

Abstract
R-loops are formed when replicative forks collide with the transcriptional machinery and can cause genomic instability. However, it is unclear how R-loops are regulated at transcription-replication conflict (TRC) sites and how replisome proteins are regulated to prevent R-loop formation or mediate R-loop tolerance. Here, we report that ATAD5, a PCNA unloader, plays dual functions to reduce R-loops both under normal and replication stress conditions. ATAD5 interacts with RNA helicases such as DDX1, DDX5, DDX21 and DHX9 and increases the abundance of these helicases at replication forks to facilitate R-loop resolution. Depletion of ATAD5 or ATAD5-interacting RNA helicases consistently increases R-loops during the S phase and reduces the replication rate, both of which are enhanced by replication stress. In addition to R-loop resolution, ATAD5 prevents the generation of new R-loops behind the replication forks by unloading PCNA which, otherwise, accumulates and persists on DNA, causing a collision with the transcription machinery. Depletion of ATAD5 reduces transcription rates due to PCNA accumulation. Consistent with the role of ATAD5 and RNA helicases in maintaining genomic integrity by regulating R-loops, the corresponding genes were mutated or downregulated in several human tumors.
INTRODUCTION
R-loops are reversible nucleic acid structures that feature an DNA–RNA hybrid and the resulting non-hybridized single-stranded DNA (ssDNA). Genome-wide analysis based on DNA–RNA immunoprecipitation (DRIP)-sequencing has shown that R-loops are enriched in promoters of actively transcribed genes and are a part of transcription terminators. R-loops play a key role in regulating gene expression at those genomic regions. In addition, R-loops are intermediates in many other cellular processes, including telomere maintenance, DNA replication and DNA repair (1,2).
R-loops temporarily form to regulate many aspects of cellular physiology. R-loop formation and resolution are regulated in at least two ways. First, RNA processing proteins limit R-loop formation by occupying RNA transcripts in order to reduce the chance of RNA invading DNA (3). Second, when R-loops have already formed, helicases unwind DNA–RNA hybrids or ribonucleases degrade the RNA, both of which remove the R-loop (4). Emerging evidence has shown that persistent R-loops make the genome vulnerable to DNA damage due to exposure of ssDNA regions and blockage of replication fork progression, leading to replication stress (5,6). Proper processing of R-loops during DNA replication and repair is therefore required to preserve genome integrity.
Transcription-replication conflicts (TRCs) interfere with DNA replication, resulting in potential threats to genome stability (1). Recent reports suggest that head-on TRCs facilitate R-loop formation while the DNA–RNA hybrid is resolved and consequently R-loop formation is reduced in co-directional TRCs (7,8). Recent reports showed that DNA–RNA hybrids, which spontaneously form independent of cell cycles stage, induce TRCs and R-loop-mediated genomic instability when the DNA–RNA hybrids are stabilized by a DNA–RNA hybrid binding protein (9) or when ATR/CHK1 DNA damage checkpoint pathway is deficient in cells (10). Additionally, the same report suggests that post-replicative ssDNA gaps and unrepaired DSBs accumulate DNA–RNA hybrids when the post‐replicative repair and DNA damage checkpoint pathways are deficient, respectively (10). Besides DNA damage response pathways, several DNA repair proteins, such as BRCA1, BRCA2 and FANCD2, are proposed to participate in R-loop resolution at TRC sites (4,11–14). However, it is not completely clear how these repair proteins prevent R-loop formation or promote R-loop turnover. There are likely effector proteins that are directly involved in R-loop tolerance at TRC sites. Sen1/Senataxin DNA/RNA helicase, which resolves R-loops at transcriptional pause sites, has been reported to promote fork progression across RNA polymerase II (RNAPII)-transcribed genes while moving together with forks (15–17). This role of Sen1/Senataxin at replication forks becomes apparent upon replication stress (17). TRCs and R-loops have been reported to each induce formation of the other (6–8). In addition, it has been shown that replication stress due to nucleotide depletion or DNA polymerase inhibition increases R-loop formation (7). However, it is not yet known how replication fork stalling increases TRCs to produce R-loops and how R-loops are removed from replication forks under normal and replication stress conditions.
DEAD-box RNA helicases play crucial roles in all steps of RNA metabolism (18). It has been recently reported that several DEAD-box RNA helicases, such as DDX1, DDX3, DDX5, DDX19, DDX21, DDX43 and DDX56, possess DNA–RNA hybrid unwinding activity in vitro (19–25). Most of these helicases have also been shown to resolve R-loops in cells (19,22,25,26). Depletion of DDX23 and DDX47 was also reported to increase R-loop levels in cells, although their DNA–RNA hybrid unwinding activity in vitro was not tested (27,28). In addition to DEAD-box RNA helicases, DHX9, a DEXH-box RNA helicase, was also reported to have a role in R-loop regulation through its DNA–RNA hybrid unwinding activity (29,30). Under replication stress, DDX19 and DDX47 were reported to function in R-loop resolution. DDX19 transiently re-localizes to the nucleus upon DNA damage in an ATR/Chk1-dependent manner to resolve R-loops (19). DDX47 processes long RNA transcripts in conjunction with heterogeneous ribonucleoprotein particle U protein under mild replication stress in a FANCD2-dependent pathway (27). However, it remains unclear whether DEAD/DEXH-box RNA helicases function at the replication fork and how they are recruited to and maintained at normal or stalled replication forks.
ATAD5, a mammalian homologue of yeast Elg1, is an AAA+ ATPase protein that forms an alternative replication factor C (RFC)-like complex (RLC) with RFC2-5 (31,32). ATAD5 is important for maintaining genomic stability and functions as a tumor suppressor based on tumor incidence in Atad5 heterozygote mutant mice and the high mutation frequency of ATAD5 gene in various human cancer patients (33,34). ATAD5 likely suppresses tumorigenesis by regulating PCNA, the eukaryotic sliding clamp for replicative polymerases, at replication forks. Specifically, ATAD5 unloads PCNA after DNA synthesis ends during normal DNA replication (32,35,36). ATAD5 also helps to deubiquitinate PCNA during the DNA damage tolerance pathway by recruiting the ubiquitin-specific protease 1 (USP1)/USP1 associated factor (UAF1) complex (37). ATAD5-depleted cells show characteristic features of replication stress, such as a slow replication rate (35). In addition, a recent study revealed that ATAD5 promotes replication fork restart under replication stress (38). Since replication stress has been reported to induce R-loop formation (7), we hypothesized that ATAD5-RLC may have a role in R-loop regulation. Here, we report that ATAD5 plays roles in both R-loop resolution and R-loop prevention. ATAD5/UAF1 increases the abundance of DEAD/DEXH-box RNA helicases at the replication forks by direct protein-protein interactions to resolve R-loops under normal and replication stress conditions, facilitating replication fork progression. In addition, PCNA unloading by ATAD5-RLC prevents the formation of new R-loops behind the replication forks by reducing the occurrence of collisions between PCNA remaining on DNA and transcription machinery.
MATERIALS AND METHODS
Cell lines and cell culture
Human embryonic kidney (HEK) 293T, HeLa, HeLa-FUCCI (fluorescent ubiquitination-based cell cycle indicator) and U2OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (GE Healthcare, Little Chalfont, UK), 100 U/ml penicillin G (Life Technologies, Carlsbad, CA, USA) and 100 μg/ml streptomycin (Life Technologies) at 5% CO2, 37°C. Transfection of plasmid DNA was performed using X-tremeGENE™ HP (Roche, Basel, Switzerland) or Lipofectamine 3000 (Invitrogen) and transfection of siRNAs, either synthetic duplexes or Dharmacon SMART pool (20 nM), was performed using RNAiMAX (Thermo Fisher Scientific) or the Dharmafect 1 transfection reagent (Dharmacon) according to the manufacturer's instructions. Cells were analyzed 48 h after transfection, unless otherwise specified in the text. To generate cell lines expressing ATAD5 in a doxycycline-inducible manner, a Lenti-X™ Tet-On® 3G inducible expression system was used following the manufacturer's protocol (Clontech Laboratories, Mountain View, CA, USA). Briefly, wild type or mutant ATAD5 cDNAs, either with a defect in UAF1 interaction or PCNA unloading (E1173K), were cloned into the pLVX-TRE3G-ZsGreen1 vector and viral particles were produced. U2OS cells expressing Tet3G were infected by viral particles and selected by puromycin to generate U2OS-TetOn-ATAD5 cell lines. To generate a cell line expressing RNaseH1-GFP in a doxycycline-inducible manner, FRT-TO-GFP-M27-RNAseH1 (wild type or D210N mutant) cDNA were cloned and stably transfected into a HeLa Flp-in cell line. U2OS-TetOn-ATAD5 and HeLa-TetOn-RNaseH1-GFP cell lines were cultured in tetracycline-free media. To induce protein expression, cells were treated with 100 ng/mL doxycycline 6 h after transfection of plasmid DNA or small interference RNAs (siRNAs) and incubated for 48 h before analysis.
Small interfering RNAs (siRNAs), reagents and antibodies
The following synthetic duplex siRNAs were purchased from Bioneer: ATAD5 3′ UTR (5′-GUAUAUUUCUCGAUGUACA-3′) (35), UAF1 (5′-AAUCAGCACAAGCAAGAUCCAUAUA-3′) (39), DDX5 (5′-CCGCAACCAUUGACGCCAU-3′) (40), DDX1 (5′-CACAAGCUGUGGAAGAGAU-3′) (Bioneer #SN-1653-1), DDX21 (5′-GCAUGUAUCUGCCUAUACUUU-3′) (22), DHX9 (5′-AAUAGCCGCCACCUCCUCUUCCCUG-3′) (41), and control siRNA (Bioneer #SN-1002). The following siRNAs were purchased from Dharmacon: si-ATAD5, si-FANCD2, and nontargeting siRNA Pool #1. The following drugs were used in this study: hydroxyurea, aphidicolin, cordycepin, indole-3-acetic acid (Sigma-Aldrich). The following antibodies were used: anti-PCNA (PC10), anti-UAF1 (E-4), anti-DDX1 (A-7), anti-DDX21 (D-8), anti-DHX9 (B-9), anti-RFC1 (B-5), anti-RFC3 (H-200) and anti-RFC4 (H-183) (Santa Cruz Biotechnology, Dallas, TX, USA); anti-p68 (DDX5, ab21696), anti-PCNA (ab18197) and anti-GFP (ab1218) (Abcam); anti–pCHK1 (S317) (Bethyl Laboratories, Montgomery, TX, USA); anti-FLAG (F7425) and anti-Actin (A3853) (Sigma-Aldrich); anti-histone H3 and anti-β actin (BA3R) (Thermo Fisher Scientific, Waltham, MA, USA); anti-ubiquityl-PCNA (Lys164) (D5C7P) (Cell signaling); anti-Myc (4A6) (Merck Millipore); anti-S9.6 (ENH001) (Kerafast); anti-pRPA2 S33 (Cedarlane) antibodies. The anti-human ATAD5 antibody was raised in rabbits against an N-terminal fragment (1–297 aa) (37).
