

Abstract
The world is currently facing the COVID-19 pandemic, for which mild symptoms include fever and dry cough. In severe cases, it could lead to pneumonia and ultimately death in some instances. Moreover, the causative pathogen is highly contagious and there are no drugs or vaccines for it yet. The pathogen, SARS-CoV-2, is one of the human coronaviruses which was identified to infect humans first in December 2019. SARS-CoV-2 shares evolutionary relationship to other highly pathogenic viruses such as Severe Acute Respiratory Syndrome (SARS) and Middle East respiratory syndrome (MERS). We have exploited this similarity to model a target non-structural protein, NSP1, since it is implicated in the regulation of host gene expression by the virus and hijacking of host machinery. We next interrogated the capacity to repurpose around 2300 FDA-approved drugs and more than 3,00,000 small molecules of natural origin towards drug identification through virtual screening and molecular dynamics. Interestingly, we observed simple molecules like lactose, previously known anti-virals and few secondary metabolites of plants as promising hits. These herbal plants are already practiced in Ayurveda over centuries to treat respiratory problems and inflammation. Disclaimer: we would not like to recommend uptake of these small molecules for suspect COVID patients until it is approved by competent national or international authorities.
Electronic supplementary material
The online version of this article (10.1007/s12038-020-00069-8) contains supplementary material, which is available to authorized users.
Keywords: Anti-virals, drug design, herbal plants, repurposing drugs, SARS-CoV-2
Introduction
Coronavirus (CoV) belongs to the family Coronaviridae and the order Nidovirales (sharing with Arteriviridae and Roniviridae). Coronaviruses are enveloped, long positive-sense single-stranded RNA viruses, which are best known for causing mild to severe respiratory and enteric infection among a vast range of hosts (Masters 2006). These are further divided into 4 groups/genera named as Alphacoronavirus (α-CoV), Betacoronavirus (β-CoV), Gammacoronavirus (γ-CoV) and Deltacoronavirus (δ-CoV), based on sequence similarities and antigenic cross-reactivity. Human-CoV belongs to group I and group II of Betacoronavirus. HCoV-OC43, HCoV-229E, SARS and MERS are some examples of Human-CoV, out of which SARS and MERS are highly pathogenic in nature (Masters 2006; Narayanan et al. 2015). Recently, a new pathogenic Human-CoV strain known as SARS-CoV2, spreading the COVID-19 infection, has emerged by December 2019 with Wuhan of Hubei province in China as the epicenter (Wu et al. 2020a, b). The origin of this virus is still under investigation but has been speculated as a zoonotic shift from bat to human. It has been shown that human ACE2 has a high predicted affinity than ACE2 from other species (Piplani et al. 2020). The outbreak of COVID-19 has spread across the globe and has taken the shape of a pandemic (Novel Coronavirus (2019-nCoV) situation reports, World Health Organization). America, Russia, United Kingdom, India, Italy, Spain, and France are among worst-hit countries. As of 14 June 2020, this has infected 7,891, 289 individuals and has caused more than 432,746 fatalities across the globe (Johns Hopkins Coronavirus Resource Center; Worldmeter. https://www.worldometers.info/coronavirus/; Novel Coronavirus (2019-nCoV) situation reports - World Health Organization). At present, there are no drugs or vaccines available against this, and patients are treated according to symptoms shown by them. Remdisivir (drug originally designed to treat Ebola), Colchicine (Deftereos et al. 2020), Chloroquine and Hydroxychloroquine (an antimalarial drug), Kevsara (an arthritis drug) and few other antiviral drugs are being considered for treatment. But they do not directly make use of the virome of SARS-CoV2 (Wang et al. 2020) (https://www.nasdaq.com/articles/8-experimental-coronavirus-treatments-to-watch-2020-03-31). Multiple vaccine trial has been going on worldwide, among which Bacillus Calmette Guerine (BCG) live attenuated vaccine and AZD1222 are in phase 2/3 of the clinical trials. (https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-vaccine-tracker)
The first genome of the COVID-19 strain was sequenced by Wu et al. (2020b) from a 41-year-old man and was found to be closely similar to SARS-CoV. The structural component of the virus consists of four proteins: Spike (S), Membrane (M), Envelope (E) and Nucleocapsid (N) protein respectively. S protein is critical for viral infection as it enables host-pathogen interaction and mediated viral entry into the host cell. M protein is a multipass transmembrane protein, a major constituent of virion envelope, and known to provide its shape. E protein, unlike the name, suggests it is a minor constituent of the envelope and 80–120 aa in length. N protein as the name suggests forms helical nucleocapsid of virion (Masters 2006).
The 5′-end of the genome encodes two open reading frames, ORF1a and ORF1b, respectively, which code for all non-structural proteins (NSP1-16) (Masters 2006; Narayanan et al. 2015). These proteins are essential for viral replication as well as infection, whereas the function of some is yet to be identified. Among these non-structural proteins within the CoV family, some are conserved in sequence, whereas others are highly diverged in nature. NSP1 (non-structural protein 1) is one of such diverged proteins which is encoded by ORF1a and varied in amino-acid length among CoV-groups (Narayanan et al. 2015). COVID-19 NSP1 consists of 180 and shows sequence similarity with SARS protein (Elbe and Buckland-Merrett 2017; Wu et al. 2020a). Despite differences in amino acid sequence and length, it has shown to be functionally highly conserved (Narayanan et al. 2015; Shen et al. 2019). SARS-CoV NSP1 is most well-studied amongst the viruses of the coronavirus family. NSP1 has shown to act as a virulence factor (Huang et al. 2011; Narayanan et al. 2015; Zst et al. 2007) and mutation in this protein results in the production of attenuated virus in vitro and in vivo (Zst et al. 2007). NSP1 deploys two strategies to inhibit host cell expression viz. Inhibition of host translation and Induction of host mRNA degradation. It inhibits host translation by forming a complex with 40s ribosome subunit, which prevents the formation of active polysome. Complex formation with 40s subunit also shown to inhibit its translational ability (Kamitani et al. 2009). It further affects host cell gene expression by inducing host mRNA degradation in a template-specific manner. Term template-specific does not imply its association with protein sequence but relates to the ability to specifically degrade capped host mRNA (Huang et al. 2011; Kamitani et al. 2006, 2009; Narayanan et al. 2008; Tanaka et al. 2012) as compared to SARS-mRNA (Kamitani et al. 2006). mRNA is hypothesized to get cleaved by unknown host endonuclease since NSP1 does not possess any endonucleolytic activity. Other than these, NSP1 is shown to cause chemokine dysregulation which correlates with high inflammation in severe patients (Law et al. 2007; Channappanavar and Perlman 2017; Wong et al. 2004). It suppresses innate immune response by degrading IFN-beta mRNA (Tanaka et al. 2012) and affecting antiviral signaling (Jauregui et al. 2013). Yeast-two hybrid assays have shown NSP1 to interact with multiple host proteins (Pfefferle et al. 2011). N-terminal region is shown to be important in immune response dysregulation (mutation studies) (Jauregui et al. 2013) and protecting viral RNA (R124, present in our dock site) (Tanaka et al. 2012). The C-terminal region is critical of transcriptional inhibition of host mRNA (Narayanan et al. 2015; Tanaka et al. 2012).
