

Abstract
DNA mismatch repair (MMR) increases replication fidelity and genome stability by correcting DNA polymerase errors that remain after replication. Defects in MMR result in the accumulation of mutations and lead to human tumor development. Germline mutations in MMR cause the hereditary cancer syndrome, Lynch syndrome. After replication, DNA is reorganized into its chromatin structure and wrapped around histone octamers. DNA MMR is thought to be less efficient in recognizing and repairing mispairs packaged in chromatin, in which case MMR must either compete for access to naked DNA before histone deposition or actively move nucleosomes to access the mispair. This article reviews studies into the mechanistic and physical interactions between MMR and various chromatin-associated factors, including the histone deposition complex CAF1. Recent Xenopus and Saccharomyces cerevisiae studies describe a physical interaction between Msh2 and chromatin-remodeling ATPase Fun30/SMARCAD1, with potential mechanistic roles for SMARCAD1 in moving histones for both mispair access and excision tract elongation. The RSC complex, another histone remodeling complex, also potentially influences excision tract length. Deletion mutations of RSC2 point to mechanistic interactions with the MMR pathways. Together, these studies paint a picture of complex interactions between MMR and the chromatin environment that will require numerous additional genetic, biochemical, and cell biology experiments to fully understand. Understanding how these pathways interconnect is essential in fully understanding eukaryotic MMR and has numerous implications in human tumor formation and treatment.
Keywords: Mismatch repair, Genome instability, Chromatin, Fun30, SMARCAD1, CAF1
1. DNA Mismatch Repair and Cancer
DNA mismatch repair (MMR) is a postreplicative repair pathway responsible for removing base-base mispairs and small insertions or deletions not corrected by the proofreading abilities of the replicative DNA polymerases (1, 2). The MMR process in eukaryotic organisms includes recognition of the mispair, followed by DNA endonuclease nicking of the daughter strand, excision of the daughter strand past the mispaired base pair and gap-filling by a DNA polymerase (Figure 1) (3, 4). In model organisms and mammalian cell culture studies, mutations in genes disrupting any of these steps leads to an increase in mutation accumulation. Defects in MMR are now known to underlie the development and progression of human tumors, presumably due to the high level of accumulating mutations in MMR deficient cells (5–8).
Figure 1. Steps in Eukaryotic Mismatch Repair.
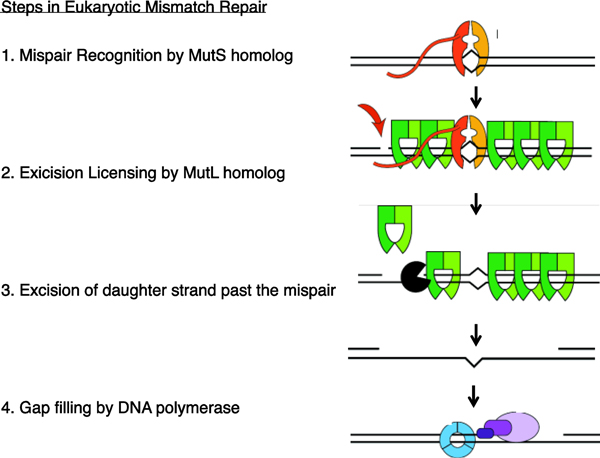
MMR occurs after replication to repair base-base mispairs and small insertion/deletion loops. Eukaryotic MMR occurs using a set of common steps: mispair recognition by the MutS homologs, recruitment of MutL homologs containing endonuclease activity, recruitment of Exo1 exonuclease and excision of the daughter strand, and gap filling by the replicative DNA polymerases.
Lynch syndrome is a hereditary cancer predisposition syndrome that increases the risks of colon, endometrial, ovarian, stomach, and other cancers (9–12). An autosomal dominant pattern of gastric and colon cancer development was originally described in the early 1900s in a large extended family. Later, the discovery of additional families by Henry Lynch and others eventually led to the identification of what was then termed hereditary nonpolyposis colon cancer (hnpcc) and later renamed Lynch syndrome (13). In 1993 it was realized that colon cancers from Lynch syndrome families harbored altered lengths in sequences that contained simple repetitive “microsatellite” sequences (13). Microsatellite instability (MSI) is a hallmark of MMR defects (14). Later the same year, Richard Kolodner cloned human Msh2 and linked Msh2 to Lynch Syndrome (5), followed by Bert Vogelstein’s group finding additional Msh2 mutations in Lynch Syndrome families (15). Other MMR genes, notably Mlh1, Pms2, and Msh6 have since been shown to be mutated in Lynch Syndrome families (16, 17).
