2 Hz, 2C), 128.8, 119.9, 116.7, 116.3, 115.3 (d, J = 21.3 Hz, 2C), 115.1 (d, J = 21.3 Hz, 2C), 114.6, 113.9, 113.0, 69.50, 69.47, 46.0. TOFMSESI m/z: 474.1525 (C28H23F2NO4 - H+ requires 474.1522).
3-((2,4-bis((4-fluorobenzyl)oxy)benzyl)amino)benzoic acid (29):
Reaction scale: 50 mg (0.36 mmol) of 3-amino benzoic acid. Compound 29 was prepared following General Procedure A with 2,4-bis((4-fluorobenzyl)oxy)benzaldehyde (0.37 mmol). The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (102 mg, 59% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.61 (s, 1H), 7.52 (dd, J = 5.7, 8.4 Hz, 2H), 7.47 (dd, J = 5.7, 8.5 Hz, 2H), 7.24 – 7.19 (m, 2H), 7.21 – 7.16 (m, 2H), 7.15 (d, J = 2.0 Hz, 1H), 7.13 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 7.7 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.75 (t, J = 1.7 Hz, 1H), 6.74 (d, J = 2.3 Hz, 1H), 6.56 (dd, J = 2.3, 8.4 Hz, 1H), 6.29 (t, J = 6.1 Hz, 1H), 5.16 (s, 2H), 5.03 (s, 2H), 4.20 (d, J = 5.3 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 161.7 (d, J = 243.9 Hz), 161.6 (d, J = 243.5 Hz), 158.3, 156.6, 148.8, 133.4 (d, J = 2.9 Hz), 133.3 (d, J = 2.8 Hz), 131.3, 129.9 (d, J = 8.2 Hz, 2C), 129.5 (d, J = 8.2 Hz, 2C), 128.8, 128.6, 119.9, 116.5, 116.3, 115.19 (d, J = 21.3 Hz, 2C), 115.17 (d, J = 21.4 Hz, 2C), 112.6, 105.8, 100.5, 68.6 (2C), 40.8. TOFMSESI m/z: 476.1663 (C28H23F2NO4 + H+ requires 476.1668).
3-((4-((4-fluorobenzyl)oxy)-3-methylbenzyl)(methyl)amino)benzoic acid (30):
Paraformaldehyde (5 mg, 0.16mmol), compound 4u (30 mg, 0.08 mmol), sodium triacetoxyborohydride (6.2 mg, 0.096 mmol) and 2 mL of ethanol were combined in a 10 mL round bottom flask and refluxed for 2 hr at 100 °C. The reaction was then cooled to 25 °C for 12 h. The solvent was removed by vacuum and the residue was dissolved in EtOAc (60 mL). The organic layer was washed with aq. 1N HCl (20 mL), water (20 mL), and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated by vacuum. The compound was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (13 mg, 44% yield). 1H NMR (600 MHz, Methanol-d4) δ 7.47 – 7.42 (m, 2H), 7.42 (d, J = 1.7 Hz, 1H), 7.37 – 7.29 (m, 1H), 7.23 (t, J = 7.9 Hz, 1H), 7.08 (t, J = 8.8 Hz, 2H), 7.01 (d, J = 2.2 Hz, 1H), 6.98 – 6.96 (m, 1H), 6.96 – 6.94 (m, 1H), 6.85 (d, J = 8.3 Hz, 1H), 4.99 (s, 2H), 4.46 (s, 2H), 3.00 (s, 3H), 2.18 (s, 3H). 13C NMR (151 MHz, Methanol-d4) δ 169.3, 162.3 (d, J = 244.4 Hz), 155.7, 149.7, 133.6 (d, J = 3.3 Hz), 131.0, 130.4, 128.9 (d, J = 8.3 Hz, 2C), 128.6, 126.7, 125.0, 117.3, 116.7, 114.7 (d, J = 21.8 Hz, 2C), 113.0, 111.39, 111.36, 68.9, 55.4, 37.4, 15.2. TOFMSESI m/z: 378.1509 (C23H22FNO3- H+ requires 378.1511).
