

SUMMARY
We describe a catalyst-free 1,2-trans-dihalogenation of alkynes with an unprecedented substrate scope and exclusive regio- and stereoselectivity. This versatile dihalogenation system—a combination of NX1S electrophile and alkali metal halide (MX2) in acetic acid—is applicable for diverse categories of alkynes (electron-rich or poor alkynes, internal and terminal alkynes, or heteroatoms such as O-, N-, S-substituted alkynes). The hydrogen bonding donor solvent acetic acid is essential for the in-situ generation of X1X2 electrophile, including ICl, IBr, BrCl, I2, and Br2.
Graphical abstract
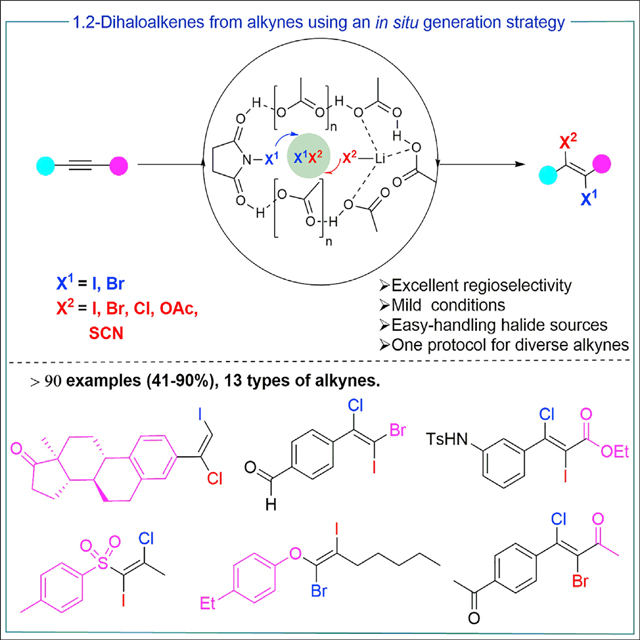
Dihaloalkenes are important raw materials for pharmaceutical and chemical industries. However, existing preparation methods suffer from a limited substrate scope as well as poor regio- and stereoselectivity. Furthermore, these methods often necessitate highly toxic reagents, such as Cl2, ICl, and BrCl. Our environmentally friendly 1,2-trans-dihalogenation is based on easy-handling halide sources, such as alkali metal halides. What is more, our method offers an unprecedented substrate scope, the regio- and stereoselectivity for the synthesis of 1,2-dihaloalkenes.
INTRODUCTION
Halogenated organic compounds are among the most important structural motifs as they are ubiquitous in natural products,1,2 materials,3 and pharmaceuticals.4 They are also among the most versatile building blocks.5,6 More specifically, 1,2-dihaloalkenes are reliable cross-coupling partners, which have been extensively utilized to construct a wide range of multisubstituted alkenes and other complex molecules.7–14 Compared with 1,2-dihaloalkenes containing only one kind of halogen (e.g., dichloroalkenes), dihaloalkenes containing two different halogens are more exciting and have broader applications.15 For example, dihaloalkenes containing two different halogens could undergo cross-coupling reactions in a step-wise manner for the installment of two different molecular fragments.7,16–27 In this regard, significant efforts have been devoted to the construction of dihaloalkenes. However, the regio- and stereoselective, yet environmentally friendly synthesis of dihaloalkenes with a broad scope is still challenging.28–34
As taught in any introductory organic chemistry course, the electrophilic addition of X2, such as Br2 or Cl2, to alkynes is a straightforward method for the synthesis of 1,2-dihaloalkenes. The electrophilic dihalogenation of alkynes suffers from limited substrate scope as well as poor regio- and stereoselectivity. Furthermore, these methods need highly toxic reagents, such as Cl2, ICl, and BrCl.35,36 For example, many electrophilic additions of permanent dipole reagents, such as ICl and Ibr, to alkynes deliver a mixture of cis- and trans-products with poor regioselectivity37,38 or, in other cases, ionic liquids39 are required (Scheme 1A). Chalifoux and co-workers23 described a trans-addition of ICl to trialkysilyl-substituted alkynes via neighboring group participation at −78 °C (Scheme 1B). However, this strategy is limited to silyl-substituted alkynes. Many other dihalogenation systems based on combinations of N-halosuccinimide (NXS),21,40 I2,41 or Cl2 42 together with TMSBr,22 TMSCl, HgCl2,43 or HCl33 have been developed (Scheme 1C). A Bu4NI/DCE system 19,20 has also been explored (Scheme 1D). All these approaches generally need harsh reaction conditions and are case-specific for a given type of alkyne substrate. Alternatively, 1,2-dihaloalkenes could also be accessed via hydrohalogenations of haloalkynes, where pre-installment of a halogen to an alkyne is needed. For instance, the nucleophilic addition of alkali metal halides to haloalkynes leads to Z-dihaloalkenes44 (Scheme 1E), but unfriendly reaction conditions are indispensable and regio-isomer formation is common45 (Scheme 1F). Zhu’s group (Scheme 1G) reported a Pa-catalyzed hydrohalogenation of haloalkynes,46 a reaction that exclusively gives anti-addition products but their method is limited to the synthesis of iodoalkenes. Recently our group developed the synthesis of (Z)- and (E)-1,2-chlorohaloalkenes via gold catalysis and hydrogen bonding catalysis, respectively47 (Scheme 1H). However, these methods were only applicable to hydrochlorination. Hitherto, the efficient regio- and stereoselective synthesis of diverse dihaloalkenes from a wide range of categories of alkynes (electron-rich or -poor alkynes, internal and terminal alkynes, heteroatoms such as O, N, S-substituted alkynes) by using a unified yet straightforward and environmentally friendly protocol remains an unsolved challenge in synthesis.
Scheme 1. Strategies for the Synthesis of 1,2-Dihaloalkenes.

