

Abstract
Water is typically considered to be insoluble in alkanes. Recently, however, monomerically dissolved water in alkanes has been shown to dramatically impact the structure of hydrogen-bonded supramolecular polymers. Here, we report that water in methylcyclohexane (MCH) also determines the outcome of combining a Michael reaction with a porphyrin-based supramolecular system. In dry conditions, the components of the reaction do not affect or destabilize the supramolecular polymer, whereas in ambient or wet conditions the polymers are rapidly destabilized. Although spectroscopic investigations show no effect of water on the molecular structure of the supramolecular polymer, light scattering and atomic force microscopy experiments show that water increases the flexibility of the supramolecular polymer and decreases the polymer length. Through a series of titrations, we show that a cooperative interaction, involving the coordination of the amine catalyst to the porphyrin and complexation of the substrates to the flexible polymers invokes the depolymerization of the aggregates. Water crucially stabilizes these cooperative interactions to cause complete depolymerization in humid conditions. Additionally, we show that the humidity-controlled interference in the polymer stability occurs with various substrates, indicating that water may play a ubiquitous role in supramolecular polymerizations in oils. By controlling the amount of water, the influence of a covalent chemical process on noncovalent aggregates can be mediated, which holds great potential to forge a connection between chemical reactivity and supramolecular material structure. Moreover, our findings highlight that understanding cooperative interactions in multicomponent noncovalent systems is crucial to design complex molecular systems.
Introduction
Over the last two decades, a profound understanding of the thermodynamic aspects of supramolecular polymers has been achieved and as a result a large variety of noncovalent structures can be synthesized.1,2 Despite being dynamic, these structures typically change their properties only as a response to an external stimulus and do not show any temporal change in structure or chemical functionality without such stimuli.3 In contrast, in natural systems, spatiotemporal control over supramolecular structure and reactivity is ubiquitous and leads to the complex behavior observed in life.4,5 Recently, chemists have endeavored to mimic these natural reaction networks to obtain out-of-equilibrium systems.6−12 Inspired by the chemistry of life, these systems are often designed in aqueous environments. Although aqueous environments provide great opportunities to control chemical reactions, it is often difficult to design and synthesize controlled supramolecular structures in these polar media.13−15
In apolar media, the well-defined character of noncovalent interactions such as charge-transfer complexation and hydrogen-bonding interactions has permitted the design of ordered and well-organized supramolecular polymers. Such structures are in thermodynamic equilibrium, giving materials that do not evolve over time.1,16 In recent years, several exciting systems have been reported that utilize kinetically trapped monomers or aggregation states to install temporal behavior in supramolecular polymers. In seminal work by Takeuchi, Sugiyasu, and co-workers, bisamidated porphyrin monomers were shown to readily assemble into small isodesmically formed J-aggregates, which gradually converted to thermodynamically stable, long H-aggregated supramolecular polymers. Upon sonication of the isodesmically formed J-aggregates, small H-aggregated seeds that act as nuclei for seeded polymerizations were formed.17 Similar behavior has been shown by groups of Sugiyasu, Würthner, Ogi, and Sánchez for various other porphyrins18−20 as well as for a wide range of other monomers.21−26 By controlling the length of the flexible spacer between the hydrogen-bonding groups at the monomer’s periphery and its rigid core, control can be achieved over the time-dependency of such living supramolecular polymerizations.27 Aida and co-workers used a similar approach to design an initiated living supramolecular polymerization. In this approach, a non-hydrogen bonded initiator can activate an intramolecularly hydrogen-bonded dormant monomer to form highly monodisperse supramolecular polymers.28 These results show that by controlling the flexibility of the monomers, great temporal and structural control over supramolecular polymerization can be obtained. However, these systems do not incorporate in situ chemical conversion that is required to obtain lifelike responses observed in chemical reaction networks and, as a result, possible applications of these systems remain limited.
To extend temporal control in supramolecular polymerizations from polymer structure to additional chemical functionality, the polymerization process has to be combined with chemical reactions.29 By enabling the combination of supramolecular polymerizations with control over chemical functionality, many new material behaviors, including feedback, adaptation, and regulation can be obtained. However, only a handful of these systems have been reported. The first examples by the groups of Ulijn, and Van Esch and Eelkema showed that the propensity of monomers to polymerize supramolecularly can be controlled in situ by reversible esterification and hydrolysis, giving rise to transient supramolecular networks.6,7,30 The groups of Hermans and George showed that also redox reactions can be used to control supramolecular aggregation.10−12,31−33 The fine temporal control over the aggregation state that these aqueous systems offer comes often at the cost of a decrease in control over polymerization mechanism and polymer morphology,7 which renders complete control over the material properties more challenging.
Since complex material responses in supramolecular systems can only be realized when both structure and chemical conversion are precisely controlled, there is a need to realize a connection between controlled chemical reactivity and supramolecular structure. In the past, we have shown that the interaction between basic amines and Zn-1 (Scheme 1a) can induce counterintuitive behavior in these multicomponent systems, such as dilution induced self-assembly,34 photoinduced depolymerization,35 and microfluidic separation of various aggregate morphologies.36 Encouraged by our previous results, where the interactions between amines and zinc center were employed to switch the aggregation of the porphyrin and the pyridines, we here combine the supramolecular polymerization with an amine-catalyzed Michael reaction. In our attempts to realize such a system, we aim to combine supramolecular polymers of Zn-1 with a chemically relevant and biologically interesting base-catalyzed Michael reaction between thiols and maleimides. In this system, the zinc metalated porphyrin molecule is particularly interesting, since the metal center offers the ability to sequester amine-based organocatalysts, thus forging a connection between supramolecular structure and chemical reactivity.
