


Abstract
We give results from a detailed analysis of human Ribosomal Protein (RP) levels in normal and cancer samples and cell lines from large mRNA, copy number variation and ribosome profiling datasets. After normalizing total RP mRNA levels per sample, we find highly consistent tissue specific RP mRNA signatures in normal and tumor samples. Multiple RP mRNA-subtypes exist in several cancers, with significant survival and genomic differences. Some RP mRNA variations among subtypes correlate with copy number loss of RP genes. In kidney cancer, RP subtypes map to molecular subtypes related to cell-of-origin. Pan-cancer analysis of TCGA data showed widespread single/double copy loss of RP genes, without significantly affecting survival. In several cancer cell lines, CRISPR-Cas9 knockout of RP genes did not affect cell viability. Matched RP ribosome profiling and mRNA data in humans and rodents stratified by tissue and development stage and were strongly correlated, showing that RP translation rates were proportional to mRNA levels. In a small dataset of human adult and fetal tissues, RP protein levels showed development stage and tissue specific heterogeneity of RP levels. Our results suggest that heterogeneous RP levels play a significant functional role in cellular physiology, in both normal and disease states.
INTRODUCTION
The human ribosome is composed of ribosomal RNA (rRNA) and 80 structural ribosomal proteins (RPs), which form its two subunits, 60S and 40S. RPs are essential in early development in complex eukaryotes (1), and are highly conserved. Furthermore, ribosomes from one species can translate mRNA from different species (2). Interestingly, a growing body of work suggests ribosome heterogeneity at many levels, in response to extra/intra cellular stimuli, such as differentiation/growth signals, tRNA abundance etc., to optimize cell/tissue-specific objectives, such as translation efficiency, selectivity and fidelity (reviewed in (3)). One of the strongest evidence for existence of ‘specialized ribosomes’ comes from a study in mice, where loss of function mutation in Rpl38 selectively perturbs translation of subsets of Hox mRNA, while maintaining global protein synthesis (4). Heterogeneity in RP mRNA levels in mouse embryonic stem cells results in differential translation of sub-pools of transcripts involved in metabolism, cell cycle and development (5). Mass spectrometry data from budding yeast and embryonic stem cells shows that RP stoichiometry varies with environmental condition (6). In complex multicellular eukaryotes, the equivalent of ‘environment’ is the tissue microenvironment. Analysis of Encode data (7) and gene-deletion data in yeast (8) shows environment/tissue-specific regulation of mRNA levels of RP genes in eukaryotes (9). In yeast, growth-defective 60S mutants increased synthesis of proteins involved in proteasome-mediated degradation, and 40S mutants had increased translation of ribosome biogenesis genes (10).
The effects of alterations in RPs have also been noted in cancers, with both germline and somatic mutations in RP genes found in 10–30% of tumors (11). Ribosomopathies, which are congenital dysfunctions in heart, bone, and kidney, derive from mutations in different RPs, and make patients susceptible to cancers later in life (12,13). The fact that specific germline RP mutations cause defects in specific tissues indicates tissue-specific roles for RP in disease. One may ask whether such heterogeneity exists only in aberrant or disease conditions, or whether such variations are also found in normal tissues. The fact that ribosomal DNA (rDNA) has copy number variation (CNV) and nucleotide differences within and among individuals, with tissue-specific allelic expressions in functional regions of the assembled ribosome (14) suggests that such tissue-specific regulatory capacity of the ribosome may indeed be found in non-diseased individuals.
Here, we asked whether there are consistent patterns in RP heterogeneity in normal and cancer samples at the mRNA and CNV level in humans. To address this, we analyzed the following large and well curated public databases:
Data from GTEx: consisting of mRNA levels for 11,688 normal samples for 53 human tissues from 714 subjects (15),
Data from TCGA: consisting of mRNA levels in 10,363 tumor samples in 33 human cancer types (https://portal.gdc.cancer.gov) and CNV data from 10,845 human tumor samples (http://gdac.broadinstitute.org),
mRNA data from 675 tumor-derived human cell lines (16),
CRISPR–Cas9 knockout data for viability/growth for 558 human cell lines (https://depmap.org),
Ribosome profiling and mRNA data from human (17–20), mouse (17,21,22) and rat (23) tissues and cell cultures,
Protein expression data from normal human adult and fetal tissues (24).
After normalizing the mRNA data for overall (and uninteresting) inter-sample variation in total RP mRNA levels, we found robust, highly tissue-specific mRNA signatures in normal and tumor samples. At least three RP mRNA signatures were necessary to capture the variation in RP levels in non-diseased brain and blood samples, and at least sixteen signatures were necessary to capture the variation in RP mRNA levels in 53 different non-diseased tissue types. Several cancer types had two RP mRNA subtypes, with significantly different prognosis and genomic characteristics. These RP subtypes mapped well to known molecular subtypes, which, in some cases, had chromosomal deletions of RP genes, which correlated with their low mRNA levels. A pan-cancer analysis of CNVs in tumors showed that loss of one or both copies of RP genes is common across cancer types. CRISPR–Cas9 knockout of RP genes in several cell lines showed that loss of several RPs does not always affect cell viability. In humans, mice and rats, RNA-seq and ribosome profiling data for RP genes in tissues and cell cultures were highly correlated, showing that RP proteins are being translated in the ribosome at levels proportional to their mRNA levels. Consistently, both mRNA data and ribosome profiling data of RP genes, normalized by total level per sample, also showed tissue-specific and development-stage–specific clusters. Finally, in a small dataset (24) of protein expression levels in adult and fetal tissues, RP protein levels, standardized per sample, were found to be both tissue- and development-stage–specific.
Overall, these results contribute to the extant literature supporting ribosomal heterogeneity and suggest that functionally heterogeneous ribosome populations may meaningfully contribute to cellular physiology, with significant heterogeneity in RP mRNA and CNV levels that are tissue- and development-stage–specific.
MATERIALS AND METHODS
Data and normalization
RNA-seq read count data was obtained for 11,688 normal tissue samples from GTEx (15) (v7p), 10,363 tumor samples from TCGA (NCI-GDC, v7), and 675 tumor-derived cell lines from a recent study (16). The data was restricted to genes that encode RPs known to be functional in humans (25). The RP genes RPS4X and RPS4Y1/RPS4Y2, located on the X and Y chromosome, were excluded, because they have sex specific expression levels. Read count for each of the 78 remaining RP genes (Supplementary Table S1a) was divided by the length of the gene in kb, and further normalized so that the sum over all 78 RP genes was the same for each sample (Supplementary Figure S1a). The reason for the second normalization is that different tissues have different total numbers of ribosomes, and hence differ in total RP mRNA levels. This trivial variation in overall RP mRNA levels among tissues is not the focus of our analysis. Instead, we are interested in potential variations in ratios of RP mRNA levels among tissues. Making the sum of reads per kb over the 78 RP genes the same for all samples removes the variation in total RP mRNA levels among samples/tissues. The data normalized as above was used as input for the t-SNE (26) and UMAP (27) analysis, but was log2 transformed and standardized (z-score) for each RP gene for the SOM analysis (28). GISTIC2 (29) copy number variation (CNV) data for the 78 RP genes in 10 845 tumors from 33 cancer types in the TCGA dataset was compiled from ‘all_thresholded.by_genes.txt’ files downloaded from Broad GDAC (http://gdac.broadinstitute.org), and the entries −2 and −1 were interpreted as double deletion (aka deep deletion) and single deletion (aka shallow deletion) respectively.
Clustering methods overview
The goal of all clustering methods applied to sample/feature data is to find ‘groups’ of samples and/or features. The t-distributed Stochastic Neighbor Embedding or t-SNE (26) method is a popular method to identify clusters in large datasets in an unsupervised manner. It projects a high dimensional dataset into two dimensions by using local Gaussian kernels so as to preserve local associations of the data points in the original high dimensional space. Given the large size (>10,000 samples) and high dimension (78 dimensions) of the datasets analyzed, t-SNE was a natural method of choice. To avoid method related biases and reduce the possibility of overfitting, it is common to validate clustering results using multiple clustering methods. The Uniform Manifold Approximation and Projection or UMAP (27) method is an alternative to t-SNE which uses a different kernel and claims to better retain the global structure of the data. Self Organized Maps or SOM (28) is another common method used to cluster data that is very different from either t-SNE or UMAP. It uses an artificial neural network to create a lower dimensional representation of the data by using competitive learning on the input data vectors, while preserving topological features of the input space. Matrix based algorithms, such as principal component analysis (PCA) or Onco-GPS-Maps (30), are effectively linear, whereas t-SNE, SOM and UMAP are non-linear. Not all algorithms are equally efficient at finding all clusters because they make different assumptions about how to assign cluster membership. However, if the clusters identified by different algorithms are consistent, they are likely to represent real groupings in the data.
