

Summary
The skin is one of the most important organs in the body, providing integrity and acting as a barrier to exclude microbes, allergens and chemicals. However, chronic skin inflammation can result when barrier function is defective and immune responses are dysregulated or misdirected against harmless or self‐antigens. During the last 15 years interleukin (IL)‐17 cytokines have emerged as key players in multiple inflammatory disorders, and they appear to be especially prominent in skin inflammation. IL‐17 cytokines produced by T cells and other cell types potently activate keratinocytes to promote inflammation in a feed‐forward loop. Given this key pathogenic role of the IL‐17 pathway in autoimmune and inflammatory disease, it has been the focus of intense efforts to target therapeutically. The inflammatory effects of IL‐17 can be targeted directly by blocking the cytokine or its receptor, or indirectly by blocking cytokines upstream of IL‐17‐producing cells. Psoriasis has been the major success story for anti‐IL‐17 drugs, where they have proven more effective than in other indications. Hidradenitis suppurativa (HS) is another inflammatory skin disease which, despite carrying a higher burden than psoriasis, is poorly recognized and under‐diagnosed, and current treatment options are inadequate. Recently, a key role for the IL‐17 pathway in the pathogenesis of HS has emerged, prompting clinical trials with a variety of IL‐17 inhibitors. In this review, we discuss the roles of IL‐17A, IL‐17F and IL‐17C in psoriasis and HS and the strategies taken to target the IL‐17 pathway therapeutically.
Keywords: hidradenitis suppurativa, IL‐17, psoriasis
A key role of IL‐17 in psoriasis has been highlighted by the remarkable success of drugs targeting the IL‐17 pathway. Recently, the emergence of a role for IL‐17 in hidradenitis suppurativa (HS) has raised the profile of this neglected skin disease, for which current treatments are inadequate. Importantly this has prompted clinical trials with anti‐IL‐17 drugs in HS.
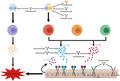
IL‐17
Interleukin (IL)‐17A is a proinflammatory cytokine first cloned in 1993 [1, 2]. Early studies indicated that T cell‐derived IL‐17 could activate a variety of cells, including rheumatoid arthritis synoviocytes, fibroblasts and keratinocytes, to release cytokines and chemokines and promote inflammation [3, 4]. More than a decade later IL‐17A was identified as the signature cytokine of the newly described T helper type 17 (Th17) cell subset [5, 6]. Interest in IL‐17 was amplified further during subsequent years, as it emerged that Th17 cells play a key pathogenic role in multiple autoimmune and inflammatory diseases. The IL‐17 pathway is now an important therapeutic target, with a range of approved drugs that inhibit IL‐17, both directly and indirectly, as outlined below (Fig. 1). For a more comprehensive review on biologicals which target the Th17 pathway, beyond the role of biologicals in inflammatory skin disease, please refer to Balato et al. [7].
Fig. 1.
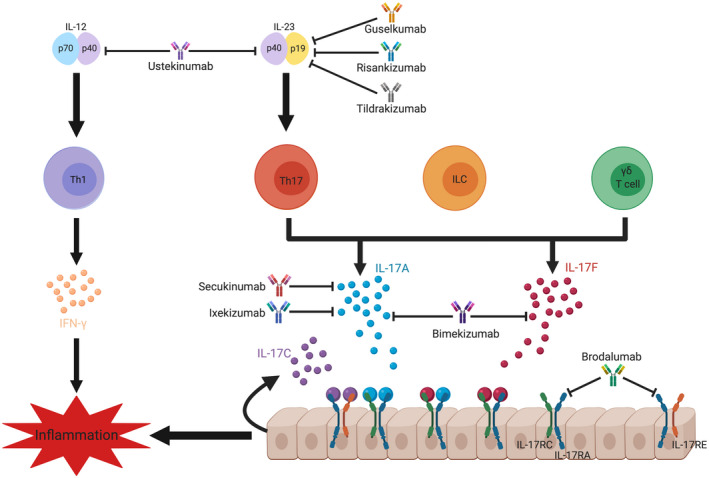
Cellular sources and targeting of interleukin (IL)‐17. IL‐17A and IL‐17F are the signature cytokines of the T helper type 17 (Th17) cell subset. They may also be produced by CD8 T cells and innate‐like lymphoid cells, including innate lymphoid cells (ILC), invariant natural killer T (iNKT) cells and γδ T cells. IL‐17 expression by myeloid cells including neutrophils and mast cells is suggested but not universally accepted. Dimers of IL‐17A, IL‐17F or IL‐17A/F bind to their IL‐17 receptor (IL‐17RA/RC), which is expressed on epithelial cells. IL‐17C is produced by epithelial cells and binds to IL‐17RA/RE. IL‐17R signalling results in the expression of anti‐microbial peptides, inflammatory cytokines and chemokines that promote inflammation via the recruitment of neutrophils and other immune cells. The inflammatory effects of IL‐17 signalling have been targeted directly via monoclonal antibody blockade of IL‐17A (secukinumab, ixekizumab), IL‐17A/F (bimekizumab) or the IL‐17 receptor IL‐17RA (brodalumab). Alternatively, the pathway can be targeted upstream of Th17 cells by blocking IL‐23 alone via its specific p19 subunit (guselkumab, tildrakizumab, risankizumab), or both IL‐23 and IL‐12 via the common p40 subunit (ustekinumab).
IL‐17A is the prototypical member of the IL‐17 family of cytokines, which consists of six members: IL‐17A–F, of which IL‐17A, C, E and F are best characterized (Table 1). IL‐17A, C and F are implicated in autoimmune inflammation. IL‐17A and IL‐17F, which are often co‐expressed, may be secreted not only by Th17 cells, but also by CD8 T cells (Tc17) [8] as well as invariant natural killer (iNK) T cells [9], gamma delta (γδ) T cells [10, 11] and type 3 innate lymphoid cells (ILC3) [12]. There is also controversial evidence regarding the ability of myeloid cells to express IL‐17 family cytokines [13, 14, 15]. In contrast, IL‐17C, which is emerging as a key player in skin inflammation, is expressed primarily by epithelial cells and keratinocytes, in response to cytokine and Toll‐like receptor (TLR) activation [16, 17, 18, 19]. Meanwhile, IL‐17E (IL‐25), which is the most divergent of the IL‐17 family, is produced by keratinocytes, but can also be secreted by endothelial cells, T cells, macrophages, myeloid cells and ILC [20]. IL‐17E is best known for its role in promoting Th2 responses and allergy [20], although it has also been implicated in psoriatic inflammation [21]. The role of IL‐17B is currently unclear; it has been shown to be pathogenic in inflammatory arthritis, lung fibrosis and some cancers [22, 23], but protective in colitis and asthma via antagonism of IL‐17E [24]. It is produced by neutrophils, B cells, neurons, stromal and epithelial cells, although not by activated T cells [22, 23]. Similarly, IL‐17D is poorly expressed by activated immune cells, but has been identified in tissue such as skeletal muscle, brain, adipose, heart and lung [22]. It has been shown to be both protective and pathogenic in the context of viral infection and pathogenic in sepsis [22, 25, 26].
Table 1.
