

SUMMARY
Human brain organoids provide unique platforms for modeling development and diseases by recapitulating the architecture of the embryonic brain. However, current organoid methods are limited by interior hypoxia and cell death due to insufficient surface diffusion, preventing generation of architecture resembling late developmental stages. Here, we report the sliced neocortical organoid (SNO) system, which bypasses the diffusion limit to prevent cell death over long-term cultures. This method leads to sustained neuro-genesis and formation of an expanded cortical plate that establishes distinct upper and deep cortical layers for neurons and astrocytes, resembling the third trimester embryonic human neocortex. Using the SNO system, we further identify a critical role of WNT/β-catenin signaling in regulating human cortical neuron subtype fate specification, which is disrupted by a psychiatric-disorder-associated genetic mutation in patient induced pluripotent stem cell (iPSC)-derived SNOs. These results demonstrate the utility of SNOs for investigating previously inaccessible human-specific, late-stage cortical development and disease-relevant mechanisms.
In Brief
Cortical organoids can be used to model human brain development and disorders. Ming and colleagues overcome the diffusion limit using a slicing method to prevent interior cell death and sustain organoid growth over long-term culture. The resulting organoids recapitulate late-stage human cortical developmental features, including formation of distinct cortical layers.
Graphical Abstract
INTRODUCTION
Human brain organoids are pluripotent stem-cell-derived self-organizing three-dimensional (3D) tissues with cell types and cytoarchitectural features resembling the embryonic human brain (Lancaster and Knoblich, 2014). Recent developments in cerebral cortex organoid technologies have demonstrated a striking recapitulation of human cortical development during early-to-mid-gestation periods at molecular, cellular, and structural levels (Qian et al., 2019). However, due to the lack of a functional circulation system, the viability of cells within large cortical organoids is restricted by the limited supply of oxygen and nutrients delivered via surface diffusion (Kadoshima et al., 2013; Kelava and Lancaster, 2016; Lancaster et al., 2017; Qian et al., 2016). Brain organoids grown as spheres in 3D suspension culture can expand up to 3 to 4 mm in diameter, but cells in the interior suffer hypoxia, and a necrotic core forms inside. Despite methods to overcome the diffusion limit and improve oxygen delivery, including using spinning bioreactors or gas-permeable culture plates, supplying higher oxygen in the incubator, or cutting organoids into smaller pieces, the healthy area of brain organoids is typically limited to a 300- to 400-μm-deep ring along the outer surface (Jacob et al., 2020; Kadoshima et al., 2013; Qian et al., 2016; Sakaguchi et al., 2015; Watanabe et al., 2017). This diffusion barrier is particularly detrimental for cortical organoids because proliferating progenitor zones are organized in the interior and eventually deplete as outer layers thicken. Depletion of neural progenitor cells (NPCs) prohibits continuous cell generation and leads to architectural disorganization, limiting the structural development of organoids (Amiri et al., 2018; Qian et al., 2016). In contrast, the developing human embryonic neocortex continues to generate neurons and glia in the third trimester to expand the cortical volume (Bystron et al., 2008; Fjell et al., 2015; Malik et al., 2013; Rakic, 2009; Samuelsen et al., 2003).
The increase in cortical plate (CP) thickness during human corticogenesis is accompanied by the establishment of six specialized cortical layers, which become distinctly separated by cytoarchitecture and layer-dependent expression of neuronal subtype markers during the perinatal period (Saito et al., 2011). During the mid-gestation stage, however, the upper (layer II–IV) and deep (layer V to VI) cortical layers are indistinguishable because the majority of CP neurons co-express markers for both upper and deep layers and the distribution of layer-dependent neuronal markers intermingles extensively (Fame et al., 2011; Ip et al., 2011; Nowakowski et al., 2017; Ozair et al., 2018; Zhong et al., 2018). A lack of accessibility to normal human embryonic brain tissues from the late gestation period has prevented systematic examination of this transition toward establishing separated cortical layers. Moreover, this prolonged period of co-expression of markers of different layers and neuronal subtypes is absent during rodent cortical development (Alsiö et al., 2013; Britanova et al., 2008; Saurat et al., 2013), indicating the presence of human/primate-specific mechanisms controlling post-mitotic cortical neuron fate specification.
Brain organoids have demonstrated tremendous promise in recapitulating human-specific developmental features, most notably exemplified by the generation of an outer subventricular zone (oSVZ)-like progenitor layer containing outer radial glia (oRG)-like cells (Bershteyn et al., 2017; Qian et al., 2016). However, interior hypoxia and cell death cause oSVZ shrinkage and radial glia scaffold disruption in prolonged cultures, prohibiting continuous neuronal production and proper inside-out migration and neuronal maturation required for cortical lamination formation (Qian et al., 2019). Thus far, despite occasionally observed rudimentary separation, no published cortical organoid models have shown consistent formation of CP structures that exhibit distinct separation between upper and deep cortical layers. Here, we report the development of cortical organoids that over-come the diffusion limit using a slicing method, which leads to formation of well-separated upper and deep cortical layers. We further demonstrate the utility of our system for investigation of molecular mechanisms regulating human cortical neuron subtype fate specification in normal development and psychiatric disorders.
RESULTS
Slicing Method Resolves Interior Hypoxia and Reduces Cell Death in Cortical Organoids
In order to generate cortical organoids to model late-stage developmental features, we aimed to sustain neurogenesis and support the expansion of cortical structures over long-term cultures. We reasoned that the diffusion limit is inevitable if the organoid is cultured as a sphere without an active circulation system. Therefore, we developed a precisely controlled slicing method to “trim” the organoid into a disk shape and expose the interior of organoids to the external culture environment (Figures 1A and S1A). We followed our previously established fore-brain organoid protocol to day 45 (day 0 refers to when human induced pluripotent stem cell [iPSC] colonies were detached to form embryoid bodies), when organoids have formed large ventricular structures and organized radial glia scaffolds (Qian et al., 2018). Forebrain organoids were then embedded in low-melting-point agarose and sectioned into 500-mm-thick slices using a vibratome (Figure 1A). Organoid slices dissociated from the agarose after sectioning spontaneously or with gentle pipetting and were collected and transferred to a 6-well plate and cultured on an orbital shaker (Figure S1A). From a spherical organoid with 1.5-mm diameter, the one or two slices in the middle plane were kept for culture. These disk-shaped organoids received oxygen and nutrients by diffusion through the exposed top and bottom surfaces, permitting growth in both horizontal (x–y) and thickness (z) directions while maintaining the organization of cortical structures (Figures 1B and 1C). They were repeatedly sliced to 500 μm every 4 weeks in the plane parallel to the first slicing (Figures 1A and S1A). From day 60 to day 140, the slicing method substantially reduced the pronounced interior hypoxia present in unsliced organoids (Figures 1D and 1E). The 500 μm section thickness was larger than the diameter of most ventricular structures formed within organoids, allowing the ventricular zone (VZ) to maintain intact adherens junctions at the apical surface (Figures 1F and S1B). Compared to unsliced forebrain organoids, these sliced neocortex organoids (hereafter referred to as SNOs) showed a drastically reduced necrotic area size, which did not increase over time (Figures S1C and S1D). Moreover, this approach also significantly reduced apoptotic cells in the outer (non-necrotic) regions (Figures 1F and 1G).
Figure 1. Sliced Neocortical Organoid (SNO) Method Resolves Interior Hypoxia and Reduces Cell Death.
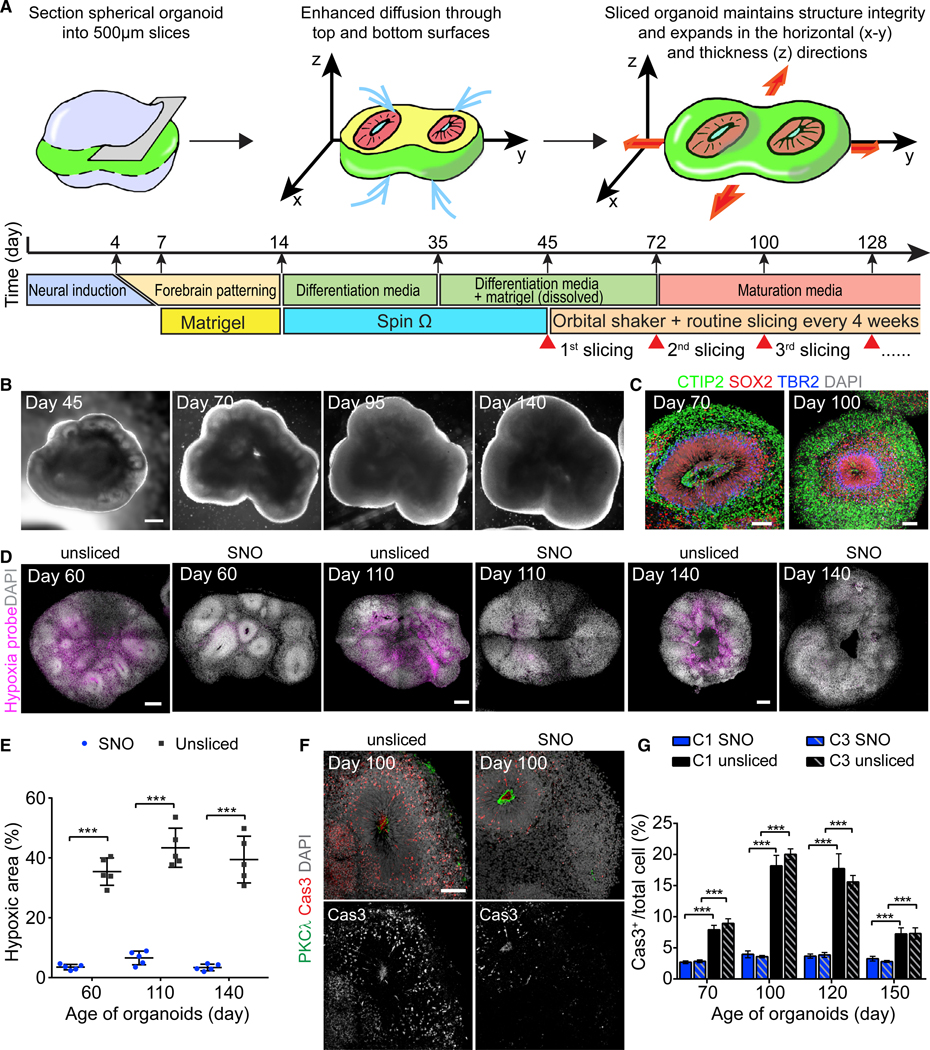
(A) Schematic illustration for the SNO protocol and culture timeline.
(B) Sample bright-field images of the same SNO captured at days 45, 70, 95, and 140. Day 0 refers to when iPSC colonies were detached to form embryoid bodies. Scale bar, 500 μm.
(C) Sample immunostaining confocal images of SNOs showing maintenance of cortical structures after slicing. Scale bars, 100 μm.
(D and E) Hypoxia assay comparing unsliced organoids and SNOs using a pimonidazole-based hypoxia probe. Shown are sample tiling confocal images with hypoxic cells in purple and DAPI in white (D). Scale bars, 200 μm. Images at the same date are shown at the same scale. Shown in (E) is the quantification of the percentage of the hypoxic area over the total organoid area. Bar values represent mean ± SD (n = 5 organoids; ***p < 0.0005; Student’s t test).
(F and G) Analyses of apoptotic cell death in SNOs and unsliced organoids. Shown are sample immunostaining confocal images for apoptosis marker cleaved caspase-3 (Cas3) and adherens junction marker PKCλ (F) and quantification of apoptotic cell death within the non-necrotic region (G). Scale bar, 100 μm. Values represent mean ± SEM (n ≥ 10 and 5 organoids for SNOs and unsliced organoids, respectively; ***p < 0.0005; Student’s t test).
See also Figure S1.
SNO Method Sustains Neurogenesis and Radial Migration of Newborn Neurons
Over long-term culture and repeated slicing, SNOs continued to grow. At days 100 and 150, the oSVZ-like structures contained a large population of HOPX+ oRG-like NPCs with radially aligned basal processes contacting the pial surface (Figures 2A and S2A). The oSVZ also contained abundant TBR2+ intermediate progenitor cells (IPCs) committed to glutamatergic neurogenesis (Figure 2B). In contrast, both KI67 and TBR2 were sparsely detected in unsliced organoids by day 150 (Figures 2A and 2B). Quantitative analysis showed that progenitor proliferation was sustained steadily in SNOs and substantially higher than unsliced organoids at days 120 and 150 (Figure 2C). The size of progenitor zones continued increasing in SNOs but decreased in unsliced organoids (Figure 2D). Sparse labeling using a GFP-expressing adenovirus showed that the majority of radial glia cells exhibited the hallmark unipolar morphology of oRGs and expressed multiple human oRG markers (Pollen et al., 2015; Figures 2E and S2B). Birth dating of newborn cells using 5-ethynyl-2′-deoxyuridine (EdU) pulse-chase labeling for 7 days confirmed sustained production of cortical neurons (Figure 2F). At 3 days post-labeling at day 95, the majority of EdU+ cells were SOX2+ and located within the oSVZ (Figures 2G–2I). At 7 days post-labeling, post-mitotic cells retaining a strong EdU signal migrated into the CP and expressed the cortical neuron marker SATB2 (Figures 2G–2I). Moreover, many EdU+ cells found within the oSVZ also expressed PRDM8 (Figure S2C), a marker for migrating neurons in the cerebral cortex (Inoue et al., 2014; Komai et al., 2009). PRDM8 was more abundantly detected in the oSVZ than in the CP (Figures S2C and S2D), suggesting that the majority of neurons in the CP have completed radial migration and reached their designated laminar positions.
Figure 2. Sustained Neurogenesis and Radial Migration of Newborn Neurons in SNOs.