Recently, we developed easily handled and reactive N,N′-Dimethylpropyleneurea (DMPU)-HX reagents that enable the regio- and stereoselective synthesis of haloal-kenes.47–51 Nevertheless, the high acidity of HX was incompatible with some functional groups.52,53 An in-situ generated reactive halogenation reagent could be advantageous in such instances.54–59 Recently, Oestreich and co-workers reported the hydroiodination of C–C multiple bonds by using in-situ-generated HI or HBr driven by the aromatization of cyclohexa-1,4-dienes.60,61 We are now glad to report a versatile dihalogenation system—a combination of NX1S and LiX2 that arises in the presence of an H-bonding solvent, such as AcOH. This system slowly generates reactive ICl, IBr, BrCl, Br2, and I2 in situ. Because of the mild conditions, our dihalogenation of alkynes has demonstrated unprecedented substrate scope, yielding products with remarkably high regio- and stereoselectivity (Scheme 1, bottom).
RESULTS
We used the iodochlorination of alkyne 1a as our model reaction (Table 1). We first evaluated this reaction by using an extensively used iodochlorination reagent—ICl (Table 1, entry 2). Not surprisingly, a mixture of regioisomers was detected; this result was consistent with the reported literature.31 Another reported dihalogeneration system (Bu4NI-dichloroethylene (DCE), 80 °C)20 gave a complex mixture of products (Table 1, entry 2). An additional dihalogenation system [N-Iodosuccinimide (NIS)-Trimethylsilyl chloride (TMSCl)]16 gave only the hydrochlorinated product (Table 1, entry 3). We found that neither NIS/PhCOCl nor NIS/AcCl gave the desirable product (Table 1, entries 4–5), suggesting that hydrochlorination occurs before iodochlorination. Remarkably, the hydrochlorination products could be inhibited when LiCl was used (Table 1, entries 6–7) but because of the limited solubility of LiCl, employing solvents like CH3CN or HFIP led to very low yields. We envisioned that a hydrogen bond donor solvent, such as HOAc not only would be able to dissolve LiCl, but would also increase the reactivity of the electrophile. As expected, a high yield of iodochlorinated product was observed with HOAc, albeit with low stereoselectivity (Table 1, entry 8). Interestingly, using dichloromethane (DCM) as the co-solvent significantly improved both the yield and the regioselectivity of the product (Table 1, entry 9). We tested another electrophilic iodide source (DIH), but it did not improve the reaction (Table 1, entry 10). Because AcOH might play an important role in the selectivity of this reaction, we investigated the reaction of Icl by using AcOH/DCM as the solvent, but we found the E/Z selectivity was still not good (Table 1, entry 11). Similarly, IOAc gave poor EZ selectivity (Table 1, entry 12). When more equivalents of LiCl were employed, we obtained a lower yield and stereoselectivity (Table 1, entry 12). Recently, Ishihara and co-workers34 reported an efficient thiourea-I2-NCS system for iodochlorination of alkenes. However, the same condition showed poor E/Z selectivity in the iodochlorination of alkyne 1a when using Ishihara and co-workers’ thiourea catalyst (Table 1, entry 14 ). Next, weswitched the solvent to DCM/HOAc, We found that it could slightly improve the stereoselectivity, but offered a lower chemical yield (We then explored the substrate scope under the optimized conditions (Figure 1). First, we evaluated the scope of terminal aromatic alkynes (Figure 1, 2–12), obtaining good yields and excellent regioselectivities. The efficiency of the reaction was not affected by electron-withdrawing groups (e.g., ester-) or electron-donating groups (e.g., MeO−, Ph). Even an unprotected phenol group was well tolerated (Figure 1, 9). Heteroaromatics such as pyridine and thiophene were not affected by the reaction conditions (Figure 1, 11 and 12). We next evaluated the scope of terminal aliphatic alkynes and found that at lower temperatures (−25 °C) the reaction showed high trans-selectivity62 (Figure 1, 13–22). It should be noted that unprotected hydroxyls (Figure 1, 14 and 17), alkene (Figure 1, 15), sulfonate (Figure 1, 18), benzotriazoles (Figure 1, 20), indazole(Figure 1, 20), thiophene (Figure 1, 21), or the nitro group (Figure 1, 22) did not hinder the efficiency of the reaction.
Table 1.
Optimization of the Reaction Conditions
| Entry | Electrophilic X+ | Nucleophilic Halogen | Solvent | Yields %a (1b:1c:1d) |
|---|---|---|---|---|
| 1 | ICl (1.2 equiv) | n/a | DCM | 95 8:1:0) |
| 2 | n/a | Bu4NI (3 equiv) | DCE (80 C) | Complexb |
| 3 | NIS | TMSCl | DCM | 89 0:0:100) |
| 4 | NIS | PhCOCl | DCM | 85 0:0:100) |
| 5 | NIS | AcCl | DCM | 45 0:0:100) |
| 6 | NIS | LiCl | CH3CN | Trace |
| 7 | NIS | LiCl | HFIP | Trace |
| 8 | NIS | LiCl | HOAc | 71 90:10:0) |
| 9c | NIS | LiCl | HOAc/DCM | 80 98:2:0) |
| 10c | DIHd | LiCl | HOAc/DCM | 81 97:3:0) |
| 11 | ICl(1.5 equiv) | n/a | HOAc/DCM | 93 88:12:0) |
| 12 | IOAc | LiCl(2 equiv) | HOAc/DCM | 66 67:33:0) |
| 13 | IOAc | LiCl (4 equiv) | HOAc/DCM | 40 61:39:0) |
| 14c | NCS | I2 | Toluene | 82 77:23:0) |
| 15d | NCS | I2, | HOAC/DCM | 75 82:18:0) |
Conditions: alkynes 1a (0.2 mmol), NIS (0.3 mmol, 1.5 equiv), Cl− reagent (0.4 mmol, 2 equiv), solvent (1 mL), 24 h.
5 mol % of thiourea catalyst34 was used.
Yields were determinated by NMR.
Both hydrochlorinated and dichlorinated products were detected.
HOAc/DCM (0.5/0.5 mL).
DIH: 1,3-diiodo-5,5-dimethylhydantion.
Figure 1.