Protein extraction, immunoprecipitation and immunoblot analysis
Isolation of a nuclear protein faction and a Triton X-100–insoluble fraction (chromatin-bound fraction), immunoprecipitation and immunoblot analysis were performed as previously described (37) with slight modifications. For nuclear protein extraction, cell pellets were resuspended in MBS-A buffer (10 mM HEPES (pH 7.5), 340 mM sucrose, 1.5 mM MgCl2, 10 mM KCl, 10% glycerol, 1 mM DTT, 0.1 M PMSF, phosphatase inhibitors and protease inhibitors) and incubated for 8 min at 4°C. After centrifugation, nuclei were further lysed in buffer X (100 mM Tris–HCl, 250 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1 M PMSF, phosphatase inhibitors and protease inhibitors) with Benzonase nuclease followed by sonication and centrifugation. For chromatin bound extraction, cell pellets were resuspended in Buffer A (100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 10 mM PIPES (pH 6.8), 1 mM EGTA, 0.2% Triton X-100, 0.1 M PMSF, phosphatase inhibitors and protease inhibitors) and incubated for 5 min at 4°C. After centrifugation, pellets were further lysed in RIPA buffer (150 mM NaCl, 50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% Na deoxycholate, 0.1 M PMSF, phosphatase inhibitors and protease inhibitors) with Benzonase nuclease for 45 min at 4°C, followed by sonication and centrifugation. For immunoblot analysis, proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The blot was incubated in Tris-buffered saline containing 0.1% Tween 20 supplemented with 5% skim milk and incubated with primary antibodies. After washing, the blot was incubated with horseradish peroxidase-conjugated secondary antibodies (Enzo Life Sciences, New York, NY, USA). The signal was detected using enhanced chemiluminescence reagent (Thermo Fisher Scientific) with an automated imaging system (ChemiDoc™; Bio-Rad Laboratories, Hercules, CA, USA).
Protein purification
Proteins were purified as described previously (32). UAF1 and ATAD5 (N400) were prepared using the Bac-to-Bac Baculovirus expression system (Thermo Fisher Scientific). Viral particles were prepared using Sf9 cells (Thermo Fisher Scientific) and proteins were expressed in Hi-5 cells (Thermo Fisher Scientific). UAF1 was N-terminally 10xHIS tagged. For ATAD5 (N400), an additional MBP tag was inserted between the N-terminal 10xHIS tag and ATAD5 (N400) to increase stability and solubility. To purify proteins, lysates were obtained and clarified by sonication and ultracentrifugation (36 000×g, 60 min) using cell lysis buffer (25 mM HEPES (pH 7.5), 300 mM potassium acetate (KOAc), 1 mM EDTA, 1 mM EGTA, 2.5 mM magnesium acetate, 10% glycerol, 1 mM ATP and 0.02% NP40) with 1× complete protease inhibitor cocktail (Roche). Proteins were subjected to sequential chromatography using complete His-Tag resin, anti-FLAG M2 agarose resin, and ion exchange chromatography. Purified proteins were frozen and stored at −80°C. The purified human recombinant MYC/FLAG-tagged DDX5 protein was purchased from Origene (TP300371).
In vitro pull-down assay
An anti-c-MYC agarose affinity gel antibody produced in rabbit (Millipore) was used for pull-down of MYC-tagged DDX5. Anti-c-MYC agarose bead was incubated with 0.2% BSA in PBS for 1 h at 4°C. Bead was washed three times with protein dilution buffer (25 mM HEPES (pH 7.5), 100 mM KoAc, 1 mM EDTA, 1 mM EGTA, 2.5 mM magnesium acetate, 10% glycerol, 1 mM ATP, and 0.02% NP40). DDX5 (1 pmol) was incubated with anti-c-MYC bead for 30 min at 4°C. Unbound proteins were washed with protein dilution buffer. Proteins of interest (1 pmol) were added to DDX5-conjugated beads and incubated for 30 min at 4°C. After washing, bead-bound proteins were collected by denaturation with 2× SDS loading buffer and resolved by SDS-PAGE and western blotting.
Immunostaining, image acquisition, and image analysis
R-loop immunostaining with the S9.6 antibody using the spread method was performed as previously described (42) with slight modification. After trypsinization, cells were slowly mixed with pre-warmed 75 mM KCl in a drop-wise manner then incubated at 37°C for 12 min. Cells were then fixed using a cold fixative solution (methanol/acetic acid 3:1). After fixation, the cell suspension was spread onto a clean slide and exposed to 90°C steam for 30 s. Slides were dried at room temperature for 1 h. Cells were incubated with blocking buffer (5% bovine serum albumin (BSA) and 0.5% Triton X-100 in PBS) for 1 h and incubated with S9.6 antibody (1:1000) in blocking buffer overnight at 4°C. After washing with wash buffer (0.1% Triton X-100 in PBS), cells were incubated with mouse AlexaFluor 488-conjugated secondary antibody (1:500) in wash buffer for 1 h at room temperature. After washing, cells were mounted using ProLong® Gold antifade reagent (Vector Laboratories). Confocal images were acquired with an LSM880 confocal microscope (Carl Zeiss). Image acquisition and analysis were performed with ZEN2.1 software.
For S9.6 epistasis analysis between ATAD5 and FACND2 following the conventional method, cells were washed with PBS, fixed with ice-cold methanol for 10 min, and then permeabilized with ice-cold acetone for 1 min. After a PBS wash, cells were blocked in 3% BSA, 0.1% Tween 20 in a 4x saline sodium citrate buffer (SSC) for 1 h at room temperature. Cells were then incubated with primary antibody S9.6 (1:500) overnight at 4°C. Cells were then washed three times in PBS and stained with mouse AlexaFluoro-568-conjugated secondary antibody (1:1000) (Life Technologies) for 1 h at room temperature, washed three times in PBS, and stained with DAPI for 5 min. Cells were imaged on LeicaDMI8 microscope at 100×. ImageJ was used for processing and quantifying nuclear S9.6 intensity in images.
Proximity ligation assay (PLA)
For the S9.6-PCNA PLA, cells were incubated in CSK buffer (10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA and 0.5% Triton X-100™) for 10 min at 4°C, fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature, blocked with 10% fetal bovine serum in PBS for 1 h at 37°C in a humidity chamber. For the RNAPII-PCNA PLA, cells grown on coverslips were washed with PBS, fixed with 4% PFA for 15 min and permeabilized with 0.2% Triton X-100 for 5 min. Cells were then blocked in 3% BSA and 0.1% Tween 20 in 4× SSC for 1 h at room temperature. After blocking, cells were incubated with primary antibody overnight at 4°C (1:250 mouse anti-S9.6 antibody (ENH001, Kerafast) and 1:250 rabbit anti-PCNA antibody (ab18197, Abcam) for S9.6-PCNA PLA; 1:500 goat anti-RNA polymerase II antibody (PLA0292, Sigma) and 1:500 rabbit anti-PCNA antibody (PLA0079, Sigma) for RNAPII-PCNA PLA). The next day, after washing with 1× PBS twice, cells were incubated with pre-mixed Duolink PLA plus and minus probes for 1 h at 37°C. The subsequent steps in proximal ligation assay were carried out using the Duolink® PLA Fluorescence kit (Sigma) according to the manufacturer's instructions. Slides were then stained with DAPI and imaged on a Zeiss LSM880 confocal microscope for S9.6-PCNA PLA and Zeiss LeicaDM18 microscope for RNAPII-PCNA PLA.
Click chemistry-based detection of nascent nucleic acid synthesis
For detecting nascent DNA synthesis, cells were labeled with 10 μM 5′-ethynyl deoxyuridine (EdU) for 30 min before harvesting and processed using the Click-iT® EdU flow cytometry assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions. In brief, cells were fixed, permeabilized and subjected to the click reaction. Cells were then incubated in PBS with 0.1 mg/ml RNase A for 1 h at 37°C and DNA was stained with 0.05 mg/ml propidium iodide. Flow cytometry was performed on a FACSVerse™ flow cytometer using the BD FACSuite™ software (BD Biosciences). Data analysis was performed by using the FlowJo software. For EU-Click analysis using microscopy, cells were fixed with 4% PFA for 15 min, permeabilized with 0.5% Triton X-100 for 5 min, and subjected to the click reaction. To inhibit transcription, cells were treated with 100 μM cordycepin (CORD) 2 h before fixation. Slides were then stained with DAPI and imaged on a Zeiss LSM880 confocal microscope.
Quantitative in situ analysis of protein interactions at DNA replication forks (SIRF) assay
The SIRF assay was performed as previously described with slight modification (43). Cells were plated on LabTek™ chamber slides (Thermo Fisher Scientific) and incubated with 100 μM EdU for 10 min. For replication stress conditions, EdU was removed and slides were washed two times with PBS before incubation in pre-warmed media with 2 mM HU for 3 h. After washing two times with PBS, cells were fixed with 4% PFA in PBS for 20 min at room temperature and permeabilized with 0.25% Triton X-100 in PBS for 30 min at room temperature. Cells were washed with PBS twice for 5 min each. After washing, the click reaction cocktail (2 mM CuSO4 (Copper sulfate), 20 μM biotin azide and 10 mM sodium ascorbate in PBS) was added to each chamber then incubated at room temperature for 1 h. Subsequent steps were carried out with the PLA methods described above using either mouse anti-biotin or rabbit anti-biotin antibodies in conjunction with the antibody for the protein of interest. Slides were stained with DAPI and imaged on a Zeiss LSM880 confocal microscope.
Isolation of protein on nascent DNA (iPOND) assay
The iPOND assay was performed as previously described (44) with slight modification. HEK293T cells were incubated with 20 μM EdU for 20 min. For a pulse-chase experiment, cells were then treated with 10 μM thymidine for 0, 15 and 30 min. Cells were subsequently fixed using 1% formaldehyde for 20 min at room temperature. The crosslinking reaction was quenched using 0.125 M glycine and cells were washed three times with PBS. Cells were incubated with 0.25% Triton X-100™ in PBS for 30 min at room temperature then pelleted. Permeabilization was stopped with 0.5% BSA in PBS. Cells were then pelleted again and washed with PBS. After centrifugation, cells were resuspended with the click reaction cocktail described in the SIRF assay methods section and incubated for 1 h at room temperature on a rotator. After centrifugation, the click reaction was stopped by resuspending cells in PBS containing 0.5% BSA. Cells were then pelleted and washed with PBS twice. Cells were resuspended in lysis buffer and sonicated. Lysates were cleared then incubated with streptavidin-agarose beads overnight at 4°C in the dark. The beads were washed once with lysis buffer, once with 1 M NaCl, then twice with lysis buffer. To elute proteins bound to nascent DNA, the 2× sodium dodecyl sulfate Laemmli sample buffer was added to packed beads (1:1, v/v). Samples were incubated at 95°C for 30 min, followed by immunoblotting.
