

Summary
The developmental trajectory of human skeletal myogenesis and the transition between progenitor and stem cell states are unclear. We employed single cell RNA-sequencing to profile human skeletal muscle tissues from embryonic, fetal and postnatal stages. In silico, we identified myogenic as well as other cell types and constructed a “roadmap” of human skeletal muscle ontogeny across development. In a similar fashion, we also profiled the heterogeneous cell cultures generated from multiple human pluripotent stem cell (hPSC) myogenic differentiation protocols, and mapped hPSC-derived myogenic progenitors to an embryonic-to-fetal transition period. We found differentially enriched biological processes and discovered co-regulated gene networks and transcription factors present at distinct myogenic stages. This work serves as a resource for advancing our knowledge of human myogenesis. It also provides a tool for a better understanding of hPSC-derived myogenic progenitors for translational applications in skeletal muscle based regenerative medicine.
Graphical Abstract
eTOC
Xi et al. developed a comprehensive view of skeletal muscle and supportive cells across human development. This atlas revealed transcriptional differences among myogenic progenitors and stem cells at distinct developmental stages. This enabled identification of the developmental status of hPSC-derived muscle cells to the embryonic-to-fetal transition period in human development.
Introduction
Skeletal myogenesis starts early during development, which initially gives rise to prenatal skeletal muscle progenitor cells (SMPCs) and later on postnatal satellite cells (SCs) (Applebaum and Kalcheim, 2015; Cerletti et al., 2008; Chal and Pourquie, 2017; Sambasivan and Tajbakhsh, 2007). Both populations are endowed with muscle stem cell properties including, in addition to the expression of the essential myogenic transcription factor (TF) PAX7, the ability to expand and fuse to generate new myofibers in vitro or in vivo (Sacco et al., 2008; Tierney et al., 2016). However, the molecular and functional differences between SMPCs and SCs are only beginning to be unveiled. In vivo, mouse SMPCs contribute to muscle establishment and growth, whereas SCs in mature muscles are typically quiescent and enter the cell cycle in the event of injury (Tierney and Sacco, 2016). In vitro, isolated mouse SMPCs proliferate and maintain PAX7 expression longer than SCs. Moreover, following transplantation after muscle injury, mouse SCs are superior to fetal SMPCs to repopulate the stem cell niche and support long-term regeneration (Tierney et al., 2016). Despite studies on developmental myogenesis in model organisms, our knowledge of muscle ontogeny in human is limited (Schiaffino et al., 2015).
Following developmental cues, we and others have developed directed differentiation protocols using human pluripotent stem cells (hPSCs) to generate myogenic cells including SMPCs or SC-like cells (Borchin et al., 2013; Chal et al., 2015; Hicks et al., 2018; Magli and Perlingeiro, 2017; Shelton et al., 2014; Xi et al., 2017; Xu et al., 2013). These cells may serve as potential sources for personalized cell replacement therapies for degenerative muscle diseases or sarcopenia. However, they have not been fully characterized and compared to in vivo human SMPCs or SCs to facilitate their proper translation to clinical usage.
Here, we performed a comprehensive single cell RNA-sequencing (scRNA-seq) analysis of myogenesis in human limb tissues across development. We identified skeletal muscle (SkM) cells as well as other supportive cell types present at distinct developmental stages. We also evaluated the myogenic and non-myogenic cell populations from three different directed differentiation strategies from hPSCs. Using the developmental trajectory built from the in vivo SMPCs and SCs, we mapped hPSC-derived progenitor cells to a developmental period corresponding to the embryonic-to-fetal transition (7–12 weeks prenatal) across all protocols. Further analysis identified gene groups differentially regulated across developmental stages and provided potential TF candidates that may regulate stage transitions. In summary, this work provides a critical resource to understand the developmental networks defining human skeletal myogenesis and can be used to aid molecular identification of myogenic cells derived from hPSCs. This work will enable the development of new approaches to mature and support the most regenerative cells from hPSCs for use in cell-based therapies.
Results
Identification of skeletal myogenic and supportive cell types using scRNA-seq across in vivo human development
To gain a comprehensive view of cell populations present during human SkM ontogeny, we used scRNA-seq to evaluate human limb muscle tissues from embryonic (week 5–8), fetal (week 9–18) as well as postnatal juvenile (year 7–11) and adult (year 34–42) stages (see STAR Methods and Table S1). To universally identify skeletal myogenic cells from different samples, we developed a computational tool called “Muscle.Score” that examines the average expression of a list of conserved genes representing myogenic cells of distinct developmental and differentiation status (PAX3, PAX7, PITX2, MYF5, MYF6, MYOD1, MYOG, NEB and MYH3). Using “Muscle.Score”, we were able to readily identify SkM cells at each developmental stage (Figure 1). Within mononucleated cells from whole limbs, SkM cells gradually increased in proportion from early embryonic (week 5–6; ~5%) to the beginning of fetal (week 9; above 20%) stage (Figures 1A–1D, 1I-1L and S1I). At early fetal stage (week 12–14; ~35%), SkM cells constituted a major cell type of the non-endothelial/hematopoietic lineages in limbs (Figures 1E, 1M and S1I). This proportion decreased during later fetal development (week 17–18; ~15%) and further dropped in postnatal juvenile and adult limb SkM tissues (below 10%) (Figures 1F–1H, 1N-1P and S1I).
Figure 1. scRNA-seq identifies dynamic cell types across human limb development. See also Figure S1.