The Scope of the Iodochlorination of Alkynesa
Haloalkynes are versatile, yet readily available, building blocks, 46,47,63–69 with which our highly regioselective iodochlorination protocol should enable to access extremely valuable trihalogenated alkenes.70 To our delight, both aromatic and aliphatic haloalkynes gave excellent regioselectivity and chemical yields of trihalogenated alkenes. Diverse functional groups, such as acid-sensitive esters (Figure 1, 24), aldehyde (Figure 1, 25), methoxyl (Figure 1, 26), and nitrile (Figure 1, 27) survived the reaction conditions. Compared with bromoalkynes and iodoalkynes (Figure 1, 28, 30), chloroalkynes (Figure 1, 31 and 32) were less reactive and required a longer time as well as more equivalents of NIS / LiCl, but the products were, never-theless, obtained in good yields and excellent trans-selectivity.
Encouraged by the broad scope of haloalkynes that tolerated our reaction conditions, we examined the scope of electron-deficient alkynes, such as alkynyl ketones (Figure 1, 33–41). The substitution pattern (ortho, meta, and para) and electronic properties of aromatic substituents (electron-deficient or electron-rich) played a small role; good yields were obtained regardless (Figure 1, 33–38). In general, substrates containing electron-rich substitution gave slightly higher yields than those containing electron-deficient substitutions (Figure 1, 36 and 37). The substitution groups attached to the carbonyl group played a negligible role in the reactivity; even the sterically hindered cyclohexyl and tertiary butyl groups were well tolerated (Figure 1, 39 and 40). A dialkyl ynone was also a suitable substrate (89%, Figure 1, 41). We were pleased to find that the same protocol worked well for alkynyl esters (Figure 1, 42–49). Moreover, phenylamine (Figure 1, 47) and alkyl alkynyl esters (Figure 1, 48 and 49) were good substrates for this reaction. It should be noted that our protocol displayed much better regioselectivities compared with the reported method.20 Our iodochlorination system could be extended to other diverse alkyne types such as highly electron-deficient sulfonyl alkynes (Figure 1, 50–57),71 electron-rich alkynyl thioethers (Figure 1, 55–57), electron-deficient alkynyl aldehydes (Figures 1, 65), electron-deficient alkynyl acid (Figure 1, 66), electron-deficient alkynyl nitrile (Figure 1, 67), electron-rich ynol ether (Figure 1, 68), and ynamide (Figure 1, 69). Most of the highly functional dihaloketenes prepared using our protocol were hitherto unknown.
Despite the wide structural diversity of the above alkynes, our dihalogenation afforded products with exclusive or very high regioselectivity. We investigated the regioselectivity of internal alkynes that exhibited small structural biases (R1 and R2) on each end of the triple bond (Figure 1, 58–64). We were pleased to find that for alkynes substituted with an aryl group and an aliphatic group (R1 = aryl, R2 = aliphatic) (Figure 1, 58–60), the reaction was regiospecific. To our delight, even very challenging unsymmetrical aryl alkynes (R1 = phenyl substituted with a withdrawing group—ester or ketone, and R2 = phenyl substituted with an electron-donating group) furnished regiospecific products (Figure 1, 61 and 62). As expected, our protocol worked very well for symmetric diaryl and dialkyl alkynes (Figure 1, 63 and 64). To corroborate the functional group tolerance and wide applicability of our methodology, we conducted late-stage dihalogenation of alkynes containing natural product motifs (Figure 1, 70–73). Alkynes tethered to complex structures such as zaltoprofen (Figure 1, 70), diacetone-D-glucose (Figure 1, 71), hiestrone (Figure 1, 72) and L-menthol (Figure 1, 73) gave desired products in excellent yields and selectivity. The structure assignments of our obtained products were further confirmed by single-crystal X-ray diffraction (see the ORTEP drawings of 10,72 29,73 51,74 and 69,75 Figure 2).
Figure 2. ORTEP Drawings of Obtained Products.

Because the iodochlorination of alkynes using ICl is the most commonly used method in the literature, we compared the reactivity of the ICl method with our new in-situ method for various types of alkynes (see Supplemental Information, Section 23). In general, possibly due to the high reactivity of ICl, a mixture of regioisomers was obtained in most cases, and we also detected dichlorinated and diiodinated products in some cases.
Having demonstrated the scope of the iodochlorination of alkynes, we switched our attention to other dihalogenation systems (Figure 3). Diiodination (NIS/LiI/AcOH), iodobromination (NIS/LiBr/AcOH), dibromination (NBS/LiBr/AcOH) and chlorobromination (NBS/LiCl/AcOH) gave the desired products with exclusive trans-stereochemistry and in good chemical yields (Figure 3). Aromatic and aliphatic terminal alkynes, haloalkynes, sulfonyl alkynes, alkynyl thioether, alkynyl ether, alkynyl ketone, alkynyl ester as well as functionalized internal alkynes proved to be suitable substrates. Moreover, acid-sensitive groups such as tert-butyldiphenylsilyl ether (Figure 3, 80), and phenolic ether (Figure 3, 83) were compatible with our mild conditions. A lower temperature ( −40 °C) was needed for chlorobromination (NBS/LiCl/AcOH), possibly because of the high reactivity of BrCl generated in situ. Our protocol is a safer and more selective alternative to literature methods that rely on the use of toxic Br2, Ibr, and BrCl. The structure assignments of our obtained products were further confirmed by single-crystal X-ray diffraction (see the ORTEP drawings of 8276 and 9177).
Figure 3. Scope of the Dihalogenation of Alkynes.

Recently, the haloacetoxylation of alkenes78–81 and alkynes82 employing electrophilic X+ and HOAc was reported. We did not detect any competitive acetate addition reactions in our experiments possibly because HOAc is a weaker nucleophile than LiX in our dihalogenation reaction. We took solace in finding that the iodoacetoxylated alkene 9883 (Scheme 2, Equation 2a) was cleanly prepared using NaOAc as the nucleophile. Encouraged by our success, we investigated other common nucleophiles. Although LiSCN delivered the desirable product 9984 in a moderate yield (Scheme 2; Equation 2b), other nucleophiles such as LiSeCN, KF, TMSCN and NaN3 only gave a trace amount of desired products (Scheme 2, Equations 2c–2f). To demonstrate the general applicability of our developed robust methods, a commercially available phenylacetylene was extended up to gram-scale under the optimized conditions, which offered desirable products in 80% yield (Scheme 2, Equation 2g), and maintain excellent selectivity ( E/Z> 20/1).