Affinity purification-mass spectrometry analysis
HEK293T cells were transfected with Strep-tag II-ATAD5 cDNA. After 48 h, nuclear extracts were prepared, and lysates were incubated with Streptactin-sepharose beads (GE Healthcare). Bead-bound proteins were washed and eluted with 2.5 mM d-Desthiobiotin (Sigma-Aldrich) in buffer X. Proteins were separated by SDS-PAGE and then visualized by staining with Coomassie blue. For LC–MS/MS analyses, gel lanes were sliced into bands and processed as follows. Briefly, the acetylated protein bands were divided into 10-mm sections and in-gel digested with trypsin. Trypsin digest products were separated by online reversed-phase chromatography using a Thermo Scientific EASY-nLC 1200 UHPLC equipped with an autosampler, a reversed-phase peptide trap Acclaim PepMap™ 100 (75-μm inner diameter, 2-cm length), and a reversed-phase analytical column PepMap™ RSLC C18 (75-μm inner diameter, 15-cm length, 3-μm particle size), both from Thermo Scientific. Samples were then subjected to electrospray ionization at a flow rate of 300 nL·min−1. The chromatography system was coupled in-line with an Orbitrap Fusion Lumos Mass Spectrometer. Obtained spectra were screened against the UniProt human database using Proteome Discoverer Sorcerer 2.1 software with a SEQUEST-based search algorithm. The comparative analysis of proteins identified in this study was performed using Scaffold 4 Q+S. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD018207
Kaplan–Meier survival analysis and log rank test
Kaplan–Meier survival analysis is a nonparametric estimation made from incomplete observations (45). The survival analysis used the characteristic ‘vital_status’ to calculate survival probability. For patients undergoing treatment, we used ‘days_to_last_followup’ and did ‘right-censoring’ compared to ‘days_to_death’ for patients who died. A log rank test was performed to identify how two groups are significantly different. For analysis of the dependency of overall survival on functional mutations in genes of interest, patients were divided into two groups: those patients in which functional mutations were detected and were not detected based on the somatic point mutation data. For gene expression-based overall survival analysis, patients were divided into two groups, 33rd-percentile high and low expression levels, based on normalized TPM values. All overall survival analyses were conducted with the survival (2.44.1.1 version) and survminer (0.4.4 version) R packages.
Data and assessment
Somatic point mutations, gene expression, and clinical information data of TCGA were downloaded from the Broad Institute TCGA Genome Data Analysis Center (GDAC) (https://gdac.broadinstitute.org). The level three data (Stddata_2016_01_28 version) documenting patterns of mutations, call and rnaseqv2 (RSEM genes normalized), were used to identify mutation status and gene expression profiles of tumor samples. Level four data (Stddata_2016_01_28 version) documenting clinical information was used to extract ‘vital_status’, ‘days_to_death’, and ‘days_to_last_followup’ for overall survival analyses.
Statistical analysis
Prism 8 (GraphPad Software) was used to generate graphs and analyze data. For all data, two-tailed paired Student's t-test was used; **** P < 0.001, *** P < 0.005, ** P<0.01, * P < 0.05 and n.s: not significant. Statistical parameters are described in the figures and the figure legends.
RESULTS
ATAD5 depletion leads to the accumulation of DNA–RNA hybrids in S/G2/M cells
We first established a detection method for DNA–RNA hybrids with S9.6 antibody in HeLa cell lines expressing a nuclease deficient form of RNaseH1 (D210N) in a doxycycline-dependent manner (Supplementary Figure S1A and B). Inhibition of DNA topoisomerase I by camptothecin treatment increased S9.6 signal, as previously reported (46), which was colocalized with green fluorescence protein (GFP)-tagged RNaseH1 (D210N) (Figure 1A). Depletion of ATAD5 increased the intensity of S9.6 signal colocalized with RNaseH1 (D210N) (Figure 1A and B). Expression of wild type RNaseH1, which specifically degrades RNA in DNA–RNA hybrids, reduced the S9.6 signal increased by the ATAD5 depletion (Figure 1C), suggesting that ATAD5 regulates DNA–RNA hybrid levels.
Figure 1.

ATAD5 depletion increases DNA–RNA hybrids predominantly in non-G1 phases. (A, B) HeLa-TetOn-RNaseH1 (D210N)-GFP cells were transfected with ATAD5 siRNA for 48 h or treated with 1 μM camptothecin (CPT) for 3 h and immunostained with the S9.6 antibody. (A) Representative images of S9.6 immunostaining. (B) The intensity of S9.6 staining was quantified and plotted. (C) HeLa-TetOn-RNaseH1-GFP cells were transfected with Ctrl or ATAD5 siRNA, treated with doxycycline to express RNaseH1 for 48 h and immunostained with the S9.6 antibody. (D, E) HeLa-FUCCI cells were transfected with ATAD5 siRNA for 48 h, sorted into G1 and S/G2/M phases using a cell sorter, and immunostained with the S9.6 antibody. (D) Representative images of a FACS sorting profile. (F) HeLa cells were transfected with a combination of ATAD5 and FANCD2 siRNAs and immunostained with the S9.6 antibody. (B, C, E, F) Three independent experiments were performed and one representative result was displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; **** P < 0.001, *** P < 0.005, ** P < 0.01, * P < 0.05 and ns, not significant.
Since ATAD5 has been reported to act primarily during the S phase (35), we tested whether the regulation of DNA–RNA hybrids by ATAD5 is dependent on the cell cycle stage. We used HeLa cells expressing the fluorescent ubiquitination-based cell cycle indicator (FUCCI) (47). This indicator contains both CDT1 fused with a monomeric version of Kusabira Orange 2 (mKO2) and Gemenin fused with a monomeric version of Azami Green (mAG). The expression levels of these components vary during the cell cycle stage, allowing for identification of the cell cycle stage of individual cells. The HeLa-FUCCI cells were transfected with ATAD5 siRNA, sorted into G1 and S/G2/M phases by fluorescence activated cell sorting (FACS), and analyzed for DNA–RNA hybrid levels (Figure 1D). ATAD5 depletion increased S9.6 signal in the S/G2/M phases, but not in the G1 phase (Figure 1E). This is consistent with previous reports that ATAD5 depletion causes defects such as PCNA accumulation on DNA during the S and G2 phases (35). In Figure 1E, S9.6 signal was similar in control cells between G1 and S/G2/M phases. On the other hand, a recent report showed that S9.6 signal was higher in the S and G2 phase (10). This might be explained in part by differences in sample preparation and inclusion of nucleolar S9.6 signal in the analysis.
Recent studies revealed that Fanconi anemia proteins, such as FANCD2 and FANCM, act as mediators for suppression of R-loop formation at sites of TRCs (14). We therefore investigated whether ATAD5 and FANCD2 show epistasis for the regulation of DNA–RNA hybrids. Depletion of either ATAD5 or FANCD2 increased S9.6 signal and depletion of both proteins further increased S9.6 signal, suggesting that ATAD5 and FANCD2 work in different pathways (Figure 1F). Taken together, these results indicate that ATAD5 is a novel regulator that suppresses DNA–RNA hybrid formation predominantly outside of the G1 phase.
ATAD5 depletion induces collisions between accumulated PCNA and the transcription machinery
Transcription-replication conflicts (TRCs) occurring during the S phase are tightly linked to the R-loop formation and genomic instability (7,48). ATAD5 unloads PCNA from chromatin during DNA replication and releases replisome proteins, which allows for establishing new replication factories (32,35). Consequently, ATAD5 depletion leads to the accumulation of PCNA and PCNA-binding proteins for an extended time on DNA behind replication forks, which could also cause a collision with the active transcription machinery. Since the DNA–RNA hybrids increased predominantly at the non-G1 phases in ATAD5-depleted cells, we hypothesized that ATAD5 depletion increased either TRCs at the replication forks or a collision between accumulated PCNA and the transcription machinery behind the replication forks. We performed a proximity-ligation assay (PLA) between RNA polymerase II (RNAPII) and PCNA, which are essential proteins in transcription and replication machineries, respectively. The RNAPII-PCNA PLA signal was increased upon ATAD5 depletion (Figure 2A and B), which disappeared with RNaseH1 expression. This suggests that ATAD5 facilitates the resolution of TRCs or prevents a collision between accumulated PCNA and the transcription machinery. RNaseH1 has been reported to release RNAPII, which is trapped by the associated R-loops, from the chromatin (49). This might explain the disappearance of the RNAPII-PCNA PLA signal by RNaseH1 expression in either cases.
Figure 2.

ATAD5 depletion increases PCNA-RNAPII PLA signals and reduces transcription rate. (A, B) HeLa cells were transfected with ATAD5 siRNA and, after 24 h, transfected again with GFP cDNA or GFP-tagged RNaseH1 cDNA. 24 h later, cells were fixed and subjected to a proximity ligation assay (PLA) between PCNA and RNA polymerase II (RNAPII). (A) Representative images of PLA. (B) Number of PLA nuclear foci was counted and plotted. (C, D, G, H) HeLa cells were transfected with ATAD5 siRNA for 48 h before harvesting or fixation. To inhibit transcription, cells were exposed to 100 μM cordycepin (CORD) for 2 h before fixation. (E, F) HeLa-TetOn-RNaseH1-GFP cells were transfected with ATAD5 siRNA, treated with doxycycline (DOX) to express RNaseH1 for 48 h before fixation. (C–F) Cells were treated with 1 mM 5-ethynyluridine (5-EU) for 10 min before fixation and subjected to EU-click reactions. (C, E) Representative images of EU staining. (G, H) Fixed cells were subjected to S9.6 immunostaining. (G) Representative images. (B, D, F, H) Three independent experiments were performed and one representative result was displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; **** P < 0.001, *** P < 0.005, ** P < 0.01 and ns, not significant.
When collisions between the transcription machinery and the replication machinery or the accumulated PCNA occur due to defects in replication, which is expected in ATAD5-depleted cells, a reduction in transcription rate is expected. We tested this possibility by measuring 5-ethynyl uridine (EU) incorporation into newly synthesized RNA. When we measured EU intensity using immunostaining, ATAD5 depletion reduced EU signal (Figure 2C and D). Treatment with the transcription inhibitor, cordycepin caused almost complete disappearance of EU signals, confirming that such EU signal represents newly transcribed RNA. Interestingly, the reduced EU signal by ATAD5 depletion was not restored by RNaseH1 expression (Figure 2E and F), suggesting that collisions between the accumulated PCNA and the transcription machinery rather than R-loop itself or R-loop-associated TRCs is a major cause of the reduced transcription rate. Unexpectedly, less EU signal was observed in control cells expressing RNaseH1 (Figure 2F). The reduction might result from a defect in transcriptional termination by RNaeH1 expression (15). Additionally, we speculated that blocking transcription would reduce the S9.6 signal increased by ATAD5 depletion if the DNA–RNA hybrids are involved in TRCs or collisions between the transcription machinery and the accumulated PCNA. As expected, cordycepin treatment lowered the S9.6 signal that is increased by ATAD5 depletion (Figure 2G and H). Taken together, these data suggest that ATAD5 depletion increases transcription-associated DNA–RNA hybrids.
