

Graphical abstract

Keywords: Terpene cyclization, QM/MM, Oxy-functionalization, Sustainable biocatalysis, Molecular dynamics
Abstract
While chemical steps involved in bioactive cembranoid biosynthesis have been examined, the corresponding enzymatic mechanisms leading to their formation remain elusive. In the tobacco plant, Nicotiana tabacum, a putative cembratriene-ol synthase (CBTS) initiates the catalytic cascade that lead to the biosynthesis of cembratriene-4,6-diols, which displays antibacterial- and anti-proliferative activities. We report here on structural homology models, functional studies, and mechanistic explorations of this enzyme using a combination of biosynthetic and computational methods. This approach guided us to develop an efficient de novo production of five bioactive non- and monohydroxylated cembranoids. Our homology models in combination with quantum and classical simulations suggested putative principles of the CBTS catalytic cycle, and provided a possible rationale for the formation of premature olefinic side products. Moreover, the functional reconstruction of a N. tabacum-derived class II P450 with a cognate CPR, obtained by transcriptome mining provided for production of bioactive cembratriene-4,6-diols. Our combined findings provide mechanistic insights into cembranoid biosynthesis, and a basis for the sustainable industrial production of highly valuable bioactive cembranoids.
1. Introduction
Terpenoids are the largest class of microbial secondary metabolites, characterized by a complex chemical architecture and a high degree of functional decoration that render their total chemical synthesis production challenging. Examples of high-value bioactive terpenoids are taxol, a potent anticancer drug, and artesiminin, an anti-malaria agent [1]. Cembranoids, represent a structurally rather diverse group of monocyclic diterpenoids (C20 terpenoids), which have been reported in soft coral and plants of the genus Pinus and Nicotiana sp. (tobacco) [2]. Most notably, cembranoids represent the major fraction of secondary metabolites detected in the secretions of tobacco plants, which indicates that they may be involved in its natural defense mechanisms. Interestingly, these hydroxylated cembranoids are reported to be of significant biomedical relevance, encompassing anti-bacterial, anti-fungal and anti-proliferative activities [2]. Almost 60% of the dry weight of the resinous substance covering the tobacco leaves contains (1S,2E,4S,6R,7E,11E)-2,7,11-cembratriene-4,6-diol (α-CBT-diol) and its epimer, (1S,2E,4R,6R,7E,11E)-2,7,11-cembratriene-4,6-diol (β-CBT-diol). These two compounds alone are reported to display a broad spectrum of antibacterial and antifungal activities. More recently, CBT-diols are flagged as potential therapeutics for neurodegenerative diseases that are increasingly prevalent in industrialized countries due to demographic change [3]. Furthermore, these compounds inhibit nicotine sensitization in rats, rendering them also interesting in treatment of nicotine addiction. The biosynthetic precursors of CBT-diols are (1S,2E,4S,7E,11E)-2,7,11-cembratriene-4-ol (α-CBT-ol, thunbergol) and (1S,2E,4R,7E,11E)-2,7,11-cembratriene-4-ol (β-CBT-ol, 4-epi-isocembrol), respectively. While these CBT-ol precursors are detected in much lower concentrations in planta, they are reported to have potent insecticidal activity specifically against aphids [4].
While the chemical steps in the biosynthesis of cembranoids have been elucidated, the detailed enzymatic mechanisms leading to the formation of this important class of natural products remains elusive [2]. The biosynthesis of CBT-ols takes place in the plastids of the glandular trichome cells of tobacco. In analogy to other plant diterpenes, the biosynthesis of cembranoids is initiated by cyclisation of the aliphatic, achiral (E,E,E)-geranylgeranyl pyrophosphate (GGPP) to form both monocyclic CBT-ol epimers. This process is catalyzed by a putative class I diterpene synthase (dTPS) termed cembratriene-ol synthase (CBTS) [3] (Fig. 1).
Fig. 1.

Cascade pathway, carbocation rearrangements, main and side-products of nCBTS and di-hydroxylated products of the nCYP/nCPR system. The black box depicts the found carbocation intermediates during the cascade pathway from GGPP->cation C. The red boxes depict the found deprotonated side-products and mono-hydroxylated main products of the nCBTS cyclization. The lower blue box depicts the di-hydroxylated products of the newly derived nCYP/nCPR system. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
A recent tobacco (N. tabacum) leaf trichome gene expression profiling resulted in annotation of a putative class I dTPS gene, which may be responsible for the cyclization of GGPP to both CBT-ols [5]. However, the definitive function of this putative dTPS gene has not yet been confirmed. Ionization dependent class I mono-, sesqui-, and di-terpene synthases contain a conserved asparagine-rich motif, and a second typical conserved “NSE/DTE” motif, involved in chelating a triade of catalytically important Mg2+- ions [6], [7]. This Mg2+ ion triade orients the GGPP pyrophosphate moiety (PPi) within the enzymes active site, followed by active site closure, and GGPP hydrolysis forming the geranylgeranyl carbocation that initiates the catalytic cycle. The structural remodeling of the initial carbocation intermediate towards the final product CBT-ol, may involve sequential hydride shifts, protonation and deprotonation reactions, which are orchestrated and chaperoned by the amino acid lining the enzymes active site [8]. Despite their low primary sequence identity, TPS share highly conserved tertiary and quaternary structural features, which are dominated by an α-helical barrel fold. The open, inactive TPS conformation, binds the linear GGPP substrate, which in turn triggers the formation of the closed, active conformation that catalyzes the reaction by stereochemical, electrostatic-, and kinetic control [8]. However, structural information of the closed, active conformation of the enzyme is required for understanding its catalytic principles. Recently the closed conformation of a dTPS was modeled and central steps of its catalytic cycle was characterized [8]. A putative mechanism for CBTS catalyzed CBTL-ol formation has also been proposed, but the cyclization cascade and formation of promiscuous side-products, remains unclear [9].
The final biocatalytic step in the biosynthesis of bioactive CBT-diols is the hydroxylation of CBT-ols at the C6 atom by a putative class II CYP450 monooxygenase (P450). Class II P450s receive electrons from a class II membrane bound FAD- and FMN- containing NADPH-dependent oxidoreductase (CPR), by a mechanism that has remained unclear. At present, it is challenging to achieve efficient hydroxylation of terpenoid macrocycles by class II P450s in conventional prokaryotic hosts due to their low solubility. A recent study, however, demonstrated that the overall production rate is highly dependent on the choice of the CPR and can be improved by transmembrane engineering in the prokaryotic host [10]. As the native CPR is often unknown, only a few examples for the successful hydroxylation of terpenoid macrocycles have been reported with class II P450s. Catalytic principles of terpene synthases have previously also been subjected to many computational studies that have highlighted key chemical transformation steps [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22].
We study here catalytic principles leading to N. tabacum derived CBT-ol and CBT-diol formation by reconstruction of the biosynthetic pathway in an engineered Escherichia coli host. Initially, we define the activity of a CBTS that is responsible for cyclisation of GGPP to CBT-ol. The catalytic principles are then explored based on homology models and quantum chemical calculations. Although the homology models do not provide as good starting point as experimentally resolved structures, they nevertheless allowed detailed insights into the cyclization cascade and identification of a catalytically relevant active site. Residues identified in the simulations were probed using experimental protein engineering approaches, which enabled us to develop an efficient de novo production of seven highly bioactive olefin, mono- and di-hydroxylated cembranoids in a metabolically engineered E. coli based microbial production host.
