

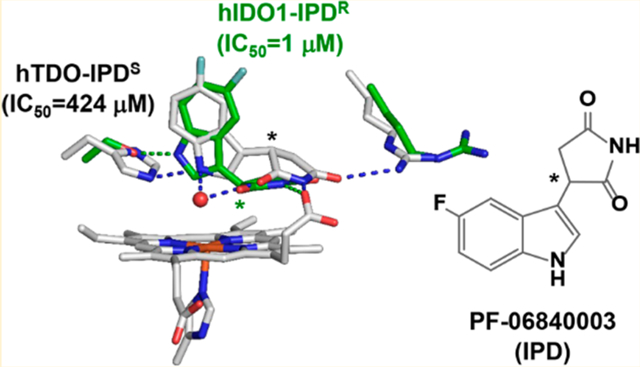
Abstract
Indoleamine 2,3-dioxygenase 1 (hIDO1) and tryptophan dioxygenase (hTDO) are two of the only three heme-based dioxygenases in humans. They have recently been identified as key cancer immunotherapeutic drug targets. While structures of hIDO1 in complex with inhibitors have been documented, so far there are no structures of hTDO-inhibitor complexes available. Here we use PF-06840003 (IPD), a hIDO1-selective inhibitor in clinical trials, as a structural probe to elucidate inhibitor-selectivity in hIDO1 versus hTDO. Spectroscopic studies show that IPD exhibits 400-fold higher inhibition activity toward hIDO1 with respect to hTDO. Crystallographic structures reveal that the binding pocket of IPD in the active site in hIDO1 is much more flexible as compared to that in hTDO, which offers a molecular explanation for the superior inhibition activity of IPD in hIDO1 with respect to hTDO. In addition to the IPD bound in the active site, a second IPD molecule was identified in an inhibitory site on the proximal side of the heme in hIDO1 and in an exosite that is ~40 Å away from the active site in hTDO. Taken together the data provide new insights into structure-based design of mono and dual inhibitors targeting hIDO1 and/or hTDO.
Graphical Abstract
INTRODUCTION
Trp is the least abundant essential amino acid. A small amount of our dietary Trp (~1%) is used to synthesize serotonin and melatonin through the serotonin (SER) pathway, while the majority of it (~95%) is metabolized through the kynurenine (KYN) pathway.1–3 The first and rate-limiting step of the KYN pathway, the degradation of Trp to N-formyl-kynurenine (NFK), is catalyzed by human tryptophan dioxygenase (hTDO) and indoleamine 2,3-dioxygenase (hIDO).1,4–6 hTDO and hIDO hence play an important role in controlling the relative Trp flux along the KYN and SER pathways. Over activation of hTDO and hIDO leads to upregulation of the KYN pathway, causing depression-associated anxiety, psychosis, and cognitive decline (due to serotonin deficiency)7–9 and contributing to neurodegenerative disorders, such as Alzheimer’s and Huntington’s disease (due to the production of neuroactive metabolites).10–15 Consequently, the two dioxygenases have been recognized as important therapeutic drug targets.
Recently it was found that hIDO is expressed in the placenta16 and in cancer cells,17,18 where the enzyme functions as an immunosuppressor by depleting Trp, the key nutrient required for T-cell activation and function, and by promoting the production of immunosuppressive kynurenine metabolites.18–23 Accordingly, hIDO was identified as a key drug target in immuno-oncology.24–27 In 2006, a second isoform of hIDO (human indoleamine 2,3 dioxygenase 2, referred to as hIDO2) was discovered in the human genome,28,29 in addition to the original isoform (human indoleamine 2,3 dioxygenase 1, referred to as hIDO1 hereinafter). Like hIDO1, hIDO2 as well as hTDO were later found to be expressed in cancer cells,30–33 where they play crucial roles in suppressing antitumor immunity. These exciting new discoveries prompted a great deal of research in drug discovery targeting the three enzymes.24,25 Accordingly, a large number of inhibitors, in particular those targeting hIDO1, have been developed.34–39
Several frontline hIDO1 inhibitors, such as epacadostat,40,41 PF-06840003 (IPD),42 navoximod,43,44 and BMS-986205,45,46 have entered clinical trials. Among them, epacadostat and IPD stand out, as epacadostat is the most advanced inhibitor that shows unprecedented outcomes in both Phase I/II trials,47,48 while IPD is the only inhibitor that is able to cross the blood-brain barrier.42 Last year, the much anticipated phase III trial of epacadostat, however, failed unexpectedly,48–51 suggesting that hIDO1 inhibition alone might not be sufficient to elicit clinical effects without simultaneously blocking hTDO and hIDO2, which also contribute to Trp depletion and kynurenine accumulation in cancer cells.30–33
Crystal structures of hIDO1 in complex with the substrate l-Trp41 and a variety of inhibitors41,42,44–46,52–54 have been reported. Most of the inhibitors bind to the active site (Sa), where they coordinate to the heme iron via a N atom, except that (i) epacadostat coordinates to the heme iron via an O atom,41 and (ii) IPD sits on top of the heme iron without coordinating to it.42 Regardless of the heme iron coordination, all the high affinity inhibitors, like the substrate Trp, possess two fragments that occupy the distinct “A” and “B” pockets in the Sa site (Figure 1). The smaller inhibitors, such as phenyl imidazole and amino triazole occupy only the “A” pocket and typically exhibit lower inhibition activities.
Figure 1.
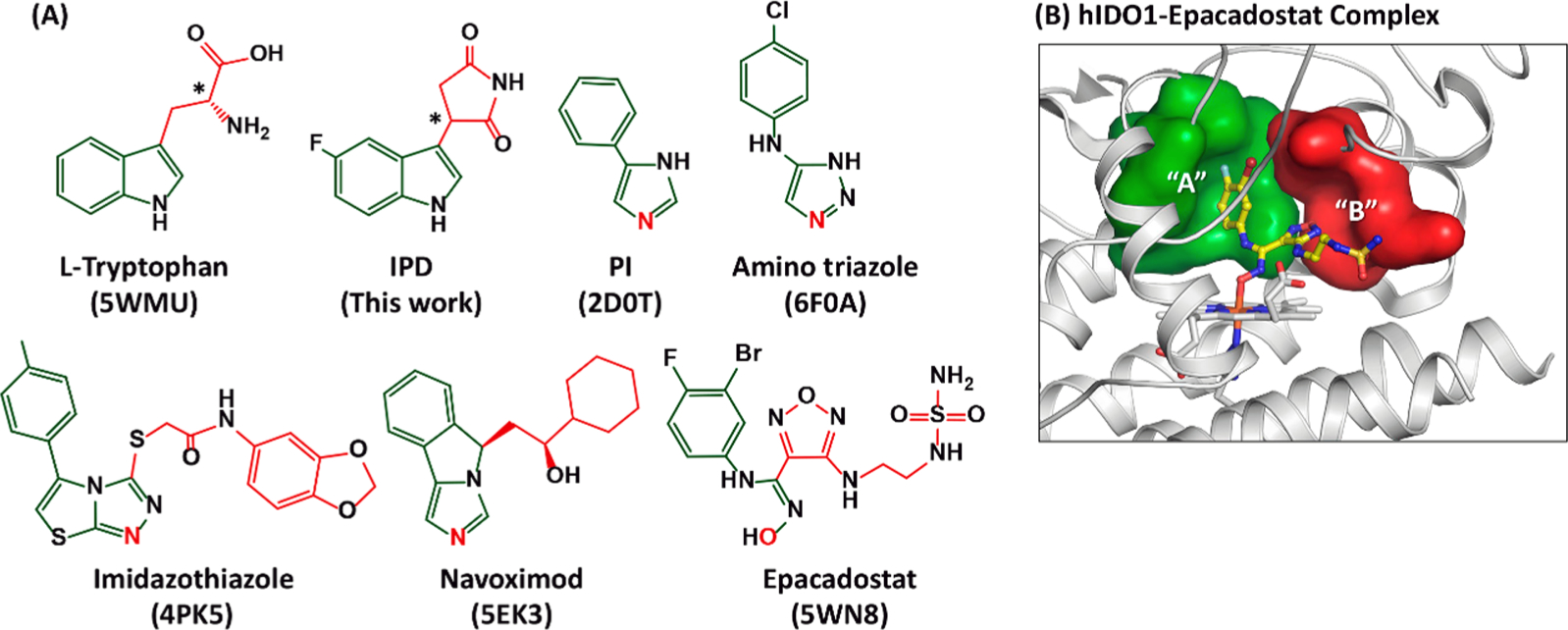
Chemical structures of hIDO1 inhibitors (A) and active site structure of hIDO1 (B). The structure of the substrate l-Trp is shown at the upper left corner as a reference. The asterisks indicate the chiral centers in l-Trp and IPD. The PDB code of hIDO1 in complex with the substrate or an inhibitor is indicated at the bottom of each structure. The atom in each inhibitor that coordinates to the heme iron as the sixth ligand is labeled in red. The fragments in each structure that occupy the “A” and “B” pockets of the Sa site are highlighted in green and red, respectively. The locations of the “A” and “B” pockets in the Sa site are elucidated in (B) based on the structure of the epacadostat complex (PDB code: 5WN8), where the bound inhibitor is shown as yellow ball-and-sticks.