PCNA remaining on DNA after replication fork passage causes R-loop formation in ATAD5-depleted cells
The reduction of EU signal, which was not restored by RNaseH1 expression, in ATAD5-depleted cells suggests that accumulated PCNA interferes with transcription machinery (Figure 2C–F). A recent study of the DNA–RNA hybrid interactome had PCNA as a top hit (30); therefore, we hypothesized that R-loops could be formed near PCNA when transcription machinery collides with PCNA accumulated on DNA in ATAD5-depleted cells. To test the hypothesis, we measured the proximity between S9.6 and PCNA by PLA analysis. ATAD5 depletion dramatically increased the number of PLA foci between S9.6 and PCNA (Figure 3A and B). Notably, either transcription inhibition or RNaseH1 expression reduced the number of S9.6-PCNA PLA foci to basal levels. PCNA accumulated on DNA by ATAD5 depletion may increase the probability of proximity with existing R-loops. However, ATAD5 depletion did not increase PLA signal between PCNA and RFC4 (Figure 3C and D). This suggests that the accumulated PCNA caused by ATAD5 depletion does not always increase PLA signal with proximal proteins and that the enhancement of S9.6-PCNA PLA foci by ATAD5 depletion was likely due to increased R-loops. The increased PLA foci between S9.6 and PCNA can be also interpreted to indicate the proximity between PCNA present at the replication fork and the abundant R-loops in ATAD5-depleted cells. However, the PLA signal between S9.6 and RFC3 was not increased in ATAD5-depleted cells (Supplementary Figures S2A and S2B), suggesting that the increased proximity is specific to PCNA accumulated on DNA behind replication fork rather than at ongoing replication forks. Next, we performed a PLA recovery experiment using U2OS-TetOn-ATAD5 cell lines capable of doxycycline-induced expression of wild type ATAD5 (ATAD5WT), ATAD5 that is defective in PCNA-unloading (ATAD5E1173K), or ATAD5 that shows defective interactions with UAF1 (ATAD5ΔUAF1) (Figure 3E and (32)). ATAD5WT and ATAD5ΔUAF1, both PCNA-unloading competent, successfully reduced the number of PLA foci between S9.6 and PCNA caused by ATAD5 depletion. However, ATAD5E1173K, which is defective in PCNA unloading, failed to fully reduce the number of PLA foci to basal levels (Figure 3F and G). These data strongly suggest that R-loops are formed at regions where PCNA unloading did not properly occur upon ATAD5 depletion.
Figure 3.

PCNA remained on DNA by ATAD5 depletion generates R-loop signal. (A, B) After transfection and doxycycline treatment, HeLa-TetOn-RNaseH1-GFP cells were treated with 100 μM cordycepin for 2 h, fixed, and subjected to a PLA for S9.6 and PCNA. (A) Representative images. (B) Number of PLA nuclear foci was counted and plotted. (C, D) U2OS cells were transfected with ATAD5 siRNA for 48 h and fixed for a PLA between PCNA and RFC4. (C) Representative PLA images. (D) Number of PLA nuclear foci was counted and plotted. (E–G) U2OS-TetOn-ATAD5 cell lines were transfected with ATAD5 siRNAs and treated with doxycycline to express wild type ATAD5 (ATAD5WT), UAF1 interaction defective mutant ATAD5 (ATAD5ΔUAF1), or PCNA unloading defective mutant ATAD5 (ATAD5E1173K). After 48 h, chromatin-bound proteins were prepared for immunoblotting (E) or fixed for a PLA between S9.6 and PCNA (F, G). (F) Representative images of PLA. (G) Number of PLA nuclear foci was counted and plotted. (H–K) After siRNA transfection, cells were exposed to 2.5 mM thymidine for 18 h (G1/S boundary) and then released to fresh media for 5 h (S phase) before fixation. Asynchronous cells (Asyn) were also analyzed (H, I). (H–J) HeLa cells. (K) HeLa-TetOn-RNaseH1-GFP cells were transfected and treated with doxycycline for 48 h. Fixed cells were subjected to a S9.6-PCNA PLA (H, I) or EU-click analysis (J, K). (L) U2OS-TetOn-ATAD5 cell lines were transfected with ATAD5 siRNAs, treated with doxycycline to express ATAD5WT or ATAD5E1173K, and fixed for S9.6 immunostaining. (A–L) Three independent experiments were performed, and one representative result was displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; **** P < 0.001, *** P < 0.005, ** P<0.01, * P < 0.05 and n.s. not significant.
Since ATAD5 depletion increased R-loop signal during S/G2/M phases (Figure 1E), we examined whether PLA foci levels between S9.6 and PCNA increase during the S phase of ATAD5-depleted cells. We arrested cells at the G1/S boundary with a single thymidine block and simultaneously released cells into the S phase (Supplementary Figure S2C). The number of S9.6-PCNA PLA foci increased after ATAD5-depleted cells entered the S phase (Figure 3H and I). Consistently, the reduction in EU signal by ATAD5 depletion occurred only after cells enter S phase (Figure 3J). Similar to the EU analysis result using asynchronous cells (Figure 2F), RNaseH1 expression did not restore the EU signal reduced by ATAD5 depletion in cells both at the G1/S boundary and the S phase (Figure 3K), suggesting that the R-loop is not the reason for the reduced transcription rate. More EU signal was observed in cells at the S phase (Figure 3J and K), which would be due to transcription of ribosomal RNA genes being maximal in the S and G2 phases (50). Taken together, these results suggest that collisions of transcription machinery with PCNA remaining on DNA generate R-loops.
Next, we performed an R-loop recovery experiment using U2OS–TetOn–ATAD5 cell lines (Figure 3L). Expression of wild type ATAD5 repressed S9.6 intensity increased by ATAD5 depletion, indicating the effect is ATAD5-specific. ATAD5E1173K expression only partially repressed the S9.6 intensity increased by ATAD5 depletion, suggesting that the PCNA unloading activity of ATAD5 is important for preventing the R-loop formation.
ATAD5 interacts with DEAD-box RNA helicases and DHX9 through the UAF1 binding domain
ATAD5E1173K partially reduced R-loop levels increased by ATAD5 depletion, although ATAD5E1173K is less efficient than ATAD5WT (Figure 3L). This suggests that ATAD5 depletion increases R-loop formation due to defects in other processes in addition to PCNA unloading. To identify any additional mechanisms of R-loop regulation by ATAD5, we identified ATAD5-interacting proteins using affinity purification-mass spectrometry (Figure 4A). We found several candidate-interacting proteins and chose to focus on DEAD-box RNA helicases such as DDX1, DDX5, DDX21 and the DEXH-box RNA helicase DHX9 since recent studies showed DNA–RNA hybrid resolution by the RNA helicases (21,22,25,29). We confirmed the protein-protein interactions between ATAD5 and these four helicases using immunoprecipitation (Figure 4B). FLAG-tagged wild type ATAD5 interacted with endogenous helicases. Interestingly, ATAD5 that is defective in UAF1 binding diminished the interaction with the helicases (Figure 4B). UAF1 depletion also reduced interactions between ATAD5 and the four helicases tested (Figure 4C). These results suggest that ATAD5 interacts with the helicases through its UAF1 binding domain. We also checked for interactions between UAF1 and the helicases. FLAG-tagged UAF1 interacted with the endogenous helicases and ATAD5 depletion lowered their levels of interaction (Figure 4D). These results suggest that both ATAD5 and UAF1 cooperate in the binding of RNA helicases. Interactions among ATAD5, UAF1 and RNA helicases were also confirmed using reverse immunoprecipitation by pulling down FLAG-tagged DDX5. DDX5 co-immunoprecipitated with ATAD5, UAF1 and RNA helicases such as DHX9 and DDX21 (Figure 4E). These interactions were also confirmed by immunoprecipitation using endogenous proteins, as immunoprecipitation of ATAD5 pulled down UAF1 and RNA helicases such as DDX5, DDX21 and DHX9 (Figure 4F and G). It was difficult to detect endogenous ATAD5 and UAF1 in immunoprecipitates of DDX5. However, when UAF1 was ectopically expressed, ATAD5 was detected in immunoprecipitates of DDX5 (Figure 4H and I), again highlighting the importance of UAF1 in binding of helicases by ATAD5. We performed an in vitro pulldown assay with purified ATAD5 N-terminus (1–400 amino acids), UAF1, and DDX5. DDX5 interacted with UAF1 but failed to interact with the ATAD5 N-terminus (Figure 4J). UAF1 has been reported to interact with USP1 using N-terminal WD40 repeats and with FANCI or RAD51AP1 using the C-terminal SUMO-Like domain (SLD) (51,52). We found that UAF1 interacted with DDX5 using the N-terminal WD40 repeats but not the C-terminal SLD domain (Figure 4K). These results strongly suggest that UAF1 mediates interaction between ATAD5 and DDX5. Since UAF1 is important for interactions between ATAD5 and the RNA helicases, UAF1 depletion might affect R-loop levels. UAF1 depletion increased S9.6 signal in an RNaseH1 expression-sensitive manner (Figure 4L). Taken together, these results suggest that ATAD5 and UAF1 cooperate in the binding of DEAD-box RNA helicases and DHX9 to regulate R-loop levels.
Figure 4.

ATAD5 interacts with DEAD-box RNA helicases and DHX9. (A) HEK293T cells were transfected with strep-Tag II-ATAD5 cDNA. After 48 h, proteins were extracted and subjected to affinity purification-mass spectrometry analysis. The numbers of peptide hits for some selected proteins from the analysis were displayed. Proteins in red indicate DEAD-box RNA helicases and DHX9. (B–E, K) After transfection of HEK293T cells as indicated, nuclear extracts were prepared for immunoprecipitation (IP) with an anti-FLAG antibody. Immunoprecipitates were eluted, resolved by SDS-PAGE, and immunoblotted using the indicated antibodies. (F–I) Nuclear extracts from HEK293T cells were immunoprecipitated with anti-ATAD5 antibody (F, G) or anti-DDX5 antibody (H, I). (H, I) Cells were transfected with UAF1. (G, I) Relative band intensities were quantified and plotted, N=4. Statistical analysis: two-tailed paired Student's t-test; **** P < 0.001, **P < 0.01, *P < 0.05, ns, not significant. (J) Purified MYC-FLAG-DDX5, His-UAF1, and His-FLAG-ATAD5-N400 (ATAD5 1–400 aa) proteins were incubated as indicated and pulled down with an anti-MYC antibody. Purified FLAG-His-MBP-TALE protein was included as a negative control. (K) HEK293T cells were transfected with FLAG-tagged DDX5 and one of S-tagged UAF1 deletion mutants (S-UAF1WD40 and S-UAF1SLD). (L) HeLa-TetOn-RNaseH1-GFP cells were transfected with UAF1 siRNA, treated with doxycycline to express RNaseH1 for 48 h and immunostained with the S9.6 antibody. The intensity of S9.6 staining was quantified and plotted. Three independent experiments were performed, and one representative result was displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; * P < 0.05 and ns, not significant.