Scheme 1.

(a) Chemical structure of Zn-1, Zn-Me, and H2-1. (b) Base-catalyzed Michael addition of thiophenol to N-propylmaleimide in MCH. (c) Various piperidine-based catalysts used in the Michael reaction.
Interestingly, we found that, in the absence of monomerically dissolved water in the MCH solvent, no interference of the amine-catalyzed reaction with the supramolecular polymer is observed. Surprisingly, when the system is studied in ambient environments, we found that small amounts of water in MCH can dramatically impact the polymer stability. With several titrations, light scattering and atomic force microscopy (AFM) experiments, we show that the polymer length and rigidity are decreased when water, reaction substrates, and catalyst are complexed to the supramolecular polymer. We propose that this effect arises from the binding of water and the reagents to the macrodipole at the chain ends of the polymer. The chain ends are subsequently stabilized by complexation of the catalyst to metal center of the porphyrin. Several other Michael substrates illustrate the generality of this effect.
Results and Discussion
Base-Catalyzed Michael Reaction and Water Depolymerize Zn-1 Polymers
To combine supramolecular polymers of Zn-1 with a chemical reaction, we set out to identify chemical conversions that occur at reasonable time scales in apolar media like MCH. Ultimately, we identified the base-catalyzed Michael addition of thiophenol, PhSH, to N-propyl maleimide, NPrMal, as a promising reaction for the desired system (Scheme 1b). Because the apolar MCH solution does not stabilize the charged reaction intermediates, no reaction occurs without the use of an organic base as catalyst. To study the effect of the catalyst on the rate of the reaction, we studied the Michael reaction with various catalysts by monitoring the decrease in absorbance of the conjugated system of NPrMal as the reaction proceeds (Scheme 1c and Figure 1). Interestingly, pyridine is not sufficiently basic to catalyze the Michael reaction and the non-nucleophilic 2,2,6,6-tetramethylpiperidine, Me4Pip, is too bulky to show high rates of conversion. By decreasing the steric bulk around the aliphatic amine, good catalytic activities were obtained, with piperidine and 2-methylpiperidine, MePip, being the most active catalysts.
Figure 1.
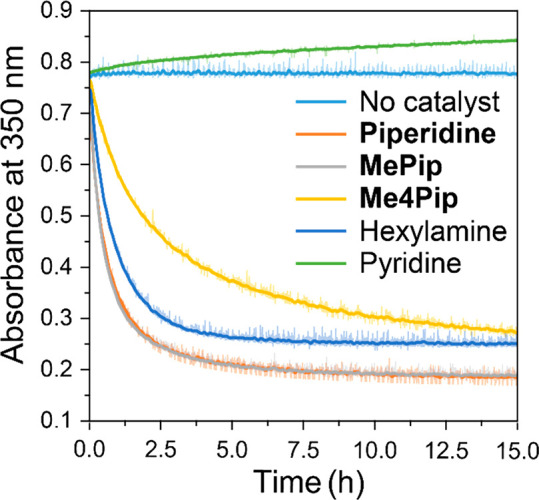
Time-dependent absorbance of NPrMal at 350 nm of reaction mixtures containing 10 mM NPrMal and 10 mM PhSH with 0.5 mM of the various catalysts at 20 °C in MCH.
To control the effect of the amine on the (de)polymerization of Zn-1 by its binding to the zinc core, the association constant of the catalyst should not be too high or too low. Therefore, an ideal catalyst has an intermediate association constant, between 104 and 105 M–1, similar to the functional ligands reported earlier.34,35 The association constants of all four investigated aliphatic amine catalysts to the nonpolymerizing Zn-Me model compound were determined through UV–vis titrations (see the Supporting Information for details). We found that the association constant of MePip (Ka = 20.6 × 103 M–1) is in the range of the previously reported functional ligands, while the association constant of piperidine (Ka = 949 × 104 M–1) is too high and those of Me2Pip and Me4Pip (Ka = 6.63 × 103 and 168 M–1, respectively) are too low. Hence, MePip does not depolymerize the polymers at the concentrations of both Zn-1 and MePip used in this work while still showing catalytic activity and is thereby an interesting candidate for the proposed multicomponent system.
After identifying a suitable chemical reaction and amine catalyst, we performed the Michael reaction in the presence of Zn-1 polymers. When studying the Zn-1 polymers in the reaction medium containing PhSH, NPrMal, and MePip, the initial results obtained were irreproducible and inexplicable. Similar to an earlier report,37 however, we serendipitously found that ppm levels of water, which amount to millimolar concentrations, play a crucial role in controlling the stability of the Zn-1 polymers in the reaction medium. This observation, which hints at a ubiquitous, yet poorly understood role of water in supramolecular polymers, prompted us to investigate this role of water in Zn-1 polymers in detail.
The low solubility of Zn-1 polymers necessitated the use of small amounts (<0.5 vol %) of CHCl3 in the MCH solvent to retain the polymers in solution, denoted here as MCH*. Consequently, all further experiments were performed in the presence of a small amount of CHCl3 cosolvent. When Zn-1, the NPrMal and PhSH reagents, and MePip catalyst are dissolved in dry MCH*, the Michael reaction occurs and the MA product is gradually formed, as indicated by the decrease in absorbance of NPrMal at 300 nm (Figure 2a,b). During the reaction, no change is observed in the CD-active absorption peak at 392 nm, which originates from the helically ordered, H-aggregated Zn-1. The absence of a change in the absorption of the Zn-1 aggregates indicates that the polymers are stable structures under these conditions (see also Figure S6a).38 Hence, reaction substrates and catalyst do not interfere with the supramolecular polymer in dry MCH*.
