

Abstract
Covalent organic frameworks (COFs) display a unique combination of chemical tunability, structural diversity, high porosity, nanoscale regularity, and thermal stability. Recent efforts are directed at using such frameworks as tunable scaffolds for chemical reactions. In particular, COFs have emerged as viable platforms for mimicking natural photosynthesis. However, there is an indisputable need for efficient, stable, and economical alternatives for the traditional platinum-based cocatalysts for light-driven hydrogen evolution. Here, we present azide-functionalized chloro(pyridine)cobaloxime hydrogen-evolution cocatalysts immobilized on a hydrazone-based COF-42 backbone that show improved and prolonged photocatalytic activity with respect to equivalent physisorbed systems. Advanced solid-state NMR and quantum-chemical methods allow us to elucidate details of the improved photoreactivity and the structural composition of the involved active site. We found that a genuine interaction between the COF backbone and the cobaloxime facilitates recoordination of the cocatalyst during the photoreaction, thereby improving the reactivity and hindering degradation of the catalyst. The excellent stability and prolonged reactivity make the herein reported cobaloxime-tethered COF materials promising hydrogen evolution catalysts for future solar fuel technologies.
Introduction
Identifying competitive alternatives to fossil-fuel-based energy constitutes one of the main research goals of this decade. Nature-inspired processes, like artificial photosynthesis, guide the way to a green and sustainable solution.1−3 Covalent organic frameworks (COFs) have been emerging as new materials in this context.4,5 COFs consist of light elements only, and their bottom-up synthesis enables high versatility and tunability on a molecular level, while benefiting from high stability and crystallinity due to covalent bonding in-plane and π–π-stacking out-of-plane.6−9 Most reports of COFs as photosensitizers for light-driven hydrogen evolution use platinum as a cocatalyst;10−12 hydrogen evolution rates up to 16.3 mmol h–1 g–1 have been reported in this context.13 Recent studies showed that the precious metal platinum can be replaced by earth-abundant molecular cocatalysts, namely, chloro(pyridine)cobaloxime and related complexes.14−16 These cocatalysts are well-known and well-defined, while offering high tunability, which facilitate their incorporation into photoactive organic and inorganic systems.17−19 Cobaloximes feature low overpotential for the hydrogen evolution reaction and have been used in heterogeneous systems with metal–organic frameworks20,21 and carbon nitrides,22,23 as well as physisorbed to COFs.14 A major drawback of molecular proton reduction catalysts physisorbed to photosensitizers is their photodeactivation over time24−26 and rate limitations due to diffusion-controlled mechanisms. While previous attempts14 used molecular cobaloxime catalysts in solution, in this work we report photocatalytic hydrogen evolution with molecular cobaloxime catalysts covalently tethered to the COF backbone, yielding unprecedented insights into the nature of the active site and the COF–cocatalyst interface. By comparison with equivalent unbound, i.e., physisorbed, systems, we show how the modification of the hydrazone-based COF-42 and attachment of functionalized chloro(pyridine)cobaloxime lead to more efficient hydrogen evolution in a water/acetonitrile mixture under visible-light illumination in the presence of a sacrificial electron donor. The structural composition of the photoreaction is verified by computational and experimental methods including advanced high-resolution solid-state NMR techniques. These results combine the advantages of fully heterogeneous systems with the tunability of molecular cocatalysts and lead the way toward true single-site COF-based photocatalytic systems with a high level of interfacial control.
Results and Discussion
In previous studies, COF-4227 has been shown to be active in photocatalytic hydrogen evolution reactions with conventional hydrogen evolution cocatalysts such as platinum nanoparticles or molecular chloro(pyridine)cobaloxime.14 At the same time, this COF is a well-known and versatile platform that is chemically robust due to its hydrazone-linked structure.28,29 In this study, we used COF-42 as a platform for covalent postsynthetic modification with cobaloxime complexes. The synthesis of COF-42 by solvothermal acid-catalyzed condensation of 1,3,5-triformylbenzene (TFB) and 2,5-diethoxyterephthalohydrazide (DETH) followed published protocols.27 In order to provide functional sites for the covalent attachment of the cocatalyst, 10 mol % of DETH was replaced by the propargyl-containing 2,5-bis(prop-2-yn-1-yloxy)terephthalohydrazide (DPTH) to obtain the propargyl-modified pCOF10. The COFs were characterized by FT-IR spectroscopy, sorption analysis, powder X-ray diffraction (PXRD), magic-angle-spinning solid-state NMR (ssNMR), and quantum-chemical calculations.
The successful transformation of the starting materials to pCOF10 was proven by the lack of a residual aldehyde stretching vibration in its FT-IR spectrum. Characteristic C=O vibrations and signals originating from the hydrazone bonds overlap at 1680 cm–1 [see Figure S13 of the Supporting Information (SI)]. New vibrations emerged at 2250 cm–1 that could be assigned to the propargyl groups, confirming the successful incorporation of DPTH building blocks into the COF backbone. This was further supported by a 1D 13C{1H} ssNMR spectrum, where 13C signals at 79 and 58 ppm can be assigned to the propargyl functional group (Figure 1C). These shifts match the corresponding chemical shift of the liquid-state NMR of the DPTH linker (see the Supporting Information for experimental details) and are also confirmed by quantum-chemical calculations (see Table S3, SI).
Figure 1.

(A) Synthesis of pCOF10 by solvothermal condensation of TFB and a 9:1 mixture of DETH and DPTH. (B) Eclipsed stacking model for pCOF10. C, N, and O atoms are represented in gray, blue, and red, respectively. H atoms are omitted, and the second and third layers are represented in orange and yellow for clarity. (C) Solid-state 1D 13C{1H} CP-MAS NMR spectrum of pCOF10 acquired at 11.7 T, 12 kHz MAS, 298 K, and using cross-polarization times of 5 ms. Spinning side bands are marked with asterisks. Calculated shifts are marked with yellow bars. The narrow signals labeled with crosses at 164, 37, and 32 ppm correspond to residual dimethylformamide. (D) Argon adsorption isotherm of pCOF10. Inlet: Pore size distribution from NLDFT calculations with cylindrical pores in equilibrium mode. The resulting main pore size is 2.3 nm. (E) PXRD pattern of pCOF10 (open, green circles), Pawley refined profile (blue line), and calculated XRD pattern for the idealized AA stacking (black line).
PXRD analysis confirmed the crystalline structure of pCOF10. The PXRD pattern shows a strong reflection at 3.3° 2θ, followed by smaller ones at 5.9°, 7.0°, and 9.1° and a very broad one at 26° 2θ. The experimental powder pattern was compared to a simulated one (see Figure 1E), and the diffraction peaks were assigned as the 100, 101, 200, 201, and 001 reflections, respectively. The peaks are broadened due to small domain sizes in the COF particles, especially in the z direction, where the interlayer interactions are defined by π–π-stacking only. Different possible orientations for the propargyl functionality as well as slightly shifted AA′ stacking modes lead to very similar powder patterns; due to broadening of the reflections in the experimental data, the different orientations cannot be distinguished. One of these possible structural models is shown in Figure 1B, featuring an AA stacking mode with an interlayer distance of 3.5 Å, which is typical for structurally similar COFs.10,30,31 Note that in the underlying structural model, one out of six DETH linkers per pore was replaced by DPTH, which results in a functionalization degree of 16.6% instead of the statistically distributed 10% in the experimentally prepared pCOF10.
