

Abstract
Microsomal epoxide hydrolase (mEH) hydrolyzes a wide range of epoxide containing molecules. Although involved in the metabolism of xenobiotics, recent studies associate mEH with the onset and development of certain disease conditions. This phenomenon is partially attributed to the significant role mEH plays in hydrolyzing endogenous lipid mediators, suggesting more complex and extensive physiological functions. In order to obtain pharmacological tools to further study the biology and therapeutic potential of this enzyme target, we describe the development of highly potent 2-alkylthio acetamide inhibitors of the human mEH with IC50 values in the low nanomolar range. These are around 2 orders of magnitude more potent than previously obtained primary amine, amide and urea-based mEH inhibitors. Experimental assay results and rationalization of binding through docking calculations of inhibitors to a mEH homology model indicate that an amide connected to an alkyl side chain and a benzyl-thio function as key pharmacophore units.
Keywords: Microsomal Epoxide Hydrolase, Enzyme Inhibitors, 2-alkylthio acetamide series, Molecular Docking, Amides, Benzyl-Thio
Graphical Abstract

Introduction
The microsomal epoxide hydrolase (mEH, EPHX1, E.C. 3.3.2.9) is a mammalian α/β - fold hydrolase enzyme, expressed in almost all tissues. It is mainly localized in the endoplasmic reticulum (ER) of eukaryotic cells.1 However, it can come off the ER, exit the cell and appear in the blood where it is termed the preneoplastic antigen2 and has been explored as a marker for liver cancer and hepatitis. It catalyzes the hydration of epoxides to the corresponding diols. It hydrolyzes a wide variety of substrates, ranging from most mono-substituted epoxides to several gem- and cis-di-substituted epoxides. trans-Disubstituted and trisubstituted epoxides are often poor substrates or even act as competitive inhibitors of mEH.3 The mEH also seems to be inactive on tetrasubstituted epoxides. Recently, oxetanes were showed to be hydrolyzed by mEH in vitro.4,5 The mEH metabolizes xenobiotics both in vitro and in vivo, where it plays either detoxification or bioactivation roles depending on the particular xenobiotic.3 Polymorphism and association studies suggest a link between the enzyme and some diseases such as preeclampsia, hypercholanemia and cancer.1
Since endogenous epoxy-fatty acids (EpFAs) are relatively poor substrates for mEH compared to soluble epoxide hydrolase (sEH), in vitro, the involvement of mEH in regulation of these beneficial lipid mediators was considered to be only marginal. Recently, however, using genetic KO mice it has been shown that mEH can play a significant role in the hydrolysis of EpFAs, such as the epoxy-eicosatrienoic acids (EETs), in vivo.6 In addition, inhibition of mEH seems to complement inhibition of sEH in improving post-ischemic functional recovery in murine hearts, suggesting that mEH inhibition may also have promising therapeutic potential.
The first reported mEH inhibitors were alternate substrates that were poorly turned over.7,8,9 Previously, we showed that primary ureas, amides and amines can inhibit mEH with a single digit micromolar range potency.10 Later, we developed stable inhibitors of the rat mEH with IC50 in the high nanomolar range.11 In order to better understand the biological roles of mEH and evaluate the therapeutic potential of mEH inhibition, we aimed to develop novel and highly potent (low nanomolar range IC50) chemical inhibitors of the human mEH.
Results and Discussion
Previously, a series of differently substituted 2-(alkylthio)propanamides was tested on the human mEH but most of the compounds were not potent inhibitors of the enzyme.12 In this study, some newly derived 2-(alkylthio)propanamides 2–5 displayed significantly improved inhibitory potencies, though none of them was more potent than the reference compound 1. Since bromoacetamides and thiols are readily available either directly from commercial sources or synthetically, optimization for potency of the 2-alkylthio acetamide class of compounds seemed very attractive in terms of potential library size and its accessibility. Therefore, the effect of substituents at the a position to the amide function and at the sulfur atom on the human mEH inhibitory potency was studied. Since the reaction between α-bromo amides and thiols often proceeds to completion, as confirmed by TLC, and the starting materials do not inhibit mEH (Table SI–1, supplementary information), most of the initial screenings were performed using crude reaction mixtures. The best inhibitors were then resynthesized, purified, characterized and tested in the pure form. As can be seen from the Table 1 very similar results were obtained for crude reaction mixtures and pure products (6, 7, 16, 17, 23, 24), validating this approach.
Table 1.
Effect of the α-substituents on the human mEH inhibitory potency of acetamides.
| # | Structure | IC50,nM | # | Structure | IC50,nM |
|---|---|---|---|---|---|
| 1 |  |
1580 ± 600 | 17 |  |
250 |
| NSPA | 140a | ||||
| 2 |  |
10400 | 18 |  |
150 |
| 3 |  |
8800 | 19 |  |
29 |
| 4 |  |
1600 | 20 |  |
140 |
| 5 |  |
5400 | 21 |  |
670 |
| 6 |  |
680 | 22 |  |
430 |
| 640a | |||||
| 7 | 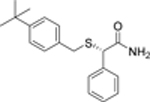 |
480 | 23 |  |
110 |
| 640a | 100a | ||||
| 8 |  |
42000 | 24 |  |
25 |
| 88a | |||||
| 9 |  |
53000 | 25 | 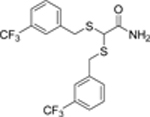 |
23 |
| 10 | 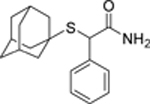 |
20000 | 26 |  |
28 |
| 11 |  |
3100 | 27 |  |
16 |
| 12 |  |
4500 | 28 |  |
19 |
| 13 |  |
2000 | 29 |  |
28000 |
| 14 |  |
500 | 30 |  |
16000 |
| 15 |  |
280 | 31 |  |
2200 |
| 16 |  |
110 | 32 |  |
>50000 |
| 110a | |||||
Reaction mixture value.
First, the effect of the alkyl/aryl-thio substituent at the α-position of phenylacetamide on the human mEH inhibition was investigated. A range of phenylacetamides with either adamantylthio-, arylthio- or benzylthio-substituents at the α-carbon (10–17) were prepared and tested (Table 1). Adamantylthio-(10) and arylthio-substituted (11) phenylacetamides were, in general, poorer inhibitors of the human mEH compared to the benzylthioacetamides (12–17), probably due to steric factors. Compounds 16 and 17 were then obtained in pure form, and were five and two-fold more effective, respectively, than the most potent inhibitors developed at that point (6, 7).
Next, the effect of the different α-alkyl substituents (18 – 28) on the mEH inhibitory potency of substituted primary amides was explored. For the series of compounds with the thioether substituent fixed to the meta-trifluoromethyl benzylthiol (4, 17–19, 24–28), the inhibitory potency increased in the range Me (4) < n-Pr (18) < n-Bu (19) and was almost two orders of magnitude higher for 19 than for 4. Replacing the n-alkyl chain by the branched chain decreased the IC50 values even further, with i-Pr side chain (24) being more potent than n-Bu (19). Among the branched side chains tested the cyclopentyl substituent (27) proved to have the largest impact on potency and shifted the IC50 value down to 16 nM. Replacement of the alkyl by a second meta- trifluoromethyl benzylthio-function resulted in compound 25 with an IC50 of 23 nM, comparable to compounds 24, 27 and 28. Since replacement of the branched alkyl side chain with meta-trifluoromethyl benzylthio-substituent did not have any major effect on inhibitory potency, we tested if the same is true for the replacement of the first meta-trifluoromethyl benzylthio-function (4, 17–19, 24–28) with alkyl function (30). Thus, 2,2-dicyclohexylacetamide 30 has been synthesized from the commercially available 2,2-dicyclohexylacetic acid. Surprisingly, amide 30 was almost three orders of magnitude less potent than the structurally related compound 28, pointing out the importance of benzylthio substituent for inhibitory potency. Interestingly, reduction of the amide function (30) to corresponding primary amine (31) resulted in almost one order of magnitude potency increase (Table 1).
