

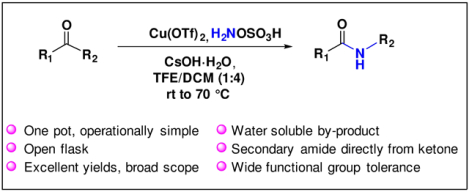
Abstract
The Beckmann Rearrangement (BKR) of ketones to secondary amides often requires harsh reaction conditions that limit its practicality and scope. Herein, we describe the Cu(OTf)2-catalyzed BKR of ketones under mild reaction conditions using hydroxylamine-O-sulfonic acid (HOSA), a commercial water soluble aminating agent. This method is compatible with most functional groups and directly provides the desired amides in good to excellent yields.
Keywords: Hydroxylamine-O-sulfonic acid (HOSA), Ketone, Beckmann Rearrangement, Cu(OTf)2, Secondary amide
Graphical Abstract
The Beckmann rearrangement (BKR) is a popular method for the formation of amides from ketones and aldehydes via an oxime intermediate.1 The conversion of an oxime into an amide was done firstly by German chemist Ernst Otto Beckmann in 1886.2 Notably, the BKR enjoys a prominent industrial role including the manufacture of monomer for polymerization into nylon-6 and nylon-12.3 Also, amides are common components in drugs, natural products, agrochemicals and functional materials (Fig. 1).4,5
Figure 1.
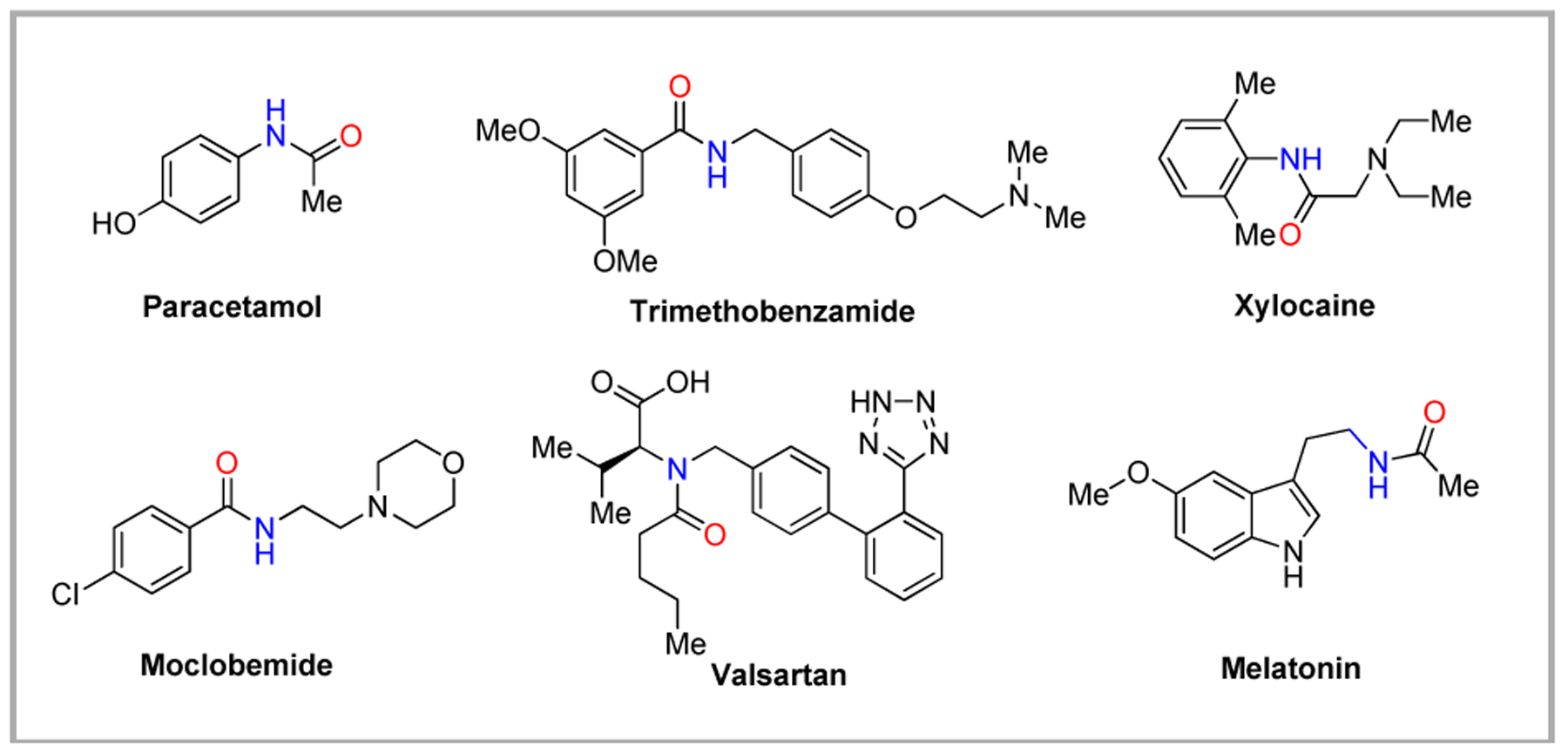
Amide bonds in drug molecules.
The traditional BKR requires harsh conditions such as high reaction temperatures and strongly acidic media, thus restricting the variety of suitable substrates; often, the process requires isolation of the oxime intermediate which can be labile and involves a cumbersome purification process (Scheme 1a,b).6 More recent modifications have addressed these limitations via catalysis with transition metals,7 calcium complexes,8 organocatalysts,9–13 inorganic Lewis acids,14 and boronic acid.15 Nevertheless, the need for a mild, inexpensive, and environmentally friendly procedure, especially for the direct conversion from ketones16, still persists.