IL‐17 cytokines and their receptors, cellular sources, target cells and functions
| IL‐17 cytokine | IL‐17R subunits | Sources | Target cells | Role in host defence and health | Role in disease |
|---|---|---|---|---|---|
| IL‐17A homodimer |
RA+RD [29] |
Th17 cells [5, 6], Tc17 cells [8], γδ T cells [9, 10, 11], iNKT cells [9], ILC3 [12] | Keratinocytes, epithelial cells, fibroblasts [38] | Defence against extracellular bacterial and fungal infections, barrier function, wound healing, microbiome [38] | Pathogenic in chronic inflammation, autoimmunity and some cancers [38] |
| IL‐17F homodimer |
RA+RC [31] RC+RC [30] |
||||
| IL‐17A+F heterodimer | RA+RC [28, 31] | Role of IL‐17A+F unclear | Role of IL‐17A+F unclear | ||
| IL‐17C homodimer | RA+RE [31] | Keratinocytes, epithelial cells, cutaneous neurones [16, 17, 18, 19, 38] | Autocrine effects on keratinocytes, epithelial cells, neurones [38] | Microbial defence, barrier maintenance, response to epithelial injury, cutaneous neuroprotection [38] | Pathogenic in inflammatory skin diseases including psoriasis [19], atopic dermatitis [112], HS [111] |
| IL‐17E homodimer (IL‐25) | RA+RB [31] | Keratinocytes, endothelial cells, epithelial cells, Th2 cells, macrophages, myeloid cells, ILC2 [20] [38] | Epithelial cells, fibroblasts, endothelial cells, Th2 cells, Th9 cells, iNKT cells, ILC2 [20] | Defence against helminths, promotes Th2 cell‐mediated immunity [38] | Pathogenic in allergy [20] and psoriasis [21] |
| IL‐17B homodimer | RB+? [31] | Neutrophils, B cells, neurons, stromal cells, epithelial cells, chondrocytes [22, 24] | Epithelial cells, fibroblasts, macrophages [24, 124] | Protective against Citrobacter rodentium infection [24] | Pathogenic in inflammatory arthritis [124], gastric and breast cancers [22], lung fibrosis [23], protective against colitis and asthma [24] |
| IL‐17D homodimer | Unknown | Skeletal muscle, brain, adipose, heart and lung tissue [22] | Endothelial cells [22], DC [25], macrophages [26] | Protective against viral infection and tumours [22] | Pathogenic in sepsis [26], pathogenic role in certain intracellular infections [25] |
IL = interleukin; Th17 = T helper type 17; iNK T = invariant natural killer T cells; ILC = innate lymphoid cells; DC = dendritic cells; HS = hidradenitis suppurativa.
The IL‐17 receptors are comprised of heterodimers of IL‐17 receptor A (IL‐17RA) together with ligand‐specific subunits (IL‐17RB–E) and are ubiquitously expressed on epithelial cells [27]. Homodimers or heterodimers of IL‐17A and IL‐17F bind to a receptor composed of the RA and RC subunits, albeit with differing affinities [28]. In addition, IL‐17RD partnered with IL‐17RA was identified as an alternative receptor for IL‐17A homodimers [29]. Recently, homodimers of IL‐17F were shown to bind homodimeric IL‐17RC, indicating the possibility of IL‐17RA‐independent IL‐17 signalling [30]. IL‐17C is recognized by a receptor consisting of RA and RE subunits, while IL‐17E signalling is mediated via a receptor of RA and RB subunits [27]. IL‐17B signals through an unidentified receptor containing IL‐17RB. The receptor for IL‐17D currently remains unknown. All IL‐17 receptors recruit actin 1 (Act1) as an adaptor molecule for downstream signalling via tumour necrosis factor receptor (TRAF6), mitogen‐activated protein kinase (MAPK) and nuclear factor kappa B (NF‐κB) [31].
Human Th17 cells differentiate and expand from naive T cells activated under the influence of cytokines, including IL‐6, IL‐1β and IL‐23 [32]. IL‐23 is a heterodimer of p19 and p40, with the p40 subunit shared with IL‐12, the key driver of Th1 cells. Thus, Th17 cell development and maintenance can be targeted by monoclonal antibodies inhibiting either IL‐12/IL‐23p40 or the specific IL‐23p19 subunit (Fig. 1). The inflammatory effects of IL‐17 can also be inhibited directly by therapies that bind to IL‐17A, IL‐17A and IL‐17F or their common receptor chain IL‐17RA (Fig. 1). These alternate blocking strategies exert different downstream effects. Targeting IL‐17A alone still allows for IL‐17F to signal through the IL‐17RA/RC receptor, whereas dual inhibition of IL‐17A and IL‐17F results in full inhibition of signalling through the receptor. Conversely, blockade of IL‐17RA inhibits the signalling induced by binding of IL‐17A, IL‐17F, IL‐17C and IL‐17E, as all these require the IL‐17RA subunit (Fig. 1). Upstream blockade of p40 inhibits differentiation of both Th1 and Th17 cells, whereas blocking IL‐23p19 specifically inhibits Th17 cells. These drugs will therefore not only inhibit the production of interferon (IFN)‐γ and IL‐17 produced by Th1 and Th17 cells, respectively, but also other inflammatory cytokines produced by these cells. In addition, more subtle distinctions in the action of some of these drugs are emerging; for example, anti‐IL‐17A therapy has been associated with exacerbation of inflammatory bowel disease [33], whereas this is not the case for anti‐IL‐23 [34]. It has been suggested that as IL‐17A promotes barrier function in the gut, perturbing this promotes gut inflammation [35]. Conversely, blocking IL‐23 inhibits Th17 cell‐derived IL‐17A but retains IL‐17A production by innate‐like cells to maintain gut barrier function [36]. IL‐23 blockade also promotes regulatory T (Treg) cells, which may further explain its improved safety profile in the context of the gut [37].
In healthy skin, IL‐17 plays a crucial role in maintaining barrier function and providing protection against extracellular bacteria and fungi by driving epithelial cell secretion of inflammatory cytokines, chemokines, matrix metalloproteinases and anti‐microbial peptides [38]. The importance of IL‐17 in fungal infection is highlighted by the fact that impaired IL‐17 immunity predisposes patients to chronic candidiasis [39]. However, dysregulation of the IL‐17 pathway perpetuates chronic inflammation in autoimmune and inflammatory conditions, such as psoriasis and hidradenitis suppurativa (HS).
IL‐17 in psoriasis
Background
Psoriasis is a chronic inflammatory skin condition affecting 1–3% of the population [40]. The disease encompasses a spectrum of clinical manifestations, including the plaque, guttate, erythrodermic and pustular subtypes. It is characterized by well‐defined red, scaly plaques affecting the arms, legs, torso and scalp [40]. Psoriasis patients also have systemic manifestations, with an increased risk of developing metabolic syndrome, Crohn’s disease, psoriatic arthritis and cardiovascular disease, while the physical affliction poses a significant emotional and social burden, with stigmatization increasing the risk of depression [41]. While its aetiology has yet to be fully elucidated, it is likely to include multiple factors, such as genetic susceptibility and environmental triggers. Clinical characteristics, including epidermal hyperplasia, led to psoriasis being initially regarded as a disorder of keratinocytes. However, further advances into its pathophysiology revealed an interplay between dysregulated keratinocyte function and aberrant immune responses.
Pathogenesis
Keratinocytes play a key role in the skin epidermis, where they provide a protective physical barrier at the skin surface. One of the hallmark features of psoriasis is epidermal modification linked to hyperproliferation and abnormal keratinocyte differentiation, which result in the psoriatic plaques that are characteristic of the disease (Fig. 2). Psoriasis is thought to be triggered in genetically susceptible individuals upon loss of T cell tolerance to particular self‐antigens. This may occur in the context of an environmental insult activating innate immune cells. For example, release of DNA from damaged cells, when complexed with the anti‐microbial peptide LL‐37, activates plasmacytoid dendritic cells (pDC) via TLR‐9 to produce IFN‐α, and licenses DC to present LL‐37 autoantigens to T cells [42]. These T cells then differentiate into Th1 and Th17 cell subsets under the influence of specific polarizing cytokines. Th17 cells produce IL‐17A, IL‐17F and IL‐22 which, alone or together with TNF, potently activate keratinocytes. These keratinocytes then secrete anti‐microbial peptides, cytokines and chemokines which recruit and activate immune cells, resulting in a feedback loop of persistent inflammation.
Fig. 2.