Since thioethers are relatively unstable in vivo and could be easily oxidized to corresponding sulfoxides or sulfones by means of either the cytochrome P45013 or flavin monooxygenase (FMO)14 family of enzymes, we tested the effect this potential oxidation products have on the inhibitory potency. Oxidation of 6 and 7 to corresponding sulfoxides 8 and 9 (mixtures of epimers at the sulfur stereogenic center) decreased their inhibitory potency by approximately two orders of magnitude (Table 1). Although the rates of thioether oxidation by CYP450 or FMO depend on stereochemical characteristics of a particular compound, the result suggests that it would be desirable to replace thioether by metabolically more stable function. Considering that 2,3-diphenylpropanamide 29 is a better inhibitor of mEH to its structurally close thioanalogue 131 (Table 4), it is likely that methylene group may serve a role of a metabolically more stable replacement function for thioether. This result is also congruent with our previous results,11 however the validity of this assumption for benzylthio-series of compounds will need to be tested.
Table 4.
Inhibition data for the compounds that showed more than 10% inhibition of mEH but less than 50% at 50 pM concentration. Blanc reactions did not inhibit mEH.
| # | Structure | Inhibition at 50 (μM,% | # | Structure | Inhibition at 50 μM,% |
|---|---|---|---|---|---|
| 77 |  |
60a | 108 |  |
15 |
| 78 | 25 | 109 |  |
12 | |
| 79 | 30 | 110 |  |
33 | |
| 80 |  |
40 | 111 |  |
18 |
| 81 |  |
40 | 112 |  |
15 |
| 82 | 38 | 113 |  |
22 | |
| 83 |  |
30 | 114 |  |
12 |
| 84 |  |
15 | 115 |  |
29 |
| 85 |  |
20 | 116 |  |
17 |
| 86 |  |
45 | 117 |  |
45 |
| 87 |  |
20 | 118 |  |
24 |
| 88 |  |
20 | 119 |  |
10 |
| 89 |  |
20 | 120 |  |
31 |
| 90 |  |
25 | 121 |  |
23 |
| 91 |  |
24 | 122 |  |
11 |
| 92 | 15 | 123 |  |
36 | |
| 93 |  |
41 | 124 |  |
41 |
| 94 |  |
31 | 125 |  |
38 |
| 95 |  |
26 | 126 |  |
19 |
| 96 |  |
32 | 128 |  |
19 |
| 97 |  |
32 | 128 |  |
19 |
| 98 | 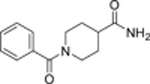 |
20 | 129 |  |
37 |
| 99 |  |
31 | 130 |  |
36 |
| 100 |  |
25 | 131 |  |
34 |
| 101 |  |
18 | 132 |  |
35 |
| 102 |  |
25 | 133 |  |
37 |
| 103 |  |
15 | 134 |  |
IC50 = 87 μM b |
| 104 |  |
11 | 135 | 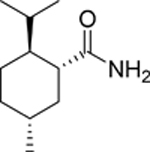 |
IC50 = 55 μM b |
| 105 |  |
15 | 136 |  |
IC50 = 96 μM b |
| 106 |  |
17 | 137 | 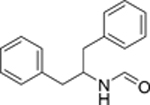 |
IC50 = 3.3 μM b |
| 107 |  |
16 | 138 |  |
IC50 = 1.9 μM |
IC50 = 33.6 μM.
apparent increase in mEH activity at low concentrations.
After optimization of the alkyl side chain, the attention was drawn to the optimization of the thioether substituent. Thus, 2-cyclopentyl-2-mercaptoacetamide 33 was synthesized from commercially available 2-cyclopentylacetyl chloride (Scheme 2) and, interestingly, it was a significantly more potent inhibitor than the parent 2-bromo-2-cyclopentylacetamide (33 versus 32). Following alkylation of mercaptoacetamide 33 with various commercially available aryl bromides, the mEH inhibitory potency of the reaction mixtures was tested, and the most potent compounds were then resynthesized, purified, characterized and the inhibitory potency of the pure compounds was tested (Table 2).
Scheme 2.

Synthesis of compounds 33 and 34–64. a) Br2, 50 °C; b) NH4OH, 0 °C; c) KSAc, DMF, 60 °C; d) NaOH, MeOH; e) RBr, K2CO3, DMF.
Table 2.
Effect of the α-alkylthio substituent on the mEH inhibitory potency of 2-cyclopentyl-acetamides.
 |
|||||
|---|---|---|---|---|---|
| # | R1 | IC50, nM | # | R1 | IC50, nM |
| 33 | H | 7650 | 49 |  |
2100 |
| 34 |  |
1466 | 50 |  |
315 |
| 35 |  |
493 | 51 |  |
32 |
| 36 |  |
1100 | 52 | 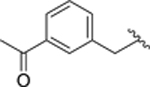 |
384 |
| 37 |  |
500 | 53 |  |
441 |
| 38 |  |
33 | 54 |  |
399 |
| 39 |  |
4940 | 55 |  |
773 |
| 40 |  |
2900 | 56 |  |
26 |
| 41 |  |
6600 | 57 |  |
11 |
| 42 |  |
839 | 58 |  |
9 |
| 43 |  |
422 | 59 |  |
18 |
| 44 |  |
433 | 60 |  |
10 |
| 45 | 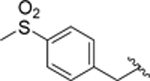 |
6960 | 61 |  |
10.1 |
| 46 | 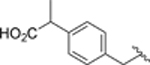 |
899 | 62 |  |
2.2 |
| 47 |  |
600 | 63 |  |
4.7 |
| 48 | 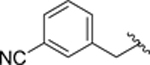 |
193 | 64 |  |
14 |
As seen in Table 2, heteroaromatic substituents mainly reduced inhibitory potency of the compounds (34–37). Compound 38 with a naphthalen-2-ylmethylthio substituent was just slightly less potent compared to the best inhibitor 27 made so far. Introduction of the polar functional groups at the para or meta positions of the benzyl substituent had mainly negative effect on the inhibitory potency (39–52), except for the nitro group in the meta position (51). Compounds with difluorobenzylthio-sidechains 53–54, as well as cyclohexylmethylthio-sidechain 55 were relatively poor inhibitors of the human mEH. Finally, compounds with bulky/lipophilic substituents in meta/para positions on phenyl ring 56–62 showed the highest inhibitory potency against the human mEH. The most potent inhibitor was 2-((3,5-bis(trifluoromethyl)benzyl)thio)-2-cyclopentylacetamide 62 which had an IC50 of 2.2 nM. It is worth noting that replacement of the 3-(trifluoromethyl)benzyl substituent in 28 with a homologous 3-(trifluoromethyl)phenethyl substituent (64) did not have any effect on inhibitory potency while its replacement with (3-(trifluoromethyl)phenyl)ethyl decreased the IC50 by over 3 times, bringing it to 4.7 nM (63).
A second assay, based on a radioactive substrate and not fluorescence,15 was conducted to validate the inhibitory potential of the most potent compound 62 for two reasons. Firstly, the IC50 obtained approached the assay limit (1.95 nM) to effectively distinguish potent inhibitors, and hence could have a degree of uncertainty associated with it. Secondly, an alternative means of IC50 determination would help verify that the observed potency was intrinsic and independent of the assay employed. The experiment yielded an IC50 of 0.94 nM, reinforcing the observation that 62 is the most potent mEH inhibitor in this series.