Scheme 1.
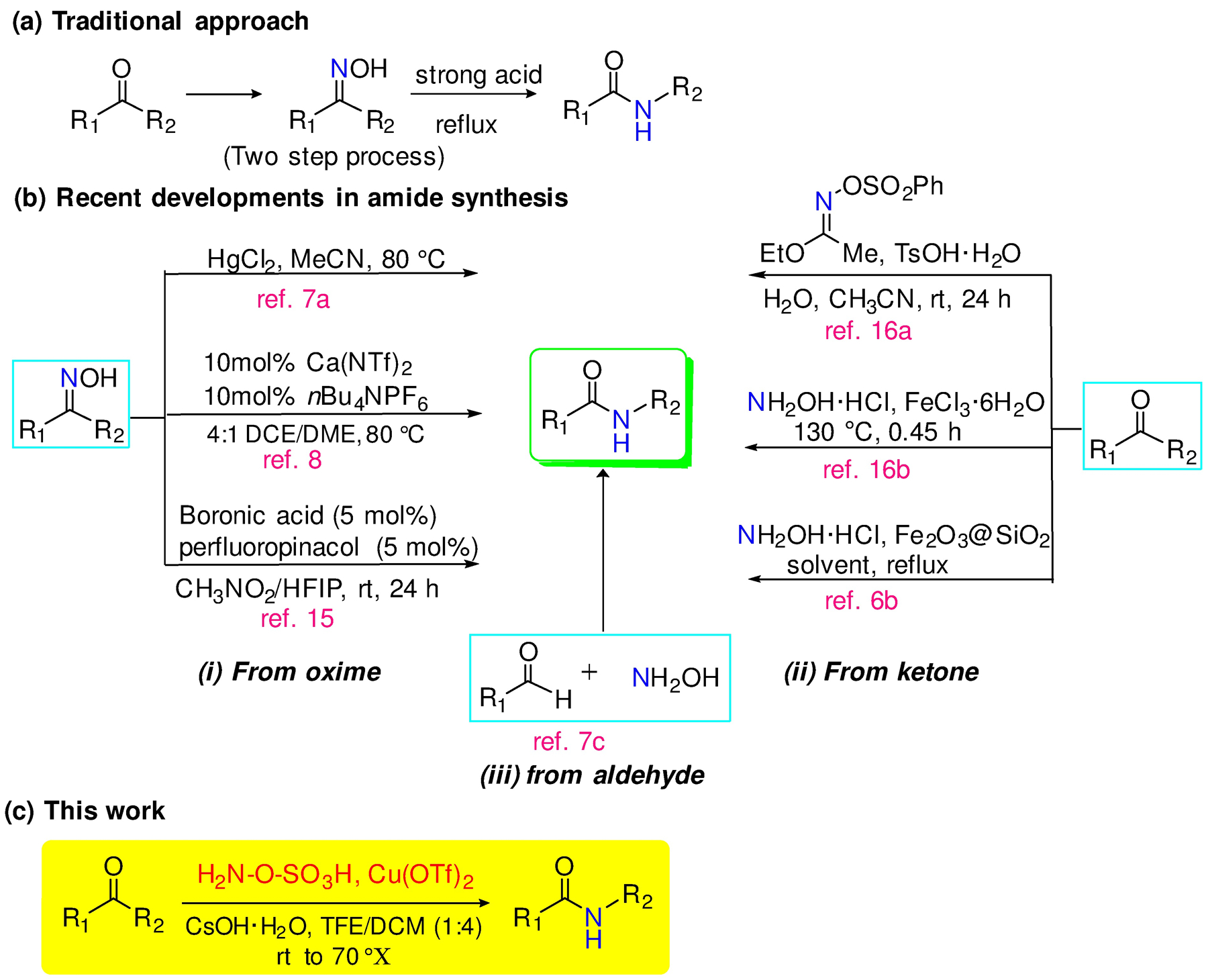
Beckmann rearrangement of ketones and recent variations.
Hydroxylamine-O-sulfonic acid (HOSA) attracted our attention for the BKR because it is commercial, inexpensive, water soluble, and readily handled.17 Indeed, it has found sporadic utility in the BKR,18 but primarily with aliphatic ketones. With aromatic ketones or hindered systems, strong acid and/or high temperatures are still required.19 Fortuitously, we had observed early transition metal salts, especially Cu(OTf)2, accelerated the condensation and rearrangement of aromatic ketones with HOSA.
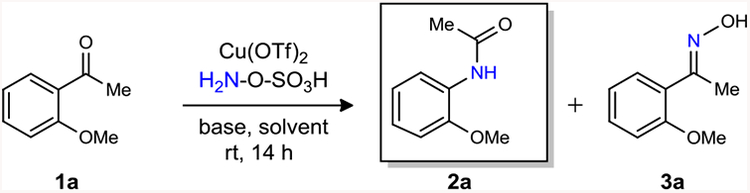
To actualize our aim, an introductory study was done using 2-methoxyacetophenone (1a) as a representative substrate along with HOSA and Cu(OTf)2 at rt (Table 1). Our investigation to optimize the reaction parameters to obtain amide 2a from 1a showed base was needed to commence the reaction. With Lithium hydroxide (LiOH), 95% of the starting material 1a was consumed and the expected amide was obtained in 85% yield along with 10% of oxime 3a (Table 1, entry 1).The most satisfactory result was achieved with cesium hydroxide monohydrate (CsOH·H2O) in which the desired amide 2a was obtained in 88% yield (entry 7) whereas other mild bases were not very effective (Table 1, entries 2–5) or in the case of K2CO3 proved ineffective (entry 6).