In contrast to hIDO1 inhibitors, none of the hTDO inhibitors have entered clinical trials. In addition, the crystal structure of hTDO has only recently been solved in a substrate-bound state,55 and no structures of hTDO-inhibitor complexes are available to date. Here we sought to use IPD, an indole derivative with a reported IC50 of ~0.4 μM and ≫50 μM for hIDO1 and hTDO, respectively,42 as a structural probe to elucidate the inhibitor selectivity in hIDO1 with respect to that in hTDO.
RESULTS AND DISCUSSION
Inhibition Activity of IPD toward hIDO1 versus hTDO.
To quantify the inhibition activity of IPD, we used optical absorption spectroscopy to follow the NFK production rate of each enzyme as a function of [IPD]. We found that IPD is able to inhibit hIDO1 and hTDO with an IC50 of 1 and 424 μM, respectively (Figure 2), consistent with the previously reported values.42 The data demonstrate that IPD inhibits hIDO1 with a ~400-fold higher potency than hTDO, confirming that IPD is a hIDO1-selective inhibitor.
Figure 2.
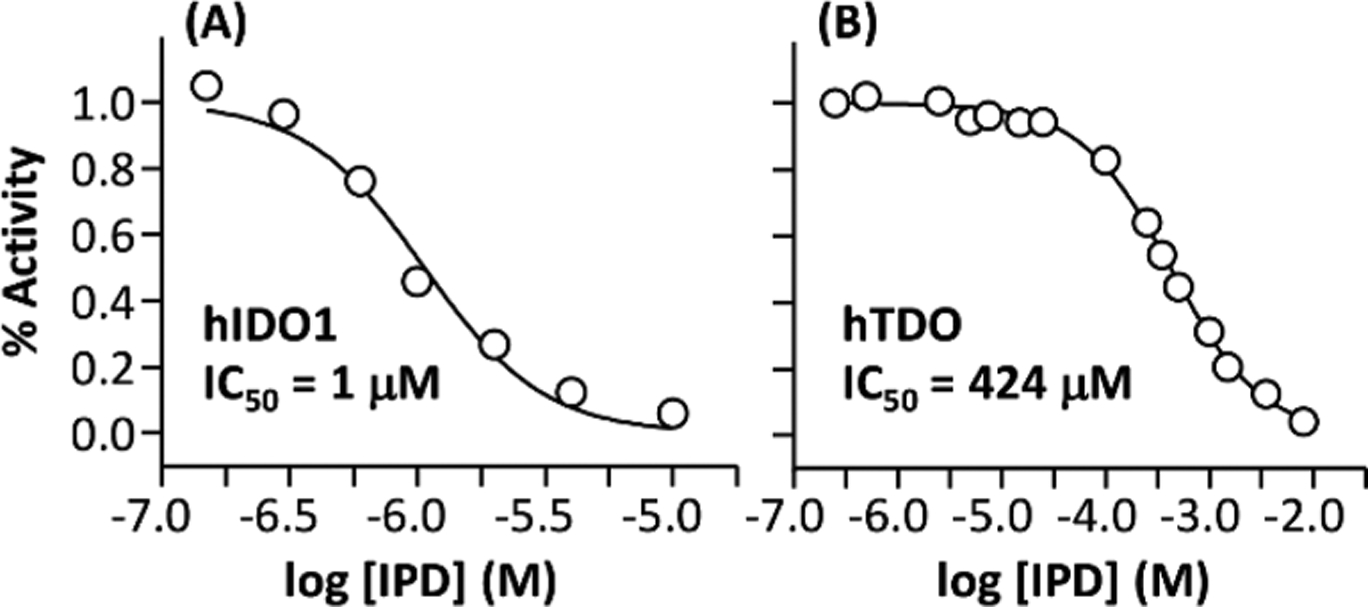
Inhibition activity of IPD toward hIDO1 (A) and hTDO (B). The activities of hIDO1 and hTDO were measured at pH 7.4 in the presence of 50 and 100 μM l-Trp, respectively.
Heme Coordination State in hIDO1 versus hTDO.
To differentiate the binding modes of IPD in the two enzymes, we used optical absorption spectroscopy to define how IPD perturbs the heme coordination state of each enzyme. As shown in Figure 3A, the ferric hIDO1 exhibits a Soret band at 404 nm and visible/charge transfer bands at 504/632 nm, suggesting a six coordinate (6C) ferric heme with a water coordinated to the heme iron as a sixth ligand.5,56–58 IPD binding leads to a dramatic reduction in the intensity of the Soret band, the broadening of the bandwidth, and the blue-shift of the peak maximum from 404 to 398 nm, as well as the appearance of two visible bands at 560/598 nm at the expense of the 504/632 nm bands, suggesting that almost all the molecules in the 6C water-bound state are converted to a five coordinate (5C) water-free state.59 The ferrous hIDO1, on the other hand, exhibits a Soret band at 426 nm and a visible band at 559 nm (Figure 3B), typical for a 5C ferrous heme.58,60 Unlike the ferric enzyme, the addition of IPD does not perturb the spectrum of the enzyme.
Figure 3.
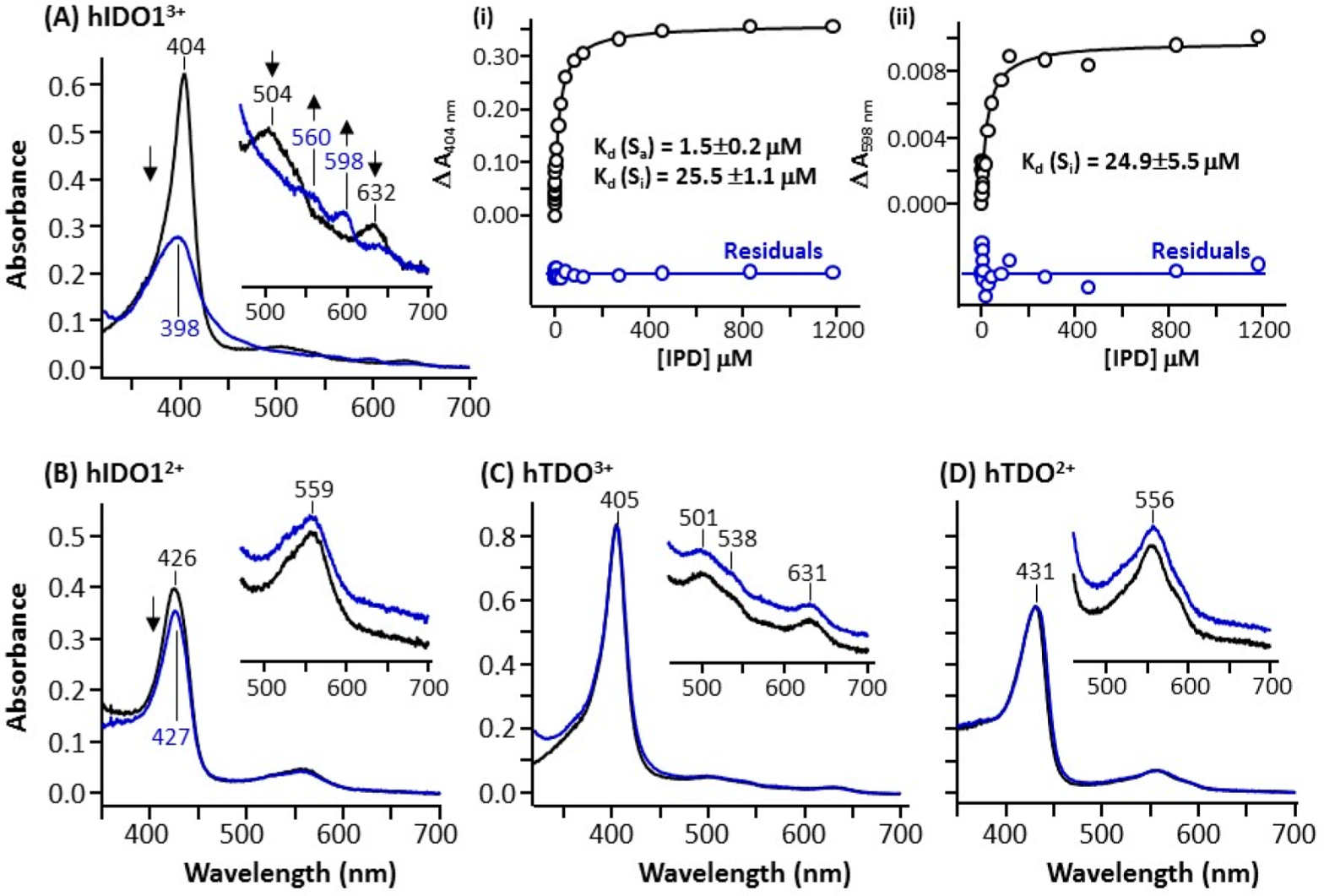
Spectral changes introduced by IPD binding to ferric and ferrous hIDO1 (A,B) and hTDO (C,D). The black and blue spectrum shown in each panel were obtained in the absence or presence of IPD, respectively. The inset in each panel shows the expanded view of the visible region of the spectra. Inset (i) and (ii) in (A) show a plot of ΔA404nm and ΔA598nm obtained as a function of [IPD] based on the difference spectra shown in Figure S1. They were fitted with a two-binding site model and one-binding site model, respectively, as described in the Methods; the residuals from the fitting (with the same scale as the data) are shown at the bottom of each plot.