Additionally, a broad panel of commercially available compounds together with some in house synthesized chemicals were screened to find new structural leads. The results of those preliminary screens are reported in Tables 3–5 and SI–1 (in supplementary information). Since simple primary amines like dodecyl amine are relatively good mEH inhibitors and can be easily functionalized at the nitrogen position, the effect of substitution at nitrogen was first studied (Table 3). Unfortunately, none of the synthesized dodecyl amine derivatives 66–76 have higher potency on mEH than the parent dodecyl amine 65. Though, N-dodecylformamide 66 and N-dodecyl-1,1,1-trifluoromethanesulfonamide 71 have potencies similar to the parent amine. Interestingly, replacement of the formyl function with larger acyl homologues resulted in compounds 72–76 that are weak inhibitors of the mEH at low concentrations but cause an apparent increase of the enzyme activity at higher concentrations. Similar activation of the mEH enzymatic activity has been observed previously for other compounds.16 Although not completely understood, this activation may be due to the binding to an allosteric site which increases catalytic activity of mEH or a change in access of the substrate to the catalytic site. Among the other compounds tested, almost all of them were poor inhibitors of human mEH (IC50 > 50 μM, Table 4 and SI–1 in supplementary information) except for 1,3-diphenylpropan-2-amine 138 and its formamide 137 that inhibited 50 % of mEH activity at 1.9 and 3.3 μM respectively. Although, the inhibitory potencies of 137 and 138 were almost an order magnitude higher than the initial dodecylamine 65, further structural optimization of those hits was considered to be difficult and low throughput due to the scarcity of the related amines, and thus was not followed here. Table 5 shows data on mEH inhibitory potency of some oxetanes. As can be seen, most of the oxetanes tested did not inhibit mEH except for 144 and 146 that inhibited 50% of mEH activity at 72 and 51 μM respectively. Considering that oxetanes could be converted to corresponding diols by mEH,4,5 it seems reasonable to assume that 144 and 146 act as slow turnover substrates, similar to the earlier reported cyclohexene oxide, 1,1,1-trichloropropane-2,3-epoxide and sterically hindered cyclodiene epoxides.7,8,9
Table 3.
Effect of N-substitution on the mEH inhibition by primary amides.
| # | Structure | IC50,μM | Inhibition at 100 μM, % |
|---|---|---|---|
| 65 |  |
12.9 | 83 |
| 66 |  |
15.1 | 80 |
| 67 |  |
61.3 | 67 |
| 68 |  |
> 100 | 5 |
| 69 |  |
> 100,100a | 0,50a |
| 70 |  |
> 100 | 40, 25a |
| 71 |  |
14.7, 12.5a | 65, 98a |
| 72 |  |
> 100 | 33,0 a,b |
| 73 |  |
> 100 | - b |
| 74 |  |
> 100 | - b |
| 75 |  |
> 100 | - b |
| 76 |  |
> 100 | -15 b |
Reaction mixture value.
increase in mEH activity.
Table 5.
mEH inhibition by oxetanes.
| # | Structure | IC50,μM | # | Structure | IC50, μM |
|---|---|---|---|---|---|
| 139 |  |
>100 | 144 | 72 | |
| 140 |  |
>100 | 145 | 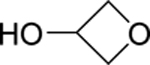 |
>100 |
| 141 |  |
>100 | 146 |  |
51 |
| 142 |  |
>100 | 147 | >100 | |
| 143 | >100 | 148 | >100 | ||
Next, homology modeling was performed to predict the mEH protein structure and docking studies were conducted with compounds 4, 27 and the most potent structure 62 to predict the preferred binding modes of the inhibitors to the active site of mEH. To ensure the charge assignments for each atom of 62 were reasonable, atomic charges were calculated with the CHelpG17 method in GAUSSIAN09.18 The charges of the ligand were then assigned to each atom and further prepared for docking. 2500 docking runs were performed, and 38 structures were retained after filtering. Two major binding modes (poses) were predicted; these are shown in Figure 1. The interface energy for the preferred pose of 62 is predicted to be −19.85 (Rosetta energy unit, REU).
Figure 1.

Two binding modes predicted by docking simulation of 62. Both shown in the same protein orientation.
For both poses, as expected,19 D226, Y299 and Y374 hydrogen bond with the amide group of the inhibitor as constrained (e.g., Figure 2, left). For pose 1 (Figure 1, left), a π-π stacking interaction between 62 and W227 was calculated (Figure 2, right), which likely contributes to favorable binding. The orientation of pose 2 (Figure 1, right) is consistent with the two dominant poses predicted for molecule 27 (vide infra). Other interactions, including S-π interactions, were predicted in some docking runs (see SI for details). To assess the inherent energies for the inhibitor conformers in poses 1 and 2, these structures were extracted and optimized with DFT (SMD(water)- ωB97XD/6–31+G(d,p)).20,21,30 The conformer in pose 1 is predicted to be 2.4 kcal/mol lower in energy than the conformer in pose 2, suggesting that both are energetically accessible.
Figure 2.

Key molecular interactions in pose 1. Left: interactions between amide group of 62 with D226, Y299 and Y374 side chain. Right: π-π stacking between 62 and W227. The two images correspond to the same pose rotated 180° to each other.
To investigate the difference in mEH inhibitory potency with different alkyl substituents alpha to the acetamide, docking simulations for molecules 4 and 27 were conducted since the experimental result shows that the latter is 100-fold more potent than the former. Among the 2500 poses generated in the docking runs for 27, 43 poses passed the filters mentioned above. The majority of these poses fall into the two binding modes shown in Figure 3A. Among the 2500 poses for 4, 41 poses passed the filters. Poses of are less converged than those for 27 (see SI for detail), which may be a result of the methyl group not filling the space in the active site as well as does the cyclopentyl group (see Figure 3B). Interface energies for the two molecules are noticeably different, with the values of −14.85 and −19.68 predicted for 4 and 27, respectively, consistent with the difference in inhibition. The two predicted preferred binding orientations of molecule 27 (Figure 3A) are consistent with pose 2 of molecule 62 (Figure 3C). Each of the trifluoromethyl groups of 62 corresponds to one of the two poses of molecule 27.
Figure 3.

(A) Two binding modes of 27 predicted by docking simulation. (B) Comparison between molecule 27 (pose 2) and 4, shown in blue and yellow respectively. (C) Comparison between molecule 27 and 62 binding orientation. Pose 2 of 62 is shown in salmon, pose 1 of molecule 27 is shown in olive.
Conclusions
In this study, a series of 2-alkylthio acetamide inhibitors of the human microsomal epoxide hydrolase, with IC50 values in the low nanomolar range (0.94–11 nM), were obtained. These are over two orders of magnitude more potent than previous primary amine, amide and urea-based mEH inhibitors (IC50 ≥ 480 nM). To better understand structural requirements for potent mEH inhibitors, docking studies were performed with a new homology model of human mEH. Both experimental and docking results point toward the importance of the aryl ring 3–4 bonds away from the acetamide function as well as a bulky aliphatic substituent at the a-position. No interaction with the thioether function was observed, suggesting it can be replaced by a potentially more metabolically stable functionalities, such as a methylene, which will be the subject of ongoing studies.
Experimental Section
Reagents and Instruments
Many compounds tested as inhibitors or required for synthesis are commercially available and were purchased from one of the following commercial sources: Sigma Aldrich Chemical Co. (Milwaukee, WI), Fisher Scientific (Houston, TX), Eanmine LLC (Monmouth Jct, NJ), Oakwood Chemical (Estill, SC), Chem-Impex Inc (Wood Dale, IL) or Combi-Blocks (San Diego, CA). All reactions were carried out under an atmosphere of dry nitrogen. All chemicals purchased from commercial sources were used as received without further purification. Analytical thin layer chromatography (TLC) was performed on Merck TLC silica gel 60 F254 plates. TLC plates predominantly indicated a single product, following exposure under 254 nm UV light or development using a potassium permanganate stain. Flash chromatography was performed on silica gel (230–400 mesh) from Macherey Nagel. NMR spectra were recorded on Varian VNMRS 600, Inova 400 or Bruker Avance III 800 MHz instruments. Multiplicity is described by the abbreviations b = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet. Chemical shifts are given in ppm. 1H NMR spectra were referenced to the residual solvent peak at δ = 7.26 (CDCl3). 13C NMR spectra were referenced to the solvent peak at δ = 77.16 (CDCl3). HRMS spectra were recorded on Thermo Electron LTQ-Orbitrap XL Hybrid MS in ESI.
Chemistry
Synthesis
General procedure of S-alkylation for synthesis of compounds 1–28. A mixture of appropriate 2-bromoacetamide (1 equiv), appropriate thiol (1.1 equiv), K2CO3 (1.5 equiv) and DMF (~2 mL/mmol) was stirred at room temperature overnight. Water was added, and the resulting precipitate was filtered. When no precipitate was formed, the product was extracted with EtOAc (3 × 20 mL). Combined organics were dried over MgSO4, filtered and evaporated. Purification by flash column chromatography (EtOAc/hexanes = 10:90 → 40:60 → 50:50) gave pure product, unless noted otherwise.
2-(naphthalen-2-ylthio)propenamide 2.