Pawley refinement of the structure in the idealized AA stacking mode suggests P2/m symmetry. For the modeled structure, the resulting cell parameters are a = 51.09 Å, b = 3.50 Å, c = 29.48 Å, α = γ = 90.00°, and β = 89.94°. Sorption analysis revealed a mesoporous structure with pore size of 2.3 nm and a Brunauer–Emmett–Teller (BET) surface area of 1839 m2 g–1, which matches the theoretically expected values of the structural model well (see Figure 1D).
For the covalent attachment of the cobaloxime catalyst to pCOF10, a postsynthetic click-chemistry approach was chosen. The copper(I)-catalyzed Huisgen-type cycloaddition of azines and alkynes is known to be broadly applicable with high yields and a high tolerance for functional groups.32−36 Therefore, the pyridine, which acts as an axial cobaloxime ligand, was functionalized with an azide group to yield the para-functionalized pyridine 1a, which forms the azide-functionalized complex [Co-1a], and likewise, the meta-functionalized analogues 1b and [Co-1b] were synthesized, as depicted in Figure 2. Additionally, the equatorially functionalized chelating ligand 2 was synthesized as described in the Supporting Information. It forms the azide-functionalized catalyst [Co-2] by metal complexation as before. Two strategies were tested for the attachment of the cobaloxime complex to pCOF10: (i) metal complexation of azide-functionalized ligands with subsequent COF modification by click-reaction with the azide-functionalized complexes, termed route I, and (ii) COF modification by click-reaction with azide-functionalized ligands with subsequent complexation, termed route II (see the Supporting Information for experimental details). The resulting COF–cobaloxime hybrid samples are labeled as follows with the respective numbering according to Figure 2: [1a]–COF for clicked ligands and [Co-1a]–COF for COF–cobaloxime hybrid samples.
Figure 2.

(A) Structure of the azide-functionalized ligands 1a, 1b, and 2 and (B) the azide-functionalized complexes Co-1a, Co-1b, and Co-2. (C) Exemplary postsynthetic COF modification toward [Co-1b]–COF. Synthesis conditions can be found in the Supporting Information.
To verify the success of the tethering of the cobaloxime and the unperturbed structural integrity of the covalently modified hybrid COF–cobaloxime systems, we performed the same systematic experimental analysis as for the intact pCOF10. PXRD shows that the crystallinity of the COF is preserved and the stacking mode does not change with respect to pCOF10 (Figure S6, SI). Sorption analysis shows the expected reduction of the surface area according to Table S1 (SI). Pore size distributions for the clicked samples were calculated from Ar sorption isotherms, as shown in Figure S5 (SI). In all samples, the 2.3 nm pore size, as found in pCOF10, is preserved with lower pore volume fraction, while additional smaller pores up to 1.9 nm occur, as seen from optimized pore models (see Figure S19, SI). FT-IR spectra display all expected vibrations of the COF, including propargyl vibrations at ca. 3300 and 2300 cm–1. These vibrations are still visible in ligand-tethered samples, which hints at partial transformation. New triazole peaks are hidden in the region around 3100 cm–1 due to low intensity. The success of the click-reaction was further confirmed by the reduced intensity of the propargyl signals relative to the other signals in the 1D 13C{1H} CP ssNMR spectrum upon addition of the azide compounds. We did not observe any additional signals arising from the clicked compound, which is probably due to signal superposition, especially in the aromatic region, and due to lower signal intensity caused by a low functionalization degree. UV–vis diffuse reflectance spectra show two additional broad absorption bands at 500 and 600 nm for the cobaloxime-containing samples (Figure S15, SI). These bands are due to the electronic transitions of the azide-functionalized cobaloximes. Depending on the reaction conditions (see the Supporting Information for more details), the cobaloxime loading can be adjusted within limits. For all samples, the total cobaloxime amount was determined by ICP analysis, and for [Co-1a]–COF, it was additionally confirmed by fast-MAS 1H-detected NMR spectra. The values range from 0.47 to 2.4 wt % for route II, while route I resulted in higher cobaloxime amounts between 1.2 and 8.5 wt %. The highest cobaloxime content was found for [Co-1a]–COF, as can be seen in Table S2 (SI). The resulting functionalization degrees ranging from 2.0 to 15% are also listed there. Scanning electron microscopy shows a flower-like morphology for all samples. Elemental mapping showed a uniform distribution of carbon, nitrogen, oxygen, and cobalt in the samples, as can be seen in the Supporting Information.
ssNMR Analysis of the COF–Cobaloxime Hybrid Systems
While powder diffraction analysis provides long-range spatial information, such as approximate interlayer separations, ssNMR provides us with short-range interatomic proximities and hints about the position of the cobaloxime inside the pore. To this end, we performed an in-depth structural analysis of the clicked samples 1a–COF and [Co-1a]–COF using 1H-detected, fast-MAS ssNMR at νrot = 55.55 kHz at 700 MHz 1H Larmor frequency (16.4 T). The samples based on [Co-1a] were chosen due to their higher molecular symmetry compared to that of [Co-1b]. Both 1a–COF and [Co-1a]–COF were studied by 1D and 2D 1H and 13C solid-state NMR techniques. All 2D measurements were 1H-detected, which significantly improved the sensitivity of the natural abundance 13C measurements. In addition to the sensitivity gain, we could exploit the 1H chemical shifts as well as the 1H–1H correlations as sources of structural information.
Figure 3B compares the 1D 1H spectra of 1a–COF (yellow) and [Co-1a]–COF (blue). The high structural order of these two-dimensional crystalline polymers is reflected in the good resolution of the 1H signals; 1H line widths vary between 800 and 1300 Hz for 1a–COF and between 1000 and 2000 Hz for [Co-1a]–COF. In the 1H spectra, we could directly observe four (1a–COF) and five ([Co-1a]–COF) distinct proton resonances, which correspond to the amide proton (10.9 ppm), aromatic protons overlapping with the olefin proton (7.2 ppm), methylene protons (3.9 ppm), and methyl protons (1.7 ppm). For [Co-1a]–COF, we also observe a well-separated, downfield-shifted, low-intensity peak that belongs to the strongly hydrogen-bonded oxime proton (19.1 ppm). Note that all 1H signals are broader in the spectrum of [Co-1a]–COF relative to that of 1a–COF, which indicates that the cobaloxime functionalization process disrupted the overall COF crystallinity to some extent. Cobaloxime contains Co(III), which is, unlike Co(II), diamagnetic; therefore, the observed line broadening of [Co-1a]–COF cannot be a consequence of paramagnetic relaxation enhancement. Also, residual CoCl2 salt is washed out during the sample preparation process. It is more likely that the postsynthetic modification reduced the crystalline domain size and increased the sample’s heterogeneity, leading to a wider range of chemical shifts for each site.