Microsatellite instability and somatic alterations in MMR genes are frequently seen in sporadic tumors of numerous origins (12, 18). In addition to MMR gene mutations, the promoter region of Mlh1 can by hypermethylated, thus silencing Mlh1 and leading to an MSI-high tumor phenotype (19). Defects in MMR, in addition to being tumor-promoting, also lead to resistance to common chemotherapeutics such as 5-fluorouracil, cisplatin and carboplatinum, and temozolomide (20). Although the mechanisms are not well understood, a functional MMR pathway is required for the recognition of certain DNA lesions and the downstream signaling that ultimately results in apoptotic cell death (21). For example, MMR is required for the recognition of O6-methylguanine mispaired with a T that can occur after treatment with an alkylating agent,.
Despite the overall resistance to chemotherapy, colon cancers with MSI-high phenotypes have a better clinical prognosis in general (22, 23). One theory is that this is due to the more favorable immune system engagement with these tumors (24). With the dawn of immunotherapy, MMR deficient/MSI-high tumor types seem to respond particularly well to anti-PD checkpoint inhibitors in general. This enhanced response is thought to be due to the increased generation of neo-antigens due to a high mutational burden (25). As advances continue to be made in immunotherapy and chemotherapy and as our ability to sequence human tumors increases, continuing to understand the detailed mechanisms of the MMR pathway is critical to our future success in treating patients.
Many details of how the MMR pathway works in eukaryotes remain to be discovered. Among these are the emerging studies into how MMR functions in a chromatin environment and how chromatin factors interplay with MMR processes. MMR is believed to follow behind the progressing replication fork, correcting errors left over by the proofreading replicative DNA polymerases, Polδ and Polε. MMR proteins should be closely coordinated with the replication machinery in order to accomplish this. However, a comprehensive understanding of this interplay between the two pathways is not fully worked out. Among the outstanding questions is whether or not MMR occurs primarily in the naked DNA environment occurring immediately after replication, or if mispair recognition also occurs in an environment containing partially or fully reconstituted nucleosomes. Furthermore, whether MMR has to accommodate a variety of chromatin states at different steps of MMR or in different chromosomal locations or cellular contexts is still a question, and if so, how are these states managed. MMR also has multiple mechanistic steps following the initial recognition of the mispair (Figure 1), and it is unclear if different steps in the process require different interactions with histones or chromatin-associated factors. A further understanding of these interactions among MMR, chromatin-associated factors, and the chromatin landscape is paramount to fully understanding the fundamental mechanisms of MMR.
2. DNA mismatch repair and chromatin assembly
MMR is spatially and temporally linked with DNA replication (26, 27). The MMR mispair recognition complexes, Msh2-Msh6 and Msh2-Msh3, bind to the mispair and can undergo a conformational change in a mispair- and ATP-dependent manner into a sliding-clamp formation that allows diffusion along the DNA (28, 29). Msh2-Msh6 physically binds to the replicative clamp loader, PCNA, through an unstructured tether on Msh6 (30, 31). This interaction can be visualized in vivo as Msh2 co-localizing with labeled replication factories (26, 32). Loss of the PCNA-Msh6 interaction results in a 10–15% decrease in MMR in the Saccharomyces cerevisiae system as a single mutation; however, in the absence of EXO1 MMR becomes absolutely dependent on the Msh6-PCNA (26, 33, 34). Together this suggests at least a portion of MMR in vivo requires a physical link to replication (26, 33, 34). MMR has been thought to preferentially take place during S-phase (27) when DNA is actively replicating. Experiments in yeast using cell cycle restricted MMR genes showed that MMR could only take place within a short window after a region has been replicated (27). Artificially restricting Msh6 expression to the G2/M cell cycle phase by fusing Msh6 with a fragment of CLB2 under the control of the cell cycle-regulated CLB2 cyclin promoter, resulted in the loss of ability for MMR to correct errors in a frameshift reversion assay that replicates in mid-S-phase. However, the movement of the assay to a late replicating region allowed repair by G2/M expressed Msh6, but not S-phase expressed Msh6 (27).