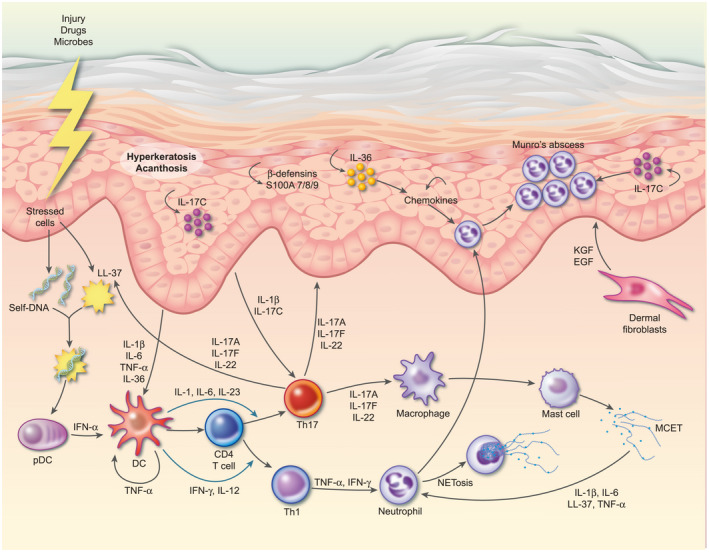
Schematic representation of psoriasis pathogenesis. Psoriasis is triggered in genetically susceptible individuals by environmental factors and/or breakdown of tolerance to self‐antigens. For example, release of self‐DNA molecules together with the anti‐microbial peptide LL‐37 activates plasmacytoid dendritic cells (pDC) to produce interferon (IFN)‐α. IFN‐α, together with tumour necrosis factor (TNF) activates myeloid DC which present self‐antigens to T cells, which then differentiate into T helper type 1 (Th1) and Th17 cells via IL‐12/IFN‐γ and IL‐23/IL‐1β/IL‐6, respectively. Th17 cells migrate to the dermis via the keratinocyte‐derived chemokines chemokine (C‐C motif) ligand 20 (CCL20) and chemokine (C‐X‐C motif) ligand (CXCL) 9/10/11, where they secrete homodimers or heterodimers of IL‐17A and IL‐17F and IL‐22. IL‐17A/F and IL‐22 bind to their receptors on both keratinocytes and fibroblasts and synergize with TNF to induce proliferation and expression of anti‐microbial peptides, such as β‐defensins and S100A7‐9, and a range of cytokines and chemokines. Chemokines such as IL‐8 and CXCL1/3/5/8 recruit neutrophils, whilst cytokines IL‐17C and IL‐36 induce further activation and proliferation of keratinocytes in an inflammatory loop. IL‐1β and TNF production by keratinocytes induces fibroblasts to release keratinocyte growth factor (KGF) and epidermal growth factor (EGF) which act back on keratinocytes, further contributing to their activation and proliferation. Meanwhile, keratinocyte‐derived IL‐1β, IL‐6 and IL‐17C feedback to further activate DC and Th17 cells.
IL‐17 in psoriasis
Accumulating evidence of increased levels of Th17 cytokines IL‐17A and IL‐22 in the sera of patients, as well as increased IL‐17 mRNA levels in psoriatic lesions [43, 44] has led to the speculation that IL‐17 plays a central role in driving psoriasis pathophysiology. Further evidence was provided in a study by Lowes et al., which found an increased frequency of IL‐17‐producing T cells and IL‐17 mRNA in the dermis of psoriatic lesions [45]. Psoriasis is now understood to be driven by multiple pathogenic IL‐17‐producing cells.
The recurrence of psoriatic plaques to previously affected sites suggests the role of tissue‐specific immunological memory in disease flare‐ups. This implicates tissue resident memory (Trm) cells, of note recently in chronic inflammatory disorders owing to their long‐term survival capabilities and lack of migration from their given site [46]. It is believed that Trm cells can be rapidly reactivated to provide an early source of IL‐17 and contribute to the recurrence of psoriasiform lesions [8, 47]. In addition, ILC3 cells are thought to be another early pool of IL‐17 contributing to psoriasis pathogenesis [12].
IL‐17A levels were found to be elevated in the lesional skin and sera of psoriasis patients [48, 49]. Furthermore, genome‐wide association studies (GWAS) have identified psoriasis‐associated variants in genes involved in IL‐17A signalling [50, 51, 52]. IL‐17A is a principle culprit in epidermal keratinocyte dysfunction and promotes their aberrant differentiation and proliferation [53]. IL‐17A also stimulates keratinocytes to release proinflammatory mediators such as chemokine (C‐C motif) ligand 20 (CCL20), IL‐8 and anti‐microbial peptides, thus promoting inflammation by indirectly facilitating recruitment of neutrophils and Th17 cells [54, 55, 56]. Furthermore, IL‐17A synergizes with other cytokines, including tumour necrosis factor (TNF) and IL‐22. Together, they up‐regulate levels of IL‐36 cytokines, members of the IL‐1 family which, in addition to activating Th17 cells, have been linked to pustular psoriasis [57, 58, 59]. IL‐17F is also elevated in sera and lesions of psoriasis patients [48, 60]. Like IL‐17A, IL‐17F is promoted by IL‐23 [61], with evidence suggesting a key role for IL‐17F in inducing IL‐6 and IL‐8 production in keratinocytes [62, 63].
IL‐17C, which is increased in psoriatic lesions [19], synergizes with TNF and induces proinflammatory cytokines and chemokines and secretion of anti‐microbial peptides [16, 19, 64]. The levels of IL‐17E are also elevated in psoriatic lesions, where it activates dermal macrophages to produce TNF and IL‐8 [65]. Mice with keratinocyte‐specific deletion of IL‐17E were resistant to imiquimod‐induced psoriasis, suggesting an important role for IL‐17E in the pathogenesis of psoriasis [21].
Therapeutic targeting
Mild psoriasis is treated with phototherapy or topical agents; however, refractory or more severe psoriasis usually requires systemic therapy including methotrexate, dimethyl fumarate or biologicals. The anti‐TNF therapies infliximab, etanercept, adalimumab, certolizumab pegol and golimumab, already approved for a range of inflammatory disorders [66], were found to improve disease scores in psoriasis patients [67, 68, 69]. Reported serious side effects include infection, lymphoma and congestive heart failure [70]. Aside from congestive heart failure, where anti‐TNF therapy is contraindicated, it is generally considered to reduce cardiovascular risk in psoriasis patients [71].
The emerging role for IL‐17A in psoriasis led to trials of the anti‐IL‐17A agent secukinumab in psoriasis and found a significant reduction in disease severity in 75% of patients. These patients had a reduction in epidermal hyperplasia and a marked reduction in IL‐17A and IL‐22 gene expression [72]. Given its efficacy, speed of onset and a favourable benefit‐risk ratio, secukinumab was the earliest biological to be approved as a first‐line treatment for plaque psoriasis [73]. Another IL‐17A inhibitor, ixekizumab, was subsequently approved to treat plaque psoriasis [74]. In addition, there have been positive results from a Phase IIb trial of the bi‐specific anti‐IL‐17A+F, bimekizumab, in psoriasis with early reports of enhanced performance over adalimumab in the Phase III BE SURE trial [75].
Brodalumab blocks the IL‐17RA subset of the IL‐17 receptor, thus inhibiting IL‐17A, IL‐17F, IL‐17C and IL‐17E, and so would be expected to have superior efficacy compared to those that inhibit IL‐17A alone. However, patients relapse soon after stopping brodalumab treatment, probably due to the persistence of active IL‐17 cytokines [76]. Furthermore, brodalumab has been linked to cases of suicide and now carries a suicide warning [77, 78].
Given its important role in Th17 cell and macrophage activation in psoriasis, targeting IL‐23 upstream of IL‐17 is now of particular focus. Ustekinumab, which blocks the p40 subunit of IL‐23, inhibits both IL‐12 and IL‐23, given its shared p40 subunit. However, while IL‐23 is known to promote disease, IL‐12 has a protective role in psoriasis, and so was collaterally neutralized by ustekinumab [79]. Therefore, specific p19 subunit inhibitors such as tildrakizumab, risankizumab and guselkumab were subsequently developed to target only IL‐23, while preserving the function of IL‐12. These p19 inhibitors were found to have superior efficacy to TNF blockade, and showed clearance in a high proportion of patients [80, 81, 82]. Furthermore, these effects were not associated with adverse events such as those seen with IL‐17 antagonists [83].