To quantify the binding affinity, we titrated the ferric enzyme with IPD and calculated the difference spectra using the IPD-free spectrum as a reference (Figure S1) and plotted ΔA404nm and ΔA598nm as a function of [IPD]. The ΔA404nm plot, shown in inset (i) in Figure 3A, shows two transitions with Kd of 1.5 and 25.5 μM, suggesting that hIDO1 possesses two IPD binding sites, despite the fact that only one IPD molecule was identified in the reported structure of the hIDO1-IPD complex.42 The ΔA598nm plot, shown in inset (ii) in Figure 3A, in contrast, is best fitted with a single Kd of ~24.9 μM, suggesting that the 598 nm band is a spectral marker for the weaker IPD binding site. On the basis of the Cheng–Prusoff equation,61 IC50 can be estimated by Kd × [1 + ([S]/KM)], where [S] is the substrate concentration used for the activity measurements and KM is the concentration of substrate at which the enzyme activity is at half maxima. The calculated IC50 associated with the strong binding site is 3 μM (with [S] = 50 μM and KM = 23 μM5), which is similar to the experimentally determined IC50 shown in Figure 2A, suggesting that the inhibition activity of IPD is dominated by its binding to the strong binding site.
hTDO exhibits spectra similar to those of hIDO1 in both the ferric and ferrous states. The ferric enzyme has a Soret band at 405 nm and visible/charge transfer bands at 501/631 nm, suggesting a 6C water-bound ferric heme, while the ferrous enzyme has a Soret band at 431 nm and a visible band at 556 nm, indicating a 5C ferrous heme. However, unlike that observed in hIDO1, the addition of IPD does not perturb the spectrum of hTDO in either the ferric or ferrous state. Taken together the spectroscopic and activity data indicate that (i) the binding of IPD to ferric hIDO1, but not hTDO, leads to the dissociation of the water ligand from the heme iron, (ii) IPD, unlike other inhibitors shown in Figure 1, does not directly coordinate to the heme iron in either hIDO1 or hTDO, and (iii) hIDO1 has two IPD binding sites.
Crystal Structure of hIDO1-IPD Complex.
To obtain the structure of the hIDO1-IPD complex, we crystallized hIDO1, soaked it with IPD and then freeze-trapped it in liquid nitrogen as a function of time (~0–9 h). The structures of the crystals were solved in a dimeric form as reported previously,41,52 although the enzyme functions as a monomer in free solution. Two types of complex structures were identified. The first structure detected at ~4 h is a homo dimer with one IPD molecule bound in the Sa site of each subunit (referred to as hIDO1-IPD adduct hereinafter). The second structure observed at a relatively longer soaking time (~9 h) is a mixed-ligand species, where one subunit is trapped in hIDO1-IPD adduct state, while the other subunit binds two IPD molecules (referred to as hIDO1-IPD2 adduct hereinafter), one in the Sa site and the other in a previously identified inhibitory site (Si)41 on the proximal side of the heme. The detection of the hIDO1-IPD2 adduct is consistent with the two IPD binding sites apparent from the solution spectrophotometric titration results shown in Figure 3A.
We refined the structure of the mixed-ligand complex, where the subunit A and B are trapped in the hIDO1-IPD and hIDO1-IPD2 adduct state, respectively, to a resolution of 2.65 Å (Table S1) (PDB Code: 6PZ1). The structure of the hIDO1-IPD adduct is similar to that published by Crosignani et al.,42 while that of the hIDO1-IPD2 adduct, shown in Figure 4, has never been reported in the past. Comparison of the structures of the two subunits reveals that the occupation of the Si site by IPD does not affect the binding pose of the IPD bound in the Sa site (Figure S2A–B) or the overall structure of the enzyme, except that local structural perturbations are evident in the Si site (vide infra). The observation that IPD binding to the Si site does not affect the structure of the Sa site, where the Trp dioxygenation reaction takes place, suggests that the inhibition activity of IPD shown in Figure 2A is a result of its binding to the Sa site, not the Si site. It, combined with the spectrophotometric titration data shown in Figure 3A, suggests that the Sa site is the high affinity IPD binding site with a Kd of 1.5 μM, while the Si site is the low affinity site with a Kd of 25.5 μM.
Figure 4.