Scheme 2. Iodo-Functionalization of Alkynes and Large-Scale Synthesis.

Figure 4 shows our proposed mechanism. We and others have reported that linear H-bond aggregates85 of (AcOH)n are the dominant species in neat AcOH or a highly concentrated AcOH solution, and (AcOH)n is a strong and tunable hydrogen bond donor system. We postulated that the interaction of (AcOH)n with NIS and LiCl led to the generation of ICl and succinimide A; the strong H-bond interaction between succinimide A, ICl and (AcOH)n was the driving force that displaced the equilibrium to the right. The textbook mechanism for dihalogenation of an alkyne is the formation of a cyclic iodonium intermediate, followed by an SN2-like backside attack of chloride nucleophile to obtain the trans-addition product.86
Figure 4. Proposed Mechanism and Geometries of Unsymmetrical Iodoniums.

LUMOs were calculated at ωB97X-D/6–311++G** level of theory.
Usually, steric and electrophilic factors determine the regioselectivity of electrophilic addition to unsymmetric alkynes. However, most literature protocols have only showcased a narrow alkyne substrate scope; thus, a comprehensive yet rational study of the regioselectivity of electrophilic additions to unsymmetric alkynes is lacking. Because our protocol works for unprecedently diverse categories of unsymmetric alkynes, we now have a rare opportunity to conduct a systematic study of the regioselectivity of the addition. We speculated that the geometry of the cyclic iodonium intermediate is the determining factor of the regioselectivity seen. For an alkyne with R1 and R2 on each end of the triple bond, an unsymmetrical iodonium intermediate will form, and the nucleophilic chloride anion will prefer to attack the carbon with the weaker (longer) bond with the iodine atom.32 As a result, we predicted that the regioselectivity would be determined by the equilibrium geometry of the corresponding iodonium, which could be easily be obtained using DFT calculations (Figure 4). From these calculations, the geometry of the unsymmetrical iodonium depended on R1 and R2’s relative ability to stabilize the neighboring positive charge, which, in turn, is determined by a combination of conjugation and hyperconjugation effects. According to the calculations (Figure 4), the neighboring positive charge stabilization ability of groups follows this approximate order: R2N-, OR, SR >> Ar > Alkyl > H> I > Br > Cl > CHO, COOR, COR > RSO2−. Thus, if the neighboring positive charge stabilization ability of R1 is higher, in an alkyne substituted with R1, and R2 that reacts with NIS/LiCl/AcOH, the nucleophilic chloride will bind to the carbon closer to the R1. From our calculations of LUMOs and geometries of unsymmetrical iodonium (Figure 4), this model explained the regioselectivity very well. We also found that the reaction system with a higher LUMO (iodonium) has a faster reaction rate in general. As a general rule, the iodine atom is added to the alkynyl carbon substituted with an electron-withdrawing group (e.g., ester and ketone) and the chlorine atom is added to the alkynyl carbon substituted with an electron-donating group (e.g., RS-, RO-, and R2N-). It should be noted that for electron-deficient alkyne substrates, the reaction might also go through a Michael addition of a chloride followed by electrophilic iodination. But we think that this pathway is relatively unlikely. For example, the addition of LiCl in acetic acid to sulfonyl alkynes needs a very high temperature (100 °C)56 and our reaction is conducted at 0 °C or lower temperatures.
To further verify our proposed mechanism, we studied the activating role of HOAc by using NMR spectroscopy. Figure 5A shows that the peak marked with Ha corresponded to the CH2 signal of NIS, located at 3.03 ppm in CDCl3. There was a small upfield shift of the methylene group, from 3.03 to 2.97 ppm, when HOAc was added (Figure 5B), indicating an H-bond interaction between the HOAc and NIS. Interestingly, the signal corresponding to CH2 was completely shifted to 2.77 ppm when LiCl was added (Figure 5C) and succinimide was detected in a quantitative yield (Figure 5D). To follow up this result, we carried out two control experiments. When NIS (0.3 mmol) was added to the mixture solvent DCM/HOAc (0.5/0.5 mL) and stirred vigorously for 10 min at room temperature, however, the bench stable NIS was undissolved (Figure 5E). Surprisingly, the reaction mixture turned to a brownish color immediately after LiCl (0.4 mmol) was added (Figure 5F); this color is very similar to the color of a solution of ICl in DCM/HOAc. The formation of succinimide and the color change was a strong indication of ICl formation in situ.
Figure 5.

(A–F) 1H NMR spectra of (A) NIS; (B) NIS and HOAc; (C) LiCl: NIS (1: 1) in HOAc after 1 h. (D) succinimide in CDCl3 and HOAc. (E) NIS In HOAc-DCM (F) Nis, LiCl in HOAc-DCM.
To illustrate the synthetic potential of the products that we obtained using our protocol, we conducted eight representative transformations of our dihaloalkene products (Scheme 3). Taking advantage of the reactivity difference between alkenyl chloride and iodide, we were able to synthesize E-enynes 100, β-chloro-α, β-unsaturated nitriles 102, phenylhexa-3,5-diyn-1-ol 103, and 2-ethynylbenzofurans 104 by using a sequence of cross-coupling reactions. Our dihaloalkene products also could be reduced to alcohol which is consistent with our reported compound 58. Moreover, the halothioalkane products enable oxidant to sulfone 101 or sulfoxide 105 with the dihaloalkene functionality untouched. The structural assignments of products 10187 and 10588 were further confirmed by single-crystal X-ray diffraction.
Scheme 3. Synthetic Applications.