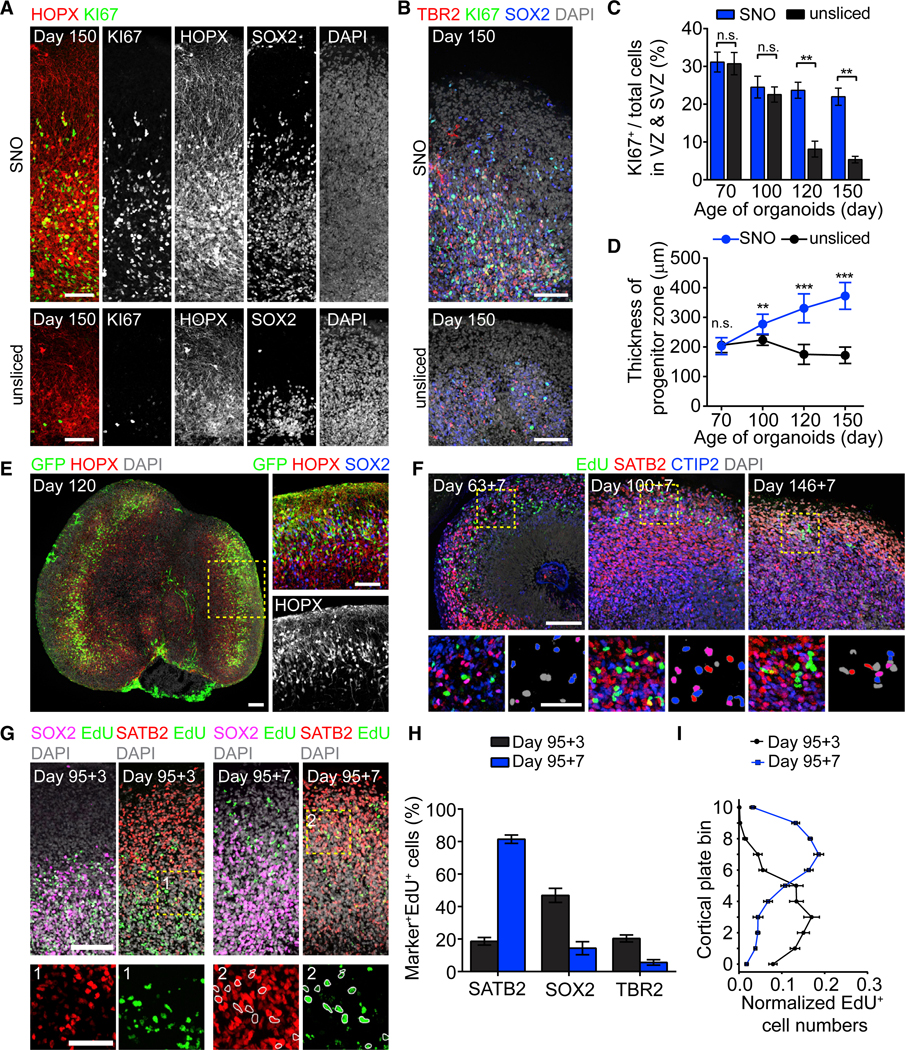
(A and B) Sample tiling confocal images showing the oSVZ of day 150 SNOs (top panel) and unsliced organoids (bottom panel), immunostaining for proliferation marker KI67, general NPC marker SOX2, oRG marker HOPX (A), and IPC marker TBR2 (B). Scale bars, 100 μm.
(C) Quantification of the percentage of KI67+ cells in progenitor zones (VZ and oSVZ). Values represent mean ± SEM (n = 5 organoids; n.s.: p > 0.05; **p < 0.005; Student’s t test).
(D) Quantification of the size of proliferative progenitor zones. Values represent mean ± SD (n ≥ 20 and 10 organoids for SNOs and unsliced organoids from 2 iPSC lines, respectively; n.s.: p > 0.05; **p < 0.005; ***p < 0.0005; Student’s t test).
(E) Preferential infection of oRG-like cells with unipolar morphologies and basal processes contacting the pial surface by GFP-expressing adenovirus. Shown on the left is a sample tiling confocal image for a day 120 SNO with two large oSVZ structures. The inset shows a magnified view with a 90-degree counter-clockwise rotation. Scale bars, 100 μm.
(F) Sample confocal images for 7-day EdU pulse-chase experiments, with newborn neurons labeled double positive for EdU and neuron marker CTIP2 or SATB2 in SNOs. Scale bars, 100 μm and 50 μm (insets). EdU+ nuclei in the insets (bottom right panels) are pseudo-colored to indicate their marker expression: gray (SATB2−/CTIP2−); red (SATB2+/CTIP2−); blue (SATB2−/CTIP2+); and purple (SATB2+/CTIP2+).
(G) Sample confocal images for 3- and 7-day EdU pulse-chase experiments, showing that EdU+ cells migrated from the SOX2-enriched progenitor zones into the SATB2-enriched CP from day 98 to day 102. The insets (box 1 and 2) show magnified views with EdU and SATB2 double-positive nuclei circled in white. Scale bars, 100 μm and 50 μm (insets).
(H) Quantification of the percentage of EdU+ cells expressing SATB2, SOX2, or TBR2 at 3 and 7 days post-EdU labeling on day 95. Values represent mean ± SEM (n = 5 SNOs for each marker).
(I) Quantifications of the localization of EdU+ cells at 3 and 7 days post-EdU labeling. The cortical structure from apical surface to basal surface is evenly divided into bins 0–10. Shown are curves representing the normalized abundance within each bin, calculated as [no. of EdU+ cells in a bin/no. of total EdU+ cells]. Values represent mean ± SEM (n = 10 SNOs). Same samples as in (H) are shown.
See also Figure S2.
Layer Expansion Persists in SNOs over Long-Term Cultures
With improved cell viability and sustained neurogenesis, the laminar structures of SNOs continuously expand over longterm cultures while maintaining the organization of progenitor zones and neuronal layers (Figure 3A). From day 70 to day 150, the cortical structures of SNOs displayed well-defined VZ, oSVZ, and CP layers. The VZ was marked by densely packed SOX2+ NPCs at the apical side; the oSVZ contained mixed populations of NPCs, IPCs, and CTIP2+ neurons; and the CP at the basal side was exclusively populated by neurons forming condensed layers. We measured the total layer thickness of cortical structures by the distance between the apical (ventricular) surface to the basal (pial) surface. Although the viable thickness in unsliced organoids was restricted to 300–400 μm and stopped increasing soon after day 70, SNOs could consistently expand far beyond this limit (Figures 3B and 3C). At day 150, the total layer thickness could reach 700 μm and the CP increased to over 300 μm on average (Figures 3B and 3C).
Figure 3. Layer Expansion in SNOs over Long-Term Cultures.
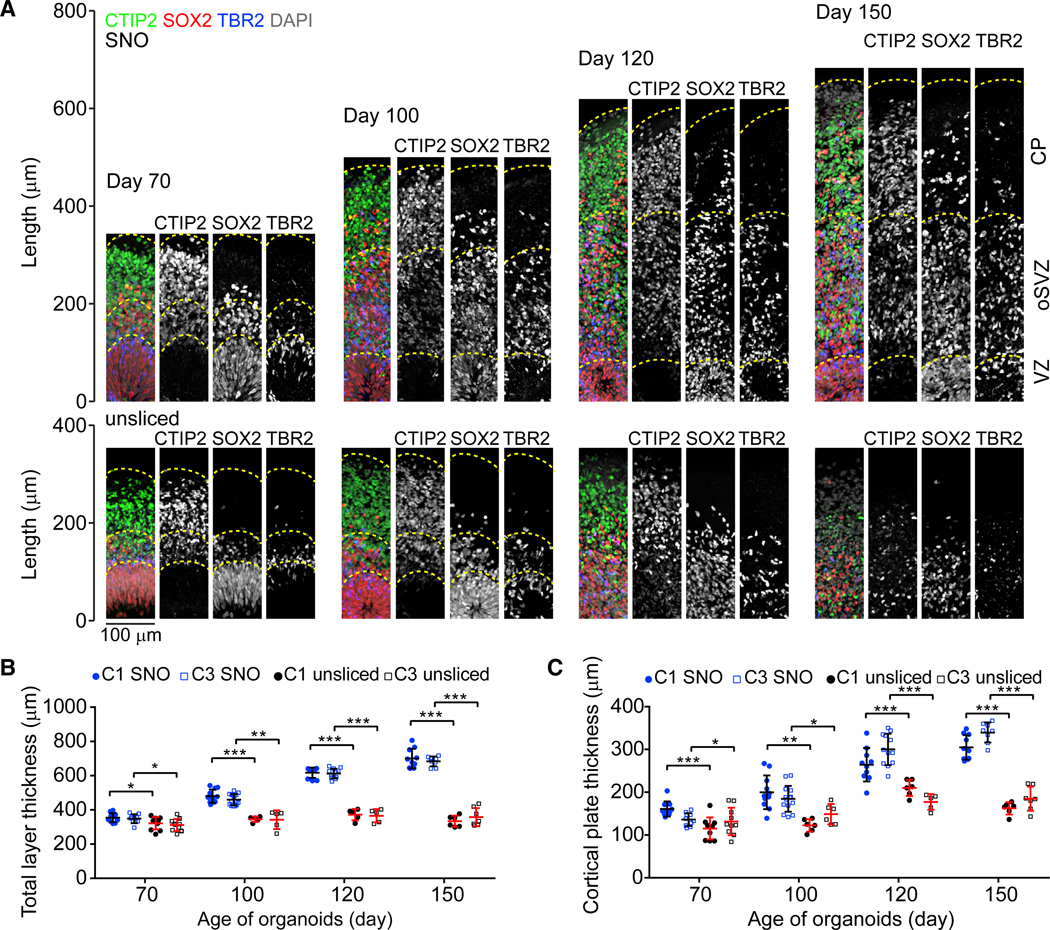
(A) Sample tiling confocal images of cortical structures in SNOs (top panel) and unsliced organoids (bottom panel), for immunostaining of CTIP2, SOX2, and TBR2. Dashed lines mark the pial surfaces and the boundaries of the VZ, oSVZ, and CP. The laminar structures in days 120 and 150 unsliced organoids are disorganized and thus not marked by dashed lines. Scale bar, 100 μm.
(B and C) Quantifications of the total thickness (B) and CP thickness (C). Values represent mean ± SD (n ≥ 10 and 5 organoids for SNOs and unsliced organoids for each iPSC line, respectively; *p < 0.05; **p < 0.005; ***p < 0.0005; Student’s t test).
SNOs Form Distinct Upper and Deep Cortical Layers
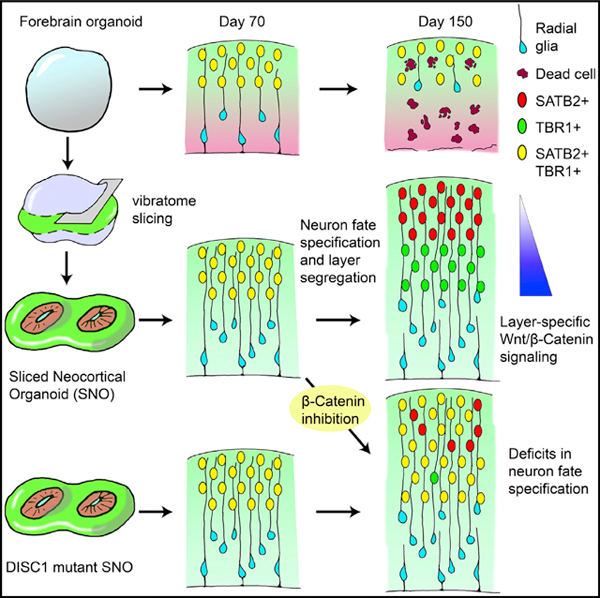
We previously reported that the CP in unsliced forebrain organoids contained cortical neurons expressing upper- and deep-layer markers that did not separate into distinguishable layers, reminiscent of developing human cortex around gestational weeks (GWs) 14–18 (Hevner, 2007; Qian et al., 2016). Similar to unsliced organoids, the CP of days 70 and 100 SNOs contained both the upper-layer intracortical (callosal) projection neuron marker SATB2 and the deep-layer subcortical projection neuron marker TBR1 without layer preferences (Figure 4A). In contrast, when SNOs were cultured to day 120 and beyond, the laminar distribution of the markers became layer specific. At days 120 and 150, the upper layers were populated exclusively by SATB2+/TBR1− neurons, whereas the deep layers were dominated by TBR1+/SATB2− neurons, separated by a defined boundary (Figure 4A; Video S1). We divided the thickness of the CP into 11 equally sized bins to quantitatively analyze the laminar expression patterns of SATB2 and TBR1. At days 70 and 100, the distributions of the two markers over-lapped with each other, but at days 120 and 150, they exhibited two separate peaks representing the upper and deep layers (Figure 4B).
Figure 4. Establishment of Separated Upper and Deep Cortical Layers and Specification of Cortical Neuron Subtypes.
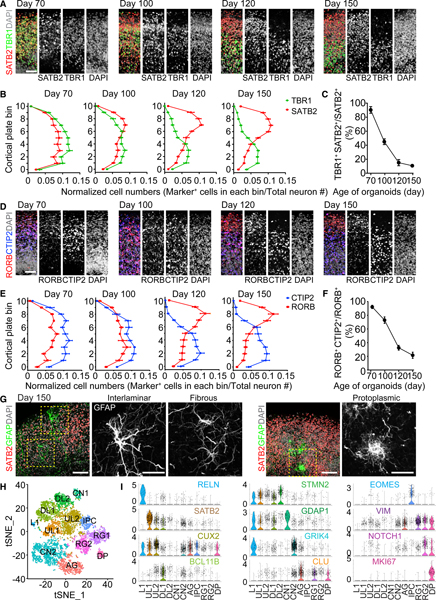
(A) Sample immunostaining confocal images of the CP of SNOs for SATB2 and TBR1. Shown are cropped 100 × 300 μm columns in the CP, and the pial surface is at the top. Scale bar, 50 μm.
(B) Quantifications of the distribution of SATB2+ and TBR1+ neurons in the CP. The CP is evenly divided into 11 bins; bins 0–10 follow the apical-to-basal direction. Shown are curves representing the normalized abundance within each bin, calculated as [no. of marker+ cells in a bin/no. of total neurons]. Values represent mean ± SEM (n = 10 SNOs from C1 and C3 iPSC lines).
(C) Quantification of the ratio of co-expression of TBR1 and SATB2 over SATB2+ cells in the CP. Same samples as in (B) are shown. Values represent mean ± SD.
(D) Sample immunostaining confocal images of the CP for RORB and CTIP2. Scale bar, 50 μm.
(E) Quantification of the distribution of RORB+ and CTIP2+ neurons in the CP. Shown is similar to (B). Values represent mean ± SEM (n = 10 SNOs from C1 and C3 iPSC lines).
(F) Quantification of the ratio of co-expression of RORB and CTIP2 over RORB+ cells in the CP. Same samples as in (E) are shown. Values represent mean ± SD.
