

Abstract
Chemotherapy-induced cardiovascular toxicity (CICT) is a well-established risk for cancer survivors and causes diseases such as heart failure, arrhythmia, vascular dysfunction, and atherosclerosis. As our knowledge of the precise cardiovascular risks of each chemotherapy agent has improved, it has become clear that genomics is one of most influential predictors of which patients will experience cardiovascular toxicity. Most recently, GWAS-led, top-down approaches have identified novel genetic variants, and their related genes that are statistically related to CICT. Importantly, the advent of human induced pluripotent stem cell (hiPSC) models provides a system to experimentally test the effect of these genomic findings in vitro, query the underlying mechanisms, and develop novel strategies to mitigate the cardiovascular toxicity liabilities due to these mechanisms. Here we review the cardiovascular toxicities of chemotherapy drugs, discuss how these can be modeled in vitro, and suggest how these models can be used to validate genetic variants that predispose patients to these effects.
Keywords: human induced pluripotent stem cell, cardio-oncology, precision medicine, cancer, cardiotoxicity, vascular toxicity
Introduction
It is well-established that many cancer treatments are toxic to the cardiovascular system. Cardiovascular toxicity is seen with traditional chemotherapy agents such as anthracyclines, as well as newer targeted therapies such as trastuzumab, tyrosine kinase inhibitors, and immunotherapies. These toxicities lead to significant morbidity and mortality amongst cancer survivors. These effects can occur immediately after the first dose of the drug or take many years to manifest, potentially due to a reduction in baseline cardiovascular function that is then exacerbated with age (1,2). As cancer treatments have become more effective and overall lifespans increase, the number of cancer survivors has also increased. There are currently nearly 17 million cancer survivors in the United States alone, and this number is projected to increase significantly in the coming years (3). As cancer survivorship increases, addressing the impact of these cardiovascular toxicities on long-term health and quality of life becomes an increasingly pressing healthcare concern.
Though many risk factors for chemotherapy-induced cardiovascular toxicity (CICT) have been identified, such as dose, patient age, BMI, and preexisting cardiovascular health, differences in interindividual incidence of toxicity exist even when risk factors are taken into account (4). This unexplained interindividual variability suggests a genomic basis to this susceptibility (5). Understanding the genomic predisposition to CICT will provide clinical tools that can be used to suggest alternative therapies for patients in addition to elucidating the CICT mechanisms in order to improve drug development for adjuvant therapy and novel chemotherapeutics.
To date, significant resources have been invested in identifying relevant genomic variants through large cohort-based candidate gene association studies (CGAS) and genome-wide association studies (GWAS). These studies are limited, however, as they do not establish causality, often fail to be replicated, and frequently have insufficient sample size to draw statistically significant conclusions (6). Furthermore, the majority of CGAS and GWAS have focused on anthracycline-induced cardiotoxicity, which represents only a fraction of all CICTs.
Human induced pluripotent stem cells (hiPSCs) offer a high-throughput, patient-specific platform for the study of CICTs. hiPSCs have been shown to faithfully recapitulate clinical phenotypes, provide a platform for genomic variant discovery and validation, and allow for mechanistic investigations (7,8). That hiPSCs recapitulate clinical phenotypes confirms the role of genomics in patient-specific predisposition to CICTs and further supports their use as a model system for CICT research.
Types of cardiovascular toxicity caused by chemotherapy
CICT manifests in a variety of ways including heart failure, arrhythmia, vascular dysfunction, and atherosclerosis. The anthracycline doxorubicin is the most well studied (9,10) and causes dose-dependent cardiotoxicity (11–13). Cardiotoxic side effects experienced with doxorubicin range from asymptomatic increases in left ventricular wall stress, to reductions in left ventricular ejection fraction (LVEF), to arrhythmias and highly symptomatic congestive heart failure, often severe enough to warrant heart transplant (14–16). Doxorubicin cardiotoxicity occurs via a number of mechanisms, including reactive oxygen species (ROS) production, DNA damage, and mitochondrial dysfunction, all of which ultimately lead to programmed cell death (7).
Trastuzumab is another chemotherapeutic with a black box warning for ventricular dysfunction and congestive heart failure. Trastuzumab is a monoclonal antibody that targets the HER2 receptor and is used in the treatment of breast cancer and some gastric cancers (17). Trastuzumab cardiotoxicity has historically been viewed as less severe and more reversible than anthracycline-mediated cardiotoxicity, though this assertion has come into question more recently (17). The trastruzumab target HER2 (ERBB2) has an integral role in cardiac development, and its inhibition leads to impaired autophagy and accumulation of ROS, which have been implicated in the mechanism by which trastuzumab leads to heart failure (18).
Chemotherapeutics are also frequently associated with conduction abnormalities including QT prolongation, torsades de pointes, arrhythmias, and sudden death. Both nilotinib and vandetanib have FDA-issued black box warnings for QT prolongation and sudden death. Nilotinib is a tyrosine kinase inhibitor (TKI) designed to inhibit the BCR-ABL1 fusion protein in chronic myeloid leukemia (19). Vandetanib is a multikinase TKI used in treatment of multiple cancers (20). Nilotinib is believed to prolong the QT interval through inhibition of a specific potassium channel, while vandetanib inhibits multiple currents in the cardiac action potential in in vitro studies (19,20). In addition to chemotherapeutics with black box warnings, many additional chemotherapeutics, including TKIs, taxanes, anthracyclines, and cyclophosphamide, have been associated with arrhythmias and QT prolongation.
CICTs go beyond the heart itself and can have significant implications for vascular health. Both the angiogenesis inhibitor bevacizumab and the multikinase inhibitor cabozantinib carry black box warnings for risk of severe hemorrhage. Bevacizumab inhibits the vascular endothelial growth factor A (VEGFA) protein, while cabozantinib inhibits multiple kinases including the VEGF receptor subtypes VEGFR1, VEGFR2, and VEGFR3 (21). The exact mechanism by which inhibition of the VEGF pathway, which is critical in angiogenesis, increases hemorrhage risk is unclear but has been hypothesized to result from endothelial cell and platelet damage or dysfunction (22). Chemotherapeutics have also been associated with arterial thrombosis, which includes fatal myocardial infarction and stroke. Ponatinib, a TKI used for chronic myeloid leukemia, has a black box warning for this CICT. The mechanism by which ponatinib causes thrombosis remains under study, but kinase inhibition studies have shown that ponatinib inhibits multiple off-target kinases, including those with critical roles in maintenance of vascular health (23).
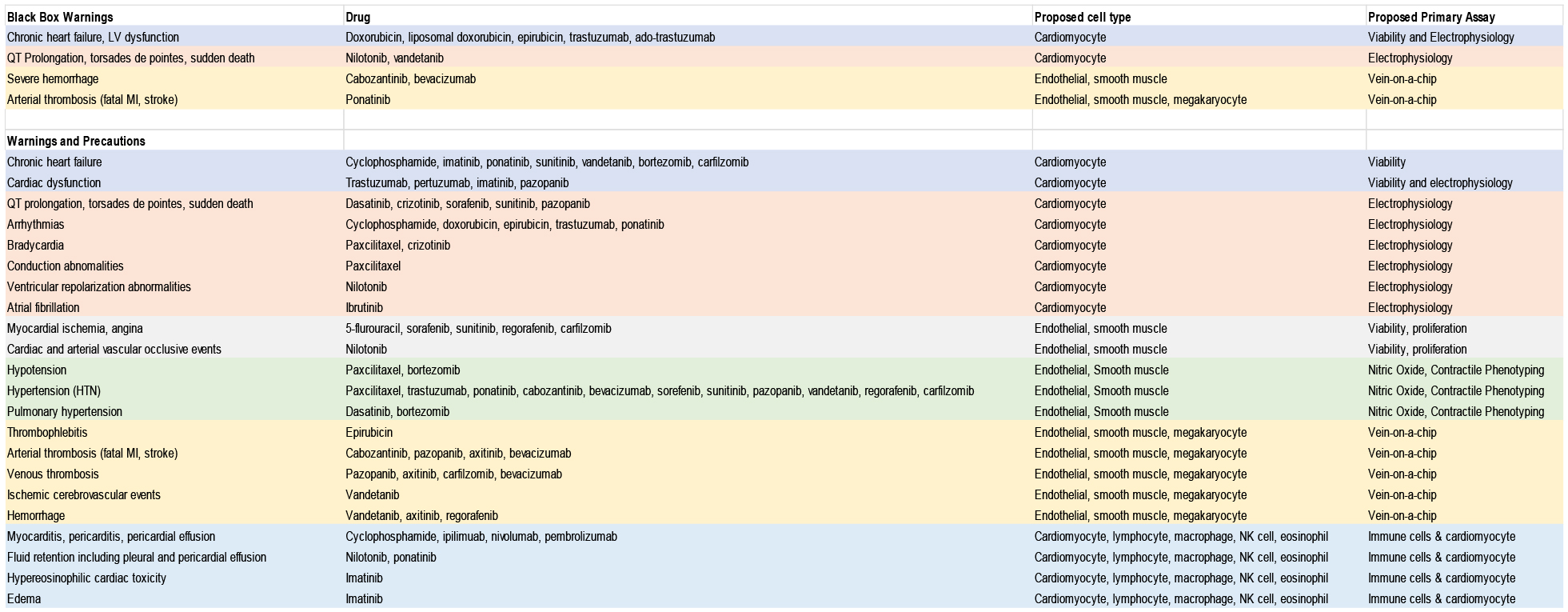
In addition to black box warnings for CICTs, FDA drug labels denote numerous additional cardiovascular toxicities associated with a number of chemotherapeutics. These CICTs include heart failure, arrhythmias, bradycardia, atrial fibrillation, angina, edema, hemorrhage, hypotension, hypertension, thrombophlebitis, arterial and venous thrombosis, ischemic cerebrovascular events, myocarditis, pleural and pericardial effusion, hypereosinophilic cardiotoxicity, and more. CICTs associated with specific chemotherapeutics are summarized in Table 1.
