

Abstract
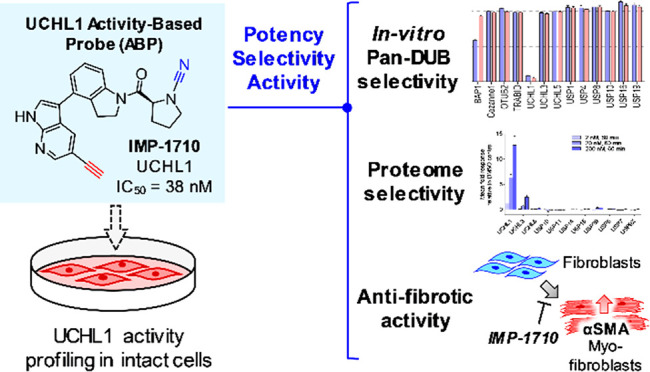
Ubiquitin carboxy-terminal hydrolase L1 (UCHL1) is a deubiquitylating enzyme that is proposed as a potential therapeutic target in neurodegeneration, cancer, and liver and lung fibrosis. Herein we report the discovery of the most potent and selective UCHL1 probe (IMP-1710) to date based on a covalent inhibitor scaffold and apply this probe to identify and quantify target proteins in intact human cells. IMP-1710 stereoselectively labels the catalytic cysteine of UCHL1 at low nanomolar concentration in cells. We further demonstrate that potent and selective UCHL1 inhibitors block pro-fibrotic responses in a cellular model of idiopathic pulmonary fibrosis, supporting the potential of UCHL1 as a potential therapeutic target in fibrotic diseases.
Targeting the ubiquitin (Ub) proteasome system is an emerging therapeutic strategy, and deubiquitylating enzymes (DUBs) have attracted increasing interest as drug targets.1 To study DUB biology and inhibition, Ub-derived activity-based probes (ABPs) have been developed that covalently bind to DUB active sites, allowing isolation or profiling in cell lysates.2 However, they are not selective between DUBs and limited to cell-free applications, highlighting a need for complementary small-molecule ABPs to profile DUB activity in intact cells.3,4
Ubiquitin carboxy-terminal hydrolase L1 (UCHL1) belongs to the UCH protease family, which feature a characteristic Cys-His-Asp catalytic triad.5 Although its biological functions are not yet fully understood, UCHL1 is abundantly expressed in the brain, where it is involved in apoptosis regulation, learning, and memory, while UCHL1 dysregulation is linked to diseases including neurodegeneration,5 cancers,6−8 and fibrosis.9
A screening and hit optimization campaign by Mission Therapeutics identified a series of novel cyanamide-containing UCHL1 inhibitors.10 Recognizing the potential of these inhibitors as covalent probes for UCHL1, we selected a potent example (1; example 27 in ref (10)) and designed analogue IMP-1710 (2) bearing an alkyne tag strategically placed in line with established structure–activity relationships (Figure 1a),10 enabling functionalization and analysis postinhibition with “capture” reagents via copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC).11,12
Figure 1.

Potency and selectivity of UCHL1 inhibitor (1), alkyne ABP (IMP-1710, 2), control compound (IMP-1711, 3), and LDN-57444. (a) Structures and UCHL1 IC50 values (Ub-Lys-TAMRA FP assay; *maximum assay concentration). (b) Selectivity profiling of 1 and 2 against DUBs in FP and FI assays (Ub-Lys-TAMRA and Ub-Rho110, respectively). See Supporting Information for 3 and LDN-57444 profiling. Data represent mean ± SEM (n = 2). (c) Immunoblot analysis of HA-Ub-VME UCHL1 labeling following HEK293T treatment with 1, 2, or 3 for 1 h. Dose-dependent competition for UCHL1 labeling occurs for active compounds, but not inactive enantiomer 3.