(G) Sample confocal images of morphologically distinct GFAP+ astrocytes in day 150 SNOs. Note localization of an interlaminar astrocyte cell body near the pial surface and protoplasmic astrocytes in the SATB2+ upper layer. Scale bars, 100 μm.
(H) Graph-based clustering of cells from day 150 SNOs by single-nucleus RNA-seq (n = 6,888 nuclei). AG, astrocyte/glia; CN, cortical neuron; DL, deep layer; DPs, dividing progenitors; IPCs, intermediate progenitor cells; L1, layer I; RG, radial glia; UL, upper layer. Cell population identities were determined by gene enrichment analysis using cell type and layer-specific marker gene sets obtained from the Allen Brain Atlas (Hawrylycz et al., 2012) and published datasets of single-cell RNA-seq of the developing and adult human cerebral cortex (Fan et al., 2018; Lake et al., 2016; Nowakowski et al., 2017; Pollen et al., 2015).
(I) Expression of selected cluster-specific marker genes used for cell type classification. Shown are violin plots overlaid on scatterplots, where the proportion of cells expressing a given gene is the highest. The color coding for the gene names indicates the cluster in which the gene is most enriched.
See also Figures S2, S3, S4, and S5, Video S1, and Table S1.
Importantly, we observed a sharp decrease in the percentage of neurons co-expressing SATB2 and TBR1 over time (Figure 4C), suggesting post-mitotic fate specification of existing cortical neurons during the establishment of separated laminar expression domains. In some cases, the CP in day 150 SNOs could be subdivided further to resemble the marker distribution and cytoarchitecture of layers I–VI in the human perinatal frontal cortex (Saito et al., 2011; Figure S2E). These dynamic progressions in layer-specific marker expression and localization closely resemble the human neocortex in the third trimester (Saito et al., 2011). In addition, another pair of markers, RORB and CTIP2 (Nakagawa and O’Leary, 2003), depicted very similar laminar expression patterns specific to the upper and deep cortical layers, respectively, after day 120 (Figures 4D and 4E). The ratio of co-expression between RORB and CTIP2 also decreased steeply over time (Figure 4F). Further analysis of laminar distribution of additional known markers for cortical neuron subtypes (Lake et al., 2016) showed that CUX1 and LAMP5 displayed exclusive upper layer localization at day 150 (Figure S2F).
In the rodent embryonic cortex, expression of SATB2 and CTIP2 directly controls axonal projections of cortical neurons (Britanova et al., 2008). Visualized by a general axon marker SMI312 (Ulfig et al., 1998), the CTIP2+ deep layer contained mostly vertically oriented axons and the SATB2+ upper layer was filled by tangentially oriented axons circling SNOs at day 150 (Figure S2G). The boundary delineated by tangential and vertical axonal patterns coincided with the boundary between SATB2-and CTIP2-enriched domains (Figure S2G).
We did not observe the formation of gyri or sulci in SNOs, and the pial surface appeared smooth. Following a recent protocol of in vitro induction of folding of the CP and pial surface in organotypic human embryonic cortical slices by embedding in an extracellular matrix (ECM) gel composed of collagen I, HAPLN1, and lumican (Long et al., 2018), we observed similar wrinkle formation in the CP of SNOs after 2–4 days (Figures S2H and S2I). Although it is unclear whether this in vitro induced phenomenon mirrors in vivo gyrification, it suggests that ECM cues induce similar mechanical responses in the CP of SNOs as in organo-typic human embryonic cortical slices.
SNOs Contain Diverse Cell Types
In addition to excitatory neurons, various subtypes of GABAergic neurons were found in SNOs, including neurons expressing somatostatin (SST), calretinin (CR), neuronal nitric oxide synthase (nNOS), neuropeptide Y (NPY), choline acetyl-transferase (ChAT), and parvalbumin (PV) (Figures S3A and S3B). Quantitative analyses revealed that the percentage of these interneurons in SNOs were much lower than in the human cerebral cortex (Figure S3C; Tremblay et al., 2016), consistent with the notion that the majority of cortical interneurons are generated ventrally and migrate to the cortex, although locally generated interneurons represent a smaller population (Radonjić et al., 2014). Moreover, immunostaining for GFAP revealed three morphologically distinct astrocyte subtypes, which displayed hallmarks of protoplasmic astrocytes, fibrous astrocytes, and primate/human-specific interlaminar astrocytes, respectively (Figures 4G, S3D, and S3E; Hodge et al., 2019; Oberheim et al., 2009). In day 150 SNOs, the subtype-dependent laminar distributions of astrocytes also resemble that of the human cerebral cortex. The cell bodies of interlaminar-like astrocytes resided exclusively in the layer I/marginal zone and extended long descending processes into the CP, although the protoplasmic and fibrous-like astrocytes were located in the CP (Figures 4G and S3E–S3G). Sustained proliferation allowed for generation of oligodendrocyte precursor cells (OPCs) and oligodendrocytes, the late-born cell types in the forebrain NPC lineage (Figures S3H and S3I). The majority of OLIG2+ OPCs co-expressed NKX2.2 at day 120, but NKX2.2 expression was much lower at day 150 (Figure S3H), suggesting a transition of OPCs toward oligodendrocyte differentiation (Zhu et al., 2014). We observed extensive overlap between the oligodendrocyte marker MBP and the OPC marker NG2, indicating that oligodendrocytes generated in day 150 SNOs were immature (Figure S3J). The specificity of our immunohistochemistry assay was validated with adult human cortical tissues (Figure S3J).
To characterize the full repertoire of cell type diversity within SNOs, we performed single-nucleus RNA sequencing (RNA-seq) analysis using SPLiT-seq method (Rosenberg et al., 2018). We analyzed 6,888 single nuclei from day 150 SNOs. Unsupervised clustering of single cells was achieved by principal-component analysis using highly variable genes and was visualized on a two-dimensional t-distributed stochastic neighbor embedding (tSNE) plot, from which we obtained 12 well-defined clusters (Figures 4H and S4A). Known layer-or subtype-specific cortical neuron markers, such as RELN (cluster L1), SATB2 (cluster UL1), CUX2 (cluster UL2), and BCL11B (CTIP2; cluster DL1), were specifically enriched in distinct clusters, and EOMES (TBR2) was exclusively expressed in the IPC cluster (Figure 4I). Radial glia cell marker VIM and dorsal forebrain NPC marker PAX6 were expressed in the radial glia cell clusters (RG1, RG2, and dividing progenitor [DP]; Figures 4I and S4C). The DP cluster is specifically enriched with MKI67 and other mitotic genes, and differential expression of NOTCH1 distinguishes RG1 and RG2 clusters (Figure 4I), as inhibition of Notch is an indicator for the differentiation state of oRGs (Hansen et al., 2010). In situ hybridization images of the adult human cortex obtained from the Allen Brain Atlas validated the layer-dependent expression patterns of selected marker genes used in analyses (Figure S4D; Table S1). We compared SNO cells with embryonic human prefrontal cortex cells from a published single-cell RNA-seq dataset (Zhong et al., 2018). Pearson correlation analyses between all SNO and fetal cell clusters showed that SNO cell types most highly correlated to the corresponding endogenous cell types in the developing human prefrontal cortex (Figure S4B). Because the fetal human dataset we used lacked a distinct astrocyte cluster, we separately compared the astroglia (AG) cluster to another published dataset of bulk RNA-seq of human fetal and postnatal brain primary astrocytes (Zhang et al., 2016). Surprisingly, the astrocytes in day 150 SNOs showed higher similarity to human postnatal astrocytes than to fetal astrocytes, suggesting that astrocyte maturation may be accelerated in the SNO culture (Figure S4E).
Next, we characterized functional properties of neurons in SNOs. Electrophysiological analysis with whole-cell recording in slices acutely prepared from SNOs showed spontaneous synaptic currents (Figures S5A and S5B). Biocytin labeling through the recording pipette further revealed the complex morphology of neurons with dendritic spine structures and long tangentially oriented axons extending for millimeters with branches into multiple regions in the CP (Figures S5C and S5D). At the circuitry level, extracellular recording of intact SNOs using a four-shank multi-electrode laminar probe revealed spontaneous firing of action potentials and coordinated burst patterns, indicative of network activity across long distances both vertically and horizontally (Figures S5E and S5F). Among sets of coordinated single-neuron spikes detected several hundred micrometers apart, the order of single-neuron recruitment and bursts showed a repeatable temporal pattern, suggesting the presence of stably established synaptic connectivity (Figures S5G–S5I).
WNT/β-Catenin Signaling Regulates Post-mitotic Fate Specification of Cortical Neurons
We next applied our SNO system to investigate molecular mechanisms regulating human neocortical development. The fate of cortical neurons is partially determined cell-autonomously during neurogenesis, but the post-mitotic fate specification that refines neuronal subtype identity and establishes layer-specific gene expression is synergistically regulated by non-cell-autonomous signaling (Fame et al., 2011; Greig et al., 2013; Molyneaux et al., 2007; Shepherd, 2013; Telley et al., 2019). The mechanisms regulating cortical neuron fate specification in humans have remained largely unclear due to the lack of an accessible and representative model. The WNT/β-catenin pathway genes, including WNT7B, are specifically enriched in the deep layers and subplate in the human embryonic cortex during the late second trimester (Abu-Khalil et al., 2004; Miller et al., 2014). WNT/β-catenin signaling regulates the specification of subplate neurons into deep-layer neurons by suppressing callosal projection fate and promoting the alternative subcortical projection fate in a layer-specific manner (Ozair et al., 2018). Similar to its localization in vivo, WNT7B was enriched in the deep CP layers of SNOs and progressively established mutually exclusive domains with the SATB2+ upper layers from day 100 to day 150 (Figure 5A). Notably, the deep-layer-specific enrichment of WNT7B was already apparent at day 100, preceding the establishment of separated cortical layers, suggesting that WNT7B expression could regulate but is not the result of cortical layer specification (Figure 5A).
Figure 5. Regulation of Cortical Neuron Fate Specification by WNT/β-Catenin Signaling.
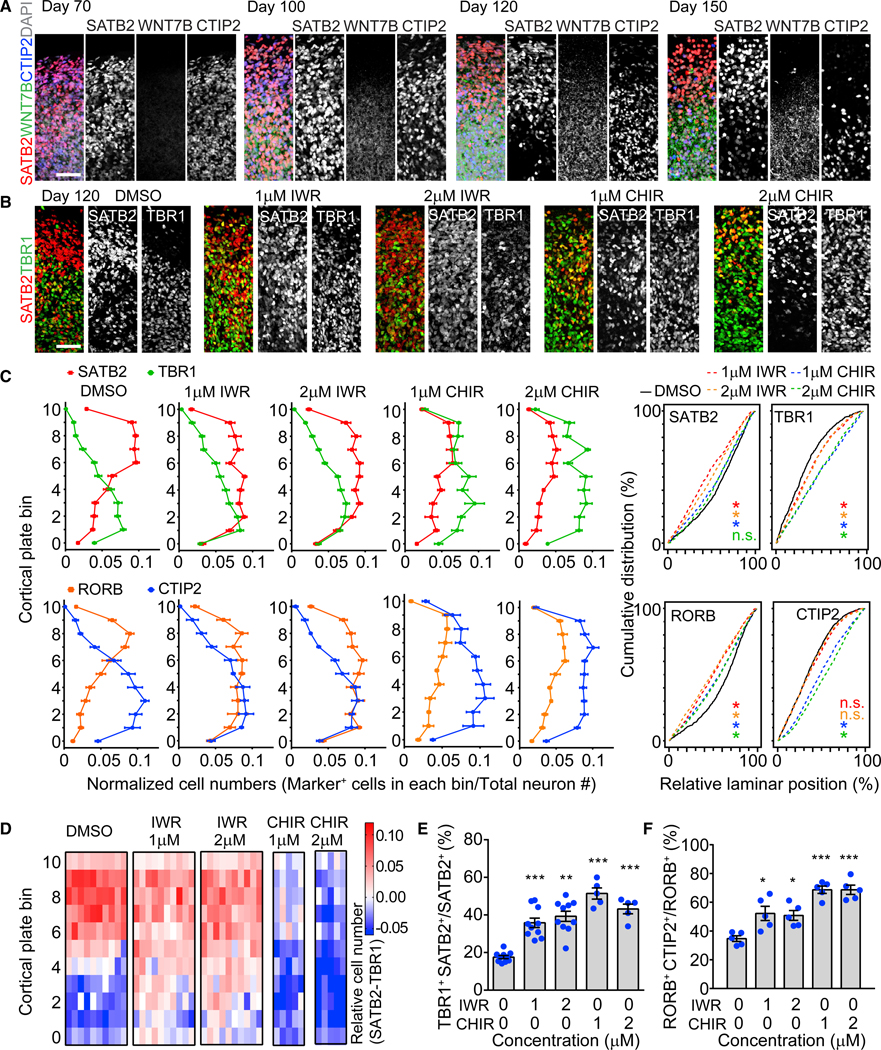
(A) Sample confocal images of the CP of SNOs with immunostaining for SATB2, CTIP2, and WNT7B. Scale bar, 100 μm.
(B) Sample confocal images showing the effects of β-catenin antagonist (IWR-1-endo [IWR]) and agonist (CHIR9902 [CHIR]) on expression patterns of SATB2 and TBR1 in the CP. Drugs were added into culture medium at the indicated concentrations from day 100 to day 120, and the analysis was performed at day 120. Scale bar, 50 μm.
(C) Quantifications of the distribution of SATB2+, TBR1+ and RORB+, CTIP2+ neurons in the CP of SNOs treated with drugs. Shown on the left are curves representing the normalized abundance within each bin, similar to Figure 4B. Values represent mean ± SEM (n = 10 SNOs from C1 and C3 iPSC lines for DMSO and IWR treatments; n = 5 SNOs for CHIR treatments). Shown on the right are cumulative distribution curves of marker-positive neurons. Kolmogorov-Smirnov tests were performed for the drug-treated conditions, indicated by the corresponding color, against the DMSO condition (n.s.: p > 0.05; *p < 0.05).
(D) Heatmap plots for the differential abundance between SATB2+ and TBR1+ nuclei in the CP of drug-treated SNOs. Each row represents one of the 11 CP bins, and each column represents an individual SNO analyzed. The differences between the abundance of SATB2+ nuclei and TBR1+ nuclei within each bin are calculated as ([normalized SATB2+ nuclei no.] – [normalized TBR1+ nuclei no.]). Red on the heatmap indicates a positive value (more SATB2), and blue indicates a negative value (more TBR1). Same samples as in (C) are shown.