Also taking place behind the replication fork is the deposition of nucleosomes and the packaging of DNA into a higher-order chromatin structure. This process is carried out in part by the histone chaperone, chromatin assembly factor 1 (CAF1) that binds to PCNA and deposits newly synthesized (H3-H4)2 tetramers onto DNA (35). The full nucleosome is later formed by the addition of H2A and H2B dimers. With both of these two processes, repair and histone deposition, taking place shortly behind the replication fork, a natural question is how these two processes influence each other. This question has been addressed by several studies utilizing elegant in vitro MMR assays that have been augmented with nucleosome deposition systems, reviewed below. Human MutSα (Msh2-Msh6) easily recognizes mispairs in vitro on naked DNA (36). The addition of nucleosome octamers added to a DNA substrate with a mispair and nucleosome positioning sites showed that MutSα binds to mispairs between nucleosome octamers, but binds to mispairs incorporated into nucleosomes to a lesser degree (37). In in vitro MMR assays utilizing human cell extracts, excess MMR-associated degradation of the nicked strand can be observed for a mispair containing plasmid substrate incubated with cytosolic HeLa cell extract (38). The same MMR associated degradation was not seen in a nuclear HeLa extract. Further investigation led to the identification of CAF1 as the nuclear factor that suppresses excess MMR provoked degradation of the discontinuous strand (38). CAF1 nick-dependent deposition of nucleosomes is hypothesized to limit the sliding of the MutS-MutL incision complex away from the mispair, thus limiting excess degradation (38). This observation that histones can block MMR protein movement on DNA is similar to the observed block to the MutSα sliding clamp movement seen by Li et al. (37). Together, this raises the hypothesis that the MMR reaction must take place before DNA is packaged into nucleosomes or otherwise displace nucleosomes.
When CAF1 nucleosome assembly is incorporated into in vitro MMR assays, MutSα can repress nucleosome assembly at that mispair and adjacent to the mispair in the plasmid substrate (38–40). A functional MMR pathway slows the overall deposition of nucleosomes (38–40). This slowing of nucleosome assembly seems to allow efficient MMR, as the addition of a CAF1 nucleosome assembly reaction, with or without the ASF1 chaperone, did not diminish MMR reactions in vitro (39, 40). This was true regardless of whether the in vitro reactions contained Exo1 or were dependent on DNA pol δ-mediated strand displacement (40). The authors also carried out reactions in which DNA Polε carried out the gap-filling step. In the DNA Pol ε reactions, CAF1 and ASF1-H3-H4 actually led to slightly enhanced MMR capacity in the in vitro assay. This was hypothesized to be due in part to the suppression of excessive degradation of the discontinuous strand (40). It is worth noting that the results of in vitro studies, at least in part, depend on the specifics of histone deposition in the system, the strength of the nucleosome-DNA interaction, and the concentration of proteins present. While Li et al. observe a block to MutSα sliding clamp movement by established nucleosomes, alternately, Javaid et al. show that a Msh2-Msh6 sliding clamp can dissociate a nucleosome from DNA in a mispair dependent manner (41). This dissociation is enhanced by H3 acetylation, suggesting interplay could depend on context.
In addition to an important role in the repair of replication-associated errors, MMR protein interactions with nucleosome deposition components may also play a role in response to cytotoxic lesions that are recognized and processed by the MMR pathway. In the Saccharomyces cerevisiae system, the deletion of either the CAC1 or CAC2 subunits of yeast CAF1resulted in an increased sensitivity to SN1-type alkylating agent MNNG (42). The authors propose this is through the excessive MMR-dependent degradation of the discontinuous strand that then results in additional conversion of the nicked DNA to doublestrand breaks (42). Deletion of Rtt106, also involved in replication-associated nucleosome deposition, does not alter MNNG sensitivity like CAF1 depletion, nor does the deletion of the non-replication associated histone chaperone, HIR (42). This may indicate redundancy in vivo as compared to the in vitro system or may be an inherent difference in how MMR and histone deposition interact in relation to replication-associated mispairs versus irreparable cytotoxic mispairs. Recent in vivo genome-wide analysis using the newly developed NMP-seq method in Saccharomyces cerevisiae showed that base excision repair of alkylation-induced N7-methylguanine and N3-methyladenine is faster in nucleosome-depleted regions. Furthermore, alkylation-induced mutations accumulate at sites of strong nucleosome positioning (43). Overall, the toxicity and mutagenicity of alkylating agents are likely closely tied with chromatin structure, regardless of whether the lesion is an MMR or base excision repair substrate.
This coordination between nucleosome assembly and MMR is likely to be facilitated by direct protein-protein interactions between the MMR protein MutSα and components of the nucleosome deposition machinery. Both CAF1 and Msh6 have known interactions with PCNA (33, 34, 44, 45). PCNA is a critical component of the MMR pathway not only for gap filling but in upstream steps, including the stimulation of the MutLα endonuclease (46–49). Interactions with PCNA seem to be especially critical in Exo1-independent pathways of MMR requiring highly active Mlh1-Pms1 (Pms2 in human) endonuclease activity (50). One possibility is that PCNA interacting with MutSα may prevent CAF1 binding, which may explain the ability of MMR to prevent nucleosome deposition near the mispair. CAF1 also directly binds MutSα, and this binding is enhanced during S-phase, in part due to enhanced Msh6 expression during S-phase (39). MutSα is capable of pulling down purified ASF1-H3-H4, although the interaction seems to be with the histone components and not the ASF chaperone components (42). Understanding how these multiple protein interactions work together is essential for fully understanding MMR occurring behind the replication fork.