We first examined biochemical UCHL1 inhibition in a fluorescence polarization (FP) assay using Ub-Lys-TAMRA.13 Compound 2 showed improved UCHL1 inhibition (IC50 38 nM, 95% CI 32–45 nM) over parent compound 1 (IC50 90 nM, 95% CI 79–100 nM), following 30 min of preincubation. However, the (R)-enantiomer of 1 (IMP-1711, 3) was >1000-fold less active than 1, demonstrating a highly stereoselective interaction with UCHL1 and providing a useful negative control (Figures 1a and S1). We further characterized inhibition kinetics for 1 (kobs/I = 7400 M–1 s–1, 95% Cl 5200–9700) and 2 (kobs/I = 11000 M–1 s–1, 95% Cl 7700–13 000 M–1 s–1); slow recovery of activity following dilution demonstrated these inhibitors are slowly reversible (Figure S2), in line with previous reports on cyanamide warheads.14 Cross-screening against 20 DUBs demonstrated exquisite selectivity for 1 and 2 for UCHL1 (Figure 1b), whereas control compound 3 was inactive against all tested DUBs (Figure S1). Cellular UCHL1 activity was demonstrated in breast cancer cells (Cal51) stably expressing FLAG-UCHL1 using a Ub-vinyl methyl ester probe (HA-Ub-VME) by a homogeneous time-resolved fluorescence (HTRF) assay.15 Compounds 1 and 2 engaged UCHL1 with an in-cell IC50 of 820 and 110 nM, respectively (Figure S3). Selective in-cell concentration-dependent competition by 1 and 2 for UCHL1 was confirmed through immunoblot analysis against HA-Ub-VME labeling (Figures 1c and S4). Cellular potencies were in line with biochemical data (Figure S1), and compound 3 did not inhibit UCHL1 in cells.
We next compared our probes to the most potent known UCHL1 inhibitor, isatin O-acyloxime LDN-57444 (Figure 1a), reported as a reversible Ub-competitive UCHL1 inhibitor with IC50 880 nM against recombinant UCHL116 and widely used as a tool inhibitor in UCHL1 studies in disease models.7,9,17,18 However, engagement of UCHL1 in intact cells has not previously been demonstrated, and we were surprised to discover that LDN-57444 failed to engage UCHL1 in a range of assays, including biochemical activity and capacity to bind UCHL1 in intact cells (Figures S1, S3, S4).
Direct target engagement by 2 was examined in HEK293 cells treated for 60 min, followed by lysis and CuAAC ligation to azide-TAMRA-biotin capture reagent (AzTB, Figure S5) (Figure 2a).11 In-gel fluorescence revealed one major target at ∼25 kDa labeled in a concentration-dependent manner and saturated at 130 nM 2, with some off-target labeling of >500 nM (Figure 2b). Labeling was confirmed by biotin pulldown and immunoblotting, with UCHL1 significantly enriched at 30 nM 2 and maximal at 130 nM, with minimal or no detectable labeling of UCHL3, UCHL5, and DUBs from other families (Figures 2c and S6). Time course experiments demonstrated maximal labeling within 60 min (Figure S7), and 2 remained stable in media for >72 h without loss of activity (Figure S8). Incubation of recombinant UCHL1 (5 μM) with 1 or 2 (13 μM) for 60 min led to single modification of UCHL1 by LC-ESI-MS (Figure 3a), occurring specifically at the catalytic cysteine (Cys90) by tryptic digest and nanoLC-MS/MS (Figure S9, Supplementary Data S1). To confirm dependence on UCHL1 catalytic activity for cellular target engagement, we overexpressed FLAG-tagged wild-type (WT) UCHL1 or C90A or C90S mutants in HeLa cells, which do not express UCHL1, and demonstrated that only WT UCHL1 is labeled by 2 (Figures 3b and S10).19 Taken together, these data demonstrate 2 is a bona fide UCHL1 ABP that rapidly targets endogenous UCHL1 in cells in a strictly activity-dependent manner.
Figure 2.

UCHL1 activity profiling using ABP 2 in HEK293 cells. (a) Chemical proteomics workflow. Cells were incubated with 2 and labeled proteins ligated via CuAAC to AzTB for in-gel fluorescence and/or affinity enrichment for immunoblotting and proteomic profiling. (b) In-gel fluorescence shows dose-dependent labeling by 2. (c) Probe-labeled protein identification by enrichment and immunoblotting.
Figure 3.

UCHL1 labeling with 1 or 2 exclusively at catalytic Cys90 in vitro and in intact cells. (a) LC-ESI demonstrating single UCHL1 covalent modification with 1 or 2. (b) HeLa cells lacking endogenous UCHL1, transfected to express UCHL1 wild-type (WT) or catalytic cysteine mutants (C90A and C90S); pulldown following treatment with 2 and AzTB functionalization confirms specific Cys90 labeling.