Figure 2.

Absorbance of 10 μM solutions of Zn-1 with 4 mM NPrMal, 4 mM PhSH, and 0.5 mM MePip in dry MCH*(a) and ambient MCH* (c) and the time dependency of the absorbance at 300, 392, and 425 nm for solutions in dry MCH* (b) and ambient MCH* (d).
Conversely, upon preparing a reaction mixture in the presence of Zn-1 in ambient MCH* containing 12.5 ppm of monomerically dissolved water,39 a strong influence of the reaction on the polymer is observed. We note here that the exact concentration of water is strongly determined by the environmental humidity. Due to equilibration of the dissolved water in MCH* with moisture from the air, precise control over the dissolved water content is hard to achieve. In the ambient environment, the Michael reaction proceeds with a slightly higher rate, as indicated by the slightly sharper decrease in absorbance of the maleimide at 300 nm (Figure 2c,d). The absorption band at 392 nm, however, shows a rapid decrease in intensity, coinciding with a strong decrease in CD intensity and an increase of the absorbance at 425 nm (Figures 2c,d and S6b). These spectral changes indicate that the helical, H-aggregated supramolecular polymers of Zn-1 depolymerize to free monomers, dimers or small aggregates in the presence of reaction substrates and catalyst.34 Interestingly, control experiments in which one or more of the components of the Michael reaction are absent, no destabilization of the supramolecular polymers was observed (Figures S8–S11). Combined, these results show that unexpected additive effects between water and one or more of the components of the Michael reaction rapidly destabilize the Zn-1 supramolecular polymers. Since these cooperative interactions crucially impact the stability of the supramolecular polymer, they must be fully understood before any additional complexity through switching of the supramolecular system can be introduced. Therefore, we set out to investigate the effect of water and the additional components on the supramolecular polymers.
Scattering and Microscopy Show That Water and Substrates Reduce Bundling
Although in the absence of one of the reaction components no change in the absorbance of the polymers is observed, this merely indicates that the number of polymerized monomers remains the same. The interactions of water and the substrates may still lead to destabilization of the polymers and a reduction in polymer length.40,41 To probe changes in polymer length and morphology, we first investigated the effect of water on the polymers through AFM experiments (Figures 3a, S12, and 13). Here, large, bundled aggregates are observed for samples that are drop-casted from 10 μM solutions of Zn-1 in dry MCH*. Conversely, only small and ill-defined particles could be observed for samples drop-casted from ambient solutions, indicating that in ambient solutions, the polymers are present as dynamic single fibers or small bundles which cannot be measured with AFM. Similar observations have been made for benzene-1,3,5-tricarboxamides, suggesting that water-induced flexibility may be a general phenomenon in supramolecular polymers.37 Thus, water appears to considerably increase the flexibility and solubility of the supramolecular polymers in the MCH* solvent.
Figure 3.

(a) AFM micrographs of samples drop-casted from solutions of 10 μM Zn-1 in dry MCH*. [H2O] = 20.0 ppm. (b) Count rates at various scattering vectors, q, of 10 μM solutions of Zn-1 obtained from SLS experiments in ambient (circles) and dry (squares symbols) MCH. Error bars indicate one standard deviation. In the case of the ambient samples, [H2O] = 28.8 ppm, and in the case of the dry samples the concentration of water was below the detection limit of the Karl Fischer titrator.
The increased rigidity and size of the Zn-1 polymers in the absence of water, as suggested by the AFM results, is corroborated by static light scattering (SLS) experiments on ambient and dry solutions of Zn-1 (Figure 3b). The results show that the samples in ambient MCH* show a weaker angle dependency of the scattering intensity, compared to the samples in dry MCH*. Since no plateau could be observed in the q-region measured, we unfortunately cannot determine the absolute length of the polymers from the SLS results. However, a comparison of the UV–vis spectra of unfiltered solutions of Zn-1 with solutions that are filtered through various filters of various pore sizes does suggest that a considerable fraction of the polymers is longer than 1 μm (Figure S16), as has previously been observed.38
The change in slope observed in the scattering results suggests that water induces flexibility and dynamics in the supramolecular polymers. The induction of flexibility is additionally supported by the poor solubility of the Zn-1 supramolecular polymers in dry, pure MCH. In the absence of water, precipitation occurs within hours, necessitating the use of a small amount of CHCl3 cosolvent in the stock solutions. In contrast, solutions of Zn-1 prepared in ambient, pure MCH are stable for weeks, which can be attributed to the better solubility of the more flexible aggregates when water is present.
Combined, the SLS and AFM results strongly suggest that water plays a pivotal role in breaking up polymer bundles to form more flexible single polymer chains. The breaking up of the polymer bundles potentially allows the complexation of the reaction components to lead to a destabilization of the polymers and a reduction of the polymer length. To further study the destabilization of the polymers by the components of the reaction, titrations in which these components are added to the polymers in varying amounts were performed.