In this study, COVID-19 NSP1 is the target protein and we hypothesized that the inhibition of NSP1 can potentially attenuate the virus and suppress adverse immune-pathology caused by it. We have generated a homology model of the NSP1 and used this model to carry out virtual screening to identify potential inhibitors and lead compounds. Our searches are directed within the database of FDA-approved drugs (DRUGBANK) and those which are derived from molecules of natural origin (SUPERNATURALDB). Finally, we performed MD simulation to ensure that there are indeed stable protein-ligand interactions when the system is visualized to undergo limited conformational freedom. We find several anti-viral compounds, few secondary metabolites of plant origin and simple compounds (like lactose) to retain the high potential to act as NSP1 inhibitors.
Methods
Sequence retrieval and analysis
The full repository of COVID-19 protein sequences was downloaded from NCBI (Brister et al. 2015; Hatcher et al. 2017). Wuhan-Hu-1 strain [Accession number: NC_045512] was among the first to be sequenced from Wuhan of Hubei province. Hence this is considered as the ‘reference genome’ in this study. NSP1 protein sequences were extracted, incomplete sequences were removed and curated sequences were then passed to SNP analyzer (utility in ViPR Database) to understand variation among the NSP1 sequences (Pickett et al. 2012). A similar analysis had been done for the Indian sequences. To understand evolutionary pressure on NSP1 protein Shannon entropy (Shannon 1948) per residue has been calculated using a python script. A set of key residues, important in suppressing host gene expression and antiviral signaling were identified. A mutation study done by another group (Jauregui et al. 2013) was used as a reference.
Homology modelling
NSP1 protein sequence [Accession number: YP_009725297] was retrieved from NCBI for Homology modelling. Blastp (Camacho et al. 2009) was used to search for the nearest structural homologue in Protein Data Bank (PDB) (Berman et al. 2003) to serve as a template for modelling. Segments of NSP1 sequence, where the association with the template was unknown, were removed. Modeller 9.12 (Eswar et al. 2006) was used for Homology modelling. Homology models were first filtered according to DOPE score. Top 3 predicted models were then subjected to structure validations (by using SAVES 5.0 (Laskowski et al. 1993) (https://servicesn.mbi.ucla.edu/SAVES/) and ProSA server (Wiederstein and Sippl 2007)). Based on DOPE score, Ramachandran plot and ProSA profile, the best predicted model was selected for virtual screening.
Virtual screening of inhibitors
FDA-approved drugs and Super Natural II database (database of natural products) were used for docking purposes. FDA-approved drugs were downloaded in SDF format (Standard Delay Format) from Drug-bank (Wishart et al. 2018) whereas supernatural compounds were obtained from supernatural database (Banerjee et al. 2015).
Ligand and protein preparation
Downloaded compounds were prepared for screening using Ligprep module in Schrodinger (Schrödinger Release 2019-4: LigPrep, Schrödinger, LLC, New York, NY, 2019). For FDA-approved drugs OPLS3e force field, targeted pH 7.4 +/−0.0, retain specified chiralities and 1 structure per ligand was specified during ligand preparation. For supernatural database we had specified pH range from 6.0 to 8.0 with maximum 32 structure per ligand. This was performed to scan and produce broad chemical and structural diversity from each molecule.
Protein was prepared for docking by using Protein-preparation wizard (Sastry et al. 2013) in Maestro Schrodinger (Schrödinger Release 2019-4: Maestro, Schrödinger, LLC, New York, NY, 2019.)
Docking site prediction
SiteMap (Halgren 2007; Halgren 2009) was used to predict the potential drug-able deep and shallow sites on the target protein. The site with high S-score, as well as D-score, was selected for ligand docking.
Receptor grid generation
Receptor-grid around docking region on the protein was generated using receptor-grid generation module in Glide, Residues from top predicted deep and shallow sites were specified and rotatable bonds across the site (if any) were checked during grid generation.
Protein–ligand docking
Using the glide docking module, a library of prepared ligands and protein with prepared receptor binding grid were docked. First, High-throughput virtual screening (HTVS) was performed. This narrowed down the list of potential ligands and the top 10 percent from this were then screened with Standard Precision (SP) mode. Finally, 10 percent of hits obtained from SP were passed to Extra precision (XP). Selection of top 10 percent compounds were done based on top dock score and binding energy (Friesner et al. 2004; Friesner et al. 2006; Halgren et al. 2004) (Schrödinger Release 2019-4: Glide, Schrödinger, LLC, New York, NY, 2019.)
Binding energy was calculated using MM-GBSA (Molecular Mechanics energies combined with the Generalized Born and Surface Area continuum solvation) tool of Schrodinger.