Depletion of ATAD5/UAF1-interacting RNA helicases increases R-loop formation in S/G2/M phases
The ATAD5-interactors DDX1, DDX5, DDX21 and DHX9 have been reported to resolve DNA–RNA hybrids and increase R-loop formation when depleted in cells (22,25,26,30). Since ATAD5 depletion increased R-loops in non-G1 phases (Figure 1E), we checked whether the depletion of ATAD5/UAF1-interacting RNA helicases affects R-loop formation in specific cell cycle phases. Depletion of these RNA helicases increased R-loop formation predominantly in non-G1 phases, similar to ATAD5 depletion, suggesting that RNA helicases interact with ATAD5 and UAF1 to resolve R-loops during S/G2/M phases (Figure 5A–H). Additionally, we found that simultaneous depletion of ATAD5 and DDX5 did not show any synergistic or additive effects on S9.6 signal intensity, suggesting that ATAD5 and DDX5 regulate R-loop resolution in the same pathway (Figure 5I and J).
Figure 5.

Depletion of ATAD5/UAF1-interacting RNA helicases increases R-loop formation predominantly at the non-G1 phases. (A–D) HeLa-FUCCI cells were transfected with siRNAs as indicated. After 48 h, cells were sorted into G1 and S/G2/M phases and subjected to S9.6 immunostaining. (A, C) Representative images. (B, D) The intensity of S9.6 staining was quantified and plotted. (E–H) Whole cell extracts from cells transfected with indicated siRNA were immunoblotted. (I, J) HeLa cells were transfected with indicated siRNAs and subjected to S9.6 immunostaining. (I) Representative images of S9.6 immunostaining. (J) The intensity of S9.6 staining was quantified and plotted. (B, D, J) Three independent experiments were performed, and one representative result is displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; **** P < 0.001, *** P < 0.005, ** P < 0.01, * P < 0.05 and ns, not significant.
We performed an in vitro DNA–RNA hybrid unwinding assay using a commercial purified DDX5 protein and a substrate of 6-carboxyfluorescein (FAM)-tagged DNA–RNA hybrids (Supplementary Figure S3A). We confirmed that DDX5 has DNA–RNA hybrid unwinding activity, as recently reported (Supplementary Figures S3B and S3C; (25)). Addition of ATPγS, a nonhydrolyzable ATP analog, blocked the DNA–RNA unwinding activity of DDX5, suggesting that DNA–RNA hybrid unwinding by DDX5 requires ATP hydrolysis (Supplementary Figure S3C).
ATAD5/UAF1-interacting RNA helicases are important for resolving R-loops under replication stress
We examined whether ATAD5/UAF1-helicase interactions are functionally important for R-loop resolution in cells. Depletion of ATAD5 and UAF1 increased S9.6 signal more than depleting only ATAD5 or UAF1 (Figure 6A). Consistently, ATAD5WT fully restored and ATAD5ΔUAF1 partially restored S9.6 intensity to basal levels (Figure 6B). ATAD5ΔUAF1 can unload PCNA (35) and consequently reduced S9.6-PCNA PLA signal increased by ATAD5 depletion (Figure 3F). Therefore, the partial decrease of S9.6 signal by ATAD5ΔUAF1 suggest that ATAD5/UAF1-helicase interactions are also important for R-loop regulation. The result is also consistent with other reports suggesting that DNA–RNA hybrids spontaneously form and acts as a blockade for replication fork progression unless they are properly resolved by the DNA/RNA helicase (9,10,17). In order to identify the specific conditions under which the ATAD5/UAF1-helicase interaction is additionally required, mild replication stress was triggered in cells by applying low doses of aphidicolin, a replicative polymerase inhibitor. Aphidicolin treatment increased R-loop signal, as reported (Figure 6C and Supplementary Figure S4A; (7)), which was lowered to the basal level when RNaseH1 was overexpressed (Supplementary Figure S4A) and was not observed in cells in the G1 phase (Figure 6D). These results strongly suggest that the S9.6 signal represents R-loop accumulated by replication stress. No changes in cell cycle profile by the low-dose aphidicolin ruled out the possibility that the increased R-loops result from an increased number of cells in the S phase (Supplementary Figure S4B). Under the replication stress conditions, expression of ATAD5WT but not ATAD5ΔUAF1 led to recovery of basal S9.6 signal (Figure 6C and D), suggesting that the interaction of ATAD5 with UAF1 is essential for R-loop regulation during replication stress. The S9.6 signal increased by aphidicolin treatment was further increased by ATAD5 depletion specifically at the S/G2/M phases, which also suggesting that the effects result from replication stress (Figure 6D). ATAD5E1173K partially recovered basal S9.6 signal under replication stress conditions (Figure 6E), similar to normal conditions (Figure 3L). The partial recovery of S9.6 intensity by ATAD5E1173K suggests that ATAD5E1173K, which can interact with UAF1, can reduce R-loops by restoring the abundance of the DEAD/DEXH box RNA helicases at the replication forks reduced by ATAD5 depletion. As expected, when UAF1 was depleted simultaneously, cells expressing ATAD5E1173K completely failed to restore S9.6 signal (Figure 6F). This data strongly suggests that ATAD5 suppresses R-loop accumulation at the replication forks through these two mechanisms; PCNA unloading and increase of the availability of DEAD/DEXH box RNA helicases at the replication forks.
Figure 6.

Depletion of ATAD5/UAF1-interacting RNA helicases increases R-loop formation under replication stress. (A) HeLa-TetOn-RNaseH1-GFP cells were transfected as indicated and subjected to S9.6 immunostaining. (B, C, E, F) U2OS-TetOn-ATAD5 cell lines were transfected with ATAD5 or UAF1 siRNAs and treated with doxycycline to express wild type or mutant ATAD5. After 48 h, cells were fixed for S9.6 immunostaining. (D) HeLa-FUCCI cells were transfected with a combination of ATAD5 siRNA and ATAD5 cDNA expression vector for 48 h, sorted into G1 and S/G2/M phases using a cell sorter, and immunostained with the S9.6 antibody. (G, H) HeLa cells were transfected with indicated siRNAs before fixation for S9.6 immunostaining. (C–E, G, H) Aphidicolin (APH) was added to cells at a concentration of 0.4 μM for 1 h before fixation. (A–H) The intensity of S9.6 staining was quantified and plotted. Three independent experiments were performed and one representative result was displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; **** P < 0.001, *** P < 0.005, **, P < 0.01, * P < 0.05 and ns, not significant.
Since interactions between ATAD5 and DEAD/DEXH-box RNA helicases are affected by UAF1 interaction (Figure 4B and C) and ATAD5 is more dependent on UAF1 interaction to regulate R-loops during replication stress (Figure 6C), we examined whether the requirement for RNA helicases in order to resolve R-loops is affected by replication stress. Aphidicolin treatment increased R-loop signal compared to untreated cells when ATAD5/UAF1-interacting RNA helicases were depleted (Figure 6G and H). Consistently, the increased S9.6 signal by the depletion of those RNA helicases was sensitive to RNaseH1 expression under both normal and replication stress conditions (Supplementary Figures S4C–F).
ATAD5 depletion reduces the protein level of DEAD/DEXH-box RNA helicases at replication forks under normal and replication stress conditions
ATAD5 primarily acts at replication forks during S phase. According to the published data from experiments using isolation of proteins on nascent DNA (iPOND)-mass spectrometry (53), the RNA helicases that interact with ATAD5 are also present at replication forks. We confirmed this using an iPOND-immunoblot assay combined with a 5-ethynyl-2-deoxyuridine (EdU)-thymidine pulse-chase. DDX1, DDX5, DDX21 and DHX9 were indeed enriched at ongoing replication forks (Figure 7A). When ATAD5 was depleted, the protein level of these helicases at the replication forks was reduced, suggesting that ATAD5 is required for recruitment or maintenance of the helicases at the replication forks (Figure 7B). The total chromatin-bound protein levels of the DEAD/DEXH-box helicases were not changed in ATAD5-depleted cells (Supplementary Figure S5A). This may be because only part of DEAD/DEXH-box helicases are regulated by ATAD5 and this regulation only occurs at certain replication forks during the S phase. It is also possible that the regulation might occur in only those replication forks that actually require R-loop resolution activity even in a temporary manner. This might explain why changes of DEAD/DEXH-box helicases were detected in iPOND experiments followed by a crosslink step but not in bulk chromatin fractionation and immunoblot.
Figure 7.
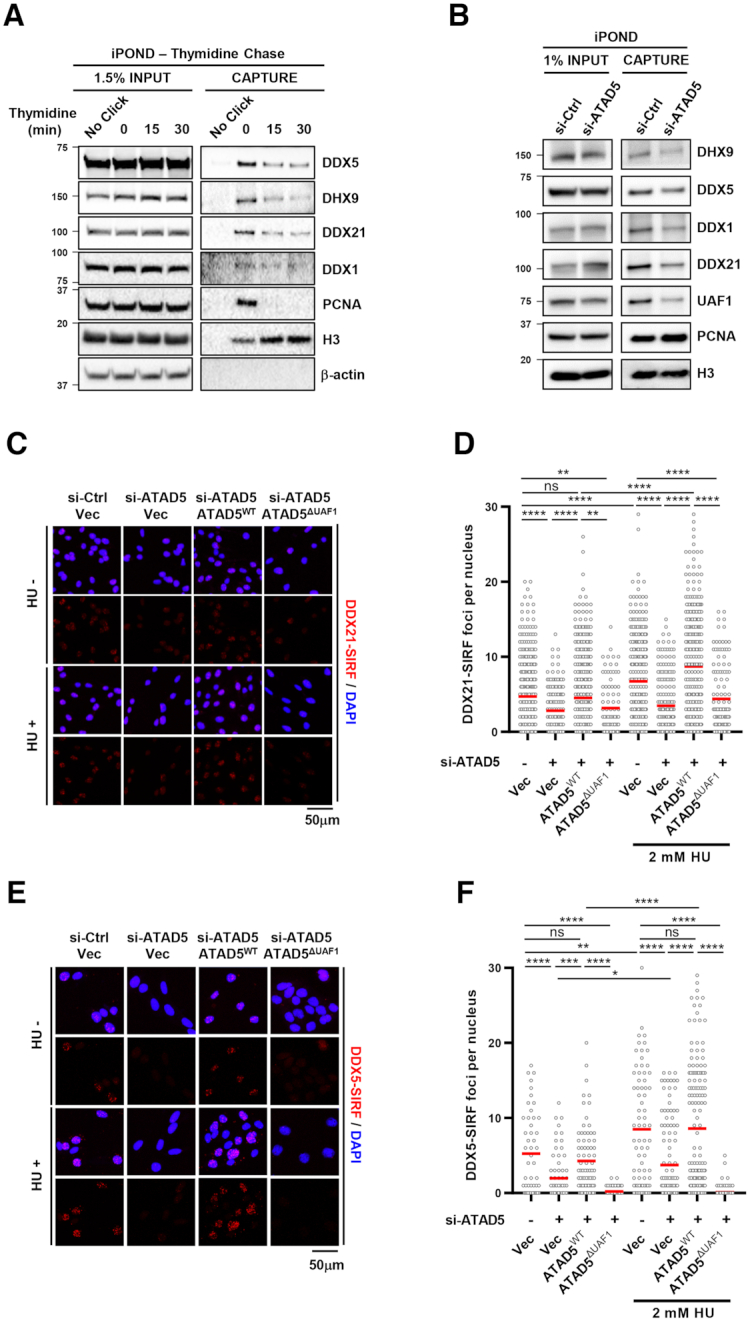
ATAD5 depletion reduces the association of RNA helicases with normal and stalled replication forks. (A) HEK293T cells were labeled with EdU for 20 min and then treated with 10 μM thymidine for indicated times. Whole cell extracts were prepared for an iPOND EdU-thymidine chase assay. (B) HEK293T cells were transfected with ATAD5 siRNA for 48 h then whole cell extracts were prepared for an iPOND assay. (C–F) U2OS-TetOn-ATAD5 cell lines were transfected with ATAD5 siRNAs and treated with doxycycline to express wild type ATAD5 (ATAD5WT) or UAF1 interaction defective mutant ATAD5 (ATAD5ΔUAF1). After 48 h, cells were fixed for a SIRF assay on DDX21 (C, D) or DDX5 (E, F). Hydroxyurea (HU) was added at a final concentration of 2 mM for 3 h before fixation. (C, E) Representative SIRF images. (D, F) Number of nuclear SIRF signal foci were counted and plotted. Three independent experiments were performed and one representative result is displayed. Red bar indicates mean value. Statistical analysis: two-tailed Student's t-test; **** P < 0.001, *** P < 0.005, **, P < 0.01, * P < 0.05 and ns, not significant.