Yield 31 %.
1H NMR (600 MHz, CDCh) 5 7.79 (m, 4H), 7.46 (m, 3H), 6.48 (bs, 1H), 5.53 (bs, 1H), 3.91 (q, J = 7.3 Hz, 1H), 1.61 (d, J = 7.3 Hz, 3H).
HRMS (ESI) calculated m/z 232.0791 (MH+), observed m/z 232.0790 (MH+)
2-((adamantan-1-yl)thio)propenamide 5.
Purified by recrystallization from EtOAc/hexanes = 40:60 mixture. Yield 49 %.
1H NMR (600 MHz, CDCh) δ 6.81 (bs, 1H), 6.19 (bs, 1H), 3.41 (q, J = 7.5 Hz, 1H), 2.00 (m, 3H), 1.86 (d, J = 11.8 Hz, 3H), 1.80 (d, J = 11.9 Hz, 3H), 1.65 (d, J = 12.4 Hz, 3H), 1.61 (d, J = 12.3 Hz, 3H), 1.43 (d, J = 7.5 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 177.9, 46.5, 43.5, 39.5, 36.1, 29.7, 20.7. HRMS (ESI) calculated m/z 240.1417 (MH+), observed m/z 302.1553 ([M+HCOONH4]+)
2-((4-bromobenzyl)thio)-2-phenylacetamide 16.
Yield 41 %.
1H NMR (600 MHz, CDCI3) δ 7.41 (d, J = 8.4 Hz, 2H), 7.34 – 7.25 (m, 5H), 7.14 (d, J = 8.4 Hz, 2H), 6.35 (bs, 1H), 6.03 (bs, 1H), 4.32 (s, 1H), 3.72 (d, J = 13.6 Hz, 1H), 3.61 (d, J = 13.6 Hz, 1H).
13C NMR (151 MHz, CDCI3) δ 172.2, 136.3, 136.2, 131.8, 130.8, 129.0, 128.4, 128.3, 1 21.3, 53.6, 36.1.
HRMS (ESI) calculated m/z 336.0052 (MH+), observed m/z 336.0029 (MH+)
2-phenyl-2-((3-(trifluoromethyl)benzyl)thio)acetamide 17.
Yield 38 %.
1H NMR (600 MHz, CDCI3) δ 7.54 – 7.42 (m, 4H), 7.37 – 7.29 (m, 5H), 6.22 (bs, 1H), 5.80 (bs, 1H), 4.36 (s, 1H), 3.85 (d, J = 13.6 Hz, 1H), 3.73 (d, J = 13.6 Hz, 1H).
13C NMR (151 MHz, CDCI3) δ 172.0, 138.2, 136.2, 132.5, 131.4, 131.2, 131.0, 130.7, 129.3, 129.2, 128.6, 128.4, 125.94, 125.92, 125.89, 125.87, 124.43, 124.41, 124.38, 124.36, 53.9, 36.4.
HRMS (ESI) calculated m/z 326.0821 (MH+), observed m/z 326.0796 (MH+)
2-((3-(trifluoromethyl)benzyl)thio)pentanamide 18.
Yield 82 %.
1H NMR (600 MHz, CDCl3) δ 7.56 (b, 1H), 7.46 (m, 2H), 7.40 (m, 1H), 6.55 (bs, 2H), 6.50 (bs, 1H), 3.78 (m, 2H), 3.13 (t, J = 7.3 Hz, 1H), 1.78 (m, 1H), 1.61 (m, 1H), 1.43 (m, 1H), 1.34 (m, 1H), 0.83 (t, J = 7.4 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 175.2, 138.5, 132.4, 131.3, 131.1, 130.9, 130.7, 129.1, 126.7, 125.74, 125.72, 125.69, 125.67, 124.9, 124.20, 124.18, 124.15, 124.13, 123.1, 121.3, 49.2, 35.6, 34.3, 20.6, 13.6.
HRMS (ESI) calculated m/z 292.0977 (MH+), observed m/z 292.0974 (MH+)
2-((3-(trifluoromethyl)benzyl)thio)hexanamide 19.
Yield 90 %.
1H NMR (600 MHz, CDCl3) δ 7.57 (s, 1H), 7.47 (m, 2H), 7.41 (m, 1H), 6.57 (bs, 1H), 6.49 (bs, 1H), 3.78 (m, 2H), 3.11 (t, J = 7.3 Hz, 1H), 1.81 (m, 1H), 1.63 (m, 1H), 1.38 (m, 1H), 1.30 (m, 1H), 1.23 (m, 2H), 0.83 (t, J = 7.3 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 175.2, 138.6, 132.4, 131.3, 131.1, 130.9, 130.7, 129.1, 126.8, 125.75, 125.72, 125.70, 125.67, 124.9, 124.21, 124.18, 124.16, 124.13, 123.1, 121.3, 77.2, 49.5, 35.6, 32.1, 29.4, 22.3, 13.8.
HRMS (ESI) calculated m/z 306.1134 (MH+), observed m/z 306.1134 (MH+)
2-((4-bromobenzyl)thio)-3-methylbutanamide 23.
Yield 93 %.
1H NMR (600 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 6.56 (bs, 1H), 6.09 (bs, 1H), 3.69 (d, J = 13.4 Hz, 1H), 3.64 (d, J = 13.4 Hz, 1H), 2.93 (d, J = 6.7 Hz, 1H), 2.11 (o, J = 6.6 Hz, 1H), 0.98 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H).
13C NMR (151 MHz, CDCI3) δ 174.4, 136.5, 131.8, 130.8, 121.3, 57.2, 35.8, 30.9, 21.0, 19.9.
HRMS (ESI) calculated m/z 302.0209 (MH+), observed m/z 302.0307 (MH+)
3-methyl-2-((3-(trifluoromethyl)benzyl)thio)butanamide 24.
Yield 68 %.
1H NMR (600 MHz, CDCl3) δ 7.57 (s, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 7.7 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 6.57 (bs, 1H), 6.26 (bs, 1H), 3.80 (d, J = 13.5 Hz, 1H), 3.75 (d, J = 13.5 Hz, 1H), 2.94 (d, J = 6.7 Hz, 1H), 2.16 – 2.10 (m, 1H), 0.99 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 174.4, 138.5, 132.5, 131.4, 131.2, 131.0, 130.8, 129.1, 126.8, 125.80, 125.77, 125.75, 125.72, 124.96, 124.29, 124.26, 124.24, 124.21, 123.2, 1 21.4, 57.4, 36.0, 30.9, 21.0, 19.8.
HRMS (ESI) calculated m/z 292.0977 (MH+), observed m/z 292.0983 (MH+)
4-methyl-2-((3-(trifluoromethyl)benzyl)thio)pentanamide 26.
Yield 74 %.
1H NMR (600 MHz, CDCl3) δ 7.54 (s, 1H), 7.41 (m, 2H), 7.33 (m, 1H), 7.02 (bs, 1H), 6.64 (bs, 1H), 3.75 (s, 2H), 3.16 (t, J = 7.8 Hz, 1H), 1.66 (m, 2H), 1.45 (m, 1H), 0.80 (d, J = 6.5 Hz, 3H), 0.71 (d, J = 6.3 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 175.6, 138.6, 132.3, 131.1, 130.9, 130.6, 130.4, 128.9, 126.6, 125.60, 125.57, 125.55, 125.52, 124.8, 123.95, 123.93, 123.90, 123.88, 123.0, 1 21.2, 47.3, 40.8, 35.2, 25.8, 22.3, 21.5.
HRMS (ESI) calculated m/z 306.1134 (MH+), observed m/z 306.1135 (MH+)
2-cyclopentyl-2-((3-(trifluoromethyl)benzyl)thio)acetamide 27.
Yield 32 %.
1H NMR (600 MHz, CDCl3) δ 7.59 (s, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 6.30 (bs, 1H), 5.64 (bs, 1H), 3.80 (s, 2H), 3.01 (d, J = 9.1 Hz, 1H), 2.16 (h, J = 8.5 Hz, 1H), 1.85 (m, 1H), 1.77 (m, 1H), 1.62 (m, 2H), 1.53 (m, 2H), 1.37 (m, 1H), 1.26 (m, 1H).