MD simulations
The conformer of protein–ligand complex, emerging from XP docking, was assembled using system builder and subject to Molecular Dynamics using the Desmond package of Schrodinger (Bowers et al. 2006). For water, the TIP4P model was specified and orthorhombic box shape was used having a buffer distance of 10 Å. Box volume was minimized. The system was neutralized and 150 mM salt (NaCl) was added. The output of the system builder was used for MD. The default relaxation protocol was used to relax the solvated system followed by production MD run for 20 nanoseconds (ns). The relaxation protocol involves energy minimization steps using the steepest descent method with a maximum of 2000 steps. The energy minimization was done with solute being restrained using 50 kcal/mol/Å force constant on all solute atoms and without restraints. Energy minimization was followed by short MD simulation steps which involve (1) Simulation for 12 picoseconds at 10 K in NVT ensemble using Berendsen thermostat with restrained non-hydrogen solute atoms, (2) Simulation for 12 picoseconds at 10 K and 1 atmospheric pressure in NPT ensemble using Berendsen thermostat and Berendsen barostat with restrained non-hydrogen solute atoms, (3) Simulation for 24 picoseconds at 300 K and 1 atmospheric pressure in NPT ensemble using Berendsen thermostat and Berendsen barostat with restrained non-hydrogen solute atoms, (4) Simulation for 24 picoseconds at 300 K and 1 atmospheric pressure in NPT ensemble using Berendsen thermostat and Berendsen barostat without restraints. After relaxation, production MD was run in NPT ensemble using OPLS 2003 force field (Harder et al. 2016). For simulations, default parameters of RESPA integrator (Humphreys et al. 1994) (2 femtoseconds time step for bonded and near non-bonded interactions while 6 femtoseconds for far non-bonded interactions) were used. The temperature and pressure were kept at 300K and 1 bar using the Nose-Hoover chain method (Martyna et al. 1992) and the Martyna-Tobias-Klein method (Martyna et al. 1994) respectively. The production MD was run for 20 nanoseconds.
Simulation analysis
MD simulation analysis was performed using the Simulation interaction diagram (SID) module of the Desmond package. The entire range of simulation time was considered for all analyses. RMSD is calculated for each frame by aligning the complex to the protein backbone of the reference frame. Significantly higher values of ‘Lig fit Prot’ than protein RMSD signifies the diffusion of ligand away from its initial binding site. Lig fit lig RMSD is calculated by aligning the ligand on the reference ligand conformation and it indicates the internal fluctuation of the ligand. Along with RMSD, the RMSF (Root Mean Square Fluctuation) was also assessed for each MD run. Protein RMSF shows the fluctuation of protein residues, highlights secondary structure (Pink: α helix; Blue: β strand) and ligand interacting residues marked by green vertical lines. Protein-ligand interactions were also monitored throughout the simulation time. Different types of protein-ligand interactions measured. are H-bond, Hydrophobic interaction, ionic interaction and water bridges. Hydrophobic interaction also includes Π-Cation and Π-Π interactions. The normalized stacked bar charts suggest the fraction of simulation time for which interaction is maintained over the course of the simulation trajectory: for example, a value of 0.6 implies that a specific interaction is maintained for 60% of the simulation time. If a protein residue makes multiple interactions of the same type with ligand then values more than 1.0 are possible.
ADME prediction
ADME: ‘absorption, distribution, metabolism, and excretion’ properties for selected compounds from supernatural database has been done using Qikprop tool of Schrodinger suite. A star is assigned if the value of the query compound falls beyond the 95% range of similar values for known drugs. Therefore, a greater number of stars indicates less drug-likeliness of the compound.
Results
Sequence Analysis identified COVID-19 NSP1 to be conserved
Around 10,000 complete NSP1 sequences of SARS-CoV2 were available in the public domain (NCBI as of 14 June 2020) and downloaded. Out of these, 6383 were sequences deposited from USA, 1829 from Australia, 446 from India and196 from Greece. Within India, 202 were sequences deposited from Ahmedabad, 40 from Vadodara and 26 from Gandhinagar. Analysis of Indian sequences between two time points, 15 May 2020 and 10 June 2020, clearly shows NSP1 to be evolving. In the dataset of 15th May, only one residue mutation (S135N) was observed. However, in the dataset corresponding to the second time point, three additional residues were found to be mutated (V38F, D147E, V167A (supplementary figure 1). Mutation analysis of the entire set of around 10,000 sequences shows mutation at multiple residues. Shannon entropy is found to be close to 0 for most of the residues (with a maximum <0.2). This indicates NSP1 is not under huge selection pressure and can be considered as highly conserved till now. In the absence of the available structure of NSP1 of SARS-CoV2, we included SARS NSP1 as a template for modeling. The major reason for this assumption is 100% query coverage and 84.44% sequence similarity of SARS-CoV2 NSP1 with SARS NSP1 (figure 1a). We performed an extensive literature survey to identify a set of key residues, important in suppressing host gene expression and antiviral signaling which are shown in figure 1b. Most of the residues among this set are found to be conserved between COVID and SARS NSP1.
Figure 1.

Sequence analysis COVID-19 (Wuhan-Hu-1) Nsp1. Represents alignment between Wuhan-Hu-1 Nsp1 and SARS Nsp1 protein sequence. Red highlights consensus sequences whereas Blue highlights difference in amino-acid sequence. Important residues shown to play role in affecting host gene expression and anti-viral signaling are highlighted in green and pink color. Green highlighting similar residues whereas Pink highlighting residues which are different in COVID-19.
Model for virtual screening generated by homology modelling
Blastp search of COVID-19 NSP1 sequence with PDB database enabled to identify 2hsx as the best template (with 68% query coverage and 86% identity). N- and C-terminal overhangs in SARS CoV-2 NSP1 have not been considered for modelling. Amino acid variations and key residues, important for function, are marked on the alignment (figure 1). Predicted models, derived using Modeller 9.22, were sorted according to the DOPE score and the top three models were validated using ProSA and SAVES5.0 sever. The best model from the above was chosen for virtual screening (figure 2a).
Figure 2.