To determine selectivity across the proteome, unbiased quantitative chemical proteomic profiling (Figure 2a) was employed in HEK293 cells treated with 2 (2, 20, or 200 nM) or vehicle (DMSO) control, for 10, 60, or 180 min. CuAAC ligation of proteins to azide-arginine-biotin (AzRB, Figure S5) capture reagent11 and enrichment on dimethylated NeutrAvidin-agarose beads were followed by on-resin digestion with LysC followed by trypsin20 and labeling with tandem mass tags (TMT) to enable relative quantification between conditions following nanoLC-MS/MS analysis. UCHL1 was significantly enriched by 2 in a concentration-dependent manner, with marginal enrichment of UCHL3 at higher concentrations and no significant enrichment of any other DUB (Figures 4a and S11); UCHL1 enrichment was similar at all time points, in line with in-gel fluorescence analysis (Figure S7). Only two proteins, UCHL1 and fibroblast growth factor receptor 2 (FGFR2), were significantly enriched at 20 nM 2, with UCHL1 by far the major target (Figure 4b, Supplementary Data S2). Full dose–response analysis by immunoblotting demonstrated that FGFR2 enrichment was barely detectable at any concentration up to 2 μM (Figure 4c), compared to UCHL1, which was strongly enriched and reached saturation at 130 nM (Figure 2c), suggesting that FGFR2 is minimally engaged by 2 at concentrations that strongly inhibit UCHL1. No significant abundance change was detected in whole proteome analysis (Figure S12, Supplementary Data S3), suggesting that 2 does not substantially alter global protein homeostasis. In-gel fluorescence and immunoblot analysis further confirmed 2 can profile activity of endogenous UCHL1 with excellent selectivity in cell types including endothelial cells (EA.hy926) and adenocarcinoma human alveolar basal epithelial cells (A549) (Figure S13).
Figure 4.

Chemical proteomic analysis of ABP 2 labeling in HEK293 cells. (a) Selected DUB quantitative profiling by 2, demonstrating dose-dependent enrichment of UCHL1. Data represent mean ± SEM (n = 3). (b) Volcano plots showing log2 difference (fold change) and significance (−log10p-value) between protein enrichment at 20 or 200 nM 2 compared to DMSO control (two sample t test, n = 3, permutation-based FDR = 0.01, S0 = 1). (c) FGFR2 shows negligible enrichment by 2, as determined by pulldown and immunoblotting. (d, e, f) Compound 1 selectively competes with ABP 2, while 3 and LDN-57444 do not. Competition for ABP 2 labeling was confirmed by (d) in-gel fluorescence; (e) pulldown and UCHL1 immunoblotting; (f) pulldown and whole proteome quantitative proteomic analysis. Data represent mean ± SEM (n = 3).
To identify selective targets of parent compound 1 and differentiate between selective labeling and nonspecific pulldown, competitive activity-based protein profiling (ABPP) was performed.21,22 HEK293 cells were treated with 1, 3, or LDN-57444 for 1 h, followed by 2 (20 nM) for 10 min. Immunoblot analysis demonstrated dose-dependent UCHL1 labeling reduction with parent compound 1 at nanomolar concentrations, whereas no competition was observed with control compound 3 or LDN-57444, confirming 3 as an effective negative control and LDN-57444 as inactive against UCHL1 in cells (Figure 4d and e). In-cell proteome-wide competitive ABPP was performed by quantitative chemical proteomics, showing that across the whole proteome UCHL1 responds strongly to 1 in a concentration-dependent manner, but does not respond with inactive control 3 or LDN-57444 (Figure 4f and S14, Supplementary Data S4). UCHL3 shows a small response to 1, while outside the DUB family FGFR2 responds (Figure S14), suggesting these proteins are possible minor off-targets of 1.
