

Summary
Type I interferon (IFN) response is commonly recognized as the main signaling activity of STING. Here, we generated the Sting1S365A/S365A mutant mouse that precisely ablated IFN-dependent while preserving IFN-independent activities of Sting. Surprisingly, StingS365A/S365A mice protected against HSV-1 infection, despite lacking STING-mediated IFN response. This challenges the prevailing view and suggests that STING controls HSV-1 infection through IFN-independent activities. Transcriptomic analysis revealed widespread IFN-independent activities of STING in macrophages and T cells, and STING activities in T cells were predominantly IFN-independent. In mouse tumor models, T cells in the tumor experienced substantial cell death that was in part mediated by IFN-independent activities of STING. We found that tumor induces STING-mediated cell death in T cells to evade immune control. Together, our data demonstrate that mammalian STING possesses widespread IFN-independent activities that are important for restricting HSV-1 infection, tumor immune evasion and likely also adaptive immunity.
Graphical Abstract
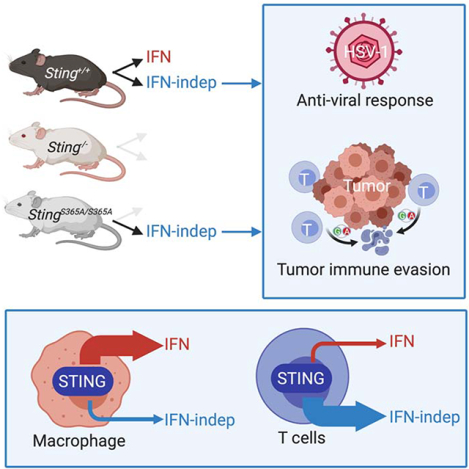
eTOC blub:
Type I interferon response was commonly believed to be the major (if not the sole) signaling activity of STING. Wu et al reveal that mammalian STING possesses widespread IFN-independent activities that are physiologically important for antiviral response, tumor immune evasion and likely also adaptive T cell immunity.
Introduction
Stimulator of interferon genes (STING) is an essential protein for innate immune defense against a wide variety of microbial pathogens. STING is a transmembrane protein on the endoplasmic reticulum (ER), where it senses cyclic dinucleotides (CDN) in the cytosol that are either mammalian 2’3’-cyclic GMP-AMP (cGAMP) produced by DNA sensor cGAS or bacterial cyclic di-AMP or cyclic di-GMP (Motwani et al., 2019b; Tan et al., 2018). After ligand binding, STING translocates from the ER to the ER-Golgi intermediate compartment (ERGIC) and the Golgi, during which time it recruits TBK1 and activates type I interferon (IFN) response via TBK1-IRF3-IFN signaling axis (Dobbs et al., 2015).
Since the initial discovery of STING, the IFN response has been the most recognized signaling activity of mammalian STING. This is in part due to myeloid cells being the primary cell type of choice in most studies. However, despite high conservation of STING protein sequence from various species across evolution, the functional motif required for IFN response is only present in mammalian and some vertebrate STINGs (Margolis et al., 2017). More ancestral species of STING do not contain the IFN motif (also known as the C-terminal tail) but do respond to CDN, and functions of these STINGs are not known. Even for mammalian STING, several recent studies have presented clear evidence for IFN-independent activities of STING playing physiologically important roles in cell death, autophagy, and cell proliferation (Cerboni et al., 2017; Gui et al., 2019; Luksch et al., 2019; Motwani et al., 2019a; Ranoa et al., 2019; Wu et al., 2019). STING-associated vasculopathy with onset in infancy (SAVI) disease pathology in mice is also independent of IFN signaling (Luksch et al., 2019; Motwani et al., 2019a; Warner et al., 2017; Wu et al., 2019).
Studying IFN-independent functions of STING in mammals is challenging due to overwhelming effects of IFN signaling when STING is activated by agonists, and IFN may mask other activities of STING. Fortunately, phosphorylation of a single serine residue at 365 position of mouse STING (S366 in human STING) is required for recruitment of IRF3 and subsequent activation of IFN signaling (Liu et al., 2015). Therefore, we mutated S365 to alanine and generated Sting1S365A/S365A mouse that should selectively inactive STING-mediated IFN signaling with the rest of STING protein as well as the innate immune system intact.
Results
Sting-S365A mutation specifically abrogates IFN signaling in mice
Sting1S365A/S365A mice were born at the expected Mendelian ratio, and adult mice were healthy compared to WT littermate controls (data not shown). We generated bone marrow-derived macrophages (BMDMs) as well as splenic T cells from wild type, Sting1−/− and Sting1S365A/S365A mice (all on C57BL/6 background), stimulated with a small molecule mouse Sting agonist 5,6-dimethylxanthenone-4-acetic Acid (DMXAA) or native mammalian ligand 2’3’-cGAMP (cGAMP), and measured innate immune response. At the mRNA level, STING-mediated induction of IFN and IFN-simulated genes (ISGs) expression (e.g. Ifnb1, Ifit1 and Cxcl10) were near completely eliminated in Sting1−/− and Sting1S365A/S365A BMDMs (Figure 1A). In contrast, NFκB target gene expression was not affected in Sting1S365A/S365A BMDMs (Figure 1A). At the protein level, IFNβ protein production was undetectable in either DNA- or cGAMP-stimulated Sting1−/− and Sting1S365A/S365A BMDMs, confirming that S365A mutation is effective at abrogating STING-mediated IFN signaling (Figure 1B). Several inflammatory cytokines and chemokines (e.g. MIP-1α, MIP-1β, IL-6 and IL-10) were also reduced or undetectable in Sting1S365A/S365A BMDMs, while other cytokines (e.g. IL-13, GM-CSF) are not affected in Sting1S365A/S365A BMDMs (Figure 1C, 1D). In T cells, we also found that S365A mutation selectively inactivated IFN, but not NFκB, signaling at both mRNA and protein levels (Figure 1E, 1F). We also performed Western blot to biochemically assess known IFN-dependent and IFN-independent activities of STING. DMXAA activated strong IFN and NFκB signaling (as indicated by TBK1, IRF3 and p65 phosphorylation) as well as autophagy (as indicated by LC3 lipidation) in wild type T cells and BMDMs (Figure 1G, Figure S1). DMXAA treatment did not cause any detectable changes in Sting1−/− cells. In contrast, only IRF3 phosphorylation is absent in Sting1S365A/S365A cells after DMXAA stimulation, while TBK1, p65 phosphorylation and LC3 lipidation were similar compared to those in wild type cells (Figure 1G, Figure S1). These data demonstrate the remarkable precision for S365A mutation to ablate only STING-mediated IFN activity in mice.
Figure 1: Sting-S365A mutation specifically abrogates IFN signaling.
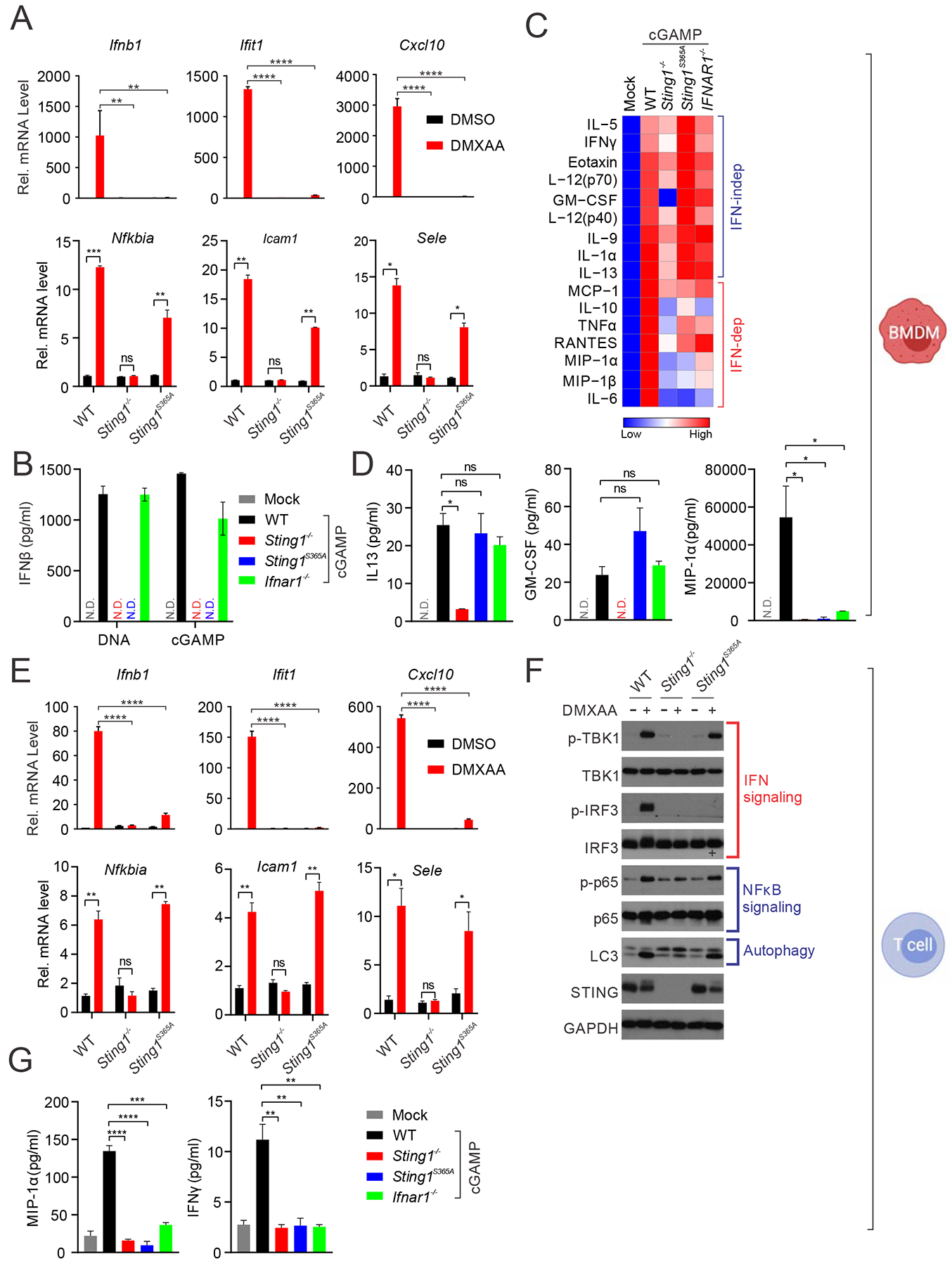
(A) Quantitative RT-PCR analysis of IFN- and NFκB-stimulated genes in BMDMs from WT, Sting1−/− or Sting1S365A/S365A mice (Sting1S365A, same throughout). Cells were stimulated with DMXAA (10 μg/ml) for 5 h in vitro followed by qRT-PCR analysis of indicated mRNA (on top). **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. Error bars, SEM. Two-way ANOVA test.