Model of COVID-19 (Wuhan-Hu-1) Nsp1 with Deep and shallow binding site predicted by SiteMap: (a) COVID-19 Nsp1 model derived using Modeller 9.22, using 2hsx as a template. Red dot represents Shallow binding site consisting region of alpha-helix and beta-sheets. Blue dots represent deep binding site present in mostly loop region. (b) Residues present in deep and shallow binding site respectively. Residue numbers are as per the structural model (Residue 1 of structure is residue 12 in the sequence)
List of potent inhibitors are identified by in silico screening of FDA-approved drugs and Supernatural Database compounds
Three deep and five shallow ligand binding sites could be recognized on the surface of COVD19-NSP1 protein. Sites were ranked according to their ability to bind various ligands which were depicted as SITEMAP site score and D-Score (please see Methods). We selected Site 1 with a site score of 0.927 and D-score 0.791 among deep sites and site 3 with site score 0.883 and D-score 1.012 among shallow sites for ligand docking (figure 2a). These sites also contain functionally important residues (figure 1) which showed their biological importance. Selected sites were then used to generate a receptor grid for molecular docking. Molecular docking for each site was carried out using a glide dock program with generated libraries of 2413 FDA-approved drugs and 3,25,287 natural compounds, respectively. The top hits from FDA-approved drug library were ranked according to their XP and MMGBSA scores. We have also considered ligands with well-known anti-viral and anti-inflammatory properties, along with top-ranked ones (entries 15–17 in table 1). The final list of compounds was taken further for the MD simulation run (table 1). The top hits from Supernatural Database compounds were ranked according to their MMGBSA score and were further selected for MD simulation runs. List of top hits, selected based on either binding energy or mode of action, for both deep and shallow binding sites are shown in table 1.
Table 1.
Top-ranking hits identified by the virtual screening and other promising small molecules
| No. | Compound | Structure | Source | Deep binding site | Shallow binding site | Comment | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| XP Score | MMGBSA score | MD | XP Score | MMGBSA score | MD | |||||
| 1 | Esculin |
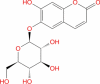
|
Drugbank | − 6.883 | − 29.24 | Stable | Vast-Protective agent, antioxidant and anti-inflammatory (Wishart et al. 2018) | |||
| 2 | Cidofovir |
|
Drugbank | − 5.776 | − 16.53 | Stable | Anti-viral (Wishart et al. 2018) | |||
| 3 | Mesalazine |
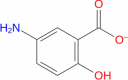
|
Drugbank | − 5.146 | − 20.74 | Not stable | Anti-inflammatory agent (Wishart et al. 2018) | |||
| 4 | Lactose |
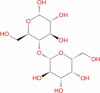
|
Drugbank | − 11.16 | − 40.72 | Not stable | It is a disaccharide of glucose and galactose. Used as a nutrient supplement (Wishart et al. 2018) | |||
| 5 | FAD |
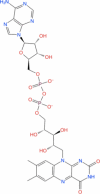
|
Drugbank | − 7.895 | − 52.47 | Not stable | − 5.643 | − 40.25 | Not stable | Used in ophthalmic treatment for vitamin B2 deficiency (Wishart et al. 2018) |
| 6 | Salmeterol |
|
Drugbank | − 5.69 | − 47.8 | Not stable | Beta-2 adrenergic receptor agonist. Used in treatment of asthma and COPD (Wishart et al. 2018) | |||
| 7 |
Zinc gluconate |
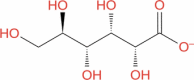
|
Drugbank | − 8.525 | − 18.19 | Not stable | Treating diarrheal episodes in children and reduced duration of common cold (Wishart et al. 2018) | |||
| 8 | Salbutamol |
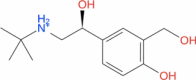
|
Drugbank | − 5.097 | − 40.86 | Not stable | A short-acting, beta-2 adrenergic receptor agonist (Wishart et al. 2018) | |||
| 9 | Fenoterol |
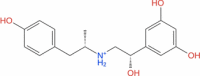
|
Drugbank | − 6.543 | − 45.18 | Not stable | Adrenergic beta-2 receptor agonist (Wishart et al. 2018) | |||
| 10 | Nelarabine |
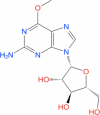
|
Drugbank | − 6.307 | − 36.86 | Not stable | Anti-neoplastic agent (Wishart et al. 2018) | |||
| 11 | Ioxilan |
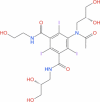
|
Drugbank | − 8.451 | − 35.28 | Not stable | Tri-iodinated diagnostic contrast agent (Wishart et al. 2018) | |||
| 12 | Edoxudine |
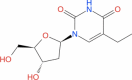
|
Drugbank | − 5.941 | − 33.63 | Stable | Deoxy-thymidine analog and Anti-viral agent [26] | |||
| 13 | Floxuridine |
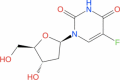
|
Drugbank | − 5.125 | − 33.27 | Not stable | Anti-neoplastic and antimetabolite agent (Wishart et al. 2018) | |||
| 14 | Brivudine |
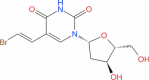
|
Drugbank | − 5.8 | − 30.61 | Stable | Anti-viral (Wishart et al. 2018) | |||
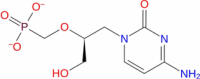
| 15 | Remdesivir |
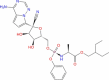
|
Drugbank | − 5.795 | − 40.01 | Stable | Anti-viral agent, a potential treatment for Ebola and shown to be effective against COVID-19 (Wang et al. 2020; Wishart et al. 2018) | |||
| 16 | Ritonavir |
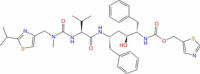
|
Drugbank | − 2.778 | − 27.75 | Not stable | − 2.25 | − 55.25 | Not stable | Anti-viral agent (Wishart et al. 2018) |
| 17 | Brincidofovir |

|
Drugbank | − 2.024 | − 27.75 | Not stable | Anti-viral agent (Wishart et al. 2018) | |||
| 18 | Galangin |
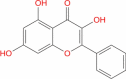
|
Natural product | − 3.532 | − 17.69 | Not stable | − 2.278 | − 22.99 | Not stable | Dietary flavonoid having anticancer properties (Chen et al. 2019) |
| 19 | Glycyrrhizic Acid |
|
Drugbank | − 4.609 | − 23.59 | Not stable | − 3.643 | − 27.06 | Stable | Anti-inflammatory, anti-diabetic, anti-oxidant, anti-tumor and anti-viral properties (Ming and Yin 2013) |
| 20 | SN00003849 |
|
Super natural database |
− 6.295 | − 51.08 | Stable | Plant product | |||
| 21 | SN00220639 |
|
Super natural database |
− 12.25 | − 50.79 | Not stable | − 10.59 | − 67.94 | Not stable | Plant product |
| 22 | SN00103215 |
|
Super natural database |