Table 1:
Description of in vitro assays that could be used for validation of SNPs associated with chemotherapy-induced cardiotoxicity. Data from drugs.fda.gov
|
Beyond the adverse effects of individual agents, CICTs are exacerbated in the context of multiple chemotherapeutics. For example, more CICTs are observed in the context of taxane treatment coadministered with HER2 inhibitors and/or anthracyclines, compared to treatment with taxanes alone (24,25). The breadth of CICTs caused by numerous chemotherapeutics and the long-term health consequences of these CICTs for cancer survivors has led to the emergence of the field of cardio-oncology. In order to move this field forward, it is crucial that we develop tools to understand who is susceptible to CICTs, determine the mechanism of action of CICTs, and identify novel chemotherapeutics and adjuvant therapies in order to prevent cardiovascular damage.
Pharmacogenomics of CICT
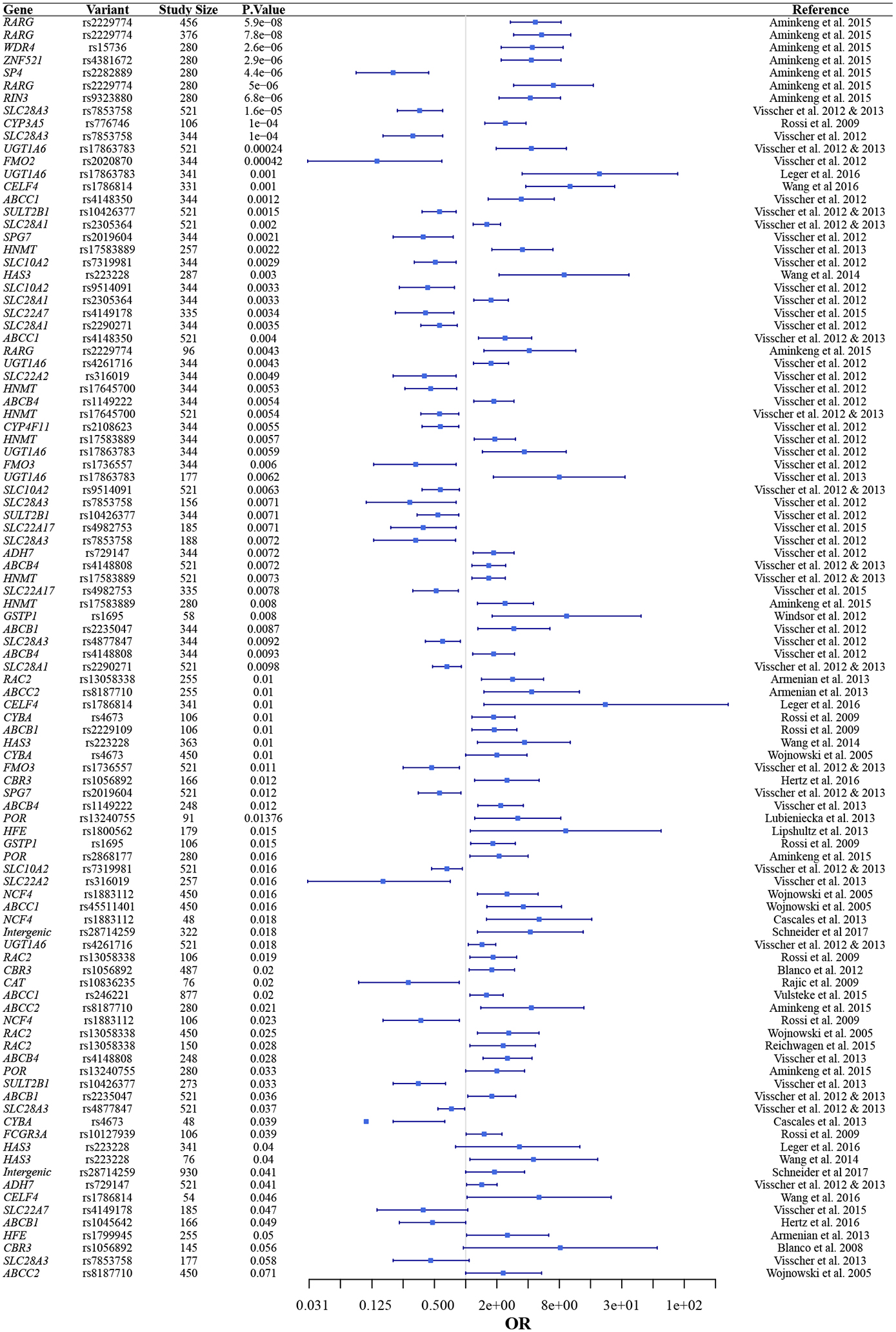
Because CICTs affect only a subset of patients and do not uniformly segregate with cardiovascular risk factors, numerous CGAS and GWAS studies have been conducted to identify potential genetic contributors to susceptibility. To date, the majority of this research has focused on anthracycline-induced cardiotoxicity. The approximately 40 CGAS and 4 GWAS that have been performed have identified a number of SNPs that are statistically correlated with DIC (26) (see Table 2). Many of these studies have included a small numbers of SNPs (i.e. <5). In these studies there is variation in the definition of cardiotoxicity, recruitment methodologies, and composition of the patient populations that likely is responsible for much of the variation of the results. Many of these studies were limited by small sample size, some including as few as seven patients with toxicity (27) with even the most thorough including only 169 toxicity patients (28). Because of these limitations, studies rarely replicate one another’s findings (29–40).
Table 2.
Forest plot showing the top 100 significant SNPs associated with CICT
|
The largest CGAS and GWAS were both multi-center programs (41,42). The first study was candidate gene-based with a 344-patient cohort and the second was genome-wide with a cohort of 562. These studies have demonstrated an association between two SNPs and a dramatically altered risk of anthracycline cardiotoxicity in both original and replication cohorts: one, rs7853758 (L461L), in SLC28A3, a concentrative transporter, and one, rs2229774 (S427L), in RARG, which encodes retinoic acid receptor–γ.
A small number of studies have examined the genomics of CICTs for non-anthracycline chemotherapeutics. Multiple CGAS, one exome association study, and one GWAS have been conducted for trastuzumab-induced cardiotoxicity. CGAS studies have consistently identified one of two variants in the trastuzumab target ERBB2, but variants in this gene were not identified in either the exome association or GWAS studies (43–50).
CGAS have also been conducted for vascular CICTs. Two CGAS have evaluated variants associated with venous thromboembolism with immunomodulator therapy and one CGAS assessed the association of a specific variation in the VEGFA untranslated region with bevacizumab-induced thrombo-hemorrhagic events (51–53).
These association studies provide clues regarding potential predictors and mechanisms of CICTs. However, they are subject to a number of limitations. Large cohorts are often required to achieve statistically significant results, and many studies are limited by small sample size (6,54). Additionally, as illustrated by genomic studies of trastuzumab-induced cardiotoxicity, studies may fail to replicate one another, leading to uncertainty regarding which variants to prioritize. Even when GWAS studies identify significant variants that remain significant in replication cohorts, the implications of the identified variant may still be unclear. For example, the rs7853758 variant in SLC28A3 was identified as significantly associated with anthracycline induced cardiotoxicity in both discovery and replication cohorts (41,55). However, this variant encodes a synonymous mutation, so it is likely that this variant is in linkage disequilibrium with the true causal variant, which is yet to be identified. When considering anthracycline-induced CICTs alone almost 50 studies have been conducted, comprising thousands of patients, and we are still without a single validated causal variant. Furthermore, anthracyclines are only one class of chemotherapeutics associated with CICTs. There have been no genomic studies on the majority of CICTs.