Cooperative Interactions between Water and Reaction Substrates Decrease Rigidity and Length
To identify which interactions are causing the depolymerization of Zn-1 by the substrates of the Michael reaction, we performed titrations of Zn-1 with the NPrMal and PhSH in the absence and presence of MePip in both dry and ambient MCH* (Figure 4). In all four cases, the CD signal at 392 nm, which indicates the presence of the polymeric H-aggregates of Zn-1, is probed at varying substrate concentration. The results show that in the absence of the catalyst, the polymers are stable in the presence of the reaction substrates (Figure 4, hollow symbols). However, in the presence of 0.5 mM MePip, the addition of NPrMal or PhSH leads to depolymerization (Figure 4, solid symbols). Moreover, the degree of depolymerization by the additives is strongly influenced by water. In ambient MCH*, containing 13.9 ppm water, the supramolecular polymers are completely destabilized when more than 8 mM NPrMal or 100 mM PhSH is present. In contrast, in dry MCH*, containing trace amounts of water that are not detectable by Karl Fischer titration, the polymers are still stable in the presence of more than 10 mM NPrMal and destabilization by PhSH is considerably less effective. Similar results are also obtained for titrations with MA. In the combined presence of PhSH and NPrMal, but absence of MePip, Zn-1 polymers only depolymerize when water is present. (Figures S17 and S18). Together, the titrations show that water is a prerequisite for the depolymerization of the supramolecular polymers by the substrates of the Michael reaction. However, the titrations do not clearly indicate a mechanism via which depolymerization occurs. To further probe the mechanism, several control experiments are required.
Figure 4.

Titrations of 10 μM solutions of Zn-1 with NPrMal (a) and PhSH (b) in dry (squares) and ambient (circles) MCH*. Hollow symbols indicate the absence of 0.5 mM MePip, and solid symbols indicate the presence of 0.5 mM MePip in the solutions. In the case of the ambient titrations, [H2O] = 13.8 ppm. In the case of the dry titrations, the concentration of water was below the detection limit of the Karl Fischer titrator.
Nucleophilicity and Bulkiness of Catalyst and Reagents Control Depolymerization
To investigate whether the basicity or nucleophilicity of the MePip catalyst causes the destabilization, we performed control titrations in dry MCH* of Zn-1 with NPrMal in the presence of either non-nucleophilic, basic Me4Pip or nucleophilic, weakly basic pyridine (Figure 5a). These titrations show that when 0.5 mM pyridine is present, a similar destabilization of the polymers by NPrMal is observed as when 0.5 mM MePip is used (see also Figure S25a–c).34 The results obtained for PhSH and MA show similar trends (Figure S26). In contrast to pyridine, the less nucleophilic Me4Pip does not induce depolymerization by NPrMal as measured by CD spectroscopy (see also Figure S25d–f). This difference indicates that the nucleophilicity rather than basicity of MePip causes the depolymerization of the Zn-1 polymers.
Figure 5.

Titrations of (a) 10 μM solutions of Zn-1 with NPrMal with 0.5 mM of either Me4Pip or pyridine in the system and (b) 10 μM solution of H2-1 with NPrMal with 0.5 mM MePip present in the system. In the titration of H2-1, the CD maximum at 390 nm is probed. The titrations were performed in dry MCH* with [H2O] below the detection limit of the Karl Fischer titrator.
The role of the nucleophilic amine in the depolymerization of Zn-1 polymers is further illustrated by a titration of H2-1, which does not contain the Zn-center that can complex nucleophiles. When H2-1 is titrated with NPrMal, PhSH, or MA in the presence of MePip, no decrease in CD intensity is observed (Figures 5b and S27). The absence of any change in CD intensity upon addition of the additives in the presence of the amine catalyst indicates that all monomers remain fully polymerized. Thus, complexation of the nucleophilic amine of the MePip catalyst to the zinc center of Zn-1 is an additional crucial factor in the destabilization of Zn-1 polymers upon addition of the substrates of the Michael reaction. Together with the water-enhanced interaction of the substrates with the polymers, the nucleophilic complexation of MePip to the zinc core of Zn-1 induces the depolymerization.
Lastly, the influence of substrate structure on the depolymerization is illustrated by titrations of Zn-1 in ambient MCH with the bulkier N-tert-butylmaleimide, tBuMal, and 4-tert-butylthiophenol, tBuPhSH, substrates in the presence of 0.5 mM MePip. These titrations show that the bulkier substrates destabilize the supramolecular polymer to a lesser extent (Figure S30). Together with the decreasing angular dependency of the scattering intensity in the presence of the substrates (Figure 4), the decreased destabilization of the bulky substrates hints at a molecular interaction of the substrates close to the porphyrin core of the supramolecular polymers. As such, the decreased destabilization upon increasing the steric bulk of the substrates corroborates that the substrates, water and the catalyst act in concert during the destabilization of the polymers.
The requirement of a nucleophilic site in the catalyst and low steric hindrance in the substrates allows us to speculate on the molecular picture of the interaction that causes the destabilization of the polymers. The SLS results observed indicate that the polymers become more flexible and shorter in the presence of water. This suggests that water can induce defects in the hydrogen-bonded backbone of the polymer. This proposed chain breakage leads to the generation of dynamic and free chain ends, where the supramolecular polymer presents polar amides to the bulk solution. The polar substrates of the Michael reaction can then bind at these chain ends, stabilizing the shorter polymer chains. Interestingly, the macrodipoles of the chain ends42 of the helical peptides are well-known to bind polar molecules,43−45 while water and the helix macrodipole are also known to play a role in collagen bundling.46,47 We hypothesize that, in an analogous fashion, the polar and monomerically dissolved water molecules39 greatly enhance the complexation of the substrates to the chain ends of the Zn-1 polymers. This interaction stabilizes the free chain ends of short polymers and oligomers, while in turn destabilizes the long polymers. The affinity of water and the additives to macrodipoles at the chain end also explains bundling of the polymer bundles in the absence of water. The bundling may shield the nonstabilized macrodipole from the aliphatic solution in the absence of water or reaction substrates or by antiparallel alignment of the polymers in the bundles, the dipoles are canceled. In the presence of the amine catalyst, the free chain ends are additionally stabilized by the complexation of the amine to the Zn-center of the porphyrin core (Figure 6). Consequently, the cooperative interactions between the water, amine catalyst, polar substrates, and helix macrodipole lead to the depolymerization of the Zn-1 polymers in the Michael reaction.