It is now clear that a dysregulated IL‐23/IL‐17 axis is central to psoriasis pathophysiology. There is no doubt that targeting this axis has been extremely successful in treating psoriasis [84]; however, the results of head‐to‐head trials will be needed to determine the relative efficacy and side effects of each of the different approaches to blocking the IL‐17 pathway.
IL‐17 in HS
Background
HS, also known as acne inversa, is a relapsing follicular disease, affecting the apocrine gland‐bearing skin in the axillae, perianal, perineal and inguinal regions [85]. Widely varying prevalence of 0·1–4% has been reported [86, 87, 88], although a recent large‐scale US study found an overall prevalence of 0·1% for women and 0·058% for men [89]. This overall variability is probably due to differences in physicians’ awareness, as well as differing susceptibility to the disease in distinct subpopulations. HS is pointedly under‐recognized, with diagnosis taking significantly longer than that of psoriasis (7·2 versus 1·6 years) [90]. Symptoms typically manifest after puberty, and the increased risk of disease in females suggests a possible role of the endocrine system [91]. This chronic inflammatory disease is characterized by the presence of painful nodules, abscesses and sinus tracts, which ultimately lead to fibrotic scar formation [85]. HS is associated with systemic inflammation and co‐morbidities which include metabolic syndrome and Crohn’s disease [92]. It is also associated with significant disability, social isolation and a general decrease in quality of life, exceeding that of psoriasis [93]. Risk factors include a family history, with ~34% of first‐degree relatives affected [94], in addition to smoking and obesity [92].
Pathogenesis of HS
The pathogenesis of HS is incompletely understood, although immune dysregulation appears to play a key role in disease aetiology (Fig. 3). An essential event in the development of HS lesions is hair follicle occlusion, induced by keratosis and follicular epithelium hyperplasia, which ultimately leads to cyst development [95]. Lesion development begins with follicular occlusion which may, in some cases, be driven by a genetic predisposition such as mutations in γ‐secretase [96], and/or exogenous factors such as the microflora, smoking, obesity and/or mechanical stress. Inherent dysfunction of keratinocytes is implicated in the initiation of HS. Keratinocytes derived from the hair follicles of HS patients had a proinflammatory phenotype when compared with healthy controls [97]. This may relate to a dysregulated cell cycle in outer root sheath stem cells, from which these keratinocytes originate, leading to replicative stress and an accumulation of cytosolic ssDNA that drives type I IFN synthesis to trigger inflammation [98]. In addition, the cutaneous microbiome in HS has been shown to be altered compared with that of healthy controls, and an imbalanced skin microbiome may precede the development of HS lesions via the triggering of an aberrant cutaneous immune response [99, 100].
Fig. 3.
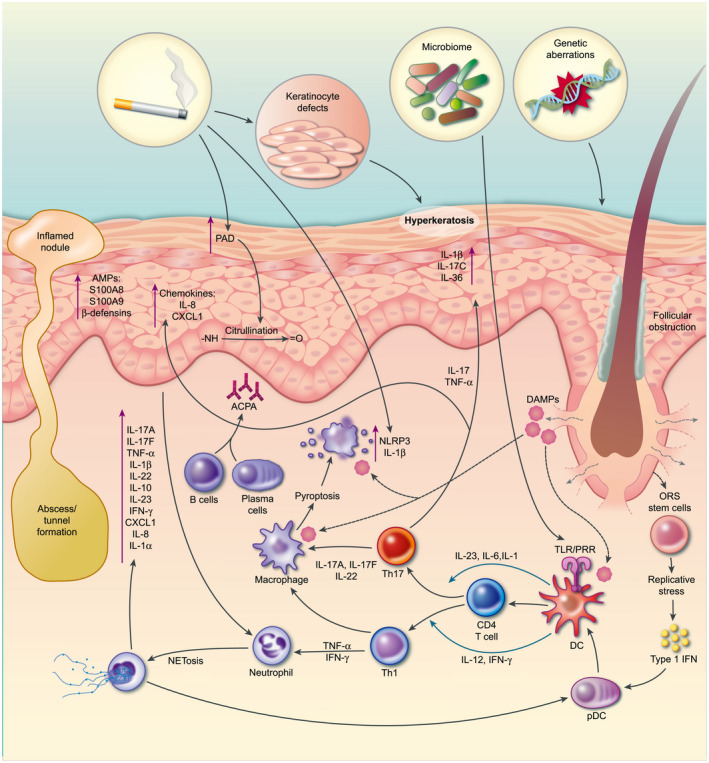
Schematic representation of hidradenitis suppurativa (HS) pathogenesis. Follicular obstruction is an essential event for the development of HS, followed by cyst formation which may rupture and trigger a potent immune response and ongoing inflammation. Genetic aberrations and/or dysfunctional keratinocyte stem cells, dysregulated microbiome, smoking, obesity and mechanical stress are thought to promote follicular occlusion and inflammation. Nucleotide‐binding domain‐like receptor protein 3 (NLRP3) activation and interleukin (IL)‐1β production are key features and may be driven by smoking, a dysregulated microbiome or damage‐associated molecular patterns (DAMP) released upon cyst rupture. Activated dendritic cells (DC) drive the differentiation of CD4 T cells into T helper type 1 (Th1) and Th17 subsets. Th1 cells can activate neutrophils to undergo NETosis, releasing proinflammatory cytokines and activating the NLRP3 inflammasome. Th17 cells, driven by IL‐1β and IL‐23, produce IL‐17A and IL‐17F, which potently activate keratinocytes. Activated keratinocytes release anti‐microbial peptides and chemokines such as IL‐8, which recruit neutrophils. In addition, cytokines, including IL‐17C and IL‐36 secreted by the keraitnocytes act in an autocrine manner to further potentiate their activation and proliferation. Smoking and inflammation cause citrullination of proteins which may be the target of a B cell response and production of anti‐citrullinated protein antibodies (ACPA) that contribute to the inflammatory response. In the later stages, keratinocytes tunnel to form sinus tracts which are susceptible to infection and can exacerbate inflammation.
Regardless of the initial cause of occlusion the follicle ultimately ruptures, releasing its contents intradermally, including keratin fibres, dermal detritus and multiple damage‐ and pathogen‐associated molecular patterns (DAMP, PAMP). Inflammatory immune pathways such as the nucleotide‐binding domain‐like receptor protein 3 (NLRP) inflammasome, TLRs and the IL‐23/IL‐17 pathway are activated [101]. This subsequently invokes a diverse immune cell infiltrate leading to cyst and abscess formation. Immune cells, including activated T cells, B cells, neutrophils and mast cells, together with multiple proinflammatory cytokines, are significantly increased in HS lesional skin [102, 103, 104, 105]. Activated keratinocytes and innate immune cells probably drive a strong Th17 response, which further activates the keratinocytes, and recruit neutrophils and other innate cells in an inflammatory loop. Recently, neutrophils in HS skin were shown to undergo NETosis, which was associated with activation of pDC to produce IFN‐α [106]. This probably adds to the type I IFN signature in HS lesional skin and promotes innate and adaptive immune responses. B cell dysregulation has also been observed in HS [105], and recently B cells specific for citrullinated proteins were identified in HS patients [106]. Interestingly, smoking, which is a risk factor for HS, can induce both citrullination of proteins and NETosis [107].
The final stage leading to severe HS is chronic inflammation and sinus tract formation. Proliferating epithelial strands, along with fibrotic factors continuously propel the immune system leading to scarring and tunnelling. These cavities may join together or burrow to distal areas. Such tracts are very hospitable to infection and biofilms, resulting in suppurative, malodorous discharge.