In summary, we have discovered an efficient method for the regio-selective dihalogenation of 14 different classes of alkynes. The in-situ generated electrophilic X1X2 reagents replaced toxic alternatives such as ICl, IBr. We transformed the dihalogenated products into synthetically useful tri- or tetra substituted alkenes via tandem cross-coupling reactions. Further exploration of the in-situ generated X1X2 electrophiles is currently under investigation in our laboratory.
Supplementary Material
The Bigger Picture.
Haloalkenes are not only commonly found in biologically active natural products but also have been used extensively in cross-coupling reactions. More specifically, 1,2-dihaloalkenes are especially important synthons because of the presence of two synthetic handles that open a broad avenue to expeditiously generate multisubstituted alkenes. Dihalogenation of alkynes is a straightforward way to prepare 1,2-dihaloalkenes. However, existing alkyne dihalogenation methods either rely on the use of toxic reagents, such as IBr and ICl, lack regio-and stereoselectivity or have limited substrate scope. Thus, the development of a widely applicable and yet efficient alkyne dihalogenation method is still highly desired. Here, we have addressed the aforementioned issues based on an in-situ-generated dihalogenation of reagents, such as ICl and Ibr, by using the readily available N-halosuccinimide (NXS) and alkali metal halides as halogen sources. Our method offers an unprecedented substrate scope, the regio- and stereoselectivity for the synthesis of 1,2-dihaloalkenes. Our simple and mild conditions might find wild applications in the preparation of high-value building blocks for medicines and materials.
HIGHLIGHTS.
Catalyst-free dihalogenation of diverse alkynes
Easy-handling halide sources—alkali metal halides
In-situ generation of ICl, IBr, BrCl, I2, and Br2
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health for financial support (R01GM121660). B.X. is grateful to the National Science Foundation of China (NSFC-21672035 and NSFC-21871046) for financial support. We acknowledge with gratitude the help provided by Dr. Mark S. Mashuta (Manager X-ray Crystallographic Facilities, Chemistry Department, University of Louisville) for his helpful insights regarding our crystallographic data.
Footnotes
DATA AND CODE AVAILABILITY
Crystal structure data of compounds 10 (CCDC: 1912785), 29 (CCDC: 1948339), 51 (CCDC: 1912792), 69 (CCDC: 1948338), 82 (CCDC: 1966560), 91 (CCDC: 1966562), 101 (CCDC: 1912791), and 105 (CCDC: 1966561) have been deposited in the Cambridge Structural Database.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chempr.020.03.011.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Gribble GW (1998). Naturally occurring organohalogen compounds. Acc. Chem. Res 31, 141–152. [Google Scholar]
- 2.Landry ML, and Burns NZ (2018). Catalytic enantioselective dihalogenation in total synthesis. Acc. Chem. Res 51, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin R, Amrute AP, and Pérez-Ramírez J. (2017). Halogen-mediated conversion of hydrocarbons to commodities. Chem. Rev 117, 4182–4247. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, and Moore BS (2017). Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chem. Rev 117, 5619–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrone DA, Ye J, and Lautens M. (2016). Modern transition-metal-catalyzed carbon– halogen bond formation. Chem. Rev 116, 8003–8104. [DOI] [PubMed] [Google Scholar]
- 6.Johansson Seechurn CCC, Kitching MO, Colacot TJ, and Snieckus V. (2012). Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 nobel prize. Angew. Chem. Int. Ed. Engl 51, 5062–5085. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Cao Y, Yuan Z, Cai H, Zheng R, Kong L, and Zhu G. (2011). Synthesis of cis-1,2-dihaloalkenes featuring palladium-catalyzed coupling of haloalkynes and α,β-unsaturated carbonyls. J. Org. Chem 76, 4071–4074. [DOI] [PubMed] [Google Scholar]
- 8.Desrat S, Remeur C, Gény C, Rivière G, Colas C, Dumontet V, Birlirakis N, Iorga BI, and Roussi F. (2014). From meiogynin A to the synthesis of dual inhibitors of Bcl-xL and Mcl-1 anti-apoptotic proteins. Chem. Commun. (Camb.) 50, 8593–8596. [DOI] [PubMed] [Google Scholar]
- 9.Zeng X, Hu Q, Qian M, and Negishi E. i. (2003). Clean inversion of configuration in the Pd-catalyzed cross-coupling of 2-bromo-1,3-dienes. J. Am. Chem. Soc 125, 13636–13637. [DOI] [PubMed] [Google Scholar]
- 10.Sun C, Camp JE, and Weinreb SM (2006). Construction of β-haloenamides via direct copper-promoted coupling of lactams with 2-chloro and 2-bromo vinyliodides. Org. Lett 8, 1779–1781. [DOI] [PubMed] [Google Scholar]
- 11.Jiang B, Tian H, Huang ZG, and Xu M. (2008). Successive copper(I)-catalyzed cross-couplings in one pot: a novel and efficient starting point for synthesis of carbapenems. Org. Lett 10, 2737–2740. [DOI] [PubMed] [Google Scholar]
- 12.Negishi E. i., Alimardanov A, and Xu C. (2000). An efficient and stereoselective synthesis of xerulin via Pd-catalyzed cross coupling and lactonization featuring (E)-iodobromoethylene as a novel two-carbon synthon. Org. Lett 2, 65–67. [DOI] [PubMed] [Google Scholar]