(E and F) Quantifications of the ratio of co-expression between TBR1 and SATB2 over SATB2+ cells (E) and between RORB and CTIP2 over RORB+ cells (F) in drug-treated SNOs. Same samples as in (C) are shown. Values represent mean ± SEM (*p < 0.05; **p < 0.005; ***p < 0.0005; Student’s t test).
See also Figure S6.
To investigate the functional role of WNT/β-catenin signaling in cortical neuron fate specification, we treated day 100 SNOs with either β-catenin inhibitor IWR-1-endo (IWR) or β-catenin activator CHIR99021 (CHIR) for 20 days between days 100 and 120, a time window with rapid changes in the laminar expression patterns of upper- and deep-layer markers (Figures 4A–4F). SNOs treated with IWR showed an increase in SATB2 expression in lower cortical bins, resulting in comparable abundance between SATB2+ neurons and TBR1+ neurons in the deep layers at day 120 (Figures 5B and 5C). On the contrary, CHIR-treated SNOs showed a decrease in SATB2 expression, accompanied by a substantial increase of TBR1 expression across the entire CP (Figures 5B and 5C). As a result, the expression patterns of SATB2 and TBR1 overlapped and lost their respective preferences to upper and deep layers in drug-treated SNOs (Figures 5C and 5D). Both IWR and CHIR treatments resulted in a significant increase in the percentage of neurons co-expressing TBR1 and SATB2, suggesting an impairment in neuronal fate specification (Figure 5E). Similarly, drug treatment altered the laminar expression patterns of RORB and CTIP2 (Figure 5C). Elevation of RORB induced by IWR in the deep layers and elevation of CTIP2 induced by CHIR in the upper layers both disrupted the respective layer specificity of the two markers, concurring with an increased proportion of double-positive cells (Figures 5C and 5F). Following the withdrawal of drug treatment at day 120, the IWR-induced effects persisted at day 150, indicating that day 100–120 may be a decisive time window for neuronal subtype fate specification (Figure S6A).
To determine whether the drug treatment affected postmitotic neurons, we performed EdU-pulse chase analysis to specifically label neurons birth dated at day 95, followed by treatment on day 103 (Figure S6B). At 20 days after drug treatment, the ratio of co-expression between SATB2 and TBR1 among SATB2+EdU+ neurons was elevated in both IWR- and CHIR- treated SNOs (Figures S6C and S6D). Because EdU is diluted in cycling cells, cells retaining EdU after 28 days of pulse chase are predominantly post-mitotic neurons born at day 95, providing direct evidence that WNT/β-catenin signaling controls post-mitotic fate specification of human cortical neurons.
Together, these results showed that, similar to the developing human cerebral cortex, SNOs rely on self-maintained layer-dependent signaling to regulate the distinct laminar expression of neuron subtype markers.
Psychiatric Disorder Patient iPSC-Derived SNOs Exhibit Cortical Neuron Subtype Fate Specification Deficits
Disorganization of cortical lamination has been hypothesized to contribute to the etiology of neurodevelopmental disorders (Kana et al., 2011; Zikopoulos and Barbas, 2013). For instance, in situ hybridization analyses revealed frequent patches of aberrant laminar expression patterns of cortical layer markers in frontal lobes of children with autism spectrum disorders (Stoner et al., 2014). The SNO system provides a platform to test the possibility that the developmental origins of other major psychiatric disorders could also involve similar abnormalities. Mutations of the DISC1 gene have been associated with schizophrenia, major depression, and autism (Brandon and Sawa, 2011). In NPCs and neurons, DISC1 protein inhibits GSK3β activity, resulting in the stabilization of β-catenin, and DISC1 loss of function consequently impairs WNT/β-catenin signaling (Mao et al., 2009; Singh et al., 2011). Previously, we derived iPSCs from psychiatric disorder patients with a heterozygous 4 bp frameshift deletion at the DISC1 carboxy (C) terminus and reported that the mutant DISC1 (mDISC1) causes an 80% reduction of wild-type DISC1 (wtDISC1) protein levels in iPSC-derived cortical neurons (Chiang et al., 2011; Wen et al., 2014). Differential expression analyses of RNA-seq data comparing mDISC1 and control cortical neurons showed a significant downregulation of WNT/β-catenin pathway genes (Figure S7A).
We generated SNOs using mDISC1 iPSCs derived from two patients with psychiatric disorders and carrying the DISC1 mutation (D2 and D3) to compare with control SNOs using iPSCs derived from a member of the same family without the DISC1 mutation (C3) and a control iPSC line from outside of the family (C1; Chiang et al., 2011; Wen et al., 2014). The mDISC1 SNOs showed normal levels of apoptotic and necrotic cell death, formed organized progenitor zones and CP, and the layer size expansion closely mirrored control SNOs at all time points examined, further supporting the reproducibility of the SNO method (Figures S7B–S7D).
The CP of mDISC1 SNOs resembled control SNOs at days 70 and 100, when the distributions of SATB2 and TBR1 were inter-mingled (Figures 6A–6C). However, unlike the control, the laminar expression of SATB2, TBR1, RORB, and CTIP2 in day 120 mDISC1 SNOs from both patient lines lost layer specificity and remained intermingled, which persisted at day 150 and therefore was unlikely due to a temporary delay in differentiation (Figures 6A–6H). Quantitative analyses revealed that SATB2 and RORB expression patterns were not restricted to the upper cortical bins but instead spanned the entire CP in mDISC1 SNOs (Figures 6B and 6E). The SATB2+ neurons outnumbered TBR1+ neurons in both upper and deep layers, preventing the establishment of mutually exclusive expression domains between the two markers (Figures 6G and 6H).
Figure 6. Aberrant Laminar Expression Patterns of Cortical Layers in mDISC1 SNOs.
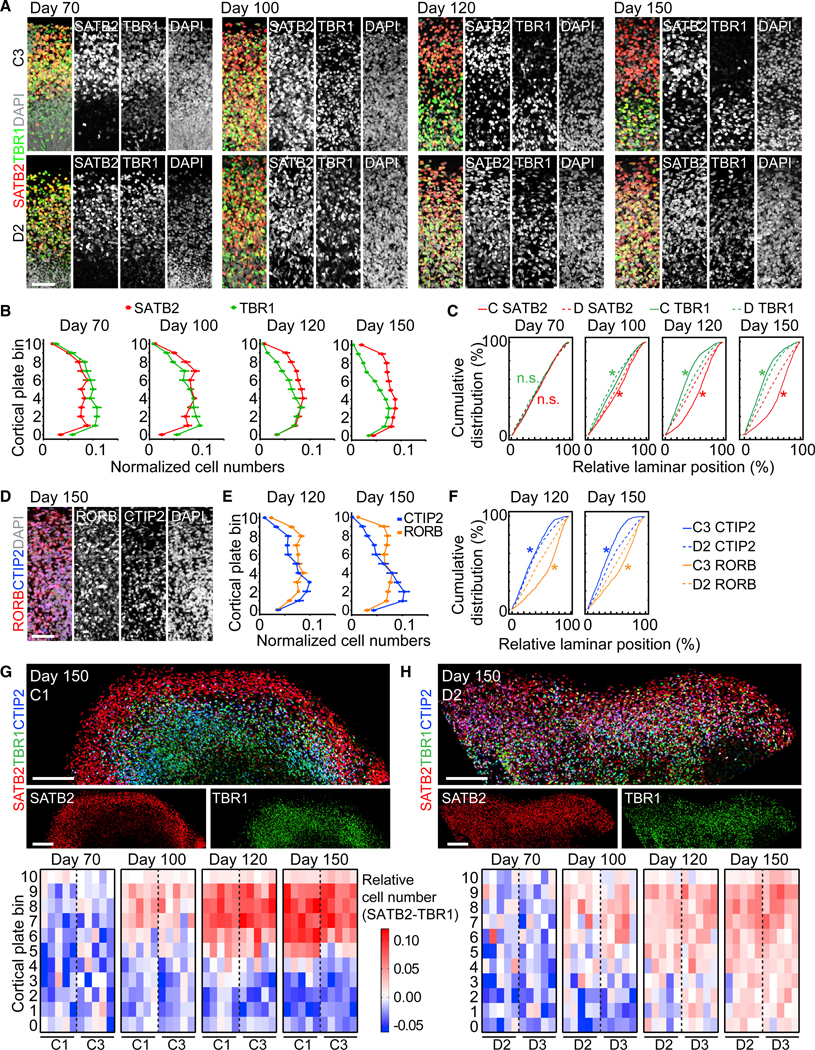
(A) Sample confocal images of the CP of control (C3; top panel) and mDISC1 (D2; bottom panel) SNOs for SATB2 and TBR1. Scale bar, 50 μm.
(B and C) Quantifications of the distribution of SATB2+ and TBR1+ neurons in mDISC1 SNOs, to be compared with Figure 4B. Values represent mean ± SEM (n = 10 SNOs from D2 and D3 iPSC lines). Also shown in (C) are cumulative distribution curves of marker-positive neurons at each time point. Kolmogorov-Smirnov tests were performed between control (C) and mDISC1 (D) SNOs for SATB2 (red) and TBR1 (green; *p < 0.05; n.s.: p > 0.05). Same samples as in (B) and Figure 4B are shown.
(D–F) Disorganized laminar distribution of RORB and CTIP2 in mDISC1 SNOs. Shown in (D) are 100 × 300 μm columns from sample confocal images at day 150. Scale bar, 50 μm. Shown in (E) are quantifications of marker distribution at days 120 and 150, to be compared with Figure 4E. Values represent mean ± SEM (n = 5 SNOs). Shown in (F) are cumulative distribution curves of marker-positive neurons at each time point. Kolmogorov-Smirnov tests were performed between control (C) and mDISC1 (D) SNOs for RORB (orange) and CTIP2 (blue) (n.s.: p > 0.05; *p < 0.05). Same samples as in (E) and Figure 4E are shown.
(G and H) Comparison of cortical layers between the control and mDISC1 SNOs. Shown at the top panel are sample tiling confocal images of the CP in day 150 SNOs. Scale bars, 200 μm. Shown at the bottom panel are heatmap plots for the differential abundance between SATB2+ and TBR1+ in control (G) and in mDISC1
(H) SNOs, similar to Figure 5D. Same samples as in (B) and Figure 4 B are shown.
See also Figure S7.
Co-expression between SATB2 and TBR1 and between RORB and CTIP2 also increased in mDISC1 SNOs, and the majority of double-positive neurons were located in the lower part of the CP (Figures 7A and 7B). These deficits of aberrant laminar distribution and elevated marker co-expression phenocopied control SNOs treated with the β-catenin antagonist IWR (Figures 5C–5F). Curiously, CUX1 and LAMP5 remained exclusively expressed in the upper layer in mDISC1 SNOs, similar to control SNOs (Figure S7E), indicating that the laminar expression abnormalities do not universally affect all cortical neuron subtypes and that LAMP5 and CUX1 expression patterns are possibly controlled independently of SATB2 and RORB. These results point to the model that the cortical disorganization observed in mDISC1 SNOs is not caused by a failure of cortical layer formation but due to specifically dysregulated cortical neuron subtype fate specification.
Figure 7. Cortical Neuron Fate Specification Deficits Caused by the DISC1 Mutation.
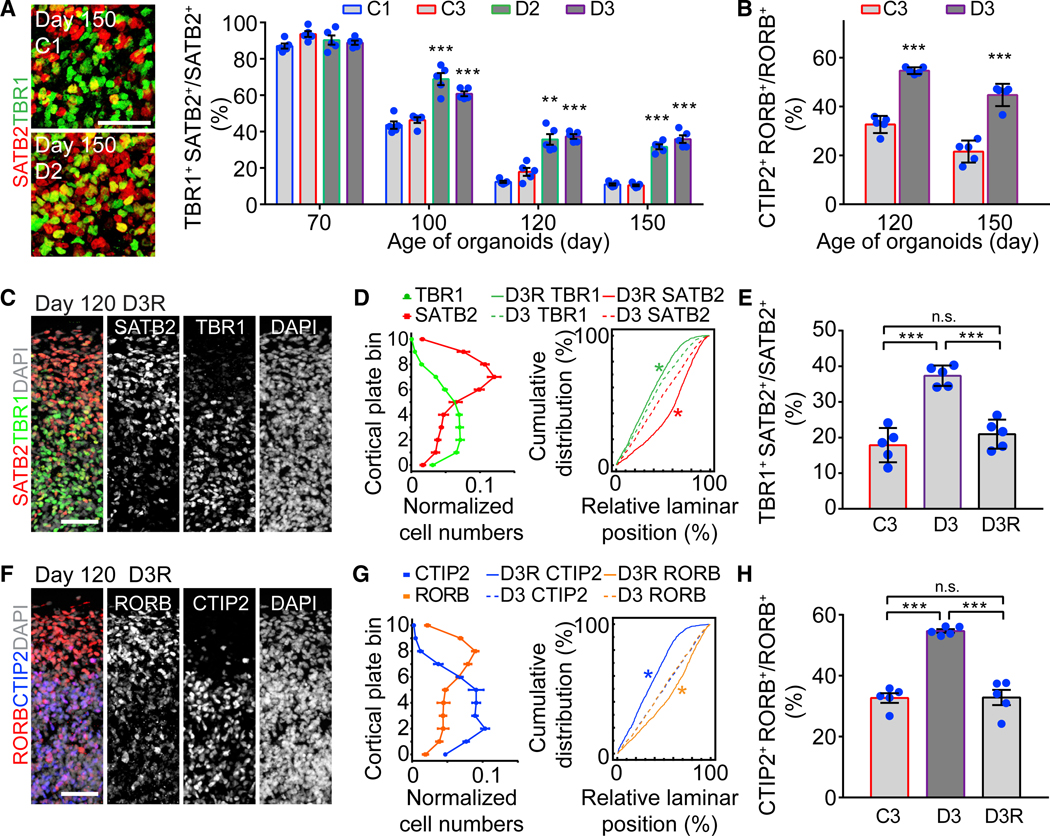
(A) Elevated co-expression between SATB2 and TBR1 in mDISC1 SNOs. Shown on the left are sample confocal images at the boundary between upper and deep layers with double-positive cells marked in yellow. Scale bar, 50 μm. Shown on the right is the quantification of the ratio of SATB2 and TBR1 co-expressing cells over SATB2+ neurons. Same sample as in Figure 6B is shown. Values represent mean ± SEM (***p < 0.0005; Student’s t test).