IL‐17 in HS
In recent years the cytokines IL‐1, IL‐23 and IL‐17 were found to be elevated in HS lesions, suggesting a role for Th17 cells in disease pathogenesis. A 30‐fold increase in IL‐17A gene expression in HS patients’ lesional skin was observed compared with healthy control skin [108]. Another study found that mRNA expression of IL‐1β was up‐regulated by 115‐fold and IL‐17A by 149‐fold in HS lesional skin compared with healthy control skin [109]. Gene array analysis showed that both IL‐17 and IFN receptor signalling were significantly up‐regulated in HS lesions [97]. IL‐17 signalling enhances the expression of proinflammatory mediators such as anti‐microbial peptides S100A8, S100A9 and human‐β‐defensin‐2 and chemokines chemokine (C‐X‐C motif) ligand 1 (CXCL1) and IL‐8, all of which are significantly up‐regulated in HS lesions [97]. It has been suggested that dysregulation of IL‐26, a Th17‐associated cytokine which exerts direct anti‐microbial activity, may contribute towards a defective anti‐microbial response in HS [110]. There is a significant infiltration of immune cells in HS lesional skin compared with uninvolved skin, and polyfunctional Th17 cells were highly enriched [104]. CD8 T cells have also been shown to be a source of IL‐17 in HS lesional skin, albeit at a much lower level than CD4 T cells [104]. In the same study, anti‐TNF therapy significantly reduced the number of IL‐17‐producing CD4 T cells in HS lesional skin compared with anti‐TNF naive HS patients, indicating synergy between TNF and IL‐17. To date, there are no reports of innate‐like IL‐17‐producing lymphocytes such as γδ T cells or ILC in HS skin. Neutrophils have been reported as the dominant IL‐17‐producing cells in HS lesional skin [102]. However, as neutrophils express large amounts of IL‐17RA, it is possible that neutrophils sequester extracellular IL‐17 via their receptors rather than being IL‐17 secretors [15] .
In HS, keratinocytes constitutively demonstrate an inflammatory profile, suggesting that they are primarily dysfunctional in HS, and rather than enhancing a tissue protective response, contribute to chronic inflammation [97]. In other IL‐17‐mediated diseases, such as psoriasis, keratinocytes are a primary target for IL‐17. Similarly, in HS, an inflammatory loop involving IL‐17 stimulation of keratinocytes induces chemokines, anti‐microbial peptides and cytokines, including IL‐1β.
Recently, IL‐17C has been implicated in the pathogenesis of HS, and HS patients have significantly elevated IL‐17C mRNA compared with healthy controls; these are comparable to levels seen in psoriasis [111]. IL‐17C is produced by keratinocytes upon stimulation with IL‐17A/F, TNF, bacterial stimulation or TLR agonists [16] and induces the production of IL‐1β, IL‐8, CXCL1 and IL‐36γ, as well as elevating IL‐17A and IL‐17F production by Th17 cells. This creates a feedback loop, driving inflammation [112].
Current treatments for HS
Treatment of HS comprises topical and systemic medications and adjuvant treatments. In some cases surgical intervention is required, aimed at excision of nodules or opening sinus tracts [113]. Antibiotics and retinoids, including tetracycline, clindamycin, rifampicin and acitretin, are systemic options, which may act on commensal bacteria and reduce secondary infection as well as possibly exerting anti‐inflammatory effects [114]. TNF‐targeting biologicals have a proven efficacy, with adalimumab the only Food and Drug Administration (FDA)‐approved biological for the treatment of moderate‐severe HS [115]. The PIONEER trials, where HS patients received adalimumab or placebo for 12 weeks, showed clinical efficacy with 50% of adalimumab‐treated HS patients maintaining a positive clinical response beyond 3 years [116]. However, at ~50% efficacy, this is far less successful than other adalimumab‐treated dermatological conditions, and 6·5% of patients generated anti‐adalimumab antibodies. Hence, there is a clear need for more effective therapies.
Targeting the IL‐17 pathway in HS
Given the accumulating evidence of a key role of the IL‐17 pathway in HS pathogenesis, there is considerable interest in therapeutic targeting of IL‐17. Indeed, the safety and efficacy of various IL‐17 inhibitors, including CJM‐112, secukinumab, bimekizumab, brodalumab and guselkumab, are currently under investigation in HS.
Secukinumab treatment in HS has been encouraging, with positive results from a series of case studies and an open‐label trial [117], and a Phase III trial (NCT03713632) is under way.
CJM‐112, a fully human monoclonal antibody targeting IL‐17A, demonstrated a significantly superior response rate compared with placebo, and was well tolerated in early proof‐of‐concept studies. CJM112 reduced the number of inflammatory lesions in HS patients, while also reducing C‐reactive protein levels in serum, demonstrating a reduced inflammatory burden [118]. Alternative strategies under investigation include dual inhibition of IL‐17A/F (bimekizumab; Phase II clinical trial NCT03248531) and IL‐17RA blockade (brodalumab; Phase II clinical trial NCT03910803); however, results are not yet available.
Despite the early promise of IL‐17 inhibition in HS, the potential adverse effects must be considered. Previously, in Phase II/III clinical trials of secukinumab in ankylosing spondylitis patients, five new cases of Crohn’s disease were reported [119]. Given the association between HS and Crohn’s disease [120], IL‐17A inhibition in HS may be problematic. It is possible that inhibiting the pathway upstream of IL‐17 could avoid this potential risk. For example, blocking IL‐23 using guselkumab has been effective in severe psoriasis, and is not shown to be associated with increased risk of Crohn’s disease. This suggests promise for the current Phase II clinical trial of guselkumab in HS (NCT03628924).
In addition, IL‐1β, which drives IL‐17 production by innate and Th17 cells, has been shown to be pathogenic in HS and represents another potential target [121]. However, results from case reports and small studies testing the IL‐1β antagonist (anakinra), anti‐IL‐1β (canakinumab) and anti‐IL‐1β receptor (MEDI8968) in HS have been mixed or negative [122]. Targeting IL‐1α appears to be more promising, with the anti‐IL‐1α monoclonal antibody bermekimab resulting in a positive clinical end‐point (HS clinical response) in 60% of moderate to severe HS patients, with no adverse events [123].
Discussion
Psoriasis and HS, both chronic inflammatory diseases of the skin, manifest differently yet share a number of pathogenic features, including dysregulation of keratinocyte function, a type I IFN signature and over‐activation of the IL‐17 pathway. A large body of research into the pathogenesis of psoriasis has led to the identification of IL‐17 as a therapeutic target, and the striking success of this strategy has underscored the importance of IL‐17 in the pathogenesis of the disease. In contrast, under‐recognition of HS and lack of research into its pathogenesis has hampered the development of new treatments for this very debilitating disease. In recent years, however, the discovery of a role for the IL‐17 pathway in HS has shed more light on the disease, and the success of anti‐IL‐17 therapies in psoriasis have paved the way for clinical trials which are currently under way. The results of these trials are eagerly awaited, and will hopefully provide better options for HS patients. In psoriasis, the results of head‐to‐head trials will provide important information regarding the risks and benefits of the various strategies to target the IL‐17 pathway.
Disclosures
All authors report no conflicts of interest.