3-fluoro-5-((4-((4-fluorobenzyl)oxy)-3-methylbenzyl)amino)benzoic acid (31):
Reaction scale: 50 mg (0.32 mmol) of 3-amino-5-fluorobenzoic acid. Compound 31 was prepared following General Procedure A with aldehyde 12b. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (104 mg, 84% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.47 (dd, J = 8.4, 5.2 Hz, 2H), 7.38 (d, J = 2.2 Hz, 1H), 7.22 (dd, J = 8.5, 2.2 Hz, 1H), 7.13 – 7.02 (m, 4H), 6.88 (d, J = 9.2 Hz, 1H), 6.48 (d, J = 11.3 Hz, 1H), 5.09 (s, 2H), 4.25 (s, 2H13C NMR (101 MHz, Methanol-d4) δ 169.5, 165.1 (d, J = 241.1 Hz), 163.8 (d, J = 244.3 Hz), 157.2, 152.1, 135.1 (d, J = 2.3 Hz), 134.3 (d, J = 10.0 Hz), 132.5, 130.9, 130.4 (d, J = 8.2 Hz), 128.1, 126.9 (2C), 116.1 (d, J = 21.6 Hz), 112.8 (2C), 111.1, 104.4 (d, J = 24.0 Hz), 103.9 (d, J = 25.8 Hz), 70.4, 47.9, 16.6. TOFMSESI m/z: 382.1254 (C22H19F2NO3 - H+ requires 382.1260).
3-((3-chloro-4-((4-fluorobenzyl)oxy)benzyl)amino)-5-fluorobenzoic acid (32):
Reaction scale: 50 mg (0.32 mmol) of 3-amino-5-fluorobenzoic acid. Compound 32 was prepared following General Procedure A with 3-chloro-4-((4-fluorobenzyl)oxy)benzaldehyde. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 99.5:0.5) to give a white amorphous solid (109 mg, 84% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.51 – 7.43 (m, 2H), 7.38 (d, J = 2.2 Hz, 1H), 7.22 (dd, J = 8.4, 2.2 Hz, 1H), 7.09 (dt, J = 5.9, 2.1 Hz, 2H), 6.92 – 6.84 (m, 1H), 6.48 (dt, J = 11.4, 2.3 Hz, 1H), 5.09 (s, 2H), 4.25 (s, 2H). 13C NMR (101 MHz, Methanol-d4) δ 169.4 (d, J = 3.4 Hz), 165.1 (d, J = 241.2 Hz), 163.9 (d, J = 244.8 Hz) 154.5, 151.8 (d, J = 11.0 Hz), 134.5, 134.3 (d, J = 3.1 Hz), 134.2 (d, J = 3.7 Hz), 130.5, 130.4, 130.1, 127.8, 124.3, 116.3, 116.1, 115.6, 111.1, 104.7 (d, J = 24.1 Hz), 103.9, 71.3, 47.3. TOFMSESI m/z: 402.1317 (C21H16ClF2NO3 - H+ requires 402.0714).
2-(3-((4-((4-fluorobenzyl)oxy)-3-methylbenzyl)amino)phenoxy)-2-methylpropanoic acid (33):
Reaction scale: 99 mg (0.22 mmol) of ethyl 2-(3-((4-((4-fluorobenzyl)oxy)-3-methylbenzyl)amino)phenoxy)-2-methylpropanoate. Compound 33 was prepared following General Procedure B. The product was purified by flash column chromatography (SiO2, CH2Cl2/CH3OH, 100:1) to give a white amorphous solid (30 mg, 32% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.41 (dd, J = 8.5, 5.5 Hz, 2H), 7.11 (d, J = 2.1 Hz, 1H), 7.09 (d, J = 6.0 Hz, 1H), 7.06 (t, J = 7.7 Hz, 2H), 6.92 (t, J = 8.0 Hz, 1H), 6.83 (d, J = 8.3 Hz, 1H), 6.30 (dd, J = 8.1, 2.1 Hz, 1H), 6.20 (t, J = 2.3 Hz, 1H), 6.17 (dd, J = 8.0, 2.3 Hz, 1H), 4.98 (s, 2H), 4.12 (s, 2H), 2.19 (s, 3H), 1.46 (s, 6H). 13C NMR (101 MHz, Methanol-d4) δ 178.2, 163.7 (d, J = 244.2 Hz), 157.7, 157.0, 151.0, 135.1 (d, J = 3.3 Hz), 133.1, 130.9, 130.3 (d, J = 8.1 Hz, 2C), 130.2, 127.9, 126.9, 116.1 (d, J = 21.7 Hz, 2C), 112.7, 109.2, 109.1, 105.7, 79.9, 70.3, 48.3, 25.8 (2C), 16.6. TOFMSESI m/z: 422.1784 (C25H26FNO4 - H+ requires 422.1773).