− 5.264 | − 47.66 | Not stable | Plant product | |||
| 23 | SN00003832 |
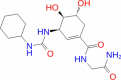
|
Super natural database |
− 6.308 | − 47.03 | Stable | Plant product | |||
| 24 | SN00216190 |
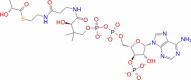
|
Super natural database |
− 7.78 | − 46.11 | Stable | Plant product | |||
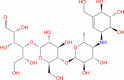
| 25 | Acarbose |
|
Drugbank | − 8.365 | − 28.35 | Stable | Anti-diabetic (Wishart et al. 2018) | |||
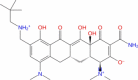
| 26 | Omadacycline |
|
Drugbank | − 6.025 | − 35.52 | Not stable | Antibiotic (Wishart et al. 2018) | |||
| 27 | Iopromide |
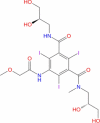
|
Drugbank | − 4.73 | − 31.58 | Stable | Used as a contrast agent (Wishart et al. 2018) | |||
| 28 | SN00037405 |
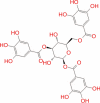
|
Super natural database |
− 9.177 | − 61.45 | Not stable | Plant product | |||
| 29 | SN00161170 |
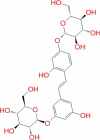
|
Super natural database | − 6.676 | − 57.94 | Not stable | Plant product | |||
| 30 | SN00038342 |
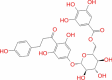
|
Super natural database |
− 6.066 | − 51 | Not stable | Plant product | |||
| 31 | SN00156190 (Gingerenone) |
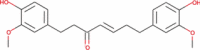
|
Super natural database |
− 4.39 | − 22.27 | Not done | − 2.578 | − 36.38 | Not done | Anti-obesity, Anti-inflammatory and antibiotic (Suk et al. 2017; Rampogu et al. 2018) |
| 32 | SN00002189 (Shogaol) |
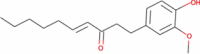
|
Super natural database |
− 2.64 | − 36.18 | Not done | Anticancer, anti-oxidant, antimicrobial, anti-inflammatory anti-allergic and antibiotic (Rampogu et al. 2018; Semwal et al. 2015) | |||
Molecules marked in bold have been discussed in the text.
MD simulation of protein–ligand complexes
The best compounds from docking analysis were further subjected to 20 ns of MD simulation to assess the stability of protein–ligand complex. The interactions between protein and ligand were designated as stable if there was less structural variations and a high percentage of hydrogen bonds or hydrophobic interactions with various residues of the protein at the docked site throughout the course of the simulations. Among the FDA-approved drugs docked at the deep site, Esculin is an example of stable complexes, while Zinc-gluconate is an example of an unstable complex. Figure 3a and supplementary figure 2a show the interaction of Esculin with NSP1 in the docking pose, where Esculin interacts mainly with Arg62, Ser63, Ala68 and His72. MD simulation of NSP1-deep-Esculin complex for 20 ns revealed the stability of the complex as assessed by the RMSD (root mean square deviation) plot. Residues in the secondary structure are expected to have fewer fluctuations than residues in the loop regions and the trend is followed for NSP1 which shows high RMSF between residues 62-76 which form a loop and also interact with Esculin (supplementary figure 2b). Arg62, Ser63, Ala68 and His72 (major interacting residues in the docking pose) interact mainly through H-bond interactions with Esculin. Met74 was also found to interact with Esculin mainly through H-bond (figure 3b). Few other residues interact with Esculin, but with less amount of simulation time. Further details of these interactions are provided in supplementary figure 2c and d.
Figure 3.

Docking and MD simulation results for NSP1-deep-Esculin. (a) Esculin-NSP1 interactions after XP docking. (b) Interaction types and Interacting residues of NSP1 with Esculin over simulation time. Normalized stacked bars indicate the fraction of simulation time for which a particular type of interaction was maintained. Values more than 1.0 suggest that the residue forms multiple interactions of the same subtype with ligand (Esculin).
Results of similarly detailed analysis for all the ligands, as in table 1, are provided in Supplementary figures S1–S35. Other promising lead compounds, among the FDA-approved ligands docked at the deep site of NSP1, are Cidofovir (supplementary figure 3), Remdesivir (the drug under investigatory group; supplementary figure 17), Brivudine (supplementary figure 16) and Edoxudine (supplementary figure 14). In the case of FDA-approved drugs docked at the shallow site, acarbose was found to be the most stable ligand. It interacts mainly with Arg32, Leu77 and Asn115 through H-bond and water bridge interactions (supplementary figure 31).
Amongst compounds from SuperNatural database docked at the deep binding site of NSP1, SN00003849 interacts mainly with Arg62, Arg66, Ala68, Gly71, His72 and Met74 (figure 4a). Further, the NSp1-SN00003849 complex was found to be stable, as suggested by RMSD plot of 20ns MD simulation (supplementary figure 25a). Residues interacting with SN00003849 are similar to that of Esculin (supplementary figure 25b). These include Arg62, Arg66, Gly71, His72 and Met74 interacting mainly through H-bond and water bridge interactions (figure 4b). Arg61, Gly71 and Met74 interact with the same atom of SN00003849 for more than 80% of simulation time (supplementary figure 25c). At any point during the simulation, the minimum number of contacts between SN00003849 and NSP1 is more than four, suggesting the strong interaction at the binding site (supplementary figure 25d). SN00003849 also has the highest binding energy as per MM-GBSA calculation (table 1). SN00003849 is structurally similar to terpene/steroid and can be classified as proto and pseudo alkaloids. SN00003832 and SN00216190 also form a stable complex with NSP1 at the deep site (supplementary figures 29 and 30).
Figure 4.

Docking and MD simulation results for NSP1-deep-SN00003849. (a) SN00003849-NSP1 interactions after XP docking. (b) Interaction types and Interacting residues of NSP1 with SN00003849 over simulation time. Normalized stacked bars indicate the fraction of simulation time for which a particular type of interaction was maintained. Values more than 1.0 suggest that the residue forms multiple interactions of the same subtype with ligand (SN00003849).