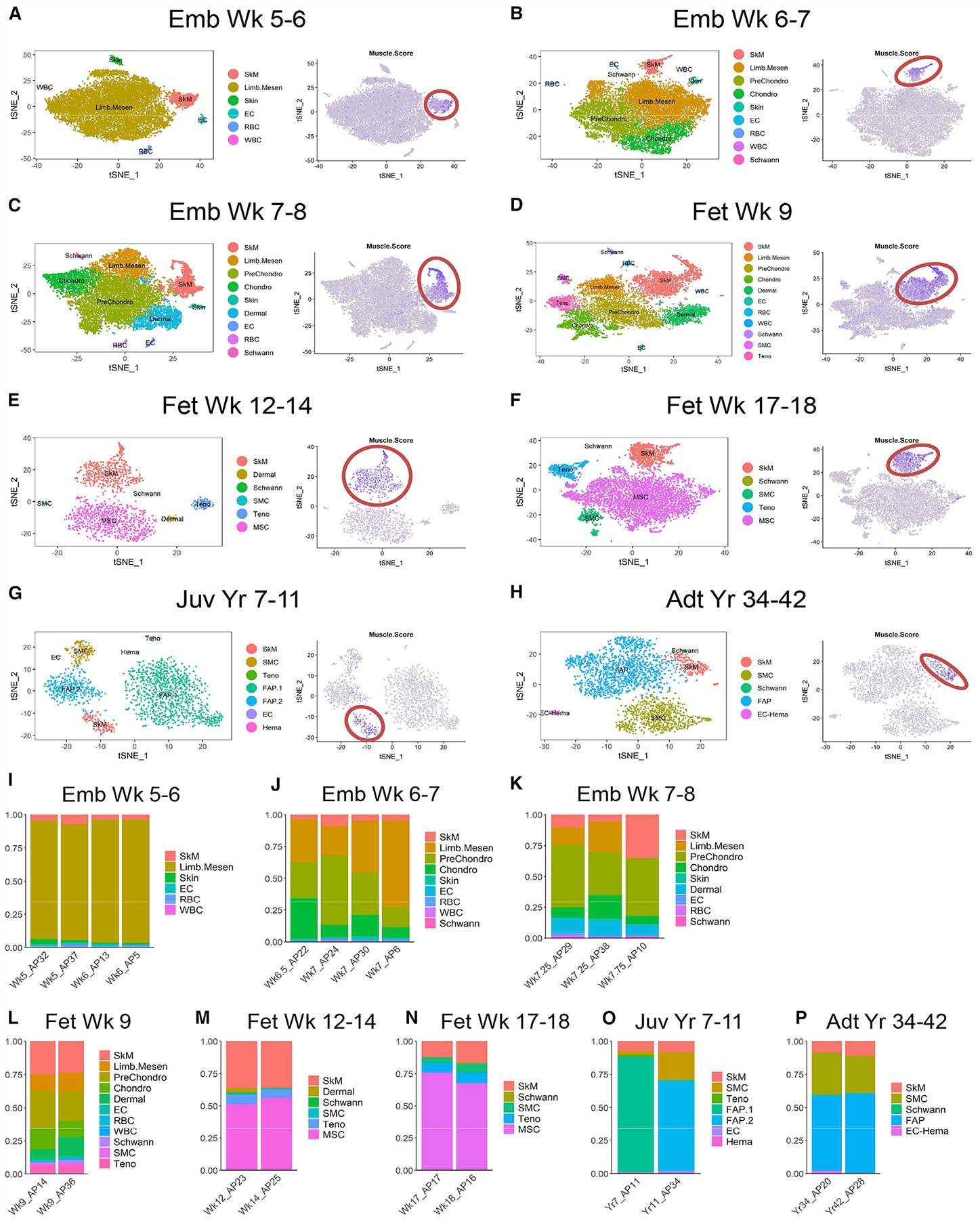
(A-H) Left panels: single cells from human biological replicates grouped by age on tSNE plots and colored by cell type. Right panels: tSNE plots showing color-scaled “Muscle.Score” (purple-to-gray: high-to-low expression). SkM populations red-circled. (I-P) Bar plots of cell type distribution in biological replicates within age groups.
In addition to SkM cells, we also found various non-myogenic populations at distinct developmental timepoints. One highly dynamic population is formed by mesenchymal cell types. Early on (week 5–6), the mesenchyme of the developing limbs was relatively homogeneous, mainly comprised of DUSP6+ multipotent limb mesenchymal progenitors (Limb.Mesen) (Gros and Tabin, 2014; Reinhardt et al., 2019) (Figures 1A, 1I and S1A). As limbs develop (week 6–9), the multipotent progenitors became more lineage restricted and SHOX2+ prechondrogenic (PreChondro) and SOX9+ chondrogenic (Chondro) progenitors became prominent (Akiyama et al., 2005; Barna and Niswander, 2007; Neufeld et al., 2014) (Figures 1B–1D, 1J-1L and S1B–1D). During fetal development (week 12–18), the mesenchymal cells expressed the mesenchymal stromal cell (MSC) marker NT5E/CD73 (Figures 1E, 1F, 1M, 1N, S1E and S1F). At postnatal stage, the mesenchymal/stromal population was highly enriched for PDGFRA, a marker for fibro-adipogenic progenitors (FAPs) found in adult mouse SkM (Joe et al., 2010; Uezumi et al., 2010) (Figures 1G, 1H, 1O, 1P, S1G and S1H). Other cell types present at various levels across limb development include dermal fibroblasts and progenitors (Dermal; TWIST2+), Schwann cells (CDH19+), smooth muscle cells (SMCs; MYLK+) and tenogenic cells (Teno; TNMD+). Skin cells (KRT19+), endothelial cells (ECs; ESAM+) and the hematopoietic (Hema; SRGN+) lineages including red and white blood cells (RBCs and WBCs; HEMGN+ and AIF1+, respectively) were detected at early stages (week 5–9) (Figures 1A–1D, 1I-1L and S1A-S1D), and only residuals of these cell types were found at later stages (week 12 and later) as they were either removed during tissue dissection (skin) or depleted during cell sorting (EC and Hema). In summary, using our scRNA-seq pipeline, we were able to identify dynamic cell populations of both myogenic and non-myogenic nature across human limb development.