(B) ELISA analysis of IFNβ production in BMDMs stimulated with DNA or 2’3’-cGAMP (cGAMP below) for 24 h.
(C, D) Cytokine array analysis of BMDMs stimulated with cGAMP for 24 h. A heatmap summarizing all the data is showing in C. Representative cytokines are showing in D.
(E) Quantitative RT-PCR analysis of IFN or NFκB-stimulated gene expression in T cell. Cells were stimulated with DMXAA (2 μg/ml) for 5 h followed by qRT-PCR analysis of indicated mRNAs (on top). **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001. Error bars, SEM. Two-way ANOVA test.
(F) Immunoblot analysis of signaling pathways activated by STING. Splenic T cells were stimulated with DMXAA (10 μg/ml) for 6 h in vitro. Cell lysates were analyzed for proteins indicated on the left. Known signaling pathways are indicated on the right.
(G) Cytokine protein levels in T cells of indicated genotype after cGAMP stimulation.
Data are representative of at least three independent experiments.independent experiments.
See also Figure S1.
Transcriptomic discovery of IFN-dependent and IFN-independent activities of STING
We next performed transcriptomic analysis to systemically identify IFN-dependent and IFN-independent activities of mammalian STING. We treated wild type, Sting1−/− and Sting1S365A/S365A BMDMs and T cells with DMXAA or DMSO (Mock) and performed RNA-seq. We first performed principle component analysis (PCA) to assess overall similarity of all expressed genes from each sample. BMDM and T cell samples grouped together by cell type as expected (Figure S2A). Interestingly, we observed drastic difference between DMXAA-treated WT vs. Sting1S365A/S365A in BMDMs but much less difference in T cells (Figure S2A). This suggests that S365 residue or STING-mediated IFN signaling plays a dominant role in BMDMs, but not in T cells.
We next analyzed the data in both cell types to determine: 1) what fraction of DEGs are IFN-dependent or IFN-independent and 2) which pathways are enriched as IFN-dependent or IFN-independent activities. We defined the IFN-dependent vs. IFN-independent DEGs or activities as follows: we compared DMXAA- vs. mock-treated wild type cells to capture all genes altered by STING activation (IFN-dependent and IFN-independent, Figure S2B). DMXAA- vs. mock-treated Sting1S365A/S365A cells will capture IFN-independent genes. Comparing DMXAA-treated wild type vs. DMXAA-treated Sting1S365A/S365A cells will capture IFN-dependent genes. Using these metrics, in BMDMs, we found 1142 DEGs up regulated, of which, 70% were IFN-dependent and 30% were IFN-independent (Figure 2A). Interestingly, we observed many more DEGs (4270) downregulated in BMDMs (after DMXAA treatment) and 78% were IFN-dependent and 22% were IFN-independent. IFN-dependent gene-downregulation has been observed before for small groups of genes and their functions are largely unknown. In T cells, we found 1313 DEGs up- and 651 DEGs down-regulated, and the majority of DEGs were IFN-independent (72% and 60%, respectively, Figure 2B). This is in sharp contrast to BMDMs and confirms our PCA analysis. T cells also express higher levels of STING protein compared to macrophages (Gulen et al., 2017; Wu et al., 2019), underscoring the importance of investigating STING activities in T cells. The amplitude of fold change in downregulated genes was less compared to that in upregulated genes in all pair-wise comparisons (Figure S2B).
Figure 2. Transcriptomic discovery of IFN-dependent and IFN-independent activities of STING.
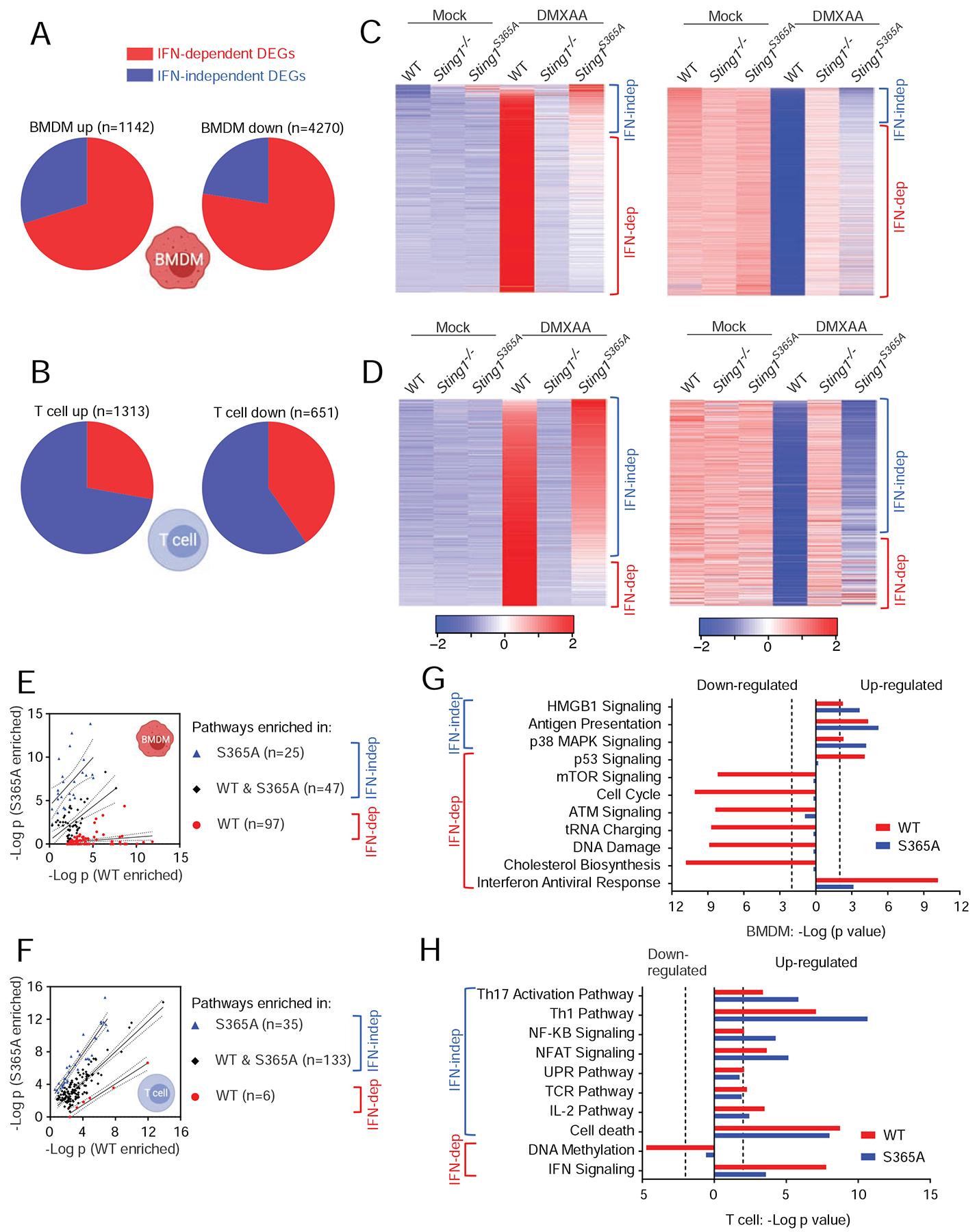
(A, B) Pie charts showing percentages of differentially expressed genes (DEGs) that are either IFN-dependent or IFN-independent in BMDMs (A) or T cells (B). Total numbers of DEGs are defined in Figure S2B. Also see STAR Methods for more details.
(C, D) Heatmaps showing gene expression patterns of up-regulated (left) or down-regulated (right) DEGs in BMDMs (C) and T cells (D).
(E, F) Scatter plots of enriched pathways in BMDMs (E) and T cells (F). Pathways enriched only in wild type DMXAA vs. Mock are considered IFN-dependent. Pathways enriched in S365A DMXAA vs. Mock or in both are considered IFN-independent (see STAR Methods). Pathways are categorized into 3 group based on enrichment p values (WT, −log10(PWT-PS365A)≥2; S365A, −log10(PWT-PS365A)≤−2; both, −2< −log10(PWT-PS365A)<2).
(G, H) Top enriched pathways in BMDMs (G) and T cells (H).
We next analyzed gene expression pattern to systemically identify IFN-independent activities of STING. DMXAA stimulation caused overwhelming changes in gene expression in wild type but not Sting1−/− BMDMs and T cells (Figure 2C, 2D). In DMXAA-stimulated Sting1S365A/S365A BMDMs, much of the gene expression changes (up or down) were lost compared to wild type BMDMs, although a small fraction of DEG remains (these should be IFN-independent, Figure 2C). Data in T cells shows an opposite trend, with the majority of DEGs unaffected by S365A mutation (Figure 2D). These data suggest that there are widespread IFN-independent activities of STING in both BMDMs and T cells, and especially in T cells, STING activities are predominantly IFN-independent.