3. DNA mismatch repair and recruitment by histone modifications
MMR components have also been found to physically interact with histone modifications. Human Msh6 has a Pro-Trp-Trp-Pro (PWWP) motif in the unstructured N-terminal domain facilitating binding to the H3K36me histone mark (51). This interaction between human MutSα and chromatin modifications that peak in G1/S-phase and diminish in G2, suggests enhanced recruitment of MMR activity during S-phase of the cell cycle. A previous special edition of DNA Repair reviewed this work (52). The common cancer-driving H3G34V/R/D mutations in the H3 subunit block the interaction with MutSα (53). Recently it has also been suggested that the MutSα interaction with the chromatin H3K36me3 marks may also play a role in promoting MMR activity at sites of transcription (54).
4. DNA mismatch repair and chromatin remodeling
In a recent study, initially designed to characterize the interaction between Msh2 and Exonuclease 1 (Exo1), a novel Msh2 interaction peptide motif (SHIP box) was newly identified (55–57). This Msh2 interaction peptide had similar characteristics to the previously identified Mlh1 interaction peptide motif (MIP box) given that it consisted primarily of two bulky hydrophobic residues in a region of Exo1 predicted to be unstructured and largely hydrophilic. However, unlike the MIP box in which two phenylalanine residues are directly adjacent to each other, the SHIP box motif consists of a phenylalanine or tyrosine spaced with one unconserved amino acid in between (57, 58). These motifs are notable in that while the intrinsically unstructured C-terminal tail of Exo1 is poorly conserved throughout evolution in length and primary amino acid sequence, the MIP and SHIP box motifs themselves are well conserved from fungi through humans, although their location within the C-terminal region varies. The MIP box motif was originally identified as the interaction site between base excision repair protein, Ntg2, and Mlh1, but has been identified as a common interaction motif in at least two other proteins, Exo1 and Sgs1(58–60). Based on the idea that the MIP box is a shared mode of binding to Mlh1 by several proteins, a bioinformatic search was performed to identify if the SHIP box may also be a shared mode of Msh2 binding and to identify potential novel SHIP box containing proteins. Using a positionspecific scoring matrix (PSSM) to measure similarity to the SHIP box motif and IUPRED analysis to determine predicted disorder, the entire yeast proteome was analyzed for potential SHIP box containing proteins (57). A subset of proteins was prioritized for validation by yeast two-hybrid assay based on high PSSM/IUPRED score and/or previous association with DNA metabolism or repair. From this study, two Saccharomyces cerevisiae SHIP box containing proteins were confirmed, the DNA polymerase ε subunit, Dpb3, and chromatin remodeling factor, Fun30. Binding of these protein partners with Msh2 is mediated by the SHIP box motif, as the mutation of the F/Y amino acids to alanine disrupted the in vivo binding (57).
Fun30, and its human homolog, SMARCAD1, is an SNF-2 family nucleosome remodeler with ATPase activity that has been shown to have roles in the control of histone turn over during replication, control of translation, and resection during homologous recombination (61–65). It is also implicated in the restoration of heterochromatin after replication and is known to bind PCNA (66, 67). In the study of FUN30 in homologous recombination end resection, FUN30 was initially selected for study out of a Saccharomyces cerevisiae screen that looked for deletion mutations that resulted in a lower frequency of homologous recombination promoted gene integration (65). Further experiments show that the deletion of FUN30 inhibited long-range resection by Exo1 5kb to 28kb away from the induced double-strand break, and both the helicase and ATPase functions of the protein were required for long-range resection. The deletion of other nucleosome remodeling complexes implicated in resection, RSC2, and to a lesser extent INO80, when combined with deletion of FUN30 had a synergistic effect on long-range resection (65). The excision step of MMR can be carried out through either exonuclease-dependent or exonuclease-independent mechanisms (4). Exo1 performs the excision step in exonuclease-dependent MMR. The binding of Msh2 to both Fun30 and Exo1 suggests this interaction could play a role in controlling excision, similar to the role of Fun30 in homologous recombination.