Methods for t-SNE (26), SOM (28) and UMAP (27)
The normalized dataset (Supplementary Figure S1a) was analyzed by t-SNE and UMAP using the distance metric  where
where 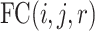 is the fold change for the corresponding RP for sample pair
is the fold change for the corresponding RP for sample pair  . The index
. The index  takes
takes 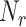 = 78 values corresponding to the number of RPs and the indices i, j range from 1 to
= 78 values corresponding to the number of RPs and the indices i, j range from 1 to  , where
, where  is the number of samples used in the analysis. In the 2D projection from t-SNE, the samples were colored by tissue of origin. To test whether the results are independent of the chosen distance metric, we repeated the t-SNE analysis on a log2 transformed version of the normalized dataset (Supplementary Figure S1a) using nine different distance metrics: ‘cityblock’ (equivalent to using our original distance metric on un-transformed data), ‘euclidean’, ‘seuclidean’, ‘chebychev’, ‘minkowski’, ‘mahalanobis’, ‘cosine’, ‘pearson’ and ‘spearman’.
is the number of samples used in the analysis. In the 2D projection from t-SNE, the samples were colored by tissue of origin. To test whether the results are independent of the chosen distance metric, we repeated the t-SNE analysis on a log2 transformed version of the normalized dataset (Supplementary Figure S1a) using nine different distance metrics: ‘cityblock’ (equivalent to using our original distance metric on un-transformed data), ‘euclidean’, ‘seuclidean’, ‘chebychev’, ‘minkowski’, ‘mahalanobis’, ‘cosine’, ‘pearson’ and ‘spearman’.
To test whether the same clustering holds when a different clustering method is used, samples were mapped to hexagonal nodes using SOM in a 2D grid using the ‘kohonen’ package in R. The mapped nodes were colored by tissue of origin using a majority rule: if >50% of the samples mapping to a node originated from a given tissue, that node was assigned the color of that tissue. Thus, a co-localization of nodes of the same color in SOM, and a clustering of samples of the same color in t-SNE, would indicate that the observed clusters are tissue-specific. The t-SNE analysis was done in matlab via the function ‘tsne’ using the ‘exact’ algorithm, and the distance metric mentioned above, with all other parameters set to default values. The SOM analysis used the ‘kohonen’ package (version 3.0.8) in R with the ‘rlen’ parameter set to 10,000 and other parameters set to default values. The UMAP analysis used the GitHub python implementation at https://github.com/lmcinnes/umap, the same data and the same distance metric as t-SNE with parameter values: number neighbors: 20, min-dist: 0.5.
Methods for matrix factorization and the Onco-GPS-Map (30)
To perform the Matrix-Factorization of the data using the Onco-GPS-Map, the RP mRNA levels of the 78 × N matrix of RPs × samples was converted to TPM (transcripts per million bases), log2 transformed after adding 1 to each entry, and standardized per sample. Finally, before each matrix factorization, each row (RP) was re-scaled to the [0, 1] interval over the samples being analyzed. In the matrix factorization procedure, the data matrix M is decomposed into the product of two matrices Mgenes × samples = Wgenes × k × Hk × samples. where W and H contain factors representing the most salient gene- and sample-specific patterns in the input matrix. In this way, for example, the blood and brain dataset was decomposed into k = 3 NMF factors (RP mRNA signatures) and the data matrix for pan-tissue analysis of all 53 tissue types was decomposed into k = 16 NMF factors. The optimal number of clusters (optimum value of k) for each data matrix was chosen as the number of clusters yielding the highest consensus-clustering cophenetic correlation (CCC) (30). The k columns of the W matrix represent the k RP signatures (directions in the space of RP mRNA levels) which best represent the data with k-factors. The columns of the H matrix correspond to samples and the entries in these k dimensional columns represent the contribution of the corresponding factors in the W matrix to the sample. The k factors and their contributions to the samples were visualized by multi-dimensionally scaling the W and H matrices to 2D, where the distances among the factors in the projection to 2D are proportional to those in the original k dimensions. The contribution of each RP in the factors and the association of the samples to the factors were visualized by projecting them onto this 2D space based on their amplitudes in the W and H matrix respectively. Finally, samples were colored by tissue type to visualize their possible associations with factors. The computer codes that implement the Onco-GPS-Map analysis are available at: https://github.com/KwatME/model_and_infer.
TuBA (31) summary
Tunable biclustering algorithm (TuBA) is an unsupervised algorithm that identifies modules of genes co-expressed in subsets of samples at relatively higher (or lower) levels compared to other samples in the dataset. TuBA requires specification of an ordered pair of parameters: (percentile cutoff, overlap significance cutoff). The ‘percentile cutoff’ is the fraction of samples in extremal sample sets for each gene, and (ii) The ‘overlap significance cutoff’ is the FDR cutoff for the association of each pair of genes to be considered relevant. For the GTEx dataset, the parameters were set to (2%, 0.01). For the analysis of the cancer subtypes, the parameters were set to (5%, 0.01) for LGG, SKCM, BLCA, PRAD, CRC and (15%, 0.01) for UVM. To determine associations among samples in the biclusters, we used the hypergeometric test. All enrichments reported are at P-value <0.001, FDR <0.01. Source code of TuBA is available at: https://github.com/KhiabanianLab/TuBA. The gene co-expression modules shown are the subset of genes forming a complete subgraph/seed, which represents the linked core of genes and samples forming a bicluster. The plots presenting the biclusters were prepared using Cytoscape (32) v3.7.0.
Identifying tissue-specific RP signatures in GTEx (15) data
Starting with the GTEx data normalized as described (Supplementary Figure S1), five tissue types (cervix–endocervix, cervix–ectocervix, fallopian tube, bladder, kidney cortex) with less than 50 samples were discarded. To avoid tissue-specific sampling biases due to unequal sample sizes among tissues, from the remaining 48 tissues, 88 samples were randomly selected per tissue without replacement, to create a dataset D of 4,224 samples. The expression of each RP in each tissue was compared to the expression of that RP in D using the Wilcoxon Rank Sum test to compute a significance P-value and log2 fold change. Using the significance cutoff P-value <0.05, those RPs which did not have a statistically significant log2 fold changes were assigned a log2 fold change of 0. This resulted in a 78 × 48 tissue-specific RP signature matrix, where each row was an RP gene, each column was a tissue, and the entries were modified log2 fold changes. This analysis identified the top differential RPs across tissue types (Supplementary Figure S6 and Table S1c). In addition, a 78 × 11,614 sample specific RP signature matrix was generated, where each row was an RP gene, each column an individual sample, and the entries were log2 fold change compared to the median expression of that RP gene in D. Spearman-Rho correlations and their P-values were computed comparing the 78 × 48 tissue-specific RP signature matrix to the 78 × 11,614 sample specific RP signature matrix, and the Spearman Rho value was set to 0 for those RPs with P-value ≥0.05. This generated a 11,614 × 48 consistency matrix where each row was an individual sample, each column was a tissue, and the entries are modified Spearman Rho values. The heatmap (Supplementary Figure S7) of this matrix shows that the RP signature of a tissue is representative of and highly correlated with the RP signatures of the samples of that tissue type.
The Kaplan–Maier recurrence/survival curve
The non-parametric Kaplan–Maier (KM) methodology (33) was developed to study rates of recurrence/death/response in situations where records were incomplete, i.e. in situations where, during the course of the study, patients either left the study, were not traceable or died from causes other than the disease in question. The data used in the KM analysis is a list of time of events (say disease recurrence) with each event labeled as a true event (recurrence) or a censoring event (loss of information for a given patient). From a defined initial time (say the end of surgery and radiation in the case of cancer patients), survival or recurrence times are measured for a cohort of patients to the occurrence of a given event, which might be the time of treatment to a recurrence/death event in a cancer patient. A survival time is called censored when the event has not yet happened but there is no more information on the patient for time points beyond this one, as may happen if a patient drops out of a study before the end of the study period. The survival or recurrence function S(t) is defined as the probability of recurring after or surviving until at least time t. The graph of S(t) against t is called the KM recurrence or survival curve. The Kaplan–Meier method can be used to estimate this curve from the observed survival times without the assumption of an underlying probability distribution. KM plots are commonly used in case/control studies, clinical trials, and pharmaceutical drug efficacy studies to compare two or more defined groups of patients.