BIOLOGICAL ASSAY PROTOCOLS
In vitro PPAR functional reporter gene assay.
Agonist activity of test compounds against human PPARα, was analyzed using commercial kits from Indigo Biosciences (hPPARα: IB00111, hPPARγ: IB00101; hPPARδ: IB00121. Protocols provided with the kit were followed for experimental execution. Briefly, a suspension of reporter cells was prepared in Cell Recovery Medium (CRM: containing 10% charcoal stripped fetal bovine serum (FBS)). 200 μL of the reporter cell suspension was dispensed into wells of white 96-well assay plates provided within the kit for a 4–6 h pre-incubation period at 37 °C and 5% CO2. At the end of the pre-incubation period, the culture media was discarded and 200 μL of Compound Screening Medium (CSM: containing 10% charcoal stripped FBS) containing the requisite concentration of derivative to be evaluated was added to representative wells. Following a 22–24 h incubation at 37 °C and 5% CO2, the treatment media were discarded and 100 μL of the provided Luciferase Detection Reagent was added to each well. Luminescence was quantified using a GloMax Explorer (Promega). Dose-response analyses of compounds were performed via non-linear curve-fitting with four parameters. Plots of relative light units vs. Log[compound, (μM)] were generated using GraphPad Prism software, which was also used to calculate EC50 and SEM values. Unless otherwise noted, each compound evaluated was tested in three separate experiments in triplicate.
Cellular thermal shift assay.
The cellular thermal shift assay (CETSA) was performed with MIO-M1 cells, a cell line derived from human retinal Müller glial cells, cultured in DMEM medium supplemented with 10% FBS. For an initial determination of the thermal melting profile of PPARα, cells were dispensed into a 96-well PCR plate in the above medium (6000 cells/well/50 μl) and subjected to a temperature gradient (40–60 °C) for 10 min. Cold non-denaturing lysis buffer (PBS supplemented with 0.1% TritonX-100 and 1x protease inhibitors) was added to the wells, and the plate was rocked, and then incubated for 15 min on ice. Subsequently, centrifugation was performed at 14,000 rpm to sediment the unstable protein content. Supernatant was collected and loaded for SDS-PAGE, and immuno-detection was performed using an anti-PPARα antibody. The PPARα band was quantified on a LI-COR C-Digit Blot Scanner, and subsequently Tagg(50) and Tagg(75) values were calculated for PPARα. In a subsequent run, cells were treated at various doses (1, 0.3, 0.1, 0.037, 0.012, 0.004 and 0.0014 μM) of FA, 4a (A91) and 4u together with DMSO controls, for 3 h. Cells were then subjected to heat shock at Tagg(75) for 10 min, and unstable protein was removed by centrifugation. Following an immuno-blotting step, bands of stable PPARα protein was quantified, normalized to loading control and plotted using GraphPad Prism software. EC50 values of FA, 4a (A91) and 4u were calculated. Data obtained from a single experiment run in duplicate.
PHARMACOKINETIC ANALYSES
Studies were carried out by Eurofins Discovery Services. Experimental protocols presented below are pulled directly from company provided SOPs. All experiments were run in duplicate.
Microsomal stability.
The following procedure is designed to determine the stability of a test compound in pooled liver microsomes from human or rat in a 96-well format. The test compound is quantified at five time points by HPLC-MS/MS analysis. The test compound was pre-incubated with pooled liver microsomes in phosphate buffer (pH 7.4) for 5 min in a 37 °C shaking water bath. The reaction was initiated by adding NADPH-generating system and incubated for 0, 15, 30, 45, and 60 min. The reaction is stopped by transferring the incubation mixture to acetonitrile/methanol. Samples are then mixed and centrifuged. Supernatants are used for HPLC-MS/MS using selected reaction monitoring. The HPLC system consists of a binary LC pump with an autosampler, a C-18 column, and a gradient. Four reference compounds are tested in each assay. Propranolol and imipramine are relatively stable, whereas verapamil and terfenadine are readily metabolized. Peak areas corresponding to the test compound are recorded. The compound remaining is calculated by comparing the peak area at each time point to time zero. The half-life is calculated from the slope of the initial linear range of the logarithmic curve of compound remaining (%) vs. time, assuming first order kinetics. In addition, the intrinsic clearance (Clint) is calculated from the half-life using the following equation: Clint = 0.693/(t1/2 x [protein]) and units are μL/min/mg protein. Human liver microsomes: mixed gender and pool of 50 donors; rat liver microsomes: male, Sprague-Dawley, pool of 100 or more.