Unsurprisingly, a single mutation deletion of FUN30 did not affect MMR capacity in vivo as measured by frameshift reversion assays. In genetic backgrounds that eliminate Exo1-independent repair through a pol30-K217E mutation (49), fun30Δ caused a small but statistically significant increase in mutation rate (57). Similar to what was seen in assays for homologous recombination excision tract length, the deletion of RSC2, seems to have a synergistic effect with the deletion of FUN30. These data point to a need for nucleosome remodeling during Exo1 excision based MMR, in at least a subset of repair reactions. Given the relatively small change in mutation rate, it can be hypothesized that either extensive excision and chromatin remodeling is not required in a majority of MMR reactions, or that additional chromatin remodeling factors play redundant roles in the process. Additional studies in vivo and in in vitro reconstitution reactions will be required to determine if Fun30 and Rsc2 change Exo1 processivity or excision tract length in the presence of nucleosomes. In a genetic background in which Exo1-independent repair occurs (exo1Δ mutation), rsc2Δ also increases the mutation rate. The rsc2Δfun30Δ double mutant, however, has a lower mutation rate than the single rsc2Δ (57). This data suggest the role of chromatin remodelers in Exo1-independent repair, carried out by either DNA polymerase δ strand displacement (68) or successive rounds of endonuclease nicking (48, 49) may be more complicated and require further studies to comprehend fully. Roles for Fun30 discovered in yeast, such as in resection after a double-strand break, seem to translate to the human homolog SMARCAD1 (64). It is likely that the same will be true for the Msh2-SMARCAD1 interaction. Human peptide analysis of potential SHIP box containing proteins using the PSSM and IUPRED analysis pipeline led to the identification of human Exo1-SHIP boxes along with a high score for the potential SMARCAD1 SHIP box. Phylogenetic analysis indicates that this motif is well conserved from yeast Fun30 to human SMARCAD1 (57).
In support of the idea that the Msh2-Fun30/SMARCAD1 interaction is significant throughout evolution, Msh2 and Xenopus SMARCAD1 physically interact, and SMARCAD1 depletion changes nucleosome exclusion around mispairs in extract-based repair assays (69). In Xenopus nucleoplasmic egg extracts, plasmids are supercoiled due to HIRA deposition of histones. A mispair in the plasmid disrupts plasmid supercoiling leading nucleosomes to be excluded in a 1kb region around the mispair. By immunodepleting the extracts of either Msh2-Msh6 or Mlh1-Pms2, it was shown that this nucleosome exclusion phenotype is Msh2-Msh6 dependent. Furthermore, the addition of Msh2 to the immunodepleted extract could displace preassembled nucleosomes (69).
Xenopus Msh2 and SMARCAD1 also directly bind. By linking the mispair containing plasmid to biotin, the authors were able to pull down associated proteins and look for proteins enriched in the presence of a mispair. SMARCAD1 and the FACT subunits, spt16 and ssrp1, were increased when a mispair was present. SMARCAD1 was confirmed to co-immunoprecipitate with Msh2 (69). Both this study and the above Saccharomyces cerevisiae study independently identified a Fun30/SMARCAD1-Msh2 interaction through unbiased approaches, strengthening the evidence that this is a significant interaction. In the Xenopus extract system, the depletion of SMARCAD1 led to more supercoiling around the mispair. An ATPase mutant of SMARCAD1 also resulted in increased supercoiling in the region around the mispair, suggesting that the ATPase function of SMARCAD1 is required (69). The presence of SMARCAD1 also facilitated MMR in the condition where nucleosomes were preassembled around the mispair in a Xenopus nucleoplasmic extract (69). This study brings up the possibility that Fun30/SMARCAD1 is an MMR accessory factor that may allow both access to naked DNA and control excision in Exo1-dependent MMR. What contribution redundant chromatin remodeling factors play is yet to be clarified.
While chromatin marks may help recruit MMR to chromatin during S-phase, carrying out efficient MMR in the context of chromatin is a challenge the cell must overcome, whether that is through inhibition of chromatin deposition or the movement of already established nucleosomes. The studies reviewed above clearly point to important and conserved interactions between Msh2-Msh6/MutSα and chromatin metabolism proteins. (Figure 2) Detailed studies into how these pathways interplay with each other are required if we want to fully understand what effect they have on genome stability and the development of human tumors.
Figure 2. Reported interactions between Msh2-Msh6 (MutSα) and chromatin remodeling proteins.
