

Abstract
Background:
The pathology of eosinophilic esophagitis (EoE) is characterized by eosinophil-rich inflammation, basal zone hyperplasia (BZH), and dilated intercellular spaces, and the underlying processes that drive the pathologic manifestations of the disease remain largely unexplored.
Objective:
We sought to investigate the involvement of the calcium-activated chloride channel anoctamin 1 (ANO1) in esophageal proliferation and the histopathologic features of EoE.
Methods:
We examined mRNA and protein expression of ANO1 in esophageal biopsy samples from patients with EoE and in mice with EoE. We performed molecular and cellular analyses and ion transport assays on an in vitro esophageal epithelial 3-dimensional model system (EPC2-ALI) and murine models of EoE to define the relationship between expression and function of ANO1 and esophageal epithelial proliferation in patients with EoE.
Results:
We observed increased ANO1 expression in esophageal biopsy samples from patients with EoE and in mice with EoE. ANO1 was expressed within the esophageal basal zone, and expression correlated positively with disease severity (eosinophils/high-power field) and BZH. Using an in vitro esophageal epithelial 3-dimensional model system revealed that ANO1 undergoes chromatin modification and rapid upregulation of expression after IL-13 stimulation, that ANO1 is the primary apical IL-13–induced Cl− transport mechanism within the esophageal epithelium, and that loss of ANO1-dependent Cl− transport abrogated esophageal epithelial proliferation. Mechanistically, ANO1-dependent regulation of basal cell proliferation was associated with modulation of TP63 expression and phosphorylated cyclin-dependent kinase 2 levels.
Conclusions:
These data identify a functional role for ANO1 in esophageal cell proliferation and BZH in patients with EoE and provide a rationale for pharmacologic intervention of ANO1 function in patients with EoE.
Keywords: Eosinophilic esophagitis, esophageal epithelium, basal cell hyperplasia, IL-13, chloride transport
Graphical Abstract

Eosinophilic esophagitis (EoE) is a recently emerging chronic inflammatory disease of the esophagus and is clinically characterized by upper gastrointestinal symptoms, including dysphagia and esophageal food impaction.1 The histopathologic manifestations involve intraepithelial infiltration of eosinophils (>15 eosinophils/high-power field [hpf]) and modifications of the esophageal epithelium, such as basal zone hyperplasia (BZH) and dilated intercellular spaces (DISs), and can lead to a narrow-caliber esophagus.2, 3
Clinical evidence reveals that EoE is characterized by immune sensitization to dietary food antigens and marked TH2-associated allergic inflammation in the esophageal mucosa that is largely refractory to acid-suppressive therapy.4-8 Indeed, during active disease, the histopathologic changes within the inflamed esophageal mucosa include the dense accumulation of type 2 immune cells, including eosinophils and mast cells.9,10 Dietary modification (ie, complete or targeted food antigen avoidance) and swallowed glucocorticoids alleviate much of the disease pathology, suggesting that adaptive immune responses are the likely driver of histopathologic manifestations of EoE.4,11-15 Consistent with this argument, the cytokine IL-13, which is generally produced by activated TH2 effector cells and highly upregulated in esophageal tissue of patients with EoE, is sufficient to alter gene expression in esophageal epithelial cells in vitro and in vivo. The IL-13–induced esophageal epithelial transcriptome changes significantly overlap with the transcriptional changes observed in the esophagi of mice engineered to overexpress IL-13, as well as esophageal biopsy samples of patients with EoE.5,6,16
With the recent advancement of whole-genome transcript expression profiling, there has been substantial progress made with regard to identifying the underlying molecular pathways involved in EoE.6,16-18 These studies have revealed a link between allergic cytokines, particularly IL-13, in esophageal epithelial proliferation and dysregulation of esophageal epithelial barrier function.5,19 Indeed, IL-13 has been shown to dysregulate the expression of a number of key epithelial barrier regulatory genes, including desmosomal cadherin desmoglein 1,19 leucine-rich repeat–containing protein 31,20 kallikrein serine proteases,20 and calpain-14,21 which have been linked to EoE and shown to alter esophageal epithelial barrier function. However, while there has been significant advancement in our understanding of the immune mechanisms that drive the allergic inflammatory response in EoE, there is a paucity of data revealing the underlying pathways that regulate epithelial basal zone (BZ) expansion in patients with EoE.
Esophageal epithelial BZ expansion in patients with EoE is defined as a basal cell layer exceeding 15% of total epithelial thickness.22 BZH is a consequence of basal cell proliferation, and the underlying pathways that regulate BZH in patients with EoE are poorly understood. The functional implication of BZH in the symptomatology of EoE is not yet fully delineated; however, BZH is thought to contribute to esophageal barrier dysfunction and chronic inflammation, which in turn could lead to an associated functional phenotype associated with EoE.
In the last several years, there has been emerging interest in the role of bioelectric circuits in the regulation of cellular proliferation.23 These studies have revealed a role for bioelectric regulation and activation of K+ currents by ATP-sensitive K+ channels and Ca2+-activated K+ channels, as well as Cl− currents by Ca2+-activated Cl− channels and chloride intracellular channels in cellular proliferation.23 Recently, we performed RNA sequencing (RNA-seq) analyses on esophageal biopsy samples from pediatric patients with EoE and found increased expression of key drivers of K+ and Cl− currents in patients with EoE, including anoctamin 1 (ANO1), which is also known as transmembrane member 16A.16,24
In this study we examined the involvement of ANO1 in esophageal epithelial proliferation and the contribution of this anion exchanger to BZ proliferation in patients with EoE. We demonstrated upregulation of ANO1 in esophageal biopsy samples from patients with EoE. ANO1 expression levels correlated with EoE severity (eosinophils/hpf) and BZH. Mechanistic analyses revealed that IL-13 stimulation of esophageal epithelial cells increased intracellular chloride concentration ([Cl−]i) levels and cellular proliferation and that this was associated with transcriptional and functional dysregulation of ANO1. Notably, induction of ANO1 was linked to increased esophageal epithelial proliferation and BZ hyperplasia, and both pharmacologic inhibition and gene silencing of ANO1 attenuated IL-13–induced esophageal epithelial proliferation. These studies identify ANO1 as a key regulator of esophageal proliferation and BZH in EoE and identify modification of ANO1 activity as a possible therapeutic strategy for attenuation of esophageal remodeling and EoE severity.
METHODS
Human subjects
Previously collected and archived paraffin-embedded and RNA samples from patients with EoE and healthy control subjects were obtained from multiple US institutions16,25-27 from 2000-2015, and the diagnosis of EoE was as per 2007 and 2011 consensus diagnostic guidelines.28,29 Healthy control subjects were defined as patients having a variety of nonspecific upper gastrointestinal complaints, including vomiting, loose stools, abdominal pain, or nausea, who underwent endoscopy and biopsy and were demonstrated to have no histologic evidence of esophageal disease and no history of EoE diagnosis and had 0 esophageal eosinophils/hpf within proximal or distal esophageal biopsy samples obtained during the same endoscopy procedure as the analyzed samples. For EoE diagnosis, patients needed to have 15 or more eosinophils in at least 1 hpf in proximal or distal esophageal biopsy samples, with other causes of esophageal eosinophilia excluded and without a response to acid suppression as per 2007 and 2011 consensus diagnostic guidelines.28,29 RNA-seq and validation quantitative RT-PCR analyses were performed on the esophageal biopsy samples (healthy subjects, n = 7; patients with EoE, n = 10), as previously described (National Center for Biotechnology Information Gene Expression Omnibus database under accession no. GSE58640; cohort 1).16 Correlative analyses of eosinophils/hpf and ANO1 mRNA studies (Fig 1) were performed on a second independent cohort (n = 166; healthy control subjects, n = 38; patients with EoE, n = 138), as previously described (cohort 2).26 RNA-seq and histologic (eosinophils/hpf and BZH quantification) studies (Fig 1, E and F) were performed on a third independent cohort (patients with EoE, n = 46), as previously described (cohort 3).27 This study was approved by the Cincinnati Children’s Hospital Medical Center (CCHMC) Institutional Review Board.
FIG 1.

ANO1 expression in patients with EoE. A, Reads per kilobase per million (RPKM) values for ANO1 from RNA-seq of esophageal biopsy samples from 10 patients with active EoE versus 7 healthy control subjects (NL). ***P = .0001, Mann-Whitney test. B, qPCR analysis of ANO1 expression in esophageal biopsy samples from independent healthy control (NL; n = 10) or active EoE (n = 10) cohorts.16 Data are represented as relative expression over HPRT. ***P = .0003, Mann-Whitney test. C, Pearson correlation coefficient analyses between eosinophil counts per hpf and normalized ANO1 expression in esophageal biopsy samples from patients with active EoE (n = 138).26 D, IF staining of esophageal biopsy samples from control subjects (NL; left panel; n = 7) and patients with active EoE (n = 7; right panel). The basal cell layer of the squamous epithelium is distinguished by a dashed line. ANO1 (in red) and CK5 or CK13 (in green) expression is shown. Solid white arrows indicate basal cells (CK5+ and CK13− cells), open white arrows indicate suprabasal cells (CK5+ CK13−; CK5+ CK13+ and CK5− CK13+), and yellow arrows indicate CK5− and CK13− cells, which possess multilobed nuclei. DAPI, 4′-6-Diamidino-2-phenylindole dihydrochloride. E and F, Pearson correlation coefficient analyses between EoE severity (expressed as number of distal eosinophils per hpf) and ANO1 expression levels (Fig 1, E) and BZH scores and ANO1 expression (Fig 1, F) in esophageal biopsy samples from pediatric patients with EoE.27 Individual data points represent 1 patient. In Fig 1, A and B, the line represents the mean value and significance of differences determined by using the Mann-Whitney test.