13C NMR (151 MHz, CDCh) 5 174.5, 138.6, 132.5, 131.4, 131.2, 131.0, 130.8, 129.2, 126.8, 125.84, 125.82, 125.79, 125.77, 125.0, 124.30, 124.27, 124.24, 124.22, 123.2, 121.4, 55.2, 42.2, 35.8, 31.1, 25.2.
HRMS (ESI) calculated m/z 318.1134 (MH+), observed m/z 318.1137 (MH+)
2-cyclohexyl-2-((3-(trifluoromethyl)benzyl)thio)acetamide 28.
Yield 42 %.
1H NMR (600 MHz, CDCl3) δ 7.59 (s, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.47 (d, J = 7.7 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 6.50 (bs, 1H), 5.80 (bs, 1H), 3.81 (d, J = 13.5 Hz, 1H), 3.75 (d, J = 13.5 Hz, 1H), 2.98 (d, J = 6.8 Hz, 1H), 1.91 – 1.58 (m, 6H), 1.28 – 0.94 (m, 5H).
13C NMR (151 MHz, CDCI3) δ 174.0, 138.6, 132.5, 131.5, 131.2, 131.0, 130.8, 129.2, 126.8, 125.84, 125.82, 125.79, 125.77, 125.0, 124.33, 124.31, 124.28, 124.26, 123.2, 121.4, 56.6, 40.3, 36.1, 31.4, 30.4, 26.20, 26.15, 26.08.
HRMS (ESI) calculated m/z 332.1290 (MH+), observed m/z 332.0029 (MH+)
2,3-diphenylpropanamide 29.
A mixture of 2,3-diphenyIpropanoic acid (403 mg, 1.78 mmol, 1 equiv) and SOCI2 (0.318 g, 2.68 mmol, 1.5 equiv) was refluxed for 3 h. The reaction mixture was cooled down to room temperature, the excess of SOCl2 was evaporated under reduced pressure and the residue was added dropwise to cooled to 0 °C saturated solution of ammonia under vigorous stirring. The resulting precipitate was filtered and dried under vacuum. Yield 0.4 g (quant).
1H NMR (600 MHz, CDCl3) δ 7.28 (m, 5H), 7.20 (t, J = 7.4 Hz, 2H), 7.15 (t, J = 7.3 Hz, 1H), 7.09 (d, J = 7.4 Hz, 2H), 5.58 (bs, 1H), 5.35 (bs, 1H), 3.63 (t, J = 7.5 Hz, 1H), 3.52 (dd, J = 13.7, 7.6 Hz, 1H), 2.98 (dd, J = 13.7, 7.4 Hz, 1H).
13C NMR (151 MHz, CDCl3) δ 175.3, 139.6, 139.5, 129.1, 128.9, 128.4, 128.2, 127.6, 126.4, 54.9, 39.5.
2,2-dicyclohexylacetamide 30.
A mixture of 2,2-dicyclohexylacetic acid (2.424 g, 10.8 mmol, 1 equiv) and SOCl2 (3.86 g, 32.5 mmol, 3 equiv) was refluxed for 3 h. The reaction mixture was cooled down to room temperature, the excess of SOCl2 was evaporated under reduced pressure and the residue was added dropwise to cooled to 0 °C saturated solution of ammonia under vigorous stirring. The formed oily precipitate was triturated and washed multiple times with water until it became solid. The solid residue was filtered and dried under vacuum. Yield 1.72 g (71 %).
1H NMR (600 MHz, CDCl3) δ 7.83 (s, 1H), 7.17 (s, 1H), 2.05 (t, J = 8.0 Hz, 1H), 1.69 (m, 12H), 1.29 – 0.92 (m, 10H).
13C NMR (151 MHz, CDCl3) δ 180.9, 57.4, 36.3, 31.4, 29.7, 26.7, 26.6, 26.5.

2-bromo-2-cyclopentylacetamide 32.
Br2 (3.6 g, 22.52 mmol, 1.1 equiv) was added to 2-cyclopentylacetyl chloride (3.01 g, 20.48 mmol, 1 equiv) at room temperature. The resulting mixture was refluxed for 2 hours, cooled down and added dropwise to the cooled to 0 °C concentrated solution of ammonium hydroxide. The resulting mixture was stirred for additional 15 min and filtered to give the product as a light-brown solid (1.745 g, 41 %).
1H NMR (600 MHz, CDCl3) δ 6.29 (bs, 1H), 6.25 (bs, 1H), 4.21 (d, J = 7.6 Hz, 1H), 2.48 (h, J = 8.1 Hz, 1H), 1.88 (m, 1H), 1.81 (m, 1H), 1.63 (m, 4H), 1.40 (m, 2H).
13C NMR (151 MHz, CDCl3) δ 171.8, 56.3, 44.3, 30.9, 30.8, 25.9, 25.3.

2-amino-1-cyclopentyl-2-oxoethyl ethanethioate 32c.
The mixture of 2-bromo-2-cyclopentylacetamide (3.96 g, 19.22 mmol, 1 equiv), AcSK (3.51 g, 30.76 mmol, 1.6 equiv) and DMF (35 mL) was stirred for two days at room temperature, then quenched with water (150 mL) and extracted with EtOAc (3 × 30 mL). Combined organics were dried over MgSO4, filtered and evaporated. The residue was chromatographed (EtOAc/hexanes = 30:70 → 40:60 → 50:50) to give pure product as a pale yellow solid (0.89 g, 23 %).
1H NMR (600 MHz, CDCl3) δ 6.40 (bs, 1H), 6.25 (bs, 1H), 3.79 (d, J = 9.9 Hz, 1H), 2.31 (m, 1H), 2.30 (s, 3H), 1.79 (m, 2H), 1.63 – 1.43 (m, 4H), 1.20 (m, 2H).
13C NMR (151 MHz, CDCl3) δ 196.4, 173.9, 51.6, 39.8, 31.3, 30.7, 30.4, 25.4, 25.1.
2-cyclopentyl-2-mercaptoacetamide 33.
A solution of 2-amino-1-cyclopentyl-2-oxoethyl ethanethioate (0.88 g, 4.38 mmol, 1 equiv) and NaOH (10 M in water, 0.88 mL, 8.76 mmol, 2 equiv) in methanol (10 mL) was stirred at room temperature overnight, concentrated to ~3 mL and quenched with water (15 mL) and few drops of hydrochloric acid to bring the pH to ~1. The resulting mixture was extracted with EtOAc (3 × 20 mL). Combined organics were dried over MgSO4, filtered and evaporated to give pure compound as pale yellow solid (0.696 g, quant).
1H NMR (600 MHz, CD3OD) δ 3.07 (d, J = 10.1 Hz, 1H), 2.18 (dp, J = 9.9, 8.0 Hz, 1H), 1.96 (m, 1H), 1.76 (m, 1H), 1.67 (m, 2H), 1.59 (m, 2H), 1.39 (m, 1H), 1.27 (m, 1H).
13C NMR (101 MHz, CDCl3) δ 175.5, 48.3, 44.4, 31.2, 30.4, 25.7, 25.3.
HRMS (ESI) calculated m/z 160.0791 (MH+), observed m/z 160.0788 (MH+)
General procedure of S-alkylation for synthesis of 34–64. A mixture of 2-cyclopentyl-2-mercaptoacetamide 33 (1 equiv), appropriate benzyl bromide (1.1 equiv), K2CO3 (1.5 equiv) and DMF (~2 mL/mmol) was stirred at room temperature overnight. Water was added, and the resulting precipitate was filtered. When no precipitate was formed, the product was extracted with EtOAc (3 × 20 mL). Combined organics were dried over MgSO4, filtered and evaporated. Purification by flash column chromatography (EtOAc/hexanes = 10:90 → 40:60 → 50:50) gave pure product, unless noted otherwise.
2-cyclopentyl-2-((3,5-dichlorobenzyl)thio)acetamide 57.
Yield 70 %.
1H NMR (400 MHz, CD3OD) 5 7.33 (m, 2H), 7.31 (m, 1H), 3.80 (d, J = 13.7 Hz, 1H), 3.75 (d, J = 13.7 Hz, 1H), 2.99 (d, J = 10.4 Hz, 1H), 2.18 (m, 1H), 1.87 (m, 1H), 1.77 (m, 1H), 1.69 – 1.48 (m, 4H), 1.29 (m, 2H).