Ribosome profiling and RNA-Seq data for humans and rodents
The ribosome profiling databases HRPDviewer (34) and RPFdb (35) were searched to identify human and rodent datasets with translation data in multiple tissue/cell-types, and the following RNA-seq (i.e. mRNA expression) data and ribosome profiling (i.e. transcripts being translated in ribosomes) data sets were downloaded from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=): human (GSE60426 (17), GSE62247 (18), GSE65885 (19,20)), mouse (GSE60426 (17), GSE41246, GSE72064 (21), GSE89108 (22), rat (GSE66715 (23)). Synonyms and annotations from NCBI (https://www.ncbi.nlm.nih.gov/gene/), Ensembl (https://www.ensembl.org/biomart/martview/), UCSC (http://genome.ucsc.edu/cgi-bin/hgTables) and UniProt (https://www.uniprot.org/uniprot/) were used to restrict the downloaded RNA-seq and ribosome profiling data to the 78 RP genes (Supplementary Table S1a) or a subset thereof (since not all datasets had data of all 78 RP genes). If a dataset had multiple rows for the same RP gene, they were aggregated so that there was only one row per RP gene. Both RNA-seq and ribosome profiling data of RP genes were normalized as previously described (Supplementary Figure S1a), i.e. corrected for gene length (if necessary) and then normalized to make the sum of reads over all RPs the same for every sample. This normalization eliminates the uninteresting and expected overall variation in total RP levels among tissues and cell-lines.
To minimize batch effects, two 48 h B-cell samples were excluded from GSE60426 as they were cultured differently from the rest (17). In GSE62247, most samples were submitted to GEO on 10 October 2014 but a few were submitted on 2 July 2015. The latter were also excluded to reduce batch effects.
RESULTS
Normal and tumor samples from humans have heterogeneous RP mRNA signatures
RNA-Seq mRNA data was obtained from (i) GTEx (15) (https://gtexportal.org/home/datasets): 11,688 normal samples from 53 normal tissue types from 714 non-diseased individuals; (ii) The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/repository): 10,688 tumor samples across 33 solid cancer types and (iii) 675 cell lines (16). The data was restricted to the 80 RPs that form the human eukaryotic ribosome (25), and the three datasets were analyzed separately to avoid batch effects. Data for the RP genes RPS4X and RPS4Y1/RPS4Y2, located on chromosomes X and Y, respectively, was excluded because of their sex-specific effects. The read counts for the remaining 78 RP genes (Supplementary Table S1a) were normalized by gene length, and then rescaled to make the sum of RP mRNA levels the same in each sample (Methods, Supplementary Figure S1a). This normalization eliminates the uninteresting variation in total RP mRNA levels among samples. Projection of the normalized data to 2D using t-SNE (26) showed multiple tissue-specific clusters for normal and tumor samples (Supplementary Figure S1b,c), indicating heterogeneity of RP mRNA signatures. In contrast, cell lines derived from many different cancer types showed only one cluster (Supplementary Figure S1d). Similar results were observed in t-SNE analysis using nine distance metrics (data not shown), showing that the results are not sensitive to the choice of distance metric.
The RP mRNA signature in normal human samples is tissue-specific
The t-SNE clusters for normal samples (Supplementary Figure S1b) mapped to different tissues (Figure 1). Brain tissue formed two clusters, one for the cerebellum and cerebellar hemisphere, and one for other brain tissues: cortex, ganglia, amygdala, hippocampus, hypothalamus, substantia nigra, and spinal cord, while pituitary, nerve, blood, transformed lymphocytes and spleen samples clustered separately (Figure 1A). Samples from the stomach, small intestine, terminal ileum and transverse colon clustered together, as did samples from esophagus-muscularis, gastro-esophageal junction, and sigmoid colon, while samples from esophagus-mucosa, liver and pancreas clustered separately (Figure 1B). Endocrine system samples (Figure 1C) had distinct clusters for pituitary, thyroid, adrenal, pancreas, ovary and testis. Soft tissue samples had distinct clusters for adipose, skin, arterial, heart, skeletal muscle, and transformed fibroblasts samples (Figure 1D). Similar results were observed in t-SNE analysis using nine distance metrics (data not shown), showing that the results are not sensitive to the choice of distance metric. Independent, unsupervised analysis of samples from each of Figure 1A–D using Self-Organized-Maps (SOM) (28) confirmed this tissue-specific clustering (Figure 1E–H). As further validation, independent analysis of the full GTEx (15) dataset using UMAP (27) again confirmed the tissue-specific clustering (Supplementary Figure S2).
Figure 1.

Normal samples have tissue-specific RP mRNA signatures. (A–D) t-SNE (26) analysis of GTEx (15) RP mRNA data for 11,688 normal samples from 53 tissue types. Each panel shows a subset of the full t-SNE plot, with samples stratified by organ system: (A) nervous and immune system, (B) digestive system, (C) endocrine system, (D) soft tissues. (E–H) Self-Organizing Maps (28) (SOM) applied to samples in A–D validate the stratification seen by t-SNE. Nodes marked NA had no tissue-specific majority and empty nodes had no samples mapped.
To study whether these tissue-specific RP clusters map to specific RP signatures, we applied the matrix factorization algorithm Onco-GPS-Map (30) (see Methods) to the GTEx data. Three RP signatures (factors) were needed to capture the variation in blood and brain samples (Figure 2A, B, Supplementary Table S1b), and 16 signatures were necessary for the 53 GTEx tissue types (Supplementary Figure S3a, b). Each tissue type mapped to combinations of RP factors (Figure 2C, Supplementary Figure S4) and the three factors for blood and brain were linear combinations of the sixteen factors for 53 normal tissue types (Supplementary Figure S3c). Figure 2D–F shows the RP genes up/down regulated among blood, cerebellum and cerebellar hemisphere and brain (rest) clusters in pairwise comparisons. Note that, although some of the fold changes in these plots are modest, the large sample sizes make them highly significant. Also note that the differentially expressed RPs shown in Figure 2D–F are consistent with the signatures F0, F1, F2 in Figure 2A and Supplementary Table S1b. As further validation of these findings, biclustering analysis of the blood-brain GTEx data by TuBA (31) identified two robust modules of RP genes with significantly higher/lower mRNA levels in subsets of samples (Supplementary Figure S5), which clearly corresponded to factors F1 and F0 for blood and cerebellum/cerebellar-hemisphere tissues respectively (Figure 2A, Supplementary Table S1b). These results show that RP mRNA signatures in normal human samples are tissue-specific, with different tissues displaying different combinations of RP mRNA signatures.
Figure 2.

RP mRNA levels in blood and brain have three distinct signatures. Matrix factorization analysis of the GTEx (15) blood and brain RP mRNA data using Onco-GPS-Map (30) showed that three RP signatures (factors) are optimal by consensus-clustering cophenetic correlation (CCC). The algorithm factorizes the 78 × N matrix M of mRNA levels of RPs × samples as M = W× H, where W is a 78 × k matrix and H is a k × N matrix (Supplementary Table S1b). (A) Heatmap of the W matrix (standardized by rows) showing the three RP signatures corresponding to three factors F0, F1, F2. (B) Heatmap of the H matrix (standardized by columns) showing the contribution of the three factors for each sample in the data. The samples for blood, cerebellar brain and rest of brain tissues use almost distinct factors. (C) A visualization of the data by multi-dimensional scaling of the W and H matrices to 2D. (D–F) Volcano plots for up/down regulated RP genes in blood, cerebellum and cerebellar hemisphere, and brain (rest) clusters pairwise; differential RPs are consistent with the signatures F0, F1, F2 in (A).
Supplementary Figure S6 (and Supplementary Table S1c) shows tissue-specific RP signatures in the GTEx data, where the highest/lowest log2 fold changes of five RPs in each tissue are shown in red/blue respectively, compared to their median expression across tissue types (details in Materials & Methods). Supplementary Figure S7 shows that the RP signature of a tissue (Supplementary Figure S6 and Table S1c) is representative of and highly correlated with the RP signatures of individual samples of that tissue type. Note also that tissue types that clustered together in Figure 1 have very similar RP signatures.
The RP mRNA signature in human tumor samples is tissue-specific
Inspection of TCGA t-SNE clusters (Supplementary Figure S1c) also showed stratification by tissue (Figure 3A–F). Nervous and immune system tumors showed separate clusters for glioblastoma (GBM), low-grade glioma (LGG), pheochromocytoma/paraganglioma (PCPG), thymoma (THYM), diffuse large B-cell lymphoma (DLBC), and acute myeloid leukemia (LAML) (Figure 3A). Digestive system tumors showed separate clusters for colorectal (CRC), gastric/esophageal (STES), pancreatic (PAAD), liver (LIHC), and head-neck squamous (HNSC) cancers (Figure 3B). Endocrine system tumors had separate clusters for thyroid (THCA), adrenocortical (ACC), pancreatic (PAAD), ovarian (OV) and testicular germ cell (TGCT) cancers (Figure 3C). Urinary system tumors stratified into bladder cancer (BLCA) and the renal cancer subtypes: clear-cell (KIRC), papillary (KIRP) and chromophobe (KICH) (Figure 3D). Cancers of the prostate (PRAD), breast (BRCA), ovary (OV), cervix (CESC), and endometrium (UCEC) formed distinct clusters (Figure 3E), while melanoma of the skin (SKCM) and eye (UVM) had two clusters each (Figure 3F). Similar results were observed in t-SNE analysis using nine distance metrics (data not shown), showing that the results are not sensitive to the choice of distance metric. Independent unsupervised analysis of each group of samples in Figure 3A–F by SOM (28) confirmed this tissue-specific clustering (Figure 3G–L). Analysis of the full TCGA dataset using UMAP (27) also confirmed the tissue-specific clustering (Supplementary Figure S8).