Air-liquid interface culture system
hTERT immortalized human esophageal keratinocytes (hTERT-EPC2) were a kind gift from Dr Anil Rustgi (University of Pennsylvania, Philadelphia, Pa).30,31 For the air-liquid interface (ALI) system, cells were grown to confluence on 0.4-μm pore size permeable Transwells (Corning, Corning, NY) in normal keratinocyte serum-free media (Life Technologies, Carlsbad, Calif) containing 0.09 mmol/L [Ca2+] followed by an additional 4 days in high-calcium keratinocyte serum-free media ([Ca2+] = 1.8 mmol/L) to induce tight junction formation. Media were removed from the inner chamber of the permeable support to create an apical ALI to induce differentiation and epithelial stratification. Five days after ALI exposure, differentiated esophageal epithelial cells were used for experiments. In some experiments EPC2-ALI cells were exposed to omeprazole (100 μg/mL) for 2 hours, followed by treatment with IL-13 (100 ng/mL) for 72 hours, and cell lysates were collected for quantitative RT-PCR analyses, as described using the primers described in Table E1 in this article’s Online Repository at www.jacionline.org.
RNA-seq and bioinformatics analysis
RNA-seq of human esophageal biopsy sample results were obtained from a previous analysis.16 For RNA-seq of EPC2-ALI cells, RNAwas isolated from cells with the RNeasy Kit (Qiagen, Germantown, Md), according to the manufacturer’s protocol. RNA quality was assessed by using the Agilent 2100 Expert Bioanalyzer (Agilent Technologies, Santa Clara, Calif), and only the samples with RNA integrity numbers of greater than 8 were processed for sequencing. RNA samples were subjected to RNA-seq at the CCHMC Gene Discovery and Genetic Variation Core, as previously described.32 Sequencing reads were aligned against the GRCh37 genome model by using TopHat2.04 with Bowtie 2.03,33,34 and the separated alignments were unified with Cuffmerge35 by using the UCSC model as a reference. Raw data were then uploaded on BioWardrobe (http://biowardrobe.com) and analyzed with the Differential Sequencing (DeSeq) tool.36
Biopsy sample preparation
Typically, 6 sections per distal esophageal biopsy sample were cut at a thickness of 5 μm from formalin-fixed, paraffin-embedded biopsy samples and stained with hematoxylin and eosin. The grade score for the epithelial BZ was performed by Dr Margaret Collins, as previously described.37 Eosinophil counts were expressed as eosinophils/hpf (400 × 0.3 mm2) and recorded as peak eosinophil counts in distal biopsy samples. The BZ of esophageal squamous epithelium is composed of closely packed small cells and normally occupies 15% or less of total epithelial thickness. The upper limit of the BZ was defined as the level at which basal epithelial cell nuclei were separated by a distance equal to or greater than the diameter of the basal cell nucleus. The grade score for BZH was based on the amount of total epithelial thickness occupied by the BZ37: 0, BZH not present; 1, BZ occupies greater than 15% but less than 33% of total epithelial thickness; 2, BZ occupies 33% to 66% of total epithelial thickness; and 3, BZ occupies greater than 66% of total epithelial thickness. The stage score for BZH was based on the amount of the biopsy sample that exhibited any BZH: 0, BZH not present; 1, BZH (any grade >0) in less than 33% of epithelium; 2, BZH (any grade >0) in 33% to 66% of epithelium; and 3, BZH (any grade > 0) in greater than 66% of epithelium.
Quantitative PCR
RNA was extracted from EPC2-ALI cells by using the RNeasy kit (Qiagen), according to the manufacturer’s protocol. Purified RNA (1 μg) was DNase treated and reverse transcribed to cDNA by using Superscript II RNase H Reverse Transcriptase (Thermo Fisher Scientific, Rockford, Ill), according to the manufacturer’s instructions. cDNA for ANO1 and hypoxanthine phosphoribosyltransferase (HPRT) was quantified by using real-time PCR with the IQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, Calif) with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories). Quantitative PCR (qPCR) analyses were performed by using Bio-Rad CFX Manager Software (version 3.1; Bio-Rad Laboratories), and the results were normalized with HPRT amplified from the same cDNA mix and expressed as fold induction. Primers used for amplification are reported in Table E1. qPCR for chromatin immunoprecipitation (ChIP) analysis was performed on a promoter sequence of peptidylprolyl isomerase A (PPIA) or B-cell lymphoma 6 (BCL6) protein as positive controls for H3K4me3 histone mark or signal transducer and activator of transcription 6 (STAT6) ChIP, respectively, whereas a promoter sequence for the myogenic differentiation 1 gene (MYOD) was used as a negative control. Genes were quantified by using real-time qPCR with the TaqMan Universal PCR Mastermix chemistry (Applied Biosystems by Life Technologies, Carlsbad, Calif) with PrimeTime 5′ 6-FAM/ZEN/3′ IBFQ TaqMan probes (IDT, Coralville, Iowa) and reported as percentage over the input sample. Primers for each sequence are reported in Table E1.
H3K4me3 histone mark and STAT6 ChIP-seq
After 5 days of ALI culture, EPC2 cells were treated with IL-13 (100 ng/mL) for 4, 12, or 48 hours for H3K4me3 or 30 minutes for STAT6 ChIP. After stimulation, cells were cross-linked with 0.8% formaldehyde for 10 minutes at room temperature (RT). Cells were collected and then lysed and sonicated for 10 minutes (peak power, 175 W; duty factor, 10%; cycles/burst, 200 count) at 1.5°C with a Covaris S series Sonicator (Covaris, Woburn, Mass), yielding chromatin fragments of 200 to 300 bp in size. Each chromatin sample was precleared with 10 μL of Dynabeads Protein G magnetic beads (Life Technologies, Carlsbad, Calif) for 45 minutes at 4°C on a rotating wheel to remove nonspecific binding. Anti-H3K4me3 (1 μL; ChIPAb+ Trimethyl-Histone H3 [Lys4]; Millipore, Temecula, Calif) or anti-STAT6 (2.5 μg; sc-621; Santa Cruz Biotechnology, Santa Cruz, Calif) were bound to 20 μL of Dynabeads Protein G magnetic beads (Life Technologies) for 1 hour at RT on a rotating wheel. Chromatin complexes (5 μg for histone mark or 25 μg for STAT6 ChIP) were then added to the antibody-coated beads and immunoprecipitated for 14 to 16 hours at 4°C on a rotating wheel. Precipitates were washed with 500 μL of each of the following buffers in succession: low-salt wash buffer (10 mmol/L Tris-HCl [pH 7.6], 1 mmol/L EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, and 150 mmol/L NaCl), high-salt wash buffer (10 mmol/L Tris-HCl [pH 7.6], 1 mmol/L EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, and 400 mmol/L NaCl), LiCl wash buffer (10 mmol/L Tris-HCl [pH 7.6], 1 mmol/L EDTA, 250 mmol/L LiCl, 0.5% sodium deoxycholate, and 0.5% Nonidet P-40), and Triton X-100 buffer (10 mmol/L Tris-HCl [pH 7.6], 1 mmol/L EDTA, and 0.2% Triton X-100).
Chromatin complexes were eluted in 100 μL of elution buffer (10 mmol/L Tris-HCl [pH 7.6], 1 mmol/L EDTA, 250 mmol/L NaCl, and 0.3% SDS) and incubated at 37°C for 1 hour with shaking. For protein degradation and reverse cross-linking, 5 μL of Proteinase K solution (Life Technologies) was added to the eluates and incubated at 37°C for 1 hour, followed by incubation at 65°C for 6 hours. DNA was purified with the QIAquick PCR Purification Kit (Qiagen, Valencia, Calif), eluted in 30 μL of molecular biology grade water (Sigma, St Louis, Mo), and processed for qPCR before sequencing. Libraries were created, as described previously,38 and sequenced by the CCHMC Gene Discovery and Genetic Variation Core.
Short circuit current
Snapwell inserts (0.33 cm2) with EPC2-ALI cell monolayers were mounted between the hemichambers of an Ussing apparatus (U2500 dual Ussing chamber; Warner Instruments, Hamden, Conn) and exposed to 10 mL of Ringer buffer at 37°C. Transepithelial potential difference was detected with 2 paired electrodes containing 4% agar in 3 mol/L KCl. The electrodes were connected to a voltage clamp amplifier (EC-800 Epithelial Voltage Clamp; Warner Instruments). The electrode potential difference and fluid resistance were compensated before mounting the cells into the chamber. Once mounted, cells were bathed for a 15-minute stabilization period before voltage clamping at 0 mV while continuously measuring the short circuit current (Isc). Changes in Isc were determined with replacement of the solution in the apical chamber with Cl−-free normal Ringer (NR) buffer to create a gradient. ANO1-mediated changes in Isc were determined with the addition of T16Ainh-A01 to the luminal reservoir. After the peak response following the gradient or inhibitor was recorded, the buffer was replaced from each side of the chamber, allowing the cells to equilibrate for 15 minutes.