Next, we performed pathway enrichment analysis using DEGs from each pair-wise comparison. Pathways that reach statistical significance (p<=0.01) were visualized by scatter plots (Figure 2E, 2F). Top pathways in each cell type were highlighted in bar graphs (Figure 2G, 2H). We also selected a few known and previously unknown IFN-dependent and IFN-independent activities of STING and presented individual gene expression as heatmaps (Figure S3). We made a few observations: 1) In wild type cells, IFN signaling stands out as one of the most significant pathways upon STING activation as expected (Figure 2G, 2H). IFN signaling was also called out by our analysis in Sting1S365A/S365A cells (albeit at less significant p values) likely due to low expression of genes that overlap with IFN pathway. 2) In BMDMs, many IFN-dependent pathways are interestingly down-regulated and not directly associated with JAK-STAT signaling, such as mTOR signaling, ATM signaling, tRNA charging, DNA damage and cholesterol biosynthesis (Figure 2G). Some of these pathways have been implicated in STING biology before but the underlying mechanisms are not well defined. For example, STING activation inhibits viral protein translation during RNA virus infection (Franz et al., 2018), thus reduced tRNA charging could be a testable hypothesis. The reduced cholesterol biosynthesis pathway as an IFN-dependent activity of STING in BMDMs is also consistent with a previous study where limiting cholesterol biosynthetic flux engages IFN signaling (York et al., 2015). We also observed a few IFN-independent pathways in BMDMs (all up-regulated), such HMGB1, antigen presentation pathway, which may be associated with macrophage maturation and activation. 3) Very few IFN-dependent pathways were called out in T cells. One of which, DNA methylation was significantly reduced upon STING activation, which could impact T cell effector functions (Scharer et al., 2013). 4) STING activates many T cell effector signaling pathways independently of IFN, such as Th17 signaling, Th1 pathway, NFκB, NFAT, UPR, IL-2 pathways and cell death (Figure 2H). We have previously shown that STING activates UPR through an evolutionarily conserved ‘UPR motif’ that is distinct from the IFN motif (Wu et al., 2019). STING activation has also been implicated in modulating Th17 response in an infection model and a chronic pancreatitis model (Dis et al., 2018; Zhao et al., 2019). Together, these data suggest that STING activation exerts widespread activities in cellular pathways, many of which are independent of S365/IFN. These data also reveal that STING plays a much broader and likely more direct role in T cell biology and adaptive immunity than previously appreciated.
STING protects mice from HSV-1 infection independently of IFN signaling
We next wanted to explore how much IFN-independent activities of STING contribute to its known physiological functions. Mammalian STING plays a critical role in controlling viral infection and cancer. In both cases, IFN signaling is widely believed to be the major (if not the sole) contributor to these functions. To more critically re-evaluate these beliefs using our new Sting1S365A/S365A mouse, we infected wild type, Sting1−/− and Sting1S365A/S365A mice with herpes simplex virus (HSV-1) in vivo. Wild type mice controlled HSV-1 infection very well, while Sting1−/− mice succumbed to infection quickly (Figure 3A). To our surprise, Sting1S365A/S365A mice controlled HSV-1 infection remarkably well, to an extent similar to that of wild type mice (Figure 3A). HSV-1 viral titer in the brain of both wild type and Sting1S365A/S365A mice were also substantially lower compared to that of Sting1−/− mice (Figure 3B). These data suggest that IFN-independent activities of STING mediate protection against HSV-1.
Figure 3. STING protects mice from HSV-1 infection independently of IFN signaling.
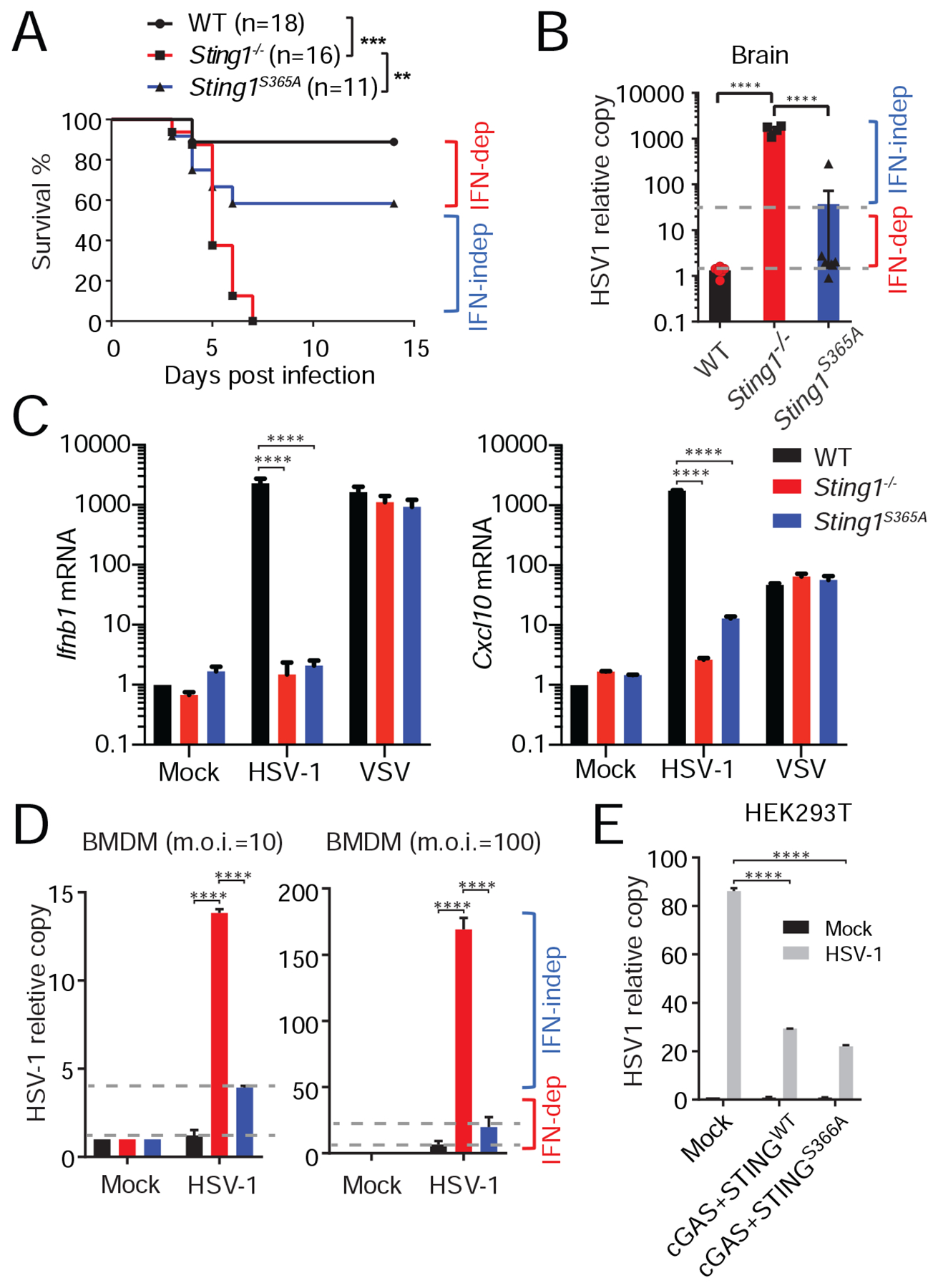
(A) Survival study of HSV-1 infection in WT, Sting1−/− or Sting1S365A/S365A mice (Sting1S365A, same throughout). Mice were infected with HSV-1 by i.v. injection, then monitored for survival. n=11–18. **, p < 0.01; ***, p < 0.001. Log-rank test.
(B) HSV-1 titer in the brain 6 d post infection. HSV-1 DNA was quantified by qPCR.
(C) Quantitative RT-PCR analysis of IFN response in WT, Sting1−/− or Sting1S365A BMDMs infected with HSV-1 or VSV. BMDMs were mock infected or infected with HSV-1 or VSV (m.o.i.=10) for 5 h followed by qRT-PCR analysis.
(D) HSV-1 replication in WT, Sting1−/− or Sting1S365A BMDMs. BMDMs were infected with HSV-1 at indicated m.o.i. for 16 h, Viral DNA copy was determined by qPCR.
(E) HSV-1 replication in HEK293T cells expressing empty Vector, cGAS and wild type human STING or cGAS and human STING-S366A mutant. Cells were transiently transfected with indicated plasmids for 12 h, then infected with HSV-1 at m.o.i.=10 for 12 h. HSV-1 DNA was quantified by qPCR. For panels B-E, ***, p < 0.001; ****, p < 0.0005. Error bars, SEM. Two-way ANOVA test. Data are representative of at least two independent experiments.
See also Figure S4.
To determine whether this IFN-independent anti-viral activity of STING is cell-intrinsic or -extrinsic, we next infected wild type, Sting1−/− and Sting1S365A/S365A BMDMs with HSV-1 in vitro. HSV-1 infection stimulated potent IFN response in wild type but not Sting1−/− and Sting1S365A/S365A BMDMs (Figure 3C). As a control, vesicular stomatitis virus (VSV, an RNA virus) infection induced similar levels of IFN response in all three BMDMs (Figure 3C). Both wild type and Sting1S365A/S365A BMDMs controlled HSV-1 replication significantly better compared to Sting1−/− BMDMs (Figure 3D). To confirm HSV-1 findings in human cells, we reconstituted HEK293T cells (that do not express cGAS nor STING) with wild type cGAS together with either wild type human STING or the STING-S366A mutant. Both wild type and S366A mutant human STING conferred similar protection against HSV-1 (Figure 3E).
Previous studies clearly demonstrated that both type I IFN and STING are important for restricting HSV-1 infection in vivo (Ishikawa et al., 2009; Leib et al., 1999), and STING-mediated IFN response was assumed to be the default mechanism. To compare Sting1−/− and Ifnar1−/− side by side, we infected WT, Sting1−/−, Sting1S365A/S365A and Ifnar1−/− BMDMs with HSV-1. We found that Ifnar1−/− BMDMs are significantly more suspectable to HSV-1 infection compared to Sting1−/− BMDMs, and both knockouts are significantly more susceptible compared to WT and Sting1S365A/S365A BMDMs (Figure S4A). This data suggests that IFN receptor signaling (downstream of multiple innate immune sensing pathways) is much more critical than STING alone in restricting HSV-1 infection, and STING-mediated restriction is mostly S365/IFN-independent. We also infected WT, Sting1−/−, Sting1S365A/S365A and Ifnar1−/− BMDMs with Vaccinia virus (VACV, another DNA virus). In contrast to HSV-1, both Sting1−/− and Sting1S365A/S365A BMDMs are more susceptible to VACV infection than WT (Figure S4B, S4C), suggesting that STING IFN-independent antiviral activity is specific for HSV-1 and does not apply to all DNA viruses. Collectively, these data demonstrate that STING controls HSV-1 infection in vitro and in vivo predominantly through IFN-independent activities.