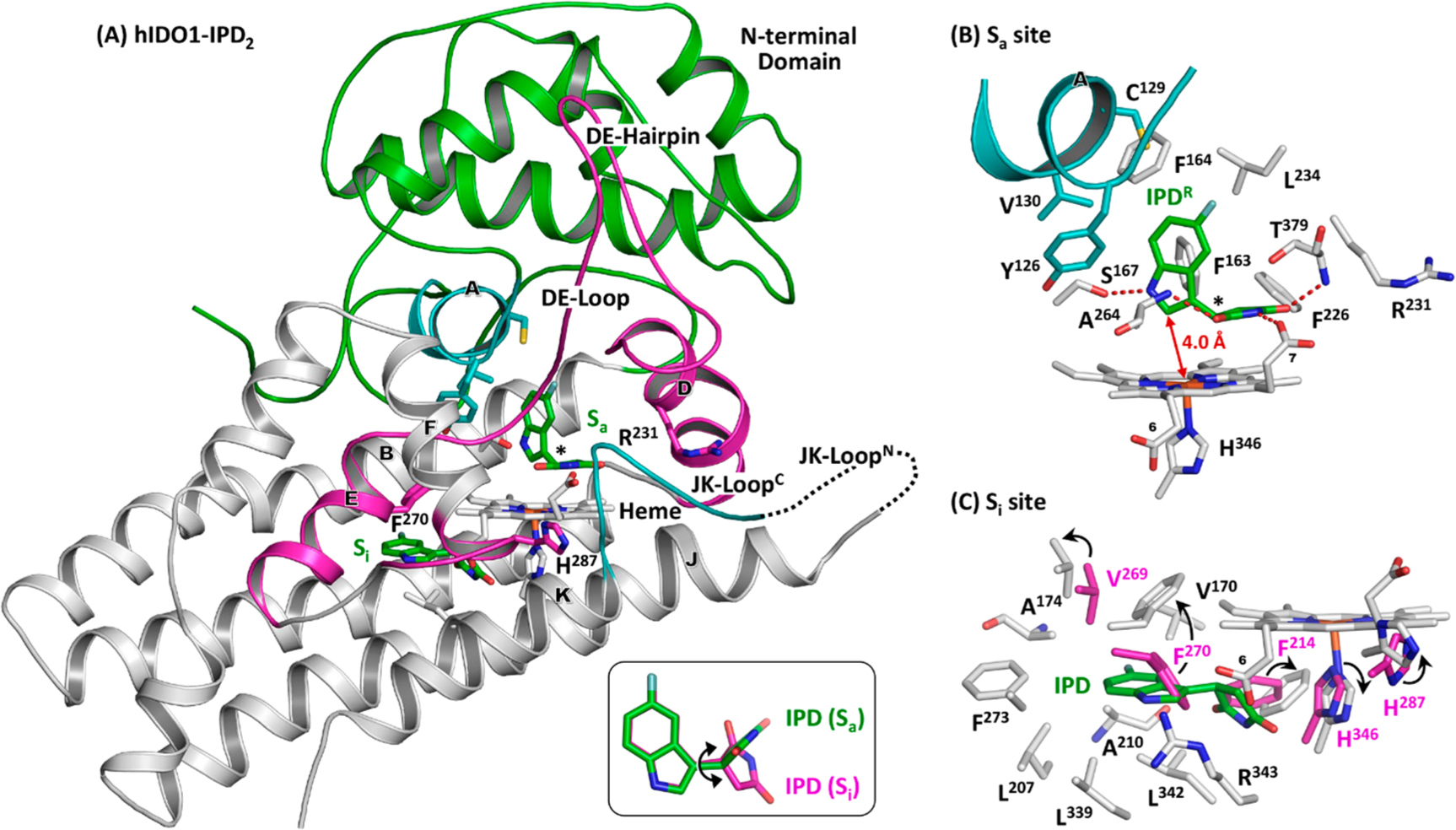
Crystal structure of hIDO1-IPD2 adduct (PDB code: 6PZ1). The bound IPD molecules are shown as green sticks. The lower right inset in (A) shows the superimposed structures of the IPD molecules bound in the Sa and Si site, where the double arrow indicates the rotation of the succinimide ring along the C–C bond linking it to the indole ring. The expanded views of the Sa and Si sites are shown in (B,C) to elucidate the protein-IPD interactions in each binding site. The chiral center in IPD bound in the Sa site is indicated by the asterisk in (B); the electron density map associated with the inhibitor is best fitted with the R enantiomer (see Figure S2A,B). The Si site structure in (C) is superimposed with those associated with the hIDO1-IPD adduct from subunit A (shown as magenta sticks) to illustrate conformational changes induced by IPD-binding to the Si site. The major conformational changes are indicated by the arrows.
hIDO1 is a two-domain α-helical protein. The large C-terminal domain contains the Sa site, where the heme prosthetic group and the substrates, Trp and O2, bind. The small N-terminal domain (colored in green), sitting on top of it, contains the A-Helix (colored in cyan), which forms the roof of the Sa site. The C-terminal domain of an active site loop, JK-Loop, adapts a reverse β-turn structure (referred to as the JK-LoopC), which shields the Sa site from the bulk solvent. The N-terminal domain of the JK-Loop (referred to as the JK-LoopN), on the other hand, is disordered.
Strong electron density associated with IPD is evident in the Sa site on the distal side of the heme (Figure S2A,B). Although a racemic mixture of IPD was used in this study, the inhibitor is best modeled with the R enantiomer (referred to as IPDR hereinafter) as reported previously.42 IPDR is surrounded by a group of hydrophobic residues (Figure 4B), including F163/ F164 from the B-Helix, F226/L234 from the D-Helix, A264 from the DE-Loop, T379 from the conserved “GTGG” domain5 in the JK-LoopC, and Y126/C129/V130 from the A-Helix. The indole ring sits in the “A” pocket, where it lies perpendicular to the heme, with its C2~4.0 Å away from the heme iron and its indoleamine H-bonded with the side chain of S167. The succinimide group occupies the “B” pocket, where it lies parallel with the heme and perpendicular to the indole ring. Its imide group H-bonds with the heme propionate-7 group and the peptide N atoms of A264 and T379. The H-bond with T379 anchors the JK-LoopC in a “closed” conformation, similar to that in the Trp complex,41 and distinct from the disordered structures detected in the substrate-free protein and all other known inhibitor complexes41,44,52–54,62
In subunit B, additional electron density associated with IPD is evident in the Si site on the proximal side of the heme (Figure S2B).41,63 The binding pose of IPD is distinct from that in the Sa site as illustrated in the right lower inset in Figure 4A. The indole ring of IPD sits in a hydrophobic pocket, while the succinimide ring stretches out toward the H346 side chain and the heme propionate-6 group. As illustrated in Figure 4C, IPD binding to the Si site introduces significant structural rearrangement to the Si site. In particular, the F270 side chain rotates up to accommodate the indole ring of the inhibitor, while the F214 and H346 side chains moves out to leave room for the succinimide group. In addition, the side chain of H287, which sits at the junction between the EF-Loop and the E-Helix, rotates by ~90°.
Crystal Structure of hTDO-IPD Complex.
Unlike hIDO1, hTDO is a homo tetramer made of a dimer of dimers, where each dimer is stabilized by domain-swapping of a ~50 residue long N-terminal fragment.5,55 To obtain the structure of the hTDO-IPD complex, we crystallized hTDO and used a soaking method, similar to that described above, to generate the complex crystals. Two distinct types of complex structures were identified. At ~30 min, we detected a species where only one IPD molecule is found in the Sa site in each subunit (referred to as hTDO-IPD adduct hereinafter), while at a longer soaking time (~4 h), we found a new species (referred to as hTDO-IPD2 adduct hereinafter) in which two IPD molecules are present in each subunit, one in the Sa site and the other in a previously identified exosite (Sexo)55 that is >40 Å away from the Sa site.
We refined the structures of the hTDO-IPD and hTDO-IPD2 complexes to a resolution of 2.02 Å (PDB Code: 6PYZ) and 2.40 Å (PDB Code: 6PYY), respectively (Table S1). Comparison of the two structures reveal that the occupation of the Sexo site by IPD does not affect the overall structure of the enzyme. The observation that IPD binding to the Sexo site does not affect the structure of the Sa site, where the dioxygenase chemistry occurs, suggests that the Sa site, not the Sexo site, is responsible for the inhibition activity of IPD shown in Figure 2B.
The structures of the four subunits of the tetramer are almost identical. Each monomer contains a core domain that binds the heme at one end and holds a helix–loop–helix domain (colored in green) at the other end (Figure 5A). The core domain shares high structural similarity with the large domain of hIDO1, while the N-terminal A-Helix from the neighboring subunit mimics the A-Helix from the small domain in hIDO1 that forms the roof of the Sa site. The helix–loop–helix domain observed in hTDO is absent in hIDO1; conversely, the N-terminal small domain, the disordered JK-LoopN, and the long DE-Hairpin present in hIDO1 is absent in hTDO.
Figure 5.