(B) Elevated co-expression ratio between RORB and CTIP2 in mDISC1 SNOs. Same sample as in Figure 6E is shown. Values represent mean ± SEM (***p < 0.0005; Student’s t test).
(C and D) Restored layer-specific expression patterns of SATB2 and TBR1 in day 120 SNOs derived from the isogenic D3R iPSC line, in which the DISC1 mutation was corrected. Shown in (C) are 100 × 300 μm columns from sample confocal image. Scale bar, 50 μm. Shown in (D) (left) is the quantification for marker distribution in the CP. Shown is similar to Figure 4B. Values represent mean ± SEM (n = 5 SNOs). Shown in (D) (right) are cumulative distribution curves. Kolmogorov-Smirnov test was performed between D3R and D3 SNOs for SATB2 (red) and TBR1 (green; *p < 0.05).
(E) Restored ratio of co-expression between SATB2 and TBR1 to a normal level in SNOs derived from the isogenic D3R iPSC line. Same samples as in (D) are shown. Values represent mean ± SD (***p < 0.0005; Student’s t test).
(F and G) Restored layer-specific expression patterns of RORB and CTIP2 in day 120 SNOs derived from the isogenic D3R iPSC line. Shown in (F) are 100 × 300 μm columns from sample confocal images. Scale bar, 50 μm. Shown in (G) (left) is the quantification for marker distribution in the CP. Values represent mean ± SEM (n = 5 SNOs). Shown in (G) (right) are cumulative distribution curves. Kolmogorov-Smirnov test was performed between D3R and D3 SNOs for RORB (orange) and CTIP2 (blue; *p < 0.05).
(H) Restored ratio of co-expression between RORB and CTIP2 to the normal level in D3R SNOs. Same samples as in (G) are shown. Values represent mean ± SD (***p < 0.0005; Student’s t test).
We previously generated an isogenic D3 iPSC line that corrected the 4-bp deletion of DISC1 (D3R iPSC line; Wen et al., 2014). At day 120, the laminar expression patterns of SATB2, TBR1, RORB, and CTIP2 were rescued in D3R SNOs, and their layer-specific distribution depicted a clear boundary between the upper and deep cortical layers (Figures 7C, 7D, 7F, and 7G). The ratio of marker co-expression was also restored to the normal level (Figures 7E and 7H). Thus, the 4-bp deletion DISC1 mutation is responsible for the fate specification deficits observed in mDISC1 SNOs.
DISCUSSION
The most remarkable feature of brain organoids over conventional in vitro 2D monolayer or 3D neurosphere models is the ability to recapitulate the elaborate architecture of the embryonic human brain via self-organization, which serves as the prerequisite for proper maintenance of the neural stem cell niche, neuronal migration, cell-cell interactions, and circuitry development. Previously published cortical organoid systems are generally most representative of early-to-mid-gestation human brain development and very useful for modeling diseases that are characterized by striking structural malformations that manifest early, such as microcephaly and lissencephaly (Bershteyn et al., 2017; Qian et al., 2016). Using a precisely controlled slicing method to overcome the diffusion barrier and prevent interior cell death, we address the fundamental limiting factor that has prohibited cortical organoids from mimicking the architecture of late-stage human cortical development. By exposing progenitor zones to the external culture environment while maintaining their structural integrity, the disk-shaped SNOs grow more healthily than traditional spherical brain organoids, allowing significantly larger cortical structures to emerge. One very recent study examined brain organoids from various published protocols and reported elevated ER stress and impaired cell subtype specification (Bhaduri et al., 2020). Although we did not specifically examine cellular stress under the SNO protocol, our method enables neuronal subtype specification and spatial segregation of upper- and deep-layer neurons, suggesting that this challenge can be overcome by improved organoid methodologies.
We showed that the SNO method is reproducible and generates consistent outcomes that can be reliably quantified for multiple iPSC lines. The protocol is simple to follow and is compatible with scaling up. The method may also be applied to organoid protocols for other brain regions or other organs as a universal approach to improve cell viability and sustain tissue expansion in 3D suspension cultures. Although the SNO method is highly effective at resolving interior hypoxia, the benefits of slicing may also include improvement of nutrient diffusion and changes in mechanical force. Hyperoxia culture has been employed in several organoid protocols (Bershteyn et al., 2017; Kadoshima et al., 2013; Watanabe et al., 2017), and it could be interesting in the future to test the combination of the slicing method with hyperoxia. Mechanical cutting has previously been employed with cortical organoids. One study used spring scissors to cut cortical organoids and reported the maintenance of structural integrity over repeated cutting and long-term culture, but the distribution of SATB2, CTIP2, and TBR1 did not exhibit upper-and deep-layer specificity at day 154 of culture (Watanabe et al., 2017). Another study used liquid-air interface culture for sectioned cerebral organoids (Giandomenico et al., 2019). Although the liquid-air interface culture significantly improved neuronal survivability, accelerated neuronal maturation, and promoted formation of aligned axonal tracks, it also resulted in flattening of the tissue and the VZ and SVZ structures were not preserved. In contrast, because we perform the slicing after the establishment of the VZ and aligned radial glia scaffolds, and return organoids immediately to a 3D suspension culture, this procedure does not disrupt the architectural integrity of the progenitor and neuronal layers.
The aforementioned protocols utilizing mechanical cutting and hyperoxia culture (Watanabe et al., 2017), or air-liquid interface culture (Giandomenico et al., 2019), were effective at reducing cellular hypoxia and improving cell viability, but the formation of distinct cortical layers was not achieved, suggesting that reducing interior cell death alone is insufficient. Through our systematic characterization of SNOs along their developmental trajectory, we propose that the formation of distinct cortical layers depends on a series of requirements fulfilled in the SNO method, but not in other protocols. First, SNOs generate enlarged oSVZs containing abundant populations of oRGs, which are the predominant NPCs producing upper-layer cortical neurons in the developing human cortex. Our analyses showed that oRGs are proliferative and actively produce neurons throughout days 70–150. Second, the organization of the ventricular structure and aligned radial glia scaffolds is preserved despite repeated slicing. The radial scaffolds are critical for the maintenance of the cytoarchitecture of both progenitor layers and CP and provide the guiding substrate for neuronal migration to reach the designated laminar location in the CP. Finally, the establishment of distinct expression patterns of upper- and deep-layer markers relies on neuronal fate specification, which is regulated both by cell-autonomous signals and layer-specific external signals. This process is slow in human neurons (Nowa-kowski et al., 2017), and thus, only after 4 to 5 months of culture can we observe a dramatic reduction in the number of neurons co-expressing upper- and deep-layer markers. However, long-term organoid cultures without organized oSVZs and radial scaffolds resulted in a gradual loss of cytoarchitecture and disrupted neuronal layers with intermingling marker expression (Velasco et al., 2019). Therefore, the SNO method distinguishes itself from existing organoid protocols by simultaneously meeting all three indispensable requirements. The laminar distribution of cortical neuron markers as well as morphologically distinct astrocyte subtypes in day 150 SNOs closely resembles their localization in the postnatal human cerebral cortex and reflects species-specific differences between human and rodents (Lake et al., 2016; Oberheim et al., 2009).
Despite the presence of OPCs and oligodendrocytes in SNOs, we did not observe wrapping of axons under transmission electron microscopy (data not shown). Three recently published organoid protocols have used additional signaling factors or genetic manipulations to specifically induce oligodendrogenesis and have successfully shown wrapping of axons by myelin using transmission electron microscopy (Kim et al., 2019; Madhavan et al., 2018; Marton et al., 2019). It is possible that the spontaneously born oligodendrocytes in our organoid cultures lack the signaling required for maturation and axonal myelination. Our approach does have the caveat of repeated slicing (once per month), which may cause some damages to axons and dendrites. However, we keep fairly thick sections (500 μm), and long-distance axonal processes and synaptic connections could be maintained in the SNO system. Our extracellular recording experiments demonstrated repeatable temporal patterns of recruitment and bursts among neurons across long distances in the CP. The robust synchronization of spontaneous firing observed in SNOs highlights the utility of this approach for investigating network properties in normal and disease-related conditions.
Because the establishment of distinct cortical layers and specification of neuron fate occur late in cortical development, the SNO method provides a unique platform to investigate phenotypes that cannot be modeled by other current organoid systems. We demonstrated the versatility of our system by identifying the critical role of WNT/β-catenin signaling in the regulation of human post-mitotic cortical neuron subtype identities and discovering potentially disease-relevant phenotypes in psychiatric disorder patient-derived organoids.
Collectively, the SNO method delivers a significant advance in brain organoid technologies and provides a platform for systematic and mechanistic studies to interrogate late-stage embryonic human brain development that is otherwise difficult to investigate in human fetal tissues or animal models. Laminar structural organization, a hallmark of the adult human cortex, is essential for brain functions, and its dysregulation has been implicated in various brain disorders. The formation of distinct cortical layers and recapitulation of cortical neuron fate specification in our organoid system opens new avenues for investigations of neuronal organization and circuitry formation during normal and disease-related human brain development.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
All unique/stable reagents and biological material generated in this study are available from the Lead Contact, Dr. Guo-li Ming (gming@pennmedicine.upenn.edu), with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation and characterization of iPSCs and adult human brain tissue
Human iPSC lines used in the current study were previously generated and fully characterized (Wen et al., 2014; Yoon et al., 2014). Skin biopsy samples were obtained from several individuals in a previously characterized American family, pedigree H (Sachs et al., 2005). C1 fibroblasts were from ATCC (CRL-2097). Generation of iPSC lines followed institutional IRB and ISCRO guidelines and was approved by Johns Hopkins University School of Medicine. Karyotyping analysis by standard G-banding technique was carried out by the Cytogenetics Core Facility at the Johns Hopkins Hospital or Cell Line Genetics. Results were interpreted by clinical laboratory specialists of the Cytogenetics Core or Cell Line Genetics. Surgical adult human brain tissues used for validation of immunostaining were obtained following institutional IRB approved by University of Pennsylvania Perelman School of Medicine.
METHOD DETAILS
Maintenance of iPSCs
All cell cultures were maintained in 5% CO2 incubators at 37○C. Human iPSCs were cultured in stem cell medium consisting of DMEM:F12 supplemented with 20% KnockOut Serum Replacement, 1X Non-essential Amino Acids, 1X Penicillin/Streptomycin, 1X 2-Mercaptoenthanol, 1X Glutamax, and 10 ng/ml FGF-2 as previously described (Yoon et al., 2014). Culture medium was changed every day. Human iPSCs were passaged every week onto a new plate pre-seeded one day in advance with irradiated CF1 mouse embryonic fibroblasts (Charles River). Human iPSCs were detached from the plate by treatment of 1 mg/ml Collagenase Type IV for 1 hr and were further dissociated into smaller pieces by manual pipetting. The iPSCs used throughout the study were below passage 50. All studies were performed with approved protocols of University of Pennsylvania.
Generation of forebrain organoids
Generation of forebrain organoids from iPSCs was performed as previously described (Qian et al., 2018; Qian et al., 2016). First, iPSC colonies were detached with Collagenase Type IV 7 days after passage and washed with fresh stem cell medium in a 15 mL conical tube. On Day 0, detached iPSC colonies were transferred to an ultra-Low attachment 6-well plate (Corning Costar), containing 3 mL of stem cell medium (without FGF-2), plus 2 μM Dorsomorphine and 2 μM A83–01. On Day 5 and Day 6, half of the medium was replaced with induction medium consisting of DMEM:F12, 1X N2 Supplement, 1X Penicillin/Streptomycin, 1X Non-essential Amino Acids, 1X GlutaMax, 1μM CHIR99021, and 1 μM SB-431542. On Day 7, organoids were embedded in Matrigel and cultured in the induction medium for 7 more days. On Day 14, embedded organoids were mechanically dissociated from Matrigel by manual pipetting in a 5 mL pipette tip. Typically, 20 organoids were transferred to each well of a 12-well spinning bioreactor (SpinU) (Qian et al., 2016) containing differentiation medium consisting of DMEM:F12, 1X N2 and B27 Supplements, 1X Penicillin/Streptomycin, 1X 2-Mercaptoenthanol, 1X Non-essential Amino Acids, 2.5 μg/ml human Insulin. From Day 35 to Day 70, extracellular matrix (ECM) proteins was supplemented in differentiation medium by dissolving Matrigel at 1% (v/v). At Day 70, differentiation medium was exchanged with maturation medium consisting of Neurobasal medium, 1X B27 Supplement, 1X Penicillin/Streptomycin, 1X 2-Mercaptoenthanol, 0.2 mM Ascorbic Acid, 20 ng/ml BDNF, 20 ng/ml GDNF, and 1 μM cAMP. Detailed step-by-step procedures for generating forebrain organoids, building a SpinU bioreactor and required 3D printing files can be found in Qian et al. (2018). For comparison between unsliced forebrain organoids with SNOs over long-term culture, forebrain organoids were transferred to ultra-Low attachment 6-well plate placed on a CO2 resistant orbital shaker (ThermoFisher) rotating at 120 rpm in the incubator after Day 45.
Generation of sliced neocortical organoids
Forebrain organoids at Day 45 were collected from the SpinU bioreactor using a cut P1000 pipette tip and immersed in melted 3% low melting point agarose dissolved in DMEM:F12 kept at 37°C in a custom cubic mold of 1.5 cm sides. Typically, up to 10 organoids were embedded in one agarose block. Before the agarose solidified, multiple organoids were spread evenly and arranged in the same horizontal plane, with minimal spacing of 1 mm in between. The agarose blocks were placed on ice for 5–10 mins to solidify. Slicing was performed using a Leica VT 1200S vibratome in ice-cold DMEM:F12 medium at 0.1 mm/s speed and 1 mm vibration amplitude. The 500 μm-thick organoid slices were separated from the agarose by gentle pipetting. The sections from the middle of an organoid were collected, while sections from the top and bottom ends were discarded. The organoid sections were transferred to 6-well plate with differentiation medium supplemented with 1% Matrigel (v/v), left to equilibrate for 1 hr in stationary culture and placed on a CO2 resistant orbital shaker rotating at 120 rpm in the incubator. After Day 70, the medium was switched to maturation medium and organoids can be maintained for up to a year with medium change every 2 days. Repeated slicing was performed every 4 weeks after initial slicing to prevent disk-shaped SNOs from growing back to a spherical or oval shape. During embedding, SNOs were carefully oriented in the agarose mold to ensure that the x–y horizontal plane (Figure 1A) is parallel to the previous slicing plane, and when necessary, the position and orientation of SNOs were adjusted under a stereo microscope (Zeiss Discovery V8). After slicing, only one 500 μm-thick section in the middle was collected and cultured following the procedures described above. After Day 35, the time schedule for SNO cultures (Figure 1A) was followed with a flexibility range of ± 3 days. SNOs were collected using a cut P1000 pipette tip when used for analyses on the indicated age (±3 days).