Figure 3.

Tumor samples have tissue-specific RP mRNA signatures. (A–F) t-SNE (26) analysis of RP mRNA data from 10 363 TCGA samples for 33 cancer types shows tissue/cancer type specific clusters. Each panel shows a subset of the full t-SNE plot with samples stratified by organ system: (A) cancers of the nervous and immune system, (B) cancers of the digestive system, (C) cancers of the endocrine system, (D) cancers of the urinary system, (E) sex-specific cancers, (F) melanomas. (G–L) Self-Organizing Maps (28) (SOM) applied to samples in A–F validate the stratification seen by t-SNE. Nodes marked NA had no tissue-specific majority and empty nodes had no samples mapped. Tumor acronyms are listed in Supplementary Table S1j.
Note that, because of the normalization that equalized the total RP mRNA level in each sample, these clusters reflect true tissue-specific variation in RP mRNA levels in tumors. These clusters are not the result of differences in total RP expression levels in tissues, which is not of interest because it would merely reflect different rates of overall ribosome activity.
Pan-cancer analysis of RP copy number variation (CNV) data for 10,845 tumor samples from TCGA by t-SNE showed no separation by tissue/cancer type (data not shown), except for kidney cancer, where the samples clustered into the three known subtypes (KIRC, KIRP and KICH) (36–38), which have distinct cells of origin in the kidney (39,40). RP genes with significantly high/low mRNA levels in KIRP or KICH versus KIRC also had significant copy number gains/losses (Supplementary Figure S9). While most KIRC tumors have single copy loss of chromosome 3p (36), ∼10% lose both copies, with complete loss of RPL14, RPL15, RPL29, RPL32, RPSA.
Six cancers have RP mRNA subtypes with distinct prognosis and genomic characteristics
Several TCGA cancer types had multiple RP mRNA sub-clusters by t-SNE (26) (Figure 3A–F), suggesting the existence of RP subtypes with distinct RP mRNA signatures. Clinical data from TCGA (41) showed that in six of these cancer types, the RP subtypes had significant disease associated survival differences: for disease specific survival in LGG, SKCM, UVM, BLCA, CRC (COAD and READ), and disease free interval in PRAD (Figure 4a) at P-value < 0.05 by two-sided log-rank tests (Supplementary Table S1d). Several RP genes (Figure 4B, Supplementary Table S1e) had significantly higher mRNA levels in the RP subtypes with worse/better prognosis. Biclustering analysis of the data for these six cancer types by TuBA (31) identified sets of co-expressed RP genes up-regulated in subsets of samples (Supplementary Figure S10), which matched the differentially expressed RP genes (Figure 4B). The samples in the biclusters also had a strong overlap with the better/worse prognosis RP subtypes (Supplementary Table S1d). These findings complement and extend results noted in recent literature (42,43).
Figure 4.

Tumors from distinct clusters for the same cancer type have significantly different prognosis. t-SNE (26) analysis of RP mRNA data for TCGA samples (Figure 3A–F), showed multiple subtypes (clusters) in several cancer types. In six of these cancers, there were significant differences in prognosis between these subtypes: low-grade glioma (LGG), skin melanoma (SKCM), eye melanoma (UVM), bladder cancer (BLCA), prostate cancer (PRAD) and colorectal cancer (CRC). (A) Kaplan–Meier plots showing significant differences in disease specific survival or disease-free interval among the RP-subtypes in LGG, SKCM, UVM, BLCA, CRC and PRAD at P < 0.05 by two-sided Wilcoxon rank-sum test. (B) Volcano plot identifies RP genes with significantly different mRNA levels among the RP-subtypes by two-sided Wilcoxon rank-sum tests. Green/red colors represent RPs upregulated in the better/worse prognosis subtypes (Figure 4a, Supplementary Table S1d). (C) In all six cancer types, the RP genes over-expressed in the better prognosis subtype (Figure 4B, Supplementary Table S1d) were highly intolerant of loss-of-function (LoF) variation, as evidenced by a high ‘probability of loss of intolerance’ (pLI) scores (44). Similarly, RP genes over-expressed in the worse prognosis RP-subtype were tolerant of loss-of-function variation, with relatively lower pLI scores.
If different RPs are indeed associated with different survival or phenotypic outcome, we should be able to see other signatures of such association in human populations. In these six cancer types, the RP genes over-expressed in the better prognosis RP subtypes (Supplementary Table S1e) were intolerant of loss-of-function (LoF) variation, as measured by the ‘probability of loss of intolerance’ (pLI) score (44) (Figure 4C). pLI scores range from 0 to 1, and estimate the probability that a given gene is haplo-insufficient. Genes with high pLI scores (e.g. Mendelian disease genes) are LoF intolerant, whereas genes with low pLI scores are LoF tolerant. The RP genes over-expressed in the worse prognosis RP subtypes in the six cancer types described above were more tolerant of loss-of-function (LoF) variation, i.e. had lower pLI scores.
Using genomic, transcriptomic, and histologic data from TCGA (45–49) and mutation data from cBioPortal (50,51) (http://www.cbioportal.org), we studied molecular differences among the RP subtypes. In LGG, SKCM and BLCA, the RP subtypes overlapped substantially with known molecular subtypes (Supplementary Figure S11a). The LGG-worse survival RP mRNA subtype was enriched in the ‘IDH-mutant 1p/19q non-co-deletion tumors’, ‘IDH-wildtype tumors’, and tumors with ‘astrocytoma histology’, while the LGG-better survival RP mRNA subtype was enriched in the ‘IDH-mutant 1p/19q co-deletion tumors’, and tumors with ‘oligodendroglioma histology’ as defined in the literature (45). The SKCM-worse survival RP mRNA subtype was enriched in the ‘keratin subtype’ while the SKCM-better survival RP mRNA subtype was enriched in ‘MITF-low subtype’ or ‘immune subtype’ as defined in the literature (46). The BLCA-worse survival RP mRNA subtype was enriched in ‘basal squamous’, ‘neuronal’, ‘luminal-infiltrated’ tumors while the BLCA-better survival RP mRNA subtype was enriched in ‘luminal papillary’ and ‘luminal’ tumors, and tumors with ‘papillary histology’ as defined in the literature (48). In LGG, UVM, BLCA, PRAD, and CRC, there were also other significant genomic differences (at P-value < 0.05 by two-sided Fisher's exact tests) among the RP mRNA subtypes (Supplementary Figure S11b).
In LGG, UVM and BLCA, the majority of differences in RP mRNA levels among the subtypes were due to CNVs in RP genes (Supplementary Figure S12a). Heatmaps of copy number data for these RPs (Supplementary Figure S12b) show that mRNA differential expressions (Supplementary Figure S12a) correlate with copy number alterations. For example, RP genes RPL5, RPL11, RPL22, RPS8 on chromosome 1p, and RPL13A, RPL18, RPL28, RPS5, RPS9, RPS11, RPS16, RPS19 on chromosome 19q are co-deleted in the better prognosis RP mRNA subtype in LGG (Supplementary Figure S12b), and these genes have significantly lower mRNA expression levels (Supplementary Figure S12a).
Double/single deletions of RPs are common in human tumors
Pan-Cancer analysis of TCGA CNV data showed that 1,272 (11.7%) tumors had double deletion of one or more of the 78 RP genes, and both copies of each of the 78 RP genes were deleted in at least one tumor in the TCGA data (Figure 5A, B). RPL13 and RPL29 had double deletions in more than 100 samples (Figure 5A, B) and both copies of up to 16 RPs were lost in one sample (Figure 5C). Double deletions of RP genes were observed in all 33 cancer types (Figure 5D), but were more frequent in some cancer types than others (Supplementary Figure S13, Table S1f). DLBC was at one extreme, where over 25% of tumors had double deletion of some RP gene, and KICH was at the other extreme, where <2% tumors had double deletion of some RP genes. Highly recurrent, double deletion of an RP gene was rare, and only one RP in DLBC (RPS12) and five RPs in KIRC (RPL14, RPL15, RPL29, RPL32, RPSA) were double deleted in >10% of tumors (Supplementary Figure S13, Table S1f).