13C NMR (101 MHz, CD3OD) δ 177.5, 143.9, 136.0, 128.7, 128.0, 55.6, 42.6, 35.7, 32.1, 26.2, 26.0.
HRMS (ESI) calculated m/z 318.0481 (MH+), observed m/z 318.0485 (MH+)
2-cyclopentyl-2-((3,5-dibromobenzyl)thio)acetamide 58.
Yield 85 %.
1H NMR (400 MHz, CDCl3) δ 7.55 (t, J = 1.8 Hz, 1H), 7.40 (d, J = 1.7 Hz, 2H), 6.23 (bs, 1H), 5.43 (bs, 1H), 3.68 (s, 2H), 3.03 (d, J = 9.2 Hz, 1H), 2.23 – 2.10 (m, 1H), 1.91 – 1.74 (m, 2H), 1.70 – 1.47 (m, 4H), 1.45 – 1.19 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 174.1, 141.6, 133.1, 130.9, 123.1, 55.2, 42.2, 35.2, 31.20, 31.19, 25.32, 25.28.
HRMS (ESI) calculated m/z 403.9325 ([M-H] −), observed m/z 441.9059 ([M+K-2H]−)
2-cyclopentyl-2-((3,5-dimethylbenzyl)thio)acetamide 59.
Yield 50 %.
1H NMR (400 MHz, CDCl3) δ 6.91 (s, 2H), 6.88 (s, 1H), 6.39 (bs, 1H), 5.45 (bs, 1H), 3.68 (s, 2H), 3.10 (d, J = 8.6 Hz, 1H), 2.29 (s, 6H), 2.19 (m, 1H), 1.91 – 1.70 (m, 2H), 1.68 – 1.23 (m, 6H).
13C NMR (101 MHz, CDCl3) 5 174.8, 138.3, 137.2, 129.1, 126.9, 55.2, 42.3, 36.5, 31.1, 31.0, 25.3, 25.3, 21.4.
HRMS (ESI) calculated m/z 278.1573 (MH+), observed m/z 278.1574 (MH+)
2-((4-chloro-3-(trifluoromethoxy)benzyl)thio)-2-cyclopentylacetamide 60.
Yield 65 %.
1H NMR (400 MHz, CDCI3) δ 7.39 (d, J = 8.2 Hz, 1H), 7.32 (s, 1H), 7.18 (dd, J = 8.2, 2.1 Hz, 1H), 6.25 (bs, 1H), 5.82 (bs, 1H), 3.72 (s, 2H), 2.96 (d, J = 9.2 Hz, 1H), 2.14 (m, 1H), 1.89 – 1.70 (m, 2H), 1.68 – 1.44 (m, 4H), 1.41 – 1.17 (m, 2H).
13C NMR (101 MHz, CDCI3) δ 174.5, 145.3, 138.4, 131.1, 128.6, 126.3, 124.4, 123.2, 121.9, 119.3, 116.7, 55.0, 42.1, 35.2, 31.19, 31.16, 25.27, 25.22.
HRMS (ESI) calculated m/z 368.0693 (MH+), observed m/z 368.0697 (MH+)
2-((3,5-bis(trifluoromethyl)benzyl) thio)-2-cyclopentylacetamide 62.
Yield quant.
1H NMR (600 MHz, CDCI3) δ 7.78 (s, 2H), 7.76 (s, 1H), 6.23 (bs, 1H), 6.11 (bs, 1H), 3.87 (s, 2H), 2.99 (d, J = 9.4 Hz, 1H), 2.15 (m, 1H), 1.85 (m, 1H), 1.78 (m, 1H), 1.67 – 1.47 (m, 4H), 1.35 (m, 1H), 1.26 (m, 1H).
13C NMR (151 MHz, CDCI3) δ 174.4, 140.4, 132.3, 132.1, 131.8, 131.6, 129.26, 129.24, 126.0, 124.2, 122.4, 121.44, 121.41, 121.39, 121.36, 121.34, 120.6, 55.29, 42.0, 35.3, 31.21, 31.19, 25.25, 25.18.
HRMS (ESI) calculated m/z 386.1008 (MH+), observed m/z 386.1007 (MH+)
2-cyclopentyl-2-((1-(3-(trifluoromethyl)phenyl)ethyl)thio)acetamide 63.
Yield 73 % (mixture of diastereomers).
1H NMR (400 MHz, CDCl3) δ 7.64 (s, 0.5H), 7.58 (s, 0.5H), 7.50 (m, 2H), 7.42 (m, 1H), 6.36 (bs, 0.5H), 6.17 (bs, 0.5H), 5.87 (bs, 0.5H), 5.62 (bs, 0.5H), 4.05 (q, J = 6.6 Hz, 0.5H), 4.00 (q, J = 7.1 Hz, 0.5H), 3.17 (d, J = 8.6 Hz, 0.5H), 2.66 (d, J = 8.6 Hz, 0.5H), 2.20 – 2.03 (m, 1H), 1.86 – 1.35 (m, 6H), 1.65 (d, J = 7.0 Hz, 1.5H), 1.56 (d, J = 7.1 Hz, 1.5H), 1.34 – 1.22 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 174.99, 174.80, 144.58, 144.37, 131.66, 131.56, 131.34, 131.24, 131.08, 131.07, 131.02, 130.92, 130.83, 130.82, 130.70, 130.60, 129.22, 129.14, 128.23, 128.19, 125.52, 125.48, 124.36, 124.35, 124.33, 124.31, 124.19, 124.15, 124.11, 124.07, 124.04, 124.00, 123.96, 122.81, 122.77, 120.10, 120.06, 55.50, 54.89, 44.64, 44.22, 42.60, 42.22, 31.05, 31.03, 30.95, 30.84, 25.27, 25.26, 25.17, 25.16, 22.95, 22.40.
HRMS (ESI) calculated m/z 332.1290 (MH+), observed m/z 332.1296 (MH+)
General procedure for the synthesis of amides in Tables 3–4 and SI–1
Mixed anhydride of the acetic and formic acid was prepared by heating the mixture of Ac2O (1 equiv) and HCOOH (2 equiv) at 60 °C for 2 hours. To accelerate the screening process, most of the synthesized amides were tested as crude products as described below. Appropriate anhydride (1 equiv) was added to the mixture of amine (1 equiv), Et3N (3 equiv) and DCM (1 mL) in 2 mL screwtop vial at room temperature. All reactions were performed on 30–100 μmol scale. The vial was vortexed and left overnight at room temperature. The solvent was evaporated under a gentle stream of nitrogen and the residue was dissolved in appropriate amount of DMSO to obtain 10 mM solution of the amide (assuming quantitative yield). The resulting solution was used in the mEH inhibition assay. Blanc reaction lacking the primary amine (i.e. all the reagents and conditions are the same, except for addition of the primary amine) performed in a similar way as described above did not show any inhibition of the mEH.
N-dodecylmethanesulfonamide 70.
Yield 60 %.
1H NMR (600 MHz, CDCl3) δ 4.19 (bs, 1H), 3.13 (q, J = 6.8 Hz, 2H), 2.95 (s, 3H), 1.57 (m, 2H), 1.38 – 1.20 (m, 18H), 0.88 (t, J = 6.9 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 43.5, 40.5, 32.1, 30.3, 29.77, 29.76, 29.69, 29.63, 29.5, 29.3, 26.7, 22.8, 14.3.
HRMS (ESI) calculated m/z 262.1846 ([M-H]−), observed m/z 340.1606 ([M+NH4Ac]−)
N-dodecyl-1,1,1-trifluoromethanesulfonamide 71.
Yield 65 %.
1H NMR (600 MHz, CDCl3) δ 4.95 (bs, 1H), 3.29 (q, J = 6.7 Hz, 2H), 1.60 (p, J = 7.2 Hz, 2H), 1.37 – 1.22 (m, 18H), 0.88 (t, J = 6.9 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 171.5, 123.0, 120.9, 118.7, 116.6, 44.7, 32.1, 30.3, 29.74, 29.64, 29.55, 29.48, 29.1, 26.4, 22.8, 14.3.