Genomic association studies conducted to date have provided important evidence to suggest a genomic contributor to CICT susceptibility. For a subset of chemotherapeutics these studies have identified potential variants to focus future mechanistic and variant validation research. However, the limitations of association studies and the breadth of chemotherapeutics that are currently unstudied point to the need for additional research methods to contribute to the identification of variants, validate a causal role for identified variants, and conduct mechanistic studies.
Model Systems for CICT
Zebrafish have been used as a model system in multiple avenues of cardiovascular research, including CICTs. Zebrafish offer a high-throughput, low cost system to study cardiovascular effects of medications, and have been well-characterized as a model system (56). In a study of anthracycline-induced cardiotoxicity, zebrafish displayed incomplete heart development, pericardial edema, and bradycardia in response to doxorubicin (57). While zebrafish in this study displayed an adverse, dose-dependent cardiovascular phenotype, it is important to note that the manifestation of this dysfunction in the zebrafish model differs significantly from the LV dysfunction and heart failure observed in patients, and thus underscores a limitation of this model. Additionally, zebrafish are not suited to patient-specific studies. Differences between the zebrafish and human genomes significantly are significantly larger than the interindividual variability that determines drug response (56).
Animal models have also been employed for CICT research and have successfully identified mechanistic contributors to CICTs. For example, Miranda et al. (58) validated the role of iron in doxorubicin cardiotoxicity by showing that HFE knockout mice exhibited greater sensitivity to doxorubicin in cardiac assays. Zhang et al. (59) validated TOP2β inhibition as a key mechanistic contributor to doxorubicin-induced cardiotoxicity in a mouse model with cardiac-specific deletion of TOP2B, where knockout mice exhibited minimal cardiotoxicity in response to doxorubicin treatment. While studies like these demonstrate the utility of animal models, particularly with respect to mechanistic studies, animal models are expensive, low-throughput, and inherently limited by interspecies variability.
hiPSCs address the limitations of previous models and offer a high-throughput, patient-specific platform to study CICT. hiPSCs provide a non-invasive, renewable source of patient-specific cells that might otherwise be difficult to obtain or limited by low passage number in culture (60). hiPSCs retain a patient’s genome and are thus suited to pharmacogenomic studies of interindividual variability in drug response (60). Additionally, hiPSCs are amenable to straightforward, specific genomic manipulation through technologies such as CRISPR/Cas9 (61,62). Genome editing facilitates mechanistic investigations, variant validation, and generation of isogenic controls in hiPSC systems.
While hiPSCs offer many advantages, important limitations exist which include maturity status of the differentiated cells, requisite time and labor, subtype specification, and limited integration of multiple tissue types. Current differentiation protocols for hiPSC-derived cardiomyocytes (hiPSC-CMs) result in cells that are more phenotypically similar to fetal rather than adult CMs (63). This presents a potential concern for accurately modeling phenotypes of the adult heart, and efforts to improve maturation status of hiPSC-CMs are ongoing. hiPSC reprogramming, culture, and differentiation require an investment of time and resources. While this does not represent a significant barrier for established hiPSC laboratories, large-scale hiPSC research may not be feasible for some groups. Subtype specification is also an ongoing effort in the field of hiPSCs; for hiPSC-CMs this involves specification to atrial versus ventricular cells and in vascular hiPSC models this includes specification between arterial and venous endothelial cells (64,65). For certain CICTs subtype specification may be crucial, as demonstrated by a study from Shafaattalab et al. (66) where ibrutinib, a drug associated with atrial fibrillation, induced a phenotype in atrial but not ventricular hiPSC-CMs. hiPSC models often include a single cell type and consequently may not capture information about interactions among tissue types, though this is addressed to some extent by co-culture models. Even with co-culture however, hiPSC models may not adequately capture phenotypes that result from structural features of tissues rather than phenotypes of individual cells. Efforts to address these limitations include the development of hiPSC-derived organoids and engineered heart tissue (67,68).
Despite these limitations, hiPSC models have been shown to recapitulate patient-specific disease. Inherited arrhythmias including Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), arrhythmia-associated calmodulinopathies, and long QT syndrome (LQTS) have been recapitulated at the cellular level in patient-specific hiPSC-CMs (69–72). Furthermore, genome editing of the variant of interest in these studies rescued the phenotype and provided validation of mechanistic contributors and causal variants for these conditions (70–72).
hiPSC confirm that chemotherapy-induced cardiovascular toxicity is a genomic disease
Multifactorial traits such as drug response are invariably more complex to model than the monogenic conditions described above. Despite this increased complexity,, multiple studies have demonstrated the ability of hiPSCs to recapitulate patient-specific drug response phenotypes. Recent work using hiPSC-CMs has shown that these cells accurately recapitulate an individual patient’s predilection to doxorubicin induced cardiotoxicity (7). Briefly, hiPSC-CMs from breast cancer patients who developed doxorubicin-induced cardiotoxicity recapitulate that increased risk in vitro, with decreased cell viability, metabolic function, contraction, sarcomeric structure, calcium handling, and increased ROS production when exposed to doxorubicin, compared to hiPSC-CMs from patients who were treated with doxorubicin but did not experience cardiotoxicity (7).
Patient-specific hiPSC-CMs have also been shown to recapitulate susceptibility to trastuzumab-induced cardiac dysfunction. Trastuzumab-exposed hiPSC-CMs demonstrated impaired contractility and calcium handling, and hiPSC-CMs from patients with severe cardiac dysfunction after trastuzumab treatment were more sensitive to the drug effects in vitro compared to hiPSC-CMs from trastuzumab-treated patients without cardiac dysfunction (73). The authors identified metabolic dysfunction in response to trastuzumab and suggested that therapies directed at normalizing metabolism may be promising adjuvant treatments (73). Importantly, hiPSC-CMs did not display differences in cell death or sarcomeric disorganization, which distinguishes this in vitro cardiotoxicity phenotype from that seen with doxorubicin (7,73). This difference mirrors the differences observed clinically in the persistence of doxorubicin-versus trastuzumab-induced heart failure after treatment cessation (74).
These patient-specific hiPSC studies demonstrate that the toxicity observed in vitro correlates with clinically relevant in vivo toxicity. To date, a priori screening in hiPSCs to predict who will develop toxicity has not been attempted, though the above studies suggest that results would correlate. Important limitations exist to the implementation of patient-specific hiPSCs for clinical screening. The reprogramming process takes several months and requires a substantial investment of labor. In addition to the potential expense of this process, patients with cancer may not reasonably be able to delay treatment for this amount of time. Thus, patient-specific hiPSC models for screening may be most high-yield in patients with rare diseases or for severe potential side effects in patients who have the ability to delay treatment. Where patient-specific hiPSC models have the potential to impact cardio-oncology more broadly is through their ability to confirm genomic underpinnings of CICTs. Patient-specific in vitro phenotyping coupled with genome sequencing and genome editing techniques can be used to identify and validate key pharmacogenomic variants that could be incorporated into a clinical screen to reduce CICTs on a larger scale and target future drug discovery efforts to bypass adverse events.
In addition to patient-specific studies, many studies have been conducted with control line hiPSCs to characterize the effects and probe the mechanisms of various CICTs. For example, Maillet et al. (8) characterized the response of control line hiPSC-CMs to doxorubicin and then used CRISPR/Cas9 to generate a TOP2B knockout line. The authors were able to demonstrate that knockout significantly reduced hiPSC-CM sensitivity to doxorubicin, corroborating previous animal studies (8). In another study, Kurokawa et al. (75) used control hiPSC-CMs to determine that trastuzumab is only cardiotoxic when ERBB2/4 signaling is activated in response to cellular stress, thus further elucidating the mechanism of this CICT. Sharma et al. (76) utilized non-patient-specific hiPSC-CMs and hiPSC-derived endothelial cells to characterize the cardiovascular toxicity of 21 TKIs. From these in vitro studies the authors were able to develop a “cardiac safety index” that corresponded with clinical phenotypes and could be used in future drug screening. These studies demonstrate the utility of hiPSCs for both patient-specific recapitulation (and consequently pharmacogenomics) as well as mechanistic research.