For shallow binding sites, none of the compounds in supernaturaldb form complex that are as stable as that for the deep binding site. Natural compounds (entries 18, 19, 32 and 33) are derived from herbal plants, well-known to treat coughs and viral fevers. Another FDA-approved compound is Glycyrrhizic acid which was ranked a bit lower for the deep site, as well as the shallow binding site during docking. The MD simulation was run for deep as well as shallow site complex of NSP1 with Glycyrrhizic acid. Glycyrrhizic acid bound at the shallow site interacts mainly with Arg32, Lys36, Arg113 and Asn115 in the docked pose (figure 5a).
Figure 5.

Docking and MD simulation results for NSP1-shallow-Glycyrrhizic acid. (a) Glycyrrhizic acid-NSP1 interactions after XP docking. (b) Interaction types and Interacting residues of NSP1 with Glycyrrhizic acid over simulation time. Normalized stacked bars indicate the fraction of simulation time for which a particular type of interaction was maintained. Values more than 1.0 suggest that the residue forms multiple interactions of the same subtype with ligand (Glycyrrhizic acid).
The MD simulation of 20 ns suggested that the complex is stable as per RMSD plot (supplementary figure 24a). NSP1-rmsf plot indicates that the few residues of α helix along with residues at the N and C-termini are also involved in an interaction with Glycyrrhizic acid (supplementary figure 24b). Major interacting residues of NSP1 are the same as those in the docking pose (figure 5b). Atom wise interactions of Glycyrrhizic acid with NSP1 has been shown in figure 24c. Similar to SN00003849, Glycyrrhizic acid also maintains at least 4 contacts with NSP1 over the entire course of simulation time (figure 24d). Glycyrrhizic acid bound at the deep site is not stable (supplementary figure 23). Interestingly, Glycyrrhizic acid is from the plant Mulethi or Liquorice (also referred to as Yashtimadhura (Glycyrrhiza glabra), which is a natural herb for cough and has expectorant properties. It can also reduce infection of the upper respiratory tract. It may reduce throat irritation and helps cases of a chronic cough.
The ADME related properties for compounds like Gingerenone, Shogaol and SN00103215 follow Lipinski’s rule of five, while others violate either one or 3 of the 4 rules of Lipinski’s rule of five (supplementary table 1). Qikprop also summarizes the drug-likeness of compounds by comparing the properties of query compounds with known drugs. Gingerenone, Shogaol and SN00103215 are observed to not retain any star (please see Methods) suggesting strong drug-likeliness of these compounds. The water solubility, a key parameter required for absorption and distribution of the compounds, ranges from − 5.101 to 0.261 and it falls within the acceptable range. Cell permeability is important for metabolism and it was found that most of the compounds have poorly predicted cell permeability. However, the cell permeability predictions are for non-active transport. Gingerenone and Shogaol also show a high percentage of oral absorption (supplementary table 1).
Discussion
COVID-19 outbreak has turned into a pandemic, which makes the identification of new target molecules, repurposing of drugs and designing vaccine an imminent necessity. Since the outbreak, many studies have been conducted along these lines (Chakraborti et al. 2020; Gordon et al. 2020; Narayanan and Nair 2020; Wu et al. 2020a) (Manfredonia et al. 2020; Quimque et al. 2020). We used NSP1 protein as our target protein. It shows 86 % identity with SARS NSP1. A model of COVID-19 NSP1 was made using SARS NSP1 as a template. Please note: During the submission process for this manuscript, the structure of NSP1 with the ribosome has been solved by another group (Thoms et al. 2020). This is not published yet, nor any mention of PDB id submissions. This preprint showed the role of NSP1 in translational shutdown and innate immune evasion). Understanding the genetic diversity of a viral gene is key in understanding evolutionary pressure and add one more dimension to virtual screening (Kasibhatla et al. 2020; Somasundaram et al. 2020). NSP1 is evolving with key residues being conserved. Virtual screening, against NSP1 protein, suggests a list of FDA-approved drugs and natural compounds against the deep and shallow binding site on NSP1. Deep and shallow binding sites include functionally important residues such as H81, H83, R124 and R43, K47, E91, R124, K125 respectively (AR Jauregui et al. 2013). R124 has shown to be important for NSP1 to interact with viral mRNA 5′-UTR region which prevents viral mRNA from NSP1 mediated mRNA degradation (Kamitani et al. 2006) (Note: Residue number in modelled structure starts with 12th residue of the sequence). Docking and MMGBSA scores suggest the binding potential of these compounds towards NSP1. Further, MD simulation of the selected compounds in complex with NSP1 ensures that some of these hits form stable interactions with NSP1.
Esculin, Cidofovir, Edoxudine, Brivudine and Remdesivir were found to form a stable complex with NSP1, among FDA-approved drugs binding at deep site of NSP1. Esculin is a glucoside and naturally occurs in barley, horse chestnut, etc. It is given to improve capillary permeability and fragility and has been reported to inhibit collagenase and hyaluronidase enzymes. This molecule has been shown to have antioxidant and anti-inflammatory activity (Wishart et al. 2018). This suggests the ability of esculin to not only inhibit NSP1 activity but also being effective against secondary symptoms such as inflammation. Cidofovir is a known anti-viral agent against CMV infection and acts via inhibition of CMV DNA Polymerase. Edoxudine is a deoxy-thymidine analog shown to be effective against herpes simplex virus type 1 and type 2. It acts as a competitive inhibitor of viral DNA polymerase in its phosphorylated form. Edoxudine is initially phosphorylated by viral thymidine kinas and it is specifically incorporated in viral DNA. Edoxudine has been discontinued. Brivudine is an organic compound and a pyrimidine 2′-deoxyribonucleosides analog. This is used in the treatment of herpes zoster, results from reactivation of varicella-zoster virus. Remdesivir is proposed as a potential antiviral drug against Ebola (Wishart et al. 2018). However, this molecule appears within the Investigational group of DRUGBANK. It is an adenosine-triphosphate analog and has shown effectivity against coronaviruses. A recent publication on COVD-19 treatment, shows it to be a potential drug along with chloroquinone (Wang et al. 2020). Remdesivir is an RNA polymerase inhibitor. Hence our study suggests an additional mechanism of action for this drug. An interesting and unexpected molecule among this list is lactose. Lactose is a disaccharide of glucose and galactose and used as nutrient supplement. Derivatives of lactose, 3′-sialyllactose have been shown to have broad-spectrum neutralization activity against avian influenza viruses in chickens (Pandey et al. 2018). Further investigation is necessary to check the anti-viral property of lactose against coronavirus.