- 13.Poulsen TB, Dickmeiss G, Overgaard J, and Jørgensen KA (2008). Organocatalytic asymmetric synthesis of versatile γ-lactams. Angew. Chem. Int. Ed. Engl 47, 4687–4690. [DOI] [PubMed] [Google Scholar]
- 14.Selina AA, Zhachkina AE, Karlov SS, Churakov AV, and Zaitseva GS (2004). Iodochlorination of silyl- and germylphenylacetylenes. Heteroatom Chem. 15, 169–174. [Google Scholar]
- 15.Gilbert N, Casault P, Ladouceur F, Ricard S, and Daoust B. (2018). 1,2-Dihaloalkenes in metal-catalyzed reactions. Synthesis 50, 3087–3113. [Google Scholar]
- 16.Yauchi Y, Ide M, Shiogai R, Chikugo T, and Iwasawa T. (2015). Regio- and stereoselective synthesis of vicinal (Z)-dihaloalkenylsilanes from silyl ethynylarenes. Eur. J. Org. Chem 2015, 938–943. [Google Scholar]
- 17.Chen D, Chen X, Lu Z, Cai H, Shen J, and Zhu G. (2011). Palladium-catalyzed dienylation of haloalkynes using 2,3-butadienyl acetates: a facile access to (1Z)-1,2-dihalo-3-vinyl-1,3-dienes. Adv. Synth. Catal 353, 1474–1478. [Google Scholar]
- 18.Chen X, Kong W, Cai H, Kong L, and Zhu G. (2011). Palladium-catalyzed highly regio-and stereoselective synthesis of (1E)- or (1Z)-1,2-dihalo-1,4-dienes via haloallylation of alkynyl halides. Chem. Commun. (Camb.) 47, 2164–2166. [DOI] [PubMed] [Google Scholar]
- 19.Lemay AB, Vulic KS, and Ogilvie WW (2006). Single-isomer Tetrasubstituted Olefins from regioselective and stereospecific palladium-catalyzed coupling of β-chloro-α-iodo-α,β-unsaturated esters. J. Org. Chem 71, 3615–3618. [DOI] [PubMed] [Google Scholar]
- 20.Ho ML, Flynn AB, and Ogilvie WW (2007). Single-isomer Iodochlorination of alkynes and chlorination of alkenes using tetrabutylammonium iodide and dichloroethane. J. Org. Chem 72, 977–983. [DOI] [PubMed] [Google Scholar]
- 21.Ide M, Yauchi Y, and Iwasawa T. (2014). Regio-, and stereoselective iodobromination of ynamides for synthesis of (E)-1-bromo-2-iodoenamides. Eur. J. Org. Chem 2014, 3262–3267. [Google Scholar]
- 22.Ide M, Yauchi Y, Shiogai R, and Iwasawa T. (2014). Regio- and stereoselective synthesis of (E)-1-bromo-2-iodoalkenes through iodobromination of internal alkynes. Tetrahedron 70, 8532–8538. [Google Scholar]
- 23.Sproul KC, and Chalifoux WA (2015). Highly regio- and diastereoselective formation of tetrasubstituted (Z)-1,2-dihaloalkenes from the halogenation of trimethylsilyl alkynes with ICl. Org. Lett 17, 3334–3337. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Yang W, Yang S, Huang L, Wu W, Sun Y, and Jiang H. (2014). Palladium-catalyzed cascade annulation to construct functionalized β- and γ-lactones in ionic liquids. Angew. Chem. Int. Ed. Engl 53, 7219–7222. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Hu W, Li C, Yang S, Wu W, and Jiang H. (2017). Palladium-catalyzed cascade reaction of haloalkynes with unactivated alkenes for assembly of functionalized oxetanes. Org. Chem. Front 4, 373–376. [Google Scholar]
- 26.Blomquist AT, Burge RE, and Sucsy AC (1952). Many-membered carbon rings. V. cyclodecyne, cis- and trans-cyclodecene and related compounds1. J. Am. Chem. Soc 74, 3636–3642. [Google Scholar]
- 27.Wang L, Zhu H, Che J, Yang Y, and Zhu G. (2014). Synthesis of cis-1,2-dichlorovinylsulfones via Fe-catalyzed regio-and stereoselective addition of sulfonyl chlorides to aromatic chloroalkynes. Tetrahedron Lett. 55, 1011–1013. [Google Scholar]
- 28.Tendil J, Verney M, and Vessiere R. (1974). Additions electrophiles sur les esters acetyleniques et alleniques—IV. Tetrahedron 30, 579–584. [Google Scholar]
- 29.Schuh (née Müller) K, and Glorius F. (2007). A domino copper-catalyzed C-N and C-O cross-coupling for the conversion of primary amides into oxazoles. Synthesis 2007, 2297–2306. [Google Scholar]
- 30.Pagni RM, Kabalka GW, Boothe R, Gaetano K, Stewart LJ, Conaway R, Dial C, Gray D, Larson S, and Luidhardt T. (1988). Reactions of unsaturated compounds with iodine and bromine on.gamma. alumina. J. Org. Chem 53, 4477–4482. [Google Scholar]
- 31.Heasley VL, Buczala DM, Chappell AE, Hill DJ, Whisenand JM, and Shellhamer DF (2002). Addition of bromine chloride and iodine monochloride to carbonyl-conjugated, acetylenic ketones: synthesis and mechanisms. J. Org. Chem 67, 2183–2187. [DOI] [PubMed] [Google Scholar]
- 32.Bellina F, Colzi F, Mannina L, Rossi R, and Viel S. (2003). Reaction of alkynes with iodine monochloride revisited. J. Org. Chem 68, 10175–10177. [DOI] [PubMed] [Google Scholar]
- 33.Barluenga J, Rodriguez MA, and Campos PJ (1990). Electrophilic additions of positive iodine to alkynes through an iodonium mechanism. J. Org. Chem 55, 3104–3106. [Google Scholar]
- 34.Horibe T, Tsuji Y, and Ishihara K. (2018). Thiourea–I2 as lewis Base–Lewis acid cooperative catalysts for Iodochlorination of alkene with in situ-generated I–Cl. ACS Catal. 8, 6362–6366. [Google Scholar]
- 35.Grushin VV (2010). The organometallic fluorine chemistry of palladium and rhodium: studies toward aromatic fluorination. Acc. Chem. Res 43, 160–171. [DOI] [PubMed] [Google Scholar]
- 36.Iodine monochloride. In Encyclopedia of Reagents for Organic Synthesis.