In vitro modeling of toxicity caused by chemotherapy drugs
The initial step in creation of any hiPSC CICT model is identification of the appropriate cell type(s) with which to conduct in vitro studies. In vivo phenotypes are complex and involve multiple cell types. For example, while in vitro studies with doxorubicin have focused on cardiomyocytes, cardiac progenitor cells, fibroblasts, and vascular cells may all play a role in the development of cardiotoxicity (77). However, hiPSC-CMs in the absence of other cell types have still proven sufficient for identifying patient-specific differences in drug susceptibility and elucidating mechanistic contributors.
With the chosen cell type the next key consideration for an hiPSC model is identification of an assay that is an appropriate readout for the given phenotype. Multiple assays are often used in conjunction with one another, as drugs may affect multiple facets of cellular health and function and identification of a phenotype across multiple assays increases confidence in the results. In the case of doxorubicin, effects can be seen across cell morphology, viability, apoptosis, ROS production, metabolic function, and calcium handling (7,8). While multiple assays can detect a phenotype with doxorubicin, the viability assay alone is sufficient to detect pharmacogenomic differences in CICT susceptibility and to identify key mechanistic contributors. Identification of a primary assay, such as viability, is necessary in order to evaluate multiple genomic or mechanistic contributors in a high-throughput manner, as all conditions can then be compared with a single readout. Identification of a primary assay also allows studies to focus on a specific output parameter, from which hypothesized effect size and consequently necessary sample size can be determined. Additional phenotypic assays can then be used to augment the primary assay and corroborate that assay, where for example, an apoptosis assay can corroborate a viability assay, or provide additional information about the phenotype. For instance, an ROS assay may provide clues about the pathways activated by a drug that ultimately causes cell death.
Identification of an assay that effectively distinguishes between affected and unaffected patients and shows a dose-response relationship with the drug of interest is a necessary early step in any hiPSC CICT investigation. Even in cases where CICT phenotypes in patients may appear similar, such as doxorubicin- and trastuzumab-induced cardiotoxicity, the primary assay to distinguish the phenotype in vitro may differ. For example, a patient-specific difference in CICT susceptibility to doxorubicin could be detected in hiPSC-CMs with a viability assay (7). Trastuzumab, on the other hand, induced no patient-specific differences in viability but instead the in vitro phenotype could be distinguished by calcium handling (73).
The majority of CICTs have yet to be studied in an hiPSC model. In the following section we outline potential cell types and assays of interest that may further research in these areas. These proposed model systems are outlined in Table 1 and summarized in Figure 1.
Figure 1.
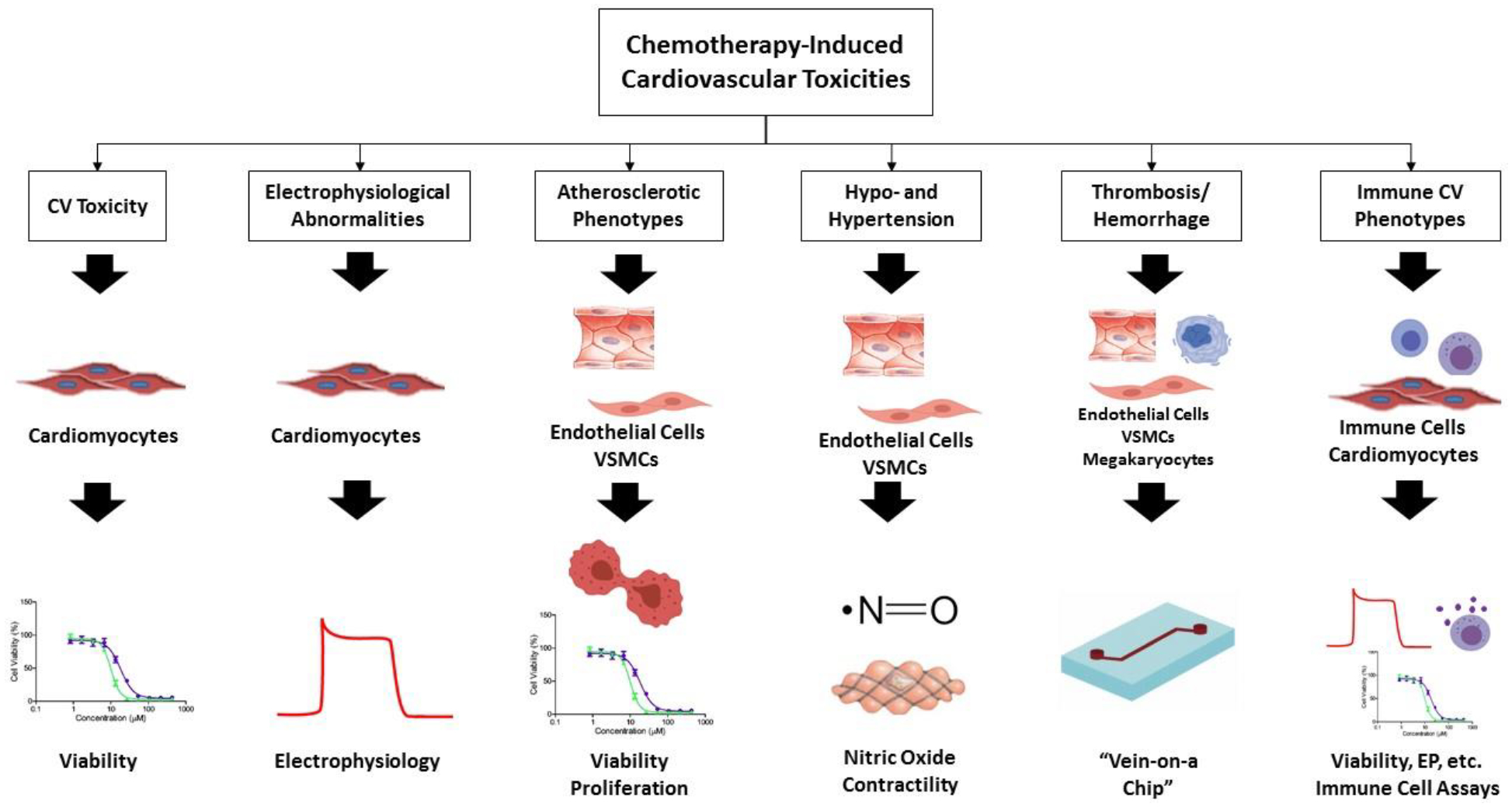
A graphical depiction of the proposed workflow for hiPSC CICT modeling organized by CICT of interest.
Cardiomyocyte Toxicity
Multiple chemotherapeutics carry either black box or general warnings of a risk of heart failure or cardiac dysfunction. In these cases one of the most straightforward assays to assess for potential use as a primary assay is viability. There are numerous ways to quantify viability but two of the most common are ATP detection luminescence assays or resazurin-based fluorescence assays (7,76,78,79). These assays use cellular metabolic function as a proxy for the number of viable cells. While there is generally a strong correlation between the level of ATP or oxidation/reduction and the number of live cells, this linear relationship is contingent on unchanged levels of metabolic activity per cell. Drugs that alter metabolic activity at the cellular level have the potential to confound interpretation of these assays (80). Fluorescence-based assays must also account for any inherent fluorescence in the drug of interest. Other assays for viability include water-soluble tetrazolium salt or cell counts that exclude nonviable cells (76,81).
Apoptosis assays are complementary to and may augment viability assays. Apoptosis assays include luminescence-based caspase assays, propidium iodide and annexin V staining, and lactate dehydrogenase or troponin release assays (7,78,79,81). These assays have specific strengths and shortcomings, and ultimately the choice of assay is contingent on the objective of the given study. For example, caspase assays assume a direct relationship between caspase activation and apoptosis, which is not always the case (82). Assays contingent on release of intracellular contents capture late stages of apoptosis and may fail to detect earlier indicators of dysfunction.
Electrophysiological Abnormalities
A significant number of chemotherapy drugs are associated with conduction abnormalities and various arrhythmias. Given the large number of arrhythmic/QT prolongation events that have been identified in approved drugs only in the post-marketing phase, drug screening in hiPSC-CMs is now required in the Comprehensive In Vitro Proarrhythmia Assay (CiPA) guidelines for cardiac drug development (83). The ability of hiPSC-CMs to recapitulate chemotherapy-specific arrhythmias was recently demonstrated in a study of ibrutinib-induced atrial fibrillation. Shafaattalab et al. (66) showed that atrial-specific hiPSC-CMs have abnormalities in voltage and calcium transients in response to ibrutinib, and this response is not seen with other drugs in the same family that are not associated with atrial fibrillation. This study demonstrates the potential of hiPSC models to capture electrophysiological phenotypes in vitro.