Measurement of EPC2 intracellular Cl− concentration
All measurements were performed in NR solution (110 mmol/L NaCl, 5 mmol/L KCl, 0.5 mmol/L MgSO4.7H2O, 1 mmol/L CaCl2.2H2O, 25 mmol/L NaHCO3, 10 mmol/L HEPES, and 4 mmol/L glucose to a pH of 7.4) at RT. EPC2 cells (100,000) were plated on an Ibidi μ-Slide 4 Well Ph chambered coverslip (80446; Ibidi, Gräfelfing, Germany) for 12 hours to equilibrate. The cells were then stimulated with 100 ng/mL rhIL-13 (PeproTech, Rocky Hill, NJ) for 12 hours and loaded with 5 mmol/L N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE; Thermo Fisher Scientific, Waltham, Mass) in NR solution for 1 hour at 37°C. Thereafter, the cells were rinsed with NR 3 times and imaged on a Nikon SpetraX inverted fluorescent microscope (Nikon, Tokyo, Japan), with excitation at 395 nm and emission at 460 nm. Twenty individual cells (region of interest [ROI] = 20) per well in duplicates per condition from 3 individual experiments were selected as ROIs by using Nikon Elements software for analysis. The mean fluorescence value from all cell cultures was used as the basis for further analysis.
Once MQAE fluorescence had been measured under physiologic conditions (in NR with a physiologic extracellular Cl− concentration), MQAE fluorescence was calibrated against [Cl−]i by incubating cell cultures in a series of gradient Cl− concentration buffers (0, 19.25, 38.5, 77, 154, and 304 mmol/L [Cl−]i.) together with 10 μmol/L tributylin (Sigma-Aldrich, St Louis, Mo) and 10 μmol/L nigericin (Life Technologies). F0 is mean fluorescence intensity at 0 mmol/L [Cl−]i. The F0/F ratio was calculated from data fitted by using the Stern–Volmer equation39:
where F is the mean ROI fluorescence value (in arbitrary units), [Cl−]i is the bath chloride concentration (in millimoles per liter), F0n is defined as F([Cl−]I = 0), and Ksv is the Stern–Volmer constant (in millimoles per liter). Mean fluorescence values of 20 individual cells (ROI) under physiologic conditions (in NR solution with an physiologic extracellular Cl− concentration) were determined. Curve fitting was performed with GraphPad Prism software (GraphPad Software, La Jolla, Calif).
Histopathologic examination
EPC2 cells were cultured in ALI conditions for 5 days and treated for 48 hours with IL-13 (100 ng/mL), T16Ainh-A01 (10 μmol/L), or both and were then fixed on Transwell support with 4% paraformaldehyde for 18 to 24 hours at 4°C. Fixed membranes were then submitted to the CCHMC Pathology Core for standard histologic processing, embedding (standard paraffin), and sectioning (5 μmol/L), and slides were stained by using standard histologic techniques for hematoxylin and eosin staining. Stained slides were imaged with the Olympus DP-72 microscope and cellSens standard software (Olympus, Semrock, New York, NY) for measurement analysis.
Immunofluorescence staining
For immunofluorescence (IF) staining, formalin- or paraformaldehyde-fixed, paraffin-embedded esophageal biopsy samples were sectioned, mounted on slides, and deparaffinized by using standard histologic procedures. Slides were then permeabilized in Tris-EDTA (1 mmol/L, pH 9.0) with 0.1% Tween-20, and antigen exposure was performed at 125°C for 30 seconds in a decloaking chamber by using a pressure cooker. Slides were then blocked by 10% normal donkey serum for 1 hour, followed by overnight incubation of primary antibodies diluted in 10% normal donkey serum at a final concentration of 1 μg/mL: anti-ANO1 (DOG-1; Thermo Fisher Scientific), anti-KRT5 (ab24647; Abcam, Cambridge, United Kingdom), and cytokeratin (CK) 13 (Invitrogen, Carlsbad, Calif). Slides were then washed and incubated with secondary antibody at RT for 1 hour. Slides were mounted with Fluoromount-G (SouthernBiotech, Birmingham, Ala) mounting solution.
Fluorescent imaging was performed with the Zeiss Apotome fluorescent microscope (Carl Zeiss Meditec, Dublin, Calif) using Nikon Elements software. For IF staining on formalin-fixed, paraffin-embedded murine esophageal tissue, paraffin-embedded murine esophagus was sectioned, mounted on slides, and deparaffinized with standard histologic procedures. Slides were then permeabilized in Tris-EDTA (1 mmol/L, pH 9.0) with 0.1% Tween-20. Antigen retrieval was performed at 125°C for 30 seconds in a decloaking chamber. Slides were then blocked with 4% normal donkey serum in 1 × PBS followed by overnight incubation of primary antibodies diluted in 4% normal donkey serum: ANO1,anti-transmembrane member 16A antibody (1:100; Abcam, and Ki-67 antibody (1:200; Thermo Fisher Scientific). Slides were then washed in 1 × PBS and incubated in secondary antibodies at RT for 2 hours. Slides were mounted with 4′-6-diamidino-2-phenylindole dihydrochloride Fluoromount-G (SouthernBiotech) mounting medium. Fluorescence imaging was performed with the Nikon E800 Epifluorescence microscope with Nikon Elements software and ImageJ software (National Institutes of Health, Bethesda, Md).
Quantification of 5-bromo-2′-deoxyuridine–positive EPC2-ALI cells
5-Bromo-2′-deoxyuridine (BrdU) was obtained from Sigma-Aldrich. EPC2 cells were cultured for 5 days in ALI conditions; treated for 48 hours with IL-13 (100 ng/mL), T16Ainh-A01 (10 μmol/L), or both; and then exposed to BrdU (10 μmol/L) in dimethyl sulfoxide for 2 hours at 37°C. Cells were then fixed on support for 2 hours with 4% paraformaldehyde at RT. Fixed membranes were then processed by the CCHMC Pathology Core for standard embedding and microscopy techniques. Cells were permeabilized in Tris-EDTA (1 mmol/L, pH 9.0) with 0.1% Tween-20 and antigen exposed at 125°C for 30 seconds in a decloaking chamber. Samples were then stained for IF detection of BrdU+ cells by using 2.5 μg/μL G3G4 anti-BrdU antibody (Developmental Studies Hybridoma Bank, Iowa City, Iowa) and 4′-6-diamidino-2-phenylindole dihydrochloride. The number of BrdU+-labeled cells were quantitated under a 20 × objective by using the Zeiss Apotome fluorescent microscope (Carl Zeiss Meditec). The BrdU+-labeled cells were quantified as the number of BrdU+-labeled cells per total linear length of filter-attached EPC2-ALI cells using Image-Pro Plus software (Media Cybernetics, Rockville, Md) and calculated based on the ratio of BrdU+ cells per millimeter of membrane. Three filters per condition from 3 individual experiments were quantitated.
Lentiviral transduction
For lentiviral short hairpin RNA (shRNA) transduction, EPC2 cells were transduced when at 60% to 70% confluence with lentiviral particles containing Mission ANO1 shRNA TRCN0000440455 or Mission nontarget control shRNA (Sigma). Both ANO1 and control shRNA lentivirus were generated by the CCHMC Viral Core using a 4-plasmid packaging system. Lentiviral particles were incubated with EPC2 cells for 6 hours at a multiplicity of infection from 0.5 to 10 in the presence of 5 μg/mL hexadimethrine bromide (Polybrene; Sigma). During the first hour of incubation, cells were spun down at 1000g for 1 hour at RT. Six hours after transduction, cells were put in fresh keratinocyte serum-free media, and 24 hours later, 1 μg/mL Puromycin (Thermo Fisher Scientific) was added for selection. Cells were grown with selection pressure and cultured as regular EPC2 cells. Stable knockdown for ANO1 was demonstrated by using qPCR.
Western blotting
EPC2-ALI cells were lysed aftr stimulation with a protein extraction reagent (10% glycerol, 20 mmol/L Tris-HCl [pH 7], 137 mmol/L NaCl, 2 mmol/L EDTA, and 1% NP-40 in H2O) with Halt protease inhibitor cocktail (Thermo Fisher Scientific). Approximately 40 μg of protein extract was separated on a 4% to 12% Bis-Tris gel and transferred to a nitrocellulose membrane (Life Technologies). For detection of phosphorylated cyclin D kinase 2, the Cell Cycle (pCDK/pHH3/Actin) Western blot cocktail (ab136810; Abcam) was used. IRDye 800 CW goat anti-rabbit IgG (H1L; LI-COR Biosciences, Lincoln, Neb) was used as a secondary antibody for detection.
Mouse models of EoE
BALB/c wild-type mice were injected with 100 μg of peanut extract (Greer Laboratories, Lenoir, NC) adsorbed to 1 mg of alum on days 0 and 14. On days 21, 23, and 25, mice were challenged with 50 μg of peanut extract in 50 μL of PBS intranasally, followed by oral gavage with 2 mg of ground peanut in 200 μL on days 27 and 29. Mice were sacrificed on day 30 and esophagi excised and analyzed.
Statistical analysis
Statistical significance of the EPC2-ALI samples was established by using unpaired t tests (2-tailed) with Welch correction, 1-way ANOVA when sample groups were greater than 2, or 2-way ANOVA with more than 1 variable. With nonnormally distributed data from patients’ biopsy specimens, the Mann-Whitney test was used, and correlation analyses were assessed with the Pearson product-moment correlation coefficient test. Graphs and statistical analyses were performed with GraphPad Prism software (GraphPad Software) or using the De-Seq algorithm on the BioWardrobe platform (http://biowardrobe.com)36.