Figure 6.
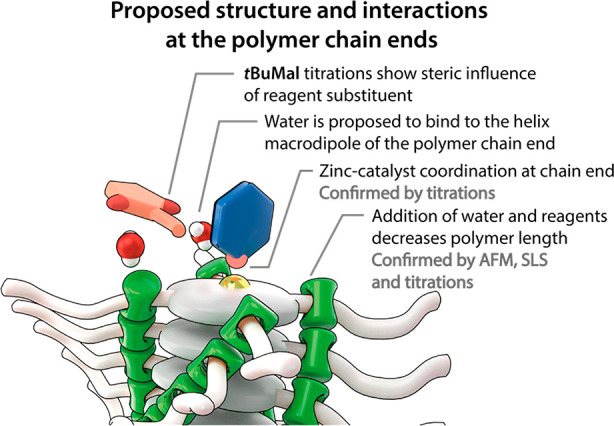
Cartoon representation of a proposed structure of the supramolecular complexation at the polymer chain ends. The cooperative interactions between water, NPrMal and the complexation of MePip to the Zn-center in the porphyrin stabilize the free chain end of the supramolecular polymer. This stabilization eventually leads to the gradual breakdown of the supramolecular polymer. Experimental evidence for the interactions is indicated by gray text.
Interference of the Reaction in Polymer Stability Observed for Various Substrates
The results above show that water plays a crucial role in controlling interference of a chemical reaction in the supramolecular polymerization of Zn-1. Changing the amount of water present in the system, which has only a marginal effect on the covalent Michael reaction, results in a strong effect on the supramolecular polymers (Figure S32). Moreover, the chemical reaction introduces a temporally controlled destabilization of the supramolecular polymer as the reagents are converted to the Michael product, which has a subtly different interaction with the polymer. Thus, by controlling the amount of water in the multicomponent supramolecular system, chemical reactivity can be coupled to or decoupled from the structural integrity of the polymer material.
Lastly, we show the generality of the influence of polar substrates of covalent reactions on the stability of noncovalent aggregates. For this, we performed Michael reactions between PhSH and 2-cyclohexen-1-one, CycHex. Although the base-catalyzed Michael reaction occurs with lower rates (Figure S33), a strong effect of water on the destabilization of Zn-1 polymers is also observed in this reaction mixture (Figure 7). In ambient MCH*, rapid depolymerization occurs when 10 mM of the CycHex and PhSH reaction substrates and 0.5 mM MePip catalyst are present, while in dry solutions no effect is observed. In addition, the spectral changes during the depolymerization are identical to the changes observed with NPrMal (Figure S34), suggesting that the molecular features of the interactions are also identical. As such, water in alkane solvents seems to play a pivotal role in the design of multicomponent systems that not only display supramolecular structure but are also coupled to covalent chemical synthesis.
Figure 7.
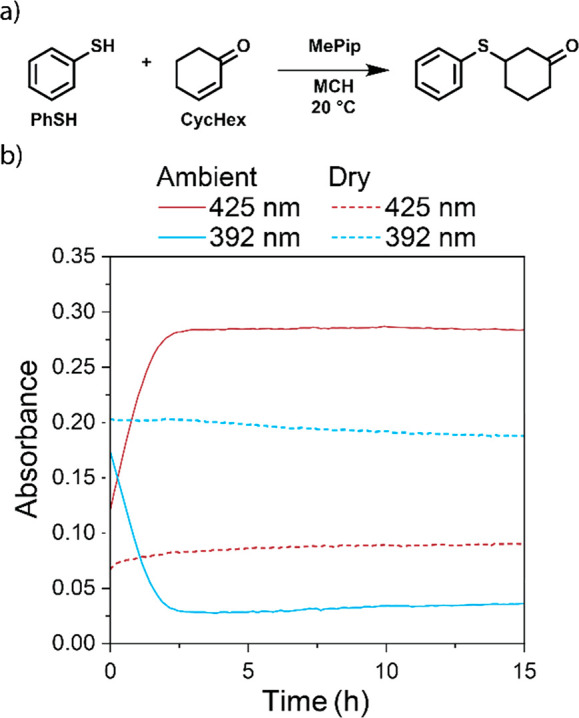
(a) MePip catalyzed Michael reaction between PhSH and CycHex in MCH*. (b) Time-dependent absorption changes of 10 μM solutions of Zn-1 containing 10 mM PhSH, 10 mM CycHex, and 0.5 mM MePip. In the ambient sample, [H2O] = 30.0 ppm, and in the dry sample [H2O] = 11.4 ppm.
Conclusions
Nature is an inexhaustible source of inspiration for complex systems where assembly processes and chemical reactions are operating in great harmony. It will take long before the stellar performance of nature can be observed using non-natural structures. However, the aim to unravel this complexity using artificial molecular systems is an important next step in chemistry.