Figure 3.
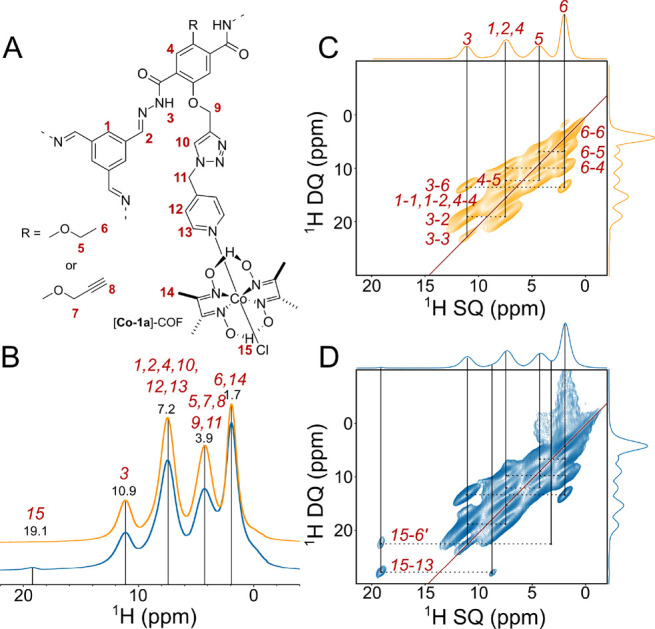
Solid-state NMR comparison of the 1H spectra of [1a]–COF (yellow) and [Co-1a]–COF (blue) measured at 700 MHz 1H Larmor frequency at νrot = 55.55 kHz. (A) Schematic structure of the subsection of [Co-1a]–COF with proton labeling. (B) 1D 1H spectra of [1a]–COF (yellow) and [Co-1a]–COF (blue). Distinct 1H resonances are given in ppm and labeled with the corresponding atom labels as displayed in part A. (C and D) 1H–1H DQ–SQ correlation spectra of [1a]–COF (yellow) and [Co-1a]–COF (blue). Horizontal dashed lines indicate the 1H–1H connectivities, and vertical solid lines reflect the individual 1H SQ resonances. Assignments are given next to the dashed lines. In part D, the assignments for only the two new connectivities are shown. The skyline projection of both dimensions is also shown.
The good 1H resolution of the fast-MAS 1H spectrum prompted us to measure 2D homonuclear correlation experiments to gain a deeper insight into the intramolecular interaction between the COF backbone and the cobaloxime cocatalyst. We probed the relative 1H–1H distances using 2D double quantum–single quantum (DQ–SQ) correlation experiments employing the R-symmetry-based R144–2 homonuclear recoupling sequence.37 The R144 is a γ-encoded symmetry sequence that suppresses all heteronuclear dipole–dipole couplings and chemical shift terms in the first-order Hamiltonian. We used a R = π0 element as the basic R-symmetry block with a nutation frequency of 97.22 kHz (3.5νrot). The homonuclear 2D 1H–1H DQ–SQ recoupling experiment relies on the generation of double-quantum coherences via homonuclear dipole–dipole coupling to obtain through-space information on nearby protons. Due to the double-quantum filter, the spectrum exhibits cross-peaks only between protons that share direct dipolar interactions with each other, and thus, no relayed magnetization transfer occurs. For protonated organic solid materials, such as the COFs of this study, the observation of a DQ peak is indicative of a proton–proton proximity that is ≤3.5 Å.38,39 The relative signal intensities could well-approximate interatomic distances.39
Parts C and D of Figure 3 show the 1H–1H DQ–SQ correlation spectra of 1a–COF (yellow) and [Co-1a]–COF (blue). The spectra reveal double-quantum correlations between both distinct and identical environments, appearing at the off-diagonal and diagonal positions, respectively. Diagonal peaks are expected for the signals of the methyl and methylene groups, as well as between the resonances of the chemically equivalent aromatic sites. However, the weak diagonal peak for the NH protons corresponds to an NH–NH autopeak, which is indicative of the dipolar interaction between COF layers; the separation of NH protons within one layer is <7 Å, while the layer-to-layer distance is 3.5 Å according to powder crystal analysis. The two spectra look almost identical, the only considerable difference being the 1H cross-peaks of the oxime 1H at 19.1 ppm with resonances at 8.7 and 3.4 ppm. In order to assign these two peaks, and thus uncover the position of cobaloxime inside the pore, we performed a detailed quantum-chemical study (vide infra). On the basis of these studies, we conclude that the resonances at 8.7 and 3.4 ppm belong to the pyridine aromatic proton (H13), as well as to a downfield-shifted methyl proton of a neighboring ethoxy group with which the cobaloxime is in close contact.
Next, we assessed the relative flexibility of the two compounds using 1D 13C NMR spectroscopy. Three different 1D 13C MAS spectra of [1a]–COF and [Co-1a]–COF are given in parts A and B of Figure 4, respectively. These spectra include 13C{1H} cross-polarization (CP) MAS and T1-weighted, direct-polarization (DP) 13C spectra recorded with short (1 s) and long (25 s) recycle delay times. These latter spectra were used to elucidate the relative mobility of certain sites in the COF samples. In the 13C spectra recorded with d1 = 1 s, those signals that have considerably shorter 13C longitudinal relaxation time constants (T1 < 1 s) are more intense, since the signal recovery is proportional to 1 – exp(−d1/T1). Such a short T1 is indicative of motions occurring on the inverse of the Larmor frequency (a few nanoseconds). The longitudinal relaxation constant depends not only on the amplitude of nanosecond time-scale motion but also on the number of directly attached protons: the more protons that are directly bound to a carbon, the faster it relaxes via heteronuclear dipolar relaxation. This is reflected in the relative change of signal intensities among the aromatic carbons. Besides, the methyl resonance relaxes rapidly due to the free rotation around the C–C axis in the ethyl group. The methyl resonance line shape in the DP spectrum of [Co-1a]–COF is markedly distorted, presenting a shoulder at lower resonances. This signal could be assigned to the methyl carbons of the cobaloxime ligand. Otherwise, the signals of the covalently tethered ligand do not show any obvious sign of increased fast time-scale flexibility, neither for [1a]–COF nor for [Co-1a]–COF. In the spectrum of [Co-1a]–COF recorded with d1 = 1 s, the intensified resonances at 128 ppm indicate rather flexible aromatic sites, but due to strong overlaps in this region, we could not identify if this signal belongs to the ligand or to some residual impurities that tend to show up more strongly in T1-weighted experiments. Selective 13C or 15N labeling at specified positions of the ligand would help us to quantify the amplitude and time-scale of the ligand motion.
Figure 4.