HRMS (ESI) calculated m/z 316.1564 ([M-H]−), observed m/z 316.1541 ([M-H]−)
2-bromopentanamide 122.
A mixture of 2-bromopentanoic acid (1.81 g, 10 mmol, 1 equiv) and SOCl2 (3.57 g, 30 mmol, 3 equiv) was heated at 70 °C for 3 hours. The reaction mixture was then cooled down and the excess of SOCl2 was evaporated under vacuum. The residue was added dropwise to the cooled to 0 °C concentrated solution of ammonium hydroxide. The resulting mixture was stirred for additional 15 min and filtered to give the product as a light-brown solid (0.61 g, 34 %).
1H NMR (600 MHz, CDCl3) δ 6.32 (bs, 1H), 5.79 (bs, 1H), 4.30 (dd, J = 8.3, 5.1 Hz, 1H), 2.10 (m, 1H), 2.00 (m, 1H), 1.56 (m, 1H), 1.48 (m, 1H), 0.96 (t, J = 7.4 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 171.6, 50.7, 37.9, 20.6, 13.4.
2-bromo-2-cyclohexylacetamide 133.
A solution of NaNO2 (3.53 g, 51.2 mmol, 1.6 equiv) in water (30 mL) was slowly added to the cooled to 0 °C mixture of 2-amino-2-cyclohexylacetic acid (5 g, 32 mmol, 1 equiv), 48% HBr in water (48.6 g, 288 mmol, 9 equiv) and water (10 mL). The reaction mixture was stirred for 2 h and extracted with EtOAc (3 × 20 mL). Combined extracts were dried over MgSO4, filtered and evaporated to give crude 2-bromo-2-cyclohexylacetic acid which was used in the next step without purification.
A mixture of crude 2-bromo-2-cyclohexylacetic acid and SOCl2 (14.28 g, 120 mmol, 3 equiv) was refluxed for 3 hours, cooled down and added dropwise to the cooled to 0 °C concentrated solution of ammonium hydroxide. The resulting mixture was stirred for additional 15 min and filtered to give the product as a light-brown solid (6.102 g, 87 %).
1H NMR (600 MHz, CDCl3) δ 6.55 (bs, 1H), 6.46 (bs, 1H), 4.18 (d, J = 5.6 Hz, 1H), 1.98 – 1.61 (m, 6H), 1.26 (m, 3H), 1.15 (m, 2H).
13C NMR (151 MHz, CDCl3) δ 171.8, 58.5, 41.8, 31.1, 29.3, 26.0, 25.9, 25.8.
Biological Evaluation
a) Measurement of mEH inhibition using a fluorometric assay
This was the primary assay utilized to determine the IC50 for all the inhibitors. It is derived from a previously described protocol used to measure sEH inhibition,15 modified and optimized for mEH. It involves an α-cyanocarbonate epoxide (CMNGC) substrate, which yields a strong fluorescent signal upon hydrolysis by mEH. The detailed, step by step protocol is described in the supplementary information.
b) Radiometric assay for mEH activity using [3H] cis-stilbene oxide (c-SO)
This was the secondary assay utilized to validate the potency of compound 62. It is derived from a previously described assay used to measure sEH activity in tissue extracts, using [3H] trans-stilbene oxide (t-SO),15 modified and optimized for mEH. It is based on the differential partitioning of the epoxide substrate and the more polar 1,2 diol product in the organic and aqueous fractions, respectively. The detailed, step by step protocol is described in the supplementary information.
Homology modeling
No crystal structure of mEH is currently available. In order to predict the protein structure of mEH, homology modeling was carried out using the RosettaCM protocol.22 While a homology model of mEH was reported previously,23 here we applied a homology modeling method that incorporates geometric constraints on catalytic residues24 in an attempt to increase the accuracy of the prediction of the active site structure.
To select templates for homology modeling, a HMMER25 search was performed and six sequences were identified as high sequence identity templates (4I19, 5F4Z,1QO7, 5BOV, 5CW2, 1EHY, see SI for more information). A multiple sequence alignment was carried out to correlate sequence position to structure using Promals3D.26 Structural fragment sets were generated and applied during the modeling to sample the unaligned regions with standard methods.27 During multi-template fragment based modeling with RosettaCM, evolutionary constraints were used to enhance sampling.28 Geometric constraints on three residues - D226, Y299 and Y374 - were applied during the homology modeling: D226 acts as the nucleophile that attacks the epoxide ring, while Y299 and Y374 are important for binding of the substrate epoxy group oxygen (i.e., they comprise an oxyanion hole).19 Thus, these residues were constrained in space during the modeling to enforce the catalytic residues to maintain a viable arrangement (see SI for details). Using this protocol, 5000 models of mEH were generated and the top scored model was selected as a starting point for ligand docking.
Docking
A library of conformations for each tested molecule was generated with Spartan ‘16 using molecular mechanics (MMFF).29 Conformers within 5 kcal/mol of the lowest energy conformer were retained for docking. These conformers were then optimized with Gaussian09 using the ωB97XD/6–31+G(d,p) density functional theory method in the gas phase.30
To investigate the binding mode of potent inhibitors, docking simulations were performed with the Rosetta Molecular Modeling Suite.31,32 The low energy homology model generated from RosettaCM was used as the protein model to which the conformer library was docked. Docking runs were carried out with chemically meaningful constraints.33,34 Two types of constraints were applied: 1) Chemistry constraints. 2) Triad Constraints. The former defines the constraints between the inhibitor with catalytic triad residues: 299Y, 374Y and 226D. The later defines constraints between catalytic residues including 404E, 431H, 374Y, 299Y, 226D. Geometric constraint values were determined using a close homologue of mEH (PDB: 3G0I, see SI for details). For each tested molecule, 2500 docking simulations were conducted, and the resulting poses were filtered on the basis of constraint satisfaction, total energy and interface energy (structures that were within one standard deviation or lower from the mean energy were retained). The top scoring models were further analyzed to evaluate the predicted intermolecular interactions at the protein-ligand interface.
Supplementary Material
Scheme 1.

General scheme for the synthesis of compounds 1–28 from Table 1.