Table 1:
Optimization of reaction conditionsa
 | ||||
|---|---|---|---|---|
| Entry | Base | Solvent | Yield (%) | |
| 2a | 3a | |||
| 1b | LiOH | TFE/CH2Cl2(1:4) | 85 | 10 |
| 2c | Na2CO3 | TFE/CH2Cl2(1:4) | 10 | 0 |
| 3d | Et3Ne | TFE/CH2Cl2(1:4) | 60 | 40 |
| 4d | Pyridine | TFE/CH2Cl2(1:4) | 60 | 40 |
| 5d | DMAPf | TFE/CH2Cl2(1:4) | 40 | 60 |
| 6g | K2CO3 | TFE/CH2Cl2(1:4) | NR | |
| 7d | CsOH·H2O | TFE/CH2Cl2(1:4) | 88 | 0 |
| 8d | CsOH·H2O | HFIP | 83 | 0 |
| 9d | CsOH·H2O | TFE | 84 | 0 |
| 10 | CsOH·H2O | MeOH | 10 | 90 |
| 11h | CsOH·H2O | EtOH | 0 | 50 |
| 12i | CsOH·H2O | THF | 0 | 30 |
| 13 | CsOH·H2O | CH2Cl2 | 10 | 90 |
| 14g | CsOH·H2O | CH3CN | NR | |
Reaction Conditions: Cu(OTf)2 (0.1equiv), HOSA (2 equiv.), CsOH·H2O (2equiv.), TFE/CH2Cl2 (1:4), rt, 14h. TFE = 2,2,2-trifluoroethanol.
5% 1a recovered.
90% 1a recovered.
100% 1a consumed.
Triethylamine.
4-Dimethylaminopyridine.
NR = no reaction.
50% 1a recovered.
70% 1a recovered.
Finally, a variety of solvents were screened including hexafluoroisopropanol (HFIP), 2,2,2-trifluoroethanol (TFE), methanol, ethyl alcohol, tetrahydrofuran (THF), acetonitrile, dichloromethane and a mixture of TFE/CH2Cl2(1:4) (Table 1). While HFIP and TFE delivered amide 2a in comparable yields (83% and 84%, respectively; entries 8 and 9), a mixture of TFE/CH2Cl2 was best for both the yield of amide 2a (88%, entry 7) and for solubilization of ketones. Only acetonitrile, despite its frequent use in amidation reactions, failed to support the BKRs (entry 14).
Having established the optimum reaction conditions, the scope of the methodology was evaluated with a range of representative ketones (Scheme 2). Generally, acetophenones with strong aryl electron donating groups reacted smoothly at rt to deliver the derived N-phenylacetamides in very good yields, e.g., phenol 2b, 4-methoxy 2c, 3,4-dimethoxy 2d, and 4-allyloxy 2e. The latter example is noteworthy for its chemoselectivity, i.e., no allylic20 or C-H amination21 or aziridination17c under the reaction conditions. In contrast, 4-tolyl 2f, phenyl 2g, and naphthyl 2h required heating for a reasonable reaction rate, although rt reactivity was restored in the homologous 2i, j. The halogenated ketones 2l-n were well behaved and provided good yields of amide, except for 4’-fluoroacetophenone which was less reactive, as it delivered the corresponding amide 2k with only 25% of yield at 70 °C in 24h. The BKR of benzophenone and 3-acetyindole furnished 2o and 2p, respectively, albeit in modest yields. Lactams 2q-t were readily obtained from the corresponding cyclic ketones in high yields. Following upon well-established migratory priorities, estrone 3-methyl ether and hex-2-one led to 2u and 2v, respectively.
Scheme 2.