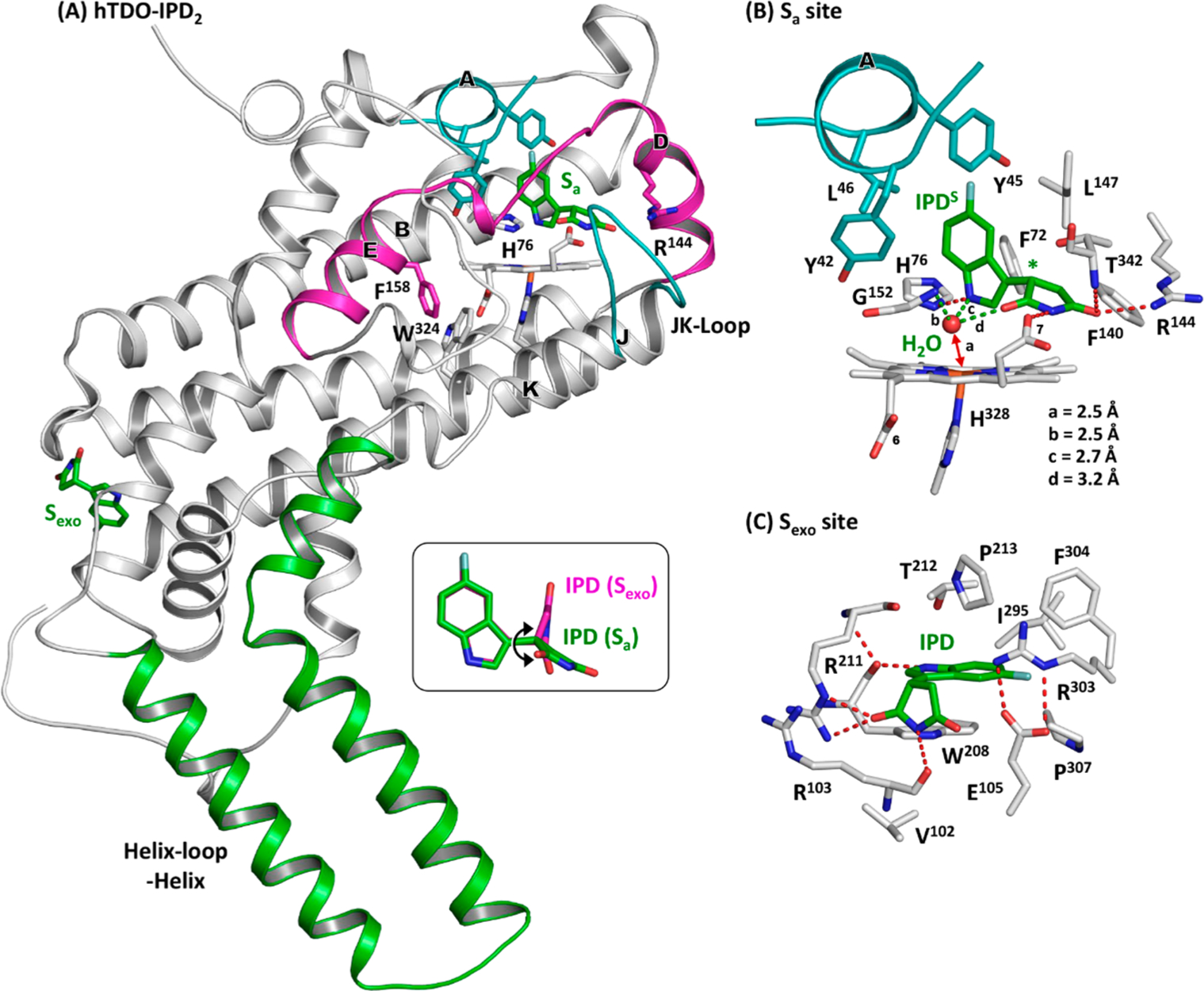
Crystal structure of hTDO-IPD2 adduct (PDB code: 6PYY). The bound IPD molecules are shown as green sticks. The inset in (A) shows the superimposed structures of the IPD molecules bound in the Sa and Sexo site, where the double arrow indicates the rotation of the succinimide ring along the C–C bond linking it to the indole ring. The expanded views of the Sa and Sexo sites are shown in (B,C) to elucidate the protein-IPD interactions in each binding site. The chiral center in IPD bound in the Sa site is indicated by the asterisk in (B); the electron density map associated with IPD is best fitted with the S enantiomer (see Figure S3B).
Clear electron density is evident in the distal heme pocket. It is surprisingly best-fitted with the S enantiomer (referred to as IPDS hereinafter) (Figure S3A,B), distinct from the R enantiomer identified in hIDO1. IPDS is surrounded by mostly hydrophobic residues, including F72 and F140/L147 from the B and D-Helix, respectively, G152 from the DE-Loop, T342 from the JK-Loop, and Y42/Y45/L46 from the A-Helix (Figure 5B). The indole group occupies the “A” pocket, where its indole ring lies perpendicular to the heme and its indoleamine group H-bonds with the side chain of H76. The succinimide group extends out into the “B” pocket, with its imide group H-bonding with (i) the heme propionate-7 group, (ii) the side chain of R144, and (iii) the peptide N atom of T342. The H-bond with T342 locks the JK-Loop in a closed conformation as that found in the Trp complex.55 Similar to that in hIDO1, IPD does not coordinate to the heme iron, but there is a water molecule that is positioned ~2.5 Å away from the heme iron in a triad formed by the peptide amine group of G152 and the indole amine and imide oxygen of IPD. The presence of the water in hTDO, but not hIDO1, is in good agreement with the spectral data shown in Figure 3.
The space equivalent to the Si site on the proximal side of the heme in hIDO1 is blocked by the bulky side chains of F158 (equivalent to F270 in hIDO1) and W324 in hTDO (Figure 5A), which prevents it from binding IPD. However, clear electron density associated with a second IPD is evident in the Sexo site (Figure S3C,D). The binding pose of IPD in the Sexo site is different from that in the Sa site, as shown in the inset in Figure 5A. The indole ring of IPD is stabilized by W208, via π-stacking with its side chain and H-bonding with its peptide carbonyl group (Figure 5C). The succinimide ring is anchored in position by H-bonding with the side chain of R211 and the peptide carbonyl group of R103.
Why Is IPD a Selective hIDO1 Inhibitor?
As highlighted in Figure 6A, the binding pose of IPD bound in the Sa site in hIDO1 is distinct from that in hTDO. Specifically, its indole ring rotates ~30° such that the indoleamine group forms a H-bond with the side chain of S167. In addition, its succinimide ring moves down and lies parallel with the heme. Together these unique structural features destabilize the water ligand of the heme iron in hIDO1, forcing it to move out of distal heme pocket.
Figure 6.
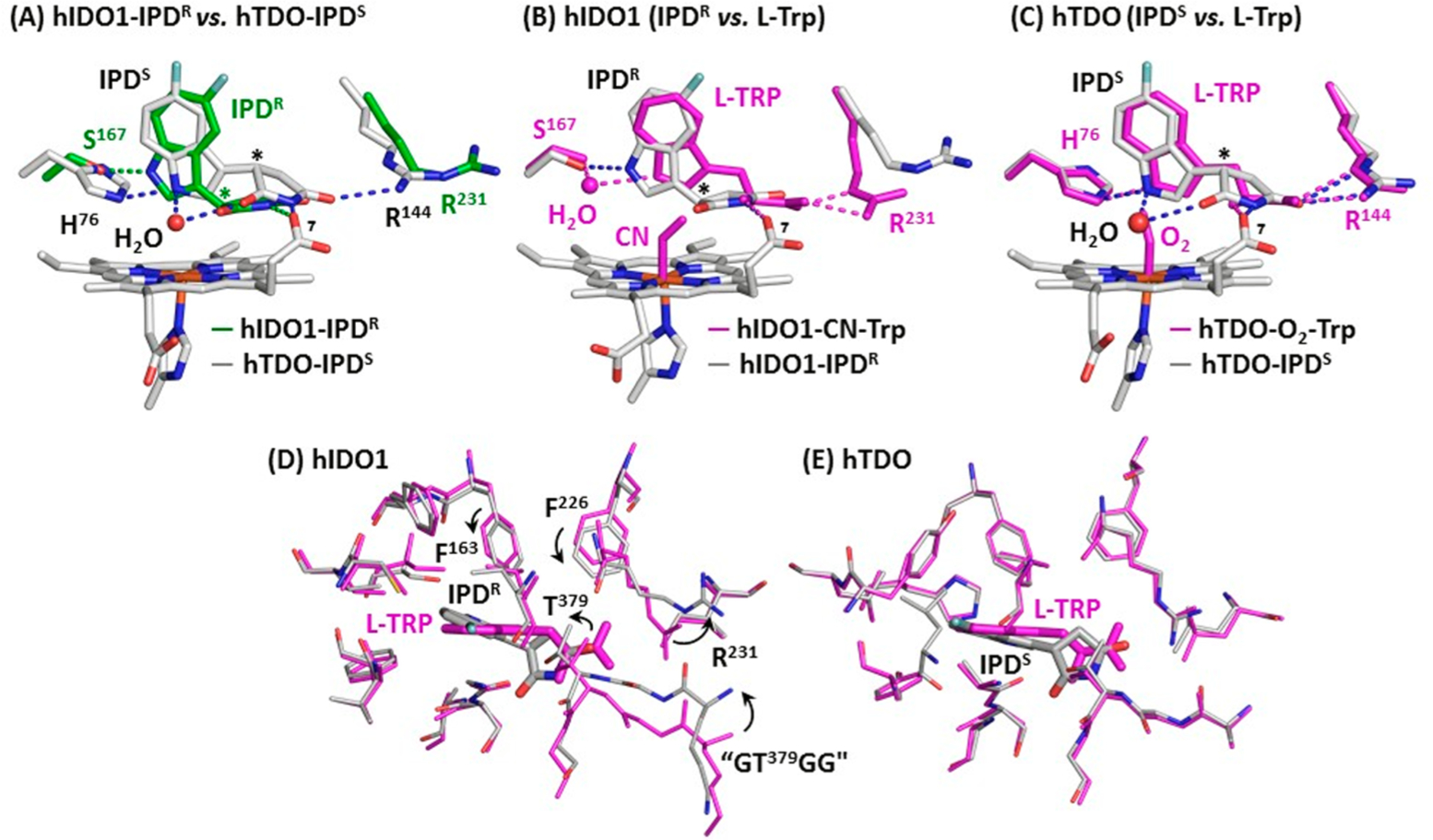
Superimposed structures of hIDO1-IPD and hTDO-IPD complexes (A) and those of IPD and Trp-bound complexes of hIDO1 (B,D) and hTDO (C,E). The asterisks in A–C indicate the chiral centers in IPD. In (D,E) the residues within ~5 Å from the bound IPD (gray) and Trp (magenta) in hIDO1 and hTDO are shown to highlight the significant protein structural rearrangement in the Sa site associated with the replacement of Trp by IPD in hIDO1 (indicated by the arrows), which are absent in hTDO. The “GTGG” motif shown in (D) is a part of the JK-Loop. The PDB codes of the hTDO-IPD and hIDO1-IPD adducts are 6PYY and 6PZ1, respectively; those of hTDO-O2-Trp and hIDO1-CN-Trp complexes are 5TI9 and 5WMU, respectively.