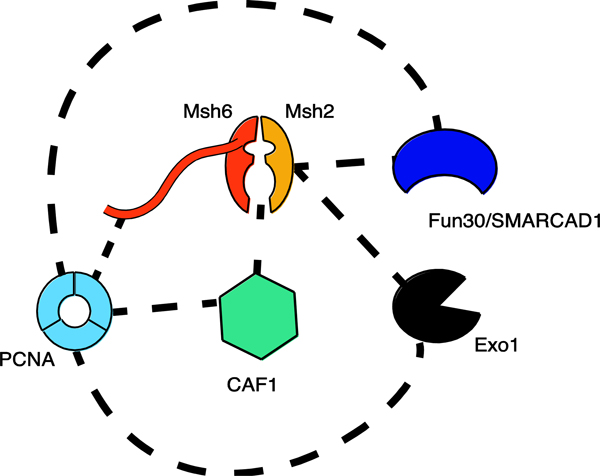
Dashed lines indicated reported physical interactions between proteins in either Saccharomyces cerevisiae, Xenopus, or Humans. There is a high level of physical interaction between the MutS homologs and chromatin modulating proteins, with PCNA being a common interacting protein. Not all interactions are confirmed in all species. Yeast Exo1 lacks the PCNA PIP-box that is contained in human Exo1.
Acknowledgments
The author would like to thank Amber Rohrberg for her diligent editing of the manuscript. Funding: This work was supported by National Institute for Health (Grant R00ES026653 to EMG) and Markey Women Strong Lexington, KY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell research 18, 85–98 [DOI] [PubMed] [Google Scholar]
- 2.Kolodner RD, and Marsischky GT (1999) Eukaryotic DNA mismatch repair. Current opinion in genetics & development 9, 89–96 [DOI] [PubMed] [Google Scholar]
- 3.Fishel R (2015) Mismatch repair. The Journal of biological chemistry 290, 26395–26403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goellner EM, Putnam CD, and Kolodner RD (2015) Exonuclease 1-dependent and independent mismatch repair. DNA repair [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, and Kolodner R (1993) The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75, 1027–1038 [DOI] [PubMed] [Google Scholar]
- 6.Fishel R, and Kolodner RD (1995) Identification of mismatch repair genes and their role in the development of cancer. Current opinion in genetics & development 5, 382–395 [DOI] [PubMed] [Google Scholar]
- 7.Durno CA, Sherman PM, Aronson M, Malkin D, Hawkins C, Bakry D, Bouffet E, Gallinger S, Pollett A, Campbell B, and Tabori U (2015) Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer 51, 977–983 [DOI] [PubMed] [Google Scholar]
- 8.Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, and Edelmann W (2004) Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer cell 6, 139–150 [DOI] [PubMed] [Google Scholar]
- 9.Kastrinos F, and Stoffel EM (2013) The History, Genetics, and Strategies for Cancer Prevention in Lynch Syndrome. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson P, and Lynch HT (1993) Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 71, 677–685 [DOI] [PubMed] [Google Scholar]
- 11.de la Chapelle A (2004) Genetic predisposition to colorectal cancer. Nature reviews. Cancer 4, 769–780 [DOI] [PubMed] [Google Scholar]
- 12.Peltomaki P (2003) Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 21, 1174–1179 [DOI] [PubMed] [Google Scholar]
- 13.Boland CR, and Lynch HT (2013) The history of Lynch syndrome. Familial cancer 12, 145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loeb LA (1994) Microsatellite instability: marker of a mutator phenotype in cancer. Cancer research 54, 5059–5063 [PubMed] [Google Scholar]
- 15.Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, and Modrich P (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75, 1227–1236 [DOI] [PubMed] [Google Scholar]
- 16.Peltomaki P, and Vasen HF (1997) Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 113, 1146–1158 [DOI] [PubMed] [Google Scholar]
- 17.Lynch HT, Snyder CL, Shaw TG, Heinen CD, and Hitchins MP (2015) Milestones of Lynch syndrome: 1895–2015. Nature reviews. Cancer 15, 181–194 [DOI] [PubMed] [Google Scholar]
- 18.Borresen AL, Lothe RA, Meling GI, Lystad S, Morrison P, Lipford J, Kane MF, Rognum TO, and Kolodner RD (1995) Somatic mutations in the hMSH2 gene in microsatellite unstable colorectal carcinomas. Human molecular genetics 4, 2065–2072 [DOI] [PubMed] [Google Scholar]
- 19.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, and Kolodner R (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer research 57, 808–811 [PubMed] [Google Scholar]
- 20.Li Z, Pearlman AH, and Hsieh P (2016) DNA mismatch repair and the DNA damage response. DNA repair 38, 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu D, Calvo JA, and Samson LD (2012) Balancing repair and tolerance of DNA damage caused by alkylating agents. Nature reviews. Cancer 12, 104–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim CG, Ahn JB, Jung M, Beom SH, Kim C, Kim JH, Heo SJ, Park HS, Kim JH, Kim NK, Min BS, Kim H, Koom WS, and Shin SJ (2016) Effects of microsatellite instability on recurrence patterns and outcomes in colorectal cancers. British journal of cancer 115, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang S, Na Y, Joung SY, Lee SI, Oh SC, and Min BW (2018) The significance of microsatellite instability in colorectal cancer after controlling for clinicopathological factors. Medicine 97, e0019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim WK, Park M, Park M, Kim YJ, Shin N, Kim HK, You KT, and Kim H (2013) Identification and selective degradation of neopeptide-containing truncated mutant proteins in the tumors with high microsatellite instability. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 3369–3382 [DOI] [PubMed] [Google Scholar]