Figure 5.

Double deletions of RP genes is common in TCGA tumors. (A) The number of tumors with double deletion of an RP gene in the TCGA dataset. Each RP gene had double deletion in at least one tumor sample. (B) Double deletion profile of the RP genes for TCGA samples with at least one RP double deletion. (C) Distribution of double deletions of RPs across tumors. Double deletion of one to sixteen RP genes is seen in the same tumor sample. (D) Distribution of RP double deletions across cancer types. All 33 cancer types in TCGA have double deletion of multiple RPs. Serous ovarian cancer (OV) had 62 RPs with double deletions in combinations across samples.
Overall, 8,910 (82.2%) tumors had single/double deletion of one or more of the 78 RP genes in the TCGA dataset. Individual RP genes had single/double deletion in 392–4,013 tumors (Supplementary Figure S14ab), and up to 55 RPs had single/double deletions in the same sample (Supplementary Figure S14c). Single/double deletions of RP genes were abundant in all 33 cancer types (Supplementary Figure S14d) but were more frequent in some cancer types than others (Supplementary Figure S15, Table S1g). OV was at one extreme, with over 99% of tumors showing single/double deletion of some RP genes, and LAML was at the other extreme, with <20% of tumors showing single/double deletion of some RP genes. Highly recurrent single/double deletion of a RP gene was quite common (Supplementary Figure S15, Table S1g), and there were even examples of RP genes single/double deleted in over 85% of tumors in a given cancer type, such as RPS15 in OV, RPL29 in LUSC, RPS24 in GBM, RPL17 in READ; and RPL14, RPL15, RPL29, RPL32, RPSA in KIRC.
In most cancer types, there was no significant association between the number of double-deleted RP genes in tumors and patient survival (Supplementary Figure S16), showing that RP loss did not reduce tumor fitness.
CRISPR–Cas9 knockout of RPs shows that loss of some RPs does not affect cell viability in several human tumor-derived cell lines
Data from CRISPR–Cas9 screens across 558 cancer cell lines (https://depmap.org) were analyzed for ‘Essentiality’ of the 74 RP genes for which data was available. ‘Essentiality’ of a gene was estimated using CERES (52), which estimates gene-Dependency Scores (gDS) (53,54), after accounting for copy number variation, using sgRNA abundance in a reference pool. ‘Strictly essential genes’, whose deletion severely affects cell viability, have gDS < −1. ‘Strictly non-essential genes’, whose deletion has no effect on cell viability, have gDS > 0. Genes with gDS between −1 and 0 are ‘potentially non-essential’, i.e. their deletion affects cell viability to some degree but is not lethal. Since the cell lines spanned cancers from over 25 tissue types, this data represents RP single-knockout effects across cellular/tissue contexts.
If all RPs are essential, their gDS should always be <−1. Instead, 73 of the 74 RPs (all except RPS20), had gDS >−1 in one or more cell lines (Figure 6A, B; Supplementary Table S1h). Every cell-line, irrespective of the cancer type, had some RP genes with gDS >−1, and the minimum/maximum number of RPs with gDS >−1 in a single cell line varied from 21 to 62, with a mode of 35 (Figure 6C). RP genes with gDS >−1 were abundant in every cancer type, as between 51 and 68 RPs had gDS >−1 in different cancer types (Figure 6D). Supplementary Figure S17 and Table S1i show the frequency (%) of gDS >−1 for each RP (shown in rows) in various cancer types (shown in columns). Unlike the corresponding data for single/double deletion (Supplementary Figure S15, Table S1g) there was very little inter-cancer variation for a given RP. Many RPs (the top ∼40% in Supplementary Figure S17) frequently have gDS >−1 independent of cancer type, showing that these RP deletions are tolerated in many different cell lines across cancer types. Conversely, many RPs (the bottom ∼45% in Supplementary Figure S17) rarely had gDS >−1, independent of cancer type, showing that these RPs are essential for growth in many different cell lines across cancer types. These differences in the essentiality of RP genes in cell lines versus RP copy number loss in tumors is in accordance with the tissue-specific clustering seen in tumors but not seen in cell lines.
Figure 6.

Essentiality of RP Genes under CRISPR-Cas9 knockout in 558 cancer cell lines. CRISPR–Cas9 essentiality screen data for 74 RP genes for 558 cancer cell lines using the computational tool CERES (52). Genes with a dependency score (53,54) (gDS) greater than −1 are considered potentially non-essential. (A) Number of cell lines with gDS >−1 for each RP. Seven RPs had gDS > −1 in every cell line and 73 RPs had gDS >−1 in at least one cell line. (B) gDS profile of the RP genes in cell lines. Blue indicates gDS >−1 in the sample. (C) Distribution of number of RP genes with gDS >−1 across cell lines. Every cell line had at least 21 RP genes with gDS >−1. While 35 RPs with gDS > −1 was the most common, as many as 62 RP genes had gDS >−1 in a gastric cancer cell line (ACH-000167). (D) Number of RPs with gDS >−1 for cell lines stratified by cancer type.
Forty-four RPs were strictly non-essential (gDS > 0) in at least one cell line. Five RPs: RPL21, RPL22, RPL35A, RPS10, RPS26, were strictly non-essential in more than 25 cell lines, and RPL22 and RPL21 were strictly non-essential in 223 and 444 cell lines respectively (Supplementary Figure S18a, b; Table S1h). While most cell lines had only 0–5 RPs with gDS > 0, 43 RPs were strictly non-essential in one gastric cancer cell line (Supplementary Figure S18c), and non-essentiality of RPs was found in cell lines from all cancer types (Supplementary Figure S18d). There was no correlation/clustering of RP gDS values and tissue of origin (data not shown), indicating that the role of RPs in survival of cell lines does not depend on tissue of origin.
Ribosome profiling and RNA-Seq data for RPs are highly correlated in human and rodent tissues and cell cultures
To address whether tissue-specific RP mRNA level differences, which represent transcribed but not translated pools of RP mRNA, actually correspond to the levels at which these mRNA pools are translated in ribosomes, we analyzed mRNA expression data from RNA-Seq, which represents transcription levels, and ribosome profiling data, which represents levels at which these transcripts are being translated in the ribosome, in matched samples across a variety of tissues and cell cultures for human (17–20), mouse (17,21,22) and rat (23) (details in Materials and Methods). Each of these datasets were normalized using the protocol in Supplementary Figure S1a.
Scatter plots of normalized translation levels of RP genes from ribosome profiling versus mRNA expression levels of RP genes from RNA-seq for the same cells/tissues for human, mouse and rat are shown in Figure 7A–G. They show highly consistent covariation of translation level of RP genes and their mRNA expression levels in every tissue/cell-type. We also see (Figure 7H) strong, consistent and statistically significant (P < 10−4) correlation between mRNA expression level and translation level of RP genes in every sample. These results show that the ribosome is translating RP transcripts at levels proportional to their mRNA levels.
Figure 7.

Comparison of mRNA levels (RNA-Seq) and ribosome profiling levels for RP genes. (A–G) Scatter plot of matched mRNA (measured by RNA-seq) and ribosome profiling levels (showing level of transcripts being translated) for RP genes in various tissues/cell-types. Median value is shown for tissue/cell-type with multiple samples. The axes are in log2 scale. RPF = ribosome protected fragment, hES = human embryonic stem cells grown in a feeder free protocol that maintains pluripotency, NDx = human embryonic stem cell grown for x days in a neural conversion protocol that induces cell differentiation. (H) Spearman Rank Correlation of mRNA and ribosome profiling levels of RP genes for paired samples shows highly consistent covariation and highly significant correlation (P < 10−4) of mRNA and ribosome profiling levels for RP genes in each individual sample.
Furthermore, the translated RP transcripts are also tissue (Supplementary Figure S19a–d), developmental-stage (Supplementary Figure S19e–g) and environment specific (Supplementary Figure S19h). As expected from Figure 7, the ribosome profiling results shown in Supplementary Figure S19 are in strong agreement with results from RP mRNA levels (Supplementary Figure S20) of the corresponding matched samples. The mRNA expression levels of RP genes in these samples are also tissue-specific (Supplementary Figure S20a–d), and developmental-stage specific (Supplementary Figure S20e–g). Note that the RNA-Seq plot corresponding to Supplementary Figure S19h is missing because RNA-seq data was available only for untreated control samples.