2. Material and methods
2.1. Bacterial strains, genes and vectors, cloning strategies, site-directed mutagenesis
The E. coli strains XL-1 Blue was used for cloning and BL21 (DE3) was used for the diterpene production. All strains and plasmids were obtained from Novagen/ Merk Millipore. Genes were synthesized by Life technologies GmbH containing the appropriate restriction sites and adjusting codon usage for E. coli. For genes, plasmids, and primer sequences used for the cembranoid production see the Supporting information materials (Supporting Tables S1–S3). All cloning procedures were performed according to standard protocols.
2.2. In vivo production of the cembranoid isomers
The in vivo approach was based on a culture volume of 3 × 1 L M9CA (M9 minimal media with 4 g/L casamino acids (Merck, Germany) was used in 5 L baffled glass flasks. The vectors pColaDuet-1 (dxp, dxs), pCDFDuet-1 (ispD/ispF, idi) and the vector pETDuet-1 (crte, cbts) were introduced into E. coli BL21 (DE3) by standard transformation procedures. For cultivation in shake flasks, single transformants were grown in 3 × 1 L M9CA medium supplemented with 10 g/L glycerol, 30 µg/mL kanamycin, 50 µg/mL streptomycin and 50 µg/mL carbenicillin. The cultures were inoculated at OD600 of 0.1 from an overnight culture (8 h cultivation 37 °C supplemented with 30 µg/mL kanamycin 50 µg/mL streptomycin and 50 µg/mL carbenicillin), grown at 37 °C and 130 rpm until OD600 of 0.8 and then cooled down to 25 °C. At a temperature of 25 °C 40 g/L glycerol was added and the cultures were induced by addition of 1 mM isopropyl β-D-1-thiogalactopyranoside. Cells were grown at 25 °C for 3 days.
2.3. Extraction and purification of the CBTS-derived cembranoid isomers
For the cembranoid isolation, cells were pelleted (15 min, 17,500 g, 4 °C). The supernatant was extracted 3 times by 200 mL ethyl acetate. Organic phases were combined, dried with MgSO4 and evaporated to dryness under continuous N2-stream. The crude extract was solved in 1 mL ethyl acetate and analyzed by GC–MS and GC-FID. For the quantification 100 µL of that resolved extract were evaporated to dryness, solved in 90 µL and 10 µL of the internal standard (α-humulene, Sigma Aldrich, final concentration 0.889 mg/L) were added. Purification of (+)-cembrene, (±)-isocembrene and (−)-casbene were carried out by flash chromatography. An isocratic 90/10 hexane/ethyl acetate silica step (Silica gel 40, Sigma-Aldrich,) was followed by a 1D thin layer chromatography (TLC) with 90/10 hexane/ethyl acetate as mobile phase. For the purification of both CBT-ol epimers by flash chromatography, the hexane/ethyl acetate solvent during the 1D-TLC run was changed to 50/50 followed by a 70/30 hexane/ethyl acetate mobile phase.
2.4. Batch bioprocess for the production of cembranoid isomers
5 L fermentations were performed in a 10 L bioreactor (LP351, Bioengineering AG, Wald, Switzerland) using M9CA medium supplemented with 10 g/L glycerol, 30 µg/mL kanamycin, 50 µg/mL streptomycin and 50 µg/mL carbenicillin. The pH was controlled at 6.8 with 4 M NH4OH and 5 M H3PO4. Oxygen saturation was constantly adjusted to 80%. 1 mg/L Antifoam B (Sigma Aldrich) was automatically added when necessary. The bioprocess was started by inoculation from 4 × 1 L overnight cultures (8 h cultivation 37 °C supplemented with 50 µg/mL kanamycin, 50 µg/mL streptomycin and 50 µg/mL carbenicillin) at OD600 = 0.25 and run 2 h at 37 °C. At a OD600 of 1.95 45 g/L glycerol and 1 mM ITPG were added aseptically, the culture was cooled down to 25 °C and grown for another 5 days. To determine optical densities and glycerol contents 10 mL aliquots were taken as triplicates. 1 mL of each triplicate was used for determination of OD600, 1 mL was used for determination of glycerol content by HPLC analysis. After the bioprocess 500 mL of culture was used for determination of cembranoid contents using ethyl-acetate extraction of the supernatant. The crude extract was resolved in 1 mL n-ethyl-acetate 100 µL of that resolved extract were evaporated to dryness under continuous N2-stream, solved in 90 µL and 10 µL of the internal standard (α-humulene, Sigma-Aldrich, final concentration 0.889 mg/L) were added. The samples were analyzed by GC–MS and GC-FID (Supporting Material and Methods).
2.5. Expression of CBTS, Δ56-CBTS and mutants for in vitro screening
Single transformations of pET28b (+) harboring wild type cbts, Δ56-cbts or mutations were cultivated for 18 h at 37 °C in LB medium (10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl) supplemented with 50 µg mL−1 kanamycin. These preinocula were used to seed fresh 500 mL LB media containing 50 µg mL−1 kanamycin at a starting OD600 of 0.1. After reaching an OD600 of 0.6 the cultures were cooled down to 16 °C, induced by 1 mM IPTG and grown at 16 °C for 24 h. Cells were harvested by centrifugation at 6.000 g for 15 min and suspended in buffer A (25 mM 3-(N-morpholino)-propane-sulfonic-acid pH 6.8, 1 mM MgCl2 and 1 mM dithiothreitol. Cells were disrupted by sonification on ice four times (60 s on and 90 s off) at medium power. Cell debris was cleared by centrifugation at 30.000 g for 1 h. The clear supernatant was applied to a pre-equilibrated HisTrap HP 5 mL column (GE Healthcare) at a flow rate of 1 mL min−1 with an ÄKTA Purifier system (GE Healthcare). The loaded column was washed one time with 5 column volumes of buffer A plus 20 mM imidazole and after that eluted with a linear gradient of 20–500 mM imidazole at a flow rate of 2.5 mL min−1. SDS-PAGE (12%) analysis was used to evaluate protein expression. Selected fractions were combined and dialysed over night at 4 °C against buffer A followed by another 6 h dialysis against fresh buffer A. Subsequently the solution was concentrated to 2 mL and subjected to isocratic size exclusion chromatography using a Superdex S75 10/300GL column (GE Healthcare) with buffer A at a flow rate of 0.8 mL min−1. Fractions were combined and concentrated to 2.5 mL. Protein concentration was determined by absorbance at 280 nm and enzyme were stored in 100 µL aliquots at −20 °C for further use.
2.6. In vitro assay using purified enzymes
100 µL of enzyme solution (1 mg mL−1) were added to 400 µL of buffer A (see above section). GGPP (Sigma Aldrich) (43.53 µM) was added to the solution. After gentle mixing, reaction solution was incubated for 12 h at 28 °C and 400 rpm. For extraction of products 750 µL ethyl-acetate was added to the reaction solution. After vigorous vortexing (2 × 20 s) phases were separated by centrifugation for 10 min, 4 °C and 12.000 rpm. Ethyl-acetate extraction was repeated twice; subsequently the organic phases were combined and evaporated to dryness under continuous N2-stream. The crude extract was resolved in 1 mL ethyl-acetate. 100 µL of that resolved extract were evaporated to dryness under continuous N2-stream, solved in 90 µL and 10 µL of the internal standard (α-humulene, Sigma-Aldrich, final concentration 0.889 mg mL−1) were added. The samples were analyzed by GC–MS.
2.7. In vivo assay for the production of CBT-diols
The four plasmid system was used, comprising pCola-, pCDF-, pET- and pACYC-Duet-1 vector (Supplemental Table S1) with the pACYC-duet-1 vector harboring the codon optimized fusion protein consisting of the membrane-engineered n-CYP450 monooxygenase (Genbank: AF166332.1, n-cyp450, synthesized and codon optimized at Thermo Fischer, GeneArt, Regensburg, Supplemental Fig. S30) and the newly assembled n-CPR (gene: n-cpr, synthesized and codon optimized at Thermo Fischer, GeneArt, Regensburg; Supplemental Fig. S31) combined via a 5‘- GSTSSGSGAS −3‘ linker peptide according to a previous report [10]. The 500 mL shake flask production was performed with M9CA medium instead of LB medium.