STING agonists show differential dependency on S365/IFN in inducing T cell death
STING activation in T cells leads to cell death (Gulen et al., 2017; Larkin et al., 2017; Wu et al., 2019). We and others recently showed that STING-mediated UPR, not IFN signaling, is important for T cell death and lung disease associated with SAVI (Wu et al., 2019). We next evaluated the capacity of various STING agonists in inducing cell death in T cells and BMDMs. We first treated wild type, Sting1−/− and Sting1S365A/S365A T cells and BMDMs with increasing dose of DMXAA in vitro and measured cell death. DMXAA did not induce cell death in any of the BMDMs or Sting1−/− T cells (Figure 4A, 4B, Figure S5A). Both wild type and Sting1S365A/S365A T cells were equally sensitive to DMXAA and rapidly committed cell death (Figure 4B). DMXAA is a cell-permeable small molecule agonist of STING, whereas CDN agonists are negatively charged thus nonpermeable to cell membrane. However, it was recently discovered that many mammalian cell types express cGAMP importers on the plasma membrane that directly import cGAMP from extracellular space to the cytosol (Lahey et al., 2020; Luteijn et al., 2019; Ritchie et al., 2019). We found that when we incubated mammalian and bacterial CDN with freshly isolated splenic T cells (without transfection), we also observed a dose-dependent killing of wild type and Sting1S365A/S365A T cells, but not Sting1−/− T cells, suggesting that CDNs were effectively taken up by the T cells into cytosol to activate STING-mediated cell death (Figure 4C–4F).
Figure 4. STING agonists show differential dependency on S365/IFN in inducing T cell death.
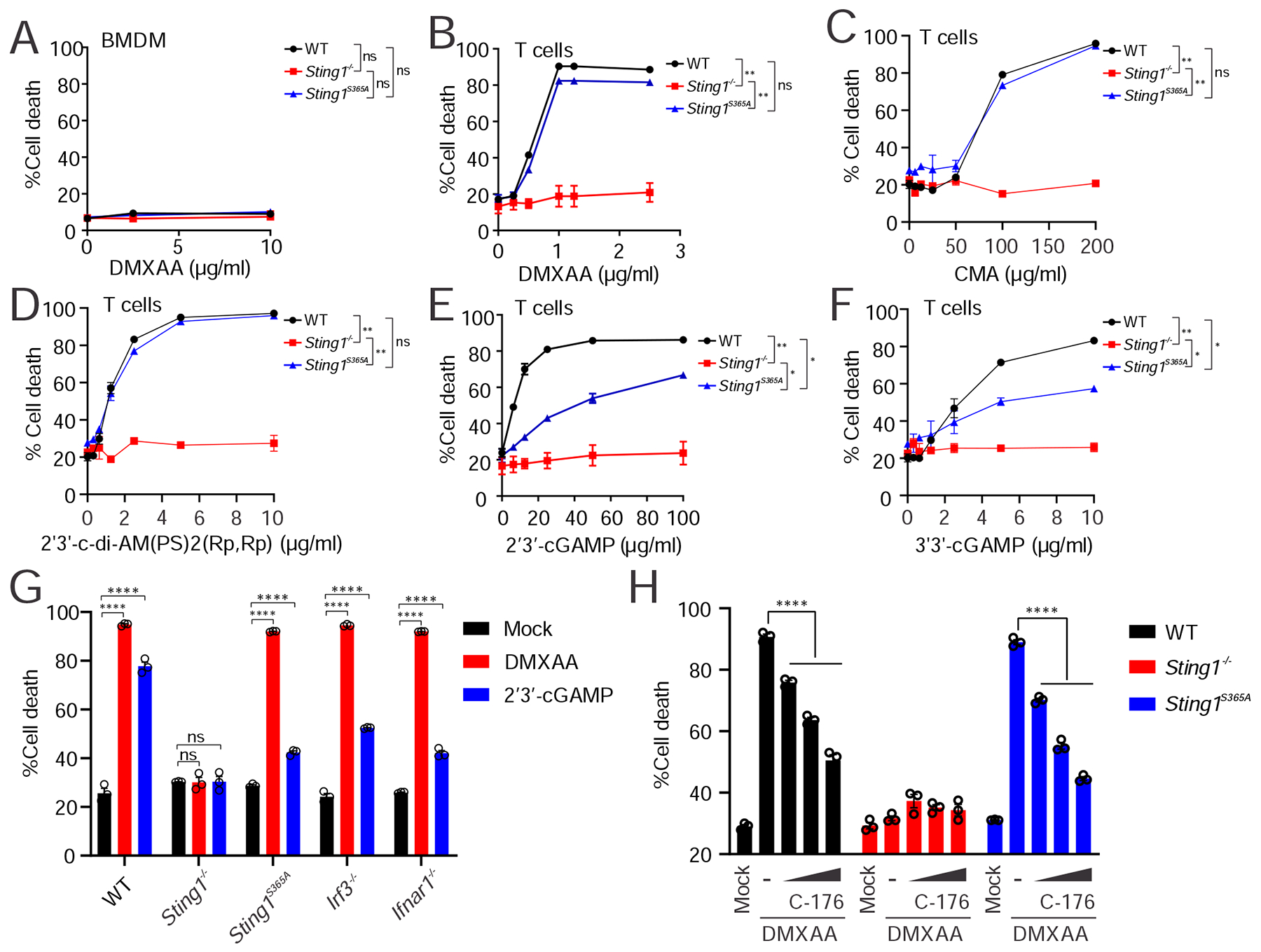
(A) Cell death analysis of BMDMs. BMDMs were treated with indicated dose of DMXAA for 16 h followed by Annexin V staining and FACS analysis of cell death.
(B-F) Cell death analysis of splenic T cells isolated from WT, Sting1−/− or Sting1S365A/S365A mice. T cells were treated with increasing dose of indicated STING agonists (bottom) for 16 h. Cell death was quantified by Annexin V staining and FACS analysis.
(G) Cell death analysis of splenic T cells from mice of different genotypes. Splenic T cell isolated from WT, Sting1−/−, Sting1S365A, Irf3−/−, Ifnar1−/− were treated with DMXAA or cGAMP followed by Annexin V staining and FACS analysis.
(H) STING palmytoilation inhibitor C-176 blocks T cell death. Splenic T cells were stimulated with DMXAA (2 μM) in the presence or absence of increasing dose of STING inhibitor C-176 (20 nm, 100 nM and 500 nM) followed by cell death analysis.
Data are representative of at least two independent experiments.
See also Figure S5.
Interestingly, although all STING agonists induced T cell death in both wild type and Sting1S365A/S365A T cells, the kill curves were qualitative different. The 5 STING agonists we tested fell into two categories. Small molecule STING agonists (e.g. DMXAA, 10-carboxymethyl-9-acridanone (CMA)) and bacteria-derived CDN mimics (e.g. 2’3’-c-di-AM(PS)2(Rp,Rp)) show identical kill curve for wild type and Sting1S365A/S365A T cells, suggesting that these STING agonists induce T cell death completely independent of S365/IFN. In contrast, 2’3’-cGAMP and 3’3’-cGAMP induced a slower kill curve in Sting1S365A/S365A compared to wild type T cells, suggesting a partial dependence of S365/IFN. 2’3’-cGAMP also induced less killing of Irf3−/− and Ifnar−/− T cells compared to WT T cells (Figure 4G, Figure S5B). These data indicate that STING-mediated T cell death may be qualitatively different depending on how STING is activated and by what type of agonist.
STING palmitoylation on the Golgi is important for activating downstream IFN signaling (Mukai et al., 2016). Small molecule inhibitors such as C-176 inhibits STING palmitoylation and IFN signaling (Haag et al., 2018). To further probe the mechanism of STING-mediated T cell death, we treated T cells with C-176. C-176 inhibited DMXAA-induced cell death in both wild type and Sting1S365A/S365A T cells in a dose-dependent manner (Figure 4H, Figure S5C). This data suggest that STING-mediated T cell death requires palmitoylation.
Tumor induces STING-mediated T cell death to evade immune control
It is unclear how and why T cells take up cGAMP, which is scarce under normal conditions in a healthy animal. Several tumors either secrete or transfer cGAMP via gap junction to surrounding dendric cells (DC) and fibroblasts then activate STING in these non-tumor cells (Marcus et al., 2018; Schadt et al., 2019). It was unclear whether T cells in the tumor are also exposed to tumor-derived cGAMP. High dose intratumor injection of STING agonist causes massive T cell death in the tumor (Sivick et al., 2018). We were thus curious how T cells respond to cGAMP naturally produced in the tumor microenvironments, and whether tumors evade immune control by inducing STING-mediated T cell death.
We first used an adoptive transfer T cell tumor control model to assess how different genotypes of T cells respond to and control implanted tumor (Figure 5A). We adoptively transferred wild type, Sting1−/− and Sting1S365A/S365A CD8+ T cells into Rag1−/− mice. One day later, we implanted B16 melanoma by subcutaneous injection and monitored tumor growth. We found that Sting1−/− CD8+ T cells controlled B16 tumor significantly better compared to wild type or Sting1S365A/S365A CD8+ T cells (Figure 5B. 5C). We also isolated CD8+ T cells from tumor and draining lymph node (dLN) then assessed cell death by FACS (Figure 5D, Figure S6A). We found that CD8+ T cells in the tumor are experiencing significant cell death compared to those from lymph nodes (64% vs 20% Figure 5E). Interestingly, Sting1−/− CD8+ T cells experienced significantly less cell death in tumors compared to wild type or Sting1S365A/S365A CD8+ T cells (Figure 5E). Intratumor CD8+ T cell percentages and numbers are significantly higher in mice transferred with Sting1−/− CD8+ T cells compared to mice transferred with wild type or Sting1S365A/S365A CD8+ T cells (Figure 5F, 5G). Immunohistochemistry analysis of T cells in tumor tissues further confirmed our findings (Figure 5H). We also performed a similar set of adoptive transfer T cell experiments against implanted MC38 tumor (colon adenocarcinoma). Again, Sting1−/− CD8+ T cells showed significantly better tumor control and less cell death compared to wild type or Sting1S365A/S365A CD8+ T cells (Figure 5I–O). CD8+ T cells from all three genotypes proliferated equally well when activated in vitro and showed no difference in cytotoxicity (Figure S6B–F). These data suggest that tumor induces STING-mediated T cell death to evade immune control.