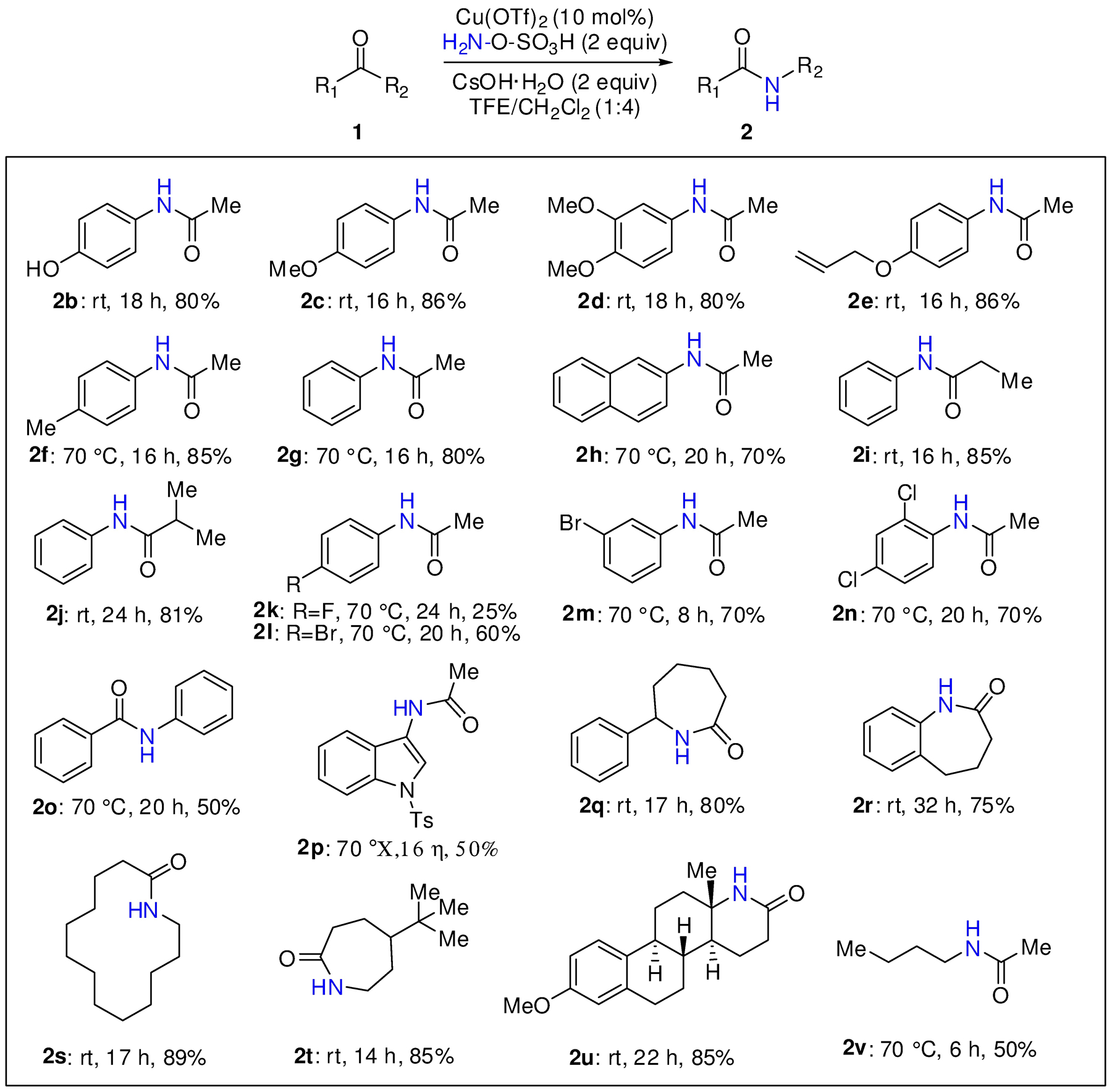
One pot synthesis of secondary amides from ketones
We propose the Cu(OTf)2 has a dual role in catalyzing the BKR (Figure 2). Firstly, as a mild Lewis acid, it assists in the formation of the transient ketoxime intermediate A that could be observed in some cases. Secondly, by way of the five membered transition state B, the copper catalyzes the migration of the R’ group to the nitrogen from which the Beckmann product is finally obtained.
In conclusion, we have developed an operationally simple, one pot BKR route to secondary amides directly from ketones using inexpensive, easily handled HOSA as aminating agent via Cu(II)-catalysis.
Reactions, unless otherwise stated, were carried out with magnetic stirring open to the atmosphere in oven dried glassware. Reagents were used as received, unless otherwise noted. TLC used precoated plates (Merck silica gel 60, F254) and was visualized with UV light and/or charring after dipping in PMA or KMnO4 solution. The compounds were purified by triturating the crude reaction mixture under hexane or by flash column chromatography using silica gel (100–200 mesh) with ethyl acetate:hexane as eluent. 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, in CDCl3 or DMSO-d6 as solvent. Chemical shifts δ are reported in parts per million (ppm) relative to residual undeuterated solvent as an internal reference (1H δ 7.26 and13C δ 77.0 for CDCl3, δ 2.50 and 39.52 for DMSO-d6respectively). The following abbreviations are used to indicate NMR peak multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and br s = broad singlet.
General procedure for synthesis of amides from ketones
To a stirring rt solution of Cu(OTf)2 (0.05 mmol, 10mol%) in TFE/CH2Cl2 (1:4) (2–3 mL) were added ketone (0.5 mmol, 1.0 equiv), HOSA (2.0 equiv) and CsOH·H2O (2.0 equiv). The reaction mixture was maintained at the temperature and for the time indicated in Table 2. After completion, the reaction mixture was diluted with CH2Cl2 (10 mL) and washed with saturated aqueous Na2CO3 (3×5 mL). The organic layer was washed with brine solution (5 mL) and dried over anhydrous Na2SO4. The crude product obtained after removal of all volatiles in vacuo was purified via SiO2 (100–200 mesh) chromatography using ethyl acetate/hexane as eluent.
N-(2-methoxyphenyl)acetamide (2a)22
Yield: 73 mg (88%); solid, mp 72–74 °C.
1H NMR (400 MHz, CDCl3): δ = 8.35 (dd, J = 7.9, 1.7 Hz, 1H), 7.77 (s, 1H), 7.03 (td, J = 7.8, 1.7 Hz, 1H), 6.95 (td, J = 7.8, 1.5 Hz, 1H), 6.87 (dd, J = 8.1, 1.5 Hz, 1H), 3.88 (s, 3H), 2.20 (s, 3H).
N-(4-Hydroxyphenyl)acetamide (2b)23
Yield: 60 mg (80%); brown solid, mp 170–171 °C.
1H NMR (400 MHz, DMSO-d6): δ = 9.64 (s, 1H), 9.13 (s, 1H), 7.33 (d, J = 8.9 Hz, 2H), 6.67 (d, J = 8.9 Hz, 2H), 1.98 (s, 3H).