RESULTS
Increased ANO1 mRNA expression in patients with EoE and correlation with disease severity
To gain insight into the potential involvement of bioelectric pathways in esophageal epithelial proliferation in patients with EoE, we analyzed gene expression levels obtained from RNA-seq analyses from patients with active EoE compared with healthy control subjects.16 ANO1, a calcium-activated chloride channel, was found to be significantly upregulated (50-fold induction) in esophageal biopsy specimens from patients with EoE when compared with levels in healthy subjects (Fig 1, A). Among ion transporters previously implicated in proliferation, the increased expression was specific to ANO1 because other ion transport molecules, such as KCNA3 and KCNA5 (Kv 1.3 and Kv 1.5), were barely expressed at baseline or in esophageal biopsy specimens from patients with EoE (results are not shown). To confirm our observation, we performed qPCR, which revealed a similarly significant increase in ANO1 expression (Fig 1, B). Notably, we observed a positive Pearson correlation coefficient between esophageal eosinophil counts (r = 0.3, P < .005) and ANO1 mRNA expression, revealing a link between ANO1 gene expression and disease severity (Fig 1, C).
IF analyses revealed weak expression of ANO1 in healthy esophageal epithelium restricted to basal cells (CK5+ and CK13−; Fig 1, D, left panels, solid white arrow). In contrast, a strong ANO1 signal was observed in biopsy samples from patients with EoE (Fig 1, D, right panels). ANO1 expression was observed within the basal layer localized to basal cells (CK5+ and CK13−; solid white arrow) and on suprabasal cells (CK5+ CK13−, CK5+ CK13+, and CK5− CK13+), which consist of cells transitioning from immature basal cells to mature squamous epithelial cells (Fig 1, D, open white arrows). We did not observe expression of ANO1 on mature squamous epithelial cells (Fig 1, D, left and right panels). Notably, ANO1 expression was occasionally observed on CK5− and CK13− cells within the esophageal epithelium of biopsy samples from patients with EoE, which were consistent with multilobed granulocytes (Fig 1, D, yellow arrows).
Localization of ANO1 to basal and suprabasal cells within the proliferative BZ led us to examine the relationship between ANO1 expression and BZH in esophageal biopsy samples from patients with EoE. In a recent study we examined the histopathologic features (BZH and DIS) and EoE-related gene transcript expression (EoE diagnostic panel) in esophageal biopsy samples from healthy subjects and patients with EoE.27 Examination of the relationship between ANO1 expression and the histologic features of EoE in this cohort revealed that ANO1 expression was significantly increased in patients with EoE and that levels of ANO1 mRNA expression positively correlated with EoE severity (eosinophils/hpf; r = 0.57, P = .003; Fig 1, E). Notably, ANO1 expression also had a positive correlation with the BZH score in our cohort subjects (r = 0.58, P < .0001; Fig 1, F). Collectively, these studies reveal a relationship between ANO1 expression and BZH in pediatric patients with EoE.
Increased esophageal ANO1 expression and epithelial proliferation in murine EoE
To determine whether there was a link between induction of the EoE phenotype and ANO1 expression, we examined the relationship between ANO1, BZH, and inflammation in a murine model of allergen-induced EoE. We show that peanut sensitization and subsequent oral challenge induced an EoE-like phenotype (increased numbers of eosinophils/hpf and esophageal proliferative [Ki-67+] response, Fig 2). Notably, numbers of esophageal proliferative response (Ki-67+) cells were associated with increased ANO1+ signal in the esophageal epithelium (Fig 2, B and C). The level of esophageal proliferation (Ki-67+) positively correlated with the level of inflammation severity (eosinophils/hpf; Fig 2, D). Collectively, these studies reveal that repetitive oral allergen challenge induced esophageal ANO1 expression and epithelial proliferation and that this correlates with inflammation severity in a murine model of EoE.
FIG 2.

ANO1 expression in murine EoE. A and B, IF staining for Ki-67 and ANO1 in the esophagi of control mice (Fig 2, A) and mice with EoE (Fig 2, B). C, Quantification of Ki-67+ epithelial cells in the esophagi of control and EoE mice. In Fig 2, A and B, ANO1 (in red) and Ki-67 (in green) and nuclei are stained with 4′-6-diamidino-2-phenylindole dihydrochloride (DAPI). D, Pearson correlation coefficient analyses between Ki-67+ cells per millimeter of epithelium and murine EoE severity (expressed as number of distal eosinophils per hpf) in mice with EoE. Individual data points represent 1 mouse. Data are shown as means ± SDs from 3 independent experiments. *P < .01, 2-way ANOVA.
ANO1 expression is upregulated by STAT6 and epigenetic modifications in EPC2-ALI cells
To gain insight into the regulation of ANO1 expression in esophageal epithelial cells in the context of EoE, we used an in vitro model developed from keratinocyte esophageal epithelial cells (EPC2) grown in ALI.19 IL-13 stimulation induced a rapid induction of ANO1 mRNA expression in mature EPC2-ALI cell cultures (Fig 3, A). Moreover, levels of ANO1 mRNA expression increased more than 4-fold within 4 hours of IL-13 stimulation and were maintained for at least 48 hours (Fig 3, A and B). STAT6 ChIP-seq analyses on EPC2-ALI cells after 30 minutes of IL-13 stimulation revealed STAT6 interaction with a genomic region located upstream of the ANO1 transcription start site (Fig 3, C, open box), indicating that IL-13 induction of ANO1 in esophageal cells involves STAT6. Consistent with this, a STAT6 γ-activated sequence motif is present in the ANO1 promoter.40 In line with the concept that IL-13 induces a STAT6-dependent transcriptional mechanism involving the ANO1 promoter, H3K4me3 ChIP-seq analyses revealed the presence of a highly trimethylated region on the ANO1 promoter after 4 hours of IL-13 treatment (Fig 3, D, open box). Bioinformatics comparative analyses of the 4-hour RNA-seq, H3K4me3 and STAT6 ChIP-seq data sets revealed that ANO1 was the only gene significantly altered under all 3 conditions (Fig 3, E). Together, these data reveal that enhanced ANO1 expression is an early event after IL-13 stimulation mediated by epigenetic modification and STAT6 promoter interaction (Fig 3, E).
FIG 3.

ANO1 is transcriptionally and epigenetically upregulated in EPC2-ALI cells through a Janus kinase/STAT6 pathway. A, Reads per kilobase per million (RPKM) values of ANO1 from EPC2-ALI cells treated for 4 or 48 hours with vehicle or IL-13. ***P = 4.04e−15 and ****P = 8.53e−23 assessed with the De-Seq algorithm. B, qPCR analysis of ANO1 expression in EPC2-ALI cells after IL-13 treatment for 1, 2, 3, 4, and 48 hours. C and D, UCSC genome browser screenshot of the ANO1 gene from STAT6 (Fig 3, C) and H3K4me3 (Fig 3, D) ChIP-seq analysis. Displayed tracks are referred to the RefSeq sequence of ANO1 (chromosome 11, bp 69900000-70040000). The area highlighted in the box is representative of the genomic region bound by STAT6 and histone mark H3K4me3. Increased H3K4me3 mark levels were detected on the 5′ region (highlighted in the box) of the gene after 4 hours of IL-13 stimulation. E, Venn diagram showing genes with STAT6 promoter/upstream regions binding after 30 minutes of incubation with IL-13 and H3K4me3 mark in promoter/upstream regions and upregulation after 4 hours of incubation with IL-13. In Fig 3, B, values are represented as means ± SDs of 3 different measurements. *P < .05, **P < .01, and ****P < .0001, 1-way ANOVA.
[Cl−]i levels and ANO1-mediated Cl− secretion are increased in EPC2-ALI cells after IL-13 treatment
To determine whether IL-13–induced ANO1 expression in EPC2-ALI cells was associated with changes in [Cl−] transport, we examined Cl− secretion of EPC2-ALI cells treated with IL-13 for 48 hours. Cholinergic carbachol-induced Isc was significantly increased in IL-13–treated EPC2-ALI cells (Fig 4, A). Basolateral-to-apical [Cl−] gradient studies (Fig 4, B) revealed that carbachol-increased Isc in IL-13–stimulated EPC2-ALI cells was primarily driven by apical outward Cl− transport. Notably, the apical outward Cl− transport was sensitive to pharmacologic inhibition with the ANO1-specific inhibitor T16Ainh-A01,41 indicating a major contribution for ANO1 in basolateral-to-apical Cl− secretion in IL-13–stimulated EPC2-ALI cells (Fig 4, C and D). Together, these data indicate that ANO1 has a major role in apical Cl− secretion in EPC2-ALI cells after IL-13 exposure. Given that ANO1 is an outward rectifying anion Cl− transporter, we predicted that IL-13 stimulation of EPC2 cells increased [Cl−]i levels. Indeed, [Cl−]i levels were significantly increased in submerged EPC2 cells after 24 hours of IL-13 stimulation (Fig 5).
FIG 4.