In hIDO1, the binding pose of IPDR significantly deviates from that of the substrate Trp (Figure 6B). In particular, the indole ring rotates toward S167, enabling their direct H-bonding interaction (instead of the water mediated H-bonding interaction in the Trp complex). Although the succinimide ring coincides well with the ammonium group of the Trp, allowing it to H-bond with the propionate-7 group of the heme, it is displaced from the carboxylate group of the Trp, preventing it from H-bonding with R231. In contrast, the binding pose of IPDS in hTDO is similar to that of Trp (Figure 6C). Specifically, the orientation of the indole ring of IPDS and its H-bond with H76 are almost identical to those associated with Trp; in addition, the succinimide ring coincides well with the ammonium/carboxylate groups of the Trp. Furthermore, the distal water superimposes well with the terminal atom of the iron-bound O2 in the hTDO-O2-Trp complex.
The unique binding pose of IPD with respect to Trp in hIDO1 is associated with significant conformational rearrangement in the Sa site (Figure 6D). In contrast, there is no noticeable protein conformational difference between the IPD and Trp complexes of hTDO (Figure 6E). The data suggest that the Sa site in hIDO1 is more flexible, which can change its conformation to optimize the inhibitor-protein interactions, thereby driving the preferential binding of the R enantiomer based on an induced-fit mechanism, while that in hTDO is more rigid, which forces it to preferentially bind the S enantiomer based on a lock-and-key mechanism. The structural flexibility of the Sa site in hIDO1 revealed here is consistent with its much broader substrate selectivity with respect to hTDO,1,5 as well as its unusual protein plasticity recently revealed in the BMS-986205 complex.46
Although a racemic mixture of IPD was employed in this work, pure R and S enantiomers were identified in the Sa sites of hIDO1 and hTDO, respectively, indicating high inhibitor stereoselectivity in each enzyme. Consistent with this notion, previous studies reported by Crosignani et al.42 showed that the efficacy of the R enantiomer of IPD in hIDO1 is ~200-fold stronger than that of the S enantiomer. While the relative efficacy of the R and S enantiomers of IPD in hTDO remains to be determined, the current data suggest that hTDO binds the S enantiomer much stronger than the R enantiomer.
CONCLUSIONS
The structures reported here reveal that the Sa site of hIDO1 is much more flexible than that of hTDO, manifesting the importance of flexible docking in rational drug design targeting the two enzymes. In addition, the structures offer the first direct evidence demonstrating that the Si site in hIDO1 and Sexo site in hTDO can be occupied by small molecules other than the substrate Trp. Previous studies show that Trp binding to the Si site in hIDO1 retards the Trp dioxygenation activity in the S site,41,63 a and that Trp binding to the Sexo site in hTDO does not affect the Trp dioxygenation activity in the Sa site, but it regulates the cellular lifetime of the enzyme.55 The data reported here suggest that these secondary sites can be occupied by potential inhibitors that directly reduce hIDO1 activity by inhibiting the Sa site activity of the enzyme, or indirectly reduce hTDO activity by promoting the cellular degradation of the enzyme. In summary, the structural data reported here open up new avenues for structure-based drug design targeting the two important immunosuppressive enzymes.
METHODS
Activity and Spectroscopic Measurements.
The steady-state activities of hIDO1 and hTDO were measured with 50 and 100 μM l-Trp, respectively, in 50 mM Tris buffer (pH 7.4) at 20 °C with standard protocols as reported previously.56,63 The initial linear velocities of the reactions as a function of the concentration of PF-06840003 (IPD) were obtained by monitoring the formation of the product, NFK, at 321 nm (ε = 3750 M−1 cm−1) as a function of time with a UV2100 spectrophotometer (Shimadzu Scientific Instruments, Inc.) with a spectral slit width of 2 nm. IPD was purchased from Advanced ChemBlocks Inc., as a racemic mixture. All the data were analyzed with Origin 6.1 software (OriginLab Corporation).
All the absorption spectra were obtained with the UV2100 spectrophotometer with a spectral slit width of 1 nm. The hIDO1 samples (4 μM) and hTDO samples (5 μM) were prepared in the absence or presence of 0.2 mM and 5 mM IPD, respectively, in 50 mM Tris buffer (pH 7.4). To determine the Kd value(s), we titrated ferric IDO1 with IPD and calculated the difference spectra using the IPD-free spectrum as a reference (see Figure S1). The ΔA404nm and ΔA598nm were then plotted as a function of [IPD] (see the insets in Figure 3A). The ΔA598nm plot is best-fitted with a one-binding site model, Y = [(A × X)/(Kd + X)], with a Kd of 24.9 ± 5.5 μM, while the ΔA404nm plot is best-fitted with a two-binding site model, Y = [(A1 × X)/(Kd(1) + X)] + [(A2 × X)/(Kd(2) + X)], with Kd(1) = 1.5 ± 0.2 μM and Kd(2) = 25.5 ± 1.1 μM.
Crystal Preparation.
hIDO1 and hTDO proteins were expressed and purified as reported previously.56,64 All the crystals were grown by using the under-oil microbatch method. The hIDO1 crystals were grown by mixing protein solutions (40 mg/mL) with the precipitant solution (100 mM sodium thiosulfate in 100 mM CAPS buffer and 20% PEG 8000 at pH 10) as reported previously.41 The crystals were then soaked with 32 mM IPD and harvested as a function of soaking time. They were then cryoprotected by supplementing the mother solution with 20% (v/v) glycerol and flash-frozen in liquid nitrogen for data collection.
The hTDO crystals were grown by mixing protein solutions (45 mg/mL) with the precipitant solution (50 mM sodium citrate, 2% Tacsimate and 5% PEG 3350 at pH 5.6) in the presence of 5 mM α-methyl tryptophan as reported previously.55 The crystals were then soaked with 10 mM IPD as a function of time before they were cryoprotected by supplementing the mother solution with 25% (v/v) ethylene glycol and flash-frozen in liquid nitrogen for data collection.
Crystallographic Data Collection and Analysis.