Multiple methodologies can be applied to assess the electrophysiological properties of hiPSC-CMs in response to drug exposure. Patch clamping is the gold standard in electrophysiology and has successfully been used to assess drug response in hiPSC-CMs (76). Higher throughput techniques include multielectrode arrays as well as voltage and calcium handling assays (84,85). These higher throughput techniques are not as informative with respect to certain parameters, such as upstroke velocity and resting membrane potential, but allow for longer-term electrophysiological assessment and substantially higher throughput than patch clamp techniques (84). Calcium and voltage sensors can either be introduced into hiPSC systems as exogenous dyes or genetically encoded reporters (7,86). In the case of genetically-encoded reporters, the reporter itself may interfere with calcium channel gating and signaling, though newer sensors are being developed to address this issue (87).
Atherosclerotic Phenotypes
The majority of CICT research to date has focused on cardiomyocyte-specific phenotypes; however, a significant number of chemotherapeutics are associated with vascular adverse events such as vascular occlusion and myocardial ischemia. These vascular phenotypes are in the atherosclerosis family and may involve two key cell types: endothelial cells (ECs) and vascular smooth muscle cells (VSMCs). EC damage is a nidus for lipid accumulation in atherosclerosis (88). In vitro EC models have demonstrated drug-specific EC toxicity and impaired angiogenesis with atherosclerosis-associated chemotherapeutics such as nilotinib (89). Atherosclerosis also involves a contractile to synthetic phenotypic switch in VSMCs that leads to extracellular matrix production and contributes significantly to vascular occlusion (90,91). Patient-specific hiPSC-VSMCs from patients with a genetic predisposition to coronary artery disease, a subtype of atherosclerosis, demonstrate altered contractility, proliferation, and migration in vitro (90).
In these cell types, viability assays can be used to assess for toxicity and are similar to those listed above for cardiomyocytes. Proliferation assays in ECs would be expected to correlate directly with EC health as proliferation is a key component in angiogenesis and vascular homeostasis (89). In VSMCs, increased proliferation would be a pathologic marker associated with the contractile to synthetic phenotypic shift observed in atherosclerosis (91). In dividing cells such as ECs and VSMCs, many viability assays can also serve as proliferation assays. In addition to the assays described above for cardiomyocyte viability, proliferation assays such as 5-bromo-2’-deoxyuridine (BrdU), 5-ethynyl-2´-deoxyuridine (EdU), or 3H-thymidine nucleoside incorporation assays, which may employ radioactive, fluorescent, or copper-catalyzed reactions for signal detection (91,92) can be used.
Migration assays can similarly be used to assess EC and VSMC phenotypes, where decreased migration is considered pathologic in ECs and increased migration is pathologic in VSMCs, consistent with the phenotypic switch (89,91). Migration can be measured through scratch assays as well as cell exclusion assays, where cells are plated in a chamber with a removable barrier. In ECs, tube formation assays are often used, as aspects of both migration and proliferation are captured in this assay of angiogenic function (89,93).
Hypo- and Hypertensive Phenotypes
Dasatinib and bortezomib are associated with development of pulmonary arterial hypertension. Pulmonary arterial hypertension involves breakdown of lung vasculature and consequently hiPSC-ECs have been incorporated into studies of this condition (94). hiPSC-ECs from patients with both heritable and idiopathic pulmonary hypertension displayed reduced adhesion, migration, tube formation, and survival compared to control subjects (94).
Numerous other chemotherapeutic drugs impact vascular tone more generally, resulting in either hypo- or hypertension. Vascular tone is regulated by multiple factors including EC nitric oxide (NO) production, VSMC contractility, and overall vascular stiffness (95–97). As in vitro models for generalized hypertension and hypotension are sparse, the appropriate assay to assess this phenotype remains to be determined. From the physiology of vascular tone regulation, we propose some potential assays below.
NO is produced and released by ECs and acts on VSMCs to increase cyclic guanosine monophosphate (cGMP) signaling, causing smooth muscle relaxation and a reduction in blood pressure (98). Intracellular NO in ECs can be quantified with the fluorescent dye 4-Amino-5 Methylamino-2’,7’-Difluorofluorescein Diacetate (DAF-FM) (99). When NO is released into cell culture media, it is quickly converted to nitrate and nitrite (100). Nitrite can be detected with the Griess reagent. To assess total NO production, nitrate can be converted to nitrite with nitrate reductase and subsequently quantified by the Griess assay (100).
VSMC contractility can be assessed with time-lapse and traction force microscopy to evaluate the contractile response to agents such as carbachol, angiotensin II, and endothelin I (92,101–103). VSMC contractility, in addition to vascular stiffness, can be affected by the contractile to synthetic phenotypic shift of VSMCs, which can be assessed through proliferation, migration, and extracellular matrix production assays as described above (91).
Thrombosis and Hemorrhage
Thrombosis is associated with a subset of chemotherapeutic drugs and can manifest as thrombophlebitis, arterial or venous thrombosis, or ischemic cerebrovascular events. The parallel plate flow chamber is a commonly used in vitro thrombosis system, and contains a hollow rectangular space with millimeter or centimeter scale dimensions, through which blood or its components can be transported to induce physiological wall shear stresses over purified proteins (such as, von Willebrand factor or tissue factor), extracellular matrix (collagen, laminin or fibronectin), or EC monolayers (104,105). Another in vitro approach to thrombosis involves microfluidic devices, which more accurately replicate blood vessel anatomy. A major advantage of microfluidic devices is that different anatomical structures (e.g. atherosclerotic plaques, bifurcations etc.) and complex flow patterns (pulsatile motion, rapid accelerations and decelerations etc.) observed in vivo can be included in these models. These systems have successfully been used to model the process of thrombosis in various diseases (106–109). A recent study demonstrated the potential to create microfluidic devices that exclusively utilize patient-derived blood outgrowth endothelial cells. These tissue-engineered blood vessels exhibit normal physiological function from healthy individuals. However, those produced with cells obtained from diabetic pigs show the diabetic phenotype in vitro, demonstrating the ability of these systems to model patient-specific features (110). Incorporation of hiPSCs into these models would allow for inclusion of multiple patient-specific cell types (some of which would otherwise be difficult to obtain) in order to investigate factors that contribute to patient-specific thrombosis in the context of chemotherapeutics.
While many chemotherapeutics are associated with thrombosis, drugs such as vandetanib, axitinib, and regorafenib, are associated with hemorrhage. In the context of bleeding disorders, microfluidic systems successfully recapitulate the clotting dysfunction and propensity towards bleeding, suggesting these systems are useful for the evaluation of both thrombosis and hemorrhage (109).
Immune-related CICTs
Immune-related CICTs occur with a variety of chemotherapeutics and manifest in myocarditis, pericarditis, pericardial and/or pleural effusion, edema, and hypereosinophilic cardiac toxicity. To date the majority of work in these conditions has been conducted in animal models (111). Human models of immune-related CICTs would be informative and bypass interspecies differences observed in animal models. However, the use of in vitro cell models for these conditions has been limited. Sharma et al. (112) developed an hiPSC-CM model of myocarditis caused by coxsackievirus. Infected hiPSC-CMs displayed erratic beating with eventual cessation of beating. Responses to known antiviral compounds in this in vitro model were consistent with the known effects of these drugs in vivo (112). Drug-induced immune CICTs often result from activation of autoimmunity (113). hiPSCs have successfully been incorporated into investigations of multiple autoimmune diseases and likely hold promise for drug-induced immune phenotypes as well (114,115). We are not aware of any current hiPSC models of drug-induced immune phenotypes, but future models would likely include the relevant tissue type (e.g. hiPSC-CMs for myocarditis) as well as relevant hiPSC-derived immune cells. Assays would include those related to the tissue type (e.g. viability, electrophysiology, etc. for hiPSC-CMs) as well as immune-cell-specific assays such as cytokine release, migration, and proliferation (116,117).
hiPSCs to Advance CICT Pharmacogenomics
Establishment of the appropriate hiPSC model system and assay(s) creates a platform with a defined readout for more and less susceptible CICT phenotypes. With this system established, whole genes and individual variants can be manipulated to determine how they affect this defined readout.
hiPSCs have confirmed a genomic contribution to CICT susceptibility. A major obstacle in CICT pharmacogenomics to date is that genome association studies are hindered by multiple shortcomings related to sample size, replication, and distinction of causality from correlation. hiPSCs are well-positioned to address this gap. hiPSCs can be easily modified through genome editing approaches to introduce or remove both whole genes and specific variants. Through genomic editing, hiPSCs allow for validation of significant variants identified in CGAS and GWAS studies. This approach has been used successfully to validate GWAS variants in the context of metabolic disease (118). Additionally, hiPSC models can contribute to novel variant discovery (119,120).