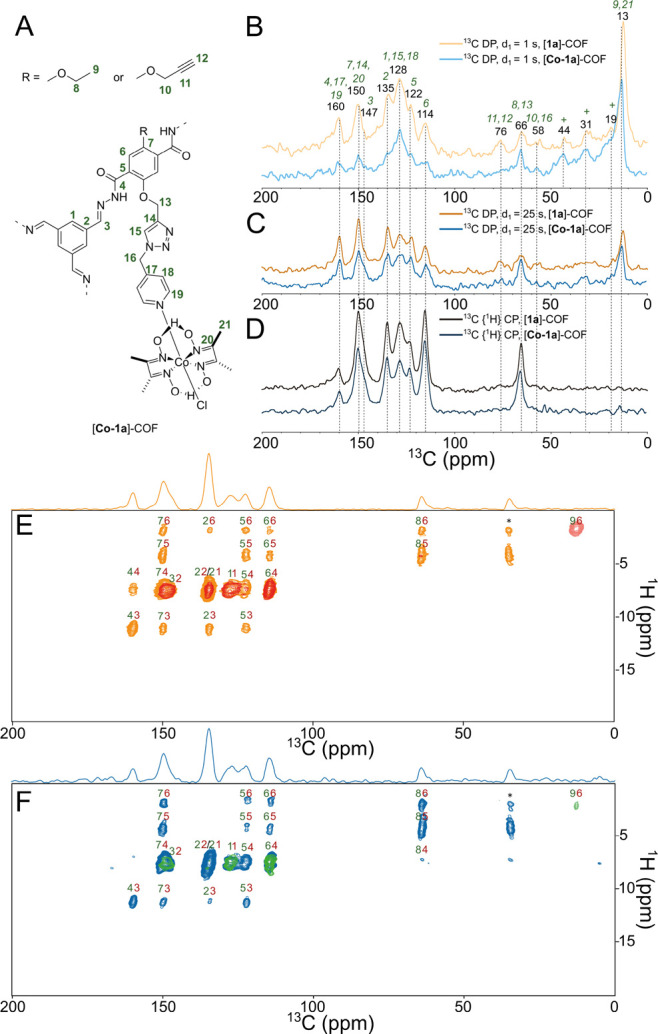
(A) Schematic structure of the subsection of [Co-1a]–COF with carbon labeling. (B–D) Comparison of the natural abundance 13C one-dimensional solid-state NMR spectra of [1a]–COF (blue shades) and [Co-1a]–COF (orange shades) measured at 700 MHz 1H Larmor frequency at νrot = 55.55 kHz. Direct polarization spectra recorded with d1 = 1 s (B) or with long d1 = 25 s (C) are compared with CP MAS spectra (D). For the CP MAS experiment, the carrier was centered at 130 ppm and the CP was optimized to transfer magnetization to the aromatic region. The CP contact time was 500 μs. Signals with short longitudinal relaxation times are enhanced in the 13C direct MAS spectrum measured with 1 s recycle delay. The assignment of the 13C resonances was obtained from 2D 1H–13C and 1H–1H correlation experiments and from the quantum-chemical calculations. The signals marked with crosses correspond to impurities, e.g., to residual solvent signals. 1H-detected 2D 1H–13C correlation spectra of [1a]–COF (E) and [Co-1a]–COF (F) recorded with 500 μs (red and green) or with 2250 μs (orange and blue) CP contact times. The CP-based spectra are overlaid with INEPT-based HSQC spectra that show only one methyl cross-peak displayed with blue (E) and magenta (F) colors. For each cross-peak, the 1H and 13C assignments are displayed with red and green colors, respectively. Signals marked with an asterisk are measurement artifacts and they do not appear in 1D 13C-detected 13C{1H} CP spectra.
The apparent lack of high-amplitude fast time-scale dynamics of the two COF frameworks was further validated by comparing 1H-detected 2D CP-based 1H–13C correlation spectra with INEPT-based 2D HSQC spectra (Figure 4D, E). High-amplitude nanosecond time-scale motion results in inherent decoupling and thus leads to increased coherent lifetimes in INEPT-based experiments and to decreased transfer efficiencies in CP-based experiments. In the HSQC spectrum of both [1a]–COF and [Co-1a]–COF, we observe only a single methyl peak, indicating that the COF backbone is generally rigid on the nanosecond time-scale.
Computational Studies
In order to provide a structural model for the position and the orientation of the covalently tethered cobaloxime cocatalyst inside the pore, we conducted a detailed in silico structural investigation of [1a]–COF and [Co-1a]–COF. Atom positions and lattices of the periodic COF structure of [1a]–COF were optimized at the RI-PBE-D3/def2-TZVP40−43 level of theory using an acceleration scheme based on the resolution of the identity (RI) technique and the continuous fast multipole method (CFMM)44−46 implemented47,48 in Turbomole ver. V7.1.49 The obtained structure for the [1a]–COF was then used to prepare parameters for molecular dynamics simulations using antechamber.50 Force field minimizations and subsequent dynamics were performed with the NAMD program package51,52 using GAFF parameters53 afterward. NMR chemical shifts were then calculated at the B97-2/pcSseg-154,55 level of theory using the FermiONs++56,57 program package, using cut models of obtained structures to compare with experimental chemical shifts and to assign the resonances.
Using this data, we prepared 200 in silico1H–1H DQ–SQ and 1H–13C 2D correlation spectra (see the Supporting Information for details) and used them to identify features that are also present in the experimentally obtained ssNMR spectra. Such features include the number of cross-peaks, especially cross-peaks of the oxime proton, their relative intensity ratios, and their peak positions. The most distinctive factor in the simulated 1H–1H DQ–SQ spectra is the presence of oxime (H15) cross-peaks with resonances at around 8.7 and 3.4 ppm, which was used to categorize the simulated spectra. These distinct chemical shifts suggest that the oxime proton is interacting with an aromatic proton (at 8.7 ppm) and with either an upfield-shifted methylene proton or with a downfield-shifted methyl proton (at 3.4 ppm). There are four different aromatic protons in [Co-1a]–COF, H1, H4, H12, and H13, out of which only H4 and H13 can get closer than 3.5 Å to H15.
To decide which resonance leads to the 3.4 ppm cross-peak with H15, we analyzed the shielding effects of the glyoxime group on the nearby ethoxy methyl and methylene protons. The approach of the glyoxime oxygen toward the ethoxy group induces a deshielding effect; consequently, both the methyl and the methylene protons resonate at higher frequencies (see Figure S16 and SI text for more details), this rules out the possibility that the cross-peak at 3.4 ppm would stem from an upfield-shifted methylene proton and leaves only a downfield-shifted methyl proton as a possible interaction partner. Besides, we excluded the possibility that the oxime proton shows a trivial intraligand cross-peak with the glyoxime methyl protons, since (i) the distance between the H15 and H16 protons is >3.5 Å and (ii) the calculated chemical shift is below 2.9 ppm.