RP protein abundance levels in humans are developmental-stage and tissue-specific
To test whether the tissue specificity of RPs at the mRNA level is also seen at the protein level, protein expression data for 77 RP genes in 17 adult tissues, 7 fetal tissues and 6 purified primary haematopoietic cells from histologically normal human samples were obtained from a recent study (24). The data was log-transformed and standardized (z-score) to eliminate global variation in overall RP protein levels among tissues. Principal component analysis (PCA) of the data cleanly distinguished fetal from adult tissues (Figure 8A). Consistently, the same tissue type had different RP protein signatures in adult and fetal tissue (Figure 8B). Using the Spearman Rho rank correlation S as a measure of similarity, the pairwise difference in RP protein signatures were compared among tissues. If the protein composition of ribosome were invariant, 1-S should be near zero for all tissue-pairs. Instead, this quantity was substantially different from zero for several tissue-pairs (Figure 8C). Consistently, both adult tissue-pairs and fetal tissue-pairs showed differential RP protein signatures (Figure 8D). These results suggest that, similar to RP translation levels measured by ribosome profiling, RP protein levels are also developmental-stage (adult versus fetus) and tissue type specific.
Figure 8.

RP protein levels in humans are developmental-stage and tissue type specific. (A) Principal component analysis (PCA) of log-transformed and standardized human RP protein levels for 30 histologically normal human samples cleanly distinguished fetal tissues (green) from adult tissues (blue/red for solid/liquid tissues). (B) Tissues had differential RP protein levels for liver and heart in adult vs fetus in humans. Dotted lines are |zx – zy| = 1, and labeled RPs have |zx – zy| > 1.25. (C) Heatmap of 1 – S, where S = Spearman Rho rank correlation, shows that although all pairs of tissues are significantly correlated (S > 0.4, P < 0.001), they are far from identical (S = 1) and can be substantially different (e.g. some tissue-pairs have 1 – S > 0.5). (D) Adult and fetal tissue from different organs show differential RP protein levels. Dotted lines are |zx – zy| = 1, and labeled RPs have |zx – zy| > 1.25.
DISCUSSION
We analyzed a variety of large datasets using several analytical methods to study the heterogeneity of mRNA expression levels and copy number variations of ribosomal proteins (RPs) in normal human tissues, cancer samples and cell lines. Our overall conclusion from this analysis, as well as from CRISPR-Cas9 knockout of RPs in human cancer cell lines, protein abundance of RPs in normal human tissues, matched ribosome profiling and mRNA data of RPs for human, mouse and rat tissues and cell cultures, is that transcriptomic, translatomic, and proteomic levels of RPs are highly variable, with strong and consistent tissue, environment, and development-stage–specific signatures. In six cancer types, there are clusters of samples with distinct RP mRNA signatures associated with differential patient survival. Finally, double copy losses of RPs in tumors, and CRISPR-Cas9 knockout of several RPs in cell lines, do not lead to loss of ribosome function.
The major question that arises from our analysis is whether the human ribosome must have the same protein composition in all cells and tissues under all conditions to be able to function? One possible but speculative and unproven explanation of our results is that ribosomal composition is variable, and depends on tissue/cell, environment, and development stage. We note that although this is the simplest explanation of our results, this hypothesis needs to be experimentally validated.
We discuss our findings below in greater detail, note their significance and shortcomings, carefully separate facts from speculation, and provide alternative explanations of our results where possible.
Fact I: TCGA CNV data showed that double deletion of each of the 78 non-sex-specific RP genes is found in one or more tumors (Figure 5A and B). Nearly 12 percent of tumors had double deletion of an RP gene while retaining ribosome functionality. Double deletions of up to 16 RPs were tolerated in the same tumor (Figure 5C), with RPL13 deleted in 159 tumor samples across various cancer types (Figure 5A and B). Furthermore, there was no correlation between number of double deleted RPs in tumors and patient survival (Supplementary Figure S16) in most cancer types, suggesting that ribosome function is not compromised by such losses of RP genes.
Speculation I: These copy number variations would suggest that at least in tumors, the ribosome can function without all its RP components, which would suggest that the ribosomal protein stoichiometry in tumors is not necessarily 1:1:1… for all RPs. An alternative explanation of these results is that the TCGA CNV data is just incorrect. This seems highly unlikely since the TCGA CNV data used in this study was inferred from Affymetrix Genome-Wide Human SNP6 Array by Broad Institute (http://gdac.broadinstitute.org) using GISTIC2 (29), a method whose accuracy has been tested and validated in real and simulated datasets (29). Although it is possible that unlike other genes RPs present some unique challenges in inferring CNV, we cannot imagine why this would be the case. Another possible explanation is that even if the RP gene is indeed deleted, the missing RP gene would be replaced by a parolog (6) or pseudogene and the protein stoichiometry of the ribosome remains 1:1:1… for all RPs.
Fact II: CRISPR–Cas9 knockout data for RPs in 558 cancer cell lines showed that only RPS20 was strictly essential in these cell lines, only 22 RPs were strictly essential in more than 500 cell lines, and only 39 were strictly essential in most cell lines (Figure 6, Supplementary Table S1h). RPL21 and RPL22 were strictly non-essential (no loss in viability or growth of cell line from knockout) in 444 and 223 cell lines respectively. These results show that not all RPs are essential in cancer cell lines, and several RPs can be knocked out without complete loss of ribosome function.
Speculation II: These results suggest that ribosome function is retained in many tumor-derived cell lines after CRISPR single knockout of several RPs. However, there are many off target effects in CRISPR knockout studies (55), so it is possible that the signal we see may be misleading. A repeat CRISPR knockout experiment along the same lines with RP genes as the only target would be a valuable test. However, such off-target activity depends on guide RNA sequences and experimental conditions. The guide RNAs used in the CRISPR knockout screens used in the study from where the data were derived were designed to minimize off-target activity (56), and the inferred dependency data was quality controlled using non-targeting controls and bench marked against a gold-standard set of core essential and non-essential genes (57). Moreover, CERES analysis corrects for association between copy number effects and sgRNA scores improving false discovery rates associated with CRISPR screens (52).
Fact III: t-SNE (26), SOM (28), and UMAP (27) analysis of RP mRNA levels for 11,688 normal samples from GTEx (15) and 10,363 tumor samples from TCGA, normalized to eliminate overall (global) variation in total RP expression level among samples (Materials and Methods), showed that the samples clustered by tissue type (Figures 1 and 3, Supplementary Figures S2 and S8). Similar analysis of 675 tumor derived cell lines (16) showed no clustering by tissue. This also agrees with the CRISPR–Cas9 deletion screen results, where there was no correlation between RP essentiality and tissue type in cell lines.
Matrix factorization using the Onco-GPS-Map (30) showed that three RP factors (signatures) are necessary to capture variation among RP mRNA levels in normal blood and brain tissues, with tissue types within the brain stratifying into cerebellar and non-cerebellar associated tissues (Figure 2A–C), and 16 RP factors are necessary to capture variations in 53 normal tissue types (Supplementary Figures S3a, b and S4). Consistently, the three factors for blood and brain were linear combinations of the 16 factors (Supplementary Figure S3c). Our results validate and build on previously observed plasticity of RP mRNA expression in cancers (42,43) and suggest that each normal tissue has a specific RP mRNA signature, which is a combination of RP signatures across tissues.
Speculation IIIa: The lack of tissue-specific clustering in cell lines suggests a universal RP mRNA signature in cell lines, possibly because of adaptation to in-vitro culture conditions.
Speculation IIIb: Whereas the mRNA data shows clear tissue-specific RP signatures in normal and cancer tissues, RPs are known to have extra ribosomal functions (3). One might wonder whether the tissue-specific mRNA signatures noted here might reflect these other functions and not the heterogeneity of ribosomal composition. This is difficult to test without isolating ribosomes from different tissues and dissecting their RP content. However, a genomic deletion or knockout would affect both structural and extra-ribosomal functions of the RPs. The ability of cancer cell lines and tumor cells to survive in the absence of certain RP genes, while not ruling out the extra ribosomal effects of RPs, might be an indication of the existence and functional viability of ribosomes with altered compositions.
Fact IV: Tissue-specificity of RP mRNA signatures was observed at both the transcription level (RNA-seq data) and the translation level (ribosome profiling data). A comparison of RP translation data from ribosome profiling (i.e. data on which RP gene transcripts are actually being translated in the ribosome) and RP mRNA expression data in matched samples showed very strong correlation between the two (Figure 7, Supplementary Figures S19 and S20). This shows that the levels of RP transcripts being translated in the ribosome are proportional to their mRNA levels. Consistently, tissue and development stage specificity of RPs were observed not only at the mRNA expression (Supplementary Figure S20) and translation (Supplementary Figure S19) levels, but also at the protein levels in a small dataset (24) of normal human tissues (Figure 8).