Skeletal myogenic subpopulations vary throughout human development
At embryonic week 5–6, the myogenic population in the developing hindlimbs was relatively homogeneous and mainly consisted of PAX3+ myogenic progenitors (MPs) (Figure 2A). Later at week 6–7, a small subset of differentiating myoblasts-myocytes (MB-MC) were observed that expressed commitment and terminal differentiation markers including MYOD1, MYOG and MYH3 (Figure 2B). At the same time, MPs increased PAX7 while decreasing PAX3 expression. The differentiating MB and MC subpopulations became more prominent during week 7–9 (Figures 2C and 2D), consistent with the rapid expansion of SkM needed to support prenatal growth. During fetal week 12–18, we found a reduction of MBs and MCs (Figures 2E and 2F), possibly due to incorporation of most differentiated myogenic cells into multi-nucleated myofibers. At postnatal stage, SkM cells were mainly comprised of PAX7+ SCs with little to no differentiating cells detected (Figures 2G and 2H).
Figure 2. Different skeletal myogenic subpopulations are present across human development. See also Figure S2.
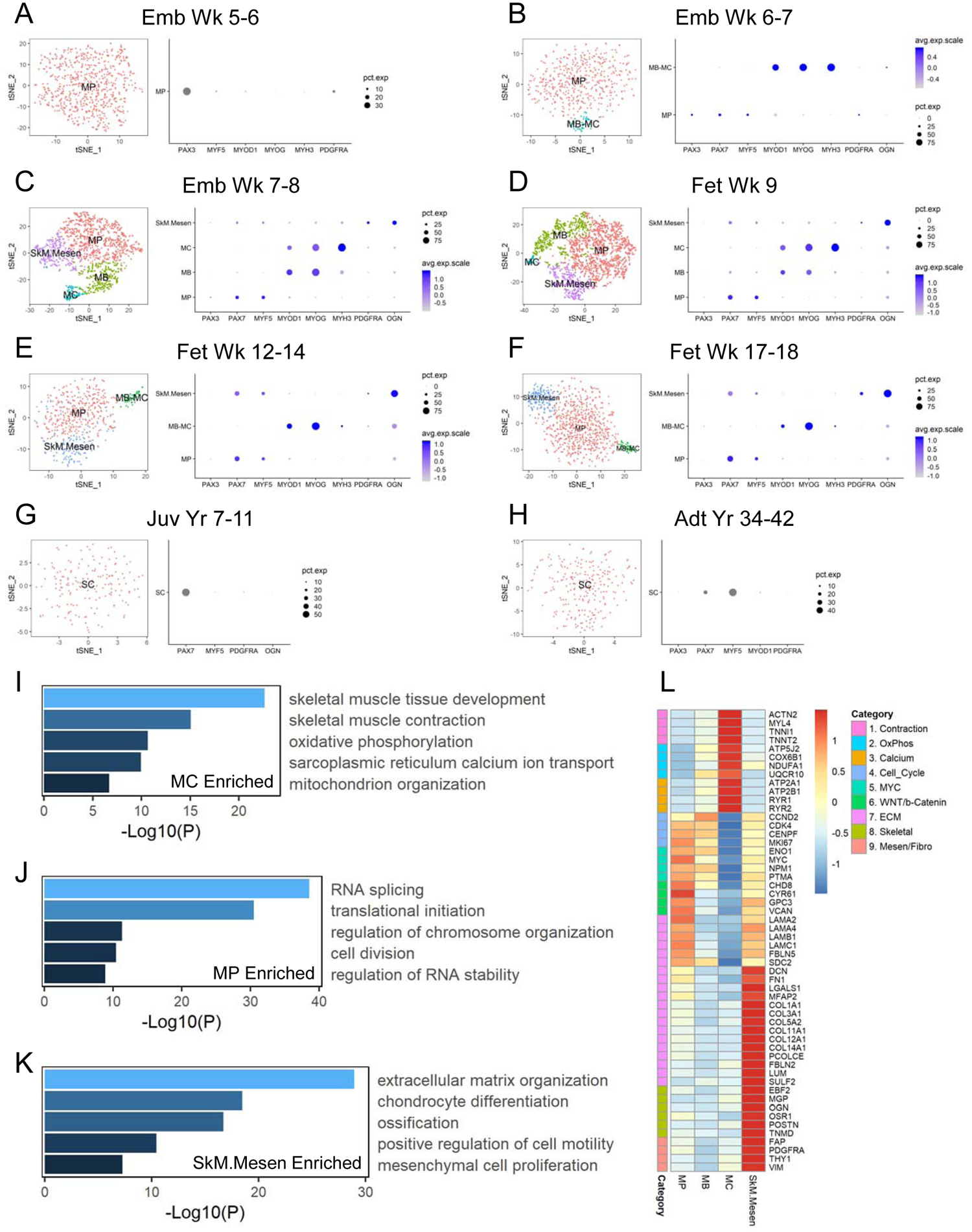
(A-H) Left panels: single cells classified as “SkM” within each age group on tSNE plots and colored by myogenic subtype. Right panels: dot plots of selected subtype markers. (I-K) Selected enriched GO terms from DEGs enriched in MC vs. MP (I), MP vs. MC (J) or SkM.Mesen vs. the main SkM subpopulations (MP, MB and MC) (K). (L) Heatmap of selected markers of different pathways across averaged SkM subpopulations.