Here, we have shown that minute amounts of water can play a crucial role in porphyrin-based multicomponent supramolecular systems in alkane solutions. In apolar MCH* solutions, supramolecular polymers of Zn-1 are highly stable against every separate component of a base-catalyzed Michael reaction of NPrMal with PhSH. However, in the combined presence of all components, including the product of the reaction, the polymers are readily destabilized and form small aggregates. Serendipitously, we found that the presence of water in the apolar solvent crucially regulates this destabilization of the polymers by the reaction substrates.
A combination of spectroscopic titrations, light scattering and AFM experiments indicates that water induces flexibility in the supramolecular polymer and breaks up bundled polymers. We propose that the destabilization of the supramolecular polymers is due to binding of water and the substrates at the chain ends and subsequent cooperative binding of the catalyst to the metal center. This destabilization consequently depolymerizes the polymers into small aggregates.
The effect of water on the interference of chemical reactions in supramolecular polymerizations is not limited to maleimides but has also been shown for other reactions. The observation of this effect for various substrates highlights the general role of water in these supramolecular systems.
We anticipate that the incorporation of chemical reactivity in supramolecular polymers will lead to a new and exciting area of adaptive materials. To accelerate the design of such materials, it is of paramount importance that all interactions in these systems are understood. As other results37 have recently shown, water seems very likely to play a crucial and ubiquitous, yet poorly understood role. By taking the interactions of water with supramolecular systems into account, it will be possible to combine great control over supramolecular polymers in apolar environments with the diversity of chemical reactions. We hope this approach will enable the design of increasingly adaptive and responsive materials that may rival the complexity of natural systems.
Acknowledgments
Ilja K. Voets is acknowledged for fruitful discussions. We acknowledge financial support from NWO (TOP-PUNT Grant 10018944) and the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04962.
Synthetic procedures and characterization of compounds, additional UV–vis and CD spectra, and AFM images (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Supramolecular Polymerization. Chem. Rev. 2009, 109 (11), 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Kulkarni C.; Meijer E. W.; Palmans A. R. A. Cooperativity Scale: A Structure–Mechanism Correlation in the Self-Assembly of Benzene-1,3,5-Tricarboxamides. Acc. Chem. Res. 2017, 50 (8), 1928–1936. 10.1021/acs.accounts.7b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Wang F.; Zheng B.; Huang F. Stimuli-Responsive Supramolecular Polymeric Materials. Chem. Soc. Rev. 2012, 41 (18), 6042. 10.1039/c2cs35091b. [DOI] [PubMed] [Google Scholar]
- Bray D. Protein Molecules as Computational Elements in Living Cells. Nature 1995, 376 (6538), 307–312. 10.1038/376307a0. [DOI] [PubMed] [Google Scholar]
- Kholodenko B. N. Cell-Signalling Dynamics in Time and Space. Nat. Rev. Mol. Cell Biol. 2006, 7 (3), 165–176. 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoven J.; Brizard A. M.; Kowlgi K. N. K.; Koper G. J. M.; Eelkema R.; van Esch J. H. Dissipative Self-Assembly of a Molecular Gelator by Using a Chemical Fuel. Angew. Chem., Int. Ed. 2010, 49 (28), 4825–4828. 10.1002/anie.201001511. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Hendriksen W. E.; Koper G. J. M.; Eelkema R.; van Esch J. H. Transient Assembly of Active Materials Fueled by a Chemical Reaction. Science 2015, 349 (6252), 1075–1079. 10.1126/science.aac6103. [DOI] [PubMed] [Google Scholar]
- Semenov S. N.; Wong A. S. Y.; van der Made R. M.; Postma S. G. J.; Groen J.; van Roekel H. W. H.; de Greef T. F. A.; Huck W. T. S. Rational Design of Functional and Tunable Oscillating Enzymatic Networks. Nat. Chem. 2015, 7 (2), 160–165. 10.1038/nchem.2142. [DOI] [PubMed] [Google Scholar]
- Maiti S.; Fortunati I.; Ferrante C.; Scrimin P.; Prins L. J. Dissipative Self-Assembly of Vesicular Nanoreactors. Nat. Chem. 2016, 8 (7), 725–731. 10.1038/nchem.2511. [DOI] [PubMed] [Google Scholar]
- Dhiman S.; Jain A.; George S. J. Transient Helicity: Fuel-Driven Temporal Control over Conformational Switching in a Supramolecular Polymer. Angew. Chem., Int. Ed. 2017, 56 (5), 1329–1333. 10.1002/anie.201610946. [DOI] [PubMed] [Google Scholar]