N-(4-Methoxyphenyl)acetamide (2c)15
Yield: 82 mg (86%); white solid, mp 129–130 °C.
1H NMR (400 MHz, CDCl3): δ = 7.41–7.35 (m, 2H), 6.87–6.81 (m, 2H), 3.78 (s, 3H), 2.14 (s, 3H).
N-(3,4-Dimethoxyphenyl)acetamide (2d)24
Yield: 78 mg (80%); white solid, mp 126–127.7 °C.
1H NMR (400 MHz, CDCl3): δ = 7.30 (d, J = 2.3 Hz, 1H), 6.87–6.70 (m, 2H), 3.86 (s, 3H), 3.85 (s, 3H), 2.15 (s, 3H).
N-(4-(Allyloxy)phenyl)acetamide(2e)25
Yield: 82 mg (86%); white solid, mp94–95 °C.
1H NMR (400 MHz, CDCl3): δ = 7.37 (d, J = 8.7 Hz, 2H), 7.14 (s, 1H), 6.87 (d, J = 8.8 Hz, 2H), 6.04 (ddd, J = 21.8, 10.4, 5.2 Hz, 1H), 5.40 (d, J = 17.3 Hz, 1H), 5.28 (d, J = 10.5 Hz, 1H), 4.51 (d, J = 5.1 Hz, 2H), 2.15 (s, 3H).
N-(p-tolyl)Acetamide (2f)23
Yield: 63 mg (85%); white solid, mp 151–152 °C.
1H NMR (400 MHz, CDCl3): δ = 7.37 (d, J = 8.3 Hz, 2H), 7.25 (br s, 1H), 7.11 (d, J = 8.1 Hz, 2H), 2.31 (s, 3H), 2.15 (s, 3H).
N-Phenylacetamide (2g)26
Yield: 54 mg (80%); white solid, mp 114–115 °C.
1H NMR (400 MHz, CDCl3): δ = 7.49 (d, J = 7.7 Hz, 2H), 7.31 (t, J = 7.9 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 2.17 (s, 3H).
N-(Naphthalen-2-yl)acetamide(2h)27
Yield: 65 mg (70%); off white solid, mp133–135 °C.
1H NMR (400 MHz, CDCl3): δ = 8.18 (br s, 1H), 7.80–7.77 (m, 3H), 7.48–7.37 (m, 4H), 2.24 (s, 3H).
N-Phenylpropionamide (2i)13
Yield: 63 mg (85%); white solid, mp 107–108 °C.
1H NMR (CDCl3, 400 MHz): δ = 7.52 (d, J = 8.0 Hz, 2H), 7.30 (t, J = 8.0 Hz, 2H), 7.26 (br, 1H), 7.10 (t, J = 7.4 Hz, 1H), 2.38 (q, J = 7.6 Hz, 2H), 1.24 (t, J = 7.6 Hz, 3H).
N-Phenylisobutyramide (2j)9
Yield: 88 mg (81%); white solid, mp 109–111 °C.
1H NMR (400 MHz, CDCl3): δ = 7.53 (d, J = 8.0 Hz, 2H), 7.31 (t, J = 7.8 Hz, 2H), 7.09 (t, J = 7.3 Hz, 1H), 2.51 (sept, J = 6.8 Hz, 1H), 1.25 (d, J = 6.8 Hz, 6H).
N-(4-Fluorophenyl)acetamide (2k)28
Yield: 19 mg(25%);white solid; mp 153–154.6 °C.
1H NMR (400 MHz, CDCl3): δ = 7.45 (dd, J = 8.8, 4.8 Hz, 2H), 7.31 (br s, 1H), 7.00 (t, J = 8.6 Hz, 2H), 2.16 (s, 3H).
N-(4-Bromophenyl)acetamide (2l)28
Yield: 65 mg (60%); white solid, mp 167–169 °C
1H NMR (400 MHz, CDCl3): δ = 7.47–7.36 (m, 4H), 7.23 (br s, 1H), 2.17 (s, 3H).
N-(3-Bromophenyl)acetamide (2m)29
Yield: 75 mg (70%); white solid, mp 81–83 °C.
1H NMR (400 MHz, CDCl3): δ = 7.81 (s, 1H), 7.69 (s, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.29 (dd, J = 16.1, 4.4 Hz, 1H), 7.20 (t, J = 7.9 Hz, 1H), 2.22 (s, 3H).
N-(2,4-Dichlorophenyl)acetamide (2n)30
Yield: 38 mg (70%); brown solid, mp 142–144 °C.
1H NMR (400 MHz, CDCl3): δ = 8.34 (d, J = 8.7 Hz, 1H), 7.55 (s, 1H), 7.38 (d, J = 1.7 Hz, 1H), 7.28–7.22 (m, 1H), 2.24 (s, 3H).
N-Phenylbenzamide (2o)13
Yield: 49mg (50%); white solid, mp 164–165 °C.
1H NMR (400 MHz, CDCl3): δ = 7.87 (d, J = 7.8 Hz, 2H), 7.80 (s, 1H), 7.64 (d, J = 8.1 Hz, 2H), 7.56 (t, J = 7.0 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 7.8 Hz, 2H), 7.16 (t, J = 7.2 Hz, 1H).
N-(1-Tosyl-1H-indol-3-yl)acetamide(2p)15
Yield: 82 mg (50%); white solid, mp193–194 °C.