2.8. Extraction and purification of CBT-diols
The cultures were centrifuged (5000 rpm, 15 min). Supernatants were discarded and the cell pellets were washed in 100 mL H2O and centrifuged again (4500 rpm, 25 min). The cells were suspended in 30 mL H2O and disrupted by sonification (4 × 2 min, 5 × 10% intensity, 50% power). The lysates were extracted twice with 20 mL ethyl acetate and centrifuged at 15000 rpm for 15 min. Organic phases were combined and evaporated to dryness using a rotary evaporator. Remaining ethyl acetate was removed under a continuous N2 stream. The powder was dissolved in 1 mL ethyl acetate and subjected to a silica phase flash column chromatography with a 95/5 hexane/ethyl acetate isocratic elution to remove olefinic and single hydroxylated compounds. The mobile phase was subsequently changed to 70/30 hexane/ethyl acetate and both major CBT-diol epimers eluted as a combined fraction. This fraction was further purified by a 2D-TLC. The sample was evaporated to dryness and solved in 100% ethyl acetate. 10% of this sample was used for the stained control plate. 90% of the sample was filled on the edge of a preparative TLC plate. The plates were first subjected to an isocratic gradient of 90/10 hexane/ethyl acetate and after the run was finished horizontally subjected to an isocratic 70/30 hexane/ethyl acetate run. Both CBT-diol spots were finally analyzed by GC/MS analysis.
2.9. Molecular dynamics simulation
Molecular dynamics (MD) simulation of our homology model of CBTS with GGPP were embedded in a TIP3P water box (water density 0.997 g/ml, water probe radius 1.4 Å) and neutralized with a 0.9 mM NaCl concentration, and simulated using periodic boundary conditions at T = 298 K using the YASARA2-force field, an 1 fs integration time, a NPT ensemble and a cutoff of 8 Å for non-bonded interactions. Long-range coulomb electrostatics were treated using the Particle-Mesh-Ewald approach (PME) with a grid spacing of < 1 Å. The size of the simulation box was 125.00 × 156.83 × 123.79 Å3. Charges and force field parameters for GGPP and cations A-C were obtained from the AutoSmiles force field parameter assignment is YASARA Structure [23]. QM-optimized structure of cation A from Hong et al. [24] was docked in CBTS using the AutoDockVina program implemented in YASARA Structure using a rectangular 14 × 14 × 14 Å3 simulation cell around residue R316, V340, N347, S451, A487, D499, A764 and Y836. The docking simulations were performed treating all atoms as rigid, and binding modes were characterized based on a cluster analysis. Cations B and C were relaxed within the active site by energy minimization and a 1 ns MD-simulation using the YASARA2 force field [23]. MD simulations were performed using YASARA Structure (version 15.8.31, YASARA Biosciences, [23]a).
2.10. Homology modeling of the open conformation of CBTS and n-CPR
Homology modeling was performed in YASARA Structure [23], based on a PSI-Blast alignment using position specific matrices obtained from the primary alignments. Secondary structure prediction, loop-modeling, and side-chain and hydrogen bond network optimization were performed in presence of explicit solvent molecules. Templates used for the homology modeling were derived from primary and secondary structure alignments from the HHpred server with default settings [25]. (4S)-limonene synthase from Mentha spicata (PDB-ID: 2ong chain A; [26]); (+)-bornyl diphosphate synthase from Salvia officinalis (PDB-ID: 1n1b chain A; [27]) 5-epi-aristolochene synthase from N. tabacum (PDB ID: 3 m00, chain A; [28]) abietadiene synthase from Abies grandis (PDB ID: 3s9v, chain A; [29]) taxadiene synthase from T. brevifolia (PDB ID: 3p5p, chain A; [30]) were employed as templates for open conformation of CBTS. NADPH-cytochrome P450 reductase from Homo sapiens (PDB ID: 3qe2, chain A; [31]) and nitric-oxide synthase reductase from Rattus norvegicus (PDB ID: 1tll, chain A; [32]) were employed as templates for modelling of n-CPR.
2.11. Homology modeling of the closed conformation of CBTS
A molecular model for the truncated CBTS (GenBank accession number: AAS46038.1) lacking the predicted amino-terminal transit sequence (N-terminal amino acids 1–56) was built in YASARA Structure [33] using the same templates as for the wild type open CBTS conformation. The refined model was structurally aligned with the closed conformation of TXS. The structure of the unfolded A-C- and J-K loops, as well as the CBTS N-terminus were replaced by their corresponding secondary structure elements from TXS [30], followed by in silico mutation to obtained the corresponding CBTS residues. The structure was minimized for 10,000 steps, followed by a 10 ns MD simulation using the simulation setup and parameters described above. A second structural alignment between the closed conformation of TXS and the minimized CBTS model were generated, and the productive GGPP and internal water molecules of TXS were integrated into the CBTS complex. A second 10,000 steps energy minimization and 10 ns MD simulation were carried out to compute the final structural model of the closed conformation of CBTS.
2.12. Quantum chemical calculations and QM/MM MD simulations
The CBT-ol formation process was studied using in 1) a small QM system of the isolated ligand, with 53 atoms, 2) a cluster model consisting of 280 atoms and 3) a hybrid QM/MM model based on the homology modeled and MD-relaxed structures of CBTS. The small QM system consisted of the geranylgeranyl cation and the conversion of this reactant to cembranyl-4-cation modelled at the B3LYP-D3/def2-TZVP//B3LYP-D3/def2-SVP level of theory in gas phase. Corresponding structures, including the reactant, intermediates, transition states (TS) and the product, were characterized by frequency calculations. For the QM cluster model calculations, residues around 4 Å from the ligand were included. The model comprised the: ligand (geranylgeranyl cation), OPP−, 3 Mg2+, 9 H2O, and protein residues R316, R492, R506, D352, D495, D499, E503, S452, A498, L571, L566, C494, M491, T577, V344, resulting in a system with 281 QM atoms. Amino acids were cut at the Cβ-positions to provide a computationally feasibly sized model system, and fixed during the structure optimization at BP86-D3 level [34], [35], by employing the multipole accelerated resolution of identity approximation (RI-MARIJ) [36], and treating the environmental dielectric with ε = 4 using the conductor-like screening model (COSMO) [37]. Single point energies for the protein model are reported at the B3LYP/def2-TZVP level. The isolated ligand was studied at the B3LYP/def2-TZVP//B3LYP/def2-SVP level of theory [38], [39]. Previous studies have also employed other functional, such as M06-2X. We however expect the employed theory level provides a balanced and accurate for both the gas phase and protein homology models, which are also subjected to approximation. Entropic and zero-point energy corrections were evaluated from the Hessian matrix. For the QM/MM MD calculations, the CBTS model was first classically relaxed in a water box for 10 ns at T = 310 K and 1 bar pressure and using an integration time step of 1 fs, an NPT ensemble, and the CHARMM27 force field [40]. The ligand was treated using electrostatic potential charges calculated at B3LYP/def2-TZVP level and Lennard-Jones parameters derived from the CHARMM27 force field. The ligand was kept fixed during the relaxation. The electrostatics were treated using the particle mesh Ewald (PME) approach with a grid spacing of 1 Å. For further QM/MM simulations, the protein model was trimmed to include a sphere 20 Å around the ligand, with everything beyond 15 Å away from the ligand was kept fixed. The QM/MM model comprised about 5200 atoms, with the ligand described at B3LYP-D3/def2-SVP level of theory. All quantum chemical calculations were performed using TURBOMOLE [41], the QM/MM calculations were performed using the CHARMM/TURBOMOLE implementation [42], and the classical relaxation using NAMD2 [43].