- Sorrenti A.; Leira-Iglesias J.; Sato A.; Hermans T. M. Non-Equilibrium Steady States in Supramolecular Polymerization. Nat. Commun. 2017, 8, 15899. 10.1038/ncomms15899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhiman S.; Jain A.; Kumar M.; George S. J. Adenosine-Phosphate-Fueled, Temporally Programmed Supramolecular Polymers with Multiple Transient States. J. Am. Chem. Soc. 2017, 139 (46), 16568–16575. 10.1021/jacs.7b07469. [DOI] [PubMed] [Google Scholar]
- Allampally N. K.; Florian A.; Mayoral M. J.; Rest C.; Stepanenko V.; Fernández G. H-Aggregates of Oligophenyleneethynylene (OPE)-BODIPY Systems in Water: Guest Size-Dependent Encapsulation Mechanism and Co-Aggregate Morphology. Chem. - Eur. J. 2014, 20 (34), 10669–10678. 10.1002/chem.201402077. [DOI] [PubMed] [Google Scholar]
- Rest C.; Mayoral M. J.; Fucke K.; Schellheimer J.; Stepanenko V.; Fernández G. Self-Assembly and (Hydro)Gelation Triggered by Cooperative π-π and Unconventional C—H···X Hydrogen Bonding Interactions. Angew. Chem., Int. Ed. 2014, 53 (3), 700–705. 10.1002/anie.201307806. [DOI] [PubMed] [Google Scholar]
- Casellas N. M.; Pujals S.; Bochicchio D.; Pavan G. M.; Torres T.; Albertazzi L.; García-Iglesias M. From Isodesmic to Highly Cooperative: Reverting the Supramolecular Polymerization Mechanism in Water by Fine Monomer Design. Chem. Commun. 2018, 54 (33), 4112–4115. 10.1039/C8CC01259H. [DOI] [PubMed] [Google Scholar]
- Sorrenti A.; Leira-Iglesias J.; Markvoort A. J.; de Greef T. F. A.; Hermans T. M. Non-Equilibrium Supramolecular Polymerization. Chem. Soc. Rev. 2017, 46 (18), 5476–5490. 10.1039/C7CS00121E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Living Supramolecular Polymerization Realized through a Biomimetic Approach. Nat. Chem. 2014, 6 (3), 188–195. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Fukui T.; Jue M. L.; Takeuchi M.; Sugiyasu K. Kinetic Control over Pathway Complexity in Supramolecular Polymerization through Modulating the Energy Landscape by Rational Molecular Design. Angew. Chem., Int. Ed. 2014, 53 (52), 14363–14367. 10.1002/anie.201407302. [DOI] [PubMed] [Google Scholar]
- Fukui T.; Kawai S.; Fujinuma S.; Matsushita Y.; Yasuda T.; Sakurai T.; Seki S.; Takeuchi M.; Sugiyasu K. Control over Differentiation of a Metastable Supramolecular Assembly in One and Two Dimensions. Nat. Chem. 2017, 9 (5), 493–499. 10.1038/nchem.2684. [DOI] [PubMed] [Google Scholar]
- Jung S. H.; Bochicchio D.; Pavan G. M.; Takeuchi M.; Sugiyasu K. A Block Supramolecular Polymer and Its Kinetically Enhanced Stability. J. Am. Chem. Soc. 2018, 140 (33), 10570–10577. 10.1021/jacs.8b06016. [DOI] [PubMed] [Google Scholar]
- Endo M.; Fukui T.; Jung S. H.; Yagai S.; Takeuchi M.; Sugiyasu K. Photoregulated Living Supramolecular Polymerization Established by Combining Energy Landscapes of Photoisomerization and Nucleation–Elongation Processes. J. Am. Chem. Soc. 2016, 138 (43), 14347–14353. 10.1021/jacs.6b08145. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Würthner F. Mechanism of Self-Assembly Process and Seeded Supramolecular Polymerization of Perylene Bisimide Organogelator. J. Am. Chem. Soc. 2015, 137 (9), 3300–3307. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Liu Y.; Wagner W.; Stepanenko V.; Ren X.; Ogi S.; Würthner F. Near-IR Absorbing J-Aggregate of an Amphiphilic BF 2 -Azadipyrromethene Dye by Kinetic Cooperative Self-Assembly. Angew. Chem., Int. Ed. 2017, 56 (21), 5729–5733. 10.1002/anie.201701788. [DOI] [PubMed] [Google Scholar]
- Wagner W.; Wehner M.; Stepanenko V.; Ogi S.; Würthner F. Living Supramolecular Polymerization of a Perylene Bisimide Dye into Fluorescent J-Aggregates. Angew. Chem., Int. Ed. 2017, 56 (50), 16008–16012. 10.1002/anie.201709307. [DOI] [PubMed] [Google Scholar]
- Valera J. S.; Gómez R.; Sánchez L. Tunable Energy Landscapes to Control Pathway Complexity in Self-Assembled N-Heterotriangulenes: Living and Seeded Supramolecular Polymerization. Small 2018, 14 (3), 1702437. 10.1002/smll.201702437. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Grzeszkiewicz C.; Würthner F. Pathway Complexity in the Self-Assembly of a Zinc Chlorin Model System of Natural Bacteriochlorophyll J-Aggregates. Chem. Sci. 2018, 9 (10), 2768–2773. 10.1039/C7SC03725B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Thein J.; Würthner F. Impact of Alkyl Spacer Length on Aggregation Pathways in Kinetically Controlled Supramolecular Polymerization. J. Am. Chem. Soc. 2016, 138 (2), 670–678. 10.1021/jacs.5b11674. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. A Rational Strategy for the Realization of Chain-Growth Supramolecular Polymerization. Science 2015, 347 (6222), 646–651. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- van Rossum S. A. P.; Tena-Solsona M.; van Esch J. H.; Eelkema R.; Boekhoven J. Dissipative Out-of-Equilibrium Assembly of Man-Made Supramolecular Materials. Chem. Soc. Rev. 2017, 46 (18), 5519–5535. 10.1039/C7CS00246G. [DOI] [PubMed] [Google Scholar]