1H NMR (400 MHz, CDCl3): δ = 8.20 (s, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.41–7.32 (m, 2H), 7.28–7.26 (m, 1H), 7.25–7.22 (m, 1H), 7.18 (d, J = 8.0 Hz, 2H), 2.31 (s, 3H), 2.24 (s, 3H).
7-Phenylazepan-2-one (2q)31
Yield: 76 mg (80%); white solid, mp 134–136 °C.
1H NMR (400 MHz, CDCl3): δ = 7.41–7.21 (m, 5H), 6.18 (br s, 1H), 4.46 (d, J = 9.3 Hz, 1H), 2.66–2.49 (m, 2H), 2.11–1.83 (m, 4H), 1.76–1.57 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 177.93, 141.92, 129.18, 128.22, 126.24, 58.85, 36.91, 36.79, 29.78, 22.91.
1,3,4,5-Tetrahydro-2H-benzo[b]azepin-2-one (2r)27
Yield: 61 mg (75%); white solid, mp 137–139 °C.
1H NMR (400 MHz, CDCl3): δ = 7.56 (br s, 1H), 7.25–7.20 (m, 2H), 7.16–7.10 (m, 1H), 6.96 (d, J = 7.6 Hz, 1H), 2.80 (t, J = 7.2 Hz, 2H), 2.36 (t, J = 7.3 Hz, 2H), 2.27–2.21 (m, 2H).
Azacyclotetradecan-2-one (2s)32
Yield: 96 mg (89%); white solid, mp 155–157 °C.
1H NMR (400 MHz, CDCl3): δ = 5.57 (br s, 1H), 3.36–3.26 (m, 2H), 2.25–2.15 (m, 2H), 1.76–1.61 (m, 2H), 1.54–1.44 (m, 2H), 1.43–1.20 (m, 16H).
13C NMR (101 MHz, CDCl3): δ = 173.10, 38.46, 36.27, 28.31, 26.49, 25.79, 25.71, 25.63, 25.41, 25.26, 23.95, 23.70, 23.13.
5-(tert-Butyl)azepan-2-one (2t)33
Yield: 73 mg (85%); semi solid.
1H NMR (400 MHz, CD3OD): δ = 3.33–3.25 (m, 2H), 2.58–2.48 (m, 1H), 2.16 (td, J = 13.4, 4.9 Hz, 1H), 2.07–1.94 (m, 2H), 1.84 (td, J = 13.9, 5.4 Hz, 1H), 1.37–1.14 (m, 3H), 0.90 (s, 9H); 13C NMR (101 MHz, CD3OD) δ 167.50, 48.46, 33.23, 32.60, 28.76, 27.91, 27.64, 26.76.
(4aS, 4bR, 10bS, 12aS)-8-methoxy-12a-methyl-3,4,4a,4b,5,6,10b,11,12,12a-decahydronaphtho[2,1-f]quinolin2(1H)-one (2u)34
Yield: 126 mg (85%); white solid, mp 220–222 °C
1H NMR (400 MHz, CDCl3): δ = 7.18 (d, J = 8.6 Hz, 1H), 6.76–6.46 (m, 3H), 3.77 (s, 3H), 2.93–2.79 (m, 2H), 2.56–2.34 (m, 4H), 2.15–1.99 (m, 2H), 1.89–1.81 (m, 1H), 1.77–1.66 (m, 1H), 1.58–1.25 (m, 5H), 1.19 (s, 3H).
13C NMR (101 MHz, CDCl3): δ = 171.96, 157.65, 137.57, 131.79, 126.17, 113.52, 111.69, 55.21, 54.49, 46.49, 43.28, 39.85, 39.34, 30.65, 29.88, 26.67, 26.21, 22.19, 19.83.
N-Butylacetamide (2v)35
Yield: 82 mg (50%); clear oil.
1H NMR (400 MHz, CDCl3): δ = 5.66 (s, 1H), 3.26–3.18 (m, 2H), 1.95 (s, 3H), 1.52–1.41 (m, 2H), 1.38–1.28 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H).
Supplementary Material
Scheme 3.
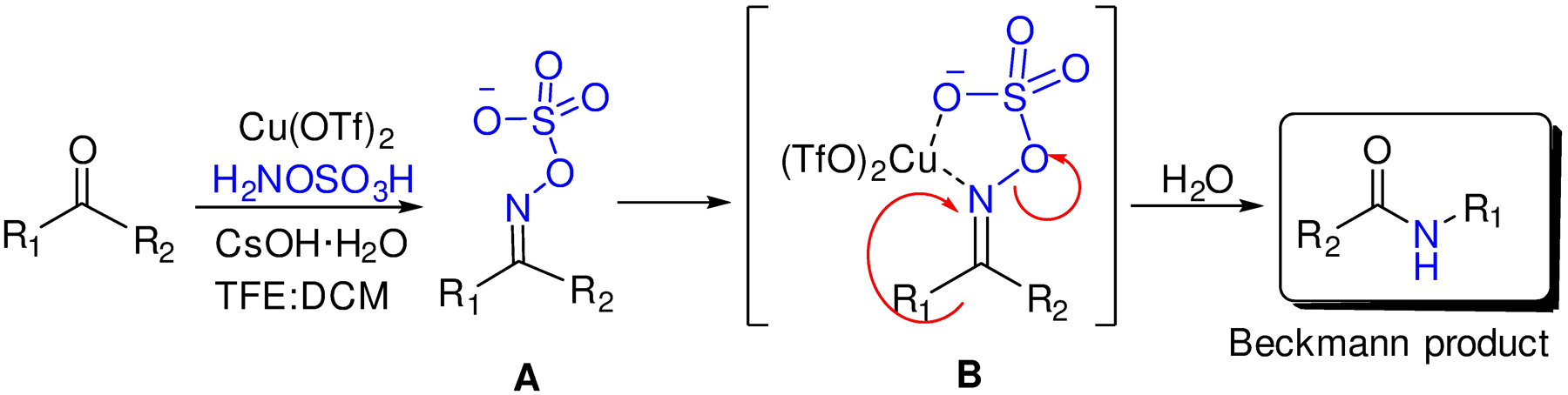
Proposed mechanism for Cu(OTf)2 catalyzed Beckmann rearrangement
Acknowledgement
J. L. J. thanks DST-SERB (YSS/2015/000838), UGC, New Delhi for UGC-BSR Grant (No.F.30-382/2017) and BBAU, Lucknow for infrastructure. S.V. expresses her thanks to CSIR, New Delhi, India for the research fellowship. J.R.F. received financial support from the Robert A. Welch Foundation (I-0011) and USPHS NIH (HL139793).