Out of the 200 simulated 1H–1H DQ–SQ spectra, 27 (13) contained two (three) oxime cross-peaks, among which 22 spectra have these peaks in the expected ppm range. By considering the relative peak intensity ratios between the oxime cross-peaks, only 15 spectra have a more intense aromatic–oxime than a methyl–oxime cross-peak. Two such spectra, together with the simulated 1H–13C spectra and corresponding structures, are displayed in Figure 5A–F. As counter-examples, Figure 5G,H and J,K displays the spectra of such structures (Figure 5I and L) where three equally intense peaks (Figure 5H) or no oxime proton cross-peak (Figure 5K) appears in the simulated DQ–SQ spectra. The possibility that, in reality, in a fraction of the [Co-1a]–COF pores the cobaloxime does not interact with the pore wall cannot be ruled out; nonetheless, our current data suggest that when it does, it gets in close contact with the nearby ethoxy group. It is also likely that this genuine interaction stabilizes the complex and restricts the cocatalyst’s degradation during the photocatalytic cycles. Note that, at this stage, both the ssNMR measurements and the in silico calculations were performed in a solvent-free environment. Future ssNMR measurements with added acetonitrile/water mixture accompanied by simulations in explicit solvent could reveal if the cobaloxime stays attached to the pore wall or wether it gains more flexibility and drifts toward the pore center.
Figure 5.

Direct comparison of quantum-chemically obtained 1H–13C (A, D, G, J) and 1H–1H DQ–SQ (B, E, H, K) 2D ssNMR spectra with corresponding structural models of [Co-1a]–COF on the right (C, F, I, L). For a better comparison, the same NMR chemical shift region is displayed as in the experimentally obtained spectra (Figures 3C,D and 4D,E). In the 1H–13C 2D spectra, blue and green colors represent 1H–13C atom pairs that are within 6 and 2 Å, respectively. In the 1H–1H DQ–SQ spectra, the orange color highlights the oxime proton cross-peaks. In parts C, F, I, and L, the Co, Cl, O, N, and H atoms are displayed in pink, lime, red, blue, and white, respectively.
To inspect the spacial arrangement inside the pore, we modeled [Co-1a]–COF including one tethered cocatalyst based on the MD-simulated structures (Figure 6). The displayed ligand has the same orientation as in Figure 5C. From the side and front views it is apparent that the ligand spreads over multiple layers and occupies a substantial portion of the pore. Due to spacial confinements, our model suggests that no more than three [Co-1a] over three layers can fit into the backbone; i.e., the maximum number of [Co-1a] per layer is one. In our case, we have 13 mol % functionalization, which translates into one [Co-1a] for every seven layers.
Figure 6.
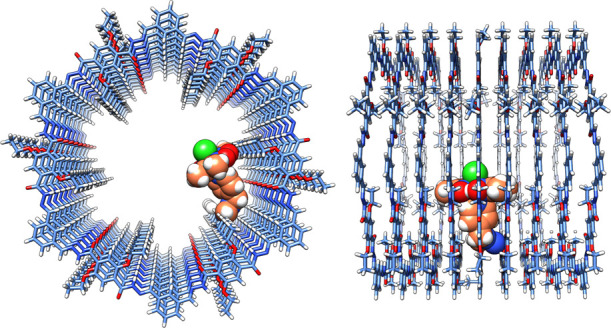
Front and side views of the MD-simulated structural model of [Co-1a]–COF showing a possible arrangement of the cocatalyst. The linker and the cobaloxime group are depicted by spheres and their carbon atoms are displayed in orange. Co, Cl, O, N, and H atoms are displayed in pink, lime, red, blue, and white, and C atoms of the backbone are light blue.
Photocatalytic Activity
To probe whether there is a possible benefit of covalent cocatalyst immobilization over simple physisorption,14,15 the COF–cobaloxime hybrid samples were tested for photocatalytic activity. In a typical photocatalysis experiment, 5 mg of COF hybrid was suspended in 10 mL of acetonitrile and water in a ratio of 4:1 at pH 8 containing 100 μL of triethanolamine (TEOA) as sacrificial donor. A housed Xe lamp was used to illuminate the suspension interface with a nominal beam spectral distribution similar to that of AM1.5G. The beam intensity before experiments was then adjusted to 100 mW cm–2. See the SI for more details. Photocatalytic hydrogen evolution reaction (HER) rates were quantified in a continuous flow reactor as previously reported15 (Figure 7A). As a reference system, we compared the hybrid systems to samples where [Co-1a] or [Co-1b] was added to the suspension and physisorbed to COF-42 with a BET surface area of 2336 m2 g–1 during photocatalysis. The maximum photonic efficiencies after in situ photoactivation of the samples ranging from 2 to 8 wt % cobaloxime catalyst according to ICP results can be found in Figure 7A. In the physisorbed samples, an increase of the photonic efficiency was found when increasing the catalyst amount from 2 to 4 wt % with a maximum efficiency of 0.06% for [Co-1a] and 0.07% for [Co-1b] at 4.0 wt %, while the efficiency is fairly constant at higher percentages (0.06%–0.08% at 5.0 and 8.0 wt %) for [Co-1b]. This behavior is expected for the system; as in the low-loading region, the photocatalytic activity scales linearly with the cocatalyst amount, while it reaches a maximum in the higher-loading region where the availability of the cocatalyst is not limiting anymore.
Figure 7.

(A) Comparison of photonic efficiencies for hybrid samples and COF-42 with physisorbed [Co-1a] and [Co-1b]. (B) Comparison of the hydrogen evolution rate of [Co-1b]–COF containing 3.2 wt % [Co-1b] and COF-42 with 4.0 wt % physisorbed [Co-1b] and coarse-grained model fits of both systems. (C) Projection of the hydrogen evolution of [Co-1b]–COF containing 3.2 wt % [Co-1b] and COF-42 with 4.0 wt % physisorbed [Co-1b] based on the coarse-grained models.