In addition to the myogenic subpopulations reflecting distinct differentiation status, we also found another subpopulation transiently present between weeks 7 and 18. This subset expressed the canonical myogenic markers, albeit at slightly lower levels. Compared to the main myogenic subpopulations (MP, MB and MC), these cells uniquely expressed genes suggesting a more mesenchymal-like nature, such as PDGFRA and OGN, and we termed them SkM mesenchymal subtype (SkM.Mesen) (Figures 2C–2F).
To better understand the molecular differences among myogenic subpopulations, we focused on fetal week 9 as an example as all four subpopulations were readily detected at this time point. We performed differential gene expression of the subpopulations (Table S2), followed by gene ontology (GO) analysis as well as Gene Set Enrichment Analysis (GSEA). As expected, MCs were enriched for muscle contraction genes compared to MPs. Moreover, MCs highly expressed genes involved in mitochondria and oxidative phosphorylation (OxPhos) as well as calcium signaling (Figures 2I, 2L and S2A). Proliferating MPs were enriched for genes regulating cell cycle progression, RNA splicing and protein translation (Figures 2J and 2L). MYC and WNT/β-catenin pathways were also enriched in MPs compared to MCs (Figures 2L and S2B). Another major category of genes enriched in MPs was the extracellular matrix (ECM), which included several members of the laminin family (Figures 2L and S2B). Interestingly, compared to the main myogenic subpopulations, SkM.Mesen cells were also highly enriched for ECM genes including collagens and regulators of collagen biosynthesis (Figures 2K, 2L and S2C). To rule out the possibility that the SkM.Mesen subtype was an artifact of misclassification of mesenchymal or skeletogenic cells into the myogenic population by using Seurat (Butler et al., 2018), we employed Monocle (Cao et al., 2019), another commonly used scRNA-seq analysis package to independently confirm this population, and found that the vast majority of SkM.Mesen cells were co-clustered with the main SkM subpopulations (Figure S2D). Although SkM.Mesen cells expressed some pro-chondrogenic genes such as COL11A1 and OGN, they barely expressed the core chondrogenic determination genes such as SOX9 and COL2A1 compared to the Chondro population (Figure S2E). Moreover, SkM.Mesen cells in general expressed higher levels of mesenchymal/fibroblastic markers (e.g., PDGFRA, DCN, and COL3A1) than the main myogenic subpopulations, but lower than the mesenchymal cell types (Limb.Mesen or PreChondro) (Figure S2E).
To better characterize SkM.Mesen cells, we first performed immunohistochemical (IHC) stainings of PAX7, along with PDGFRA which is enriched in the SkM.Mesen subpopulation (Figures 2C–2F). We found in human embryonic and fetal limb sections that a subset of PAX7-expressing myogenic cells were co-stained with PDGFRA (Figures 3A and 3B), corroborating the presence of this myogenic subpopulation in vivo. To further explore myogenic subpopulations, we examined cell surface markers enriched in the SkM lineage over other cell types and identified CDH15 as a potential surface marker to isolate the total SkM population from human embryonic and fetal limbs (Figure 3C). Next, we performed flow cytometry analysis of CDH15 and PDGFRA (Figure 3D) and sorted cell fractions based on these two markers. In freshly sorted cells, myogenic genes PAX7, MYOD1 and MYOG were upregulated in both CDH15+ fractions compared to the CDH15− ones, which validated the usage of this marker for enriching the total myogenic cells. Interestingly, compared to the CDH15+PDGFRA− (15+P−) cells, the CDH15+PDGFRA+ (15+P+) cells showed lower expression of myogenic genes but higher expression of genes involved in osteogenesis (RUNX2 and COL1A1) as well as mesenchyme and ECM (PDGFRA, OGN and DCN) (Figure 3E). When subjected to myogenic and osteogenic differentiation in vitro, respectively, 15+P− cells showed more prevalent formation of MyHC+ myotubes and higher expression of terminal myogenic differentiation genes (Figures 3F and 3G), while 15+P+ cells displayed increased Alizarin Red S-stained calcium depots and higher expression of osteogenic differentiation markers (Figures 3H and 3I). By focusing on SkM cells in the developing human hindlimbs, we were able to detect myogenic subpopulations representing not only various commitment status but also unique myogenic/osteogenic bipotential differentiation properties.
Figure 3. Prospective isolation and in vitro differentiation potential of the SkM.Mesen subpopulation in human embryonic and fetal limbs. See also Figure S2.