Since replication stress increases R-loop formation (Supplementary Figure S4A; (7)), it is possible that replication stress also increases the abundance of DEAD-box RNA helicases and DHX9 at the replication forks. To test this possibility, we performed an assay called the quantitative in situ analysis of protein interactions at DNA replication forks (SIRF) assay, which examines the association of a protein and EdU-labeled nascent DNA at the single cell level (43,54). Replication stress was induced by hydroxyurea (HU) which depletes intracellular nucleotide pools. HU treatment increased R-loop signal, as reported, which was lowered to basal levels when RNaseH1 was overexpressed (Supplementary Figure S4A; (15)). In addition, a cell cycle profile was not changed by our HU treatment condition (Supplementary Figure S4B). The association of DDX21 or DDX5 with nascent DNA was increased by HU treatment, suggesting higher recruitment or retention of helicases at the replication forks under replication stress (Figure 7C-F). The specificity of the antibodies used for the SIRF assay was confirmed by disappearance of immunofluorescence signals with the corresponding siRNAs (Supplementary Figures S5B and C). HU-induced higher association of DDX21 or DDX5 with nascent DNA was reduced in ATAD5-depleted cells and was fully restored by expression of ATAD5WT, but not ATAD5ΔUAF1, under both normal and replication stress conditions. Taken together, the results suggest that the abundance of the RNA helicases at the replication forks is maintained in an ATAD5/UAF1-dependent manner under normal and replication stress conditions, which was reinforced by UAF1 under replication stress.
It is also possible that the association of ATAD5 with replication forks is dependent on the DEAD/DEXH-box helicases that interact with ATAD5/UAF1. To test the possibility, we performed the SIRF assay for ATAD5 after depletion of DDX1 or DDX21. Depletion of neither DDX1 nor DDX21 changed the ATAD5-SIRF signals while ATAD5 depletion significantly reduced the signals (Supplementary Figures S5D–F).
Depletion of DEAD/DEXH-box RNA helicases reduces replication fork progression under normal and replication stress conditions
ATAD5 depletion reduces replication rates (35). Since ATAD5/UAF1 is important for maintaining DEAD/DEXH-box RNA helicases at the replication fork, it is possible that less DNA–RNA hybrid unwinding activity at replication forks can hinder replication fork progression. Consistent with this hypothesis, depletion of ATAD5/UAF1-interacting RNA helicases reduced replication rates measured by EdU incorporation (Figure 8A and B). Low-dose aphidicolin further reduced replication rate in all cells tested. Importantly, expression of RNaseH1 restored the replication rates to normal levels (Figure 8C), suggesting that reduction of replication rates by DDX5 depletion is due to defects in R-loop resolution during fork progression. Depletion of any helicases did not show additive or synergistic effects with ATAD5 depletion on replication rates under normal conditions (Figure 8D and E). Moreover, ATAD5ΔUAF1 unable to interact with DDX1, DDX5, DDX21 and DHX9 RNA helicases could not fully restore the replication rate reduced by ATAD5 depletion (Figure 8F). Taken together, these results suggest that R-loop resolution by the ATAD5/UAF1-interacting RNA helicases is important for replication fork progression during both normal and replication stress conditions.
Figure 8.

Depletion of ATAD5/UAF1-interacting RNA helicases reduces DNA replication rate. (A, B, D, E) U2OS cells were transfected with indicated siRNAs. (A, B) Cells were pre-treated with 0.1 μM aphidicolin for 30 min before EdU labeling. (C) HeLa-TetOn-RNaseH1-GFP cells were transfected with DDX5 siRNA and treated with doxycycline to express RNaseH1. (F) U2OS-TetOn-ATAD5 cell lines were transfected with ATAD5 siRNAs and treated with doxycycline to express wild type ATAD5 (ATAD5WT) or UAF1 interaction defective mutant ATAD5 (ATAD5ΔUAF1). (A–F) After 48 h, 10 μM EdU was added for 30 min before fixation for EdU-click reactions and propidium iodide (PI) staining. (A) Representative FACS results. (B–F) The geometric mean of EdU signal was quantified and plotted. Statistical analysis: two-tailed paired Student's t-test; *** P < 0.005, **, P < 0.01, * P < 0.05. (B) N = 4. (C) N = 4. (D) N = 6. (E) N = 5. (F) N = 3.
Kaplan-Meier overall survival curves were dependent on the mutation or down-regulation of ATAD5/UAF1-interacting DEAD/DEXH-box RNA helicases
Given the known associations between ATAD5 and cancer, we analyzed data from The Cancer Genome Atlas (TCGA) to examine whether the mutation status or gene expression profiles of DEAD/DEXH-box RNA helicases DDX1, DDX5, DDX21 and DHX9 significantly correlate with cancer survival. For analysis, we organized the four helicases into Group I (Supplementary Figure S6A). Patients with head-neck squamous cell carcinomas (HNSC) and glioblastomas (GBM) that had at least one mutation in a DEAD/DEXH-box RNA helicase gene had shorter survival lengths compared to patients lacking mutations (Supplementary Figure S6B). Kaplan-Meier analysis indicated that patients with low expression of DDX1 or DDX21 in GBM and low-grade gliomas (LGG) had shorter survival lengths compared to patients with high expression of DDX1 or DDX21 (Supplementary Figure S6C). UAF1 expression was also low in patients with HNSC and LGG with short survival times. Taken together, the cancer survival analysis suggests a significant association between the mutation status or gene expression profiles of ATAD5/UAF1-interacting DEAD/DEXH-box RNA helicases and cancer patient survival. However, since ATAD5/UAF1-interacting RNA helicases play additional roles besides R-loop regulation, further studies will be needed to specifically correlate cancer patient survival with R-loop regulation by the ATAD5/UAF1-interacting RNA helicases.
DISCUSSION
R-loops possess dual opposing functions, as they are necessary for many normal cellular processes but threaten genomic stability upon dysregulation. To reduce unnecessary R-loops, cells use two major mechanisms: resolving R-loops and preventing new R-loop formation. In this study, we found that ATAD5 regulates R-loop levels at replication forks by both mechanisms (Figure 9). Under normal conditions (Figure 9, i), RNA helicases such as DDX1, DDX5, DDX21 and DHX9 migrate with replication forks in a process dependent on interactions with ATAD5 and UAF1. When replication forks encounter R-loops that already exist or are newly generated by the head-on TRCs in the genome, the RNA helicases resolve these R-loops and allow the replication forks to continue moving. Replication stress causes stalled replication forks, which can also result in TRCs and R-loop formation (Figure 6 and (1)). Under replication stress (Figure 9, ii), additional ATAD5/UAF1-interacting helicases are recruited to the replication forks, resolve the R-loops, and facilitate replication fork progression (Figure 8). The interactions between ATAD5/UAF1 and the RNA helicases are also important for increasing the abundance of the helicases at the replication forks under replication stress. The interactions might help recruit more helicases or their retention recruited by other mechanisms.
Figure 9.

Graphical model for R-loop regulation by ATAD5 and ATAD5/UAF1-interacting RNA helicases. (i) During normal replication, ATAD5-RLC and ATAD5/UAF1-interacting DNA/RNA helicases migrate with a replication fork. ATAD5-RLC unloads PCNA after Okazaki fragment synthesis is completed. ATAD5/UAF1-interacting DNA/RNA helicases resolve R-loops ahead of the replication fork and facilitate replication fork progression. (ii) Upon replication stress, stalled replication forks increase TRCs and unscheduled R-loop formation. In this case, additional ATAD5/UAF1-interacting DNA/RNA helicases are recruited to the replication fork and resolve R-loops to ensure faithful replication fork progression. In both normal and replication stress conditions (i and ii), the abundance of the helicases at the replication fork are dependent on ATAD5/UAF1 interaction. (iii) In ATAD5-depleted cells, the reduced R-loop resolution by ATAD5/UAF1-interacting DNA/RNA helicases leads to defects in replication fork progression. In addition, PCNA and its interacting proteins accumulated on DNA in the lagging strand behind forks collide with transcription machineries, which consequently increase R-loop formation at the collision site. (iv) In ATAD5ΔUAF1-complemented cells, abundance of the helicases decreases at the replication forks and consequently R-loops ahead of replication fork cannot be efficiently resolved in both normal and replication stress conditions. (v) In ATAD5E1173K-complemented cells, collisions between accumulated PCNA with its interacting proteins and transcription machinery increase R-loop formation. The abundance of ATAD5/UAF1-interacting DNA/RNA helicases are restored at the replication fork via the UAF1-bining domain of ATAD5E1173K, but they cannot resolve the newly formed R-loop due to spatial and temporal gaps between the replication fork and the collision site.
R-loops are present throughout the genome, predominantly near gene promoters. Accumulating evidence suggests that R-loops can be generated at sites of stalled RNA polymerase complexes and block DNA-based processes, promoting genomic instability (9,10,55–57). Head-on TRCs (7,8) have been also reported to facilitate R-loop formation. RNaseH1 expression increases replication rates or replication fork progression even without external replication stress (Figure 8C and (58)), suggesting that R-loops act as obstacles for efficient fork progression and that there is likely a mechanism for R-loop removal during replication fork movement (10,55). Sen1/Senataxin DNA/RNA helicase has been reported to move together with forks and promote fork progression across RNAPII-transcribed genes (17). Recently, two groups also reported that DDX19 and DDX47 RNA helicases resolve R-loops formed during replication stress (19,27). Here, using nascent DNA-associated protein analyses and cell cycle-based approaches, we provided strong evidence that the activity of the DEAD/DEHX-box RNA helicases actually occurs at DNA replication forks during S phase. We did not provide data to distinguish the DNA–RNA hybrids that were already present before the cells enter the S phase from the newly generated DNA–RNA hybrids at the S phase by head-on TRCs or replication stress. We propose that DNA–RNA hybrids formed at either the G1 or S phase would be resolved by DNA/RNA helicases including ATAD5/UAF1-interacting helicases during the S phase. Given the presence of R-loops throughout the genome, the activity of DEAD/DEXH-box helicases and Sen1/Senataxin helicases, which move with replication forks, seems to be required during general DNA replication to remove DNA–RNA hybrids that may become obstacles.