Footnotes
Supporting Information: Copies of 1H and 13C NMR spectra.
References
- (1).(a) Vinnik MI; Zarakhani NG Russ. Chem. Soc 1967, 36, 51. [Google Scholar]; (b) Krow GR Tetrahedron 1981, 37, 1283. [Google Scholar]; (c) Gawley RE Org. React 1988, 35, 1. [Google Scholar]; (d) Katritzky AR; Monteux DA; Tymoshenko DO Org. Lett 1999, 1, 577. [Google Scholar]; (e) Smith MB; March J In Advance Organic Chemistry, 5th ed.; John Willey and Sons: New York, 2001; p 1415.and references therein. [Google Scholar]; (f) Chandrasekhar S In Comprehensive Organic Synthesis II. Knochel P; Molander GA Eds: Elsevier: Amsterdam, 2014, 7, 770. [Google Scholar]; (g) Holth TAD; Hutt OE; Georg GI Rojas CM, Eds.; John Wiley & Sons, Inc.:2015, 111. [Google Scholar]; (h) Zhang W; Yang S; Lin Q; Cheng H; Liu JJ Org. Chem 2019, 84, 851. [DOI] [PubMed] [Google Scholar]
- (2).(a) Beckmann E Ber. Dtsch. Chem. Ges 1886, 19, 988. [Google Scholar]; (b) Blatt AH Chem. Rev 1933, 12, 215. [Google Scholar]
- (3).(a) Luedeke VD; Mcketta JJ Ed.; Marcel Dekker; In Encyclopedia of Chemical Processing and Design. New York: 1978,72. [Google Scholar]; (b) Rademacher H In Ullmann’s Encyclopedia of Industrial Chemistry, 5th ed.; Gerhartz W, Ed.; Wiley: New York: 1987, A8, 201. [Google Scholar]; (c) Weber JN In Kirk-Othmer Encyclopedia of Chemical Technology, 4th ed.; Kroschwitz JI, Ed.; Wiley: New York, 1990, 19, 500. [Google Scholar]; (d) Palmer RJ Encyclopedia of Polymer Science and Technology. 4th ed.; John Wiley & Sons, Inc; 2001 [Google Scholar]; (e) Wessermel K; Arpe H-J Industrial Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2003, 239. [Google Scholar]; (f) Owston NA; Parker AJ; Williams JM J. Org. Lett 2007, 9, 3599. [DOI] [PubMed] [Google Scholar]; (g) You K; Mao L; Yin D; Liu P; Luo H Catal.Commun 2008, 9, 1521. [Google Scholar]
- (4).(a) Zabicky J The Chemistry of Amides, Wiley-Interscience, New York, 1970. [Google Scholar]; (b) Greenberg A; Breneman CM; Liebman JF John Wiley & Sons, New York, 2000. [Google Scholar]; (c) Carey JS; Laffan D; Thomson C; Williams MT Org. Biomol. Chem 2006, 4, 2337. [DOI] [PubMed] [Google Scholar]; (d) Cupido T; Tulla-Puche J; Spengler J; Albericio F Curr. Opin. Drug Discovery Dev 2007, 10, 768. [PubMed] [Google Scholar]; (e) Pathare SP; Jain AKH; Akamanchi KG RSC Adv. 2013, 3, 7697. [Google Scholar]; (f) Rao SN; Mohana DC; Adimurthy S RSC Adv. 2015, 5, 95313. [Google Scholar]
- (5).(a) Humphrey JM; Chamberlin AR Chem. Rev 1997, 97, 2243. [DOI] [PubMed] [Google Scholar]; (b) Larock RC Comprehensive Organic Transformatins, 2nd ed. Wiley-CH: Weinheim: 1999, 1234. [Google Scholar]; (c) Bode JW Curr. Opin. Drug Discovery Dev 2006, 9, 765. [PubMed] [Google Scholar]; (d) Selvamurugan S; Ramachandran R; Prakash G; Viswanathamurthi P; Malecki JG; Endo AJ Organomet. Chem 2016, 803, 119. [Google Scholar]
- (6).(a) Kalia J; Raines RT Angew. Chem. Int. Ed 2008, 47,7523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jain PU; Samant SD ChemistrySelect 2018, 3, 1967. [Google Scholar]; see also refs.7–18..