Figure 5. Tumor induces STING-mediated T cell death to evade immune control in the adoptive transfer T-cell model.
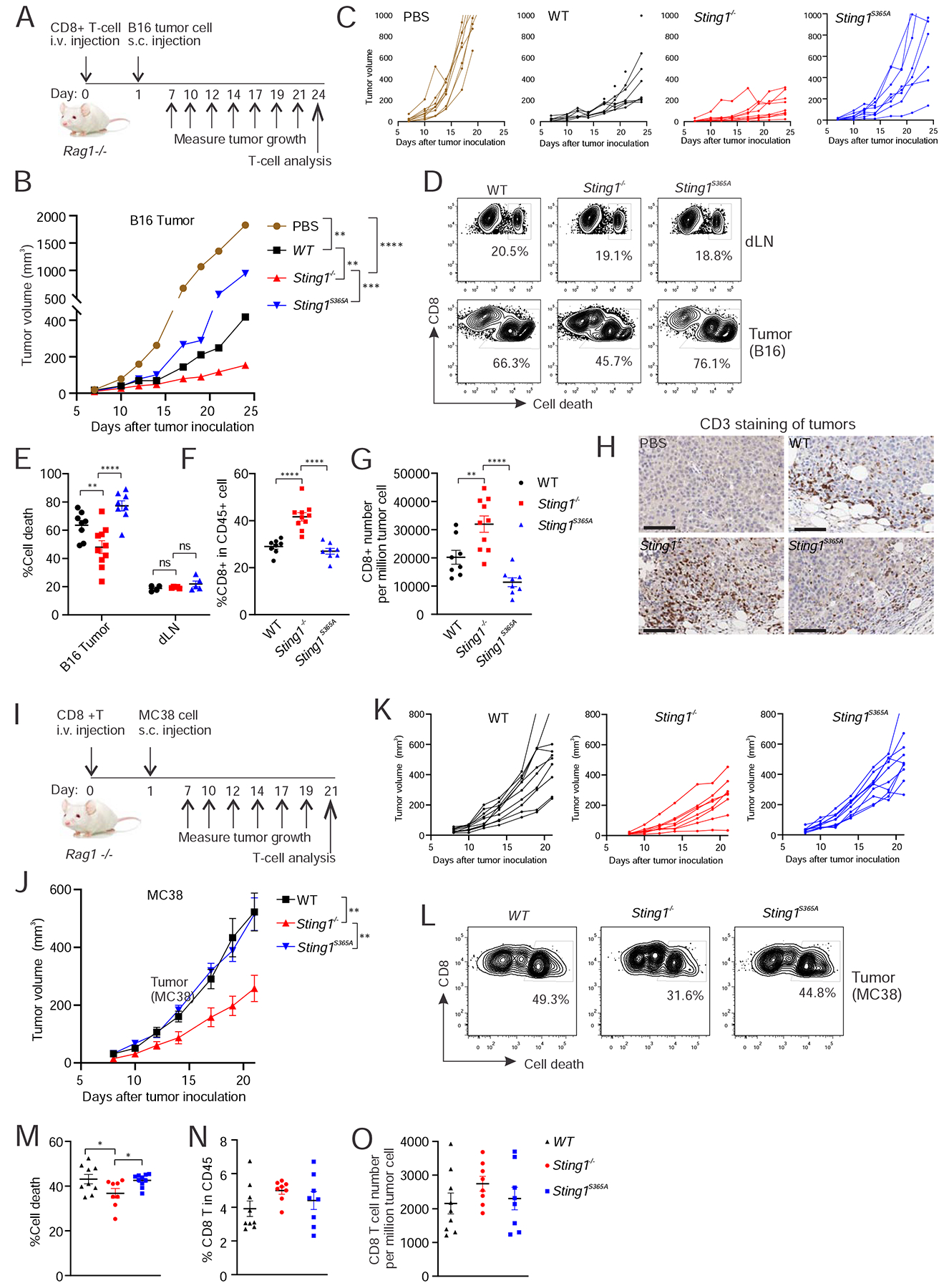
(A) A diagram of the B16 tumor experiment in C-H (also see STAR Methods). n=8–10. B and C, B16 tumor growth curves in Rag1−/− mice adoptively transferred with CD8+ T cell of indicated genotype. Average growth curve (B) and individual tumor growth curves (C) are shown. D-G, FACS analysis of T cells in the tumor and draining lymph node (dLN). Single cell suspension from B16 tumor or dLN were stained with cell death marker and T cell surface antibody followed by FACS analysis (see STAR Method). Gating strategy are shown in Figure S6A. Representative FACS plots are shown in D. Dot plots are shown for percentage of CD8+ T cell death (E), percentage of CD8+ T cells in CD45+ population (F) and number of CD8+ T cells in tumor (G). H, immunohistochemistry analysis of T cells (CD3 staining) in tumor. Scale bar, 100 μm. (I) A diagram of the MC38 tumor experiment in J-O. n=8. J and K, MC38 tumor growth curves in Rag1−/− mice adoptively transferred with CD8+ T cell of indicated genotype. Average growth curve (J) and individual tumor growth curves (K) are shown. L-O, FACS analysis of T cells in the tumor. Single cell suspension from MC38 tumor were stained with cell death marker and T cell surface antibody followed by FACS analysis. Representative FACS plots are shown in L. Dot plots are shown for percentage of CD8+ T cell death in tumor (M), percentage of CD8+ T cells in CD45+ population (N) and number of CD8+ T cells in tumor (O). *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001. ns, not significant. Error bars, SEM. Two-way ANOVA test.
Data are reprehensive of at least three independent experiments.
See also Figure S6.
We next tested whether endogenous T cells responding to implanted syngeneic tumors would also experience STING-mediated cell death (Figure 6A). We injected MC38 cells subcutaneously in wild type, Sting1−/− or Sting1S365A/S365A mice. Fourteen days later, we similarly isolated T cells from both tumor and draining lymph node and assessed cell death. We again observed significantly reduced T cell death in tumors implanted in Sting1−/− mice compared to tumors implanted in wild type or Sting1S365A/S365A mice (Figure 6B, 6C). We note that the total number of tumor infiltrating CD8+ T cell are low in both Sting1−/− and Sting1S365A/S365A mice compared to wild type mice (Figure 6D), which is likely due to the fact that T cell recruitment to the tumor requires IFN response in tumor microenvironment. Collectively, these data suggest that T cell recruitment to the tumor is largely IFN-dependent, once they reach the tumor, T cells experience STING-mediated cell death independently of IFN signaling.
Figure 6. Tumor induces STING-mediated T cell death in the endogenous T-cell model.
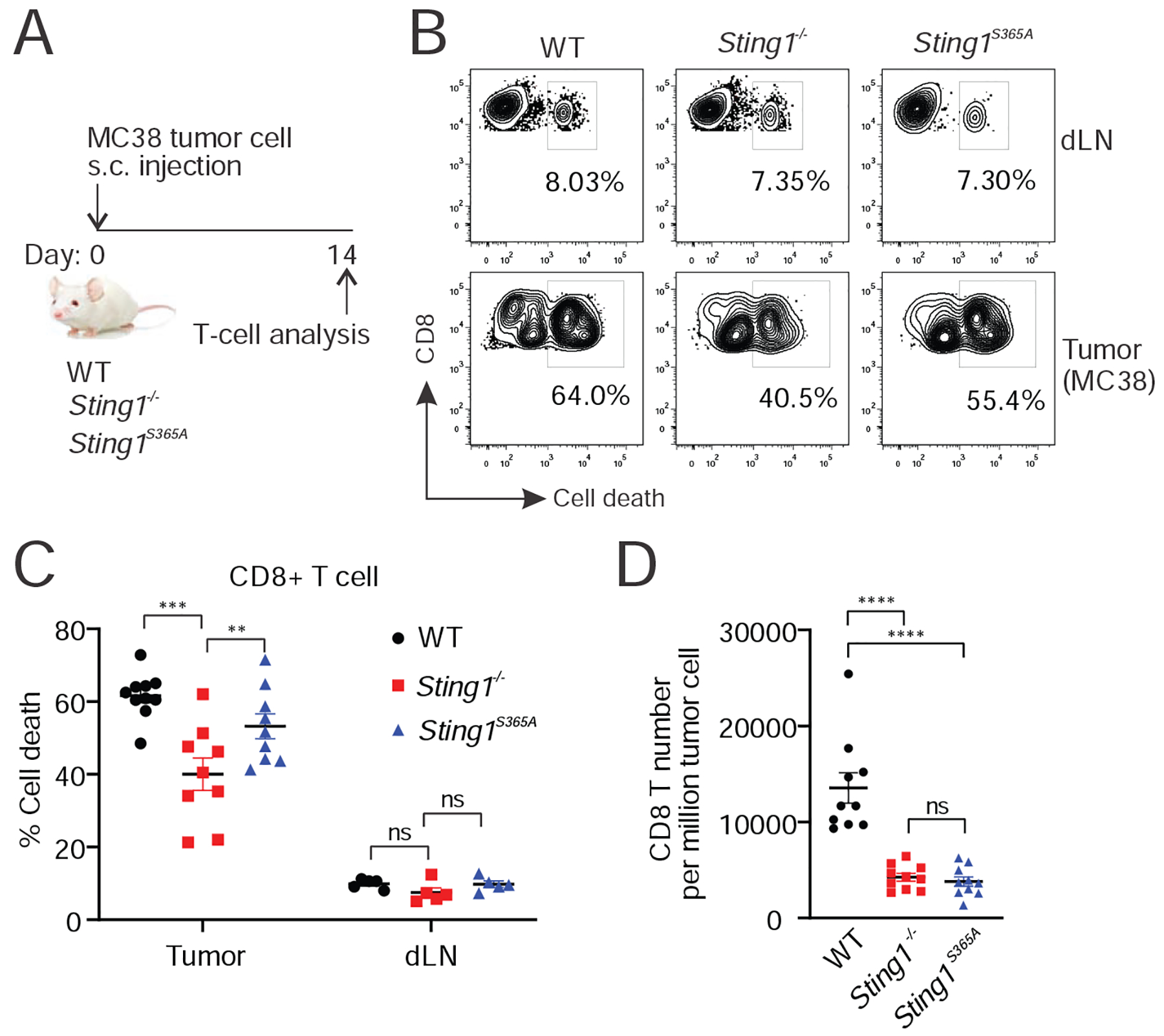
(A) A diagram of the experiment. n=8–10. (B-D) FACS analysis of T cells in the tumor and dLN. MC38 cells were implanted subcutaneously in WT, Sting1−/− or Sting1S365A/S365A mice for 14 d. T cells in tumor and dLN were analyzed by FACS for cell death. Representative FACS plot are shown in B. Dot plots are shown for percentage of CD8+ T cell death (C) and number of CD8+ T cells in tumor (D). *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001. ns, not significant.