Dysregulation of apical ANO1 activity in EPC2-ALI cells after IL-13 stimulation. A, Change in Isc (ΔIsc) of 48-hour IL-13–stimulated (100 ng/mL) or control-stimulated EPC2-ALI cells after basolateral carbachol (100 μmol/L) stimulation. B, Description of Cl− gradient analyses of EPC2-ALI cells. C, Change in Isc (ΔIsc) of control- and IL-13–stimulated EPC2-ALI cells in the presence of a basolateral-to-apical Cl− concentration gradient (Baso → Ap [Cl−]) ± apical exposure to T16Ainh-A01 (10 μmol/L). D, Decrease in ΔIsc in vehicle- and IL-13–stimulated EPC2-ALI cells after apical exposure to T16Ainh-A01 (10 μmol/L) in the presence of a basolateral-to-apical Cl− gradient. Values are expressed as means ± SDs of 8 different samples (Fig 4, A) and 4 different samples (Fig 4, C and D) from 2 independent experiments. Fig 4, A and D: ***P < .001, unpaired t test. Fig 4, C: **P < .01 and ****P < .0001, 2-way ANOVA. Inh, Inhibitor.
FIG 5.
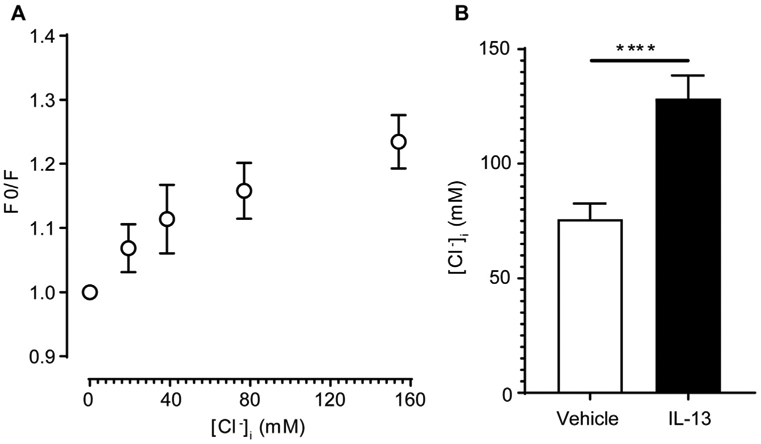
Intracellular Cl− concentration in EPC2-ALI cells following IL-13 exposure. A, Calibration of [Cl−]i in EPC2 cells. B, Calculated [Cl−]i in EPC2 cells after 24 hours of vehicle or IL-13 stimulation. Data are representative of 20 individual cells from 3 independent experiments. Data are shown as means ± SDs. ****P < .0001, 2-way ANOVA.
ANO1 upregulation is involved in the IL-13–mediated increase in proliferation rate of EPC2-ALI cells
To determine the link between ANO1 expression and activity and BZH in esophageal cells, we performed a BrdU pulse chase in EPC2-ALI cells following IL-13 stimulation in the presence and absence of T16Ainh-A01 (10 μmol/L). IL-13 exposure of EPC2 cells leads to basal cell proliferation, as evidenced by BrdU+ staining (Fig 6, A-C). IL-13 exposure induced an approximately 2.5-fold increase in numbers of BrdU+ EPC2 cells (Fig 6, A-C). Notably, the increased number of BrdU+ cells was associated with increased thickness and expansion of the suprabasal layer (Fig 6, A-D), whereas pharmacologic inhibition of ANO1 attenuated this effect (Fig 6, A-D).
FIG 6.

ANO1 inhibition blocks IL-13–mediated increased proliferation in the EPC2-ALI basal layer. A and B, Representative hematoxylin and eosin staining (Fig 6, A) and BrdU (green) and 4′-6-diamidino-2-phenylindole dihydrochloride (DAPI; blue) IF staining (Fig 6, B). C and D, Quantification of BrdU+ cells (Fig 6, C) and esophageal epithelial monolayer thickness of vehicle- and IL-13–stimulated EPC2-ALI monolayers (Fig 6, D) after 48 hours of exposure to 10 μmol/L T16Ainh-A01. E, qPCR analysis of ANO1 expression and (F) quantification of BrdU+ cells in scrambled shRNA (CTRL)– and shRNA-ANO1 knockdown (ANO1 KD)–transduced EPC2-ALI cells after vehicle or IL-13 stimulation. In Fig 6, B, merged BrdU (green) and DAPI-stained (blue) EPC2-ALI monolayers are shown. No BrdU+ cells were identified in the vehicle-treated group. Data are shown as means ± SDs representative of 6 different sections from 3 independent experiments (Fig 6, A-D) and of 4 different sections from 2 independent experiments (Fig 6, E and F). Fig 6, A–D: **P < .01 and ****P < .001, 2-way ANOVA; **P < .01 and ***P < .001, 2-way ANOVA. CTRL, Control; DMSO, dimethyl sulfoxide.
To confirm specificity, we used an independent approach using lentiviral shRNA gene silencing. Stably transduced EPC2 cells demonstrated approximately 80% reduction in ANO1 expression (Fig 6, E). EPC2-ALI cells transduced with either scrambled (CTRL) shRNA or ANO1-shRNA were cultured in ALI conditions, and stimulated with IL-13 for 48 hours. IL-13 stimulation of CTRL-shRNA–transduced EPC2-ALI cells increased cellular proliferation (Fig 6, F). Notably, the proliferative capacity in the basal state of ANO1-shRNA–transduced EPC2-ALI cells was reduced; furthermore, the IL-13–induced proliferative response was significantly attenuated compared with that in the CTRL-shRNA–transduced EPC2-ALI cells (Fig 6, F). These studies demonstrate that IL-13–induced proliferation in EPC2-ALI cells is dependent on ANO1 expression and activity.
ANO1 modulates the proliferation-regulating cell cycle and TP63 levels
Transit-amplifying cells in the basal layer of epithelial tissues express TP63,42 a key transcription factor that controls the switch between epithelial differentiation and proliferation.43-46 Notably, TP63 is upregulated in both biopsy samples from patients with EoE (Fig 7, A) and in EPC2-ALI cells after 48 hours of IL-13 stimulation (Fig 7, B), and pharmacologic blockade of ANO1 functional activity significantly decreased TP63 expression, suggesting an interaction between ANO1 function and TP63 expression in esophageal cells (Fig 7, C).
FIG 7.

ANO1 regulates cell cycle–modulating TP63 expression. A, Individual reads per kilobase per million (RPKM) values for TP63 from RNA-seq of esophageal biopsy samples from patients with active EoE and healthy control subjects. B, qPCR analysis of TP63 expression in EPC2-ALI cells treated for 48 hours with vehicle or IL-13 (100 ng/mL). C, qPCR analysis of TP63 expression in EPC2-ALI cells stimulated for 48 hours with T16Ainh-A01 (10 μmol/L) or dimethyl sulfoxide (DMSO) as a vehicle. D, Western blot analysis of p-CDK2 levels in vehicle-treated (−) or TMEM16inhA01 inhibitor–treated EPC2-ALI cells after vehicle or IL-13 (100 ng/mL) stimulation for 48 hours. E and F, Densitometric quantification of p-CDK2 levels in vehicle- or TMEM16inhA01 inhibitor–treated EPC2-ALI cells (Fig 7, E) or scrambled shRNA Ctrl– or shRNA Ano1–transduced EPC2-ALI cells (Fig 7, F) after vehicle or IL-13 (100 ng/mL) stimulation for 48 hours. Values are represented as means ± SEMs of 6 different measurements (Fig 7, B) and 3 different measurements (Fig 7, C). *P < .05. Three Western blots from 3 independent experiments (Fig 7, D) and 3 different measurements from 3 independent experiments (Fig 7, E and F) are shown. Fig 7, A-C: *P < .05 and **P < .01, unpaired t test with Welch correction. Fig 7, E and F: *P < .05 and **P < .01, 2-way ANOVA.
As decreased TP63 expression has been associated with cell-cycle arrest in the G0/G1 phase and loss of proliferative capacity,47 we hypothesized that ablation of ANO1-dependent Cl− transport in EPC2 cells would lead to decreased TP63 expression, causing cell-cycle arrest. To test this, we examined phosphorylation of the G1/S phase marker cyclin-dependent kinase 2 (p-CDK2) in IL-13–stimulated EPC2-ALI cells in the presence and absence of T16Ainh-A01. IL-13 stimulation of EPC2-ALI cells induced an increase in p-CDK2 levels (Fig 7, D), which was associated with proliferation (Fig 6, C). Notably, in the presence of T16Ainh-A01, IL-13 induced p-CDK2, and proliferation in EPC2-ALI cells was attenuated (Fig 6, C, and Fig 7, D and E). Consistent with this observation, the IL-13–induced increase in p-CDK2 in CTRL-shRNA–transduced EPC2-ALI cells was ablated in ANO1-shRNA–transduced EPC2-ALI cells (Fig 7, F). Collectively, these data suggest that ANO1 regulation of EPC2-ALI proliferative potential is associated with modulation of TP63 expression and cell-cycle activity.
DISCUSSION
Here we demonstrate increased expression of the Ca2+-activated chloride transporter ANO1 in the BZ and suprabasal zone of the esophageal epithelium in patients with EoE. Moreover, we show that ANO1 expression correlated with EoE severity and BZH. Using an established esophageal epithelial culture system, we show that (1) ANO1 mRNA and protein expression is rapidly induced by an IL-13–STAT6 mechanism; (2) IL-13 induced an increase in [Cl−]i levels in esophageal cells; (3) ANO1 is the predominant transport pathway of IL-13–induced Cl− efflux in esophageal cells; and (4) IL-13–induced ANO1-mediated Cl− transport is required for esophageal epithelial proliferation. Mechanistic analyses revealed a requirement for IL-13–induced ANO1 expression and function in p-CDK2 and transition through the G1/S phase cell-cycle checkpoint to permit esophageal epithelial proliferation.