Designed 2-alkylthio acetamide inhibitors of microsomal epoxide hydrolase, low nanomolar IC50 (0.94–11 nM)
Novel series, 2 orders of magnitude more potent than previous amines, amides and ureas (IC50 ≥ 480 nM)
Best inhibitors possess amide with bulky a-substituent and, 3–4 bonds away, phenyl ring with lipophilic meta groups
Docking simulations validate findings, favorable active site interactions (amide H-bonding, π-π stacking)
Acknowledgements and Funding
This study was supported by grants from the National Institute of Health (R35 ES030443, R01 DK107767 and R01 GM076324–11), the National Institute of Environmental Health Sciences Superfund Research Program P42 ES004699, the National Science Foundation (award numbers 1827246, 1805510 and 1627539) and the Rosetta Commons. DJT and YZ gratefully acknowledge computation support from the US National Science Foundation’s XSEDE program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Václavìková R; Hughes DJ; Souček P. Microsomal Epoxide Hydrolase 1 (EPHX1): Gene, Structure, Function, and Role in Human Disease. Gene 2015, 571 (1), 1–8. https://Doi.Org/10.1016/j.Gene.2015.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hammock BD; Loury DN; Moody DE; Ruebner B; Baselt R; Milam KM; Volberding P; Ketterman A; Talcott R. A methodology for the analysis of the preneoplastic antigen. Carcinogenesis. 1984, 5 (11), 1467–1473 https://Doi.Org/10.1093/carcin/5.11.1467 [DOI] [PubMed] [Google Scholar]
- (3).El-Sherbeni AA; El-Kadi AO The Role of Epoxide Hydrolases in Health and Disease. Arch Toxicol. 2014, 88, 2013–2032. 10.1007/s00204-014-1371-y. [DOI] [PubMed] [Google Scholar]
- (4).Toselli F; Fredenwall M; Svensson P; Li X-Q; Johansson A; Weidolf L; Hayes MA Oxetane Substrates of Human Microsomal Epoxide Hydrolase. Drug Metab. Dispos. 2017, 45 (8), 966–973. 10.1124/dmd.117.076489. [DOI] [PubMed] [Google Scholar]
- (5).Toselli F; Fredenwall M; Svensson P; Li X-Q; Johansson A; Weidolf L; Hayes MA Hip to Be Square: Oxetanes as Design Elements to Alter Metabolic Pathways. J. Med. Chem. 2019, 62, 7383–7399 10.1021/acs.jmedchem.9b00030. [DOI] [PubMed] [Google Scholar]
- (6).Edin ML; Hamedani BG; Gruzdev A; Graves JP; Lih FB; Arbes SJ; Singh R; Leon ACO; Bradbury JA; DeGraff LM; et al. Epoxide Hydrolase 1 (EPHX1) Hydrolyzes Epoxyeicosanoids and Impairs Cardiac Recovery after Ischemia. J. Biol. Chem. 2018, 293 (9), 3281–3292. 10.1074/jbc.RA117.000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brooks GT; Harrison A; Lewis SE Cyclodiene epoxide ring hydration by microsomes from mammalian liver and houseflies. Biochem Pharmacol. 1970, 19 (1), 255–273. 10.1016/0006-2952(70)90346-1. [DOI] [PubMed] [Google Scholar]
- (8).Oesch F. Mammalian Epoxide Hydrases: Inducible Enzymes Catalysing the Inactivation of Carcinogenic and Cytotoxic Metabolites Derived from Aromatic and Olefinic Compounds. Xenobiotica. 1973, 3 (5), 305–340. 10.3109/00498257309151525. [DOI] [PubMed] [Google Scholar]
- (9).Brooks GT Epoxide hydratase as a modifer of biotransformation and biological activity. Gen Pharmacol. 1977, 8 (4), 221–226. 10.1016/0306-3623(77)90016-7. [DOI] [PubMed] [Google Scholar]
- (10).Morisseau C; Newman JW; Dowdy DL; Goodrow MH; Hammock BD Inhibition of Microsomal Epoxide Hydrolases by Ureas, Amides, and Amines. Chem. Res. Toxicol. 2001, 14 (4), 409–415. 10.1021/tx0001732. [DOI] [PubMed] [Google Scholar]
- (11).Morisseau C; Newman JW; Wheelock CE; Hill T; Morin D; Buckpitt AR; Hammock BD Development of Metabolically Stable Inhibitors of Mammalian Microsomal Epoxide Hydrolase. Chem. Res. Toxicol. 2008, 21 (4), 951–957. 10.1021/tx700446u. [DOI] [PubMed] [Google Scholar]
- (12).Kamita SG; Yamamoto K; Dadala MM; Pha K; Morisseau C; Escaich A; Hammock BD Cloning and Characterization of a Microsomal Epoxide Hydrolase from Heliothis Virescens. Insect Biochem. Mol. Biol. 2013, 43 (3), 219–228. https://doi.org/10.1016Zj.ibmb.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meunier B; de Visser SP; Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104 (9), 3947–3980. 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- (14).Krueger SK; Williams DE Mammalian Flavin-Containing Monooxygenases: Structure/Function, Genetic Polymorphisms and Role in Drug Metabolism. Pharmacol. Ther. 2005, 106 (3), 357–387. 10.1016/J.PHARMTHERA.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Morisseau C; Hammock BD Measurement of Soluble Epoxide Hydrolase (SEH) Activity In Current Protocols in Toxicology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Vol. 33, pp 4.23.1–4.23.18. 10.1002/0471140856.tx0423s33. [DOI] [PubMed] [Google Scholar]
- (16).Spiegelstein O; Kroetz DL; Levy RH; Yagen B; Hurst SI; Levi M; Haj-Yehia A; Bialer M. Structure Activity Relationship of Human Microsomal Epoxide Hydrolase Inhibition by Amide and Acid Analogues of Valproic Acid. Pharm. Res. 2000, 17 (2), 216–221. 10.1023/A:1007577600088. [DOI] [PubMed] [Google Scholar]
- (17).Breneman CM; Wiberg KB Determining Atom-centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11 (3), 361–373. 10.1002/jcc.540110311. [DOI] [Google Scholar]
- (18).Cossio MLT; Giesen LF; Araya G; Pérez-Cotapos MLS; Vergara RL; Manca M; Tohme RA; Holmberg SD; Bressmann T; Lirio DR; et al. “Face Recognition: A Literature Survey”. Uma ética para quantos? 2012, XXXIII (1), 81–87. 10.1007/s13398-014-0173-7.2. [DOI] [Google Scholar]
- (19).Morisseau C; Hammock BD EPOXIDE HYDROLASES: Mechanisms, Inhibitor Designs, and Biological Roles. Annu. Rev. Pharmacol. Toxicol. 2004, 45 (1), 311–333. 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- (20).Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113 (18), 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- (21).Chai J. Da; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10 (44), 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- (22).Song Y; Dimaio F; Wang RYR; Kim D; Miles C; Brunette T; Thompson J; Baker D. High-Resolution Comparative Modeling with RosettaCM. Structure 2013, 21 (10), 1735–1742. 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Saenz-Méndez P; Katz A; Pérez-Kempner ML; Ventura ON; Vázquez M. Structural Insights into Human Microsomal Epoxide Hydrolase by Combined Homology Modeling, Molecular Dynamics Simulations, and Molecular Docking Calculations. Proteins Struct. Funct. Bioinforma. 2017, 85 (4), 720–730. 10.1002/prot.25251. [DOI] [PubMed] [Google Scholar]
- (24).Bertolani SJ; Siegel JB A New Benchmark Illustrates That Integration of Geometric Constraints Inferred from Enzyme Reaction Chemistry Can Increase Enzyme Active Site Modeling Accuracy. PLoS One 2019, 14 (4), e0214126. 10.1371/journal.pone.0214126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Potter SC; Luciani A; Eddy SR; Park Y; Lopez R; Finn RD HMMER Web Server: 2018 Update. Nucleic Acids Res. 2018, 46 (W1), W200–W204. 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pei J; Grishin NV PROMALS3D: Multiple Protein Sequence Alignment Enhanced with Evolutionary and Three-Dimensional Structural Information. Methods Mol. Biol. 2014, 1079, 263–271. 10.1007/978-1-62703-646-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gront D; Kulp DW; Vernon RM; Strauss CEM; Baker D. Generalized Fragment Picking in Rosetta: Design, Protocols and Applications. PLoS One 2011, 6 (8), e23294. 10.1371/journal.pone.0023294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Thompson J; Baker D. Incorporation of Evolutionary Information into Rosetta Comparative Modeling. Proteins Struct. Funct. Bioinforma. 2011, 79 (8), 2380–2388. 10.1002/prot.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shao Y; Molnar LF; Jung Y; Kussmann J; Ochsenfeld C; Brown ST; Gilbert ATB; Slipchenko LV; Levchenko SV; O’Neill DP; et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package Physical Chemistry Chemical Physics. The Royal Society of Chemistry; 2006, 3172–3191. 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- (30).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; et al. Gaussian 03, revision C. 02; Gaussian, Inc; 2013, 1–20. [Google Scholar]
- (31).Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman KW; Renfrew PD; Smith CA; Sheffler W; et al. Rosetta3 In Methods in Enzymology; Academic Press, 2011; Vol. 487, 545–574. 10.1016/b978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Alford RF; Leaver-Fay A; Jeliazkov JR; O’Meara MJ; DiMaio FP; Park H; Shapovalov MV; Renfrew PD; Mulligan VK; Kappel K; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13 (6), 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).O’Brien TE; Bertolani SJ; Tantillo DJ; Siegel JB Mechanistically Informed Predictions of Binding Modes for Carbocation Intermediates of a Sesquiterpene Synthase Reaction. Chem. Sci. 2016, 7 (7), 4009–4015. 10.1039/c6sc00635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).O’Brien TE; Bertolani SJ; Zhang Y; Siegel JB; Tantillo DJ Predicting Productive Binding Modes for Substrates and Carbocation Intermediates in Terpene Synthases - Bornyl Diphosphate Synthase As a Representative Case. ACS Catal. 2018, 8 (4), 3322–3330. 10.1021/acscatal.8b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.