All the crystallographic data were collected by the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline staff at Sector 31 of the Advanced Photon Source. The diffraction images were indexed, integrated, and scaled with XDS65 and Aimless.66 The Karplus–Diederichs method67 was used to find a proper resolution cutoff for each structure. Molecular replacement was conducted with Phaser68 through the CCP4i graphic interface69 using hIDO1-CN-Trp complex structure (PDB code: 5WMU) and the hTDO-Trp complex structure (PDB code: 5TIA) as the search model for hIDO1 and hTDO, respectively. Further model building was performed using COOT.70 Structure refinements were performed using Refmac5.69,71,72 Data processing and refinement statistics are summarized in Table S1. The structural models were displayed with PyMOL (http://www.pymol.org/).
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Denis L. Rousseau for helpful discussions. The structural data were collected by the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline staff at Sector 31 of the Advanced Photon Source. This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. This work was supported by National Institute of Health Grant GM115773 and GM126297 to S.-R.Y.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08871.
Table S1; Figures S1–S3 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Sono M; Roach MP; Coulter ED; Dawson JH Heme-Containing Oxygenases. Chem. Rev 1996, 96 (7), 2841–2888. [DOI] [PubMed] [Google Scholar]
- (2).Leklem JE Quantitative aspects of tryptophan metabolism in humans and other species: a review. Am. J. Clin. Nutr 1971, 24 (6), 659–72. [DOI] [PubMed] [Google Scholar]
- (3).Takikawa O Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem. Biophys. Res. Commun 2005, 338 (1), 12–9. [DOI] [PubMed] [Google Scholar]
- (4).Raven EL A short history of heme dioxygenases: rise, fall and rise again. JBIC, J. Biol. Inorg. Chem 2017, 22 (2–3), 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lewis-Ballester A; Pham KN; Liao M; Correia MA; Yeh S-R Structure, Function and Regulation of Human Heme-based Dioxygenases In Dioxygen-Dependent Heme Enzymes; The Royal Society of Chemistry, 2019; Chapter 9, pp 181–221. [Google Scholar]
- (6).Geng J; Liu A Heme-dependent dioxygenases in tryptophan oxidation. Arch. Biochem. Biophys 2014, 544, 18–26. [DOI] [PubMed] [Google Scholar]
- (7).Oxenkrug G Serotonin-kynurenine hypothesis of depression: historical overview and recent developments. Curr. Drug Targets 2013, 14 (5), 514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Oxenkrug GF Genetic and hormonal regulation of tryptophan kynurenine metabolism: implications for vascular cognitive impairment, major depressive disorder, and aging. Ann. N. Y. Acad. Sci 2007, 1122, 35–49. [DOI] [PubMed] [Google Scholar]
- (9).Miura H; Ozaki N; Sawada M; Isobe K; Ohta T; Nagatsu T A link between stress and depression: shifts in the balance between the kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress 2008, 11 (3), 198–209. [DOI] [PubMed] [Google Scholar]
- (10).Guillemin GJ; Brew BJ Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. Redox Rep. 2002, 7 (4), 199–206. [DOI] [PubMed] [Google Scholar]
- (11).Baran H; Jellinger K; Deecke L Kynurenine metabolism in Alzheimer’s disease. J. Neural Transm 1999, 106 (2), 165–81. [DOI] [PubMed] [Google Scholar]
- (12).Chen Y; Guillemin GJ Kynurenine pathway metabolites in humans: disease and healthy States. Int. J. Tryptophan Res 2009, 2, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Heyes MP; Saito K; Crowley JS; Davis LE; Demitrack MA; Der M; Dilling LA; Elia J; Kruesi MJ; Lackner A; Larsen SA; Lee K; Leonard HL; Markey SP; Martin A; Milstein S; Mouradian MM; Pranzatelli MR; Quearry BJ; Salazar A; Smith M; Strauss SE; Sunderland T; Swedo SW; Tourtellotte WW Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115 (5), 1249–1273. [DOI] [PubMed] [Google Scholar]
- (14).Campesan S; Green EW; Breda C; Sathyasaikumar KV; Muchowski PJ; Schwarcz R; Kyriacou CP; Giorgini F The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol 2011, 21 (11), 961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ogawa T; Matson WR; Beal MF; Myers RH; Bird ED; Milbury P; Saso S Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42 (9), 1702–6. [DOI] [PubMed] [Google Scholar]
- (16).Munn DH; Zhou M; Attwood JT; Bondarev I; Conway SJ; Marshall B; Brown C; Mellor AL Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281 (5380), 1191–3. [DOI] [PubMed] [Google Scholar]
- (17).Friberg M; Jennings R; Alsarraj M; Dessureault S; Cantor A; Extermann M; Mellor AL; Munn DH; Antonia SJ Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101 (2), 151–5. [DOI] [PubMed] [Google Scholar]
- (18).Uyttenhove C; Pilotte L; Theate I; Stroobant V; Colau D; Parmentier N; Boon T; Van den Eynde BJ Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med 2003, 9 (10), 1269–74. [DOI] [PubMed] [Google Scholar]
- (19).Prendergast GC Cancer: Why tumours eat tryptophan. Nature 2011, 478 (7368), 192–4. [DOI] [PubMed] [Google Scholar]
- (20).Prendergast GC; Smith C; Thomas S; Mandik-Nayak L; Laury-Kleintop L; Metz R; Muller AJ Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother 2014, 63 (7), 721–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mellor AL; Munn DH IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol 2004, 4 (10), 762–74. [DOI] [PubMed] [Google Scholar]
- (22).Mellor AL; Munn DH Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today 1999, 20 (10), 469–73. [DOI] [PubMed] [Google Scholar]
- (23).Platten M; Wick W; Van den Eynde BJ Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012, 72 (21), 5435–40. [DOI] [PubMed] [Google Scholar]
- (24).Mullard A Immunotherapy interest drives IDO deals. Nat. Rev. Drug Discovery 2015, 14, 373. [Google Scholar]
- (25).Sheridan C IDO inhibitors move center stage in immunooncology. Nat. Biotechnol 2015, 33 (4), 321–2. [DOI] [PubMed] [Google Scholar]
- (26).Huggett B Biotech’s wellspring—a survey of the health of the private sector in 2014. Nat. Biotechnol 2015, 33, 470–7. [DOI] [PubMed] [Google Scholar]
- (27).Muller AJ; DuHadaway JB; Donover PS; Sutanto-Ward E; Prendergast GC Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med 2005, 11 (3), 312–9. [DOI] [PubMed] [Google Scholar]
- (28).Ball HJ; Sanchez-Perez A; Weiser S; Austin CJ; Astelbauer F; Miu J; McQuillan JA; Stocker R; Jermiin LS; Hunt NH Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 2007, 396 (1), 203–13. [DOI] [PubMed] [Google Scholar]
- (29).Metz R; Duhadaway JB; Kamasani U; Laury-Kleintop L; Muller AJ; Prendergast GC Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007, 67 (15), 7082–7. [DOI] [PubMed] [Google Scholar]
- (30).Pilotte L; Larrieu P; Stroobant V; Colau D; Dolusic E; Frederick R; De Plaen E; Uyttenhove C; Wouters J; Masereel B; Van den Eynde BJ Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (7), 2497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lob S; Konigsrainer A; Zieker D; Brucher BL; Rammensee HG; Opelz G; Terness P IDO1 and IDO2 are expressed in human tumors: levo-but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother 2009, 58 (1), 153–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Witkiewicz AK; Costantino CL; Metz R; Muller AJ; Prendergast GC; Yeo CJ; Brody JR Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. J. Am. Coll Surg 2009, 208 (5), 781–787 discussion 787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nevler A; Muller AJ; Sutanto-Ward E; DuHadaway JB; Nagatomo K; Londin E; O’Hayer K; Cozzitorto JA; Lavu H; Yeo TP; Curtis M; Villatoro T; Leiby BE; Mandik-Nayak L; Winter JM; Yeo CJ; Prendergast GC; Brody JR Host IDO2 Gene Status Influences Tumor Progression and Radiotherapy Response in KRAS-Driven Sporadic Pancreatic Cancers. Clin. Cancer Res 2019, 25 (2), 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Prendergast GC; Malachowski WP; DuHadaway JB; Muller AJ Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77 (24), 6795–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rohrig UF; Majjigapu SR; Vogel P; Zoete V; Michielin O Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J. Med. Chem 2015, 58 (24), 9421–37. [DOI] [PubMed] [Google Scholar]