During replication stress, additional DDX5 and DDX21 proteins are recruited to replication forks (Figure 7C–F). Since these helicases interact with ATAD5, it is possible that the number of interactions between these helicases and ATAD5 would increase. However, there was no significant change in interactions between ATAD5/UAF1-interacting helicases and ATAD5 under replication stress (data not shown). Replication stress might generate a situation where more R-loop resolution activity is required. However, even in that case, the abundance of proteins at replication forks is dependent on the UAF1 interaction domain of ATAD5, and there appears to be a UAF1-dependent mechanism that increases the RNA helicases at replication forks under replication stress.
It has been reported that DEAD/DEXH-box RNA helicases function in complexes. DDX1, DDX21 and DHX36 form a complex with the adaptor molecule TRIF to detect double-stranded RNA in myeloid dendritic cells (59). Interactions between DDX5 and DHX9 have also been reported (44,60). In addition, ATAD5 interacts with RNA helicases including DDX1, DDX5, DDX21 and DHX9 and maintains their abundance at the replication forks through its UAF1 binding domain (Figures 4 and 7). Since DDX5 immunoprecipitates with DHX9 and DDX21 (Figure 4E and G), it is possible that at least these three helicases are recruited as a complex to single replication forks. However, depletion of any RNA helicases increases R-loop formation (Figure 5) and decreases replication rates (Figure 8). Therefore, it is possible that each RNA helicase is recruited to a different fork, or that the helicases serve different functions at stalled forks depending on context.
The 5′ to 3′ DNA–RNA hybrid unwinding activity is probably required to resolve intracellular R-loops. ATAD5-interacting RNA helicases, DDX1, DDX5, DDX21 and DHX9, have been reported to have the DNA–RNA hybrid unwinding activity in vitro (21,22,25,29). Among them, DDX1 has proven to have a 5′ to 3′ polarity. DHX9 has been reported to have a 3′ to 5′ polarity but the 5′ to 3′ polarity has not been tested yet. On the other hand, in the in vitro assay for DDX5 and DDX21, the substrates used were unable to identify polarity of the helicases. This also applies to our DDX5 data (Supplementary Figures S3B and C). However, we suspect that the DNA–RNA hybrid unwinding by DDX5 and DDX21 would come from 5′ to 3′ unwinding activity both the in vitro assay and in cells since increased S9.6 signal by depletion of either proteins was observed by many studies including ours.
The other mechanism of R-loop regulation by ATAD5 depends on PCNA unloading activity. When ATAD5 is depleted in cells, PCNA cannot be unloaded from the chromatin and consequently accumulates on DNA in the lagging strand behind replication forks (Figure 9, iii; (35)). In addition, in ATAD5-depleted cells, PCNA and many PCNA-binding proteins remain there for a long time, and in some cases after cells enter the G2 phase (35). This situation can provide an opportunity for the transcription machinery to collide with the accumulated PCNA at any time before the PCNA is unloaded by the residual ATAD5 activity. Accordingly, such situation reduces the transcription rate specifically in S phase in ATAD5-depleted cells (Figure 3J and K). This provides new evidence that proteins non-covalently accumulated on DNA can collide with RNAPII leading to R-loop generation. There is a spatial and temporal gap between an ongoing or stalled replication fork and a collision site. We do not know the exact or even the approximate number of PCNA molecules remaining on DNA in ATAD5-depleted cells. However, based on the published PCNA immunoblot and imaging data (32,35,36) and considering the very short length (∼160 bp) of eukaryotic Okazaki fragments (61), significant numbers of PCNA would occupy the lagging strand away from the replication fork. The spatial and temporal gap between a replication fork and the collision sites behind replication forks could explain why ATAD5E1173K cannot reduce the increased number of PLA foci between S9.6 and PCNA by ATAD5 depletion (Figure 3F and G). Even though the abundance of RNA helicases at the replication fork can be restored by ATAD5E1173K via the UAF1 interaction domain, these helicases cannot act on the R-loop generated by the collision between accumulated PCNA and RNAPII due to the spatial and temporal separation between the replication fork and the R-loop (Figure 9, v).
In our study, PCNA remaining on DNA upon ATAD5 depletion increases the PLA signal between PCNA and RNAPII, thus increasing R-loop formation near PCNA (Figures 2A and 3). This raises the following question: what is the main source of conflicts between RNAPII and PCNA? As described above, it is strongly suggested that accumulated PCNA stalls the RNA polymerase complex leading to new R-loop generation at the collision sites in ATAD5-depleted cells. Alternatively, the RNAPII-PCNA PLA signal in ATAD5-depleted cells may result from conflicts between transcription machinery and PCNA on leading strands. When replication forks encounter R-loops under normal conditions, active PCNA unloading from the leading strand may be important to resolve conflicts. The reduction of RNAPII-PCNA PLA signal by RNaseH1 expression in ATAD5-depleted cells (Figure 2A) partially supports this possibility, although questions remain about whether active PCNA unloading actually occurs and how PCNA that remains on leading strands in ATAD5-depleted cells generates conflicts.
The S9.6 antibody recognizes any DNA–RNA hybrids larger than six nucleotides in length in the genome (62). Therefore, the RNaseH1-sensitive S9.6 signal detected in ATAD5-depleted cells may correspond to the DNA–RNA hybrid generated during DNA damage repair or Okazaki fragments synthesis in addition to the R-loop. Depletion of DDX5 or DDX21 has been reported to increase DNA damage (22,63), which could lead to S9.6 signal. However, since DNA damages and damage checkpoint signals were not observed in the ATAD5-transiently depleted cells (35), it appears that the reduced ATAD5-interacting RNA helicases at the replication forks caused by ATAD5 depletion does not produce DNA damage, at least in the time course of our experiments. In our study, the co-depletion of ATAD5 and DDX5 showed a similar level of S9.6 signal compared to the single depletion of ATAD5 (Figure 5J). This suggests that the contribution of DNA damage to the S9.6 signal increased by DDX5 depletion may be cell context-dependent. The replication fork rate and the number of origins fired did not change in ATAD5-depleted cells (35). This implies that almost the same numbers of Okazaki fragment compared as a control cell are present in an ATAD5-depleted cell. However, when ATAD5 is depleted, the cell population in S phase increased (35). It is possible that the increased S9.6 signal might simply indicate an increase of S phase cells in which Okazaki fragments are formed. However, S9.6 signal was increased when ATAD5 was depleted in cells enriched at the S/G2/M phases (Figure 1E), which rules out the possibility. We also found that S9.6-PCNA PLA signal, but not S9.6-RFC3 PLA signal, was increased in ATAD5-depleted cells (Figure 3A, B and Supplementary Figures S2A, B). These data strongly suggest that the cause of S9.6 signal is the accumulated PCNA behind replication forks rather than Okazaki fragment at the ongoing replication forks. All these data strongly support that the RNaseH1-sensitive S9.6 signal detected in ATAD5-depeled cells corresponds to the R-loop rather than to the DNA–RNA hybrid during DNA damage repair or Okazaki fragments synthesis.
In the present study, we provided evidence that ATAD5/UAF1-interacting helicases clear DNA–RNA hybrids during DNA fork progression. It has been reported that these helicases also have roles beyond acting at replication forks. DDX1 promotes clearance of DNA–RNA duplexes formed at the I-SceI-induced DNA double strand break sites to facilitate homologous recombination (26,64). DHX9 promotes R-loop suppression at transcription termination regions and prevents R-loop-associated DNA damage in cells treated with camptothecin (30). Interestingly, recent studies have shown the opposite role for DDX1 and DHX9 in R-loop regulation. DDX1 converts RNA G-quadruplex structures into R-loops to promote Immunoglobin H (IgH) class switch recombination (65). Such post-transcriptionally-formed R-loops appear to have a distinct R-loop tolerance mechanism compared to R-loops formed co-transcriptionally by RNAPII (65,66). DHX9 helicase promotes R-loop formation at the β-actin locus with impaired RNA splicing (49). This suggests that ATAD5/UAF1-interacting helicases play different roles depending on genomic context and can actually promote R-loop formation in certain genomic loci. Future studies will be required to identify regulatory mechanisms that direct helicase activity to properly deal with various genomic contexts.
DATA AVAILABILITY
ATAD5 interactome data are available via ProteomeXchange with identifier PXD18207. All original data are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members in the Center for Genomic Integrity, IBS for helpful discussions and comments on the manuscript. GFP or nuclear-targeting GFP-RNaseH1 DNA was a gift from Dr R. Crouch.
Contributor Information
Sangin Kim, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea; Department of Biological Sciences, School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Korea.
Nalae Kang, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea.
Su Hyung Park, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea.
James Wells, Terry Fox laboratory, BC Cancer Agency, Vancouver, Canada.
Taejoo Hwang, Department of Biomedical Engineering, School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Korea.
Eunjin Ryu, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea; Department of Biological Sciences, School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Korea.
Byung-gyu Kim, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea.
Sunyoung Hwang, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea.
Seong-jung Kim, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea; Department of Biological Sciences, School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Korea.
Sukhyun Kang, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea.
Semin Lee, Department of Biomedical Engineering, School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Korea.
Peter Stirling, Terry Fox laboratory, BC Cancer Agency, Vancouver, Canada; Department of Medical Genetics, University of British Columbia, Vancouver, Canada.
Kyungjae Myung, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea; Department of Biological Sciences, School of Life Sciences, Ulsan National Institute of Science and Technology, Ulsan, Korea.