Speculation IVa: The simplest conclusion from these data is that the relative amounts of RP proteins made from the mRNA are proportional to their mRNA levels and would reflect in the amounts of these RP proteins used when the ribosome is assembled. However, it is still possible that when the ribosome is actually assembled, it always uses the full complement of 80 RPs and its stoichiometry remains 1:1:1… Finally, RPs undergo post-translational modifications (PTM) which cannot be captured by ribosome profiling or even by many other protein-estimation methodologies. Such PTMs could buffer the RP variability seen in our study, or, possibly, further contribute to the heterogeneity of RP function (58).
Speculation IVb: Interestingly, actual tissues obtained from mouse (e.g. hippocampus, adipose) and corresponding cell cultures grown in the lab (e.g. hippocampal neuron culture, adipocyte culture) were cleanly distinguishable in both ribosome profiling (Supplementary Figure S19f–g) and mRNA expression (Supplementary Figure S20f–g) data of RP genes, suggesting that there is a difference in the amounts of RP mRNA that are transcribed and translated in ribosomes in actual tissues versus their corresponding cell cultures. This would suggest that the RP protein needs of cells in culture are different from those same cells in-vivo, which is an interesting fact in itself.
Fact V: Kidney cancer subtypes, which are known to arise from different cells of origin (39,40), had distinct RP mRNA and RP CNV signatures (Figure 3d, Supplementary Figure S9). Several cancer types had RP subtypes with distinct RP mRNA signatures (Figure 3). In six of these, the RP-subtypes exhibited differential patient survival (Figure 4a), and distinct molecular characteristics that mapped well onto known genomic, histologic, or transcriptomic subtypes for these cancers (45–49) (Supplementary Figure S11). In LGG, UVM, and BLCA, the majority of the difference in RP mRNA levels among the RP subtypes were due to copy number alteration of RP genes (Supplementary Figure S12). Significantly, the RP genes RPL5, RPL11, RPL22, RPS8 on chromosome 1p, and RPL13A, RPL18, RPL28, RPS5, RPS9, RPS11, RPS16, RPS19 on chromosome 19q were co-deleted in the better prognosis RP subtype in LGG.
Speculation Va: The fact that deletions of RP genes on chromosome 1p and chromosome 19q did not abrogate ribosome function in these LGG samples, again points to the ability of the ribosome to function with loss of some RPs. These results are important by themselves in the biology of these tumors. At a minimum, as previously reported (42,43), the RP subtype signatures are useful biomarkers of prognosis for current treatment methods.
Speculation Vb: If the ribosome can retain its function despite the loss of many structural proteins, perhaps there are many functionally equivalent but structurally distinct ribosome types, which can perform the job of translating mRNA into protein with almost equal efficiency and fidelity. Some evidence for this conclusion comes from the CNV and CRISPR deletion data, which show that deletion of both copies or knockout of RP genes does not abrogate ribosome function. If this view is correct then cells/tissues might be choosing a combination of the available ribosome types to optimize its functions in a dynamic manner.
Speculation VI: An interesting question that is beyond the scope of this paper is to understand the mechanisms that regulate ribosome structural heterogeneity in various cells, tissues, environments, and development stages. Since RPs are essential for early development (1) in complex eukaryotes, this regulation is probably established during embryogenesis. This is also consistent with our observation that RP protein levels are both tissue and development-stage–specific in normal human fetal and adult tissues.
Speculation VII: In most cancers, loss of both copies of some RPs seems to have no effect on survival (Supplementary Figure S16). This suggests that when tumors alter the genomic landscape to survive and grow, some ribosome types are lost, and the tumor compensates by using others, without losing the ability to translate the proteins it needs. A more interesting possibility is that cancers may deliberately alter the choice of which ribosome types they use. They might even invent novel ribosome types, by altering RP compositions to enhance translation speed or fidelity of proteins that they need. If this last possibility is true, then there may exist ribosome types that are in cancers but not in normal tissue. This would mean that in a given tissue, a RP gene may be essential for the tumor but redundant for the normal tissue. In such a situation, at least in principle, drugs similar to antibiotics, which target bacterial ribosomes, might be designed to target cancer specific ribosomes. Functional inhibition of tumor specific ribosomes by locally delivering such drugs may be a novel way to target cancer in a highly tissue-specific and potentially toxicity free manner.
Speculation VIII: While the existence of ‘structurally distinct ribosomes’ can be validated only upon isolation, and structural, biochemical, and cellular characterization of individual ribosomes, our study contributes to a growing body of evidence suggesting the possibility of ‘specialized ribosomes’ (3,58). Our findings also suggest the existence of a novel regulatory layer of control, which determines tissue and development-stage–specific ribosome compositions either by establishing them during embryogenesis and/or by adapting them to the dynamic needs of the tissue over time.
DECLARATION REGARDING DATA ACCESS
The data used in this paper was all derived from public sources and the links to these are included in the paper. We will make any result data and associated software generated by us in this paper available to the community on request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr W. Kimryn Rathmell and Dr Aguirre A. de Cubas for many helpful conversations. We also thank Venkatesh Deshpande and Sudheshna Vemula for analytical help. G.B. was partly supported by grants from M2GEN/ORIEN, DoD/ KRCP (KC180159) and NIH/NCI (1R01CA243547-01A1). He thanks Professors Pablo Tamayo and Jill Mesirov for their kind hospitality at UC San Diego during his sabbatical year 2019–2020.
Author contributions: A.P.: Idea development, all analysis of mRNA, CNV, ribosome profiling and protein data, all figures/tables, manuscript writing, editing, formatting. A.Y.: Idea development, analysis of CRISPR data, manuscript writing. HY: Analysis of mRNA data by GPS-Map. A. Singh: TuBA analysis. M.B.: Idea development, t-SNE, SOM analysis. M.L.: UMAP analysis. A. Schulz: t-SNE, UMAP analysis. T.K.: Assistance with analysis of GTEx mRNA data. S.D.: Idea development. H.K.: Idea development, TuBA analysis. S.G.: Idea development, interpretation of results. P.T.: Idea development, GPS-Map analysis. G.B.: Idea development, interpretation of results, manuscript writing, editing, formatting.
Contributor Information
Anshuman Panda, Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA.
Anupama Yadav, Center for Cancer Systems Biology (CCSB), Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Genetics, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA; Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Huwate Yeerna, Moores Cancer Center, University of California San Diego, La Jolla, CA 92103, USA.
Amartya Singh, Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA.
Michael Biehl, Bernoulli Institute for Mathematics, Computer Science and Artificial Intelligence, University of Groningen, Nijenborgh 9, NL-9747 AG Groningen, The Netherlands.
Markus Lux, Cognitive Interaction Technology (CITEC), Bielefeld University, Inspiration 1, D-33619 Bielefeld, Germany.
Alexander Schulz, Cognitive Interaction Technology (CITEC), Bielefeld University, Inspiration 1, D-33619 Bielefeld, Germany.
Tyler Klecha, Department of Molecular Biology and Biochemistry, Rutgers University, Piscataway, NJ,08854, USA.
Sebastian Doniach, Department of Applied Physics, Stanford University, Palo Alto, CA 94305, USA.
Hossein Khiabanian, Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA.
Shridar Ganesan, Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA.
Pablo Tamayo, Moores Cancer Center, University of California San Diego, La Jolla, CA 92103, USA; School of Medicine, University of California San Diego, La Jolla, CA 92093, USA.
Gyan Bhanot, Rutgers Cancer Institute of New Jersey, New Brunswick, NJ 08903, USA; Moores Cancer Center, University of California San Diego, La Jolla, CA 92103, USA; Department of Molecular Biology and Biochemistry, Rutgers University, Piscataway, NJ,08854, USA; Department of Physics and Astronomy, Rutgers University, Piscataway, NJ 08854, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A.P. is supported by a post-doctoral fellowship [DFHS18PPC022] from the New Jersey Commission on Cancer Research (NJCCR); A.Y. is supported by NHGRI [U01CA232161, U41HG001715, P50HG004233 to M.V.] at DFCI; S.G. is supported by grants from NCI [P30CA072720, R01CA202752]; DoD, Hugs For Brady and the Val Skinner Foundation; A.P., G.B., S.D. and M.B. thank the Aspen Center for Physics for their hospitality; P.T. and H.Y. are supported by NIH [U01-CA217885, P30-CA023100, R01-HG009285, R01-GM074024, R01-CA172513, U24-CA194107, U24-CA220341]; Many ideas for this work came from a summer study group at the Aspen Center for Physics, which is supported by National Science Foundation [PHY-1607611]; A. Schulz thanks BMBF Germany for support [01IS18053E] within the MechML project. Funding for open access charge: NCI [P30CA072720, R01CA202752]; DoD, Hugs For Brady and the Val Skinner Foundation.
Conflict of interest statement. S.G. has consulted for Foghorn Therapeutics, Roche, Foundation Medicine, Novartis, Inspirata and spouse is an employee of Merck.