References
- 1. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA‐8, cloned from an activated T cell, bearing AU‐rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol 1993; 150:5445–56. [PubMed] [Google Scholar]
- 2. Yao Z, Painter SL, Fanslow WC et al Human IL‐17: a novel cytokine derived from T cells. J Immunol 1995; 155:5483–6. [PubMed] [Google Scholar]
- 3. Chabaud M, Durand JM, Buchs N et al Human interleukin‐17: A T cell‐derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum 1999; 42:963–70. [DOI] [PubMed] [Google Scholar]
- 4. Fossiez F, Banchereau J, Murray R, Van Kooten C, Garrone P, Lebecque S. Interleukin‐17. Int Rev Immunol 1998; 16:541–51. [DOI] [PubMed] [Google Scholar]
- 5. Langrish CL, Chen Y, Blumenschein WM et al IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harrington LE, Hatton RD, Mangan PR et al Interleukin 17‐producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6:1123–32. [DOI] [PubMed] [Google Scholar]
- 7. Balato A, Scala E, Balato N et al Biologics that inhibit the Th17 pathway and related cytokines to treat inflammatory disorders. Expert Opin Biol Ther 2017; 17:1363–74. [DOI] [PubMed] [Google Scholar]
- 8. Cheuk S, Wiken M, Blomqvist L et al Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 2014; 192:3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venken K, Jacques P, Mortier C et al RORgammat inhibition selectively targets IL‐17 producing iNKT and gammadelta‐T cells enriched in spondyloarthritis patients. Nat Commun 2019; 10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Laggner U, Di Meglio P, Perera GK et al Identification of a novel proinflammatory human skin‐homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. J Immunol 2011; 187:2783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai Y, Shen X, Ding C et al Pivotal role of dermal IL‐17‐producing gammadelta T cells in skin inflammation. Immunity 2011; 35:596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teunissen MBM, Munneke JM, Bernink JH et al Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol 2014; 134:2351–60. [DOI] [PubMed] [Google Scholar]
- 13. Lin AM, Rubin CJ, Khandpur R et al Mast cells and neutrophils release IL‐17 through extracellular trap formation in psoriasis. J Immunol 2011; 187:490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noordenbos T, Blijdorp I, Chen S et al Human mast cells capture, store, and release bioactive, exogenous IL‐17A. J Leukoc Biol 2016; 100:453–62. [DOI] [PubMed] [Google Scholar]
- 15. Tamassia N, Arruda‐Silva F, Calzetti F et al A reappraisal on the potential ability of human neutrophils to express and produce IL‐17 family members in vitro: failure to reproducibly detect it. Front Immunol 2018; 9:795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramirez‐Carrozzi V, Sambandam A, Luis E et al IL‐17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 2011; 12:1159–66. [DOI] [PubMed] [Google Scholar]
- 17. Johansen C, Riis JL, Gedebjerg A, Kragballe K, Iversen L. Tumor necrosis factor alpha‐mediated induction of interleukin 17C in human keratinocytes is controlled by nuclear factor κB. J Biol Chem 2011; 286:25487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roth SA, Simanski M, Rademacher F, Schroder L, Harder J. The pattern recognition receptor NOD2 mediates Staphylococcus aureus‐induced IL‐17C expression in keratinocytes. J Invest Dermatol 2014; 134:374–80. [DOI] [PubMed] [Google Scholar]
- 19. Johnston A, Fritz Y, Dawes SM et al Keratinocyte overexpression of IL‐17C promotes psoriasiform skin inflammation. J Immunol 2013; 190:2252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu M, Dong C. IL‐25 in allergic inflammation. Immunol Rev 2017; 278:185–91. [DOI] [PubMed] [Google Scholar]
- 21. Xu M, Lu H, Lee YH et al An interleukin‐25‐mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity 2018; 48:787–98.e4. [DOI] [PubMed] [Google Scholar]
- 22. Brembilla NC, Senra L, Boehncke WH. The IL‐17 family of cytokines in psoriasis: IL‐17A and beyond. Front Immunol 2018; 9:1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang D, Chen X, Wang J et al Dysregulated lung commensal bacteria drive interleukin‐17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity 2019;50:692–706.e7. [DOI] [PubMed] [Google Scholar]
- 24. Reynolds JM, Lee YH, Shi Y et al Interleukin‐17B antagonizes interleukin‐25‐mediated mucosal inflammation. Immunity 2015; 42:692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee Y, Clinton J, Yao C, Chang SH. Interleukin‐17D promotes pathogenicity during infection by suppressing CD8 T cell activity. Front Immunol 2019; 10:1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan X, Tu H, Liu Y, Chen T, Cao J. Interleukin‐17D aggravates sepsis by inhibiting macrophage phagocytosis. Crit Care Med 2020; 48:e58–65. [DOI] [PubMed] [Google Scholar]
- 27. Yao Z, Spriggs MK, Derry JM et al Molecular characterization of the human interleukin (IL)‐17 receptor. Cytokine 1997; 9:794–800. [DOI] [PubMed] [Google Scholar]
- 28. Wright JF, Bennett F, Li B et al The human IL‐17F/IL‐17A heterodimeric cytokine signals through the IL‐17RA/IL‐17RC receptor complex. J Immunol 2008; 181:2799–805. [DOI] [PubMed] [Google Scholar]
- 29. Su Y, Huang J, Zhao X et al Interleukin‐17 receptor D constitutes an alternative receptor for interleukin‐17A important in psoriasis‐like skin inflammation. Sci Immunol 2019; 4:eaau9657. [DOI] [PubMed] [Google Scholar]
- 30. Goepfert A, Lehmann S, Blank J, Kolbinger F, Rondeau JM. Structural analysis reveals that the cytokine IL‐17F forms a homodimeric complex with receptor IL‐17RC to drive IL‐17RA‐independent signaling. Immunity 2020; 52:499–512.e5. [DOI] [PubMed] [Google Scholar]
- 31. Gaffen SL. Structure and signalling in the IL‐17 receptor family. Nat Rev Immunol 2009; 9:556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilson NJ, Boniface K, Chan JR et al Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol 2007; 8:950–7. [DOI] [PubMed] [Google Scholar]
- 33. Hueber W, Sands BE, Lewitzky S et al Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012; 61:1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakamura M, Lee K, Jeon C et al Guselkumab for the treatment of psoriasis: a review of Phase III trials. Dermatol Ther 2017; 7:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maxwell JR, Zhang Y, Brown WA et al Differential roles for interleukin‐23 and interleukin‐17 in intestinal immunoregulation. Immunity 2015; 43:739–50. [DOI] [PubMed] [Google Scholar]
- 36. Lee JS, Tato CM, Joyce‐Shaikh B et al Interleukin‐23‐independent IL‐17 production regulates intestinal epithelial permeability. Immunity 2015; 43:727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Izcue A, Hue S, Buonocore S et al Interleukin‐23 restrains regulatory T cell activity to drive T cell‐dependent colitis. Immunity 2008; 28:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGeachy MJ, Cua DJ, Gaffen SL. The IL‐17 family of cytokines in health and disease. Immunity 2019; 50:892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puel A, Cypowyj S, Bustamante J et al Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin‐17 immunity. Science 2011; 332:65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parisi R, Symmons DP, Griffiths CE, Ashcroft DM. Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) Project Team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 2013; 133:377–85. [DOI] [PubMed] [Google Scholar]
- 41. Takeshita J, Grewal S, Langan SM et al Psoriasis and comorbid diseases: Implications for management. J Am Acad Dermatol 2017; 76:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lande R, Botti E, Jandus C et al The antimicrobial peptide LL37 is a T‐cell autoantigen in psoriasis. Nat Commun 2014; 5:5621. [DOI] [PubMed] [Google Scholar]
- 43. Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin‐17 and interferon‐gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol 1998; 111:645–9. [DOI] [PubMed] [Google Scholar]