Time-dependent CYP inhibition.
The following procedure is designed to assess if a test compound displays time-dependent inhibition on cytochrome P450 activity in pooled human liver microsomes in a 96-well plate format. Test compounds were assessed at 10 μM with 0.1% DMSO. The test compound was pre-incubated with human liver microsomes (mixed gender, pool of 50 donors, 1 mg/mL) in phosphate buffer (pH 7.4) for 30 min in a 37 °C shaking water bath. The pre-incubation is performed in two sets: in the presence and absence of NADPH-generating system, respectively. The reaction was then initiated by adding one volume of the pre-incubation mixture to nine volumes of a mixture of the substrate and the NADPH-generating system. The incubation was allowed for 10 min and stopped by transferring the reaction mixture to acetonitrile/methanol. Samples were mixed and centrifuged and supernatants used for HPLC-MS/MS analysis of the respective metabolite. Reference time dependent inhibitors were tested in each assay at multiple concentrations to obtain IC50 (concentration causing a half-maximal inhibition of the control value) values for both reaction conditions (±NADPH). Peak areas corresponding to the metabolite were recorded. The percent of control activity was calculated by comparing the peak area in the presence of the test compound to the control samples containing the same solvent. Subsequently, the percent inhibition was calculated by subtracting the percent control activity from 100. The IC50 value was determined by non-linear regression analysis of the concentration-response curve using the Hill equation. Reference compounds: phenacetin (CYP1A), diclofenac (CYP2C9), omeprazole (CYP2C19), dextromethorphan (CYP2D6), and midazolam (CYP3A).
hERG Automated Patch-Clamp.
The automated whole cell patch-clamp (Qpatch 48) technique was used to record outward potassium currents from a single cell. CHO-K1 (Chinese Hamster Ovary) cells stably transfected with human hERG cDNA were used. The cells were harvested by trypsinization and maintained in Serum Free Medium at room temperature before recording. The cells were washed and re-suspended in Extracellular Solution before being applied to the automated patch-clamp sites. The test solutions are prepared in the Extracellular Solution on the day of patch-clamp assay. The assay can tolerate up to 1% DMSO. Test compounds were assessed at three concentrations (0.1, 1 and 10 μM). Intracellular Solution: 130 mM KCl, 10 mM NaCl, 1 mM MgCl2, 10 mM EGTA, 5 mM MgATP, 10 mM HEPES (pH adjusted to 7.2 with KOH) Extracellular Solution: 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM D(+)-Glucose, 10 mM HEPES (pH adjusted to 7.4 with NaOH) After whole cell configuration was achieved, the cell was held at −80 mV. A 50 ms pulse to −40 mV was delivered to measure the leaking current, which was subtracted from the tail current on-line. Then the cell was depolarized to +20 mV for 2 s, followed by a 1 s pulse to −40 mV to reveal the hERG tail current. This paradigm was delivered once every 5 s to monitor the current amplitude. The assay was conducted at room temperature. The Extracellular Solution (control) was applied first and the cell was stabilized in the solution for 5 min. Then the test compound was applied from low to high concentrations sequentially on the same cell. The cells were incubated with each test concentration for 5 min. The reference compound E-4031 was tested concurrently at multiple concentrations to obtain an IC50 value. The percent inhibition of hERG channel was calculated by comparing the tail current amplitude before and after application of the compound (the current difference was normalized to control).
In vivo retinal vascular permeability assay.