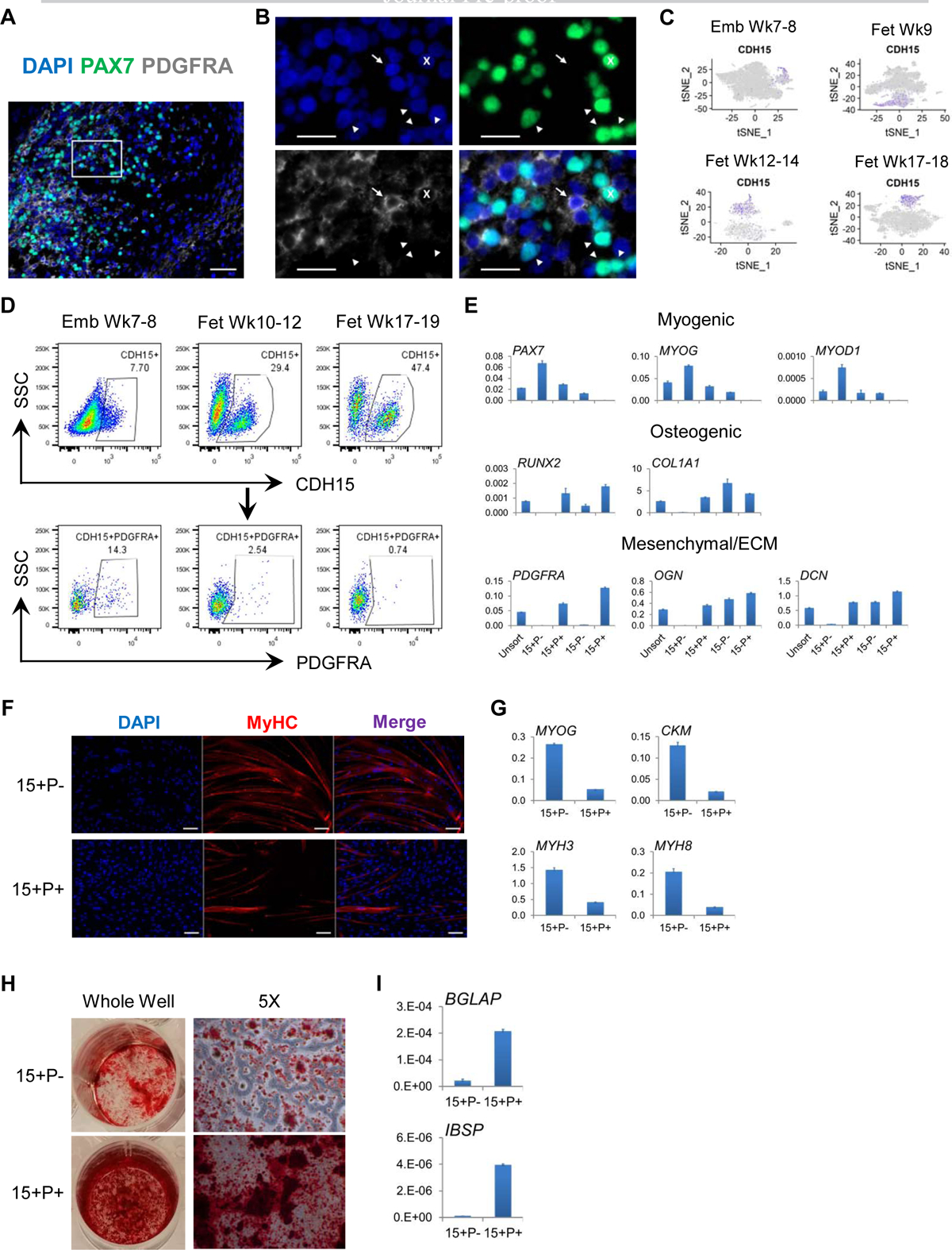
(A and B) IHC staining of PAX7 and PDGFRA in human limb sections. Images in (B) show enlarged area of the boxed region in (A). Cross (x), PAX7+PDGFRA+; arrow, PAX7−PDGFRA+; arrowhead, PAX7+PDGFRA−. Scalebars represent 50 (A) or 20 (B) μm. Representative images are shown from 4 week 7–17 human embryonic and fetal limbs. (C) tSNE plots of CDH15 (purple-to-gray: high-to-low expression). (D) Flow cytometry analysis of CDH15 and PDGFRA co-expression. Representative FACS plots are shown from 3–4 samples for each age group. (E) Freshly sorted CDH15 (15) and PDGFRA (P) subpopulations were subjected to qRT-PCR for myogenic, osteogenic as well as mesenchymal and ECM gene expression. (F-I) Sorted 15+P− and 15+P+ cells subjected to myotube fusion followed by IF of MyHC (F) and qRT-PCR of myogenic commitment genes (G), or osteogenic conditions followed by Alizarin Red S staining (H) and qRT-PCR of osteogenic differentiation markers (I). Scalebars in (F) represent 100 μm. Data shown in (E-I) are representative of 2–3 human fetal limbs. Data of qRT-PCR are normalized to RPL13A as mean+SD of technical triplicates.
Skeletal muscle progenitor and stem cells at distinct stages of human development exhibit different gene expression programs
We next isolated SMPCs (only the MP subpopulations from prenatal samples) and SCs (from postnatal samples) in silico and subjected them to trajectory analysis. These cells formed a developmental trajectory in the diffusion map (DM) space (Haghverdi et al., 2015) consistent with the ages of individual human samples. Unbiased clustering divided the trajectory into 5 major stages (Figures 4A, 4B and S3A). Stage 1 mainly consisted of week 5–6 early embryonic SMPCs, while stage 2 harbored the majority of cells beyond embryonic week 6 to early week 7. Late week 7–8 embryonic SMPCs and those from week 9 in fetal development distributed relatively equally between stages 2 and 3. During fetal development of week 12–18, cells gradually progressed from stage 3 to 4. We observed some degree of overlap among sample ages and computationally calculated “stages”, suggesting early prenatal myogenic development is a continuous process. Postnatal SCs from both juvenile and adult muscles comprised stage 5, and they diverged from the prenatal SMPCs on a separate trajectory (Figures 4A, 4B and S3A).
Figure 4. Skeletal myogenic progenitor and stem cells display dynamic gene expression signatures across human development. See also Figure S3.