- (7).(a) Ramalingan C; Park Y-T J. Org. Chem 2007,72, 4536. [DOI] [PubMed] [Google Scholar]; (b) Crochet P; Cadierno V Chem.Comm 2015, 51, 2495. [DOI] [PubMed] [Google Scholar]; (c) Martinez-Asencio A; Yus M; Ramon DJ Tetrahedron Lett. 2012, 68, 3948. [Google Scholar]
- (8).Kiely-Collins HJ; Sechi I; Brennan PE; McLaughlin MG Chem. Commun 2018, 54, 654. [DOI] [PubMed] [Google Scholar]
- (9).Furuya Y; Ishihara K; Yamamoto HJ Am. Chem. Soc 2005, 127, 11240. [DOI] [PubMed] [Google Scholar]
- (10).Betti C; Landini D; Maia A; Pasi M Synlett 2008, 6, 908. [Google Scholar]
- (11).Hashimoto M; Obora Y; Sakaguchi S; Ishii Y J. Org. Chem 2008, 73, 2894. [DOI] [PubMed] [Google Scholar]
- (12).Zhu M; Cha C; Deng W-P; Shi X-X Tetrahedron Lett. 2006, 47, 4861. [Google Scholar]
- (13).Pi H-J; Dong J-D; An N; Du W; Deng W-P Tetrahedron 2009, 65, 7790. [Google Scholar]
- (14).Zicmanis A; Katkevica S; Mekss P Catal. Commun 2009, 10, 614. [Google Scholar]
- (15).Mo X; Morgan TDR; Ang HT; Hall DG J. Am. Chem. Soc 2018, 140, 5264. [DOI] [PubMed] [Google Scholar]
- (16).(a) Hyodo K; Hasegawa G; Oishi N; Kuroda K; Uchida K J. Org. Chem 2018, 83, 13080. [DOI] [PubMed] [Google Scholar]; (b) Mahajan S; Sharma B; Kapoor KK Tetrahedron Lett. 2015, 56, 1915. [Google Scholar]
- (17).(a) Wallace RG AldrichimicaActa 1980, 13, 3. [Google Scholar]; (b) Hydroxylamine-O-sulfonic acid: Erdik E, Saczewski J, eEROSEncyclopedia of Reagents for Organic Synthesis, Wiley, Hoboken, 2013, 1. [Google Scholar]; (c) Ma Z; Zhou Z; Kürti L Angew. Chem. Int. Ed 2017, 56, 9886. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sabir S; Kumar G; Jat JL Org. Biomol. Chem 2018, 16, 3314. [DOI] [PubMed] [Google Scholar]
- (18).Olah GA; Fung AP Synthesis 1979, 7, 537. [Google Scholar]
- (19).(a) Sanford JK; Blair FT; Arroya J; Sherk KW J. Am. Chem. Soc 1945, 67, 1941. [Google Scholar]; (b) Freeman JP J. Org. Chem 1961, 26, 3507. [Google Scholar]
- (20).Du Bois J Org. Process Res. Dev 2011, 15, 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Munnuri S; Anugu RR; Falck JR Org. Lett 2019, 21, 1926. [DOI] [PubMed] [Google Scholar]
- (22).Srivastava VP; Patel R; Garima.; Yadav LDS Chem. Comm 2010, 46, 5808. [DOI] [PubMed] [Google Scholar]
- (23).Gao Y; Liu J; Li Z; Guo T; Xu S; Zhu H; Wei F; Chen S; Gebru H; Guo KJ Org. Chem 2018, 83, 2040. [DOI] [PubMed] [Google Scholar]
- (24).Schulz L; Enders M; Elsler B; Schollmeyer D; Dyballa KM; Franke R; Waldvogel SR Angew. Chem. Int. Ed 2017, 56, 4877. [DOI] [PubMed] [Google Scholar]
- (25).Schmidt B; Wolf F J. Org. Chem 2017, 82, 4386. [DOI] [PubMed] [Google Scholar]
- (26).Stuart DR; Bertrand-Laperle M; Burgess KMN; Fagnou K J. Am. Chem. Soc 2008, 130, 16474. [DOI] [PubMed] [Google Scholar]
- (27).Luca LD; Giacomelli G; Porcheddu AJ Org. Chem 2002, 67, 6272. [DOI] [PubMed] [Google Scholar]
- (28).Mahajan PS; Humne VT; Tanpure SD; Mhaske SB Org. Lett 2016, 18, 3450. [DOI] [PubMed] [Google Scholar]
- (29).Pialat A; Liegault B; Taillefer M Org. Lett 2013, 15, 1764. [DOI] [PubMed] [Google Scholar]
- (30).Singh H; Sen C; Sahoo T; Ghosh SC Eur. J. Org. Chem 2018, 4748. [DOI] [PubMed] [Google Scholar]
- (31).Tokuyama H; Itabashi S; Shimomura M; Sato M; Azuma H; Okano K; Sakata J; Hidetoshi T Synlett 2018, 29, 1786. [Google Scholar]
- (32).Mudiyanselage AY; Viamajala S; Varanasi S; Yamamoto K ACS Sustainable Chem. Eng 2014, 2, 2831. [Google Scholar]
- (33).Aube J; Wang Y; Hammond M; Tanol M; Takusagawa F; Velde DV J. Am. Chem. Soc 1990, 112, 4879. [Google Scholar]
- (34).Liu Z-J; Lu X; Wang G; Li L; Jiang W-T; Wang Y-D; Xiao B; Fu Y J. Am. Chem. Soc 2016, 138, 9714. [DOI] [PubMed] [Google Scholar]
- (35).Steffel LR; Cashman TJ; Reutershan MH; Linton BR J. Am. Chem. Soc 2007, 129, 12956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.