All in vivo experiments were performed in compliance with the NIH guidelines for the Care and Use of Animals and approved by the Institutional Animal Care and Use Committees at the University of Oklahoma Health Sciences Center. Brown Norway rats were purchased from Charles Rivers Laboratories (Wilmington, MA). Diabetes was induced in male Brown Norway rats (at the ages of 8 weeks) using an intraperitoneal injection of streptozotocin (STZ, 55 mg/kg of body weight). Diabetes was confirmed 3 days following STZ injection and rats with blood glucose levels higher than 350 mg/dl were used as diabetic rats. Two weeks following the STZ injection, the animals received daily injections of the compound for another 4 weeks, with the same volume vehicle as control. The rats were then used for retinal vascular permeability assay using Evan blue as tracer as described previously30 and described as follows. At the study end-point, the animals were anesthetized by intraperitoneal injection of Ketamine (75 mg/kg, ZetamineTM, MWI, Boise, ID) and Xylazine (10 mg/kg, AnaSedTM, LA, MWI, Boise, ID). Evans blue (30 mg/ml) (Sigma-Aldrich, St. Louis, MO) was administrated over 10 s through the femoral vein using 30G needle under microscopic inspection at the dosage of 1 μl/g body weight. The animals were placed on a warm pad for 2 hrs to ensure the complete circulation of Evans blue. The animals were perfused via the left ventricle with 1% paraformaldehyde in citrate buffer (pH 4.2), which was pre-warmed to 37°C to prevent vasoconstriction. Immediately after perfusion, the eyeball was enucleated, and the retina was carefully dissected under an operating microscope. The retinal homogenate was incubated with formamide for 18 hrs at 70℃ to extract the Evans blue. Then the extract was ultra-centrifuged (Optima TLX Ultracentrifuge, Beckman Coulter Inc., Brea, CA) at 80,000 rpm and 4℃ for 30 minutes. The supernatant was used for measurement of absorbance at 620 nm with a spectrophotometer (DU-800 UV/Vis Spectrophotometer, the Lab World Group, Woburn, MA). Concentrations of Evans blue in the extracts were calculated based on a standard curve of Evans blue in formamide. The concentration of Evans blue was normalized by total protein concentration in the supernatant. The result of retinal vascular permeability was expressed in μg of Evans blue/mg of proteins. All values are express as mean ± SD. Student’s t-test was used to compare 2 groups, and P values less than 0.05 were considered statistically significant.
DOCKING PROTOCOLS
Molecular modelling and docking procedures were executed using the standard protocol implemented in Maestro Molecule Builder of Schrödinger,19, 31 version 11.9. File preparation and docking protocol was completed as follows:
Protein Preparation.
The desired crystal structure (PDB: 2P54) was obtained from the Protein Data Bank and prepared with Schrödinger’s Protein Preparation Wizard software. Proteins were preprocessed by assigning bond orders using the ccd database, adding hydrogens, creating zero-bond order to metals, creating disulfide bonds, filling missing side chains and loops using Prime, deleting water molecules beyond 5 Å, and generating Het state using Epik pH 7 ± 2. Het -COOH was modified to COO-. Structural refinement was done employing H-bond assignment by sampling water orientations, minimizing hydrogens of altered species at pH 7.0, and restraining minimization based on the OPLS3 force field. All remaining parameters were kept as default settings.
Ligand Preparation.
SMILES files of each ligand were loaded into Schrödinger’s LigPrep software. Ligand preparation employed the OPLS3 force field. Ionization states for pH 7.0 ± 2 were generated with Epik. Ligands were desalted and tautomers were generated. Chirality specified in the ligand input file was retained.
Receptor Grid Generation.
i) Receptor tab: select the native ligand and show markers, set the Van der Waals scaling factor to 1.0 with a partial charge cut off of 0.25. In advanced settings, select read from the input structure file and Force field OPLS3. ii) Site tab: Select centroid of workspace ligand, and dock ligand with length < 10 Å. In advanced settings set the box length = 10 Å. iv) Constraints tab: H-Bond Tyr 464, His-440, Tyr-314 and Ser-280; v) Rotatable Groups tab: select all in the grid box.
Ligand Docking.
Docking studies of compounds were performed using Schrödinger’s Glide docking module from suite 2017–4. Docking was executed with the following parameters: Using partial charge input, scaling of Van der Waal radii with a scaling factor of 0.8 and partial charge cut off of 0.15, with XP precision, sample nitrogen inversion, sample ring conformation, bias sampling of torsion for amide group only, add Epik state penalties to docking score, OPLS3 force field with no constraints selected.
Metabolism Prediction.