3. Results
3.1. Defining the enzyme activity of a putative N. tabacum CBTS by recombinant expression in an engineered E. coli host
The N. tabacum derived CBTS gene (GenBank: AAS46038.1) was introduced into a previously reported three-plasmid based diterpenoid production system optimized for E. coli [8]. Typically, plant class I dTPS contain an N-terminal chloroplast leader sequence. To date, the CBTS protein has not been structurally characterized, hence the complete gene sequence encoding for a 589 amino acid protein was introduced into the diterpenoid producing strain. Three day shake flask cultures provided four unidentified diterpenoid compounds detected via gas chromatography/mass spectrometry (GC/MS) analysis (Fig. 2 and Supplemental Fig. S1). After sequential column purifications, three of the four diterpenoid compounds have subsequently been characterized by nuclear magnetic resonance (NMR) spectroscopy. The two most abundant diterpenoids compounds derived from the E. coli CBTS extract could initially be identified by NMR as (+)-cembrene (Supplemental Fig. S2 and Supplemental Table S4) and (−)-casbene (Supplemental Fig. S3 and Supplemental Table S5) using previously reported NMR reference spectra [44], [45]. Interestingly, NMR based identification of the third substance could only be accomplished after TMS (trimethylsilyl) derivatization and comparison to a TMS-silylated thunbergol standard. The third substance is a mixture of the CBT-ol epimers, termed α-CBT-ol and β-CBT-ol (Supplemental Fig. S4, S5, and S6) as revealed by comparison to recorded reference data (Supplemental Fig. S7 and S8 and Table S6 and S7). In three day shake flask cultures, the relative abundance of α- versus β-CBT-ol (0.3:1) generated by the CBTS was determined using 1H NMR spectroscopy [46]. In analogy to previous reports, the fourth compound that could not be separated from (+)-cembrene upon purification was assigned according to its mass spectrum as (±)-isocembrene (Supplemental Fig. S1b) [47]. The cumulative evidence confirms CBTS to be an active CBT-ol synthase.
Fig. 2.
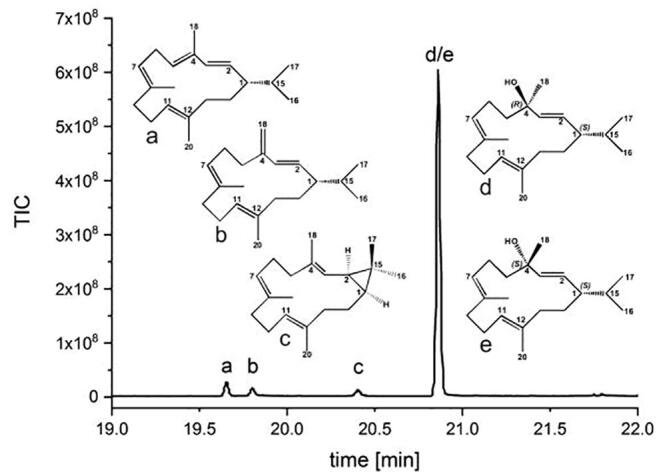
GC chromatogram of the ethyl acetate extract of the three day shake flask experiments. a: (+)-cembrene, b: (±)-isocembrene, c: (−)-casbene as well as d: α- CBT-ol and e: β-CBT-ol.
To optimize CBT-ol production, the fermentation was scaled to 5 L in a controlled, stirred tank bioreactor. The refined five day batch fermentation (Supplemental Fig. S9) provided final concentrations of 120 mg/L CBT-ol (α-CBT-ol: 54 mg/L, β-CBT-ol: 66 mg/L), 6.23 mg/L (+)-cembrene, 3.74 mg/L (±)-isocembrene and 1.88 mg/L (−)-casbene referenced against α-humulene as an internal standard (Supplemental Fig. S10).
3.2. Structural and mechanistic characterization of the CBTS from N. tabacum
Initially, we intended to gain structural information of CBTS by crystallization and X-ray crystallography. Since we failed in obtaining crystals of CBTS, we constructed a CBTS homology model from N. tabacum based on its reported primary sequence and the five X-ray structures of class I TPSs in order to structurally and mechanistically characterize CBTS. The resulting model displays the typical α-barrel fold of mono-functional plant class I terpene synthases, comprising both the signature α class I and the β class I domains (Supplemental Fig. S11). This structure represents the open, catalytically inactive conformation together with an unfolded N-terminal peptide. Comparison of the CBTS structure against taxadiene synthase (TXS), a structurally well characterized class I plant terpene synthases model, indicated that CBTS lacks the presence of a chloroplast leader sequence and a transmembrane region. This result is consistent with recombinant expression of the full length CBTS in the pET28b-vector, showing a good expression rate for CBTS in E. coli (Supplemental Fig. S12). The unfolded N-terminal region in CBTS comprises the first 56 amino acids, whereas TXS exhibits an N-terminal unfolded peptide that consists of 79 residues. However, TXS does not seem to rely on these 79 residues, as a genetic truncation retains catalytic activity [30].
Hence, for modeling of the closed CBTS complex, we excluded the first 56 N-terminal residues. To ensure that the generated Δ56-CBTS model still represents a soluble and active form of the enzyme, we expressed the truncated protein in E. coli and probed its activity by in vitro addition of GGPP. Both native and Δ56-CBTS proteins show identical expression levels (Supplemental Fig. S12), in vitro activities and product profiles.
Subsequently, we constructed the CBTS closed complex model as described previously [8]. The structural alignment of the α class I domain of the closed complex of Δ79-TXS with that of Δ56-CBTS exhibits a root mean square deviation (RMSD) of 0.31 Å over 321 residues with 27% primary sequence identity (Supplemental Fig. S11). The TXS/CBTS superposition suggests, that the two CBTS magnesium binding motifs are spatially located on α-helical secondary structure elements, that correspond to those reported for TXS [30]. Using the closed TXS complex containing GGPP in its productive conformation as a template [8], we constructed a putative closed complex of CBTS with GGPP coordinated by the three Mg2+ ions and corresponding water molecules. Water molecules and amino acid mediated bonding of productive GGPP resembles that of the productive closed conformation of TXS-GGPP template. Interestingly, the closed CBTS-GGPP complex exhibits a similar hydrogen-bonding donor network as observed in the TXS-GGPP template and other pro- and eukaryotic class I terpene synthases [6]. Specifically, this hydrogen bonding network is characterized by three arginine residues R316, R492 and R506. In TXS, this conserved triade, stabilizes PPi during the catalytic cascade. More importantly, the CBTS closed complex lacks the tyrosine residues, Y89 and Y835, which in closed TXS-GGPP model are important for binding of a water molecule adjacent to the PPi moiety upon active site closure. Instead, CBTS harbors two non-polar residues (F65, L571) in the analogous positions. These residues are chemically not able to coordinate water molecules, and may thus shield the active site. Furthermore, in contrast to TXS, where N757 coordinates the terminal oxygen-atom of PPi via a hydrogen bond, the analogous position in CBTS is occupied by an aspartate residue (D495) that does not form a similar coordinating bond. Instead, our obtained CBTS closed complex structure suggests that D495 might coordinate a water molecule, which originates from the bulk solvent upon active site closure (Fig. 3B). Interestingly, this water molecule is in much closer contact to the carbocation intermediate in comparison to the “protected” TXS-active site. Therefore, the water molecule in CBTS may experience enhanced mobility, as the coordination with Y835 in TXS is not present. The enhanced water mobility in the CBTS active site and its close proximity to the carbocation intermediate may facilitate premature carbocation quenching that could facilitate the formation of monocyclic cembranes as the main product. This hypothesis was examined using in silico guided CBTS protein engineering.