- 25.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, and Diaz LA Jr. (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hombauer H, Campbell CS, Smith CE, Desai A, and Kolodner RD (2011) Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147, 1040–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hombauer H, Srivatsan A, Putnam CD, and Kolodner RD (2011) Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science (New York, N.Y 334, 1713–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gradia S, Subramanian D, Wilson T, Acharya S, Makhov A, Griffith J, and Fishel R (1999) hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Molecular cell 3, 255–261 [DOI] [PubMed] [Google Scholar]
- 29.Mendillo ML, Mazur DJ, and Kolodner RD (2005) Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. The Journal of biological chemistry 280, 22245–22257 [DOI] [PubMed] [Google Scholar]
- 30.Johnson RE, Kovvali GK, Guzder SN, Amin NS, Holm C, Habraken Y, Sung P, Prakash L, and Prakash S (1996) Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. The Journal of biological chemistry 271, 27987–27990 [DOI] [PubMed] [Google Scholar]
- 31.Clark AB, Valle F, Drotschmann K, Gary RK, and Kunkel TA (2000) Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. The Journal of biological chemistry 275, 36498–36501 [DOI] [PubMed] [Google Scholar]
- 32.Kleczkowska HE, Marra G, Lettieri T, and Jiricny J (2001) hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes & development 15, 724–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shell SS, Putnam CD, and Kolodner RD (2007) The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Molecular cell 26, 565–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flores-Rozas H, Clark D, and Kolodner RD (2000) Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nature genetics 26, 375–378 [DOI] [PubMed] [Google Scholar]
- 35.Groth A, Rocha W, Verreault A, and Almouzni G (2007) Chromatin challenges during DNA replication and repair. Cell 128, 721–733 [DOI] [PubMed] [Google Scholar]
- 36.Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, and Modrich P (2004) A defined human system that supports bidirectional mismatch-provoked excision. Molecular cell 15, 31–41 [DOI] [PubMed] [Google Scholar]
- 37.Li F, Tian L, Gu L, and Li GM (2009) Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. The Journal of biological chemistry 284, 33056–33061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadyrova LY, Blanko ER, and Kadyrov FA (2011) CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. Proceedings of the National Academy of Sciences of the United States of America 108, 2753–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schopf B, Bregenhorn S, Quivy JP, Kadyrov FA, Almouzni G, and Jiricny J (2012) Interplay between mismatch repair and chromatin assembly. Proceedings of the National Academy of Sciences of the United States of America 109, 1895–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodriges Blanko E, Kadyrova LY, and Kadyrov FA (2016) DNA Mismatch Repair Interacts with CAF-1- and ASF1A-H3-H4-dependent Histone (H3-H4)2 Tetramer Deposition. The Journal of biological chemistry 291, 9203–9217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Javaid S, Manohar M, Punja N, Mooney A, Ottesen JJ, Poirier MG, and Fishel R (2009) Nucleosome remodeling by hMSH2-hMSH6. Molecular cell 36, 1086–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadyrova LY, Dahal BK, and Kadyrov FA (2016) The Major Replicative Histone Chaperone CAF-1 Suppresses the Activity of the DNA Mismatch Repair System in the Cytotoxic Response to a DNA-methylating Agent. The Journal of biological chemistry 291, 27298–27312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mao P, Brown AJ, Malc EP, Mieczkowski PA, Smerdon MJ, Roberts SA, and Wyrick JJ (2017) Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome research 27, 1674–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shibahara K, and Stillman B (1999) Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96, 575–585 [DOI] [PubMed] [Google Scholar]
- 45.Moggs JG, Grandi P, Quivy JP, Jonsson ZO, Hubscher U, Becker PB, and Almouzni G (2000) A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Molecular and cellular biology 20, 1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kadyrov FA, Dzantiev L, Constantin N, and Modrich P (2006) Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126, 297–308 [DOI] [PubMed] [Google Scholar]