Error bars, SEM. Two-way ANOVA test.
Data are representative of at least three independent experiments.
Discussion
The Sting1S365A/S365A mouse is defective in STING-mediated IFN response, thus enabled systematic discovery of IFN-independent activities of mammalian STING in an organism setting. The most surprising finding is that Sting1S365A/S365A mice protected against HSV-1 infection to an extent close to that in wild type mice, which challenges the current prevailing assumption that STING-mediated IFN response is the major anti-viral activity against HSV-1 infection. Type I IFN plays a clear role in host defense against HSV-1 infection (Leib et al., 1999). HSV-1 activates several other innate immune receptors besides cGAS-STING that could also lead to IFN signaling (Kurt-Jones et al., 2017). Thus, our data demonstrate that the portion of IFN coming from STING activation is dispensable for host restriction of HSV-1. STING-mediated protection against HSV-1 infection appears to be cell-intrinsic (at least to macrophages) and is conserved in both mouse and human cells. STING was recently shown to induce a non-canonical autophagy pathway that is IFN-independent and has anti-viral activity in vitro against HSV-1 infection (Gui et al., 2019). Thus, STING-mediated autophagy is a strong candidate. Other IFN-independent activities we discovered in transcriptomic analysis of BMDMs are also possibilities.
Another important finding is that STING activates many signaling pathways in T cells and the majority of them are IFN-independent. Previous studies of STING biology have largely focused on myeloid cells and innate immunity. However, STING protein levels in lymphoid cells are much higher than those in myeloid cells (Gulen et al., 2017; Wu et al., 2019). Despite normal development of the lymphoid compartment in Sting1−/− mice, gain-of-function mutations of Sting cause severe T-cell immunodeficiency in mice and in human SAVI patients (Bouis et al., 2018; Liu et al., 2014; Motwani et al., 2019a; Warner et al., 2017; Wu et al., 2019). The mechanism by which STING induces T cell death requires further investigation. We showed here that different STING agonists activate qualitatively different T cell death that are either not or partially dependent on S365/IFN. It has been reported that different STING agonists stimulate different extent of calcium leakage from the ER when STING exits the ER (Kim et al., 2017). We also showed previously that calcium homeostasis plays a critical role in STING-N153S-mediated T cell death (Wu et al., 2019). STING interacts with and regulates the activity of the ER calcium sensor STIM1 (Srikanth et al., 2019). Thus, it is possible that STING agonists induce qualitatively different STING activation of the cell death pathway depending on their capacity to alter calcium response. Nonetheless, IFN-independent activities of STING discovered here involve many T cell effector functions, which should guide future studies of STING in T cell biology and adaptive immunity.
Lastly, we discovered a novel mechanism used by tumors to evade T cell-mediated immune control. On the adaptive immunity level, tumors hijack PD-1:PD-L1 interaction to inactivate surrounding T cells, and this can be therapeutically targeted by checkpoint inhibitor antibodies to reinvigorate T cell attack on tumors. We found that T cells in the tumor also experience considerable cell death compared to T cells in the dLN, and this tumor-induced T cell death is in part dependent on STING IFN-independent activities in T cells. Thus, STING-mediated T cell death pathway represents an ‘innate immune checkpoint’ mechanism that is also hijacked by tumors to compromise immune control. We showed that inhibitors targeting STING palmitoylation is effective at blocking STING-mediated T cell death in vitro. However, unless targeted delivery to T cells, STING inhibitors could also dampen STING-mediated IFN signaling in other cell types (e.g. DCs) and jeopardize other arms of anti-tumor immunity. The same pros and cons consideration also apply to STING agonists treatment of tumors. The clinical candidate STING agonist ADU-S100 induces favorable anti-tumor immunity at low dose, but when administered at high dose, causes substantial T cell death and compromised anti-tumor immunity (Sivick et al., 2018), presumably due to STING-mediated T cell death. As more STING agonists going into clinical trials, we propose that dosing optimization to balance pros (e.g. IFN response, DC:T cell priming) and cons (e.g. T cell death, tissue toxicity) of STING activation and management of T cell death via combination therapy should be important considerations.
In summary, our study revealed a new and exciting tumor immune evasion mechanism that is aimed at inactivating surrounding T cells, which resembles another ‘checkpoint’ that should be considered when designing next generation cancer immunotherapy. Further studies, especially in humans, are needed to elucidate how STING-mediated anti-tumor and pro-tumor effects balance out in human cancers and/or during STING agonist treatment.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for supporting data, resources, and reagents should be directed to and will be fulfilled upon request by the Lead Contact, Nan Yan (nan.yan@utsouthwestern.edu).
Materials Availabilty
Reagents from this study are available upon request.
Data and Code Availability
Original RNA-seq data is deposited at NCBI GEO (accession number GSE149744)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The Sting1S365A/S365A mutant mouse was generated by CRISPR/Cas9 technology at UT Southwestern Transgenic Mouse Facility on the C57BL/6 background. Sting1−/− mice (C57BL/6 background) were obtained from Glen Barber (Univ. of Miami). Wild type and Rag1−/− mice were purchased from UTSW Mouse Breeding Core. Irf3−/− mice were obtained from Kate Fitzgerald (Univ. of Mass Medical School) with permission from Tadatsugu Taniguchi. Ifnar1−/− mice were purchased from Jackson Labs. Both male and female mice were used. Most studies used 6–8 week old mice. All mice were maintained in pathogen-free barrier facilities and were used in accordance with protocols approved by the Institutional Animal Care and Use Committee at University of Texas Southwestern Medical Center.
Cell lines
HEK293T cells were purchased from ATCC. B16-F10 and MC-38 cell lines were obtained from Yang-Xin Fu (UT Southwestern). Sex of cell line information is not available. These cells were cultured in the DMEM with 10% (vol/vol) FBS, 2 mM L-glutamine, 10 mM Hepes, and 1 mM sodium pyruvate with the addition of 100 U/ml penicillin and 100 mg/ml streptomycin and cultured at 37°C with 5% CO2.
Viruses
HSV-1 (strain 17) were obtained from David Leib (Dartmouth Univ.). Vaccinia virus (B5R-GFP) was obtained from Bernard Moss (NIH). VSV-GFP was obtained from Asit Pattnaik (Univ. Nebraska).
METHOD DETAILS
Antibodies
The following antibodies were used for immunoblotting analysis: anti-STING (D2P2F; Cell Signaling), anti-TBK1 (D1B4; Cell Signaling), anti-pTBK1 (D52C2; Cell Signaling), anti-IRF3 (D83B9, Cell Signaling), anti-pIRF3 (4D4G, Cell Signaling), anti-p-p65 (93H1, Cell Signaling), anti-p65 (D14E12, Cell Signaling), anti-LC3 (Novus). The following reagents were used in this study: CFSE (ThermoFisher, C34554), Pan T Cell Isolation Kit II (Miltenyi, Order no. 130-095-130), Naive CD8a+ T Cell Isolation Kit (Miltenyi, Order no: 130-096-543), Trizol (Sigma, T9424). STING palmytoilation inhibitor C-176 was purchased from BioVision (B2341).
Primary cell preparation
For BMDM preparation, femurs and tibias were dissected from WT, Sting1−/−, Sting1S365A/S365A mice. Bone marrow was flushed from bones, and red blood cells were lysed with ACK lysis buffer. Debris was removed by passing cells through a 70-μM strainer and cells were counted via Countess II FL Automated Cell Counter. Approximately 5 × 106 bone marrow cells were cultured in 100-mm dishes in DMEM complete medium with 20% M-CSF-containing conditional medium from L929 cells for 6 days. The medium was changed on day 3 and day 5. Differentiated BMDMs were directly used for experiment.
For the T cell proliferation assay, the spleen was isolated, crashed, and pass through a 70-μM strainer to get the single cell suspension. Red blood cells were lysed by ACK lysis buffer. Pan-T cells were then isolated with the Pan T Cell Isolation Kit II (Miltenyi, Order no. 130-095-130) according manufacture’s instruction. T cells were stained with CFSE at 5 μM for 10 minutes at 37 degrees, then wash with RPMI complete medium twice. Labeled cells were cultured in cell culture plates pre-coated with anti-CD3 (145–2C11, 5 μg/ml) and anti-CD28 (37.51; both from eBioscience, 5 μg/ml) antibodies. After 3 days, T cells were stained with percp/cy5.5-anti-CD4 and APC-anti-CD8 antibodies and analyzed by FACS. T cell proliferation was determined by CFSE dilution. Flow cytometry data were analyzed using FlowJo.
For the T cell death analysis, pan-T cells were isolated and cultured in the 96-well plate pre-coated with anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) antibody in the presence of 20U/ml IL2 for 1 day. Next day STING agonists such as 2’3’-cGAMP, 3’3’-cGAMP, 2’3’-c-di-AM, DMXAA or CMA was added at indicated concentration. After 16 h treatment, the cells were harvested and stained with Annexin V Apoptosis Detection Kit (Biolegend) according to the manufacture’s instruction.