Pharmacological UCHL1 inhibition was next investigated in primary human lung cells derived from idiopathic pulmonary fibrosis (IPF) patients based on the potential of UCHL1 as a therapeutic target in fibrosis.9,23 Fibroblast–myofibroblast transition (FMT) was stimulated by transforming growth factor beta 1 (TGF-β1) using alpha-smooth muscle actin (αSMA) as a disease-relevant marker for transition (Figure 5a)24,25 and validated by high content imaging analysis (HCA) of three donor cell lines alongside response to 1 h pretreatment with 1 μM TGF-β1 receptor kinase inhibitor SB525334 (Figure S15).26 Donor cells were treated with compounds 1, 2, 3, LDN-57444, or the FDA-approved IPF drug nintedanib,27 and response to TGF-β1 and cell viability were measured after 3 days by staining and HCA quantification of αSMA and nuclei (DAPI), respectively.
Figure 5.
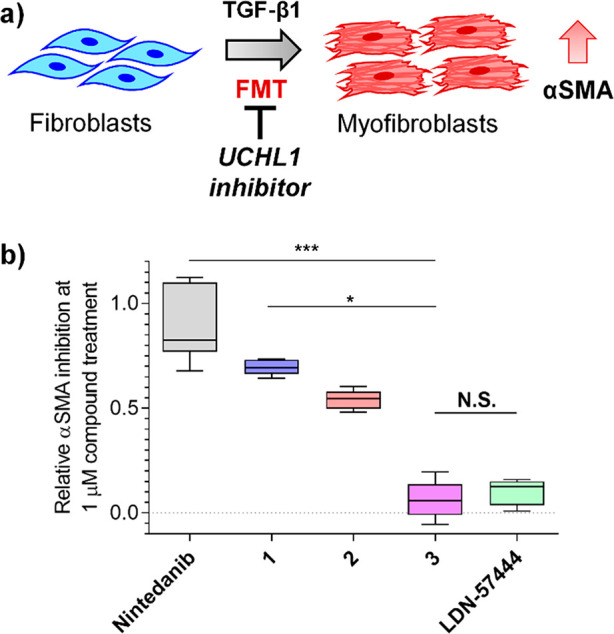
Phenotypic effects of selective UCHL1 inhibition in idiopathic pulmonary fibrosis (IPF). (a) Schematic of TGF-β1-mediated fibroblast-to-myofibroblast transition in primary human lung fibroblasts, increasing alpha-smooth muscle actin (αSMA) transdifferentiation marker. (b) Primary fibroblasts from IPF donors were preincubated with 1 μM 1, 2, 3, LDN-57444, or IPF approved drug (nintedanib) for 1 h followed by TGF-β1 treatment for 3 days. αSMA expression was analyzed by high content imaging, demonstrating 1, 2, and nintedanib, but not 3 or LDN-57444, inhibit transdifferentiation (N.S. nonsignificant, *P ≤ 0.05, ***P ≤ 0.01). Plots represent median values (center lines) and 25th/75th percentiles (box limits) with Tukey whiskers.
Compounds 1 and 2 (1 μM) demonstrated >50% FMT inhibition (IC50 100 and 740 nM, respectively), with comparable potency to nintedanib (Figure 5b), and inactive control compound 3 showed minimal αSMA inhibition compared to 1 and 2. While the dose–response observed for inhibition suggests a potentially complex role for UCHL1 in FMT, nuclear count remained stable at <5 μM, providing a good window between inhibition and cytotoxicity (Figure S16). Although LDN-57444 showed evidence for weak αSMA inhibition, this was concurrent with increased cytotoxicity over the same concentration range, suggesting that LDN-57444 toxicity may drive decreased αSMA (Figure S16).
In summary, we report the discovery and characterization of the most potent and selective small-molecule DUB ABP (2) to date, enabling robust detection of UCHL1 activity in living cells at low nanomolar concentrations. The only previously reported UCHL1 ABP is >150-fold less potent than 2 and has multiple significant off-targets,14 with no selectivity over UCHL3. UCHL1 labeling by 2 is strictly activity-dependent, occurring only at the catalytic cysteine and providing a new chemical tool to examine UCHL1 activity in various intact cell types. ABPP demonstrated that parent compound 1 is a potent UCHL1 inhibitor that targets the active site cysteine residue with impressive selectivity in cells and that UCHL1 inhibition is highly stereoselective, providing an ideal control compound (3) for future studies. Further, evidence from multiple assays (biochemical, cellular, proteomics) demonstrates that LDN-57444 showed negligible inhibition compared to 1 and 2.16,28 Previous studies with multiple batches of LDN-57444 suggest that its reported biochemical activity may be assay-dependent29 and further support reinterpretation of previous cellular studies using this compound. Finally, we show that selective UCHL1 inhibitors can suppress fibrotic phenotypes in IPF cellular models without substantial cytotoxicity. While the precise role of UCHL1 in fibrosis remains to be determined, UCHL1 was recently identified as a potential target in triple-negative breast cancer (TNBC), where it promotes TGF-β1 signaling8 and suppresses estrogen receptor expression.301 and 2 represent powerful and selective probes to explore UCHL1 activity with potential application to substrate identification, mode of action studies, and cellular target profiling, which can accelerate future development of UCHL1 inhibitors as potential therapeutics.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository31 with the data set identifier PXD015825.