Metabolism prediction studies were performed using Schrödinger’s P450 Site of Metabolism module. After conducting Ligand Preparation (see above), compound 4b was subjected to metabolism prediction studies with 2C9, 2D6, and 3A4. For these calculations, default parameters were held constant. The algorithm calculates reactivity of each ligand based on Hammett and Taft linear free energy schemes for these promiscuous P450 enzymes, which are believed to be the most independent on structural restrictions. The software then subjects the ligand of interest to induced-fit docking (2C9 and 2D6) to identify possible sites of metabolism based on Fe-accessibility. Intrinsic reactivity and site of metabolism data is then output as 1) Fe-accessibility, 2) Intrinsic reactivity, 3) and overall SOM score (the linear combination of the accessibility and the intrinsic reactivity), which identifies the major predicted sites of P450 metabolism.
Supplementary Material
Table 8.
Thermodynamic Binding Profiles of Selected Ligands with the Ligand Binding Domain of PPARα.a
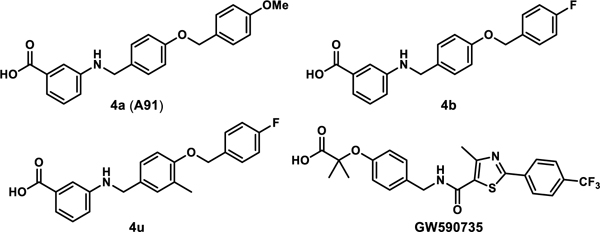 | |||||
|---|---|---|---|---|---|
| Compound | EC50 (μM) | Kd (μM) | ΔG (kcal/mol) |
ΔH (kcal/mol) |
−TΔS (kcal/mol) |
| 4a (A91) | 4.43 ± 0.01 | 16.90 ± 1.50 | −6.50 ± 0.05 | −12.20 ± 1.40 | 5.71 ± 1.31 |
| 4b | 0.77 ± 0.03 | 5.32 ± 0.78 | −7.20 ± 0.09 | −8.18 ± 0.20 | 0.98 ± 0.27 |
| 4u | 0.037±0.005 | 0.14 ± 0.05 | −9.39 ± 0.17 | −7.75 ± 0.26 | −1.65 ± 0.37 |
| GW590735 | 0.015 ± 0.002 | 0.17 ± 0.03 | −9.24 ± 0.09 | −14.10 ± 0.50 | 4.90 ± 0.50 |
Buffer: 20 mM Hepes, 150 mM NaCl, pH 7.4. Ligand solution: 200–1000 μM ligand in buffer with 0.8–10% DMSO. Protein solution: 30–85 μM PPARα LBD in buffer with same concentration of DMSO as corresponding ligand solution. Thermodynamic parameters reported as mean ± S.D. of at least three separate experiments. All experiments were performed at 25 °C. N.D. = not determined.
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the University of Oklahoma Growth Fund (A.S.D., H.S., J.X.M.) and the National Eye Institute of the National Institutes of Health under award numbers R21EY028279 (A.S.D.), R01EY019309, R01EY018659, and R01EY012231 (J.X.M.). We acknowledge the use of the Protein Production Core (PPC) at the University of Oklahoma, Norman. PPC is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103640. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors are grateful to Cameron D. Siler and Will Drover for enlightening discussions and Eurofins for conducting the PK studies.
ABBREVIATIONS
- CETSA
cellular thermal shift assay
- DR
diabetic retinopathy
- FenoFA
fenofibric acid
- Feno
fenofibrate
- ITC
isothermal titration calorimetry
- NV
neovascularization
- POC
proof of concept
- SI
selectivity index
- STZ
streptozotocin
- Tagg
protein aggregation temperature
- VEGF
vascular endothelial growth factor
Footnotes
ASSOCIATED CONTENT
The Supporting Information is avilable free of charge on the ACS Publications website at DOI:
Molecular Formula Strings, two-concentration luciferase data, PPARα expression and purification, cellular thermal shift western blots, LDH cytotoxicity data, in silico metabolic predictions, isothermal titration calorimetry thermograms and curve fits, 1H and 13C NMR spectra for final compounds, HPLC traces for select leads, and pharmacokinetic data sets.