Tissue preparation
Whole organoids were fixed in 4% Paraformaldehyde in Phosphate Buffered Saline (PBS) for 30 mins at room temperature. Organoids were washed 3 times with PBS and then immersed in 30% sucrose solution overnight. Organoids were embedded in tissue freezing medium and sectioned with a cryostat (Leica) at 30 μm thickness unless otherwise specified. The disk-shaped SNOs were oriented carefully in the tissue freezing medium to ensure that the x–y horizontal plane (Figure 1A) is parallel to the section plane. The first 5 and last 5 sections for each SNO were not used for any immunohistochemistry analysis.
Immunohistochemistry and microscopy
For immunostaining, cryosectioned slides were washed with PBS before permeabilization with 0.5% Triton-X in PBS for 1 hr. Tissues were blocked with blocking medium consisting of 10% donkey serum in PBS with 0.05% Triton-X (PBST) for 30 mins. Primary antibodies diluted in blocking solution were applied to the sections overnight at 4°C. The primary antibodies used and their dilution are summarized in the Key Resources Table. After washing with PBST for a minimum of 5 times, secondary antibodies diluted in blocking solution were applied to the sections for 1–4 hr at room temperature or overnight at 4°C. Finally, sections were washed with PBST for a minimum of 5 times before mounting. Secondary antibodies were: AlexaFluor 488, 546, 594, or 647 -conjugated donkey antibodies (Invitrogen) used at 1:500 dilution. Images were captured by a confocal microscope (Zeiss LSM 800). Sample images were prepared in ImageJ (NIH) and Photoshop (Adobe) software.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BRN2 | Santa Cruz | sc-6029 |
| Calretinin | Swant | 7697 |
| ChAT | Chemicon International | AB144P-200UL |
| Cleaved Caspase-3 (Asp175) | Cell Signaling | #9661 |
| CTIP2 | Abcam | ab18465 |
| CUX1 | Santa Cruz | sc-514008 |
| CUX2 | Abcam | ac130395 |
| FAM107A | Sigma | HPA055888 |
| GFP | Aves | GFP-1020 |
| GFAP | DaKO | Z0334 |
| HOPX | Santa Cruz | sc-30216 |
| KI67 | BD | 550609 |
| Laminin | Abcam | ab11575 |
| LAMP5 | Sigma | HPA070765-100UL |
| MBP | Millipore | MAB395-1mL |
| Nestin | Aves | NES |
| Neurofilament (SMI312) | BioLegend | 837904 |
| NG2 | Millipore | AB5320 |
| NKX2.2 | DSHB | 74.5A5 |
| nNOS | Invitrogen | 61–7000 |
| NPY | Santa Cruz | sc-28943 |
| OLIG2 | Millipore | AB9610 |
| PAX6 | BD | 561664 |
| PRDM8 | Sigma | HPA044298 |
| PTPRZ1 | Sigma | HPA015103 |
| Parvalbumin | Swant | PVG213 |
| RORB | Sigma | HPA008393–100UL |
| SATB2 | Abcam | ab51502 |
| Somatostatin | Santa Cruz | sc-7819 |
| SOX2 | Santa Cruz | sc-17320 |
| Synaptophysin | R&D | AF5555 |
| TBR1 | Abcam | ab31940 |
| TBR2 | Sigma | HPA028896–100UL |
| WNT7B | Abcam | ab94915 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM:F12 | Invitrogen | cat. # 11330032 |
| Neurobasal medium | GIBCO | cat. # 21103049 |
| Phosphate Buffered Saline | GIBCO | cat. # 10010023 |
| KnockOut Serum Replacement | GIBCO | cat. # 10828028 |
| Non-essential Amino Acids | GIBCO | cat. # 11140050 |
| Penicillin/Streptomycin | GIBCO | cat. # 15140122 |
| 2-Mercaptoenthanol | GIBCO | cat. # 21985023 |
| Glutamax | GIBCO | cat. # 35050061 |
| Collagenase Type IV | Invitrogen | cat. # 17104019 |
| FGF-2 | Peprotech | cat. # 100–18B |
| Dorsomorphine | StemCell Technologies | cat. # 72102 |
| A83–01 | StemCell Technologies | cat. # 72022 |
| N2 Supplement | GIBCO | cat. # 17502048 |
| B27 Supplements | GIBCO | cat. # 17504044 |
| CHIR99021 | StemCell Technologies | cat. # 72052 |
| SB-431542 | StemCell Technologies | cat. # 72232 |
| IWR-1-endo | StemCell Technologies | cat. # 72564 |
| Matrigel | Corning | cat. # 354230 |
| Human Insulin solution | Sigma | cat. # I0516 |
| Ascorbic Acid | Sigma | cat. # 1043003 |
| BDNF | Peprotech | cat. # 450–02 |
| GDNF | Peprotech | cat. # 450–10 |
| Cyclic AMP | Sigma | cat. # A9501 |
| UltraPure low melting point agarose | Invitrogen | cat. # 16520100 |
| Paraformaldehyde | Polysciences | cat. # 18814–10 |
| Sucrose | Sigma | cat. # S5016 |
| Tissue freezing medium | General Data | cat. # TFM-5 |
| Triton-X | Sigma | cat. # T9284 |
| donkey serum | Millipore | cat. # S30 |
| BrainPhys neuronal medium | StemCell Technologies | cat. # 05790 |
| NeuroCult SM1 supplement | StemCell Technologies | cat. # 05711 |
| Recombinant human HAPLN1 | R&D | cat. # 2608-HP-025 |
| Recombinant human lumican | R&D | cat. # 2846-LU-050 |
| human collagen I-based hydrogel (Cellmatrix type I-A) | Kyowa chemical products | N/A |
| HB buffer (1 mM DTT) | Sigma | cat. # D0632 |
| Spermine | Sigma | cat. # S4264-1G |
| Spermidine | Sigma | cat. # S0266-1G |
| EDTA-free protease inhibitor | Roche | cat. # 11836170001 |
| IGEPAL-630 | Sigma | cat. # I8896-50ML |
| MgCl2 | Thermo Fisher | cat. # AM9530G |
| Tricine-KOH | Sigma | cat. # T5816-100G |
| RNase Inhibitor | Enzymatics | cat. # Y924L |
| EvaGreen® Dye, 20X in Water | Biotium | cat. # 31000 |
| Kapa Pure Beads | KAPA Biosystems | cat. # KK8000 |
| T4 DNA Ligase | New England Biolabs | cat. # M0202S |
| Critical Commercial Assays | ||
| RNeasy mini kit | QIAGEN | cat. # 74104 |
| RNase-Free DNase Set | QIAGEN | cat. # 79254 |
| Click-iT® EdU Alexa Fluor® 488 Imaging kit | ThermoFisher | cat. # C10337 |
| NEBNext® Ultra RNA Library Prep Kit for Illumina | New England Biolabs | cat. # E7530L |
| Nextera XT DNA Library Preparation Kit | Illumine | cat. # FC-131-1024 |
| Dynabeads MyOne Streptavidin C1 Kit | Thermo Fisher | cat. # 65001 |
| Kapa HiFi HotStart Master Mix | KAPA Biosystems | cat. # KK2600 |
| Recombinant DNA | ||
| Ad-CMV-eGFP | Vector Biolabs | cat. # 1060 |
| Deposited Data | ||
| Single-nucleus RNA-sequencing data of SNO | This paper | GSE137941 |
| Experimental Models: Cell Lines | ||
| Human WT iPSC line “C1–2” derived from healthy male fibroblasts | ATCC fibroblasts (CRL-2097); (Wen et al., 2014) | C1–2 |
| Human WT iPSC line “C3–1” derived from healthy female fibroblasts from the “pedigree H” | Johns Hopkins University School of Medicine (Wen et al., 2014) | C3–1 |
| Human Schizophrenia patient iPSC line “D2–1” derived from female fibroblasts from the “pedigree H,” carrying heterozygous 4-bp deletion mutation of DISC1 gene. | Johns Hopkins University School of Medicine (Wen et al., 2014) | D2–1 |
| Human Major Depression patient iPSC line “D3–2” derived from female fibroblasts from the “pedigree H,” carrying heterozygous 4-bp deletion mutation of DISC1 gene. | Johns Hopkins University School of Medicine (Wen et al., 2014) | D3–2 |
| Isogenic human iPSC line derived from “D3–2,” with the DISC1 mutation corrected using transcription activator-like effector nuclease (TALEN) | Johns Hopkins University School of Medicine (Wen et al., 2014) | D3-R |
| Software and Algorithms | ||
| Zeiss Zen (Blue edition) | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| NextSeq System Suite v2.0 | Illumine | https://support.illumina.com/downloads/nextseq-system-suite-v2–0.html |
| Neuralynx Cheetah | Neuralynx | https://neuralynx.com/software/cheetah |
| Axon AxoScope 10.4 | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/18779/~/axon%E2%84%A2pclamp%E2%84%A2-10-electrophysiology-data-acquisition-%26-analysis-software-download |
| ImageJ (Fiji) | NIH | https://imagej.nih.gov/ij/docs/guide/146–2.html |
| GraphPad Prism (8.0) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/p/excel/cfq7ttc0k7dx?activetab=pivot%3aoverviewtab |
| Adobe Photoshop(CC) | Adobe | https://www.adobe.com/products/photoshop.html |
| Adobe Illustrator (CC) | Adobe | https://www.adobe.com/products/illustrator.html |
| TopHat (2.0) | Johns Hopkins University | https://ccb.jhu.edu/software/tophat/index.shtml |
| R, R studio with R package Seurat (v 2.3.4) | RStudio | https://cran.r-project.org/web/packages/Seurat/index.html |
| R studio with edgeR (v2.15) package | RStudio | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4023662/ |
| R package ClustifyR | RStudio | https://github.com/rnabioco/clustifyr |
| Axon CLAMPfit | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/18779/~/axon%E2%84%A2pclamp%E2%84%A2–10-electrophysiology-data-acquisition-%26-analysis-software-download |
| KlustaKwik (1.5) | Klusta | http://klustakwik.sourceforge.net/ |
| Neuralynx SpikeSort3D | Neuralynx | https://neuralynx.com/software/spikesort-3d |
| MathWorks MATLAB | MathWorks | https://www.mathworks.com/ |
| SpinU 12-well Spinning Bioreactor | (Qian et al., 2016), (Qian et al., 2018) | N/A |
| Bulk RNA sequencing data of mutant DISC1 neurons | (Wen et al., 2014) | GSE57821 |
Analysis of hypoxia
Detection of hypoxic cells in unsliced organoids and SNOs was performed using Hypoxyprobe Kit (HPI). Pimonidazole HCL was added to culture medium at 200 μM working concentration, and incubated with organoids for 2 hr before fixation for analysis. Immunochemical detection of hypoxic cells containing pimonidazole was performed following the manufacturer’s instructions. SNOs and unsliced organoids were analyzed at Days 60, 110 and 140. All three dates were 2 weeks after a routine slicing for SNOs to ensure consistency. Only the cryosections in the very middle of the organoids were used for this analysis. Images were captured by a confocal microscope (Zeiss LSM 800) using the same acquisition parameters (laser intensity and gain), and “tile”/ “stitching” functions in the Zen software (Zeiss) were used when necessary. To quantify the percentage of hypoxic area in sliced and unsliced organoids, the area labeled by the hypoxyprobe was measured using ImageJ software and divided by the total organoid area marked by DAPI.
Analyses of cell death
For quantification of the necrotic area in SNOs and unsliced organoids, organoid cryosections were immunostained with Cleaved-Caspase-3 (Cas3) and SOX2. Only sections in the middle were analyzed. The entire organoid section was imaged using a confocal microscope, and “tile”/ “stitching” functions in the Zen software (Zeiss) were used when necessary. The necrotic area was defined as the interior portion of the organoid marked by: (1) diffusive non-cell-specific signals of Cas3 immunoreactivity; (2) absence of SOX2+ cells and (3) fragmented nuclei morphology stained by DAPI. The total area and necrotic area of an organoid was manually drawn and measured using ImageJ software. For quantification of the percentage of apoptotic cells, random cortical structures outside the necrotic area were imaged under a confocal microscope at single z-plane. A randomly selected 400×400 μm area containing both the progenitor and neuronal layers was analyzed. The number of cells positive for Cas3 were counted and divided by the total cell number labeled by DAPI using ImageJ software.
Analyses of layer thickness
Analyses of progenitor zone thickness, cortical plate/subplate (CP) thickness and total thickness were performed similarly to previously described methods (Qian et al., 2016). Only cryosections near the middle of each organoid were used for analyses. SNOs and unsliced organoids were immunostained for SOX2, TBR2 and CTIP2. Random cortical structures were imaged under a confocal microscope and 15 μm z stacks were projected with maximum intensity in ImageJ software. The ventricular zone (VZ) was defined by exclusive SOX2 immunoreactivity and neural tube-like morphology. The outer subventricular zone (oSVZ) was defined by the region containing mixed populations of SOX2+, CTIP2+ and TBR2+ nuclei outside the VZ. The CP was defined by the region from the boundary of the oSVZ to the outer surface containing exclusively CTIP2+ nuclei. After defining the boundaries of each layer, the layer thickness was measured in the direction with maximum radial distance from the ventricular (apical) surface to the pial (basal) surface using ImageJ software. For old organoids (typically > 120 day) where the VZ shrunk in size and became difficult to define morphologically, the apical surface was defined at the center of the progenitor zone. The progenitor zone thickness was the sum of VZ and oSVZ, and the total thickness was the sum of progenitor zone and CP thickness.
Analyses of progenitor cell proliferation
For quantification of cell proliferation, SNOs and unsliced organoids were immunostained for SOX2 and KI67. Random cortical structures were imaged using a confocal microscope. Fan-shaped regions from the apical surfaces of the VZ to the boundary between the oSVZ and CP were cropped for analyses. KI67+ nuclei were counted and divided by the total number of nuclei stained by DAPI in the region.