Beyond traditional CGAS and GWAS studies, more recent studies have examined the association between the genome and mRNA levels, themselves quantitative traits, in expression quantitative trait locus (eQTL) mapping (121–123). Traditional eQTL analysis has been performed on primary patient samples (~60), comparing gene expression with genotypes. hiPSC models permit comparison of gene expression both with and without the chemotherapeutic of interest to establish differential eQTL (deQTL) related to the variation between patients in the gene expression response to drug. Using primary patient samples, the drug and disease status of an individual could confound the association of gene expression and their genotype. hiPSCs are not influenced by a patient’s disease status and would thus allow for a true recapitulation of the effect of their genetic variation. Though deQTL mapping has only been demonstrated a small number of times (124,125), these studies have confirmed that deQTL mapping is uniquely powerful and that a majority of genome-wide findings would not have been uncovered via analysis of untreated (i.e. non-differential) samples.
Conclusions and Future Directions
Chemotherapeutics are increasingly effective at allowing patients to outlive their cancer. As the number of cancer survivors increases, we are developing a greater realization of the long term adverse cardiovascular effects of some of these treatments. Understanding who is most susceptible to these adverse effects and the mechanisms by which they occur will allow providers to select alternative medications (where possible) and will promote drug discovery with respect to both adjuvant protective therapies and novel cancer therapies with fewer cardiovascular effects.
Researchers have invested substantial resources into chemotherapy side effect pharmacogenomics, but we are still without consensus regarding which variants are critical in determining patient susceptibility, and the vast majority of chemotherapeutics have yet to be studied. Additionally, the mechanisms of many side effects remain unexplained, which impairs our ability to identify potential protective strategies.
hiPSC models represent a path forward for CICT research. By providing a high-throughput, patient-specific system that is amenable to genome editing, hiPSCs will allow the field to model numerous CICTs, enabling us to better understand these side effects and identify susceptible patients in order to avert long-term cardiovascular health effects of cancer treatment.
As the field of cardio-oncology continues to grow, hiPSCs will be an indispensable tool to study drug effects and develop targeted solutions that ultimately translate into more effective patient care.
Funding:
This study was funded by NIH NCI grant R01 CA220002, American Heart Association Transformational Project Award 18TPA34230105 and the Fondation Leducq (P.W.B).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest: The authors declare they have no conflict of interest.
References
- 1.Mishra T, Shokr M, Ahmed A, Afonso L. Acute reversible left ventricular systolic dysfunction associated with 5-fluorouracil therapy: a rare and increasingly recognised cardiotoxicity of a commonly used drug. BMJ Case Rep 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volkova M, Russell R 3rd. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev 2011;7:214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Cancer Institute. Statistics. 2019.
- 4.Lotrionte M, Biondi-Zoccai G, Abbate A et al. Review and meta-analysis of incidence and clinical predictors of anthracycline cardiotoxicity. Am J Cardiol 2013;112:1980–4. [DOI] [PubMed] [Google Scholar]
- 5.Magdy T, Burridge PW. The future role of pharmacogenomics in anticancer agent-induced cardiovascular toxicity. Pharmacogenomics 2018;19:79–82. [DOI] [PubMed] [Google Scholar]
- 6.Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet 2003;361:865–72. [DOI] [PubMed] [Google Scholar]
- 7.Burridge PW, Li YF, Matsa E et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med 2016;22:547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maillet A, Tan K, Chai X et al. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci Rep 2016;6:25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudson MM, Ness KK, Gurney JG et al. Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 2013;309:2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Dalen EC, Raphael MF, Caron HN, Kremer LC. Treatment including anthracyclines versus treatment not including anthracyclines for childhood cancer. Cochrane Database Syst Rev 2014:CD006647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 1973;32:302–14. [DOI] [PubMed] [Google Scholar]
- 12.Von Hoff DD, Layard MW, Basa P et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 1979;91:710–7. [DOI] [PubMed] [Google Scholar]
- 13.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 2003;97:2869–79. [DOI] [PubMed] [Google Scholar]
- 14.Shakir DK, Rasul KI. Chemotherapy induced cardiomyopathy: pathogenesis, monitoring and management. J Clin Med Res 2009;1:8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein D, Burridge P. Patient-Specific Pluripotent Stem Cells in Doxorubicin Cardiotoxicity: A New Window Into Personalized Medicine. Prog Pediatr Cardiol 2014;37:23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipshultz SE, Cochran TR, Franco VI, Miller TL. Treatment-related cardiotoxicity in survivors of childhood cancer. Nat Rev Clin Oncol 2013;10:697–710. [DOI] [PubMed] [Google Scholar]
- 17.Mohan N, Jiang J, Dokmanovic M, Wu WJ. Trastuzumab-mediated cardiotoxicity: current understanding, challenges, and frontiers. Antibody therapeutics 2018;1:13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohan N, Shen Y, Endo Y, ElZarrad MK, Wu WJ. Trastuzumab, but Not Pertuzumab, Dysregulates HER2 Signaling to Mediate Inhibition of Autophagy and Increase in Reactive Oxygen Species Production in Human Cardiomyocytes. Mol Cancer Ther 2016;15:1321–31. [DOI] [PubMed] [Google Scholar]
- 19.Xu Z, Cang S, Yang T, Liu DJHR. Cardiotoxicity of tyrosine kinase inhibitors in chronic myelogenous leukemia therapy. 2009;1. [Google Scholar]
- 20.Lee HA, Hyun SA, Byun B, Chae JH, Kim KS. Electrophysiological mechanisms of vandetanib-induced cardiotoxicity: Comparison of action potentials in rabbit Purkinje fibers and pluripotent stem cell-derived cardiomyocytes. PLoS One 2018;13:e0195577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacal PM, Graziani G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol Res 2018;136:97–107. [DOI] [PubMed] [Google Scholar]
- 22.Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens 2018;12:409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moslehi JJ, Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol 2015;33:4210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giordano SH, Booser DJ, Murray JL et al. A detailed evaluation of cardiac toxicity: a phase II study of doxorubicin and one- or three-hour-infusion paclitaxel in patients with metastatic breast cancer. Clin Cancer Res 2002;8:3360–8. [PubMed] [Google Scholar]
- 25.Pentassuglia L, Timolati F, Seifriz F, Abudukadier K, Suter TM, Zuppinger C. Inhibition of ErbB2/neuregulin signaling augments paclitaxel-induced cardiotoxicity in adult ventricular myocytes. Exp Cell Res 2007;313:1588–601. [DOI] [PubMed] [Google Scholar]
- 26.Magdy T, Burmeister BT, Burridge PW. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol Ther 2016;168:113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Tokhy MA, Hussein NA, Bedewy AM, Barakat MR. XPD gene polymorphisms and the effects of induction chemotherapy in cytogenetically normal de novo acute myeloid leukemia patients. Hematology 2014;19:397–403. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Liu W, Sun CL et al. Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: a report from the children’s oncology group. J Clin Oncol 2014;32:647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanco JG, Leisenring WM, Gonzalez-Covarrubias VM et al. Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer 2008;112:2789–95. [DOI] [PubMed] [Google Scholar]
- 30.Chugh R, Griffith KA, Davis EJ et al. Doxorubicin plus the IGF-1R antibody cixutumumab in soft tissue sarcoma: a phase I study using the TITE-CRM model. Ann Oncol 2015;26:1459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reichwagen A, Ziepert M, Kreuz M et al. Association of NADPH oxidase polymorphisms with anthracycline-induced cardiotoxicity in the RICOVER-60 trial of patients with aggressive CD20(+) B-cell lymphoma. Pharmacogenomics 2015;16:361–72. [DOI] [PubMed] [Google Scholar]
- 32.Reinbolt RE, Patel R, Pan X et al. Risk factors for anthracycline-associated cardiotoxicity. Support Care Cancer 2016;24:2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vivenza D, Feola M, Garrone O, Monteverde M, Merlano M, Lo Nigro C. Role of the renin-angiotensin-aldosterone system and the glutathione S-transferase Mu, Pi and Theta gene polymorphisms in cardiotoxicity after anthracycline chemotherapy for breast carcinoma. Int J Biol Markers 2013;28:e336–47. [DOI] [PubMed] [Google Scholar]