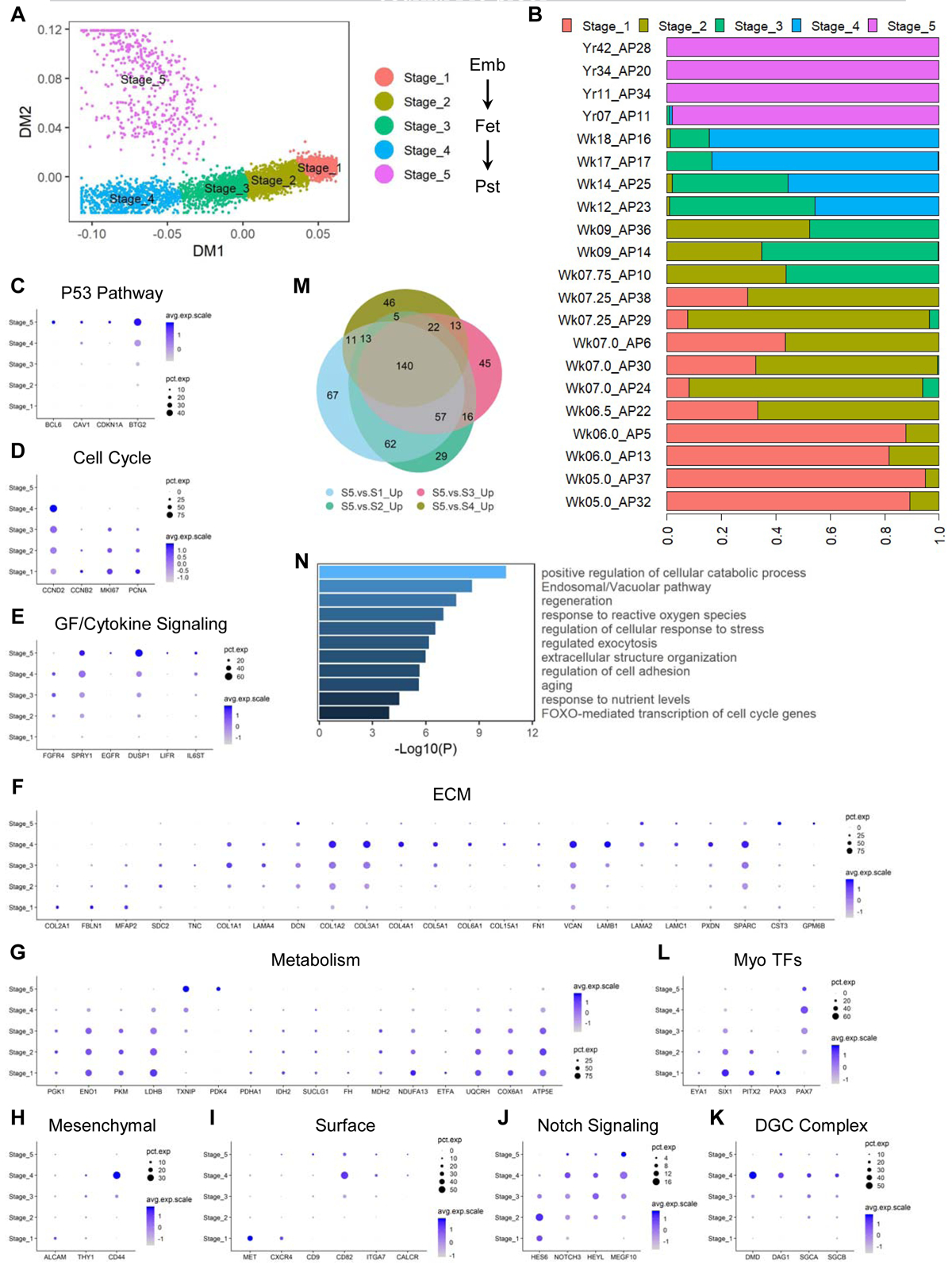
(A) DM plot of single cells of in vivo SMPCs and SCs computationally clustered into 5 major stages. (B) Proportions of cells from each biological sample assigned to each computational stage. (C-L) Dot plots of selected markers for each labelled category. Pst, postnatal (including juvenile and adult). (M) Venn diagram of upregulated genes in stage 5 SCs compared to each stage of SMPCs from stage 1–4. (N) Selected enriched pathways from the 140 genes (M) commonly upregulated in SCs compared to each stage of SMPCs.
Although SMPCs and SCs share some common molecular markers and functionalities (Sacco et al., 2008; Tierney et al., 2016), our developmental trajectory analysis indicates that they display significant differences at the transcriptomic level. To further investigate this, we examined differentially expressed genes (DEGs) between distinct stages (Table S3) and found multiple biological processes and pathways differentially regulated across development. Postnatal SCs were enriched for P53 pathway components (Figure 4C) while expressing virtually no cell cycle promoting genes (Figure 4D), consistent with their quiescent state in homeostatic SkM tissues (Flamini et al., 2018). Nevertheless, several growth factor/cytokine signaling genes were enriched in SCs (Figure 4E), suggesting SCs use specific pathways to actively maintain their quiescence (Price et al., 2014; Shea et al., 2010; Tierney et al., 2014). Two other major differentially regulated biological processes were ECM and cellular metabolism (Figures 4F and 4G). Multiple ECM components showed dynamic expression patterns including collagens and laminins (Figure 4F). For example, while COL2A1 was uniquely enriched in early embryonic SMPCs (stage 1), COL5A1 gradually increased up to later fetal period (stage 4) and was virtually undetectable at postnatal stage 5. Interestingly, genes facilitating major cellular metabolic pathways (e.g., glycolysis, TCA cycle and OxPhos) were progressively downregulated from early to late developmental stages, while metabolic inhibitors such as TXNIP and PDK4 were increased (Figure 4G). Other dynamically expressed gene sets included mesenchymal-like markers, myogenic cell surface molecules and Notch signaling components (Figures 4H–4J). Interestingly, genes encoding the major components of the dystrophin-glycoprotein complex (DGC) including dystrophin, dystroglycan and sarcoglycans, were increased along prenatal development with the highest expression at fetal week 17–18, and then decreased postnatally (Figure 4K).