In the hybrid samples, an activity maximum rather than a constant behavior is found for each hybrid type. For the para-functionalized [Co-1a], the highest photonic efficiency was found at 4.1 wt %, while for the meta-functionalized [Co-1b] the maximum was found at 3.2 wt %. As before, a linear increase of the photonic efficiency in the low-loading regime was observed. However, a further increase in cobaloxime loading resulted in lower activity in the immobilized samples. We attribute this to a predominant pore-clogging effect of the active sites with increasing functionalization. In general, the highest photonic efficiency was achieved with [Co-1a]–COF at 0.14% followed by [Co-1b]–COF at 0.11%. Compared to the physisorbed samples with the corresponding cobaloxime content, the activity doubles for both systems. Additionally, to emphasize the role of the complex environment of the cobaloxime over the pure presence of Co(II), we performed a measurement where we added CoCl2 to a suspension of pCOF10 and triethanolamine in the photocatalysis medium, as well as experiments where one of the components (COF, TEOA) was excluded (see Supporting Information). None of the reference samples showed hydrogen evolution after several hours of irradiation. For the hybrid samples, the close contact between the cobaloxime and the COF pore wall—revealed by representative solid-state NMR and computational studies (vide supra) with [Co-1a]–COF—might facilitate charge transfer to the cobaloxime catalyst from the COF pore wall, as also observed from photoluminescence measurements (see Figures S11 and S12, SI) where [Co-2]–COF shows a significantly lower activity in CH3CN/H2O, which is a known effect for cobaloximes that lack equatorial protons. The protonation of the oxime oxygen, which is necessary for the catalytic process, is hindered in those cases.58,59 The catalytic activity could not be improved by lowering the pH to 4. In this case, different acids (ascorbic acid, acetic acid, and citric acid) were tested that simultaneously served as sacrificial electron donors instead of the amine base TEOA. Even though the stability of [Co-2]–COF is predicted to be higher than that for the other tested cobaloximes, the complex proved not to be appropriate in our case. We compared the best performing [Co-1b]–COF sample (containing 3.2 wt % cobaloxime) to COF-42 physisorbed with [Co-1b]. A sample with the same amount of physisorbed cobaloxime was qualitatively active in photocatalytic hydrogen evolution, but for precise quantification, we increased the catalyst amount to 4.0 wt %. Even though it contained 20% less catalyst, the hybrid sample was 47% more active than the physisorbed one (163 vs 111 μmol h–1 g–1) (see Figure 7A). Additionally, the long-term stability increased significantly. After 20 h, the physisorbed sample shows 52% of its initial activity, while the hybrid sample maintains 80% of its initial activity. To get an estimate of the longevity of the systems, we fitted the hydrogen evolution rates of both samples with a coarse-grained model (Figure 7C) that was established in an earlier study on photocatalysis with COFs and a nickel-based oligomer as cocatalyst.15 The model resulted in very precise fitting for the physisorbed catalyst because of similarities to the original nickel-based system from where the coarse-grain fitting model was obtained, while the hybrid sample showed a more complex behavior that is not perfectly mapped with this simplified model. On the basis of the coarse-grained fits, we projected the total amount of hydrogen evolved by the samples at full depletion (see Figure 7C). After 780 h, the projection of the physisorbed sample reaches 35 μmol of hydrogen evolved, while the value is 59 μmol for the hybrid sample, which is a gain of 69%. Comparing the estimated turnover numbers (TONs) of both systems, the deviation gets even more obvious. While the TON after 780 h is simulated to be 81 for the physisorbed sample, it increases by 110% to a value of 170 in the hybrid sample. We attribute this activity enhancement to the local confinement in the COF hybrid samples, as supported by MD simulations.
Cobaloximes are known to slowly decompose under photocatalytic conditions. The labile axial pyridine ligand decoordinates in the catalytic cycle due to a square-planar Co(II) transition state. The catalyst in solution can then possibly be reduced, which limits its stability. Due to the confinement between the ligand and catalyst in the COF pores, the recoordination might be enhanced, hence counteracting degradation, which leads to reactivation of the catalyst. Additionally, charge transfer is favored in the case of the spatial proximity of the cocatalyst and the pore wall. Both effects result in higher overall activity as well as longevity. Interestingly, the activation period for the hybrid samples is significantly longer than for the physisorbed ones. This may be attributed to the time-delayed accessibility of the catalyst in the pores. Both limitations could be addressed via a method that was recently published by Thomas and co-workers,60,61 where silica spheres were used to create an inverse-opal architecture in the COF material. The so created macropores could serve as channels for reagents and products. Also, immobilization of the cocatalyst in a COF with larger pores might have a similar effect.
Conclusion
In summary, we have developed a platform derived from COF-42 as a support for the immobilization of cobaloxime catalysts. The postsynthetic modification of propargyl-functionalized COF-42 enabled the covalent tethering of three different cobaloximes to form COF–cobaloxime hybrid systems. This tethering significantly enhanced the photocatalytic activity of the system by more than 100% compared to that of the physisorbates with the corresponding cobaloxime amount. The high crystallinity of our materials allowed for an in-depth solid-state 2D NMR characterization using fast MAS and proton detection. In the 1D 1H spectrum of [Co-1a]–COF, we could clearly identify the resonance corresponding to the oxime proton on the basis of its highly downfield-shifted resonance. The 2D 1H–1H DQ–SQ experiment showed two cross-peaks for the oxime proton, consistent with the incorporation of the cocatalyst into the COF material. MD simulations with subsequent quantum-chemical NMR chemical shift calculations allowed us to locate the position of the tethered ligand inside the pore on the basis of the experimentally observed oxime proton cross-peaks. Our analysis suggests that the cobaloxime in [Co-1a]–COF closely interacts with the pore wall. We surmise that this interaction is responsible both for the improved photocatalytic activity and for the prolonged activity of the hybrid samples with respect to the physisorbed variant. We anticipate that larger pore channels or the addition of dedicated transport pores will further improve the pore accessibility and prevent back-reaction via local confinement of the products, thereby increasing the hydrogen evolution activity of the system even further.
Acknowledgments
Financial support is gratefully acknowledged from the Max Planck Society, an ERC Starting Grant (project COF Leaf, grant number 639233), the Deutsche Forschungsgemeinschaft (DFG) via the SFB 1333 (project A03), the Cluster of Excellence e-conversion, and the Center for Nanoscience. P.R. acknowledges the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB 1309-325871075, project A3, and Fonds der Chemischen Industrie. We thank Prof. T. Bein and Prof. W. Schnick (University of Munich, LMU) for granting access to the XRD facility and V. Duppel and M.-L. Schreiber for the assistance with material analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c02155.