REFERENCES
- 1.Nentwich MM; Ulbig MW, Diabetic retinopathy - ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai X; McGinnis JF, Diabetic retinopathy: animal models, therapies, and perspectives. J. Diabetes Res 2016, 2016, No. 3789217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chew EY; Ambrosius WT; Davis MD; Danis RP; Gangaputra S; Greven CM; Hubbard L; Esser BA; Lovato JF; Perdue LH; Goff DC Jr.; Cushman WC; Ginsberg HN; Elam MB; Genuth S; Gerstein HC; Schubart U; Fine LJ, Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med 2010, 363, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keech AC; Mitchell P; Summanen PA; O’Day J; Davis TME; Moffitt MS; Taskinen MR; Simes RJ; Tse D; Williamson E; Merrifield A; Laatikainen LT; d’Emden MC; Crimet DC; O’Connell RL; Colman PG, Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 2007, 370, 1687–1697. [DOI] [PubMed] [Google Scholar]
- 5.Noonan JE; Jenkins AJ; Ma JX; Keech AC; Wang JJ; Lamoureux EL, An update on the molecular actions of fenofibrate and its clinical fffects on diabetic retinopathy and other microvascular end points in patients with diabetes. Diabetes 2013, 62, 3968–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simo R; Roy S; Behar-Cohen F; Keech A; Mitchell P; Wong TY, Fenofibrate: a new treatment for diabetic retinopathy. Molecular mechanisms and future perspectives. Curr. Med. Chem 2013, 20, 3258–3266. [DOI] [PubMed] [Google Scholar]
- 7.Wei Shen YG, Therapeutic potential of topical fenofibrate eyedrops in diabetic retinopathy and AMD rat models. J. Clin. Exp. Ophthalmol 2014, 5, No. 1000347. [Google Scholar]
- 8.Wong TY; Simo R; Mitchell P, Fenofibrate - a potential systemic treatment for diabetic retinopathy? Am. J. Ophthalmol 2012, 154, 6–12. [DOI] [PubMed] [Google Scholar]
- 9.Berger J; Moller D, The mechanisms of action of PPARs. Annu. Rev. Med 2002, 53, 409–435. [DOI] [PubMed] [Google Scholar]
- 10.Feige JN; Gelman L; Michalik L; Desvergne B; Wahli W, From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res 2006, 45, 120–159. [DOI] [PubMed] [Google Scholar]
- 11.Michalik L; Wahli W, Peroxisome proliferator-activated receptors: three isotypes for a multitude of functions. Curr. Opin. Biotechnol 1999, 10, 564–570. [DOI] [PubMed] [Google Scholar]
- 12.Fruchart JC, Selective peroxisome proliferator-activated receptor alpha modulators (SPPARM alpha): the next generation of peroxisome proliferator-activated receptor alpha-agonists. Cardiovas. Diabetol 2013, 12, No. 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirat C; Farce A; Lebegue N; Renault N; Furman C; Millet R; Yous S; Speca S; Berthelot P; Desreumaux P; Chavatte P, Targeting peroxisome proliferator-activated receptors (PPARs): development of modulators. J. Med. Chem 2012, 55, 4027–4061. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y; Hu Y; Lin M; Jenkins AJ; Keech AC; Mott R; Lyons TJ; Ma JX, Therapeutic effects of PPAR agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013, 62, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y; Chen Y; Ding L; He X; Takahashi Y; Gao Y; Shen W; Cheng R; Chen Q; Qi X; Boulton ME; Ma JX, Pathogenic role of diabetes-induced PPAR-alpha down-regulation in microvascular dysfunction. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 15401–15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearsall EA; Cheng R; Zhou K; Takahashi Y; Matlock HG; Vadvalkar SS; Shin Y; Fredrick TW; Gantner ML; Meng S; Fu Z; Gong Y; Kinter M; Humphries KM; Szweda LI; Smith LEH; Ma JX, PPAR is essential for retinal lipid metabolism and neuronal survival. BMC Biol 2017, 15, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou XZ; Nath D; Shin Y; Ma JX; Duerfeldt AS, Structure-guided evolution of a 2-phenyl-4-carboxyquinoline chemotype into PPAR-alpha selective agonists: new leads for oculovascular conditions. Bioorg. Med. Chem. Lett 2018, 28, 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernardes A; Souza PCT; Muniz JRC; Ricci CG; Ayers SD; Parekh NM; Godoy AS; Trivella DBB; Reinach P; Webb P; Skai MS; Polikarpov I, Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: a new mode of ligand recognition and receptor stabilization. J. Mol. Biol 2013, 425, 2878–2983. [DOI] [PubMed] [Google Scholar]