- 34.Barac A, Lynce F, Smith KL et al. Cardiac function in BRCA1/2 mutation carriers with history of breast cancer treated with anthracyclines. Breast Cancer Res Treat 2016;155:285–93. [DOI] [PubMed] [Google Scholar]
- 35.Cascales A, Sanchez-Vega B, Navarro N et al. Clinical and genetic determinants of anthracycline-induced cardiac iron accumulation. Int J Cardiol 2012;154:282–6. [DOI] [PubMed] [Google Scholar]
- 36.Cascales A, Pastor-Quirante F, Sanchez-Vega B et al. Association of anthracycline-related cardiac histological lesions with NADPH oxidase functional polymorphisms. Oncologist 2013;18:446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lubieniecka JM, Liu J, Heffner D et al. Single-nucleotide polymorphisms in aldo-keto and carbonyl reductase genes are not associated with acute cardiotoxicity after daunorubicin chemotherapy. Cancer Epidemiol Biomarkers Prev 2012;21:2118–20. [DOI] [PubMed] [Google Scholar]
- 38.Pearson EJ, Nair A, Daoud Y, Blum JL. The incidence of cardiomyopathy in BRCA1 and BRCA2 mutation carriers after anthracycline-based adjuvant chemotherapy. Breast Cancer Res Treat 2017;162:59–67. [DOI] [PubMed] [Google Scholar]
- 39.Volkan-Salanci B, Aksoy H, Kiratli PO et al. The relationship between changes in functional cardiac parameters following anthracycline therapy and carbonyl reductase 3 and glutathione S transferase Pi polymorphisms. J Chemother 2012;24:285–91. [DOI] [PubMed] [Google Scholar]
- 40.Vinodhini MT, Sneha S, Nagare RP et al. Evaluation of a polymorphism in MYBPC3 in patients with anthracycline induced cardiotoxicity. Indian Heart J 2018;70:319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visscher H, Ross CJ, Rassekh SR et al. Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr Blood Cancer 2013;60:1375–81. [DOI] [PubMed] [Google Scholar]
- 42.Aminkeng F, Bhavsar AP, Visscher H et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet 2015;47:1079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beauclair S, Formento P, Fischel JL et al. Role of the HER2 [Ile655Val] genetic polymorphism in tumorogenesis and in the risk of trastuzumab-related cardiotoxicity. Ann Oncol 2007;18:1335–41. [DOI] [PubMed] [Google Scholar]
- 44.Gomez Pena C, Davila-Fajardo CL, Martinez-Gonzalez LJ et al. Influence of the HER2 Ile655Val polymorphism on trastuzumab-induced cardiotoxicity in HER2-positive breast cancer patients: a meta-analysis. Pharmacogenet Genomics 2015;25:388–93. [DOI] [PubMed] [Google Scholar]
- 45.Lemieux J, Diorio C, Cote MA et al. Alcohol and HER2 polymorphisms as risk factor for cardiotoxicity in breast cancer treated with trastuzumab. Anticancer Res 2013;33:2569–76. [PubMed] [Google Scholar]
- 46.Roca L, Dieras V, Roche H et al. Correlation of HER2, FCGR2A, and FCGR3A gene polymorphisms with trastuzumab related cardiac toxicity and efficacy in a subgroup of patients from UNICANCER-PACS 04 trial. Breast Cancer Res Treat 2013;139:789–800. [DOI] [PubMed] [Google Scholar]
- 47.Udagawa C, Nakamura H, Ohnishi H et al. Whole exome sequencing to identify genetic markers for trastuzumab-induced cardiotoxicity. Cancer Sci 2018;109:446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stanton SE, Ward MM, Christos P et al. Pro1170 Ala polymorphism in HER2-neu is associated with risk of trastuzumab cardiotoxicity. BMC Cancer 2015;15:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boekhout AH, Gietema JA, Milojkovic Kerklaan B et al. Angiotensin II-Receptor Inhibition With Candesartan to Prevent Trastuzumab-Related Cardiotoxic Effects in Patients With Early Breast Cancer: A Randomized Clinical Trial. JAMA Oncol 2016;2:1030–7. [DOI] [PubMed] [Google Scholar]
- 50.Serie DJ, Crook JE, Necela BM et al. Genome-wide association study of cardiotoxicity in the NCCTG N9831 (Alliance) adjuvant trastuzumab trial. Pharmacogenet Genomics 2017;27:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson DC, Corthals S, Ramos C et al. Genetic associations with thalidomide mediated venous thrombotic events in myeloma identified using targeted genotyping. Blood 2008;112:4924–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bagratuni T, Kastritis E, Politou M et al. Clinical and genetic factors associated with venous thromboembolism in myeloma patients treated with lenalidomide-based regimens. Am J Hematol 2013;88:765–70. [DOI] [PubMed] [Google Scholar]
- 53.Di Stefano AL, Labussiere M, Lombardi G et al. VEGFA SNP rs2010963 is associated with vascular toxicity in recurrent glioblastomas and longer response to bevacizumab. J Neurooncol 2015;121:499–504. [DOI] [PubMed] [Google Scholar]
- 54.Hewitt JK. Editorial policy on candidate gene association and candidate gene-by-environment interaction studies of complex traits. Behav Genet 2012;42:1–2. [DOI] [PubMed] [Google Scholar]
- 55.Visscher H, Ross CJ, Rassekh SR et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol 2012;30:1422–8. [DOI] [PubMed] [Google Scholar]
- 56.Zakaria ZZ, Benslimane FM, Nasrallah GK et al. Using Zebrafish for Investigating the Molecular Mechanisms of Drug-Induced Cardiotoxicity. BioMed research international 2018;2018:1642684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han Y, Zhang JP, Qian JQ, Hu CQ. Cardiotoxicity evaluation of anthracyclines in zebrafish (Danio rerio). J Appl Toxicol 2015;35:241–52. [DOI] [PubMed] [Google Scholar]
- 58.Miranda CJ, Makui H, Soares RJ et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood 2003;102:2574–80. [DOI] [PubMed] [Google Scholar]
- 59.Zhang S, Liu X, Bawa-Khalfe T et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012;18:1639–42. [DOI] [PubMed] [Google Scholar]
- 60.Musunuru K, Sheikh F, Gupta RM et al. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ Genom Precis Med 2018;11:e000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu M, Liu S, Gao Y et al. Conditional gene knockout and reconstitution in human iPSCs with an inducible Cas9 system. Stem Cell Res 2018;29:6–14. [DOI] [PubMed] [Google Scholar]
- 62.Kamiya A, Chikada H, Ida K et al. An in vitro model of polycystic liver disease using genome-edited human inducible pluripotent stem cells. Stem Cell Res 2018;32:17–24. [DOI] [PubMed] [Google Scholar]
- 63.Jiang Y, Park P, Hong S-M, Ban K. Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells: Current Strategies and Limitations. Mol Cells 2018;41:613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arora S, Yim EKF, Toh Y-C. Environmental Specification of Pluripotent Stem Cell Derived Endothelial Cells Toward Arterial and Venous Subtypes. Front Bioeng Biotechnol 2019;7:143–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JH, Protze SI, Laksman Z, Backx PH, Keller GM. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017;21:179–194.e4. [DOI] [PubMed] [Google Scholar]
- 66.Shafaattalab S, Lin E, Christidi E et al. Ibrutinib Displays Atrial-Specific Toxicity in Human Stem Cell-Derived Cardiomyocytes. Stem cell reports 2019;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eder A, Vollert I, Hansen A, Eschenhagen T. Human engineered heart tissue as a model system for drug testing. Adv Drug Del Rev 2016;96:214–224. [DOI] [PubMed] [Google Scholar]
- 68.Nugraha B, Buono MF, von Boehmer L, Hoerstrup SP, Emmert MY. Human Cardiac Organoids for Disease Modeling. Clin Pharmacol Ther 2019;105:79–85. [DOI] [PubMed] [Google Scholar]
- 69.Ross SB, Fraser ST, Semsarian C. Induced pluripotent stem cell technology and inherited arrhythmia syndromes. Heart Rhythm 2018;15:137–144. [DOI] [PubMed] [Google Scholar]
- 70.Liang P, Sallam K, Wu H et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J Am Coll Cardiol 2016;68:2086–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Limpitikul WB, Dick IE, Tester DJ et al. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ Res 2017;120:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamamoto Y, Makiyama T, Harita T et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum Mol Genet 2017;26:1670–1677. [DOI] [PubMed] [Google Scholar]
- 73.Kitani T, Ong SG, Lam CK et al. Human-Induced Pluripotent Stem Cell Model of Trastuzumab-Induced Cardiac Dysfunction in Patients With Breast Cancer. Circulation 2019;139:2451–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol 2015;12:547–58. [DOI] [PubMed] [Google Scholar]
- 75.Kurokawa YK, Shang MR, Yin RT, George SC. Modeling trastuzumab-related cardiotoxicity in vitro using human stem cell-derived cardiomyocytes. Toxicol Lett 2018;285:74–80. [DOI] [PubMed] [Google Scholar]
- 76.Sharma A, Burridge PW, McKeithan WL et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Angelis A, Urbanek K, Cappetta D et al. Doxorubicin cardiotoxicity and target cells: a broader perspective. 2016;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verheijen M, Schrooders Y, Gmuender H et al. Bringing in vitro analysis closer to in vivo: Studying doxorubicin toxicity and associated mechanisms in 3D human microtissues with PBPK-based dose modelling. Toxicol Lett 2018;294:184–192. [DOI] [PubMed] [Google Scholar]