Fig. 3.

(A) Structural alignment of the closed form of TXS (blue) with the modeled closed form of Δ56-CBTS (red-ochre). Structural alignment of the closed form of TXS harbouring GGPP (blue) with the modelled closed form of Δ56-CBTS harbouring GGPP (red). Magnesium ions are shown in yellow (TXS) or green (CBTS). Important carbons of GGPP are shown in red (TXS) or magenta (CBTS). Corresponding amino acid residues are shown in blue (TXS) or red (CBTS). Mg2+ ions are coloured in red (TXS) or yellow (CBTS) and labelled A to C. (B) Inner view of the modelled productive closed form of CBTS, same view as in panel a. The Mg2+ coordinating motifs (D351, D352 and D355) and (D495, D499 and E503) are shown. GGPP is coloured in blue (C02, C15 in magenta, O01 of PPi in yellow), Mg2+ ions are coloured in green and bulk water is coloured in cyan. The H-bond donor network of CBTS is shown (yellow dotted lines), containing R316, R492 and R506 in analogy to TXS. CBTS harbours a phenylalanine residue F65 and a leucine residue L571 instead of the two tyrosine residues Y89 and Y835 in TXS that protect the active site of premature water quenching. (C) 2D diagram of CBTS-substrate interaction generated with LigPlot [49]. Same color-coding as in panel (A). For the sake of clarity water molecules have been omitted. Polar amino acids interacting with Mg2+ cations are shown in stick representation. Residues in hydrophobic contact with the substrate are represented by red semicircles with radiating spokes.–(D) Inner view of the modelled productive form of CBTS with the docked geranylgeranyl cation A. An identical colouring as in panel b is used. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Initially, we examined whether the identified CBTS hydrogen-bonding network displays a similar function as in TXS. Therefore, we substituted the amino acid network surrounding R316, R492 and R506 with aliphatic, aromatic, and other polar residues (Supplemental Table S3). Additionally, we substituted D495 and S452, which may be important for water coordination within the active site, to other small aliphatic and polar residues. Previously, we reported that in the TXS closed complex carbocation intermediates are stabilized by π-π and cation-π interaction [8], which coordinates the reaction cascade and determines the specific product outcome. In analogy, we replaced all residues with aromatic character, which decorate the CBTS active site, with aliphatic, polar and other aromatic residues and tested their reactivity in vitro.
As expected, single substitutions of the three essential arginine residues (R316, R492 and R506) resulted in inactive CBTS variants (Supplemental Table S10), consistent with the results for TXS [8]. Moreover, substitution of the aromatic CBTS residues W323, F570 and Y577 by aliphatic and non-aromatic polar residues resulted in inactive variants (Supplemental Table S10). By contrast, the amino acids substitutions W323Y, F570Y and Y577F resulted in CBTS variants with similar activity and product profiles as the wild type enzyme. The CBTS-GGPP closed complex model suggests that residues C491, L566 and V344 are in close proximity to the substrate and the corresponding carbocation intermediates generated during catalysis (Supplemental Fig S13). Although expression rates of CBTS variants L566H/W/A/S, V344K/M/L/S, C491A/S/Y, S452A/T/D and D495T/A/E were similar to that of the wild type enzyme, these amino acid substitutions resulted in inactive enzyme variants. Notably, only the CBTS C491L mutant showed a similar product profile as the wild type enzyme, while the D495N substitution does result in a significantly altered product profile were hydroxylated cembranoids are absent (Supplemental Table S10). Nonetheless, the generated concentrations of mono- and bi-cyclic hydrocarbons remains comparable to wild type CBTS. This cumulative data strongly suggests that D495 is involved in the control of water-assisted quenching of carbocation intermediates during the catalytic cycle. The in silico model of the D495N mutant shows that the amide group of the asparagine coordinates the terminal γ-oxygen of PPi via a hydrogen bridge instead of bulk water coordination (Supplemental Fig. S14), which leads to a displacement of the water molecule from the carbocation intermediate, and might thus explain the lack of hydroxylated hydrocarbon products observed in this mutant.
To probe possible water-quenching mechanism and the energetics of the discrete cyclization pathway, we performed quantum chemical density functional theory (DFT) and hybrid QM/MM-MD calculations of the isolated substrates and on the closed CBTS model. As a starting model for these calculations, we employed the closed conformation of CBTS harboring the docked geranylgeranyl cation A (Supplemental Table S9). We expect that the ca. 30% overall sequence identity of the homology model provides a sufficiently accurate starting point for exploration of the chemical principles.
3.3. Elucidation of the discrete cyclization pathway and the molecular basis for the amino acid-assisted water capture
We first studied the energetics in the gas-phase at the density functional theory level (B3LYP-D3/def2-TZVP//B3LYP-D3/def2-SVP), using the isolated ligand, with 53 atoms, as a model system. We find that the conversion of the geranylgeranyl cation A (Fig. 4A and B) into the first cyclic intermediate B is exothermic by −10.8 kcal mol−1, consistent with the expected stability of a tertiary carbocation, but the overall free energy for this step is 0.3 kcal mol−1, since the ring formation has a negative entropic contribution. Hong et al. [24] previously found a reaction energy of −2.9 kcal mol−1 (by including electronic energies and zero point corrections at the MPWB1K/6–31 + G(d,p)//B3LYP/6–31 + G(d,p) level), and a barrierless cyclization step, consistent with our QM/MM MD simulations (see below). Our calculations further suggest that formation of the cembra-4-yl cation (C) from cation B could occur via a stepwise mechanism involving the formation of the intermediate BC, via two low-energy transition states, TS1 and TS2, with barriers of 1.5 and 3.5 kcal mol−1, respectively, whereas the concerted route is energetically highly unfavorable by ca. 40 kcal mol−1, and is thus unlikely to occur. Our DFT calculations further suggest that formation of the cation C is exergonic by −6.5 kcal mol−1 in the gas phase (Fig. 4A).
Fig. 4.

(A) Energetics for the stepwise hydrogen shift from the geranylgeranyl cation (A) to cembranyl-4-cation (C) at the B3LYP-D3/def2-TZVP//B3LYP-D3/def2-SVP level of theory. Electronic energies (in purple) and Gibbs free energies (in black) are shown. Formation of the cationic intermediate C occurs via a two-step mechanism that yield two distinct intermediates, the cyclic products, B and BC. (B) Quantum chemical DFT models, comprising 281 atoms, of the CBTS active site with GGPP. Energetics (in kcal mol−1) of CBT-ol formation at the B3LYP-D3/def2-TZVP level in the protein and for the isolated ligand in gas phase. Only electronic energies are reported. (C) QM/MM MD simulations (B3LYP-D3/def2-SVP//CHARMM27) with the dynamics of the C1-C2 (red) and C1-C7 (black) bonds, and D495-O to H (in red) during 5 ps simulations QM/MM-MD simulations with D495 in its deprotonated (D495) and protonated (neutral) (D495+) states. (D) The central α-CBT-ol and β-CBT-ol structures, showing the hydrogen bonded network involving a water molecule and D495 that lead to a better stabilization of the β-CBT-ol product. Carbon, oxygen, nitrogen, and hydrogen atoms are colored in gray, red, blue, and white, respectively. (E) Central atoms of the structures are shown for the D495+ system. Carbon, oxygen, nitrogen, and hydrogen atoms are shown in gray, red, blue, and white, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Next we examined the hydride shift reaction, as well as formation of α-CBT-ol (PR) and β-CBT-ol (PS) within the active site of the CBTS. Our model consists of first and second sphere protein surroundings of the active site (about 280 atoms) that were constructed based on the homology models (see Methods, Supplemental Fig. S15A). We optimized these cluster models to obtain the reaction mechanism elements we found in our gas phase calculations. The formation of the cation C is exothermic by −16.2 kcal mol−1, similar to the gas-phase exothermicity of −18.3 kcal mol−1. Moreover, formation of the β-CBT-ol (PS) is about 10 kcal mol−1 more favorable than the α-CBT-ol (PR), in agreement with the experimentally observed product (Fig. 2).