- 44. Li J, Li D, Tan Z. The expression of interleukin‐17, interferon‐gamma, and macrophage inflammatory protein‐3 alpha mRNA in patients with psoriasis vulgaris. J Huazhong Univ Sci Technolog Med Sci 2004; 24:294–6. [DOI] [PubMed] [Google Scholar]
- 45. Lowes MA, Kikuchi T, Fuentes‐Duculan J et al Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 2008; 128:1207–11. [DOI] [PubMed] [Google Scholar]
- 46. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med 2015; 7:269rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gallais Serezal I, Classon C, Cheuk S et al Resident T cells in resolved psoriasis steer tissue responses that stratify clinical outcome. J Invest Dermatol 2018; 138:1754–63. [DOI] [PubMed] [Google Scholar]
- 48. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin‐17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 2009; 160:319–24. [DOI] [PubMed] [Google Scholar]
- 49. Fotiadou C, Lazaridou E, Sotiriou E et al IL‐17A, IL‐22, and IL‐23 as markers of psoriasis activity: a cross‐sectional, hospital‐based study. J Cutan Med Surg 2015; 19:555–60. [DOI] [PubMed] [Google Scholar]
- 50. Cargill M, Schrodi SJ, Chang M et al A large‐scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis‐risk genes. Am J Hum Genet 2007; 80:273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsoi LC, Spain SL, Knight J et al Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012; 44:1341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sonder SU, Paun A, Ha HL, Johnson PF, Siebenlist U. CIKS/Act1‐mediated signaling by IL‐17 cytokines in context: implications for how a CIKS gene variant may predispose to psoriasis. J Immunol 2012; 188:5906–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gutowska‐Owsiak D, Schaupp AL, Salimi M et al IL‐17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp Dermatol 2012; 21:104–10. [DOI] [PubMed] [Google Scholar]
- 54. Ivanov S, Linden A. Interleukin‐17 as a drug target in human disease. Trends Pharmacol Sci 2009; 30:95–103. [DOI] [PubMed] [Google Scholar]
- 55. Nograles KE, Zaba LC, Guttman‐Yassky E et al Th17 cytokines interleukin (IL)‐17 and IL‐22 modulate distinct inflammatory and keratinocyte‐response pathways. Br J Dermatol 2008; 159:1092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liang SC, Tan XY, Luxenberg DP et al Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006; 203:2271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chiricozzi A, Guttman‐Yassky E, Suarez‐Farinas M et al Integrative responses to IL‐17 and TNF‐α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 2011; 131:677–87. [DOI] [PubMed] [Google Scholar]
- 58. Carrier Y, Ma HL, Ramon HE et al Inter‐regulation of Th17 cytokines and the IL‐36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol 2011; 131:2428–37. [DOI] [PubMed] [Google Scholar]
- 59. Capon F. IL36RN mutations in generalized pustular psoriasis: just the tip of the iceberg? J Invest Dermatol 2013; 133:2503–4. [DOI] [PubMed] [Google Scholar]
- 60. Soderstrom C, Berstein G, Zhang W et al Ultra‐sensitive measurement of IL‐17A and IL‐17F in psoriasis patient serum and skin. AAPS J 2017; 19:1218–22. [DOI] [PubMed] [Google Scholar]
- 61. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin‐23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin‐17. J Biol Chem 2003; 278:1910–4. [DOI] [PubMed] [Google Scholar]
- 62. Watanabe H, Kawaguchi M, Fujishima S et al Functional characterization of IL‐17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol 2009; 129:650–6. [DOI] [PubMed] [Google Scholar]
- 63. Fujishima S, Watanabe H, Kawaguchi M et al Involvement of IL‐17F via the induction of IL‐6 in psoriasis. Arch Dermatol Res 2010; 302:499–505. [DOI] [PubMed] [Google Scholar]
- 64. Song X, Zhu S, Shi P et al IL‐17RE is the functional receptor for IL‐17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol 2011; 12:1151–8. [DOI] [PubMed] [Google Scholar]
- 65. Senra L, Stalder R, Alvarez Martinez D, Chizzolini C, Boehncke WH, Brembilla NC. Keratinocyte‐derived IL‐17E contributes to inflammation in psoriasis. J Invest Dermatol 2016; 136:1970–80. [DOI] [PubMed] [Google Scholar]
- 66. Taylor PC. Anti‐tumor necrosis factor therapies. Curr Opin Rheumatol 2001; 13:164–9. [DOI] [PubMed] [Google Scholar]
- 67. Reich K, Nestle FO, Papp K et al, Express Study Investigators. Infliximab induction and maintenance therapy for moderate‐to‐severe psoriasis: a phase III, multicentre, double‐blind trial. Lancet 2005; 366:1367–74. [DOI] [PubMed] [Google Scholar]
- 68. Paller AS, Siegfried EC, Langley RG et al, Etanercept Pediatric Psoriasis Study Group . Etanercept treatment for children and adolescents with plaque psoriasis. N Engl J Med 2008; 358:241–51. [DOI] [PubMed] [Google Scholar]
- 69. Urdaneta M, Jethwa H, Sultan R, Abraham S. A review on golimumab in the treatment of psoriatic arthritis. Immunotherapy 2017; 9:871–89. [DOI] [PubMed] [Google Scholar]
- 70. Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatolog Treat 2004; 15:280–94. [DOI] [PubMed] [Google Scholar]
- 71. Caiazzo G, Fabbrocini G, Di Caprio R et al Psoriasis, cardiovascular events, and biologics: lights and shadows. Front Immunol 2018; 9:1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hueber W, Patel D d, Dryja T et al Effects of AIN457, a fully human antibody to interleukin‐17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2010; 2:52ra72. [DOI] [PubMed] [Google Scholar]
- 73. Langley RG, Elewski BE, Lebwohl M et al, Erasure Study Group, Fixture Study Group . Secukinumab in plaque psoriasis – results of two phase 3 trials. N Engl J Med 2014; 371:326–38. [DOI] [PubMed] [Google Scholar]
- 74. Griffiths CE, Reich K, Lebwohl M et al, UNCOVER 2 and UNCOVER 3 investigators . Comparison of ixekizumab with etanercept or placebo in moderate‐to‐severe psoriasis (UNCOVER‐2 and UNCOVER‐ 3): results from two phase 3 randomised trials . Lancet 2015;386:541–51. [DOI] [PubMed] [Google Scholar]
- 75. Papp KA, Merola JF, Gottlieb AB et al Dual neutralization of both interleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12‐week randomized, double‐blinded, placebo‐controlled phase 2b trial. J Am Acad Dermatol 2018; 79:277–86.e10. [DOI] [PubMed] [Google Scholar]
- 76. Masson Regnault M, Konstantinou MP, Khemis A et al Early relapse of psoriasis after stopping brodalumab: a retrospective cohort study in 77 patients. J Eur Acad Dermatol Venereol 2017; 31:1491–6. [DOI] [PubMed] [Google Scholar]
- 77. Mullard A. New plaque psoriasis approval carries suicide warning. Nat Rev Drug Discov 2017; 16:155. [DOI] [PubMed] [Google Scholar]
- 78. Lebwohl MG, Papp KA, Marangell LB et al Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J Am Acad Dermatol 2018; 78:81–9.e5. [DOI] [PubMed] [Google Scholar]
- 79. Kulig P, Musiol S, Freiberger SN et al IL‐12 protects from psoriasiform skin inflammation. Nat Commun 2016; 7:13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gordon KB, Strober B, Lebwohl M et al Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet 2018; 392:650–61. [DOI] [PubMed] [Google Scholar]
- 81. Gordon KB, Armstrong AW, Foley P et al Guselkumab efficacy after withdrawal is associated with suppression of serum IL‐23‐regulated IL‐17 and IL‐22 in psoriasis: VOYAGE 2 study. J Invest Dermatol 2019; 139:2437–46.e1. [DOI] [PubMed] [Google Scholar]
- 82. Reich K, Armstrong AW, Foley P et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J Am Acad Dermatol 2017; 76:418–31. [DOI] [PubMed] [Google Scholar]
- 83. Reich K, Warren RB, Iversen L et al Long‐term efficacy and safety of tildrakizumab for moderate‐to‐severe psoriasis: pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2) through 148 weeks. Br J Dermatol 2019; 182:605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rompoti N, Katsimbri P, Kokkalis G et al Real world data from the use of secukinumab in the treatment of moderate‐to‐severe psoriasis, including scalp and palmoplantar psoriasis: a 104‐week clinical study. Dermatol Ther 2019; 32:e13006. [DOI] [PubMed] [Google Scholar]