Experimental procedures, COF synthesis, and details of molecular dynamic simulations, quantum-chemical calculations, and additional measurements (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Li L.; Cai Z.; Wu Q.; Lo W.-Y.; Zhang N.; Chen L. X.; Yu L. Rational Design of Porous Conjugated Polymers and Roles of Residual Palladium for Photocatalytic Hydrogen Production. J. Am. Chem. Soc. 2016, 138, 7681–7686. 10.1021/jacs.6b03472. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Mao F.; Wang L.; Yuan H.; Liu P. F.; Yang H. G. Recent Advances in Photocatalysis over Metal–Organic Frameworks-Based Materials. Solar RRL 2020, 4, 1900438. 10.1002/solr.201900438. [DOI] [Google Scholar]
- Diercks C. S.; Liu Y.; Cordova K. E.; Yaghi O. M. The role of reticular chemistry in the design of CO2 reduction catalysts. Nat. Mater. 2018, 17, 301–307. 10.1038/s41563-018-0033-5. [DOI] [PubMed] [Google Scholar]
- Vyas V. S.; Lau V. W.-h.; Lotsch B. V. Soft Photocatalysis: Organic Polymers for Solar Fuel Production. Chem. Mater. 2016, 28, 5191–5204. 10.1021/acs.chemmater.6b01894. [DOI] [Google Scholar]
- Sick T.; Hufnagel A. G.; Kampmann J.; Kondofersky I.; Calik M.; Rotter J. M.; Evans A.; Döblinger M.; Herbert S.; Peters K.; Böhm D.; Knochel P.; Medina D. D.; Fattakhova-Rohlfing D.; Bein T. Oriented Films of Conjugated 2D Covalent Organic Frameworks as Photocathodes for Water Splitting. J. Am. Chem. Soc. 2018, 140, 2085–2092. 10.1021/jacs.7b06081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté A. P.; Benin A. I.; Ockwig N. W.; O'Keeffe M.; Matzger A. J.; Yaghi O. M. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166. 10.1126/science.1120411. [DOI] [PubMed] [Google Scholar]
- Tilford R. W.; Mugavero S. J.; Pellechia P. J.; Lavigne J. J. Tailoring Microporosity in Covalent Organic Frameworks. Adv. Mater. 2008, 20, 2741–2746. 10.1002/adma.200800030. [DOI] [PubMed] [Google Scholar]
- Ding S.-Y.; Wang W. Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 2013, 42, 548–568. 10.1039/C2CS35072F. [DOI] [PubMed] [Google Scholar]
- Lohse M. S.; Bein T. Covalent Organic Frameworks: Structures, Synthesis, and Applications. Adv. Funct. Mater. 2018, 28, 1705553. 10.1002/adfm.201705553. [DOI] [Google Scholar]
- Stegbauer L.; Schwinghammer K.; Lotsch B. V. A hydrazone-based covalent organic framework for photocatalytic hydrogen production. Chem. Sci. 2014, 5, 2789–2793. 10.1039/C4SC00016A. [DOI] [Google Scholar]
- Haase F.; Banerjee T.; Savasci G.; Ochsenfeld C.; Lotsch B. V. Structure–property–activity relationships in a pyridine containing azine-linked covalent organic framework for photocatalytic hydrogen evolution. Faraday Discuss. 2017, 201, 247–264. 10.1039/C7FD00051K. [DOI] [PubMed] [Google Scholar]
- Stegbauer L.; Zech S.; Savasci G.; Banerjee T.; Podjaski F.; Schwinghammer K.; Ochsenfeld C.; Lotsch B. V. Tailor-Made Photoconductive Pyrene-Based Covalent Organic Frameworks for Visible-Light Driven Hydrogen Generation. Adv. Energy Mater. 2018, 8, 1703278. 10.1002/aenm.201703278. [DOI] [Google Scholar]
- Wang X.; Chen L.; Chong S. Y.; Little M. A.; Wu Y.; Zhu W.-H.; Clowes R.; Yan Y.; Zwijnenburg M. A.; Sprick R. S.; Cooper A. I. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 2018, 10, 1180–1189. 10.1038/s41557-018-0141-5. [DOI] [PubMed] [Google Scholar]
- Banerjee T.; Haase F.; Savasci G.; Gottschling K.; Ochsenfeld C.; Lotsch B. V. Single-Site Photocatalytic H2 Evolution from Covalent Organic Frameworks with Molecular Cobaloxime Co-Catalysts. J. Am. Chem. Soc. 2017, 139, 16228–16234. 10.1021/jacs.7b07489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswal B. P.; Vignolo-González H. A.; Banerjee T.; Grunenberg L.; Savasci G.; Gottschling K.; Nuss J.; Ochsenfeld C.; Lotsch B. V. Sustained Solar H2 Evolution from a Thiazolo[5,4-d]thiazole-Bridged Covalent Organic Framework and Nickel-Thiolate Cluster in Water. J. Am. Chem. Soc. 2019, 141, 11082–11092. 10.1021/jacs.9b03243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee T.; Gottschling K.; Savasci G.; Ochsenfeld C.; Lotsch B. V. H2 Evolution with Covalent Organic Framework Photocatalysts. ACS Energy Letters 2018, 3, 400–409. 10.1021/acsenergylett.7b01123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey J. L.; Brunschwig B. S.; Winkler J. R.; Gray H. B. Hydrogen Evolution Catalyzed by Cobaloximes. Acc. Chem. Res. 2009, 42, 1995–2004. 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- Artero V.; Chavarot-Kerlidou M.; Fontecave M. Splitting Water with Cobalt. Angew. Chem., Int. Ed. 2011, 50, 7238–7266. 10.1002/anie.201007987. [DOI] [PubMed] [Google Scholar]
- Muresan N. M.; Willkomm J.; Mersch D.; Vaynzof Y.; Reisner E. Immobilization of a Molecular Cobaloxime Catalyst for Hydrogen Evolution on a Mesoporous Metal Oxide Electrode. Angew. Chem., Int. Ed. 2012, 51, 12749–12753. 10.1002/anie.201207448. [DOI] [PubMed] [Google Scholar]
- Nasalevich M. A.; Becker R.; Ramos-Fernandez E. V.; Castellanos S.; Veber S. L.; Fedin M. V.; Kapteijn F.; Reek J. N. H.; van der Vlugt J. I.; Gascon J. Co@NH2-MIL-125(Ti): cobaloxime-derived metal–organic framework-based composite for light-driven H2 production. Energy Environ. Sci. 2015, 8, 364–375. 10.1039/C4EE02853H. [DOI] [Google Scholar]
- Gao L.-F.; Zhu Z.-Y.; Feng W.-S.; Wang Q.; Zhang H.-L. Disentangling the Photocatalytic Hydrogen Evolution Mechanism of One Homogeneous Cobalt-Coordinated Polymer. J. Phys. Chem. C 2016, 120, 28456–28462. 10.1021/acs.jpcc.6b09767. [DOI] [Google Scholar]
- Cao S.-W.; Liu X.-F.; Yuan Y.-P.; Zhang Z.-Y.; Fang J.; Loo S. C. J.; Barber J.; Sum T. C.; Xue C. Artificial photosynthetic hydrogen evolution over g-C3N4 nanosheets coupled with cobaloxime. Phys. Chem. Chem. Phys. 2013, 15, 18363. 10.1039/c3cp53350f. [DOI] [PubMed] [Google Scholar]
- Li X.; Masters A. F.; Maschmeyer T. Photocatalytic Hydrogen Evolution from Silica-Templated Polymeric Graphitic Carbon Nitride-Is the Surface Area Important?. ChemCatChem 2015, 7, 121–126. 10.1002/cctc.201402567. [DOI] [Google Scholar]
- Lazarides T.; McCormick T.; Du P.; Luo G.; Lindley B.; Eisenberg R. Making Hydrogen from Water Using a Homogeneous System Without Noble Metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. 10.1021/ja903044n. [DOI] [PubMed] [Google Scholar]
- Lakadamyali F.; Reisner E. Photocatalytic H2 evolution from neutral water with a molecular cobalt catalyst on a dye-sensitised TiO2 nanoparticle. Chem. Commun. 2011, 47, 1695. 10.1039/c0cc04658b. [DOI] [PubMed] [Google Scholar]