For the CD8 T cell cytotoxicity analysis, naïve CD8 T cells were isolated from spleen with Naïve CD8 T cell isolation kit (Miltenyi, Order no: 130-096-543). The cells were then cultured in plates pre-coated with anti-CD3 and anti-CD28 antibodies in the presence of 20U/ml IL2 for 2 days. For intracellular cytokine staining, 5μg/ml BFA was added 4 hours before cell collection. The cells were then stained with PE/Dazzle-IFNγ (1:200), FITC-TNFα (1:200), PE-Perforin (1:200) and APC-Granzyme B (1:200) with BD Fixation/Permeabilization Solution Kit according to manufacturer’s instruction.
RNA-seq
BMDMs and pan-T cells isolated from WT, Sting1−/−, Sting1S365A/S365A mice were treated with DMSO or DMXAA (10 μg/ml) for 16 h. Total RNA was isolated with TRIZOL and analyzed by bio-analyzer by the UT Southwestern Microarray Core Facility. RNA-seq was performed by BGI Americas. Gene expression levels were quantified as Fragments Per Kilobase Million (FPKM). Gene expression with FPKM value ≥ 0.5 in at least one sample were selected and Log2-transformed for the Differential Gene Expression (DEGs) analysis using iDEP (http://bioinformatics.sdstate.edu/idep/). DEGs were defined by 2-fold changes within each indicated paired-comparison and then used for pathway analysis. Total DEGs including both IFN-dependent and IFN-independent genes were defined by comparing WT DMX vs. WT M group. DEGs containing only IFN-independent genes were defined by comparing S365A DMX vs. S365A M group. DEGs containing only IFN-dependent genes were defined by comparing WT DMX vs. S365A DMX.
For heatmaps, DEGs from WT DMX vs. WT Mock were selected. The FPKM value for each gene were Log2-transformed and mean center normalized with indicated z-score. Heatmaps was plotted with “heatmap app” integrated into VisR software (https://visrsoftware.github.io/). For the MA plot, the Log2-transformed data were used to calculate gene average expression (x-axis) and Log2-fold change (y-axis), then data were plotted with GraphPad. DEGs were also used for Ingenuity Pathway Analysis (IPA, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). For the IPA analysis, the core analysis and comparison analysis were performed. P-values generated by the comparison analysis were used to plot pathway comparison between different samples. P-values were transformed into the −Log10(p-value) as the relative pathway enrichment score. Pathway enriched score ≥ 2 (reflect 0.01 difference in p value) was defined as significant. Total pathways were divided into three groups: WT pathway, −Log10(pWT-pS365A) ≥2; S365A pathway, −Log10(pWT-pS365A) ≤2; both, −2< Log10(pWT-pS365A)<2. Data was visualized with scatter plot with linear regression analysis to modeling the trend line of each pathway group. Top pathways were also grouped and plotted by bar graphs and selected pathways were further presented in heatmaps showing individual genes.
Tumor experiments
In the adoptive transfer T cell tumor control experiments, 5 × 106 naive CD8+ T cells were isolated from the spleens of WT, Sting1−/−, Sting1S365A/S365A donor mouse with the Naive CD8a+ T Cell Isolation Kit (Miltenyi, Order no: 130-096-543) and transferred into Rag1−/− recipient mice by i.v. injection. One day later, 5 × 105 B16-F10 cells or MC38 were s.c. injected on the flank of mice. Tumor size was measured by length (a), width (b) and height (c), and tumor volume was calculated as ½*a*b*c. Tumor immunohistology analysis was performed by HistoWiz (Brooklyn, New York).
For the B16 tumor, tumors were harvested, minced, and filtered through a 70-μm strainer to obtain single-cell suspensions. Red blood cells were lysed with ACK lysis buffer. After pelleting, cells were stained with a mixture of antibodies FITC-CD8 (1:200), PE-CD3 (1:200), APC-CD45 (1:200) and Zombie Aqua™ Fixable Viability cell death dye (1:500), then analyzed by BD LSRII flow cytometry. For the MC38 tumor, the tumors were harvested and minced, then digested in the RPMI medium supplemented with 1mg/ml Collagenase IV and 0.1 mg/ml DNase I for 40 min at 37 degree with shaking. The digest reaction was quenched with addition of 10 mM EDTA, and then the red blood cells were lysed with ACK lysis buffer. After twice wash the cells were pass through 70-μm strainer to obtain single-cell suspension. The single cells were stained with FITC-CD8 (1:200), PE-CD3 (1:200), APC-CD45 (1:200) and Zombie Aqua™ Fixable Viability cell death dye (1:500) for tumor infiltrating T cell analysis. For the draining lymph nodes, the collected lymph nodes were crashed and pass through the 70-μm strainer to obtain single-cell suspension. Red blood cells were removed by ACK lysis buffer treatment. The cell staining and cell death analysis was performed as indicated above. In the endogenous T cell tumor control experiment, 1 × 106 MC-38 cells were s.c. injected on the flank of WT, Sting1−/− or Sting1S365A/S365A mice. Fourteen days later, T cells from tumor or draining lymph node were stained with anti-CD45, anti-CD3, anti-CD8 and Zombie Aqua™ Fixable cell death dye analyzed for cell death by FACS.
RNA isolation and qRT-PCR
Total RNA was isolated with TRI reagent according to the manufacturer’s instructions (Sigma-Aldrich). cDNA was synthesized with an iScript cDNA synthesis kit (Bio-Rad). iTaq Universal SYBR Green Supermix (Bio-Rad) and an CFX96 Touch Real-Time PCR Detection System (Bio-Rad) were used for q-RT-PCR analysis. Hprt and Gapdh were used as housekeeping genes for data normalization.
Cytokine multiplex array and ELISA
WT, Sting1−/−, Sting1S365A/S365A, Ifnar1−/− BMDMs were transfected with cGAMP (5 μg/ml) for 24 h, the medium was collected for cytokine array analysis using the Bio-Plex Pro Mouse Cytokine Group I Panel 23-Plex Assay kit (Bio-Rad). The BioPlex Pro Assay was performed by the University of Texas Southwestern Genomics and Microarray Core according to the manufacturer’s protocol. IFNβ ELISA was performed separately from the array. For the IFNβ ELISA, 96 well Nunc MaxiSorp™ flat-bottom (Thermofisher #44-2404-21) plates were coated with the of IFNβ capture antibody (2 μg/mL) (BioLegend) diluted in sodium carbonate coating buffer (pH 9.5) and incubated overnight at 4 degrees Celsius. Plates were washed four times with 0.05% PBS-Tween-20 solution 4 times using a BioTek Microplate 405 LS Washer. ELISA plates were blocked using 1% BSA in PBS and incubated 1 hr at room temperature shaking on an orbital plate shaker at 250 rpm. Plates were washed 4 times. The IFNβ standard (BioLegend) was serially diluted seven times 1:1 in assay diluent composed of 1% Bovine Serum Albumin (BSA) in PBS starting with 1000 pg/mL down to 15.6 pg/mL to create a standard curve. Samples and standards were plated at 100 μL per well in duplicate and incubated at room temperature for 2 hrs shaking at 250 rpm. Plates were washed and incubated with biotinylated IFNβ detection antibody (BioLegend) at 2 μg/mL diluted in 1% BSA/PBS at room temperature for 1 hr shaking at 250 rpm. Plates were washed five times and then incubated with HRP-Avidin diluted at 1:1000 in 1% BSA in PBS for 30 minutes at room temperature and covered with foil to block out light. Plates were developed using the TMB substrate from BioLegend following manufacterer’s directions incubating for 30 minutes at room temperature not shaking, and enzymatic reactions were stopped using 2N sulfuric acid. Plates were read in our BioTek Synergy Plate reader at 450 nM and 570 nM. Results were calculated using a 5-parameter logistics curve-fitting algorithum at www.elisaanalysis.com.
Viral infection
Age and sex-matched WT, Sting1−/−, Sting1S365A/S365A mice were infected with wild type HSV-1 (stain 17, obtained from David Leib, Dartmouth Univ.) at sub-lethal dose 1 × 107 pfu per mouse via i.v. injection and monitored for survival. In a separate experiment (5 × 106 pfu per mouse), mouse brains were harvested at 6 d post infection, homogenized and total DNA was isolated by DNeasy Blood & Tissue Kits (Qiagen, Cat No./ID: 69504) according to manufacturer’s instruction. Viral DNA level was determined by qPCR with primers for HSV-1 polymerase: Forward, 5’-CATCACCGACCCGGAGAGGGAC-3’; Reverse, 5’-GGGCCAGGCGCTTGTTGGTGTA-3’.