- 47.Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O’Donnell M, Kunkel TA, and Modrich P (2007) Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. The Journal of biological chemistry 282, 37181–37190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith CE, Mendillo ML, Bowen N, Hombauer H, Campbell CS, Desai A, Putnam CD, and Kolodner RD (2013) Dominant Mutations in S. cerevisiae PMS1 Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway. PLoS genetics 9, e1003869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goellner EM, Smith CE, Campbell CS, Hombauer H, Desai A, Putnam CD, and Kolodner RD (2014) PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Molecular cell 55, 291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goellner EM, Putnam CD, and Kolodner RD (2015) Exonuclease 1-dependent and independent mismatch repair. DNA repair 32, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, and Li GM (2013) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153, 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li GM (2014) New insights and challenges in mismatch repair: getting over the chromatin hurdle. DNA repair 19, 48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang J, Huang Y, Mao G, Yang S, Rennert G, Gu L, Li H, and Li GM (2018) Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proceedings of the National Academy of Sciences of the United States of America 115, 9598–9603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Y, Gu L, and Li GM (2018) H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. The Journal of biological chemistry 293, 7811–7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tishkoff DX, Amin NS, Viars CS, Arden KC, and Kolodner RD (1998) Identification of a human gene encoding a homologue of Saccharomyces cerevisiae EXO1, an exonuclease implicated in mismatch repair and recombination. Cancer research 58, 5027–5031 [PubMed] [Google Scholar]
- 56.Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, and Kolodner RD (1997) Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proceedings of the National Academy of Sciences of the United States of America 94, 7487–7492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goellner EM, Putnam CD, Graham W. J. t., Rahal CM, Li BZ, and Kolodner RD (2018) Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-binding partners. Nature structural & molecular biology 25, 650–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dherin C, Gueneau E, Francin M, Nunez M, Miron S, Liberti SE, Rasmussen LJ, Zinn-Justin S, Gilquin B, Charbonnier JB, and Boiteux S (2009) Characterization of a highly conserved binding site of Mlh1 required for exonuclease I-dependent mismatch repair. Molecular and cellular biology 29, 907–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gellon L, Werner M, and Boiteux S (2002) Ntg2p, a Saccharomyces cerevisiae DNA N-glycosylase/apurinic or apyrimidinic lyase involved in base excision repair of oxidative DNA damage, interacts with the DNA mismatch repair protein Mlh1p. Identification of a Mlh1p binding motif. The Journal of biological chemistry 277, 29963–29972 [DOI] [PubMed] [Google Scholar]
- 60.Gueneau E, Dherin C, Legrand P, Tellier-Lebegue C, Gilquin B, Bonnesoeur P, Londino F, Quemener C, Le Du MH, Marquez JA, Moutiez M, Gondry M, Boiteux S, and Charbonnier JB (2013) Structure of the MutLalpha C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nature structural & molecular biology 20, 461–468 [DOI] [PubMed] [Google Scholar]
- 61.Awad S, Ryan D, Prochasson P, Owen-Hughes T, and Hassan AH (2010) The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. The Journal of biological chemistry 285, 9477–9484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee J, Choi ES, Seo HD, Kang K, Gilmore JM, Florens L, Washburn MP, Choe J, Workman JL, and Lee D (2017) Chromatin remodeller Fun30(Fft3) induces nucleosome disassembly to facilitate RNA polymerase II elongation. Nature communications 8, 14527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taneja N, Zofall M, Balachandran V, Thillainadesan G, Sugiyama T, Wheeler D, Zhou M, and Grewal SI (2017) SNF2 Family Protein Fft3 Suppresses Nucleosome Turnover to Promote Epigenetic Inheritance and Proper Replication. Molecular cell 66, 50–62 e56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, van Attikum H, and Llorente B (2012) The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489, 581–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, and Ira G (2012) The Fun30 nucleosome remodeller promotes resection of DNA doublestrand break ends. Nature 489, 576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rowbotham SP, Barki L, Neves-Costa A, Santos F, Dean W, Hawkes N, Choudhary P, Will WR, Webster J, Oxley D, Green CM, Varga-Weisz P, and Mermoud JE (2011) Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Molecular cell 42, 285–296 [DOI] [PubMed] [Google Scholar]
- 67.Mermoud JE, Rowbotham SP, and Varga-Weisz PD (2011) Keeping chromatin quiet: how nucleosome remodeling restores heterochromatin after replication. Cell Cycle 10, 4017–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kadyrov FA, Genschel J, Fang Y, Penland E, Edelmann W, and Modrich P (2009) A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proceedings of the National Academy of Sciences of the United States of America 106, 8495–8500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terui R, Nagao K, Kawasoe Y, Taki K, Higashi TL, Tanaka S, Nakagawa T, Obuse C, Masukata H, and Takahashi TS (2018) Nucleosomes around a mismatched base pair are excluded via an Msh2-dependent reaction with the aid of SNF2 family ATPase Smarcad1. Genes & development 32, 806–821 [DOI] [PMC free article] [PubMed] [Google Scholar]