Acknowledgments
The authors thank L. Haigh (Department of Chemistry Mass Spectrometry Facility, Imperial College London) for assistance in acquiring nanoLC-MS/MS, LC-ESI, and high-resolution mass spectrometry (HRMS) data, C. Das (Purdue University) for a UCHL1 DNA construct, M. Morgan (Imperial College London) for assistance with structural studies, D. Conole (Imperial College London) for insightful comments on the manuscript, Charles River (Leiden, Netherlands) for FMT data, and R. Williams, N. Jama, and C. Stead (Mission Therapeutics) for technical assistance. This study was supported by Cancer Research UK (Programme Foundation Award C29637/A20183 to E.W.T.), Royal Thai Government scholarship (PhD studentship to N.P.), Swiss National Science Foundation (Early Postdoc Mobility Fellowship P2ELP2_175069 to A.G.), and the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001097, FC010636), the UK Medical Research Council (FC001097, FC010636), and the Wellcome Trust (FC001097, FC010636).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04527.
Additional results and materials and methods for protein production, biochemical assay, cell-based experiment, chemical proteomic sample preparation and data analysis, and FMT assay; detailed chemical synthesis for all synthesized compounds; 1H, 13C NMR spectra and HRMS data (PDF)
Data files of protein identification and quantification from the proteomic experiments (ZIP)
Author Present Address
∥ Liver Disease Laboratory, CIC bioGUNE, Bizkaia Technology Park, Derio, 48160, Spain.
The authors declare the following competing financial interest(s): K.M., S.E., L.M.S., and J.A.H. are current or previous employees of Mission Therapeutics Ltd; E.W.T. is a Director and shareholder in Myricx Pharma Ltd.
Supplementary Material
References
- Harrigan J. A.; Jacq X.; Martin N. M.; Jackson S. P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discovery 2018, 17 (1), 57–78. 10.1038/nrd.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Flygare J. A.; Bogyo M.; Wertz I. E. Activity-based probes for the ubiquitin conjugation-deconjugation machinery: new chemistries, new tools, and new insights. FEBS J. 2017, 284 (10), 1555–1576. 10.1111/febs.14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. A.; McLellan L.; Stockley M.; Gibson K. R.; Whitlock G. A.; Knights C.; Harrigan J. A.; Jacq X.; Tate E. W. Quantitative Chemical Proteomic Profiling of Ubiquitin Specific Proteases in Intact Cancer Cells. ACS Chem. Biol. 2016, 11 (12), 3268–3272. 10.1021/acschembio.6b00766. [DOI] [PubMed] [Google Scholar]
- Conole D.; Mondal M.; Majmudar J. D.; Tate E. W. Recent Developments in Cell Permeable Deubiquitinating Enzyme Activity-Based Probes. Front. Chem. 2019, 7, 876–882. 10.3389/fchem.2019.00876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop P.; Rocca D.; Henley J. M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473 (16), 2453–2462. 10.1042/BCJ20160082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst-Kennedy J.; Chin L. S.; Li L. Ubiquitin C-terminal hydrolase l1 in tumorigenesis. Biochem. Res. Int. 2012, 2012, 123706. 10.1155/2012/123706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Zeng L.; Yeom C. J.; Zhu Y.; Morinibu A.; Shinomiya K.; Kobayashi M.; Hirota K.; Itasaka S.; Yoshimura M.; Tanimoto K.; Torii M.; Sowa T.; Menju T.; Sonobe M.; Kakeya H.; Toi M.; Date H.; Hammond E. M.; Hiraoka M.; Harada H. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat. Commun. 2015, 6, 6153–6165. 