- 19.Capelli D; Cerchia C; Montanari R; Loiodice F; Tortorella P; Laghezza A; Cervoni L; Pochetti G; Lavecchia A, Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep 2016, 6, No. 34792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gangwal RP; Damre MV; Das NR; Sharma SS; Sangamwar AT, Biological evaluation and structural insights for design of subtype-selective peroxisome proliferator activated receptor-alpha (PPAR-alpha) agonists. Bioorg. Med. Chem. Lett 2015, 25, 270–275. [DOI] [PubMed] [Google Scholar]
- 21.Li J; Kennedy LJ; Shi Y; Tao S; Ye XY; Chen SY; Wang Y; Hernandez AS; Wang W; Devasthale PV; Chen S; Lai Z; Zhang H; Wu S; Smirk RA; Bolton SA; Ryono DE; Zhang H; Lim NK; Chen BC; Locke KT; O’Malley KM; Zhang L; Srivastava RA; Miao B; Meyers DS; Monshizadegan H; Search D; Grimm D; Zhang R; Harrity T; Kunselman LK; Cap M; Kadiyala P; Hosagrahara V; Zhang L; Xu C; Li YX; Muckelbauer JK; Chang C; An Y; Krystek SR; Blanar MA; Zahler R; Mukherjee R; Cheng PT; Tino JA, Discovery of an oxybenzylglycine based peroxisome proliferator activated receptor alpha selective agonist 2-((3-((2-(4-chlorophenyl)-5-methyloxazol-4-yl)methoxy)benzyl)(methoxycarbonyl)amino)acetic acid (BMS-687453). J. Med. Chem 2010, 53, 2854–2864. [DOI] [PubMed] [Google Scholar]
- 22.Mizuno CS; Ma G; Khan S; Patny A; Avery MA; Rimando AM, Design, synthesis, biological evaluation and docking studies of pterostilbene analogs inside PPARalpha. Bioorg. Med. Chem 2008, 16, 3800–3808. [DOI] [PubMed] [Google Scholar]
- 23.Nomura M; Tanase T; Ide T; Tsunoda M; Suzuki M; Uchiki H; Murakami K; Miyachi H, Design, synthesis, and evaluation of substituted phenylpropanoic acid derivatives as human peroxisome proliferator activated receptor activators. discovery of potent and human peroxisome proliferator activated receptor alpha subtype-selective activators. J. Med. Chem 2003, 46, 3581–3599. [DOI] [PubMed] [Google Scholar]
- 24.Sierra ML; Beneton V; Boullay AB; Boyer T; Brewster AG; Donche F; Forest MC; Fouchet MH; Gellibert FJ; Grillot DA; Lambert MH; Laroze A; Le Grumelec C; Linget JM; Montana VG; Nguyen VL; Nicodeme E; Patel V; Penfornis A; Pineau O; Pohin D; Potvain F; Poulain G; Ruault CB; Saunders M; Toum J; Xu HE; Xu RX; Pianetti PM, Substituted 2-[(4-aminomethyl)phenoxy]-2-methylpropionic acid PPARalpha agonists. 1. Discovery of a novel series of potent HDLc raising agents. J. Med. Chem 2007, 50, 685–695. [DOI] [PubMed] [Google Scholar]
- 25.Xu HE; Lambert MH; Montana VG; Plunket KD; Moore LB; Collins JL; Oplinger JA; Kliewer SA; Gampe RT Jr.; McKee DD; Moore JT; Willson TM, Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao S; Kanno Y; Li W; Sasaki T; Zhang X; Wang J; Cheng M; Koike K; Nemoto K; Li H, Identification of Picrasidine C as a Subtype-Selective PPAR Agonist. J. Nat. Prod 2016, 79, 3127–3133. [DOI] [PubMed] [Google Scholar]
- 27.Schonherr H; Cernak T, Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem. Int. Ed. Engl 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- 28.Molina DM; Jafari R; Ignatushchenko M; Seki T; Larsson EA; Dan C; Sreekumar L; Cao Y; Nordlund P, Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [DOI] [PubMed] [Google Scholar]
- 29.Babic I; Kesari S; Nurmemmedov E, Cellular target engagement-a new paradigm in drug discovery. Future Medicinal Chemistry 2018, 10, 1641–1644. [DOI] [PubMed] [Google Scholar]
- 30.Zhang SX; Ma JX; Sima J; Y., C.; Hu MS; Ottlecz A; Lambrou GN, Genetic difference in susceptibility to the blood-retina barrier breakdown in diabetes and oxygen-induced retinopathy. Am J. Pathol 2005, 166, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.