When examining the canonical TFs involved in myogenesis, we also found distinct expression patterns at each developmental stage (Figure 4L). EYA1, SIX1 and PITX2 showed gradually decreased expression as development progresses. PAX3 progressively decreased while PAX7 increased along development. To corroborate our in silico findings, we performed IHC stainings of PAX3 and PAX7 proteins. At week 6, developing human hindlimbs contain only PAX3+ and not PAX7+ SMPCs, and no MyHC+ myofibers could be detected (Figure S3B). At week 7, both PAX3 and PAX7 were detected in limbs, with the proximal region containing PAX7 single positive cells while distal region harboring SMPCs transitioning from PAX3 to PAX7 expression. At this stage, thin myofibers were present with single or low number of myonuclei (Figure S3C). In later fetal and adult stage muscles examined (quadriceps), myofibers continued to grow in size and SMPCs and SCs were exclusively PAX7+ (Figures S3D and S3E). These results confirmed the findings of PAX3 and PAX7 transcript changes across development from our scRNA-seq analysis.
To explore the common features distinguishing between postnatal SCs and prenatal SMPCs, we performed differential gene expression analysis comparing stage 5 SCs to each individual stage SMPCs from stage 1–4 (Table S3). We intersected the upregulated genes in stage 5 SCs from each of the above comparisons and generated a list of 140 genes commonly enriched in SCs compared to SMPCs (Figure 3M). GO analysis showed several biological processes and signaling pathways were significantly overrepresented, including metabolic and nutrient regulation, intracellular trafficking, ECM organization and cell adhesion as well as FOXO-mediated cell cycle regulation (Figure 3N). Interestingly, FOXO3 has been shown to promote quiescence of adult SCs in mice (Gopinath et al., 2014), suggesting that the FOXO family and related signaling pathways might play an important role in regulating the transition of proliferative prenatal SMPCs to quiescent postnatal SCs.
Although we have identified CDH15 as a cell surface marker capable of isolating SkM cells from embryonic week 7 to fetal week 19 human limbs, this marker was not shown in our scRNA-seq dataset to be enriched in the myogenic population in embryonic week 5–6 limb tissues (Figure S4F), and no prospective markers for myogenic cell isolation have been established for this developmental stage. Thus, we performed differential gene expression analysis between myogenic and non-myogenic cells and found some known SkM cell surface markers enriched in myogenic vs. non-myogenic populations, such as MET and CXCR4 (Bareja et al., 2014; Yin et al., 2013). However, other markers were not expressed at this stage (e.g., CD82) (Alexander et al., 2016; Uezumi et al., 2016) or not distinguishing between myogenic and other cells (e.g., NCAM1 and ITGB1) (Figure S3G) (Castiglioni et al., 2014; Xu et al., 2015). Next, we examined co-expression of PAX3 and MET proteins in week 5–6 human limbs using IHC (Figure S3H). We found nearly overlapping expression patterns of these two proteins at the ventral or dorsal level, but there was a condensed population of PAX3− cells expressing low levels of MET across a small length at the central level. When co-stained with CDH2 (Hayashi and Ozawa, 1995; Yajima et al., 1999), these central cells were found to be PAX3− METlow/+CDH2− (Figure S3G; right panel mosaic images). Thus, we used MET and CDH2 to sort cells from human week 5–6 limbs (Figure S3I), and found the MET+CDH2+ (M+C+) fractions highly enriched for PAX3 and LBX1 transcripts compared to the MET− fractions or unsorted cells (Figure S3J). When cultured in vitro, only M+C+ cells were supported by the myogenic growth medium and expressed PAX3 proteins, and they could form MyHC+ myotubes after switching to fusion conditions (Figure S3K).
Taken together, we mapped SMPCs and SCs from different in vivo stage human samples onto a developmental trajectory, and unequivocally demonstrated the highly dynamic gene expression profiles of these cells across development. We showed striking differences in expression of genes regulating cellular processes, including ECM and metabolism, and confirmed the observed in silico differences of the classical PAX3 and PAX7 myogenic TFs at the protein levels in human tissues. We also identified cell surface markers that enabled prospective isolation of the earliest PAX3+ myogenic population from week 5–6 developing human limbs.
Directed myogenic differentiation of hPSCs generate heterogeneous cell types including both myogenic and non-myogenic cells
Although there are numerous reports describing generation of SkM cells from hPSCs, there is often a large variation in efficiency and consistency in directed differentiation protocols (Kim et al., 2017). We reasoned that by using scRNA-seq, we could identify the different cell types present across representative protocols (See STAR Methods and Table S4). The balance of myogenic and non-myogenic populations may modulate the effectiveness of each differentiation towards SMPCs or SC-like cells. Using our recently published protocol (termed HX protocol) (Xi et al., 2017), we differentiated hPSCs towards the SkM lineage and profiled all live mononuclear cells in culture from 3–8 weeks of differentiation. To better track PAX7+ cells during differentiation, we used CRISPR-Cas9 directed homologous recombination to construct an endogenous PAX7-driven GFP reporter in hPSC cell lines (Figures S4A and S4B). These reporter cells were validated to enrich for PAX7 when GFP+ cells were sorted after artificial activation of the PAX7 locus by the dCas9-VPR system (Figures S4C–S4E) or from directed myogenic differentiation (Figures S4F–S4I).