REFERENCES
- 1. Luo H., Lin Y., Gao F., Zhang C.-T., Zhang R.. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2013; 42:D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brenner S., Jacob F., Meselson M.. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature. 1961; 190:576–581. [DOI] [PubMed] [Google Scholar]
- 3. Genuth N.R., Barna M.. The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell. 2018; 71:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kondrashov N., Pusic A., Stumpf C.R., Shimizu K., Hsieh A.C., Xue S., Ishijima J., Shiroishi T., Barna M.. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011; 145:383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi Z., Fujii K., Kovary K.M., Genuth N.R., Röst H.L., Teruel M.N., Barna M.. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs Genome-wide. Mol. Cell. 2017; 67:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Slavov N., Semrau S., Airoldi E., Budnik B., Oudenaarden A.V.. Differential stoichiometry among core ribosomal proteins. Cell Rep. 2015; 13:865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R. et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hillenmeyer M.E., Fung E., Wildenhain J., Pierce S.E., Hoon S., Lee W., Proctor M., Onge R.P.S., Tyers M., Koller D. et al.. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008; 320:362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yadav A., Radhakrishnan A., Panda A., Singh A., Sinha H., Bhanot G.. The modular adaptive ribosome. PLoS One. 2016; 11:e0166021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng Z., Mugler C.F., Keskin A., Hodapp S., Chan L.Y., Weis K., Mertins P., Regev A., Jovanovic M., Brar G.A.. Small and large ribosomal subunit deficiencies lead to distinct gene expression signatures that reflect cellular growth rate. Mol. Cell. 2019; 73:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sulima S.O., Hofman I.J.F., Keersmaecker K.D., Dinman J.D.. How ribosomes translate cancer. Cancer Discov. 2017; 7:1069–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Narla A., Ebert B.L.. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010; 115:3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farley K.I., Baserga S.J.. Probing the mechanisms underlying human diseases in making ribosomes. Biochem. Soc. Trans. 2016; 44:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parks M.M., Kurylo C.M., Dass R.A., Bojmar L., Lyden D., Vincent C.T., Blanchard S.C.. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv. 2018; 4:eaao0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N. et al.. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klijn C., Durinck S., Stawiski E.W., Haverty P.M., Jiang Z., Liu H., Degenhardt J., Mayba O., Gnad F., Liu J. et al.. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2014; 33:306–312. [DOI] [PubMed] [Google Scholar]
- 17. Eichhorn S.W., Guo H., McGeary S.E., Rodriguez-Mias R.A., Shin C., Baek D., Hsu S.-H., Ghoshal K., Villén J., Bartel D.P.. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell. 2014; 56:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Werner A., Iwasaki S., McGourty C.A., Medina-Ruiz S., Teerikorpi N., Fedrigo I., Ingolia N.T., Rape M.. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature. 2015; 525:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji Z., Song R., Regev A., Struhl K.. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife. 2015; 4:e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ji Z., Song R., Huang H., Regev A., Struhl K.. Transcriptome-scale RNase-footprinting of RNA-protein complexes. Nat. Biotechnol. 2016; 34:410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cho J., Yu N.-K., Choi J.-H., Sim S.-E., Kang S.J., Kwak C., Lee S.-W., Kim J.-I., Choi D.I., Kim V.N. et al.. Multiple repressive mechanisms in the hippocampus during memory formation. Science. 2015; 350:82–87. [DOI] [PubMed] [Google Scholar]
- 22. Reid D.W., Xu D., Chen P., Yang H., Sun L.. Integrative analyses of translatome and transcriptome reveal important translational controls in brown and white adipose regulated by microRNAs. Sci. Rep.-UK. 2017; 7:5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ori A., Toyama B.H., Harris M.S., Bock T., Iskar M., Bork P., Ingolia N.T., Hetzer M.W., Beck M.. Integrated transcriptome and proteome analyses reveal Organ-Specific proteome deterioration in old rats. Cell Syst. 2015; 1:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim M.-S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S. et al.. A draft map of the human proteome. Nature. 2014; 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ben-Shem A., Loubresse N.G.d., Melnikov S., Jenner L., Yusupova G., Yusupov M.. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011; 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 26. Maaten L.V.D., Hinton G.. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 27. McInnes L., Healy J., Melville J.. UMAP: uniform manifold approximation and projection for dimension reduction. 2018; arXiv:06 December 2018, preprint: not peer reviewedhttps://arxiv.org/abs/1802.03426. [Google Scholar]
- 28. Kohonen T. Self-organized formation of topologically correct feature maps. Biol. Cybern. 1982; 43:59–69. [Google Scholar]
- 29. Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G.. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011; 12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim J.W., Abudayyeh O.O., Yeerna H., Yeang C.-H., Stewart M., Jenkins R.W., Kitajima S., Konieczkowski D.J., Medetgul-Ernar K., Cavazos T. et al.. Decomposing oncogenic transcriptional signatures to generate maps of divergent cellular states. Cell Syst. 2017; 5:105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh A., Bhanot G., Khiabanian H.. TuBA: tunable biclustering algorithm reveals clinically relevant tumor transcriptional profiles in breast cancer. Gigascience. 2019; 8:giz064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaplan E.L., Meier P.. Nonparametric estimation from incomplete observations. J. Am. Statist. Assoc. 1958; 53:457–481. [Google Scholar]
- 34. Wu W.-S., Jiang Y.-X., Chang J.-W., Chu Y.-H., Chiu Y.-H., Tsao Y.-H., Nordling T.E.M., Tseng Y.-Y., Tseng J.T.. HRPDviewer: human ribosome profiling data viewer. Database. 2018; 2018:bay074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H., Yang L., Wang Y., Chen L., Li H., Xie Z.. RPFdb v2.0: an updated database for genome-wide information of translated mRNA generated from ribosome profiling. Nucleic Acids Res. 2018; 47:D230–D234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Creighton C.J., Morgan M., Gunaratne P.H., Wheeler D.A., Gibbs R.A., Robertson A.G., Chu A., Beroukhim R., Cibulskis K., Signoretti S. et al.. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. The Cancer Genome Atlas Research Network Linehan W.M., Spellman P.T., Ricketts C.J., Creighton C.J., Fei S.S., Davis C., Wheeler D.A., Murray B.A., Schmidt L. et al.. Comprehensive molecular characterization of papillary Renal-Cell carcinoma. N. Engl. J. Med. 2015; 374:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davis C.F., Ricketts C.J., Wang M., Yang L., Cherniack A.D., Shen H., Buhay C., Kang H., Kim S.C., Fahey C.C. et al.. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014; 26:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V. et al.. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018; 173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen F., Zhang Y., Bossé D., Lalani A.-K.A., Hakimi A.A., Hsieh J.J., Choueiri T.K., Gibbons D.L., Ittmann M., Creighton C.J.. Pan-urologic cancer genomic subtypes that transcend tissue of origin. Nat. Commun. 2017; 8:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V. et al.. An integrated TCGA Pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018; 173:400–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guimaraes J.C., Zavolan M.. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016; 17:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dolezal J.M., Dash A.P., Prochownik E.V.. Diagnostic and prognostic implications of ribosomal protein transcript expression patterns in human cancers. BMC Cancer. 2018; 18:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ceccarelli M., Barthel F.P., Malta T.M., Sabedot T.S., Salama S.R., Murray B.A., Morozova O., Newton Y., Radenbaugh A., Pagnotta S.M. et al.. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016; 164:550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. The Cancer Genome Atlas Network Akbani R., Akdemir K.C., Aksoy B.A., Albert M., Ally A., Amin S.B., Arachchi H., Arora A., Auman J.T. et al.. Genomic classification of cutaneous melanoma. Cell. 2015; 161:1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robertson A.G., Shih J., Yau C., Gibb E.A., Oba J., Mungall K.L., Hess J.M., Uzunangelov V., Walter V., Danilova L. et al.. Integrative analysis identifies four molecular and clinical subsets in Uveal melanoma. Cancer Cell. 2017; 32:204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R. et al.. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017; 171:540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. The Cancer Genome Atlas Research Network Abeshouse A., Ahn J., Akbani R., Ally A., Amin S., Andry C.D., Annala M., Aprikian A., Armenia J. et al.. The molecular taxonomy of primary prostate cancer. Cell. 2015; 163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S. et al.. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017; 49:1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown Kevin R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S. et al.. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015; 163:1515–1526. [DOI] [PubMed] [Google Scholar]
- 54. Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M. et al.. Defining a cancer dependency Map. Cell. 2017; 170:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D.. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. et al.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hart T., Brown K.R., Sircoulomb F., Rottapel R., Moffat J.. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol. 2014; 10:733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dinman J.D. Pathways to specialized ribosomes: the Brussels lecture. J. Mol. Biol. 2016; 428:2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.