We show that ANO1 expression is significantly increased in both esophageal biopsy samples from patients with EoE and in IL-13–stimulated EPC2-ALI cells. Consistent with this, we provide evidence of increased transcriptional activation through chromatin remodeling (H3K4me3) and demonstrate STAT6 interaction with the ANO1 upstream region of EPC2-ALI cells after IL-13 stimulation, strongly supporting the notion that ANO1 is a primary early target of IL-13–mediated transcriptional response. STAT6 ChIP analyses revealed the presence of a STAT6 binding site on the ANO1 5′ region within approximately 15 kb of the ANO1 transcription start site. Recent studies have revealed the presence of a γ-activated sequence motif STAT6 binding domain upstream of the human ANO1 transcription start site within the ANO1 promoter.40 Notably, polymerase II ChIP, 5′-RACE analyses, and in vitro promoter studies revealed that this region was an active and functional ANO1 promoter possessing regulatory regions, including putative CCAAT enhancer–binding proteins, SPI, and STAT6 binding sites.40 These studies demonstrated that IL-4 receptor–mediated induction of ANO1 mRNA expression was dependent on STAT6 because mutation of the STAT6 consensus sequence or small interfering RNA–mediated knockdown of STAT6 ablated IL-4 receptor–mediated induction of ANO1.40 Our bioinformatic comparative analyses of the 4-hour RNA-seq, H3K4me3, and STAT6 ChIP-seq data sets revealed that ANO1 is the only altered gene, strongly supporting the notion that ANO1 is a primary early target of the IL-13–induced transcriptional response in esophageal cells.
We show that ANO1 expression was significantly increased in patients with EoE, and levels positively correlated with BZH scores. Importantly, IF analyses localized ANO1 to the predominant proliferative zone (basal cells and the CK13+ suprabasal zone), indicating a relationship between ANO1 expression and esophageal epithelial proliferation. We have previously reported no significant change in ANO1 mRNA expression in patients with gastroesophageal reflux disease.24 Notably, ANO1 expression BZH and lamina propria papillae elongation are more common in patients with EoE compared with those with gastroesophageal reflux disease,48 supporting the link between ANO1 expression and the esophageal proliferative response in patients with EoE. Using an esophagus-based EoE diagnostic panel, we have previously demonstrated that both EoE and proton pump inhibitor–responsive esophageal eosinophilia (PPI-REE) are characterized by an overlapping IL-13–driven common transcriptome, including increased expression of ANO1,24 suggesting that IL-13 is a driver of BZH in patients with EoE, possibly through an ANO1-dependent mechanism. Consistent with this concept, treatment of patients with EoE with anti–IL-13 mAb leads to a reduction in epithelial remodeling, including basal cell hyperplasia, which was associated with a significant reduction in ANO1 mRNA expression.49 We have previously reported increased ANO1 mRNA expression in esophageal biopsy samples from patients with PPI-REE before proton pump inhibitor (PPI) therapy.24 Notably, an 8-week course of PPI therapy significantly decreased ANO1 mRNA expression in these subjects.25 These studies suggest that PPI therapy can reduce ANO1 expression; however, it is currently unclear whether this is due to a direct effect of PPIs on ANO1 gene expression or alternatively through suppression of esophageal inflammation.
We examined the effect of PPIs on IL-13–induced ANO1 mRNA expression in EPC2-ALI cells in vitro and showed that PPI exposure significantly reduced IL-13–mediated ANO1 mRNA expression (see Fig E1 in this article’s Online Repository at www.jacionline.org). This was not specific to ANO1 because PPI also inhibited IL-13–mediated CCL26 mRNA expression (see Fig E1). Collectively, these studies suggest that PPIs can act directly on esophageal epithelial cells and downregulate IL-13–induced ANO1 mRNA expression and suggest that these effects are independent on esophageal inflammation. These data are consistent with those of Wen et al25 using the eosinophilic diagnostic panel, which demonstrated a reduction in ANO1 expression in patients with PPI-REE after PPI therapy. Notably, PPI trial in patients with PPI-REE led to a significant reduction in ANO1 expression; however, ANO1 mRNA levels remained significantly greater than those observed in healthy control subjects,25 indicating that ANO1 expression is in part responsive to PPI therapy.
The role of ANO1 in cellular proliferation is not yet fully understood. Previous studies have demonstrated that the ANO1 gene is located within the 11q13 amplicon, one of the most frequently amplified chromosomal regions in patients with cancer,50-60 and that inhibition of ANO1 activity in interstitial cells of Cajal and tumor cell lines inhibited cell proliferation,61,62 indicating an important potential contribution for this transporter in cell proliferation. Interestingly, a key requirement of G1/S phase and G2/M phase transition and cellular proliferation is increased cell volume, which is regulated in part by ion channel regulation of the membrane potential.63-69 The increased membrane potential across the plasma membrane osmotically drives water influx from the extracellular to the intracellular space, leading to cell swelling and promotion of G1/S and G2/M phase progression.23
Ion channels commonly observed to be associated with control of proliferation include Ca2+-activated Cl− channels, such as ANO1.23 ANO1 is a calcium-activated Cl− transporter primarily expressed on the apical side of several epithelia, including the colon and tracheal epithelia.70,71 We show that ANO1 is the primary apical Cl− transporter in EPC2-ALI cells and that blockade of ANO1 expression and function reduced the level of IL-13–induced EPC2 cell proliferation, suggesting that ANO1-dependent Cl− efflux might be important in EPC2 cell proliferation. Interestingly, in vitro studies in D54-MG glioma and human cervical cancer cells have revealed a relationship between Cl− flux, cell-volume dynamics, and proliferation.72,73
Moreover, transient changes in Cl− levels have been associated with specific phases of cell cycle and division. Before cell mitosis (before the M phase of the cell cycle), dividing cells condense their cytoplasm, and this is associated with a rapid decrease (~40%) in intracellular Cl− levels. We observed an association between transient changes in [Cl−]i levels and proliferation in EPC2 cells after IL-13 stimulation. We speculate that in esophageal cells ANO1 is the primary transporter involved in IL-13–induced Cl− efflux and responsible for reducing [Cl−]i, which is required for transition through the cell-cycle checkpoint, and permitting cell division and proliferation. Consistent with this argument, we show that the IL-13–induced increase in phosphorylation of the key G1/S phase transition protein CDK2 in EPC2-ALI cells was inhibited by blockade ANO1-dependent Cl− efflux. CDK2 is required for alleviating retinoblastoma protein-mediated inhibition of E2F and S phase entry and DNA synthesis,74,75 and loss of CDK2 activity leads to cell-cycle arrest at the G1/S checkpoint.76,77 Notably, decreased ANO1 activity and proliferation were also associated with reduced levels of TP63, a key transcription factor crucial for maintaining proliferative potential in basal keratinocytes through cell-cycle regulation.78 Expression of TP63 correlates with keratinocyte-proliferative capacity and is thought to serve as a marker for keratinocyte stems cells and denotes proliferative capacity.43 Consistent with this, genetic ablation of TP63 in mature human keratinocytes leads to cell-cycle arrest.78 Currently, we do not know the relationship between ANO1 function and TP63 activity. It is possible that a consequence of loss of ANO1 function and decreased proliferation is loss of proliferative potential in basal keratinocytes, leading to terminal differentiation. Alternatively, ANO1 can directly inhibit TP63 expression and decrease proliferative capacity, an area of current investigation. Intriguingly, increased expression of ANO1 has been observed in patients with esophageal squamous cell carcinoma, and levels of expression correlated with progression of precancerous lesions,79 linking ANO1 to esophageal cancer progression. Although we and others have reported increased ANO1 expression and function in patients with EoE, there is no association observed between esophageal carcinoma and EoE.80 We predict that ANO1 activity is likely a general requirement for esophageal epithelial proliferation, regulating critical steps in cell-cycle progression and division, and any process by which esophageal epithelial proliferation occurs irrespective of whether this involves normal cell growth and dysplasia could be associated with increased ANO1 expression and activity.
The functional relevance of BZH and ANO1 expression and the relationship to human EoE is unclear. Recently, Shoda et al81 used an EoE endotype-prediction algorithm and molecular, clinical, and histopathologic parameters from a multicenter patient cohort to identify different EoE endotypes. Despite no differences in peak eosinophil levels or length of time from diagnosis of EoE, they were able to identify 3 EoE endotypes.81 Notably, EoE endotype 3 was associated with a fibrostenotic (rings, narrowing, or strictures) phenotype and the greatest degree of endoscopic and histologic severity. Molecular analyses revealed that ANO1 was one of 2 discriminatory genes (ANO1 and UPK1A) to provide 98% positive predictive value of EoE endotype 3, suggesting a link between ANO1 and the fibrostenotic and histologic phenotype.81 We speculate that ANO1’s contribution to functional outcomes in patients with EoE, such as fibrostenosis, is not through directly driving esophageal dysfunction but rather indirectly through driving chronicity of inflammation and subsequent development of secondary associated histopathologic manifestations, such as BZH and DIS formation. There is strong clinical and experimental evidence showing that BZH and DISs contribute to esophageal barrier dysfunction.82,83 Esophageal barrier dysfunction is permissive to food allergen accumulation in the esophageal epithelium, which is likely to perpetuate inflammation and dysfunction associated with EoE. In support of this concept, Koutlas and Dellon84 reported that some patients with EoE at diagnosis had esophageal inflammation but very little evidence of fibrostenosis. However, left untreated for approximately 8 years, subjects had fibrostenosis, including strictures, a small-caliber esophagus, and a majority requiring esophageal dilation. Similarly, a retrospective analysis of adults with EoE revealed that delayed diagnosis of EoE was associated with increased stricture risk, supporting the concept that chronicity of esophageal inflammation can lead to functional EoE outcomes.85 Collectively, these studies suggest that use of ANO1 antagonists might be a therapeutic approach for reduction in esophageal epithelial proliferation and BZH and indirectly associated esophageal function in patients with EoE.