The QM calculations suggest that D495 and S452 could stabilize the reaction product by formation of a hydrogen-bond with a nearby water molecule (Fig. 4D). D495 may thus indeed function as a primary proton acceptor for the water molecule, providing a precursor for the hydroxylation at C4 (see Fig. 1 for the numbering) that precedes the formation of α-CBT-ol and β-CBT-ol. In addition to this hydrogen-bonded network, β-CBT-ol is also in close contact with the OPP4−, suggesting that formation of the β-isomer is energetically preferred. Although the stabilization of the CBT-ol product is strongly stabilized by the hydrophilic amino acids and the OPP4− network, the formation of the cyclic carbocation intermediates could be controlled by the hydrophobic cage within the active site (Fig. 3). We therefore probed how the protein environment may control the formation of the first cycloadduct B. To this end, we modeled all possible monocyclic products, which could form via cation-π interactions involved in cation A using the isolated ligand model (Supplemental Table S9). We suggest that formation of a covalent bond between C7 and C2 is somewhat more favorable than our natural product (B) in CBTS (Supplemental Table S9). This alternative product involves a 6-membered ring, which is energetically more stable than a 19 membered cycle (B), but this structure might be sterically hindered from forming in the protein environment (Supplemental Fig. S15B). In order to probe the formation of possible cyclization products in the protein active site, we performed hybrid QM/MM MD simulations starting from compound A. We performed two independent 5 ps, simulations, with the D495 in its deprotonated (D495, Fig. 4C) and protonated (D495+; Fig. 4C) states. When D495 is modeled in its protonated state (D495+), we observe a spontaneous formation of B around in about 3 ps, as indicated by the decrease in the C1-C2 distance from 3 Å to 1.6 Å (Fig. 4C and E). This rapid cyclization process is consistent with the previously suggested barrierless ring formation in gas-phase [24]. We further find that the 6-membered ring or the other possible species are not formed in the protein due to the unique orientation of A within the active site in the homology model (Supplemental Table S9j). Conformational changes that are difficult to model could, however, in principles also allow for such side pathways. The active site of the CBTS contains several charged and polar residues, as well as the OPP4− and its Mg2+ counter ion that may favor the deprotonation of water. Considering the proximity and hydration energetics (Fig. 4), D495 may serve as proton acceptor, and providing a site for the further attack of the hydroxyl with the carbocation to form CBT-ol.
3.4. Functional reconstitution of class II P450 hydroxylase through in-silico indentification of a cognate N. tabacum reductase aimed at the biotechnological production of bioactive CBT-diols
Taxa-4,5-diene hydroxylation at C5 has been achieved in E. coli using protein engineering of the transmembrane region of the native taxadiene 5-α-ol hydroxylase (t-P450) and a cognate reductase (t-CPR) from Taxus cuspidate [10]. By contrast, the native class II CPR of CBT-diols-forming P450 monooxygenase (GenBank: AAD47832.1, n-P450) is, however, not available. We therefore employed several bioinformatic approaches to search for a suitable CPR in the genome of N. tabacum using the t-CPR as a template. This task was complicated as the whole genome sequence of N. tabacum is not available. In contrast, N. tabacum transcriptome sequence information is in form of expressed sequence tags (ESTs) available. Homology-based searches in the EST database with t-CPR as template provided several hits (Supplementary Fig. S16). However, the identified ESTs did not cover the entire t-CPR sequence space. Instead, only parts of t-CPR could be aligned with differing N. tabacum ESTs (Supplementary Fig. S17). According to a previous report, the identified parts resemble the domains that are responsible for FAD, FMN and NADPH binding [48]. Based on this information, the N. tabacum CPR (n-CPR) was assembled from these differing EST hits, since all of four TBLASTN hits constituted overlapping sequences. As expressed sequence tags did not display any transmembrane consensus motifs, the assembled n-CPR does not contain a transmembrane spanning α-helix, which renders this reductase naturally soluble in our E. coli production system. However, to establish CBT-diol production in our E. coli system, also the n-P450 had to be solubly expressed. This was accomplished by transmembrane engineering of the n-P450, in analogy to the transmembrane engineering of the 5α-ol hydroxylase from T. cuspidate (t-P450), which resulted in production of taxadiene-5α-ol [10].
To establish optimal expression conditions in E. coli, the assembled CPR (n-CPR) was genetically fused to the n-P450 via an N-terminal n-CPR GSTSSGSGAS-linker peptide, analogous to the genomic construct used for a soluble expression of the T. cuspidate class II P450 system [10]. Three day, 500 mL shake flask production in M9CA medium with glycerol as the sole carbon source resulted in measurable production of CBT-diols (Supplemental Fig. S18 and S19), which confirms the functional reconstitution of n-P450 using the assembled n-CPR. Subsequent purification steps using flash column chromatography, in combination with 1D- und 2D-TLC allowed the separation of the two major CBT-diol products from the ethyl acetate extract of the shake flask culture. The subsequent GC–MS analysis of the culture extracts identified GC peaks at 22.37 min and 22.33 min as α- and ß-CBT-diol respectively, when compared to authentic control standards (Supplemental Fig. S19). For NMR-spectroscopic identification of the generated CBT-diols the yields were unfortunately too low. However, GC–MS data indicated that α-CBT-diol seems to be the preferred substrate of the P450 monooxygenase from N. tabacum (Supplemental Fig. S20), as the relative titer of α-CBT-diol is increased compared to the β-CBT-diol titer.
4. Discussion and conclusions
In this study, we have explored catalytic principles of the cembranoid-centered biosynthetic enzymes CBTS and a specific P450-CPR system, whose functional reconstitution allowed design of an efficient biotechnological production route for bioactive cembranoids in E. coli. We could confirm the activity of CBTS as cembratriene-ol synthase and provide a possible cyclisation mechanism of this enzyme by combination of molecular simulation with site-directed mutagenesis experiments. The non-functionalized CTBS products ((+)-cembrene, (±)-isocembrene and (−)-casbene) have been shown to exhibit cytotoxic activity against various human cancer cell lines, whereas the mono-functionalized CBT-ols have been shown to significantly enhance the aphid resistance of tobacco plants. The bi-functionalized CBT-diols, which are generated by hydroxylation of primary diterpene macrocycles via a functionally reconstituted class II P450-CRP system (Fig. 1), display a high variety of biological activities including cytotoxicity, neuro-protectivity, antimicrobial and antifungal as well as insecticidal activity. The latter reconstitution of the P450-CRP system required in silico identification and assembly of a cognate CRP extracted from fragmented transcriptomic data of N. tabacum.
Using a simple E. coli production system, we produced non-functionalized and mono-functionalized cembranoid bioactives. Moreover, our study demonstrated that the high titer production of CBT-ols with up to 120 mg L−1 yield using glycerol as the sole carbon source is a good sustainable alternative compared to in planta production. Remarkably, we could also demonstrate that CBTS is able to produce five different cembranoid bioactives as major products or as side-products. In silico characterization of the N. tabacum CBTS by homology modeling and quantum chemical calculations further allowed us to understand the structural basis for three major non-functionalized by-products and a possible basis for the quenching process from GGPP to α- and β-CBT-ol.