For HSV-1 infection of BMDMs, the BMDMs from WT, Sting1−/−, Sting1S365A/S365A, Ifnar1−/− mice were infected with HSV-1 at the indicated MOI. The cells were harvested at 24 h post infection, and the total intracellular DNA was exacted with DNeasy Blood & Tissue Kits, the HSV-1 DNA level was determined by qPCR as indicated above. For the VACV and VSV infection, the BMDMs were infected with virus at indicated MOI for 12 h. The cells were then harvested, fixed and analyzed by FACS.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the mean ± SEM. Prism 8 (GraphPad) was used for statistical analysis. Statistical tests performed are indicated in figure legends. *, p < 0.05; **, p < 0.01; ***, p < 0.001; and ****, p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-TBK1/NAK (Ser172) (D52C2) XP® Rabbit mAb | Cell signaling | Cat#5483 |
| TBK1/NAK (D1B4) Rabbit mAb | Cell signaling | Cat#3504 |
| Phospho-IRF-3 (Ser396) (4D4G) Rabbit mAb | Cell signaling | Cat #4947 |
| Phospho-NF-κB p65 (Ser536) (93H1) Rabbit mAb | Cell signaling | Cat#3033 |
| NF-κB p65 (D14E12) XP® Rabbit mAb | Cell signaling | Cat#8242 |
| STING (D1V5L) Rabbit mAb (Rodent Preferred) | Cell signaling | Cat#50494 |
| GAPDH (D16H11) XP® Rabbit mAb | Cell signaling | Cat#5174 |
| LC3B Antibody | Novus | Cat#NB100–2220 |
| PE anti-mouse CD3 Antibody | Biolegend | Cat#100206 |
| FITC anti-mouse CD8a Antibody | Biolegend | Cat#100706 |
| APC anti-mouse CD45 Antibody | Biolegend | Cat#103112 |
| PE/Dazzle™ 594 anti-mouse IFN-γ Antibody | Biolegend | Cat#505845 |
| FITC anti-mouse TNF-α Antibody | Biolegend | Cat#506304 |
| PE anti-mouse Perforin Antibody | Biolegend | Cat#154305 |
| APC-Granzyme B | Invitrogen | Cat#17-8898-80 |
| CD3e Monoclonal Antibody (145–2C11) | Invitrogen | Cat#16-0031-81 |
| CD28 Monoclonal Antibody (37.51) | Invitrogen | Cat#16-0281-82 |
| Purified anti-mouse IFN-β Antibody | BioLegend | Cat#519202 |
| Biotin anti-mouse IFN-β Antibody | BioLegend | Cat#508105 |
| Chemicals, and Recombinant Proteins | ||
| 2’3’-cGAMP | Invivogen | Cat#tlrl-nacga23–02 |
| 3’3’-cGAMP | Invivogen | Cat#tlrl-nacga |
| 2’3’-c-di-AM(PS)2 (Rp,Rp) | Invivogen | Cat#tlrl-nacda2r-01 |
| DMXAA | Invivogen | Cat#tlrl-dmx |
| 9-Oxo-10(9H)-acridineacetic acid (CMA) | Sigma | Cat#17927 |
| CellTrace™ CFSE Cell Proliferation Kit | Invivogen | Cat#C34554 |
| Annexin V Apoptosis Detection Kit | Biolegend | Cat#640914 |
| DNeasy Blood & Tissue Kits | QIAGEN | Cat#69506 |
| Protease Inhibitor Cocktail | Roche | Cat#11873580001 |
| Lipofectamine 2000 | ThermoFisher scientific | Cat#11668019 |
| TRI Reagent | Sigma | Cat#T9424 |
| Annexin V Binding Buffer | Biolegend | Cat#422201 |
| RIPA Lysis Buffer | Millipore | Cat#20–188 |
| ACK Lysing Buffer | ThermoFisher scientific | Cat#A1049201 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher scientific | Cat#23225 |
| Polybrene | Sigma | Cat#TR-1003 |
| DNase I | Sigma | Cat#11284932001 |
| Collagenase | Sigma | Cat#C5138 |
| iScript™ Reverse Transcription Supermix | Bio-Rad | Cat#1708840 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | Cat#1725120 |
| Naive CD8a+ T Cell Isolation Kit | Miltenyi Biotec | Cat#130-096-543 |
| Pan T Cell Isolation Kit II | Miltenyi Biotec | Cat#130-095-130 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#E1960 |
| Recombinant Murine IL-2 | PEPROTECH | Cat#212–12 |
| C-176 STING inhibitor | BioVision | Cat#B2341 |
| Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| HRP Avidin | BioLegend | Cat#405103 |
| Recombinant Mouse IFN-β (ELISA Std.) | BioLegend | Cat#581309 |
| TMB Substate Set | BioLegend | Cat#421101 |
| Sulfuric acid (ELISA stop solution) | Millipore Sigma | Cat#160313 |
| Bacterial and Virus Strains | ||
| HSV-1 stain 11 | Dr. David Leib | N/A |
| VSV-GFP | Dr. Asit Pattnaik | N/A |
| Vaccinia virus (VACV) B5R-GFP | Dr. Bernard Moss | N/A |
| Recombinant DNA | ||
| Flag-cGAS PCMV | PMID:30886058 | N/A |
| Flag-STING PCMV | PMID:30886058 | N/A |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216 |
| B16-F10 | Dr. Yang-Xin Fu | N/A |
| MC38 | Dr. Yang-Xin Fu | N/A |
| Experimental Models: Organisms/Strains | ||
| C56BL/6J | Jackson Laboratory | JAX: 000664 |
| Sting1S365A/S365A C56BL/6J | This study | N/A |
| Rag1−/− C56BL/6J | Jackson Laboratory | JAX:002216 |
| Sting1−/− C51BL/6J | Dr. Glen Barber | N/A |
| Irf3−/− C51BL/6J | Dr. Tada Taniguchi | N/A |
| Ifnar1−/− C51BL/6J | Jackson Laboratory | JAX:028288 |
| Software and Algorithms | ||
| Graphpad Prism | Graphpad | https://www.graphpad.com/ |
| FlowJo | FlowJo | https://www.flowjo.com |
| iDEP90 | iDEP | http://bioinformatics.sdstate.edu/idep/ |
| Ingenuity Pathway Analysis (IPA) | IPA | https://digitalinsights.qiagen.com/product-login/ |
| VisR | VisR | https://visrsoftware.github.io/ |
| ELISA Analysis | ELISA | https://elisaanalysis.com/ |
| Deposited data | ||
| RNA-seq data | BGI | NCBI GEO (accession number GSE149744) |
Highlights:
Sting-S365A mutation in mice specifically abrogates IFN signaling.
STING has many IFN-independent activities that are physiologically important.
STING restricts HSV-1 infection via IFN-independent activities.
Tumor induces STING-mediated T cell death to evade immune control.
Acknowledgements
We thank Glen Barber (Univ. of Miami) for Sting1−/− mice, David Leib (Dartmouth Univ.) for wild type HSV-1 (strain 17), Bernard Moss (NIH) for Vaccinia virus (B5R-GFP), Ryan Huang, Robert Maples and Gustavo Torres-Ramirez for mouse breeding and members of the Yan lab for helpful discussions. This work was supported by National Institutes of Health (AR067135, AI134877, AI151708 to N.Y.), Cancer Prevention and Research Institute of Texas (CPRIT, RP180288) and the Burroughs Wellcome Fund (N.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors have no financial interests to declare.
References
- Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, Ebel C, Jacobs H, Knapp A-M, Jeremiah N, et al. (2018). Severe combined immunodeficiency in Sting V154M/WT mice. The Journal of allergy and clinical immunology. [DOI] [PubMed] [Google Scholar]
- Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, Picard C, Neven B, Fischer A, Amigorena S, et al. (2017). Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytesSTING inhibits T cell proliferation. The Journal of Experimental Medicine 214, jem.20161674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dis EV, Sogi KM, Rae CS, Sivick KE, Surh NH, Leong ML, Kanne DB, Metchette K, Leong JJ, Bruml JR, et al. (2018). STING-Activating Adjuvants Elicit a Th17 Immune Response and Protect against Mycobacterium tuberculosis Infection. Cell reports 23, 1435 1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, and Yan N (2015). STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 18, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz KM, Neidermyer WJ, Tan Y-J, Whelan SPJ, and Kagan JC (2018). STING-dependent translation inhibition restricts RNA virus replication. Proceedings of the National Academy of Sciences of the United States of America 12, 201716937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, and Chen ZJ (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat Commun 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, and Ablasser A (2018). Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Koch P, Li L, Peshkin L, and Mitchison TJ (2017). Evidence for a role of calcium in STING signaling. bioRxiv, 145854. [Google Scholar]
- Kurt-Jones EA, Orzalli MH, and Knipe DM (2017). Cell Biology of Herpes Viruses. 49–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahey LJ, Wen X, Mardjuki RE, Böhnert V, Ritchie C, Carozza JA, Hess GT, Maduke M, Bassik MC, and Li L (2020). The LRRC8A:C Heteromeric Channel Is a cGAMP Transporter and the Dominant cGAMP Importer in Human Vasculature Cells. bioRxiv, 2020.2002.2013.948273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, and Poltorak A (2017). Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. The Journal of Immunology 199, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, and Virgin HW (1999). Interferons Regulate the Phenotype of Wild-type and Mutant Herpes Simplex Viruses In Vivo. The Journal of Experimental Medicine 189, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014). Activated STING in a vascular and pulmonary syndrome. The New England journal of medicine 371, 507 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksch H, Stinson WA, Platt DJ, Qian W, Kalugotla G, Miner CA, Bennion BG, Gerbaulet A, Rosen-Wolff A, and Miner JJ (2019). STING-associated lung disease in mice relies on T cells but not type I interferon. J Allergy Clin Immunol 144, 254–266 e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, McWhirter SM, Katibah GE, Corn JE, Woodward JJ, and Raulet DH (2019). SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis SR, Wilson SC, and Vance RE (2017). Evolutionary Origins of cGAS-STING Signaling. Trends Immunol 38, 733–743. [DOI] [PubMed] [Google Scholar]
- Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, Burkly L, Marshak-Rothstein A, and Fitzgerald KA (2019a). Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proceedings of the National Academy of Sciences of the United States of America 74, 201818281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani M, Pesiridis S, and Fitzgerald KA (2019b). DNA sensing by the cGAS-STING pathway in health and disease. Nature Reviews Genetics, 1–18. [DOI] [PubMed] [Google Scholar]
- Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, and Taguchi T (2016). Activation of STING requires palmitoylation at the Golgi. Nature communications 7, 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, Bolt MJ, Arina A, Parry R, Kron SJ, et al. (2019). STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res 79, 1465–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie C, Cordova AF, Hess GT, Bassik MC, and Li L (2019). SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell 75, 372–381 e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, Yagita H, Guggisberg E, Saba S, Nascakova Z, et al. (2019). Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep 29, 1236–1248 e1237. [DOI] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Youngblood BA, Ahmed R, and Boss JM (2013). Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function. The Journal of Immunology 191, 3419–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, et al. (2018). Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep 25, 3074–3085 e3075. [DOI] [PubMed] [Google Scholar]
- Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, Rice L, Seo GJ, Calmettes G, Ramakrishna C, et al. (2019). The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol 20, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Sun L, Chen J, and Chen ZJ (2018). Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annual review of microbiology 72, 447 478. [DOI] [PubMed] [Google Scholar]
- Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, Sakai T, Gonugunta VK, Wu J, Platt DJ, et al. (2017). STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med 214, 3279–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, and Yan N (2019). STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med 216, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, et al. (2015). Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 163, 1716 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Manohar M, Wei Y, Pandol SJ, and Habtezion A (2019). STING signalling protects against chronic pancreatitis by modulating Th17 response. Gut 68, 1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original RNA-seq data is deposited at NCBI GEO (accession number GSE149744)