- 37.Mukherjee A, Pati K, and Liu RS (2009). A convenient synthesis of tetrabenzo [de,hi,mn,qr]naphthacene from readily available 1,2-di(phenanthren-4-yl)ethyne. J. Org. Chem 74, 6311–6314. [DOI] [PubMed] [Google Scholar]
- 38.Mao W, Zhang J, Li X, Li C, and Tian H. (2017). Regioisomerically pure multiaryl coronene derivatives: highly efficient synthesis via bay-extended perylene tetrabutylester. Chem. Commun. (Camb.) 53, 5052–5055. [DOI] [PubMed] [Google Scholar]
- 39.Bortolini O, Bottai M, Chiappe C, Conte V, and Pieraccini D. (2002). Trihalide-based ionic liquids. Reagent-solvents for stereoselective iodination of alkenes and alkynes. Green Chem. 4, 621–627. [Google Scholar]
- 40.Endo N, Kanaura M, and Iwasawa T. (2016). Elucidation of reaction process through β-halogen elimination in CuCN-mediated cyanation of (E)-1-bromo-2-iodoalkene. Tetrahedron Lett. 57, 483–486. [Google Scholar]
- 41.Uemura S, Miyoshi H, and Okano M. (1979). Regio- and stereospecific Z-iodo- and Z-bromochlorination of alkylphenylacetylenes via Z-chlorotelluration. Chem. Lett 8, 1357–1358. [Google Scholar]
- 42.Zefirov NS, Sereda GA, Sosonuk SE, Zyk NV, and Likhomanova TI (1995). Versatile iodination of olefins by potassium dichloroiodate(I). Synthesis 1995, 1359–1361. [Google Scholar]
- 43.Barluenga J, Martínez-Gallo JM, Nájera C, and Yus M. (1987). Stereoselective bifunctionalization of alkynes by means of the mercury(II) salt–iodine combination. J. Chem. Soc. Perkin Trans 1, 1017–1019. [Google Scholar]
- 44.Chen Z, Jiang H, Li Y, and Qi C. (2010). Highly efficient two-step synthesis of (Z)-2-halo-1-iodoalkenes from terminal alkynes. Chem. Commun 46, 8049–8051. [DOI] [PubMed] [Google Scholar]
- 45.efirov NS, Samsoniya NS, Kutateladze TG, and Zhdankin VV (1991). (Difluoroiodo) benzene - boron trifluoride as a reagent for the preparation of 1,4-diketones from O-silyl derivatives of enols. Zh. Organ. Khim 27, 220–222. [Google Scholar]
- 46.Zhu G, Chen D, Wang Y, and Zheng R. (2012). Highly stereoselective synthesis of (Z)-1,2-dihaloalkenes by a Pd-catalyzed hydrohalogenation of alkynyl halides. Chem. Commun 48, 5796–5798. [DOI] [PubMed] [Google Scholar]
- 47.Zeng X, Liu S, Hammond GB, and Xu B. (2018). Hydrogen-bonding-assisted Bronsted acid and gold catalysis: access to both (E)- and (Z)-1,2-haloalkenes via hydrochlorination of haloalkynes. ACS Catal. 8, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng X, Lu Z, Liu S, Hammond GB, and Xu B. (2017). Metal-free, Regio-, and stereo-controlled hydrochlorination and hydrobromination of Ynones and Ynamides. J. Org. Chem 82, 13179–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okoromoba OE, Han J, Hammond GB, and Xu B. (2014). Designer HF-based fluorination reagent: highly regioselective synthesis of fluoroalkenes and gem-difluoromethylene compounds from alkynes. J. Am. Chem. Soc 136, 14381–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Ebule R, Kostyo J, Hammond GB, and Xu B. (2017). HBr–DMPU: the first aprotic organic solution of hydrogen bromide. Chemistry 23, 12739–12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ebule R, Liang S, Hammond GB, and Xu B. (2017). Chloride-tolerant gold(I)-catalyzed regioselective hydrochlorination of alkynes. ACS Catal. 7, 6798–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Compain G, Jouvin K, Martin-Mingot A, Evano G, Marrot J, and Thibaudeau S. (2012). Stereoselective hydrofluorination of ynamides: a straightforward synthesis of novel α-fluoroenamides. Chem. Commun. (Camb.) 48, 5196–5198. [DOI] [PubMed] [Google Scholar]
- 53.Fosu SC, Hambira CM, Chen AD, Fuchs JR, and Nagib DA (2019). Site-selective C–H functionalization of (hetero)arenes via transient, non-symmetric iodanes. Chem 5, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gray EE, Nielsen MK, Choquette KA, Kalow JA, Graham TJA, and Doyle AG (2016). Nucleophilic (radio)fluorination of α-diazocarbonyl compounds enabled by copper-catalyzed H–F insertion. J. Am. Chem. Soc 138, 10802–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai W, Geib SJ, and Curran DP (2019). Ring opening reactions of NHC-boriranes with in situ generated HCl: synthesis of a new class of NHC-boralactones. J. Am. Chem. Soc 141, 3623–3629. [DOI] [PubMed] [Google Scholar]
- 56.Chen B, Xia X, Zeng X, and Xu B. (2018). Hydrogen bonding network assisted regio-and stereo-controlled hydrohalogenations of sulfonyl alkynes. Tetrahedron Lett. 59, 3950–3954. [Google Scholar]
- 57.Schevenels FT, Shen M, and Snyder SA (2017). Isolable and readily handled halophosphonium pre-reagents for hydro- and deuteriohalogenation. J. Am. Chem. Soc 139, 6329–6337. [DOI] [PubMed] [Google Scholar]
- 58.Derosa J, Cantu AL, Boulous MN, O’Duill ML, Turnbull JL, Liu Z, De La Torre DM, and Engle KM (2017). Palladium(II)-catalyzed directed anti-hydrochlorination of unactivated alkynes with HCl. J. Am. Chem. Soc 139, 5183–5193. [DOI] [PubMed] [Google Scholar]
- 59.Zeng X, Li J, Ng CK, Hammond GB, and Xu B. (2018). Radio)fluoroclick reaction enabled by a hydrogen-bonding cluster. Angew. Chem. Int. Ed. Engl 57, 2924–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W, Walker JCL, and Oestreich M. (2019). Metal-free transfer hydroiodination of C–C multiple bonds. J. Am. Chem. Soc 141, 1135–1140. [DOI] [PubMed] [Google Scholar]