Kyoo-young Lee, Center for Genomic Integrity, Institute for Basic Science, Ulsan, Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institute for Basic Science [IBS-R022-D1]; UNIST research fund [1.180063, in part]; P.C.S. was supported by a Canadian Institutes of Health Research project grant and the Canadian Cancer Society [705750]. Funding for open access charge: Institute for Basic Science [IBS-R022-D1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gomez-Gonzalez B., Aguilera A.. Transcription-mediated replication hindrance: a major driver of genome instability. Genes Dev. 2019; 33:1008–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crossley M.P., Bocek M., Cimprich K.A.. R-loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dominguez-Sanchez M.S., Barroso S., Gomez-Gonzalez B., Luna R., Aguilera A.. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011; 7:e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stirling P.C., Hieter P.. Canonical DNA repair pathways influence r-loop-driven genome instability. J. Mol. Biol. 2017; 429:3132–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aguilera A., Garcia-Muse T.. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012; 46:115–124. [DOI] [PubMed] [Google Scholar]
- 6. Gan W., Guan Z., Liu J., Gui T., Shen K., Manley J.L., Li X.. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011; 25:2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lang K.S., Hall A.N., Merrikh C.N., Ragheb M., Tabakh H., Pollock A.J., Woodward J.J., Dreifus J.E., Merrikh H.. Replication-transcription conflicts generate R-Loops that orchestrate bacterial stress survival and pathogenesis. Cell. 2017; 170:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garcia-Rubio M., Aguilera P., Lafuente-Barquero J., Ruiz J.F., Simon M.N., Geli V., Rondon A.G., Aguilera A.. Yra1-bound RNA-DNA hybrids cause orientation-independent transcription-replication collisions and telomere instability. Genes Dev. 2018; 32:965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barroso S., Herrera-Moyano E., Munoz S., Garcia-Rubio M., Gomez-Gonzalez B., Aguilera A.. The DNA damage response acts as a safeguard against harmful DNA–RNA hybrids of different origins. EMBO Rep. 2019; 20:e47250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhatia V., Barroso S.I., Garcia-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A.. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014; 511:362–365. [DOI] [PubMed] [Google Scholar]
- 12. Bhatia V., Herrera-Moyano E., Aguilera A., Gomez-Gonzalez B.. The role of replication-associated repair factors on R-Loops. Genes (Basel). 2017; 8:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L. et al.. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015; 57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwab R.A., Nieminuszczy J., Shah F., Langton J., Lopez Martinez D., Liang C.C., Cohn M.A., Gibbons R.J., Deans A.J., Niedzwiedz W.. The fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell. 2015; 60:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skourti-Stathaki K., Proudfoot N.J., Gromak N.. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011; 42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mischo H.E., Gomez-Gonzalez B., Grzechnik P., Rondon A.G., Wei W., Steinmetz L., Aguilera A., Proudfoot N.J.. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell. 2011; 41:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alzu A., Bermejo R., Begnis M., Lucca C., Piccini D., Carotenuto W., Saponaro M., Brambati A., Cocito A., Foiani M. et al.. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell. 2012; 151:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Linder P., Jankowsky E.. From unwinding to clamping - the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011; 12:505–516. [DOI] [PubMed] [Google Scholar]
- 19. Hodroj D., Recolin B., Serhal K., Martinez S., Tsanov N., Abou Merhi R., Maiorano D.. An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 2017; 36:1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kikuma T., Ohtsu M., Utsugi T., Koga S., Okuhara K., Eki T., Fujimori F., Murakami Y.. Dbp9p, a member of the DEAD box protein family, exhibits DNA helicase activity. J. Biol. Chem. 2004; 279:20692–20698. [DOI] [PubMed] [Google Scholar]
- 21. Li L., Monckton E.A., Godbout R.. A role for DEAD box 1 at DNA double-strand breaks. Mol. Cell. Biol. 2008; 28:6413–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song C., Hotz-Wagenblatt A., Voit R., Grummt I.. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017; 31:1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Talwar T., Vidhyasagar V., Qing J., Guo M., Kariem A., Lu Y., Singh R.S., Lukong K.E., Wu Y.. The DEAD-box protein DDX43 (HAGE) is a dual RNA-DNA helicase and has a K-homology domain required for full nucleic acid unwinding activity. J. Biol. Chem. 2017; 292:10429–10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang Q., Jankowsky E.. The DEAD-box protein Ded1 unwinds RNA duplexes by a mode distinct from translocating helicases. Nat. Struct. Mol. Biol. 2006; 13:981–986. [DOI] [PubMed] [Google Scholar]
- 25. Mersaoui S.Y., Yu Z., Coulombe Y., Karam M., Busatto F.F., Masson J.Y., Richard S.. Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 2019; 38:e100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li L., Germain D.R., Poon H.Y., Hildebrandt M.R., Monckton E.A., McDonald D., Hendzel M.J., Godbout R.. DEAD Box 1 facilitates removal of RNA and homologous recombination at DNA double-strand breaks. Mol. Cell. Biol. 2016; 36:2794–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okamoto Y., Abe M., Itaya A., Tomida J., Ishiai M., Takaori-Kondo A., Taoka M., Isobe T., Takata M.. FANCD2 protects genome stability by recruiting RNA processing enzymes to resolve R-loops during mild replication stress. FEBS J. 2019; 286:139–150. [DOI] [PubMed] [Google Scholar]
- 28. Sridhara S.C., Carvalho S., Grosso A.R., Gallego-Paez L.M., Carmo-Fonseca M., de Almeida S.F.. Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-dependent DDX23 phosphorylation. Cell Rep. 2017; 18:334–343. [DOI] [PubMed] [Google Scholar]
- 29. Chakraborty P., Grosse F.. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair (Amst.). 2011; 10:654–665. [DOI] [PubMed] [Google Scholar]
- 30. Cristini A., Groh M., Kristiansen M.S., Gromak N.. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-Loop-Associated DNA damage. Cell Rep. 2018; 23:1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yao N.Y., O’Donnell M.. The RFC clamp loader: structure and function. Subcell. Biochem. 2012; 62:259–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang M.S., Ryu E., Lee S.W., Park J., Ha N.Y., Ra J.S., Kim Y.J., Kim J., Abdel-Rahman M., Park S.H. et al.. Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat. Commun. 2019; 10:2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bell D.W., Sikdar N., Lee K.Y., Price J.C., Chatterjee R., Park H.D., Fox J., Ishiai M., Rudd M.L., Pollock L.M. et al.. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLos Genet. 2011; 7:e1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sikdar N., Banerjee S., Lee K.Y., Wincovitch S., Pak E., Nakanishi K., Jasin M., Dutra A., Myung K.. DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle. 2009; 8:3199–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee K.Y., Fu H., Aladjem M.I., Myung K.. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013; 200:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kubota T., Nishimura K., Kanemaki M.T., Donaldson A.D.. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell. 2013; 50:273–280. [DOI] [PubMed] [Google Scholar]
- 37. Lee K.Y., Yang K., Cohn M.A., Sikdar N., D’Andrea A.D., Myung K.. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J. Biol. Chem. 2010; 285:10362–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park S.H., Kang N., Song E., Wie M., Lee E.A., Hwang S., Lee D., Ra J.S., Park I.B., Park J. et al.. ATAD5 promotes replication restart by regulating RAD51 and PCNA in response to replication stress. Nat. Commun. 2019; 10:5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kee Y., Yang K., Cohn M.A., Haas W., Gygi S.P., D’Andrea A.D.. WDR20 regulates activity of the USP12 x UAF1 deubiquitinating enzyme complex. J. Biol. Chem. 2010; 285:11252–11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jalal C., Uhlmann-Schiffler H., Stahl H.. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Res. 2007; 35:3590–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawai S., Amano A.. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J. Cell Biol. 2012; 197:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wan Y., Zheng X., Chen H., Guo Y., Jiang H., He X., Zhu X., Zheng Y.. Splicing function of mitotic regulators links R-loop-mediated DNA damage to tumor cell killing. J. Cell Biol. 2015; 209:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roy S., Luzwick J.W., Schlacher K.. SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J. Cell Biol. 2018; 217:1521–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hegele A., Kamburov A., Grossmann A., Sourlis C., Wowro S., Weimann M., Will C.L., Pena V., Luhrmann R., Stelzl U.. Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell. 2012; 45:567–580. [DOI] [PubMed] [Google Scholar]
- 45. Dinse G.E., Lagakos S.W.. Nonparametric estimation of lifetime and disease onset distributions from incomplete observations. Biometrics. 1982; 38:921–932. [PubMed] [Google Scholar]
- 46. Sordet O., Redon C.E., Guirouilh-Barbat J., Smith S., Solier S., Douarre C., Conti C., Nakamura A.J., Das B.B., Nicolas E. et al.. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009; 10:887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H. et al.. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008; 132:487–498. [DOI] [PubMed] [Google Scholar]
- 48. Garcia-Muse T., Aguilera A.. Transcription-replication conflicts: how they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016; 17:553–563. [DOI] [PubMed] [Google Scholar]
- 49. Chakraborty P., Huang J.T.J., Hiom K.. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018; 9:4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Klein J., Grummt I.. Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl Acad. Sci. U.S.A. 1999; 96:6096–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang K., Moldovan G.L., Vinciguerra P., Murai J., Takeda S., D’Andrea A.D.. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011; 25:1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang F., Longerich S., Miller A.S., Tang C., Buzovetsky O., Xiong Y., Maranon D.G., Wiese C., Kupfer G.M., Sung P.. Promotion of RAD51-Mediated homologous DNA pairing by the RAD51AP1-UAF1 complex. Cell Rep. 2016; 15:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dungrawala H., Rose K.L., Bhat K.P., Mohni K.N., Glick G.G., Couch F.B., Cortez D.. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell. 2015; 59:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petruk S., Sedkov Y., Johnston D.M., Hodgson J.W., Black K.L., Kovermann S.K., Beck S., Canaani E., Brock H.W., Mazo A.. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell. 2012; 150:922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Santos-Pereira J.M., Aguilera A.. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015; 16:583–597. [DOI] [PubMed] [Google Scholar]
- 56. Shivji M.K.K., Renaudin X., Williams C.H., Venkitaraman A.R.. BRCA2 regulates transcription elongation by RNA Polymerase II to prevent R-Loop accumulation. Cell Rep. 2018; 22:1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang X., Chiang H.C., Wang Y., Zhang C., Smith S., Zhao X., Nair S.J., Michalek J., Jatoi I., Lautner M. et al.. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017; 8:15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parajuli S., Teasley D.C., Murali B., Jackson J., Vindigni A., Stewart S.A.. Human ribonuclease H1 resolves R-loops and thereby enables progression of the DNA replication fork. J. Biol. Chem. 2017; 292:15216–15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Z., Kim T., Bao M., Facchinetti V., Jung S.Y., Ghaffari A.A., Qin J., Cheng G., Liu Y.J.. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011; 34:866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wan C., Borgeson B., Phanse S., Tu F., Drew K., Clark G., Xiong X., Kagan O., Kwan J., Bezginov A. et al.. Panorama of ancient metazoan macromolecular complexes. Nature. 2015; 525:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith D.J., Whitehouse I.. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012; 483:434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Phillips D.D., Garboczi D.N., Singh K., Hu Z., Leppla S.H., Leysath C.E.. The sub-nanomolar binding of DNA–RNA hybrids by the single-chain Fv fragment of antibody S9.6. J. Mol. Recognit. 2013; 26:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mersaoui S.Y., Yu Z., Coulombe Y., Karam M., Busatto F.F., Masson J.Y., Richard S.. Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 2019; 38:e100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aguilera A., Gomez-Gonzalez B.. DNA–RNA hybrids: the risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017; 24:439–443. [DOI] [PubMed] [Google Scholar]
- 65. Ribeiro de Almeida C., Dhir S., Dhir A., Moghaddam A.E., Sattentau Q., Meinhart A., Proudfoot N.J.. RNA helicase DDX1 converts RNA G-Quadruplex structures into R-Loops to promote IgH class switch recombination. Mol. Cell. 2018; 70:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roy D., Yu K., Lieber M.R.. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 2008; 28:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ATAD5 interactome data are available via ProteomeXchange with identifier PXD18207. All original data are available upon request.