Our quantum chemical calculations suggested that although the hydride shift and cyclization reaction of the geranylgeranyl cation have low barriers and are highly favorable in gas-phase, CBTS provides and important surroundings that favor ring closure at the correct C1-C2 position. We could observe spontaneous ring closure in the QM/MM MD simulations on picosecond timescales, particularly after protonation of D495. Moreover, our DFT cluster models further suggested that hydroxylation of cation C leading to formation of β-CBT-ol (-26.9 kcal/mol−1 Fig. 4B, PS) is energetically favored over α-CBT-ol formation (-17.4 kcal/mol−1 Fig. 4B, PS), due to the hydrogen-bonding network formed between D495 and S452, consistent with our NMR experiments and site-directed mutagenesis experiments showing that both residues are crucial for the catalytic activity of CBTS. These results further corroborate, that in absence of a crystal structure CBTS, our in silico modeled structures provide important insights into the function of the enzyme. Based on the computational data, we could further design targeted mutagenesis studies to demonstrate that the hydrogen-bonded donor network stabilizes PPi similar to its function in TXS. We were not, however, able to enhance production of one or more of the side-products in our targeted protein engineering experiments. Homologous substitutions of the respective amino acids resulted in a similar product profile, whereas non-homologous substituted enzymes showed no GGPP cyclization activity. This, together with the results obtained from our QM/MM-MD simulations indicate that the native decorative coating of the active site of CBTS is exclusively designed to produce monocyclic cembranoid structures.
Our combined data indicates that (+)-cembrene, (±)-isocembrene and (−)-casbene could derive from initial GGPP misfolding, imprecise barrier crossing and substrate tumbling during the cyclization cascade in CBTS, in line with structural investigations of TXS [8]. Our closed complex model of CBTS harboring cations B and C suggest that, in analogy to TXS, the bi-functional CBTS-motif depicted by R316-PPi, is the prime candidate for the premature deprotonation of the non-hydroxylated cembranoids (Fig. 5). Premature deprotonation by this bi-functional motif at cation B stage is expected to lead to the formation of (−)-casbene (Fig. 1). Cation tumbling during the cascade and/or initial GGPP misfolding would result in the premature deprotonation at C4 or C20 of cation C by R316-PPi that lead to the formation of (+)-cembrene or (±)-isocembrene instead of formation of the CBT-ols (Fig. 1).
Fig. 5.

(A) Closed conformation of CBTS harboring cation B, according to Fig, 4A. Coloring is according to Fig. 3. (B) Closed conformation of CBTS harboring (−)-casbene that could be derived from cation B of Fig. 5A, if premature deprotonation at H02 by the R316-PPi bi-functional motif occurs, due to GGPP misfolding or cation tumbling. A subsequent H14 -> C02 hydride shift accompanied with nucleophilic attack of cationic C15 at the then cationic C14 would enable the formation of the dimethyl-cyclopropyl moiety of (−)-casbene. (C) Closed conformation of CBTS harboring cation C. Premature deprotonation of H05 upon cation tumbling by the R316-PPi bi-functional motif could lead to the formation of (+)-cembrene, premature deprotonation of H20 upon cation tumbling during the cascade by this motif could lead to the formation of (±)-isocembrene.
We could show that the D495N substitution only produced non-hydroxylated cembranoids in a similar distribution compared to the wild type CBTS (Supplemental Fig. S21), supporting the putative role that this residue is catalysis. The closed complex model of the D495N mutant suggested that the amide group of N495 is directly engaged in ion pairing with PPi due to its close proximity and is thus presumably not able to act in the coordination of a remaining bulk water molecule. Combined with results from our QM calculations, this suggests that the residue is involved in an amino acid-assisted quenching reaction for functionalized cembranoids in CBTS.
In addition to the biotechnological production of non- and mono-functionalized cembranoids, the four plasmid system containing the n-P450-n-CPR redox couple, we were able to produce di-hydroxylated cembranoids, and provides a first report of a biotechnological production route for these bioactive compounds. Application of trans-membrane signal engineering and bioinformatics were crucial to produce α- and β-CBT-diols. Nevertheless, these compounds were produced only in minor amounts. A few examples for diterpenoid hydroxylations using class II P450 systems have been achieved. In all cases, production rates are low compared to the production rate of the non-functionalized precursors. Therefore, the choice of a eukaryotic host in combination with a more detailed trans-membrane engineering and the balancing of the metabolic flux through the up- and downstream pathway of cembranoid biosynthesis may result in an efficient biotechnological production of the two CBT-diol epimers. Our integrated experimental and computational approach could provide a basis for the rational design of engineered cembranoid enzymes.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgement
This work was in part supported by the German BMBF Grant SysBioTerp (support code 031A305A) to T.B. and the German BMBF Grant ELP (support code 1340/68351/3/11) to T.B. and P.S. We acknowledge support by the Open Access Publication Fund of the Freie Universität Berlin. We thank Ronja Driller and Max Hirte for support in preparing figures.
Author contributions
T.B. and P.S. designed the study, planned and supervised experiments. P.S. conducted metabolic engineering of E. coli and enzyme mutagenesis experiments. P.S. conducted fermentation experiments, NMR data collection and verified produced structures, and the homology modelling of CBTS. I.U., S.K. and V.R.I.K. performed the quantum chemical calculations. P.S., V.R.I.K., B.L., and T.B. wrote the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.06.030.
Contributor Information
Bernhard Loll, Email: loll@chemie.fu-berlin.de.
Ville R.I. Kaila, Email: ville.kaila@dbb.su.se.
Thomas Brück, Email: brueck@tum.de.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Brück T, Kourist R., Loll B. Production of macrocyclic sesqui- and diterpenes in heterologous microbial hosts: a systems approach to harness nature‘s molecular diversity. ChemCatChem. 2014;6:1142–1165. [Google Scholar]
- 2.Yan N., Du Y., Liu X., Zhang H., Liu Y., Zhang P. Chemical structures, biosynthesis, bioactivities, biocatalysis and semisynthesis of tobacco cembranoids: An overview. Ind Crops Prod. 2016;83:66–80. [Google Scholar]
- 3.El Sayed K.A., Sylvester P.W. Biocatalytic and semisynthetic studies of the anticancer tobacco cembranoids. Expert Opin Invest Drugs. 2007;16:877–887. doi: 10.1517/13543784.16.6.877. [DOI] [PubMed] [Google Scholar]
- 4.Mischko W., Hirte M., Fuchs M., Mehlmer N., Bruck T.B. Identification of sesquiterpene synthases from the Basidiomycota Coniophora puteana for the efficient and highly selective beta-copaene and cubebol production in E. coli. Microb Cell Fact. 2018;17:164. doi: 10.1186/s12934-018-1010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ennajdaoui H., Vachon G., Giacalone C., Besse I., Sallaud C., Herzog M. Trichome specific expression of the tobacco (Nicotiana sylvestris) cembratrien-ol synthase genes is controlled by both activating and repressing cis-regions. Plant Mol Biol. 2010;73:673–685. doi: 10.1007/s11103-010-9648-x. [DOI] [PubMed] [Google Scholar]