- 61.Chen W, and Oestreich M. (2019). Metal-free transfer hydrobromination of C–C triple bonds. Org. Lett 21, 4531–4534. [DOI] [PubMed] [Google Scholar]
- 62.Herges R, Papafilippopoulos A, Hess K, Chiappe C, Lenoir D, and Detert H. (2005). cis-bromination of alkynes without cationic intermediates. Angew. Chem. Int. Ed. Engl 44, 1412–1416. [DOI] [PubMed] [Google Scholar]
- 63.Wu W, and Jiang H. (2014). Haloalkynes: a powerful and versatile building block in organic synthesis. Acc. Chem. Res 47, 2483–2504. [DOI] [PubMed] [Google Scholar]
- 64.Hein JE, Tripp JC, Krasnova LB, Sharpless KB, and Fokin VV (2009). Copper(I)-catalyzed cycloaddition of organic azides and 1-iodoalkynes. Angew. Chem. Int. Ed. Engl 48, 8018–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trofimov A, Chernyak N, and Gevorgyan V. (2008). Dual role of alkynyl halides in one-step synthesis of alkynyl epoxides. J. Am. Chem. Soc 130, 13538–13539. [DOI] [PubMed] [Google Scholar]
- 66.Jiang G, Fang S, Hu W, Li J, Zhu C, Wu W, and Jiang H. (2018). Palladium-catalyzed sequential C(sp2)-H alkynylation/annulation of 2-phenylphenols with haloalkynes using phenolic hydroxyl as the traceless directing group. Adv. Synth. Catal 360, 2297–2302. [Google Scholar]
- 67.Xie L, Wu Y, Yi W, Zhu L, Xiang J, and He W. (2013). Gold-catalyzed hydration of Haloalkynes to α-halomethyl ketones. J. Org. Chem 78, 9190–9195. [DOI] [PubMed] [Google Scholar]
- 68.Zou H, He W, Dong Q, Wang R, Yi N, Jiang J, Pen D, and He W. (2016). First catalyzed hydration of halo-alkynes by a recyclable catalytic-system. Eur. J. Org. Chem 2016, 116–121. [Google Scholar]
- 69.Zeng X, Liu S, Shi Z, and Xu B. (2016). Hydrogen bonding cluster-enabled addition of sulfonic acids to haloalkynes: access to both (E)- and (Z)-alkenyl sulfonates. Org. Lett 18, 4770–4773. [DOI] [PubMed] [Google Scholar]
- 70.Barluenga J, Rodriguez MA, Campos PJ, and Asensio G. (1988). Synthesis of 2-functionalized 1,1-diiodo-1-alkenes. Generation and reactions of 1-iodo-1-lithio-1-alkenes and 1,1-dilithio-1-alkenes. J. Am. Chem. Soc 110, 5567–5568. [Google Scholar]
- 71.Yang Z, Chen X, Kong W, Xia S, Zheng R, Luo F, and Zhu G. (2013). An operationally simple approach to (E)-α-halo vinyl sulfides and their applications for accessing stereodefined trisubstituted alkenes. Org. Biomol. Chem 11, 2175–2185. [DOI] [PubMed] [Google Scholar]
- 72.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1912785).
- 73.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1948339).
- 74.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1912792).
- 75.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1948338).
- 76.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1966560).
- 77.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1966562).
- 78.Pimenta LS, Gusevskaya EV, and Alberto EE (2017). Intermolecular halogenation/esterification of alkenes with N-halosuccinimide and acetic acid catalyzed by 1,4-diazabicyclo[2.2.2]octane. Adv. Synth. Catal 359, 2297–2303. [Google Scholar]
- 79.Ribeiro R.d.S., Esteves PM, and de Mattos MCS (2007). Triiodoisocyanuric acid: a new and convenient reagent for regioselective coiodination of alkenes and enolethers with oxygenated nucleophiles. Tetrahedron Lett. 48, 8747–8751. [Google Scholar]
- 80.Monenschein H, Sourkouni-Argirusi G, Schubothe KM, O’Hare T, and Kirschning A. (1999). Reactions of alkenes, alkynes, and alkoxyallenes with new polymer-supported electrophilic reagents. Org. Lett 1, 2101–2104. [Google Scholar]
- 81.Rao DS, Reddy TR, Babachary K, and Kashyap S. (2016). Regioselective vicinal functionalization of unactivated alkenes with sulfonium iodate(i) reagents under metal-free conditions. Org. Biomol. Chem 14, 7529–7543. [DOI] [PubMed] [Google Scholar]
- 82.Hokamp T, Storm AT, Yusubov M, and Wirth T. (2018). Iodine monoacetate for efficient oxyiodinations of alkenes and alkynes. Synlett 29, 415–418. [Google Scholar]
- 83.Xia XF, Gu Z, Liu W, Wang N, Wang H, Xia Y, Gao H, and Liu X. (2014). Selective oxygenation of alkynes: a direct approach to diketones and vinyl acetate. Org. Biomol. Chem 12, 9909–9913. [DOI] [PubMed] [Google Scholar]
- 84.Lu L-H, Zhou S-J, Sun M, Chen J-L, Xia W, Yu X, Xu X, and He W-M (2019). Metal-and solvent-free ultrasonic multicomponent synthesis of (Z)-β-iodo vinylthiocyanates. ACS Sustainable Chem. Eng 7, 1574–1579. [Google Scholar]
- 85.Henkel S, Misuraca MC, Troselj P, Davidson J, and Hunter CA (2018). Polarisation effects on the solvation properties of alcohols. Chem. Sci 9, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith M, and March J. (2007). March’s Advanced Organic Chemistry Reactions, Mechanisms, and Structure, Sixth Edition (Wiley-Interscience; ). [Google Scholar]
- 87.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1912791).
- 88.Its crystallographic data have been deposited to Cambridge Crystallographic Data Centre (CCDC 1966561).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.