- 79.Cohen JD, Babiarz JE, Abrams RM et al. Use of human stem cell derived cardiomyocytes to examine sunitinib mediated cardiotoxicity and electrophysiological alterations. Toxicol Appl Pharmacol 2011;257:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Posimo JM, Unnithan AS, Gleixner AM et al. Viability assays for cells in culture. Journal of visualized experiments : JoVE 2014:e50645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsu WT, Huang CY, Yen CYT, Cheng AL, Hsieh PCH. The HER2 inhibitor lapatinib potentiates doxorubicin-induced cardiotoxicity through iNOS signaling. Theranostics 2018;8:3176–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hyman BT. Caspase activation without apoptosis: insight into Abeta initiation of neurodegeneration. Nat Neurosci 2011;14:5–6. [DOI] [PubMed] [Google Scholar]
- 83.Gintant G, Fermini B, Stockbridge N, Strauss D. The Evolving Roles of Human iPSC-Derived Cardiomyocytes in Drug Safety and Discovery. Cell Stem Cell 2017;21:14–17. [DOI] [PubMed] [Google Scholar]
- 84.Sala L, Ward-van Oostwaard D, Tertoolen LGJ, Mummery CL, Bellin M. Electrophysiological Analysis of human Pluripotent Stem Cell-derived Cardiomyocytes (hPSC-CMs) Using Multi-electrode Arrays (MEAs). Journal of visualized experiments : JoVE 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang Y, Zhou Y, Bao X et al. An Ultrasensitive Calcium Reporter System via CRISPR-Cas9-Mediated Genome Editing in Human Pluripotent Stem Cells. iScience 2018;9:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shinnawi R, Huber I, Maizels L et al. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem cell reports 2015;5:582–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang Y, Liu N, He Y et al. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nature communications 2018;9:1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473:317–25. [DOI] [PubMed] [Google Scholar]
- 89.Hadzijusufovic E, Albrecht-Schgoer K, Huber K et al. Nilotinib-induced vasculopathy: identification of vascular endothelial cells as a primary target site. Leukemia 2017;31:2388–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lo Sardo V, Chubukov P, Ferguson W et al. Unveiling the Role of the Most Impactful Cardiovascular Risk Locus through Haplotype Editing. Cell 2018;175:1796–1810.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang J, McIntosh BE, Wang B et al. A Human Pluripotent Stem Cell-Based Screen for Smooth Muscle Cell Differentiation and Maturation Identifies Inhibitors of Intimal Hyperplasia. Stem cell reports 2019;12:1269–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheung C, Bernardo AS, Trotter MW, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol 2012;30:165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gover-Proaktor A, Granot G, Pasmanik-Chor M et al. Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib differentially affect the vascular molecular pathways and functionality of human endothelial cells. Leuk Lymphoma 2019;60:189–199. [DOI] [PubMed] [Google Scholar]
- 94.Sa S, Gu M, Chappell J et al. Induced Pluripotent Stem Cell Model of Pulmonary Arterial Hypertension Reveals Novel Gene Expression and Patient Specificity. Am J Respir Crit Care Med 2017;195:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khaddaj Mallat R, Mathew John C, Kendrick DJ, Braun AP. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit Rev Clin Lab Sci 2017;54:458–470. [DOI] [PubMed] [Google Scholar]
- 96.Tykocki NR, Boerman EM, Jackson WF. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Comprehensive Physiology 2017;7:485–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Safar ME. Arterial stiffness as a risk factor for clinical hypertension. Nat Rev Cardiol 2018;15:97–105. [DOI] [PubMed] [Google Scholar]
- 98.Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: Beyond eNOS. J Pharmacol Sci 2015;129:83–94. [DOI] [PubMed] [Google Scholar]
- 99.Namin SM, Nofallah S, Joshi MS, Kavallieratos K, Tsoukias NM. Kinetic analysis of DAF-FM activation by NO: toward calibration of a NO-sensitive fluorescent dye. Nitric Oxide 2013;28:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Verdon CP, Burton BA, Prior RL. Sample pretreatment with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduces nitrate while avoiding interference by NADP+ when the Griess reaction is used to assay for nitrite. Anal Biochem 1995;224:502–8. [DOI] [PubMed] [Google Scholar]
- 101.Biel NM, Santostefano KE, DiVita BB et al. Vascular Smooth Muscle Cells From Hypertensive Patient-Derived Induced Pluripotent Stem Cells to Advance Hypertension Pharmacogenomics. Stem Cells Transl Med 2015;4:1380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patsch C, Challet-Meylan L, Thoma EC et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol 2015;17:994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kinnear C, Chang WY, Khattak S et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med 2013;2:2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rennier K, Ji JY. Effect of shear stress and substrate on endothelial DAPK expression, caspase activity, and apoptosis. BMC Res Notes 2013;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Van Kruchten R, Cosemans JM, Heemskerk JW. Measurement of whole blood thrombus formation using parallel-plate flow chambers - a practical guide. Platelets 2012;23:229–42. [DOI] [PubMed] [Google Scholar]
- 106.Tsai M, Kita A, Leach J et al. In vitro modeling of the microvascular occlusion and thrombosis that occur in hematologic diseases using microfluidic technology. J Clin Invest 2012;122:408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jain A, Barrile R, van der Meer AD et al. Primary Human Lung Alveolus-on-a-chip Model of Intravascular Thrombosis for Assessment of Therapeutics. Clin Pharmacol Ther 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jain A, Barrile R, van der Meer AD et al. Primary Human Lung Alveolus-on-a-chip Model of Intravascular Thrombosis for Assessment of Therapeutics. Clin Pharmacol Ther 2018;103:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jain A, Graveline A, Waterhouse A, Vernet A, Flaumenhaft R, Ingber DE. A shear gradient-activated microfluidic device for automated monitoring of whole blood haemostasis and platelet function. Nature communications 2016;7:10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mathur T, Singh KA, NK RP et al. Organ-on-chips made of blood: endothelial progenitor cells from blood reconstitute vascular thromboinflammation in vessel-chips. Lab Chip 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blyszczuk P Myocarditis in Humans and in Experimental Animal Models. Frontiers in cardiovascular medicine 2019;6:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharma A, Marceau C, Hamaguchi R et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ Res 2014;115:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Semper H, Muehlberg F, Schulz-Menger J, Allewelt M, Grohe C. Drug-induced myocarditis after nivolumab treatment in a patient with PDL1-negative squamous cell carcinoma of the lung. Lung Cancer 2016;99:117–9. [DOI] [PubMed] [Google Scholar]
- 114.Thomas CA, Tejwani L, Trujillo CA et al. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017;21:319–331.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Joshi K, Elso C, Motazedian A et al. Induced pluripotent stem cell macrophages present antigen to proinsulin-specific T cell receptors from donor-matched islet-infiltrating T cells in type 1 diabetes. Diabetologia 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Montel-Hagen A, Crooks GMJEh. From pluripotent stem cells to T cells. 2018. [DOI] [PubMed] [Google Scholar]
- 117.Bernarreggi D, Pouyanfard S, Kaufman DSJEh. Development of innate immune cells from human pluripotent stem cells. 2019;71:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Warren CR, O’Sullivan JF, Friesen M et al. Induced Pluripotent Stem Cell Differentiation Enables Functional Validation of GWAS Variants in Metabolic Disease. Cell Stem Cell 2017;20:547–557 e7. [DOI] [PubMed] [Google Scholar]
- 119.Pashos EE, Park Y, Wang X et al. Large, Diverse Population Cohorts of hiPSCs and Derived Hepatocyte-like Cells Reveal Functional Genetic Variation at Blood Lipid-Associated Loci. Cell Stem Cell 2017;20:558–570 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Knowles DA, Burrows CK, Blischak JD et al. Determining the genetic basis of anthracycline-cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stranger BE, Forrest MS, Dunning M et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007;315:848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stranger BE, Montgomery SB, Dimas AS et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet 2012;8:e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stranger BE, Nica AC, Forrest MS et al. Population genomics of human gene expression. Nat Genet 2007;39:1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Qiu W, Rogers AJ, Damask A et al. Pharmacogenomics: novel loci identification via integrating gene differential analysis and eQTL analysis. Hum Mol Genet 2014;23:5017–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Powell JE, Henders AK, McRae AF et al. Genetic control of gene expression in whole blood and lymphoblastoid cell lines is largely independent. Genome Res 2012;22:456–66. [DOI] [PMC free article] [PubMed] [Google Scholar]