- 6.Christianson D.W. Structural and chemical biology of terpenoid cyclases. Chem Rev. 2017;117:11570–11648. doi: 10.1021/acs.chemrev.7b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aaron J.A., Christianson D.W. Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl Chem. 2010;82:1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schrepfer P., Buettner A., Goerner C., Hertel M., van Rijn J., Wallrapp F. Identification of amino acid networks governing catalysis in the closed complex of class I terpene synthases. Proc Natl Acad Sci USA. 2016;113:E958–E967. doi: 10.1073/pnas.1519680113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo W. Biosynthesis of cembratrienols in cell-free extracts from trichomes of Nicotiana tabacum. Plant Sci. 1995;110:1–10. [Google Scholar]
- 10.Ajikumar P.K., Xiao W.H., Tyo K.E., Wang Y., Simeon F., Leonard E. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science. 2010;330:70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Major D.T., Freud Y., Weitman M. Catalytic control in terpenoid cyclases: multiscale modeling of thermodynamic, kinetic, and dynamic effects. Curr Opin Chem Biol. 2014;21:25–33. doi: 10.1016/j.cbpa.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Freud Y., Ansbacher T., Major D.T. Catalytic control in the facile proton transfer in taxadiene synthase. ACS Catal. 2017;7:7653–7657. [Google Scholar]
- 13.O'Brien T.E., Bertolani S.J., Zhang Y., Siegel J.B., Tantillo D.J. Predicting productive binding modes for substrates and carbocation intermediates in terpene synthases-bornyl diphosphate synthase as a representative case. ACS Catal. 2018;8:3322–3330. doi: 10.1021/acscatal.8b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Major D.T. Electrostatic control of chemistry in terpene cyclases. ACS Catal. 2017;7:5461–5465. [Google Scholar]
- 15.Shoemaker S.R., Zhang Y., O'Brien T., Tantillo D., Siegel J.B. Hybrid quantum mechanics/rosetta modeling mechanistic study of a terpene synthase. Biophys J. 2020;118:304a-a. [Google Scholar]
- 16.van Rijn J.P.M., Escorcia A.M., Thiel W. QM/MM study of the taxadiene synthase mechanism. J Comput Chem. 2019;40:1902–1910. doi: 10.1002/jcc.25846. [DOI] [PubMed] [Google Scholar]
- 17.Escorcia A.M., van Rijn J.P.M., Cheng G.J., Schrepfer P., Bruck T.B., Thiel W. Molecular dynamics study of taxadiene synthase catalysis. J Comput Chem. 2018;39:1215–1225. doi: 10.1002/jcc.25184. [DOI] [PubMed] [Google Scholar]
- 18.Tantillo D.J. Biosynthesis via carbocations: theoretical studies on terpene formation. Nat Prod Rep. 2011;28:1035–1053. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- 19.Dixit M., Weitman M., Gao J.L., Major D.T. Chemical control in the battle against fidelity in promiscuous natural product biosynthesis: the case of trichodiene synthase. ACS Catal. 2017;7:812–818. doi: 10.1021/acscatal.6b02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen N., Zhou J., Li J., Xu J., Wu R. Concerted cyclization of lanosterol C-ring and D-ring under human oxidosqualene cyclase catalysis: an ab initio QM/MM MD study. J Chem Theory Comput. 2014;10:1109–1120. doi: 10.1021/ct400949b. [DOI] [PubMed] [Google Scholar]
- 21.Tian B., Poulter C.D., Jacobson M.P. Defining the product chemical space of monoterpenoid synthases. PLoS Comput Biol. 2016;12 doi: 10.1371/journal.pcbi.1005053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Driller R., Janke S., Fuchs M., Warner E., Mhashal A.R., Major D.T. Towards a comprehensive understanding of the structural dynamics of a bacterial diterpene synthase during catalysis. Nat Commun. 2018;9:3971. doi: 10.1038/s41467-018-06325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong Y.J., Tantillo D.J. The taxadiene-forming carbocation cascade. J Am Chem Soc. 2011;133:18249–18256. doi: 10.1021/ja2055929. [DOI] [PubMed] [Google Scholar]
- 25.Söding J., Biegert A., Lupas A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyatt D.C., Youn B., Zhao Y., Santhamma B., Coates R.M., Croteau R.B. Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc Natl Acad Sci USA. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whittington D.A., Wise M.L., Urbansky M., Coates R.M., Croteau R.B., Christianson D.W. Bornyl diphosphate synthase: structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc Natl Acad Sci USA. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noel J.P., Dellas N., Faraldos J.A., Zhao M., Hess B.A., Jr., Smentek L. Structural elucidation of cisoid and transoid cyclization pathways of a sesquiterpene synthase using 2-fluorofarnesyl diphosphates. ACS Chem Biol. 2010;5:377–392. doi: 10.1021/cb900295g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou K., Gao Y., Hoy J.A., Mann F.M., Honzatko R.B., Peters R.J. Insights into diterpene cyclization from structure of bifunctional abietadiene synthase from Abies grandis. J Biol Chem. 2012;287:6840–6850. doi: 10.1074/jbc.M111.337592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koksal M., Jin Y., Coates R.M., Croteau R., Christianson D.W. Taxadiene synthase structure and evolution of modular architecture in terpene biosynthesis. Nature. 2011;469:116–120. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia C., Panda S.P., Marohnic C.C., Martasek P., Masters B.S., Kim J.J. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc Natl Acad Sci USA. 2011;108:13486–13491. doi: 10.1073/pnas.1106632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcin E.D., Bruns C.M., Lloyd S.J., Hosfield D.J., Tiso M., Gachhui R. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 33.Land H., Humble M.S. YASARA: a tool to obtain structural guidance in biocatalytic investigations. Methods Mol Biol. 2018;1685:43–67. doi: 10.1007/978-1-4939-7366-8_4. [DOI] [PubMed] [Google Scholar]
- 34.Perdew J.P. Density-functional approximation for the correlation-energy of the inhomogeneous electron-gas. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 35.Grimme S., Antony J., Ehrlich S., Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132 doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 36.Sierka M., Hogekamp A., Ahlrichs R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J Chem Phys. 2003;118:9136–9148. [Google Scholar]
- 37.Klamt A., Schuurmann G. Cosmo - a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perk T. 1993;2:799–805. [Google Scholar]
- 38.Schäfer A., Horn H., Ahlrichs R. Fully optimized contracted gaussian-basis sets for atoms Li to Kr. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 39.Weigend F., Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. PCCP. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 40.MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 41.Ahlrichs R., Bär M., Häser M., Horn H., Kölmel C. Electronic-structure calculations on workstation computers - the program system turbomole. Chem Phys Lett. 1989;162:165–169. [Google Scholar]
- 42.Riahi S., Rowley C.N. The CHARMM-TURBOMOLE interface for efficient and accurate QM/MM molecular dynamics, free energies, and excited state properties. J Comput Chem. 2014;35:2076–2086. doi: 10.1002/jcc.23716. [DOI] [PubMed] [Google Scholar]
- 43.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai S., Jain M. 1H and 13C assignments of five cembrenes from guggul. Magn Reson Chem. 2008;46:791–793. doi: 10.1002/mrc.2252. [DOI] [PubMed] [Google Scholar]
- 45.Crombie L., Kneen G., Pattenden G., Whybrow D. Total synthesis of the macrocyclic diterpene (–)-casbene, the putative biogenetic precursor of lathyrane, tigliane, ingenane, and related terpenoid structures. J Chem Soc Perkin Trans. 1980;1:1711–1717. [Google Scholar]
- 46.Wahlberg W, Narbonne, Nishida, Enzell. Note on the Stereostructures of Thunbergol (Isocembrol) and 4-Epiisocemb. Acta Chemica Scandinavica. 197;35:65-8.
- 47.Chow S.Y., Williams H.J., Huang Q., Nanda S., Scott A.I. Studies on taxadiene synthase: interception of the cyclization cascade at the isocembrene stage with GGPP analogues. J Org Chem. 2005;70:9997–10003. doi: 10.1021/jo0517489. [DOI] [PubMed] [Google Scholar]
- 48.Yamada T., Imaishi H., Oka A., Ohkawa H. Molecular cloning and expression in Saccharomyces cerevisiae of tobacco NADPH-cytochrome P450 oxidoreductase cDNA. Biosci Biotechnol Biochem. 1998;62:1403–1411. doi: 10.1271/bbb.62.1403. [DOI] [PubMed] [Google Scholar]
- 49.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.