Viral infection
For sparse labeling of radial glia cells and astrocytes with GFP, AV-CMV-GFP adenovirus (Vector Biolabs, titer = 1×1010 PFU/ml) was added into maturation medium at 1:2000 dilution, and incubated with SNOs for 24 hr before being washed away. SNOs were analyzed 3–5 days after exposure to virus, as indicated in figure legends, and immunohistochemistry against GFP was used to enhance the fluorescence signals.
EdU labeling and quantification of cell identity
For Figure 2F, SNOs at Days 63, 100 or 146 were pulsed with 10 μM EdU for 1 hr. The media was then replaced and SNOs were washed 3 times with fresh media to remove residual EdU. After 7 days, SNOs were fixed for immunohistochemistry and EdU detection using Click-iT® EdU Alexa Fluor® 488 Imaging Kit according to the manufacturer’s manual. Cortical structures were randomly imaged under a confocal microscope at the single z-plane. Each EdU+ nucleus in the insets was traced in Photoshop software (Adobe), and manually pseudo-colored to indicate whether it expresses SATB2 or CTIP2, or both.
For Figure 2G, Day 95 SNOs were pulsed with 10 μM EdU for 24 hr and analyzed 3 or 7 days later. The number of EdU+ cells expressing SOX2, SATB2, and TBR2 were counted in ImageJ software. The positions of all EdU+ nuclei were manually marked using the “Cell Counter” plugin in ImageJ. Their y-coordinates on the image were recorded and normalized to the total thickness from apical to basal surfaces of cortical structures to measure their relative laminar positions. The frequency distributions of the relative vertical positions in 11 evenly divided bins for each marker were calculated and plotted in Prism software (GraphPad) (Figure 2I).
For Figures S6B–S6D, Day 95 SNOs were pulsed with 10 μM EdU for 24 hr and analyzed 28 days later (Day 123). SNOs were treated with DMSO, IWR or CHIR from Day 103 to Day 123. The number of EdU+ cells expressing TBR1, SATB2, or co-expressing both was counted in ImageJ software. The ratio of co-expression was calculated by [#TBR1+SATB2+EdU+ nuclei/#SATB2+EdU+ nuclei]. Each EdU+ nucleus in the insets was traced in Photoshop software (Adobe), and manually pseudo-colored to indicate whether it expresses SATB2 or TBR1, or both (Figure S6C).
Analyses of cortical neuron distribution
SNOs were immunostained for SATB2 and TBR1 and random cortical structures were imaged on a confocal microscope in a single z-plane. From the pial surface, radial columns of 100 μm width and 200–500 μm length covering the full thickness of the CP layer were cropped for analyses. The positions of all SATB2+, TBR1+ and double-positive nuclei were separately marked using the “Cell Counter” plugin in ImageJ. Their y-coordinates on the image were recorded and normalized to the full thickness of the CP to measure their relative laminar positions in the CP. The frequency distributions of the relative vertical positions in 11 evenly divided bins for each marker were calculated and plotted in Prism software (GraphPad). The ratio of co-expression was calculated by [#TBR1+ SATB2+ nuclei/#SATB2+ nuclei]. For highlighting the differences between the relative cell numbers of SATB2+ nuclei and TBR1+ nuclei within each bin, a heatmap was plotted in Prism software to represent [(normalized SATB2+ nuclei #) – (normalized TBR1+ nuclei #)], with red indicating a positive value (more SATB2) and blue indicating a negative value (more TBR1). The analyses for RORB and CTIP2, and additional neuron markers were performed similarly. Kolmogorov-Smirnov tests were performed using a web-based tool (http://www.physics.csbsju.edu/stats/KS-test.n.plot_form.html). The relative locations of all marker+ nuclei were used to plot the cumulative distribution curve and calculate the P values.
For the β-Catenin agonist/antagonist treatment experiments, CHIR99021 or IWR-1-endo was added to the maturation medium at the specified concentrations to SNOs at Day 100 with medium change every 2 days. After 20 days (Day 120), SNOs were collected for analysis while the remaining SNOs were washed and cultured in fresh medium without the drugs till Day 150 for analyses.
In vitro induction of cortical folding using ECM factors
The induction of wrinkles in the CP in slice-cultured SNOs was performed following published methods for human cortical slices with some modifications (Long et al., 2018). SNOs at Day 95, 110 or 125 were further sliced into 200 μm thick sections using a vibratome following similar procedures described above. Each section was cut in half at the midline with a scalpel to mimic the shape of an organotypic cortical slice used in the published study (Long et al., 2018). Sections were resuspended in a human collagen I-based hydrogel supplemented with 5 μg/mL recombinant human HAPLN1 and 5 μg/ml recombinant human lumican for the “LHC” condition, the equivalent volume of PBS was added to the hydrogel solution for the control condition. Droplets of hydrogel solution each containing one SNO section was placed on a glass-bottom Petri dish (Matek) and incubated in the incubator for 30 mins before 3 mL of maturation medium was added to the dish. The embedded sections were then cultured in the stationary culture for an additional 2 to 4 days before analyses. Fixed sections were cryosectioned in the parallel direction for immunohistochemistry analyses. The CP of SNO sections was imaged using a confocal microscope and the gyrification index was calculated by [inner length/outer length].
Quantification of interneuron subtypes and OPCs
For the quantitative analyses in Figures S3C and S3I, immunostaining was performed for the corresponding marker on random Day 150 SNOs and imaged under confocal microscopy. For each SNO, three 500 × 500 μm areas were imaged under 20X magnification without Z stack. The number of marker positive cells and total cells stained by DAPI were manually counted in ImageJ software. The total counts of three areas were summed for each SNO to calculate the percentage of marker positive cells, and represented as one data point in the plot.
Quantification of astrocyte subtypes
For the quantitative analyses in Figure S3G, immunostaining of GFAP was performed on random Day 150 SNOs and imaged under confocal microscopy. For each SNO, three 500 × 500 μm areas were imaged under 20X magnification with a Z stack depth of 20 μm. The morphology of GFAP+ cells after Z-projection was used to determine the subtype identity of astrocytes. Astrocytes with multiple long fiber-like processes were labeled as fibrous astrocytes, and astrocytes with complex processes branching from thicker stem branches are labeled as protoplasmic astrocytes. The primate-specific interlaminar astrocytes were identified by cell soma in the marginal zone and long, descending processes into the cortical plate layers. Because we performed this analysis on single cryosections, we could not visualize the complete morphology of every GFAP+ astrocyte as some glial processes were inevitably cut during the sectioning. We categorized these astrocytes lacking complete morphology as “undefined” in our analysis. For each subtype, the counts of three areas were summed for each SNO to calculate the proportion in total GFAP+ cells, and represented as one data point in the plot.
Single-nucleus RNA-seq and data analyses
Single-nucleus RNA sequencing was performed following the SPLiT-seq method with minor modifications (Rosenberg et al., 2018). Nuclei isolated from fresh SNOs was performed as previously described (Su et al., 2017). Briefly, tissue was minced with a razor blade and homogenized using a tissue grinder in a 1 mL of HB buffer (1 mM DTT), 0.15 mM spermine, 0.5 mM spermidine, EDTA-free protease inhibitor, 0.3% IGEPAL-630, 0.25 M sucrose, 25 mM MgCl2, 20 mM Tricine-KOH) for 5 to 10 strokes, then filtered through a 40 μm strainer, underlayer with a cushion buffer (0.5 mM MgCl2, 0.5 mM DTT, EDTA-free protease inhibitor, 0.88 M sucrose) and centrifuged at 2800 g for 10 minutes in a swinging bucket centrifuge at 4°C. Nuclei were collected as pellets.
Nuclei were then centrifuged for 3 mins at 500 g at 4°C. The pellet was resuspended in 1 mL of cold PBS-RI (1x PBS + 0.05 U/μl RNase Inhibitor). The nuclei were passed through a 40 μm strainer. 3 mL of cold 1.33% formaldehyde solution was then added to 1 mL of cells. Nuclei were fixed for 10 mins before adding 160 μL of 5% Triton X-100. Nuclei were then permeabilized for 3 mins and centrifuged at 500 g for 3 mins at 4○C. Nuclei were resuspended in 500 μL of PBS-RI before adding 500 μL of cold 100 mM Tris-HCl pH 8. Then, nuclei were spun down at 500 g for 3 mins at 4°C and resuspended in 300 μL of cold 0.5 X PBS-RI. Finally, nuclei were again passed through a 40 μm strainer and then counted on a hemocytometer, diluted to 1,000,000 cells/ml.
mRNA from single nuclei were tagged 3 rounds with barcoded primers, with in-cell ligations using T4 DNA ligase. Plates were incubated for 30 mins at 37°C with gentle shaking (50 rpm) to allow hybridization and ligation to occur. The ligation products were purified with Dynabeads MyOne Streptavidin C1 beads. After washing beads once with 10 mM Tris and 0.1% Tween-20 solution and once with water, beads were resuspended into a solution containing 110 μL of 2X Kapa HiFi HotStart Master Mix, 8.8 μL of 10 μM stocks of primers BC_0062 and BC_0108, and 92.4 μL of water. PCR thermocycling was performed as follows: 95°C for 3 mins, then five cycles at 98°C for 20 s, 65°C for 45 s, 72°C for 3 mins. After these five cycles, Dynabeads beads were removed from PCR solution and EvaGreen dye was added at a 1X concentration. Samples were again placed in a qPCR machine with the following thermocycling conditions: 95°C for 3 mins, cycling at 98°C for 20 s, 65°C for 20 s, and then 72°C for 3 mins, followed by a single 5 mins at 72°C after cycling. Once the qPCR signal began to plateau, reactions were removed.
PCR reactions were purified using a 0.8X ratio of KAPA Pure Beads and cDNA concentration was measured using a qubit. For tagmentation, a Nextera XT Library Prep Kit was used. 600 pg of purified cDNA was diluted in water to a total volume of 5 μl. 10 μL of Nextera TD buffer and 5 μL of Amplicon Tagment enzyme were added to bring the total volume to 20 μl. After mixing by pipetting, the solution was incubated at 55°C for 5 mins. A volume of 5 μL of neutralization buffer was added and the solution was mixed before incubation at room temperature for another 5 mins. PCR was then performed with the following cycling conditions: 95°C for 30 s, followed by 12 cycles of 95°C for 10 s, 55°C for 30 s, 72°C for 30 s, and 72°C for 5 mins after the 12 cycles. 40 μL of this PCR reaction was removed and purified with a 0.7X ratio of SPRI beads to generate an Illumina-compatible sequencing library.
Sequencing was performed with 50 bp paired end sequencing by Illumina NextSeq 550. Data processing was preprocessed using Drop-seq-1.13 (Macosko et al., 2015) with modifications. Briefly, after mapping the reads to the human genome (hg38, Gencode release V28), both exonic and intronic reads mapped to the predicted strands of annotated genes were retrieved for the cell type classification. Uniquely mapped reads were grouped by cell barcode. To digitally count gene transcripts, a list of UMIs in each gene, within each nucleus, was assembled, and UMIs within ED = 1 were merged together. The total number of unique UMI sequences was counted, and this number was reported as the number of transcripts of that gene for a given nucleus. Raw digital expression matrices were generated for the 1 sequencing runs. The raw digital expression matrices were combined and loaded into the R package Seurat (v 2.3.4) (Butler et al., 2018). For normalization, UMI counts for all nuclei were scaled by library size (total UMI counts), multiplied by 10,000 and transformed to log space. As a result, 6,888 nuclei were kept for further analysis.
For clustering, the highly variable genes were identified using the function FindVariableGenes with the parameters: x.low.cutoff = 0.01, x.high.cutoff = 3 and y.cutoff = 0.8 in Seurat, resulting in 2,677 highly variable genes. The expression level of 2,677 genes in the nuclei was scaled and centered along each gene for PCA analysis. We selected different cut-offs of the number of PCs and empirically found that downstream clustering analyses were optimized when using a 25-PC cutoff. The first 25 PCs were selected and used for two-dimension t-distributed stochastic neighbor embedding (tSNE), implemented by the Seurat software with default parameters. Based on the tSNE map, 12 clusters were identified using the function FindCluster in Seurat with the resolution parameter of 0.6. To identify marker genes, differential expression analysis was performed by the function FindAllMarkers in Seurat with likeli-hood-ratio test. Differentially expressed genes that were expressed in at least 10% cells within the cluster and with a fold change more than 0.5 (log scale) were considered to be marker genes.
To compare cell-type classifications between SNO data and single-cell RNA-seq data of second trimester human fetal prefrontal cortex, we first downloaded the raw UMI count matrix from Zhong et al. (2018). Using identical clustering parameters in Seurat defined above for the SNO analysis, we identified 11 clusters in the human fetal cortex dataset that were annotated based on the expression and enrichment of known biologically-relevant marker genes. Using the R package ClustifyR, we converted the fetal cortex dataset Seurat object into a cluster-specific average expression matrix using the function “use_seurat_comp”and considered only the highly variable genes in the output expression matrix. Using the function clustify in clustifyR with the argument compute_method = “pearson,” we computed the cluster-specific Pearson correlation coefficients between the SNO dataset and the Zhong et al. average expression matrix.
To compare the astroglial cells in SNOs to fetal and postnatal human primary astrocytes, we obtained bulk RNA-seq datasets from a published study (Zhang et al., 2016). Differential expression analysis was performed using edgeR (v2.15.0), from which we generated two lists of top 50 genes highly expressed by fetal and postnatal astrocytes. The relative expression of these genes was plotted for the single nuclei in AG cluster from our SNO dataset (Figure S4E).
Whole-cell recording in SNO slices
SNOs were sliced into 200 μm thick sections on a vibratome following procedures described above. Slices were placed in maturation medium in the incubator to recover for 30 mins and then transferred into a recordings chamber perfused with ACSF solution consisted of the following (in mM): 130 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 1 MgCl2, and 2 CaCl2 (pH 7.2–7.4 when saturated with 95% O2/5% CO2). The osmolarity of all solutions was 305–315 mOsm. Slices were viewed using infrared differential interference contrast optics under an upright microscope (Slice Scope Pro, Scientifica) with a 40x water-immersion objective. Whole-cell postsynaptic patch-clamp recordings were made using glass pipettes of 4–8 MU resistance, filled with an internal recording solution of the following (in mM): 145 K-Gluconate, 2 MgCl2, 0.1 BAPTA, 2.5 KCl, 2.5 NaCl, 10 HEPES, 0.5 GTP. Tris, 2 Mg-ATP, 2 QX-314, pH: 7.2, 280–290 mOsm. In combination with the electrophysiological recordings we added 0.2% biocytin to the internal solution in order to label the patched neurons. Recordings were digitized at 20 kHz with Digidata 150A (AxonInstruments/Molecular Devices UnionCity, CA). Access resistance and leak currents were monitored and data were discarded if either parameter changed by > 25% over the course of data acquisition. Spontaneous excitatory postsynaptic potentials currents (sEPSCs) were recorded at a holding potential of —70 mV for 10 mins. Data were analyzed offline using Clampfit (Molecular Devices) and Prism 7.0 (GraphPad).