In summary, we identified a relationship between ANO1 expression and BZH in biopsy samples from patients with EoE. Mechanistically, we identified a previously undescribed role for ANO1 in esophageal epithelial proliferation and provided data to indicate that ANO1 counterregulates the basal keratinocyte–specific proliferative protein TP63 and the cell-cycle protein CDK2. Collectively, these studies provide a rationale for the therapeutic use of ANO1 antagonists for reduction in BZH and reduction in associated esophageal dysfunction in patients with EoE.
Supplementary Material
Key messages.
SLC26A4 (ANO1) expression is increased within the basal layer of esophageal biopsy specimens from patients with EoE, and this expression correlated positively with disease severity (eosinophils/hpf) and BZH.
IL-13 rapidly induces ANO1 expression by means of epigenetic modification and STAT6 promoter interaction.
ANO1 is the primary apical IL-13–induced Cl− transport mechanism.
Loss of ANO1-dependent Cl− transport abrogated esophageal epithelial proliferation.
ANO1-dependent regulation of basal cell proliferation was associated with modulation of TP63 expression and p-CDK2 levels.
Acknowledgments
This work was supported by National Institutes of Health grants DK090119, AI112626, AI140133, and AI138177; Food Allergy Research & Education (FARE); the Crohn’s Colitis Foundation of America; and the Mary H. Weiser Food Allergy Center (to S.P.H).
Abbreviations used
- ALI
Air-liquid interface
- ANO1
Anoctamin 1
- BrdU
5-Bromo-2′-deoxyuridine
- BZ
Basal zone
- BZH
Basal zone hyperplasia
- CCHMC
Cincinnati Children’s Hospital Medical Center
- CDK2
Cyclin-dependent kinase 2
- ChIP
Chromatin immunoprecipitation
- CK
Cytokeratin
- CTRL
Scrambled
- DIS
Dilated intercellular space
- EoE
Eosinophilic esophagitis
- hpf
High-power field
- HPRT
Hypoxanthine phosphoribosyltransferase
- IF
Immunofluorescence
- Isc
Short circuit current
- MQAE
N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide
- NR
Normal Ringer
- PPI
Proton pump inhibitor
- PPI-REE
Proton pump inhibitor–responsive esophageal eosinophilia
- qPCR
Quantitative PCR
- RNA-seq
RNA sequencing
- ROI
Region of interest
- RT
Room temperature
- shRNA
Short hairpin RNA
- STAT6
Signal transducer and activator of transcription 6
Footnotes
Disclosure of potential conflict of interest: M. E. Rothenberg is a consultant for Immune Pharmaceuticals, NKT Therapeutics, PulmOne, Celgene, Shire, AstraZeneca, and Novartis; has an equity interest in the first 3 companies listed and royalties from reslizumab (Teva Pharmaceuticals); is an inventor of several patents owned by Cincinnati Children’s Hospital Medical Center and a set of these patents relates to molecular diagnostics. M. Chehade received research funding from the National Institutes of Health (NIH), Patient-Centered Outcomes Research Institute (PCORI), American Partnership for Eosinophilic Disorders (APFED), and the American Academy of Allergy, Asthma & Immunology (AAAAI); clinical trial funding from Regeneron, Shire, and Allakos; consulting fees from Shire, Allakos, and Adare; lecture fees from Danone and the Annenberg Center for Health Sciences at Eisenhower; and serves (no fees) on the medical advisory boards for APFED and the Campaign Urging Research for Eosinophilic Diseases (CURED). The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Simon D, Straumann A, Simon HU. Eosinophilic esophagitis and allergy. Dig Dis 2014;32:30–3. [DOI] [PubMed] [Google Scholar]
- 2.Collins MH. Histopathologic features of eosinophilic esophagitis. Gastrointest Endosc Clin N Am 2008;18:59–71, viii-ix. [DOI] [PubMed] [Google Scholar]
- 3.Collins MH. Histopathology of eosinophilic esophagitis. Dig Dis 2014;32:68–73. [DOI] [PubMed] [Google Scholar]
- 4.Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol 2007;120:1292–300. [DOI] [PubMed] [Google Scholar]
- 5.Blanchard C, Stucke EM, Burwinkel K, Caldwell JM, Collins MH, Ahrens A, et al. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J Immunol 2010;184:4033–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanchard C, Stucke EM, Rodriguez-Jimenez B, Burwinkel K, Collins MH, Ahrens A, et al. A striking local esophageal cytokine expression profile in eosinophilic esophagitis. J Allergy Clin Immunol 2011;127:208–17.e1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 2003;125: 1419–27. [DOI] [PubMed] [Google Scholar]
- 8.Zuo L, Fulkerson PC, Finkelman FD, Mingler M, Fischetti CA, Blanchard C, et al. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13R alpha 2-inhibited pathway. J Immunol 2010;185: 660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abonia JP, Blanchard C, Butz BB, Rainey HF, Collins MH, Stringer K, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol 2010;126:140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanchard C, Mingler MK, McBride M, Putnam PE, Collins MH, Chang G, et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal Immunol 2008;1:289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora AS, Perrault J, Smyrk TC. Topical corticosteroid treatment of dysphagia due to eosinophilic esophagitis in adults. Mayo Clin Proc 2003;78:830–5. [DOI] [PubMed] [Google Scholar]
- 12.Faubion WA Jr, Perrault J, Burgart LJ, Zein NN, Clawson M, Freese DK. Treatment of eosinophilic esophagitis with inhaled corticosteroids. J Pediatr Gastroenterol Nutr 1998;27:90–3. [DOI] [PubMed] [Google Scholar]
- 13.Kelly KJ, Lazenby AJ, Rowe PC, Yardley JH, Perman JA, Sampson HA. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology 1995;109:1503–12. [DOI] [PubMed] [Google Scholar]
- 14.Liacouras CA, Ruchelli E. Eosinophilic esophagitis. Curr Opin Pediatr 2004;16: 560–6. [DOI] [PubMed] [Google Scholar]
- 15.Liacouras CA, Wenner WJ, Brown K, Ruchelli E. Primary eosinophilic esophagitis in children: successful treatment with oral corticosteroids. J Pediatr Gastroenterol Nutr 1998;26:380–5. [DOI] [PubMed] [Google Scholar]
- 16.Sherrill JD, Kiran K, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014;15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006;116:536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherrill JD, Blanchard C. Genetics of eosinophilic esophagitis. Dig Dis 2014;32: 22–9. [DOI] [PubMed] [Google Scholar]
- 19.Sherrill JD, Kc K, Wu D, Djukic Z, Caldwell JM, Stucke EM, et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol 2014;7:718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Mello RJ, Caldwell JM, Azouz NP, Wen T, Sherrill JD, Hogan SP, et al. LRRC31 is induced by IL-13 and regulates kallikrein expression and barrier function in the esophageal epithelium. Mucosal Immunol 2016;9:744–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis BP, Stucke EM, Khorki ME, Litosh VA, Rymer JK, Rochman M, et al. Eosinophilic esophagitis-linked calpain 14 is an IL-13-induced protease that mediates esophageal epithelial barrier impairment. JCI Insight 2016;1:e86355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noffsinger AE. Update on esophagitis: controversial and underdiagnosed causes. Arch Pathol Lab Med 2009;133:1087–95. [DOI] [PubMed] [Google Scholar]
- 23.Blackiston DJ, McLaughlin KA, Levin M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle 2009;8:3527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen T, Dellon ES, Moawad FJ, Furuta GT, Aceves SS, Rothenberg ME. Transcriptome analysis of proton pump inhibitor-responsive esophageal eosinophilia reveals proton pump inhibitor-reversible allergic inflammation. J Allergy Clin Immunol 2015;135:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen T, Dellon ES, Moawad FJ, Furuta GT, Aceves SS, Rothenberg ME. Transcriptome analysis of proton pump inhibitor-responsive esophageal eosinophilia reveals PPI-reversible allergic inflammation. J Allergy Clin Immunol 2015;135:187–97.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen T, Stucke EM, Grotjan TM, Kemme KA, Abonia JP, Putnam PE, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology 2013;145:1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin LJ, Franciosi JP, Collins MH, Abonia JP, Lee JJ, Hommel KA, et al. Pediatric Eosinophilic Esophagitis Symptom Scores (PEESS® v2.0) identify histologic and molecular correlates of the key clinical features of disease. J Allergy Clin Immunol 2015;135:1519–28.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furuta GT, Liacouras CA, Collins MH, Gupta SK, Justinich C, Putnam PE, et al. Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 2007;133: 1342–63. [DOI] [PubMed] [Google Scholar]
- 29.Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol 2011;128:3–22.e6. [DOI] [PubMed] [Google Scholar]
- 30.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem 2003;278:1824–30. [DOI] [PubMed] [Google Scholar]
- 31.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res 2003;1:729–38. [PubMed] [Google Scholar]
- 32.Lu TX, Sherrill JD, Wen T, Plassard AJ, Besse JA, Abonia JP, et al. MicroRNA signature in patients with eosinophilic esophagitis, reversibility with glucocorticoids, and assessment as disease biomarkers. J Allergy Clin Immunol 2012;129:1064–75.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods 2011; 8:469–77. [DOI] [PubMed] [Google Scholar]