10.1038/ncomms7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Gonzalez-Prieto R.; Zhang M.; Geurink P. P.; Kooij R.; Iyengar P. V.; van Dinther M.; Bos E.; Zhang X.; Le Devedec S. E.; van de Water B.; Koning R. I.; Zhu H. J.; Mesker W. E.; Vertegaal A. C. O.; Ovaa H.; Zhang L.; Martens J. W. M.; Ten Dijke P. Deubiquitinase Activity Profiling Identifies UCHL1 as a Candidate Oncoprotein That Promotes TGFbeta-Induced Breast Cancer Metastasis. Clin. Cancer Res. 2020, 26 (6), 1460–1473. 10.1158/1078-0432.CCR-19-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. L.; Murphy L. B.; Leslie J.; Kendrick S.; French J.; Fox C. R.; Sheerin N. S.; Fisher A.; Robinson J. H.; Tiniakos D. G.; Gray D. A.; Oakley F.; Mann D. A. Ubiquitin C-terminal hydrolase 1: A novel functional marker for liver myofibroblasts and a therapeutic target in chronic liver disease. J. Hepatol. 2015, 63 (6), 1421–1428. 10.1016/j.jhep.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp M.; Stockley M.; Jones A.. Cyanopyrrolidines as Dub Inhibitors for the Treatment of Cancer. WO 2017009650 (A1), 2017/01/19/, 2017.
- Broncel M.; Serwa R. A.; Ciepla P.; Krause E.; Dallman M. J.; Magee A. I.; Tate E. W. Multifunctional reagents for quantitative proteome-wide analysis of protein modification in human cells and dynamic profiling of protein lipidation during vertebrate development. Angew. Chem., Int. Ed. 2015, 54 (20), 5948–5951. 10.1002/anie.201500342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storck E. M.; Morales-Sanfrutos J.; Serwa R. A.; Panyain N.; Lanyon-Hogg T.; Tolmachova T.; Ventimiglia L. N.; Martin-Serrano J.; Seabra M. C.; Wojciak-Stothard B.; Tate E. W. Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nat. Chem. 2019, 11 (6), 552–561. 10.1038/s41557-019-0237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurink P. P.; El Oualid F.; Jonker A.; Hameed D. S.; Ovaa H. A general chemical ligation approach towards isopeptide-linked ubiquitin and ubiquitin-like assay reagents. ChemBioChem 2012, 13 (2), 293–297. 10.1002/cbic.201100706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabill A. D.; Chen H.; Hussain S.; Feng C.; Abdullah A.; Das C.; Aryal U. K.; Post C. B.; Wendt M. K.; Galardy P. J.; Flaherty D. P. Ubiquitin C-Terminal Hydrolase L1: Biochemical and Cellular Characterization of a Covalent Cyanopyrrolidine-Based Inhibitor. ChemBioChem 2020, 21 (5), 712–722. 10.1002/cbic.201900434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki N.; Watanabe S.; Kudoh T.; Nihira S.; Miyamato C. Application of homogeneous time-resolved fluorescence (HTRFTM) to monitor poly-ubiquitination of wild-type p53. Comb Chem. High Throughput Screen 1999, 2 (5), 279–287. [PubMed] [Google Scholar]
- Liu Y.; Lashuel H. A.; Choi S.; Xing X.; Case A.; Ni J.; Yeh L. A.; Cuny G. D.; Stein R. L.; Lansbury P. T. Jr. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 2003, 10 (9), 837–846. 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Yan C.; Huo H.; Yang C.; Zhang T.; Chu Y.; Liu Y. Ubiquitin C-Terminal Hydrolase L1 regulates autophagy by inhibiting autophagosome formation through its deubiquitinating enzyme activity. Biochem. Biophys. Res. Commun. 2018, 497 (2), 726–733. 10.1016/j.bbrc.2018.02.140. [DOI] [PubMed] [Google Scholar]
- Kobayashi E.; Hwang D.; Bheda-Malge A.; Whitehurst C. B.; Kabanov A. V.; Kondo S.; Aga M.; Yoshizaki T.; Pagano J. S.; Sokolsky M.; Shakelford J. Inhibition of UCH-L1 Deubiquitinating Activity with Two Forms of LDN-57444 Has Anti-Invasive Effects in Metastatic Carcinoma Cells. Int. J. Mol. Sci. 2019, 20 (15), 3733–3750. 10.3390/ijms20153733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen C. N.; Price J. S.; Wilkinson K. D. Substrate binding and catalysis by ubiquitin C-terminal hydrolases: identification of two active site residues. Biochemistry 1996, 35 (21), 6735–6744. 10.1021/bi960099f. [DOI] [PubMed] [Google Scholar]