- Debnath S.; Roy S.; Ulijn R. V. Peptide Nanofibers with Dynamic Instability through Nonequilibrium Biocatalytic Assembly. J. Am. Chem. Soc. 2013, 135 (45), 16789–16792. 10.1021/ja4086353. [DOI] [PubMed] [Google Scholar]
- Leira-Iglesias J.; Sorrenti A.; Sato A.; Dunne P. A.; Hermans T. M. Supramolecular Pathway Selection of Perylenediimides Mediated by Chemical Fuels. Chem. Commun. 2016, 52 (58), 9009–9012. 10.1039/C6CC01192F. [DOI] [PubMed] [Google Scholar]
- Leira-Iglesias J.; Tassoni A.; Adachi T.; Stich M.; Hermans T. M. Oscillations, Travelling Fronts and Patterns in a Supramolecular System. Nat. Nanotechnol. 2018, 13 (11), 1021–1027. 10.1038/s41565-018-0270-4. [DOI] [PubMed] [Google Scholar]
- Jalani K.; Dhiman S.; Jain A.; George S. J. Temporal Switching of an Amphiphilic Self-Assembly by a Chemical Fuel-Driven Conformational Response. Chem. Sci. 2017, 8 (9), 6030–6036. 10.1039/C7SC01730H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmich F.; Lee C. C.; Nieuwenhuizen M. M. L.; Gielen J. C.; Christianen P. C. M.; Larsen A.; Fytas G.; Leclère P. E. L. G.; Schenning A. P. H. J.; Meijer E. W. Dilution-Induced Self-Assembly of Porphyrin Aggregates: A Consequence of Coupled Equilibria. Angew. Chem., Int. Ed. 2010, 49 (23), 3939–3942. 10.1002/anie.201000162. [DOI] [PubMed] [Google Scholar]
- Hirose T.; Helmich F.; Meijer E. W. Photocontrol over Cooperative Porphyrin Self-Assembly with Phenylazopyridine Ligands. Angew. Chem., Int. Ed. 2013, 52 (1), 304–309. 10.1002/anie.201205085. [DOI] [PubMed] [Google Scholar]
- Helmich F.; Meijer E. W. Controlled Perturbation of the Thermodynamic Equilibrium by Microfluidic Separation of Porphyrin-Based Aggregates in a Multi-Component Self-Assembling System. Chem. Commun. 2013, 49 (18), 1796–1798. 10.1039/C2CC36887K. [DOI] [PubMed] [Google Scholar]
- Van Zee N. J.; Adelizzi B.; Mabesoone M. F. J.; Meng X.; Aloi A.; Zha R. H.; Lutz M.; Filot I. A. W.; Palmans A. R. A.; Meijer E. W. Potential Enthalpic Energy of Water in Oils Exploited to Control Supramolecular Structure. Nature 2018, 558 (7708), 100–103. 10.1038/s41586-018-0169-0. [DOI] [PubMed] [Google Scholar]
- Mabesoone M. F. J.; Markvoort A. J.; Banno M.; Yamaguchi T.; Helmich F.; Naito Y.; Yashima E.; Palmans A. R. A.; Meijer E. W. Competing Interactions in Hierarchical Porphyrin Self-Assembly Introduce Robustness in Pathway Complexity. J. Am. Chem. Soc. 2018, 140 (25), 7810–7819. 10.1021/jacs.8b02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenden R.; Radzicka A. On the Probability of Finding a Water Molecule in a Nonpolar Cavity. Science 1994, 265 (5174), 936–937. 10.1126/science.8052849. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Moore J. S. Nucleation-Elongation: A Mechanism for Cooperative Supramolecular Polymerization. Org. Biomol. Chem. 2003, 1 (20), 3471–3491. 10.1039/B308788C. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A. A.; Schaefer C.; De Greef T. F. A. F. A.; Meijer E. W. W. Controlling Chemical Self-Assembly by Solvent-Dependent Dynamics. J. Am. Chem. Soc. 2012, 134 (32), 13482–13491. 10.1021/ja305512g. [DOI] [PubMed] [Google Scholar]
- Kulkarni C.; Bejagam K. K.; Senanayak S. P.; Narayan K. S.; Balasubramanian S.; George S. J. Dipole-Moment-Driven Cooperative Supramolecular Polymerization. J. Am. Chem. Soc. 2015, 137 (11), 3924–3932. 10.1021/jacs.5b00504. [DOI] [PubMed] [Google Scholar]
- Hol W. G. J.; van Duijnen P. T.; Berendsen H. J. C. The α-Helix Dipole and the Properties of Proteins. Nature 1978, 273 (5662), 443–446. 10.1038/273443a0. [DOI] [PubMed] [Google Scholar]
- Shoemaker K. R.; Kim P. S.; York E. J.; Stewart J. M.; Baldwin R. L. Tests of the Helix Dipole Model for Stabilization of α-Helices. Nature 1987, 326 (6113), 563–567. 10.1038/326563a0. [DOI] [PubMed] [Google Scholar]
- Aqvist J.; Luecke H.; Quiocho F. A.; Warshel A. Dipoles Localized at Helix Termini of Proteins Stabilize Charges. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (5), 2026–2030. 10.1073/pnas.88.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao K.; Mizuno K.; Murashima M.; Kita Y.; Hongo C.; Okuyama K.; Norisuye T.; Bachinger H. P. Chain Dimensions and Hydration Behavior of Collagen Model Peptides in Aqueous Solution: [Glycyl-4(R)-Hydroxyprolyl-4(R)-Hydroxyproline]n [Glycylprolyl-4(R)-Hydroxyproline]n and Some Related Model Peptides. Macromolecules 2008, 41 (19), 7203–7210. 10.1021/ma800790w. [DOI] [Google Scholar]
- Shoulders M. D.; Raines R. T. Interstrand Dipole-Dipole Interactions Can Stabilize the Collagen Triple Helix. J. Biol. Chem. 2011, 286 (26), 22905–22912. 10.1074/jbc.M110.199984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.