Acarbose, Iopromide and Glycyrrhizic Acid form stable interactions with the shallow binding site of NSP1. Acarbose is an alpha-glucosidase inhibitor and administered to patients with non-insulin-dependent diabetes mellitus (Wishart et al. 2018). As the death rate among COVD-19 patients with diabetes is high, the anti-diabetic nature of acarbose can be highly useful in the treatment regime. Iopromide is a contrast agent, used in radiographic studies. Glycyrrhizic acid is a plant product obtained form Mulethi or Liquorice (also referred as Yashtimadhura (Glycyrrhiza glabra)). It has been shown to have anti-inflammatory, anti-diabetic, anti-oxidant, anti-tumor and anti-viral properties (Ming and Yin 2013). These properties suggest Glycyrrhizic acid to be of high importance in COVID-19 treatment.
We next pursued virtual screening against supernaturaldb – a database of 3,25,287 natural small molecules (giving rise to 5,03,604 confirmations). Virtual screening for the shallow site also predicted natural products with high medicinal value such as Gingerenone A (SN00156190) and Shogaol (SN00002189), but with lower docking score (table 1). Gingerenone A has anti-obesity, anti-inflammatory and antibiotic properties (Rampogu et al. 2018; Suk et al. 2017), whereas Shogaol is anticancer, anti-oxidant, antimicrobial, anti-inflammatory anti-allergic and antibiotic in nature (Rampogu et al. 2018; Semwal et al. 2015). MD simulation was not performed for the above two because of their lower rank but can be tested further. Molecules like Galangin, Gingerenone and Shaogaol are reported in high quantities in the medicinal plant, Sitharathai (Alpinia Officinarum; a form of ginger, also referred as ‘Kulanjan’ (Chen et al. 2019) which has been used for bronchial infections, as a carminative and recently recognized for its antiviral properties (Pillai and Young 2017). Extracts from herbal plants provide a host of secondary metabolites which could have a combinatorial effect to reduce the viral load, once consumed in the proper manner.
Other hits from supernaturaldb include compounds SN00003849, SN00003832 and SN00216190, which were found to have stable interactions with the deep binding site of NSP1 as suggested by docking and MD simulation. Therefore, along with FDA-approved drugs which will constitute the treatment by repurposing, these new natural compounds can also be tested for their activity against COVID-19.
Conclusion
Virtual screening helps in the identification of novel drug candidates and repurposing of known drugs. The current pandemic caused by SARS-Cov2. In order to assist in the development of a cure, we have targeted NSP1 protein of this virus and screened known drugs and natural compounds against it. In this effort, we have identified known antiviral compounds like Remdesivir and Edoxudine. Other drugs, like Esculin and Acarbose which are not antiviral, but are used as anti-inflammatory and antidiabetic (respectively) were also identified. These FDA-approved drugs can be considered as potential candidates for drug repurposing. Natural compounds like Glycyrrhizic acid (entry 19 in table 1) from Liquorice and Galangan, Gingeronone and Shogaol (entries 18, 32 and 33 in table 1) from Sitharathai, were also found to be interacting with NSP1. These compounds can be considered as novel drug candidates against COVID-19. We find these results to be encouraging and hopefully useful immediately to the community and follow-up validation by other researchers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank NCBS (TIFR) for infrastructural facilities. The authors thank Dr. Radhika Venkatesan for useful discussions. RS would like to acknowledge her JC Bose fellowship (JC Bose fellowship (SB/S2/JC-071/2015)) from the Science and Engineering Research Board, India.
Abbreviations
- NSP1
Non-structural protein 1
- SARS-CoV2
Severe acute respiratory syndrome coronavirus 2
Footnotes
This article is part of the Topical Collection: COVID-19: Disease Biology & Intervention.
Corresponding editor: Sreenivas Chavali
Joint first authors
References
- Banerjee P, Erehman J, Gohlke B-O, Wilhelm T, Preissner R, et al. Super Natural II—a database of natural products. Nucleic Acids Res. 2015;43:D935–D939. doi: 10.1093/nar/gku886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, et al. 2006 Scalable algorithms for molecular dynamics simulations on commodity clusters. SC ‘06: Proceedings of the 2006 ACM/IEEE conference on Supercomputing 10.1145/1188455.1188544
- Brister JR, Ako-Adjei D, Bao Y, Blinkova O. NCBI viral Genomes resource. Nucleic Acids Res. 2015;43:D571–D577. doi: 10.1093/nar/gku1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, et al. BLAST+: Architecture and applications. BMC Bioinform. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017;39:529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborti S, Bheemireddy S and Srinivasan N 2020 Repurposing drugs against main protease of SARS-CoV-2: mechanism based insights supported by available laboratory and clinical data. 10.26434/chemrxiv.12057846.v2 [DOI] [PubMed]
- Chen CY, Lin CL, Kao CL, Yeh HC, Li HT, et al. Secondary Metabolites from the Rhizomes of Alpinia officinarum. Chem. Nat. Compounds. 2019;55:1176–1178. doi: 10.1007/s10600-019-02927-w. [DOI] [Google Scholar]
- Deftereos S, Giannopoulos G, Vrachatis DA, Siasos G, Giotaki SG, et al. Colchicine as a potent anti-inflammatory treatment in COVID-19: can we teach an old dog new tricks? Eur Heart J Cardiovasc: Pharmacother; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, et al. Comparative protein structure modeling using modeller. Curr: Protocols Bioinform; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K et al. 2020 A SARS-CoV-2-Human protein-protein interaction map reveals drug targets and potential drug-repurposing. bioRxiv10.1101/2020.03.22.002386
- Halgren T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Design. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- Halgren TA. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inform. Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, et al. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Hatcher EL, Zhdanov SA, Bao Y, Blinkova O, Nawrocki EP, et al. Virus Variation Resource-improved response to emergent viral outbreaks. Nucleic Acids Res. 2017;45:D482–D490. doi: 10.1093/nar/gkw1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, et al. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: Viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathogens. 2011;7:e1002433. doi: 10.1371/journal.ppat.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys DD, Friesner RA, Berne BJ. A multiple-time-step molecular dynamics algorithm for macromolecules. J. Phys. Chem. 1994;98:6885–6892. doi: 10.1021/j100078a035. [DOI] [Google Scholar]
- Jauregui AR, Savalia D, Lowry VK, Farrell CM, Wathelet MG. Identification of residues of SARS-CoV nsp1 that differentially affect inhibition of gene expression and antiviral signaling. PLoS ONE. 2013;8:1–11. doi: 10.1371/journal.pone.0062416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus NSP1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W, Narayanan K, Huang C, Lokugamage K, Ikegami T, et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Nat. Acad. Sci. USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasibhatla SM, Kinikar M, Limaye S, Kale MM, Kulkarni-Kale U. Understanding evolution of SARS-CoV-2: A perspective from analysis of genetic diversity of RdRp gene. J. Med. Virol. 2020 doi: 10.1002/jmv.25909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Law AHY, Lee DCW, Cheung BKW, Yim HCH, Lau ASY. Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J. Virol. 2007;81:416–422. doi: 10.1128/JVI.02336-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredonia I, Nithin C, Ponce-Salvatierra A, Ghosh P, Wirecki TK et al. 2020 Genome-wide mapping of therapeutically-relevant SARS-CoV-2 RNA structures. bioRxiv10.1101/2020.06.15.151647 [DOI] [PMC free article] [PubMed]
- Martyna GJ, Klein ML and Tuckerman M 1992 Nos \’ e -Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys.
- Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994 doi: 10.1063/1.467468. [DOI] [Google Scholar]
- Masters PS. The molecular biology of coronaviruses. Adv. Virus Res. 2006;65:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming LJ, Yin ACY. Therapeutic effects of glycyrrhizic acid. Nat. Prod. Commun. 2013;8(3):415–418. [PubMed] [Google Scholar]
- Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, et al. Severe Acute Respiratory Syndrome Coronavirus nsp1 suppresses host gene expression, including that of type i interferon, in infected cells. J. Viro. 2008;82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K, Ramirez SI, Lokugamage KG, Makino S. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015;202:89–100. doi: 10.1016/j.virusres.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan N and Nair DT 2020 Vitamin B12 may inhibit RNA-dependent-RNA polymerase activity of nsp12 from the SARS-CoV-2 virus. Preprints 10.20944/preprints202003.0347.v1 [DOI] [PMC free article] [PubMed]
- Pandey RP, Kim DH, Woo J, Song J, Jang SH, et al. Broad-spectrum neutralization of avian influenza viruses by sialylated human milk oligosaccharides: In vivo assessment of 3′-sialyllactose against H9N2 in chickens. Sci. Rep. 2018;8:2563. doi: 10.1038/s41598-018-20955-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferle S, Schpf J, Kgl M, Friedel CC, Mller MA, et al. The SARS-Coronavirus-host interactome: Identification of cyclophilins as target for pan-Coronavirus inhibitors. PLoS Pathogens. 2011 doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40:D593–D598. doi: 10.1093/nar/gkr859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai MK, Young DJ. Therapeutic potential of Alpinia officinarum. Mini Rev. Med. Chem. 2017;18:1220–1232. doi: 10.2174/1389557517666171002154123. [DOI] [PubMed] [Google Scholar]
- Piplani S, Singh PK, Winkler DA and Petrovsky N 2020 In silico comparison of spike protein-ACE2 binding affinities across species; significance for the possible origin of the SARS-CoV-2 virus. arXiv:2005.06199
- Quimque MTJ, Notarte KIR, Fernandez RAT, Mendoza MAO, Liman RAD, et al. Virtual screening-driven drug discovery of SARS-COV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1776639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampogu S, Baek A, Gajula RG, Zeb A, Bavi RS, et al. Ginger (Zingiber officinale) phytochemicals-gingerenone-A and shogaol inhibit SaHPPK: Molecular docking, molecular dynamics simulations and in vitro approaches. Ann. Clin. Microbiol. Antimicrob. 2018;17:16. doi: 10.1186/s12941-018-0266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Semwal RB, Semwal DK, Combrinck S, Viljoen AM. Gingerols and shogaols: Important nutraceutical principles from ginger. Phytochemistry. 2015;117:554–568. doi: 10.1016/j.phytochem.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Shannon CE. A mathematical theory of communication. The Bell System Technical Journal. 1948;27:379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- Shen Z, Wang G, Yang Y, Shi J, Fang L, et al. A conserved region of nonstructural protein 1 from alphacoronaviruses inhibits host gene expression and is critical for viral virulence. Journal of Biological Chemistry. 2019;294:13606–13618. doi: 10.1074/jbc.RA119.009713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram K, Mondal M and Lawarde A 2020 Genomics of Indian SARS-CoV-2: Implications in genetic diversity, possible origin and spread of virus. 10.1101/2020.04.25.20079475
- Suk S, Kwon GT, Lee E, Jang WJ, Yang H, et al. Gingerenone A, a polyphenol present in ginger, suppresses obesity and adipose tissue inflammation in high-fat diet-fed mice. Mol. Nutr. Food Res. 2017 doi: 10.1002/mnfr.201700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. Severe Acute Respiratory Syndrome Coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J. Virol. 2012;86:11128–11137. doi: 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T et al. 2020 Structural basis for translational shutdown and immune evasion by the NSP1 protein of SARS-CoV-2. bioRxiv10.1101/2020.05.18.102467 [DOI] [PMC free article] [PubMed]
- Wang M, Cao R, Zhang L, Yang X, Liu J, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004;136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceut: Sinica B; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zhao S, Yu B, Chen Y-M, Wang W, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zst R, Cervantes-Barragn L, Kuri T, Blakqori G, Weber F, et al. Coronavirus non-structural protein 1 is a major pathogenicity factor: Implications for the rational design of coronavirus vaccines. PLoS Pathogens. 2007;3:e109. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.