- 85. Martorell A, Garcia‐Martinez FJ, Jimenez‐Gallo D et al An update on hidradenitis suppurativa (Part I): epidemiology, clinical aspects, and definition of disease severity. Actas Dermosifiliogr 2015; 106:703–15. [DOI] [PubMed] [Google Scholar]
- 86. Vazquez BG, Alikhan A, Weaver AL, Wetter DA, Davis MD. Incidence of hidradenitis suppurativa and associated factors: a population‐based study of Olmsted County, Minnesota. J Investigative Dermatol 2013; 133:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Revuz JE, Canoui‐Poitrine F, Wolkenstein P et al Prevalence and factors associated with hidradenitis suppurativa: results from two case‐control studies. J Am Acad Dermatol 2008; 59:596–601. [DOI] [PubMed] [Google Scholar]
- 88. Jemec GB, Heidenheim M, Nielsen NH. A case–control study of hidradenitis suppurativa in an STD population. Acta Derm Venereol 1996; 76:482–3. [DOI] [PubMed] [Google Scholar]
- 89. Garg A, Kirby JS, Lavian J, Lin G, Strunk A. Sex‐ and age‐adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol 2017; 153:760–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Saunte DM, Boer J, Stratigos A et al Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol 2015; 173:1546–9. [DOI] [PubMed] [Google Scholar]
- 91. Garg A, Lavian J, Lin G, Strunk A, Alloo A. Incidence of hidradenitis suppurativa in the United States: a sex‐ and age‐adjusted population analysis. J Am Acad Dermatol 2017; 77:118–22. [DOI] [PubMed] [Google Scholar]
- 92. Miller IM, McAndrew RJ, Hamzavi I. Prevalence, risk factors, and comorbidities of hidradenitis suppurativa. Dermatol Clin 2016; 34:7–16. [DOI] [PubMed] [Google Scholar]
- 93. Hamzavi IH, Sundaram M, Nicholson C et al Uncovering burden disparity: a comparative analysis of the impact of moderate‐to‐severe psoriasis and hidradenitis suppurativa. J Am Acad Dermatol 2017; 77:1038–46. [DOI] [PubMed] [Google Scholar]
- 94. Fitzsimmons JS, Guilbert PR. A family study of hidradenitis suppurativa. J Med Genet 1985; 22:367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. von Laffert M, Helmbold P, Wohlrab J, Fiedler E, Stadie V, Marsch WC. Hidradenitis suppurativa (acne inversa): early inflammatory events at terminal follicles and at interfollicular epidermis. Exp Dermatol 2010; 19:533–7. [DOI] [PubMed] [Google Scholar]
- 96. Wang B, Yang W, Wen W et al Gamma‐secretase gene mutations in familial acne inversa. Science 2010; 330:1065. [DOI] [PubMed] [Google Scholar]
- 97. Hotz C, Boniotto M, Guguin A et al Intrinsic defect in keratinocyte function leads to inflammation in hidradenitis suppurativa. J Invest Dermatol 2016; 136:1768–80. [DOI] [PubMed] [Google Scholar]
- 98. Orvain Cindy, Lin Yea‐Lih, Jean‐Louis Francette et al Hair follicle stem cell replication stress drives IFI16/STING‐dependent inflammation in hidradenitis suppurativa [published online ahead of print, 2020 Apr 2]. J Clin Invest. 2020; 10.1172/jci131180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ring HC, Bay L, Kallenbach K et al Normal skin microbiota is altered in pre‐clinical hidradenitis suppurativa. Acta Derm Venereol 2017; 97:208–13. [DOI] [PubMed] [Google Scholar]
- 100. Ring HC, Thorsen J, Saunte DM et al The follicular skin microbiome in patients with hidradenitis suppurativa and healthy controls. JAMA Dermatol 2017; 153:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vossen A, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol 2018; 9:2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lima AL, Karl I, Giner T et al Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br J Dermatol 2016; 174:514–21. [DOI] [PubMed] [Google Scholar]
- 103. van der Zee HH, de Ruiter L, Boer J et al Alterations in leucocyte subsets and histomorphology in normal‐appearing perilesional skin and early and chronic hidradenitis suppurativa lesions. Br J Dermatol 2012; 166:98–106. [DOI] [PubMed] [Google Scholar]
- 104. Moran B, Sweeney CM, Hughes R et al Hidradenitis suppurativa Is characterized by dysregulation of the Th17 : Treg cell axis, which is corrected by anti‐TNF therapy. J Invest Dermatol 2017; 137:2389–95. [DOI] [PubMed] [Google Scholar]
- 105. Musilova J., Moran B., Sweeney C.M. et al Enrichment of plasma cells in the peripheral blood and skin of patients with hidradenitis suppurativa. J Invest Dermatol. 2020;140: 5:1091–1094.e2. [DOI] [PubMed] [Google Scholar]
- 106. Byrd AS, Carmona‐Rivera C, O’Neil LJ et al Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci Transl Med 2019; 11:eaav5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Qiu SL, Zhang H, Tang QY et al Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax 2017; 72:1084–93. [DOI] [PubMed] [Google Scholar]
- 108. Schlapbach C, Hanni T, Yawalkar N, Hunger RE. Expression of the IL‐23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol 2011; 65:790–8. [DOI] [PubMed] [Google Scholar]
- 109. Kelly G, Hughes R, McGarry T et al Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br J Dermatol 2015; 173:1431–9. [DOI] [PubMed] [Google Scholar]
- 110. Scala E, Di Caprio R, Cacciapuoti S et al A new T helper 17 cytokine in hidradenitis suppurativa: antimicrobial and proinflammatory role of interleukin‐26. Br J Dermatol 2019; 181:1038–45. [DOI] [PubMed] [Google Scholar]
- 111. Navrazhina K, Frew JW, Krueger JG. Interleukin 17C is elevated in lesional tissue of hidradenitis suppurativa. Br J Dermatol 2019;182:1045–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Guttman‐Yassky E, Krueger JG. IL‐17C: a unique epithelial cytokine with potential for targeting across the spectrum of atopic dermatitis and psoriasis. J Invest Dermatol 2018; 138:1467–9. [DOI] [PubMed] [Google Scholar]
- 113. Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA 2017; 318:2019–32. [DOI] [PubMed] [Google Scholar]
- 114. Deckers IE, Prens EP. An update on medical treatment options for hidradenitis suppurativa. Drugs 2016; 76:215–29. [DOI] [PubMed] [Google Scholar]
- 115. Haslund P, Lee RA, Jemec GB. Treatment of hidradenitis suppurativa with tumour necrosis factor‐alpha inhibitors. Acta Derm Venereol 2009; 89:595–600. [DOI] [PubMed] [Google Scholar]
- 116. Kimball AB, Okun MM, Williams DA et al Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med 2016; 375:422–34. [DOI] [PubMed] [Google Scholar]
- 117. Casseres RG, Prussick L, Zancanaro P et al Secukinumab in the treatment of moderate to severe hidradenitis suppurativa: results of an open‐label trial. J Am Acad Dermatol. 2020; 82:1524–6. [DOI] [PubMed] [Google Scholar]
- 118. Kimball Aea . Novel anti‐IL17 antibody (CJM112) reduces inflammation of hidradenitis suppurativa patients in a placebo‐controlled trial. Exp Dermatol 2019; 28:5–55.30723943 [Google Scholar]
- 119. Schreiber S, Colombel JF, Feagan BG et al Incidence rates of inflammatory bowel disease in patients with psoriasis, psoriatic arthritis and ankylosing spondylitis treated with secukinumab: a retrospective analysis of pooled data from 21 clinical trials. Ann Rheum Dis 2019; 78:473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Parkes GC, Whelan K, Lindsay JO. Smoking in inflammatory bowel disease: impact on disease course and insights into the aetiology of its effect. J Crohns Colitis 2014; 8:717–25. [DOI] [PubMed] [Google Scholar]
- 121. Witte‐Handel E, Wolk K, Tsaousi A et al The IL‐1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction. J Invest Dermatol 2019; 139:1294–305. [DOI] [PubMed] [Google Scholar]
- 122. Lim SYD, Oon HH. Systematic review of immunomodulatory therapies for hidradenitis suppurativa. Biologics 2019; 13:53–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kanni T, Argyropoulou M, Spyridopoulos T et al MABp1 targeting IL‐1α for moderate to severe hidradenitis suppurativa not eligible for adalimumab: a randomized study. J Invest Dermatol 2018; 138:795–801. [DOI] [PubMed] [Google Scholar]
- 124. Yamaguchi Y, Fujio K, Shoda H et al IL‐17B and IL‐17C are associated with TNF‐alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol 2007; 179:7128–36. [DOI] [PubMed] [Google Scholar]