- 36.Kartashov AV, Barski A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol 2015;16:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collins MH, Martin LJ, Alexander ES, Boyd JT, Sheridan R, He H, et al. Newly developed and validated eosinophilic esophagitis histology scoring system and evidence that it outperforms peak eosinophil count for disease diagnosis and monitoring. Dis Esophagus 2017;30:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007;129:823–37. [DOI] [PubMed] [Google Scholar]
- 39.Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol Cell Physiol 1990;259:C375–88. [DOI] [PubMed] [Google Scholar]
- 40.Mazzone A, Gibbons SJ, Bernard CE, Nowsheen S, Middha S, Almada LL, et al. Identification and characterization of a novel promoter for the human ANO1 gene regulated by the transcription factor signal transducer and activator of transcription 6 (STAT6). FASEB J 2015;29:152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem 2011;286:2365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998;2:305–16. [DOI] [PubMed] [Google Scholar]
- 43.Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A 2001;98:3156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parsa R, Yang A, McKeon F, Green H. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Invest Dermatol 1999; 113:1099–105. [DOI] [PubMed] [Google Scholar]
- 45.Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol 2003;23:2264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King KE, Ponnamperuma RM, Yamashita T, Tokino T, Lee LA, Young MF, et al. deltaNp63alpha functions as both a positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Oncogene 2003;22: 3635–44. [DOI] [PubMed] [Google Scholar]
- 47.Wu N, Sulpice E, Obeid P, Benzina S, Kermarrec F, Combe S, et al. The miR-17 family links p63 protein to MAPK signaling to promote the onset of human keratinocyte differentiation. PLoS One 2012;7:e45761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parfitt JR, Gregor JC, Suskin NG, Jawa HA, Driman DK. Eosinophilic esophagitis in adults: distinguishing features from gastroesophageal reflux disease: a study of 41 patients. Mod Pathol 2006;19:90–6. [DOI] [PubMed] [Google Scholar]
- 49.Rothenberg ME, Wen T, Greenberg A, Alpan O, Enav B, Hirano I, et al. Intravenous anti-IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J Allergy Clin Immunol 2015;135:500–7. [DOI] [PubMed] [Google Scholar]
- 50.Schuuring E The involvement of the chromosome 11q13 region in human malignancies: cyclin D1 and EMS1 are two new candidate oncogenes—a review. Gene 1995;159:83–96. [DOI] [PubMed] [Google Scholar]
- 51.Ormandy CJ, Musgrove EA, Hui R, Daly RJ, Sutherland RL. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat 2003; 78:323–35. [DOI] [PubMed] [Google Scholar]
- 52.Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1, that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci U S A 2002;99:11369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 2004;165: 107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang X, Godfrey TE, Gooding WE, McCarty KS Jr, Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer 2006;45:1058–69. [DOI] [PubMed] [Google Scholar]
- 55.Ayoub C, Wasylyk C, Li Y, Thomas E, Marisa L, Robe A, et al. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br J Cancer 2010;103: 715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A 2013;110: E1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu W, Lu M, Liu B, Huang Y, Wang K. Inhibition of Ca(2+)-activated Cl(−) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett 2012;326:41–51. [DOI] [PubMed] [Google Scholar]
- 58.Sauter DR, Novak I, Pedersen SF, Larsen EH, Hoffmann EK. ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflugers Arch 2015; 467:1495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sui Y, Sun M, Wu F, Yang L, Di W, Zhang G, et al. Inhibition of TMEM16A expression suppresses growth and invasion in human colorectal cancer cells. PLoS One 2014;9:e115443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruiz C, Martins JR, Rudin F, Schneider S, Dietsche T, Fischer CA, et al. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One 2012;7:e43265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stanich JE, Gibbons SJ, Eisenman ST, Bardsley MR, Rock JR, Harfe BD, et al. Ano1 as a regulator of proliferation. Am J Physiol Gastrointest Liver Physiol 2011;301:G1044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guan L, Song Y, Gao J, Gao J, Wang K. Inhibition of calcium-activated chloride channel ANO1 suppresses proliferation and induces apoptosis of epithelium originated cancer cells. Oncotarget 2016;7:78619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.MacFarlane SN, Sontheimer H. Modulation of Kv1.5 currents by Src tyrosine phosphorylation: potential role in the differentiation of astrocytes. J Neurosci 2000;20:5245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacFarlane SN, Sontheimer H. Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia 2000;30:39–48. [DOI] [PubMed] [Google Scholar]
- 65.Habela CW, Olsen ML, Sontheimer H. ClC3 is a critical regulator of the cell cycle in normal and malignant glial cells. J Neurosci 2008;28:9205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ouadid-Ahidouch H, Le Bourhis X, Roudbaraki M, Toillon RA, Delcourt P, Prevarskaya N. Changes in the K1 current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-gogo K1 channel. Receptors Channels 2001;7:345–56. [PubMed] [Google Scholar]
- 67.Ouadid-Ahidouch H, Roudbaraki M, Ahidouch A, Delcourt P, Prevarskaya N. Cell-cycle-dependent expression of the large Ca2+-activated K+ channels in breast cancer cells. Biochem Biophys Res Commun 2004;316:244–51. [DOI] [PubMed] [Google Scholar]
- 68.Ouadid-Ahidouch H, Roudbaraki M, Delcourt P, Ahidouch A, Joury N, Prevarskaya N. Functional and molecular identification of intermediate-conductance Ca(2+)-activated K(+) channels in breast cancer cells: association with cell cycle progression. Am J Physiol Cell Physiol 2004; 287:C125–34. [DOI] [PubMed] [Google Scholar]
- 69.Woodfork KA, Wonderlin WF, Peterson VA, Strobl JS. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. J Cell Physiol 1995;162:163–71. [DOI] [PubMed] [Google Scholar]
- 70.Ousingsawat J, Martins JR, Schreiber R, Rock JR, Harfe BD, Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J Biol Chem 2009;284:28698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rock JR, O’Neal WK, Gabriel SE, Randell SH, Harfe BD, Boucher RC, et al. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl− secretory channel in mouse airways. J Biol Chem 2009;284:14875–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Habela CW, Ernest NJ, Swindall AF, Sontheimer H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J Neurophysiol 2009;101:750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shen MR, Chou CY, Hsu KF, Hsu YM, Chiu WT, Tang MJ, et al. KCl cotransport is an important modulator of human cervical cancer growth and invasion. J Biol Chem 2003;278:39941–50. [DOI] [PubMed] [Google Scholar]
- 74.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 2000;14:2393–409. [DOI] [PubMed] [Google Scholar]
- 75.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol 2000;2:E65–7. [DOI] [PubMed] [Google Scholar]
- 76.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999;98:859–69. [DOI] [PubMed] [Google Scholar]
- 77.Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol 2008;9:910–6. [DOI] [PubMed] [Google Scholar]
- 78.Truong AB, Khavari PA. Control of keratinocyte proliferation and differentiation by p63. Cell Cycle 2007;6:295–9. [DOI] [PubMed] [Google Scholar]
- 79.Shang L, Hao JJ, Zhao XK, He JZ, Shi ZZ, Liu HJ, et al. ANO1 protein as a potential biomarker for esophageal cancer prognosis and precancerous lesion development prediction. Oncotarget 2016;7:24374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Syed A, Maradey-Romero C, Fass R. The relationship between eosinophilic esophagitis and esophageal cancer. Dis Esophagus 2017;30:1–5. [DOI] [PubMed] [Google Scholar]
- 81.Shoda T, Wen T, Aceves SS, Abonia JP, Atkins D, Bonis PA, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross-sectional study. Lancet Gastroenterol Hepatol 2018;3:477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Warners MJ, van Rhijn BD, Verheij J, Smout A, Bredenoord AJ. Disease activity in eosinophilic esophagitis is associated with impaired esophageal barrier integrity. Am J Physiol Gastrointest Liver Physiol 2017;313:G230–8. [DOI] [PubMed] [Google Scholar]
- 83.Warners MJ, Vlieg-Boerstra BJ, Verheij J, van Hamersveld PHP, van Rhijn BD, Van Ampting MTJ, et al. Esophageal and small intestinal mucosal integrity in eosinophilic esophagitis and response to an elemental diet. Am J Gastroenterol 2017;112:1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koutlas NT, Dellon ES. Progression from an inflammatory to a fibrostenotic phenotype in eosinophilic esophagitis. Case Rep Gastroenterol 2017;11:382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schoepfer AM, Safroneeva E, Bussmann C, Kuchen T, Portmann S, Simon HU, et al. Delay in diagnosis of eosinophilic esophagitis increases risk for stricture formation in a time-dependent manner. Gastroenterology 2013;145:1230–6.e1-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.