Extracellular recording and analyses
Intact SNOs were transferred from maturation medium to BrainPhys neuronal medium (Bardy et al., 2015) supplemented with SM1, 10 ng/ml GDNF, 10 ng/ml BDNF and 1X Penicillin/Streptomycin and cultured for at least 7 days prior to the recordings. Day 170 ± 5 SNOs were randomly selected and transferred to a Slotted Bath Oocyte Recording Chamber (Warner Instruments) containing BrainPhys medium. Throughout the recording, SNOs were constantly perfused with medium (roughly 2 ml/min) heated using a temperature controller (Warner Instruments) to maintain a constant temperature of 37°C (±2°C) in the recording chamber. A 4-shank, 64-channel, Buzsaki style electrode (E-Series, Cambridge NeuroTech; 300 μm electrode coverage, 250 μm shank spacing) was slowly inserted into the SNO using a micromanipulator (Kopf) until all contacts were in the SNO. Wide bandwidth signals from the electrodes were amplified using a headstage (Neuralynx), digitized with a Digital Lynx 4SX control system (Neuralynx) at 32 kHz, and bandpass filtered at 0.1 to 9 kHz with Cheetah acquisition and recording software (Neuralynx). In parallel, electrode sites were artificially grouped into tetrodes and multi-unit activity was visualized on each tetrode online in the Cheetah software. Briefly, signals from each electrode site were filtered from 0.6–6 kHz online using a tap finite impulse response (FIR) filter, and a threshold was set (20–25 μV in our setup) to be just above noise levels in the system. Anytime a signal crossed the user-defined threshold value, the next inflection point in the filtered signal was found and set as the alignment point. A 1 ms recording (comprising of 32 samples) from all 4 electrodes that made up the tetrode was obtained such that the sample alignment point made up the 8th of 32 samples. Once multiunit activity was observed in the Cheetah software, a baseline recording of at least 20 mins was obtained and stored for later analysis.
Single unit clusters were initially isolated from the 1 ms filtered, thresholded recordings using an unsupervised masked expectation-maximization (EM) algorithm that uses principle component analysis of spike features to sort spikes (KlustaKwik) (Kadir et al., 2014). Clusters were further refined by merging or hand-cutting output clusters from KlustaKwik based on differential spike shape between electrodes within a tetrode using SpikeSort3D software (Neuralynx). Once single units were separated, data were imported into MATLAB software (MathWorks) or NeuroExplorer software (Plexon) and analyzed using custom and built-in routines. Inter-spike interval histograms (20 bins per decade) and auto-correlograms (bin size = 5 ms) were created to visualize interactions within neurons, and cross-correlograms (bin size = 5 ms) were created to visualize interactions between different neurons.
Transcriptomic analyses of iPSC-derived neurons
Previously published bulk RNA sequencing data from our group (Wen et al., 2014) on cortical neurons derived from the same D2 and C3 iPSC lines used in the current study was re-analyzed to evaluate the differences in the WNT/β-Catenin signaling pathway genes in relatively pure cortical neuron populations. Reads covering gene coding regions were counted with BEDTools and count data were analyzed for differential expression using edgeR (v2.15.0). The list of WNT pathway related genes was obtained from KEGG pathway (https://www.genome.jp/kegg/pathway/hsa/hsa04310.html).
QUANTIFICATION AND STATISTICAL ANALYSIS
Individual organoids are treated as biological replicates, unless otherwise indicated in the Figure Legends. Data are presented as mean ± S.E.M., or mean ± S.D., unless otherwise indicated in the Figure Legends. Statistical analyses were performed using the Student’s t test in Excel or Prism software. Significance was defined by P value < 0.05. Organoid samples were randomly taken from the culture for experiments and analyses. Sample sizes were determined empirically. The sample sizes were designed to account for the variability between organoids and human iPSC cell lines and match current standards in human brain organoid-related studies. Other statistical details of experiments can be found in the Figure Legends. Data analyses comparing control and disease individual-derived organoids were performed blindly. Data analyses comparing sliced and unsliced organoids were not performed blindly because the visual difference between the two groups is striking, and blinding was not possible to an informed researcher. No data were excluded.
DATA AND CODE AVAILABILITY
Normalized gene expression values from single-nucleus RNA-seq were deposited into the GEO database (GSE137941). The data that support the findings of this study are available from the lead contact, Dr. Guo-li Ming (gming@pennmedicine.upenn.edu) upon reasonable request.
Supplementary Material
Highlights.
SNOs maintain growth and laminar expansion over long-term culture
SNOs exhibit separated upper and deep cortical layers
Layer-specific WNT/β-catenin signaling regulates neuronal fate specification
DISC1 mutation causes deficits in cortical neuron fate specification
ACKNOWLEDGMENTS
We thank members of Ming and Song laboratories for suggestions, B. Temsamrit and E. LaNoce for technical support, and J. Schnoll for lab coordination. This work was supported by grants from National Institutes of Health (U19AI131130 and R35NS097370 to G.M. and U19MH106434 and R37NS047344 to H.S.) and by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G.M.). U19MH106434 is part of the National Cooperative Reprogrammed Cell Research Groups (NCRCRG) to Study Mental Illness. S.L. was partially supported by Nantong University.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2020.02.002.
REFERENCES
- Abu-Khalil A, Fu L, Grove EA, Zecevic N, and Geschwind DH (2004). Wnt genes define distinct boundaries in the developing human brain: implications for human forebrain patterning. J. Comp. Neurol. 474, 276–288. [DOI] [PubMed] [Google Scholar]
- Alsiö JM, Tarchini B, Cayouette M, and Livesey FJ (2013). Ikaros promotes early-born neuronal fates in the cerebral cortex. Proc. Natl. Acad. Sci. USA 110, E716–E725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, Pochareddy S, Shin Y, Safi A, Song L, et al. ; PsychENCODE Consortium (2018). Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362, eaat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardy C, van den Hurk M, Eames T, Marchand C, Hernandez RV, Kellogg M, Gorris M, Galet B, Palomares V, Brown J, et al. (2015). Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc. Natl. Acad. Sci. USA 112, E2725–E2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, and Kriegstein AR (2017). Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 20, 435–449.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, et al. (2020). Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, and Sawa A (2011). Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 12, 707–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, et al. (2008). Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 57, 378–392. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystron I, Blakemore C, and Rakic P (2008). Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 9, 110–122. [DOI] [PubMed] [Google Scholar]
- Chiang CH, Su Y, Wen Z, Yoritomo N, Ross CA, Margolis RL, Song H, and Ming GL (2011). Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol. Psychiatry 16, 358–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, MacDonald JL, and Macklis JD (2011). Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 34, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Dong J, Zhong S, Wei Y, Wu Q, Yan L, Yong J, Sun L, Wang X, Zhao Y, et al. (2018). Spatial transcriptomic survey of human embryonic cerebral cortex by single-cell RNA-seq analysis. Cell Res. 28, 730–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Grydeland H, Krogsrud SK, Amlien I, Rohani DA, Ferschmann L, Storsve AB, Tamnes CK, Sala-Llonch R, Due-Tønnessen P, et al. (2015). Development and aging of cortical thickness correspond to genetic organization patterns. Proc. Natl. Acad. Sci. USA 112, 15462–15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, Sutcliffe M, Boulanger J, Tripodi M, Derivery E, et al. (2019). Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 22, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, and Macklis JD (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 14, 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, and Kriegstein AR (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561. [DOI] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF (2007). Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J. Neuropathol. Exp. Neurol. 66, 101–109. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Kuroda T, Honda A, Komabayashi-Suzuki M, Komai T, Shinkai Y, and Mizutani K (2014). Prdm8 regulates the morphological transition at multipolar phase during neocortical development. PLoS ONE 9, e86356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip BK, Bayatti N, Howard NJ, Lindsay S, and Clowry GJ (2011). The corticofugal neuron-associated genes ROBO1, SRGAP1, and CTIP2 exhibit an anterior to posterior gradient of expression in early fetal human neocortex development. Cereb. Cortex 21, 1395–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, Thokala R, Sheikh S, Saxena D, Prokop S, et al. (2020). A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadir SN, Goodman DF, and Harris KD (2014). High-dimensional cluster analysis with the masked EM algorithm. Neural Comput. 26, 2379–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, and Sasai Y (2013). Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 110, 20284–20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kana RK, Libero LE, and Moore MS (2011). Disrupted cortical connectivity theory as an explanatory model for autism spectrum disorders. Phys. Life Rev. 8, 410–437. [DOI] [PubMed] [Google Scholar]
- Kelava I, and Lancaster MA (2016). Stem cell models of human brain development. Cell Stem Cell 18, 736–748. [DOI] [PubMed] [Google Scholar]
- Kim H, Xu R, Padmashri R, Dunaevsky A, Liu Y, Dreyfus CF, and Jiang P (2019). Pluripotent stem cell-derived cerebral organoids reveal human oligodendrogenesis with dorsal and ventral origins. Stem Cell Reports 12, 890–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai T, Iwanari H, Mochizuki Y, Hamakubo T, and Shinkai Y (2009). Expression of the mouse PR domain protein Prdm8 in the developing central nervous system. Gene Expr. Patterns 9, 503–514. [DOI] [PubMed] [Google Scholar]
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, et al. (2016). Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, and Knoblich JA (2014). Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, Otani T, Livesey FJ, and Knoblich JA (2017). Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 35, 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KR, Newland B, Florio M, Kalebic N, Langen B, Kolterer A, Wimberger P, and Huttner WB (2018). Extracellular matrix components HAPLN1, lumican, and collagen I cause hyaluronic acid-dependent folding of the developing human neocortex. Neuron 99, 702–719.e6. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan M, Nevin ZS, Shick HE, Garrison E, Clarkson-Paredes C, Karl M, Clayton BLL, Factor DC, Allan KC, Barbar L, et al. (2018). Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Vinukonda G, Vose LR, Diamond D, Bhimavarapu BB, Hu F, Zia MT, Hevner R, Zecevic N, and Ballabh P (2013). Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J. Neurosci. 33, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, et al. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton RM, Miura Y, Sloan SA, Li Q, Revah O, Levy RJ, Huguenard JR, and Pașca SP (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, and Macklis JD (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, and O’Leary DD (2003). Dynamic patterned expression of orphan nuclear receptor genes RORalpha and RORbeta in developing mouse forebrain. Dev. Neurosci. 25, 234–244. [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozair MZ, Kirst C, van den Berg BL, Ruzo A, Rito T, and Brivanlou AH (2018). hPSC modeling reveals that fate selection of cortical deep projection neurons occurs in the subplate. Cell Stem Cell 23, 60–73.e6. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, et al. (2015). Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. (2016). Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Jacob F, Song MM, Nguyen HN, Song H, and Ming GL (2018). Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 13, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Song H, and Ming GL (2019). Brain organoids: advances, applications and challenges. Development 146, dev166074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radonjić NV, Ayoub AE, Memi F, Yu X, Maroof A, Jakovcevski I, Anderson SA, Rakic P, and Zecevic N (2014). Diversity of cortical interneurons in primates: the role of the dorsal proliferative niche. Cell Rep. 9, 2139–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs NA, Sawa A, Holmes SE, Ross CA, DeLisi LE, and Margolis RL (2005). A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol. Psychiatry 10, 758–764. [DOI] [PubMed] [Google Scholar]
- Saito T, Hanai S, Takashima S, Nakagawa E, Okazaki S, Inoue T, Miyata R, Hoshino K, Akashi T, Sasaki M, et al. (2011). Neocortical layer formation of human developing brains and lissencephalies: consideration of layer-specific marker expression. Cereb. Cortex 21, 588–596. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, Kadoshima T, Soen M, Narii N, Ishida Y, Ohgushi M, Takahashi J, Eiraku M, and Sasai Y (2015). Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 6, 8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsen GB, Larsen KB, Bogdanovic N, Laursen H, Graem N, Larsen JF, and Pakkenberg B (2003). The changing number of cells in the human fetal forebrain and its subdivisions: a stereological analysis. Cereb. Cortex 13, 115–122. [DOI] [PubMed] [Google Scholar]
- Saurat N, Andersson T, Vasistha NA, Molná r Z, and Livesey FJ (2013). Dicer is required for neural stem cell multipotency and lineage progression during cerebral cortex development. Neural Dev. 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM (2013). Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 14, 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, De Rienzo G, Drane L, Mao Y, Flood Z, Madison J, Ferreira M, Bergen S, King C, Sklar P, et al. (2011). Common DISC1 polymorphisms disrupt Wnt/GSK3β signaling and brain development. Neuron 72, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, Colamarino SA, Lein ES, and Courchesne E (2014). Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 370, 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci. 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L, Agirman G, Prados J, Amberg N, Fiè vre S, Oberst P, Bartolini G, Vitali I, Cadilhac C, Hippenmeyer S, et al. (2019). Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, eaav2522. [DOI] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfig N, Nickel J, and Bohl J (1998). Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res. 291, 433–443. [DOI] [PubMed] [Google Scholar]
- Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, Paulsen B, Nguyen L, Adiconis X, Regev A, et al. (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Buth JE, Vishlaghi N, de la Torre-Ubieta L, Taxidis J, Khakh BS, Coppola G, Pearson CA, Yamauchi K, Gong D, et al. (2017). Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep. 21, 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, Kim NS, Yoon KJ, Shin J, Zhang C, et al. (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim NS, Wen Z, Makri G, Nauen D, Shin JH, Park Y, et al. (2014). Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, Zhang H, Li L, Sun L, Pan N, et al. (2018). A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Zhao X, Zheng K, Li H, Huang H, Zhang Z, Mastracci T, Wegner M, Chen Y, Sussel L, and Qiu M (2014). Genetic evidence that Nkx2.2 and Pdgfra are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development 141, 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, and Barbas H (2013). Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci. 7, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Normalized gene expression values from single-nucleus RNA-seq were deposited into the GEO database (GSE137941). The data that support the findings of this study are available from the lead contact, Dr. Guo-li Ming (gming@pennmedicine.upenn.edu) upon reasonable request.