- Goya Grocin A.; Serwa R. A.; Morales Sanfrutos J.; Ritzefeld M.; Tate E. W. Whole Proteome Profiling of N-Myristoyltransferase Activity and Inhibition Using Sortase A. Mol. Cell. Proteomics 2019, 18 (1), 115–126. 10.1074/mcp.RA118.001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalesh K. A.; Clulow J. A.; Tate E. W. Target profiling of zerumbone using a novel cell-permeable clickable probe and quantitative chemical proteomics. Chem. Commun. (Cambridge, U. K.) 2015, 51 (25), 5497–5500. 10.1039/C4CC09527H. [DOI] [PubMed] [Google Scholar]
- Clulow J. A.; Storck E. M.; Lanyon-Hogg T.; Kalesh K. A.; Jones L. H.; Tate E. W. Competition-based, quantitative chemical proteomics in breast cancer cells identifies new target profiles for sulforaphane. Chem. Commun. (Cambridge, U. K.) 2017, 53 (37), 5182–5185. 10.1039/C6CC08797C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. C.; Tseng C. P.; Liao M. H.; Peng C. Y.; Yu J. S.; Chuang P. H.; Huang J. T.; Chen J. J. W. Activation of hepatic stellate cells by the ubiquitin C-terminal hydrolase 1 protein secreted from hepatitis C virus-infected hepatocytes. Sci. Rep. 2017, 7 (1), 4448–4459. 10.1038/s41598-017-04259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepparanta O.; Sens C.; Salmenkivi K.; Kinnula V. L.; Keski-Oja J.; Myllarniemi M.; Koli K. Regulation of TGF-beta storage and activation in the human idiopathic pulmonary fibrosis lung. Cell Tissue Res. 2012, 348 (3), 491–503. 10.1007/s00441-012-1385-9. [DOI] [PubMed] [Google Scholar]
- Saito A.; Horie M.; Nagase T. TGF-beta Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19 (8), 2460–2477. 10.3390/ijms19082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grygielko E. T.; Martin W. M.; Tweed C.; Thornton P.; Harling J.; Brooks D. P.; Laping N. J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313 (3), 943–951. 10.1124/jpet.104.082099. [DOI] [PubMed] [Google Scholar]
- Richeldi L.; du Bois R. M.; Raghu G.; Azuma A.; Brown K. K.; Costabel U.; Cottin V.; Flaherty K. R.; Hansell D. M.; Inoue Y.; Kim D. S.; Kolb M.; Nicholson A. G.; Noble P. W.; Selman M.; Taniguchi H.; Brun M.; Le Maulf F.; Girard M.; Stowasser S.; Schlenker-Herceg R.; Disse B.; Collard H. R.; Investigators I. T. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370 (22), 2071–2082. 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- D’Arcy P.; Wang X.; Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Kemp M. Recent Advances in the Discovery of Deubiquitinating Enzyme Inhibitors. Prog. Med. Chem. 2016, 55, 149–192. 10.1016/bs.pmch.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. S.; Wang K. S.; Guo W.; Li L. Y.; Yu P.; Sun X. Y.; Wang H. Y.; Guan Y. D.; Tao Y. G.; Ding B. N.; Yin M. Z.; Ren X. C.; Zhang Y.; Chen C. S.; Ye Y. C.; Yang J. M.; Cheng Y. UCH-L1-mediated Down-regulation of Estrogen Receptor alpha Contributes to Insensitivity to Endocrine Therapy for Breast Cancer. Theranostics 2020, 10 (4), 1833–1848. 10.7150/thno.39814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Rios D.; Dianes J. A.; Sun Z.; Farrah T.; Bandeira N.; Binz P. A.; Xenarios I.; Eisenacher M.; Mayer G.; Gatto L.; Campos A.; Chalkley R. J.; Kraus H. J.; Albar J. P.; Martinez-Bartolome S.; Apweiler R.; Omenn G. S.; Martens L.; Jones A. R.; Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32 (3), 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.