- (36).Platten M; von Knebel Doeberitz N; Oezen I; Wick W; Ochs K Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol 2015, 5, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liu X; Shin N; Koblish HK; Yang G; Wang Q; Wang K; Leffet L; Hansbury MJ; Thomas B; Rupar M; Waeltz P; Bowman KJ; Polam P; Sparks RB; Yue EW; Li Y; Wynn R; Fridman JS; Burn TC; Combs AP; Newton RC; Scherle PA Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115 (17), 3520–30. [DOI] [PubMed] [Google Scholar]
- (38).Cheong JE; Sun L Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy - Challenges and Opportunities. Trends Pharmacol. Sci 2018, 39 (3), 307–325. [DOI] [PubMed] [Google Scholar]
- (39).Prendergast GC; Malachowski WJ; Mondal A; Scherle P; Muller AJ Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol 2018, 336, 175–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Beatty GL; O’Dwyer PJ; Clark J; Shi JG; Bowman KJ; Scherle PA; Newton RC; Schaub R; Maleski J; Leopold L; Gajewski TF First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin. Cancer Res 2017, 23, 3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lewis-Ballester A; Pham KN; Batabyal D; Karkashon S; Bonanno JB; Poulos TL; Yeh SR Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun 2017, 8 (1), 1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Crosignani S; Bingham P; Bottemanne P; Cannelle H; Cauwenberghs S; Cordonnier M; Dalvie D; Deroose F; Feng JL; Gomes B; Greasley S; Kaiser SE; Kraus M; Negrerie M; Maegley K; Miller N; Murray BW; Schneider M; Soloweij J; Stewart AE; Tumang J; Torti VR; Van Den Eynde B; Wythes M Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1H-indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate. J. Med. Chem 2017, 60 (23), 9617–9629. [DOI] [PubMed] [Google Scholar]
- (43).Nayak A; Hao Z; Sadek R; Dobbins R; Marshall L; Vahanian N; Ramsey J; Kennedy E; Mautino M; Link C; Lin R; Royer-Joo S; Morrissey K; Mahrus S; McCall B; Pirzkall A; Munn D; Janik J; Khleif S Phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of GDC-0919 in patients with recurrent/advanced solid tumors. Eur. J. Cancer 2015, 51, S69. [Google Scholar]
- (44).Peng YH; Ueng SH; Tseng CT; Hung MS; Song JS; Wu JS; Liao FY; Fan YS; Wu MH; Hsiao WC; Hsueh CC; Lin SY; Cheng CY; Tu CH; Lee LC; Cheng MF; Shia KS; Shih C; Wu SY Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J. Med. Chem 2016, 59 (1), 282–93. [DOI] [PubMed] [Google Scholar]
- (45).Nelp MT; Kates PA; Hunt JT; Newitt JA; Balog A; Maley D; Zhu X; Abell L; Allentoff A; Borzilleri R; Lewis HA; Lin Z; Seitz SP; Yan C; Groves JT Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (13), 3249–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Pham KN; Yeh SR Mapping the Binding Trajectory of a Suicide Inhibitor in Human Indoleamine 2,3-Dioxygenase 1. J. Am. Chem. Soc 2018, 140 (44), 14538–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mitchell TC; Hamid O; Smith DC; Bauer TM; Wasser JS; Olszanski AJ; Luke JJ; Balmanoukian AS; Schmidt EV; Zhao Y; Gong X; Maleski J; Leopold L; Gajewski TF Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol 2018, JCO.2018.78.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Long GV; Dummer R; Hamid O; Gajewski TF; Caglevic C; Dalle S; Arance A; Carlino MS; Grob JJ; Kim TM; Demidov L; Robert C; Larkin J; Anderson JR; Maleski J; Jones M; Diede SJ; Mitchell TC Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20 (8), 1083–1097. [DOI] [PubMed] [Google Scholar]
- (49).Muller AJ; Manfredi MG; Zakharia Y; Prendergast GC Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin. Immunopathol 2019, 41 (1), 41–48. [DOI] [PubMed] [Google Scholar]
- (50).Ledford H Cancer drug’s stumbles prompt calls to rethink how immune therapies are tested. Nature 2018, DOI: 10.1038/d41586-018-07445-3. [DOI] [Google Scholar]
- (51).Garber K A new cancer immunotherapy suffers a setback. Science 2018, 360 (6389), 588. [DOI] [PubMed] [Google Scholar]
- (52).Sugimoto H; Oda S; Otsuki T; Hino T; Yoshida T; Shiro Y Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (8), 2611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Alexandre JAC; Swan MK; Latchem MJ; Boyall D; Pollard JR; Hughes SW; Westcott J New 4-Amino-1,2,3-Triazole Inhibitors of Indoleamine 2,3-Dioxygenase Form a Long-Lived Complex with the Enzyme and Display Exquisite Cellular Potency. ChemBioChem 2018, 19 (6), 552–61. [DOI] [PubMed] [Google Scholar]
- (54).Tojo S; Kohno T; Tanaka T; Kamioka S; Ota Y; Ishii T; Kamimoto K; Asano S; Isobe Y Crystal Structures and Structure-Activity Relationships of Imidazothiazole Derivatives as IDO1 Inhibitors. ACS Med. Chem. Lett 2014, 5 (10), 1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lewis-Ballester A; Forouhar F; Kim SM; Lew S; Wang Y; Karkashon S; Seetharaman J; Batabyal D; Chiang BY; Hussain M; Correia MA; Yeh SR; Tong L Molecular basis for catalysis and substrate-mediated cellular stabilization of human tryptophan 2,3-dioxygenase. Sci. Rep 2016, 6, 35169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Batabyal D; Yeh SR Human tryptophan dioxygenase: a comparison to indoleamine 2,3-dioxygenase. J. Am. Chem. Soc 2007, 129 (50), 15690–701. [DOI] [PubMed] [Google Scholar]
- (57).Lu C; Mukai M; Lin Y; Wu G; Poole RK; Yeh SR Structural and functional properties of a single domain hemoglobin from the food-borne pathogen Campylobactor jejuni. J. Biol. Chem 2007, 282 (35), 25917–28. [DOI] [PubMed] [Google Scholar]
- (58).Terentis AC; Thomas SR; Takikawa O; Littlejohn TK; Truscott RJ; Armstrong RS; Yeh SR; Stocker R The heme environment of recombinant human indoleamine 2,3-dioxygenase. Structural properties and substrate-ligand interactions. J. Biol. Chem 2002, 277 (18), 15788–94. [DOI] [PubMed] [Google Scholar]
- (59).Egawa T; Yeh SR Structural and functional properties of hemoglobins from unicellular organisms as revealed by resonance Raman spectroscopy. J. Inorg. Biochem 2005, 99 (1), 72–96. [DOI] [PubMed] [Google Scholar]
- (60).Lu C; Egawa T; Mukai M; Poole RK; Yeh SR Hemoglobins from Mycobacterium tuberculosis and Campylobacter jejuni: a comparative study with resonance Raman spectroscopy. Methods Enzymol. 2008, 437, 255–86. [DOI] [PubMed] [Google Scholar]
- (61).Yung-Chi C; Prusoff WH Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22 (23), 3099–3108. [DOI] [PubMed] [Google Scholar]
- (62).Wu Y; Xu T; Liu J; Ding K; Xu J Structural insights into the binding mechanism of IDO1 with hydroxylamidine based inhibitor INCB14943. Biochem. Biophys. Res. Commun 2017, 487 (2), 339–343. [DOI] [PubMed] [Google Scholar]
- (63).Lu C; Lin Y; Yeh SR Inhibitory substrate binding site of human indoleamine 2,3-dioxygenase. J. Am. Chem. Soc 2009, 131 (36), 12866–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Samelson-Jones BJ; Yeh SR Interactions between nitric oxide and indoleamine 2,3-dioxygenase. Biochemistry 2006, 45 (28), 8527–38. [DOI] [PubMed] [Google Scholar]
- (65).Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–26. [DOI] [PubMed] [Google Scholar]
- (66).Evans PR; Murshudov GN How good are my data and what is the resolution? Acta Crystallogr., Sect. D: Biol. Crystallogr 2013, 69 (7), 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Karplus PA; Diederichs K Linking crystallographic model and data quality. Science 2012, 336 (6084), 1030–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40 (4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67 (4), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (71).Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67 (4), 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53 (3), 240–255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.