- Yin M.; Ma S.; Wu C.; Fan Y. A noble-metal-free photocatalytic hydrogen production system based on cobalt(iii) complex and eosin Y-sensitized TiO2. RSC Adv. 2015, 5, 1852–1858. 10.1039/C4RA10767E. [DOI] [Google Scholar]
- Uribe-Romo F. J.; Doonan C. J.; Furukawa H.; Oisaki K.; Yaghi O. M. Crystalline Covalent Organic Frameworks with Hydrazone Linkages. J. Am. Chem. Soc. 2011, 133, 11478–11481. 10.1021/ja204728y. [DOI] [PubMed] [Google Scholar]
- Gottschling K.; Stegbauer L.; Savasci G.; Prisco N. A.; Berkson Z. J.; Ochsenfeld C.; Chmelka B. F.; Lotsch B. V. Molecular Insights into Carbon Dioxide Sorption in Hydrazone-Based Covalent Organic Frameworks with Tertiary Amine Moieties. Chem. Mater. 2019, 31, 1946–1955. 10.1021/acs.chemmater.8b04643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Shen X.; Feng X.; Xia H.; Mu Y.; Liu X. Covalent organic frameworks as pH responsive signaling scaffolds. Chem. Commun. 2016, 52, 11088–11091. 10.1039/C6CC05748A. [DOI] [PubMed] [Google Scholar]
- Chen X.; Addicoat M.; Jin E.; Xu H.; Hayashi T.; Xu F.; Huang N.; Irle S.; Jiang D. Designed synthesis of double-stage two-dimensional covalent organic frameworks. Sci. Rep. 2015, 5, 14650. 10.1038/srep14650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.-J.; Ding S.-Y.; Xue H.-D.; Cao W.; Wang W. Synthesis of – C=N– linked covalent organic frameworks via the direct condensation of acetals and amines. Chem. Commun. 2016, 52, 7217–7220. 10.1039/C6CC00947F. [DOI] [PubMed] [Google Scholar]
- Himo F.; Lovell T.; Hilgraf R.; Rostovtsev V. V.; Noodleman L.; Sharpless K. B.; Fokin V. V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hein C. D.; Liu X.-M.; Wang D. Click Chemistry, A Powerful Tool for Pharmaceutical Sciences. Pharm. Res. 2008, 25, 2216–2230. 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amblard F.; Cho J. H.; Schinazi R. F. Cu(I)-Catalyzed Huisgen Azide-Alkyne 1,3-Dipolar Cycloaddition Reaction in Nucleoside, Nucleotide, and Oligonucleotide Chemistry. Chem. Rev. 2009, 109, 4207–4220. 10.1021/cr9001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W. Cu-Catalyzed Azide-Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Levitt M. H.Symmetry-Based Pulse Sequences in Magic-Angle Spinning Solid-State NMR. In eMagRes; Wiley, 2007; pp 1–31. [Google Scholar]
- Brown S. P.; Lesage A.; Elena B.; Emsley L. Probing Proton-Proton Proximities in the Solid State: High-Resolution Two-Dimensional 1H-1H Double-Quantum CRAMPS NMR Spectroscopy. J. Am. Chem. Soc. 2004, 126, 13230–13231. 10.1021/ja045461p. [DOI] [PubMed] [Google Scholar]
- Bradley J. P.; Tripon C.; Filip C.; Brown S. P. Determining relative proton–proton proximities from the build-up of two-dimensional correlation peaks in 1H double-quantum MAS NMR: insight from multi-spin density-matrix simulations. Phys. Chem. Chem. Phys. 2009, 11, 6941. 10.1039/b906400a. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Eichkorn K.; Weigend F.; Treutler O.; Ahlrichs R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. 10.1007/s002140050244. [DOI] [Google Scholar]
- Burow A. M.; Sierka M.; Mohamed F. Resolution of identity approximation for the Coulomb term in molecular and periodic systems. J. Chem. Phys. 2009, 131, 214101. 10.1063/1.3267858. [DOI] [PubMed] [Google Scholar]
- Grajciar L. Low-memory iterative density fitting. J. Comput. Chem. 2015, 36, 1521–1535. 10.1002/jcc.23961. [DOI] [PubMed] [Google Scholar]
- Burow A. M.; Sierka M. Linear Scaling Hierarchical Integration Scheme for the Exchange-Correlation Term in Molecular and Periodic Systems. J. Chem. Theory Comput. 2011, 7, 3097–3104. 10.1021/ct200412r. [DOI] [PubMed] [Google Scholar]
- Łazarski R.; Burow A. M.; Sierka M. Density Functional Theory for Molecular and Periodic Systems Using Density Fitting and Continuous Fast Multipole Methods. J. Chem. Theory Comput. 2015, 11, 3029–3041. 10.1021/acs.jctc.5b00252. [DOI] [PubMed] [Google Scholar]
- Łazarski R.; Burow A. M.; Grajciar L.; Sierka M. Density functional theory for molecular and periodic systems using density fitting and continuous fast multipole method: Analytical gradients. J. Comput. Chem. 2016, 37, 2518–2526. 10.1002/jcc.24477. [DOI] [PubMed] [Google Scholar]
- TURBOMOLE, ver. 7.1; TURBOMOLE GmbH, 2016. Available at http://www.turbomole.com.
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006, 25, 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Lu R.; Madej B.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Nguyen H. T.; Omelyan I.; Onufriev A.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Botello-Smith W. M.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Xiao L.; Kollman P. A.. AMBER 2016; University of California: San Francisco, CA, 2016. [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wilson P. J.; Bradley T. J.; Tozer D. J. Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials. J. Chem. Phys. 2001, 115, 9233–9242. 10.1063/1.1412605. [DOI] [Google Scholar]
- Jensen F. Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding. J. Chem. Theory Comput. 2015, 11, 132–138. 10.1021/ct5009526. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Preselective Screening for Linear-Scaling Exact Exchange-Gradient Calculations for Graphics Processing Units and General Strong-Scaling Massively Parallel Calculations. J. Chem. Theory Comput. 2015, 11, 918–922. 10.1021/ct501189u. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Pre-selective screening for matrix elements in linear-scaling exact exchange calculations. J. Chem. Phys. 2013, 138, 134114. 10.1063/1.4796441. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A.; Andreiadis E. S.; Chavarot-Kerlidou M.; Fontecave M.; Field M. J.; Artero V. A Computational Study of the Mechanism of Hydrogen Evolution by Cobalt(Diimine-Dioxime) Catalysts. Chem. - Eur. J. 2013, 19, 15166–15174. 10.1002/chem.201301860. [DOI] [PubMed] [Google Scholar]
- Kaeffer N.; Chavarot-Kerlidou M.; Artero V. Hydrogen Evolution Catalyzed by Cobalt Diimine–Dioxime Complexes. Acc. Chem. Res. 2015, 48, 1286–1295. 10.1021/acs.accounts.5b00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Pachfule P.; Li S.; Langenhahn T.; Ye M.; Schlesiger C.; Praetz S.; Schmidt J.; Thomas A. Macro/Microporous Covalent Organic Frameworks for Efficient Electrocatalysis. J. Am. Chem. Soc. 2019, 141, 6623–6630. 10.1021/jacs.9b01226. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Pachfule P.; Li S.; Langenhahn T.; Ye M.; Tian G.; Schmidt J.; Thomas A. Silica-Templated Covalent Organic Framework-Derived Fe–N-Doped Mesoporous Carbon as Oxygen Reduction Electrocatalyst. Chem. Mater. 2019, 31, 3274–3280. 10.1021/acs.chemmater.9b00204. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.