At week 3, the earliest differentiation time point examined, we detected very few SkM cells in dissociated and live-sorted cultures by our scRNA-seq approach. When the reporter cells were used to enrich for PAX7-GFP+ cells at this time point, the sorted populations mainly consisted of the neural lineage including neural progenitor cells (NPCs; SOX2+) and differentiated neurons (DCX+), while no skeletal myogenic cells could be detected (Figure 5A). Interestingly, the proportion of SkM cells dramatically increased one week later at week 4 in live-sorted populations. At the same time, SkM cells increased to close to half of the PAX7-GFP+-sorted populations, which was accompanied by a significant decrease in the proportion of neural cells (Figures 5B and S5C). During week 5–6 of differentiation, the proportions of SkM cells in live-sorted populations were relatively stable, and they represented the major cell type in PAX7-GFP+-sorted populations (Figures 5C, 5D and S5C). The SkM cell proportions at week 8 of differentiation were slightly decreased in both live- and PAX7-GFP+-sorted populations (Figures 5E and S5C). Our scRNA-seq approach also confirmed the enrichment of SkM cells by using a combination of surface markers recently published by our group (Hicks et al., 2018) (Figure 5C).
Figure 5. scRNA-seq identifies skeletal myogenic populations as well as other cell types during hPSC differentiation. See also Figures S4 and S5.
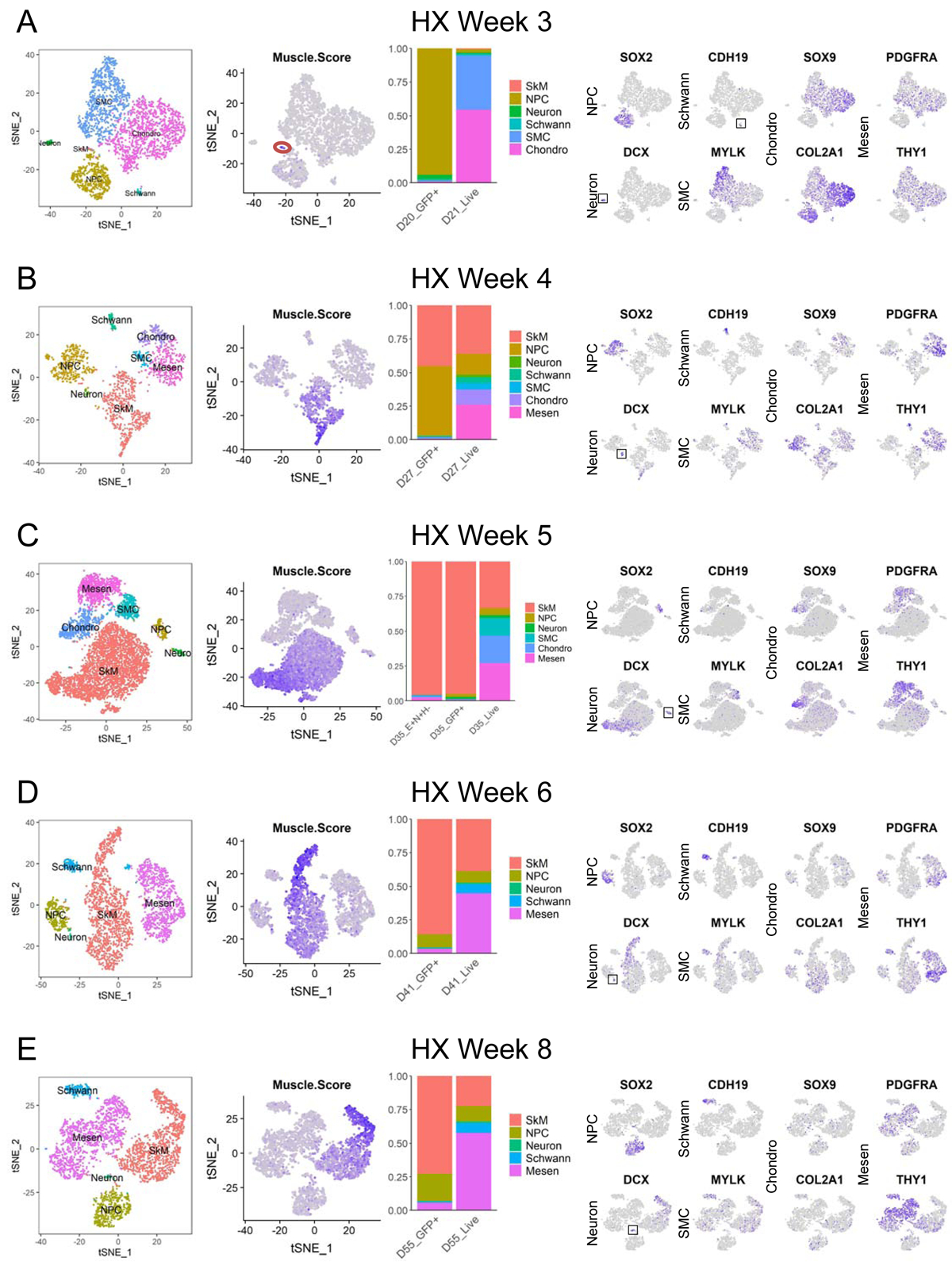
(A-E) From left to right. First panels: single cells from hPSC-derived samples using HX protocol grouped by differentiation time on tSNE plots and colored by cell type. Second panels: tSNE plots showing color-scaled “Muscle.Score” (purple-to-gray: high-to-low expression). The tiny SkM population at week 3 is red-circled. Third panels: bar plots of cell type distribution in enriched or unenriched samples at similar differentiation time points. Fourth panels: tSNE plots of selected cell type markers (purple-to-gray: high-to-low expression). Small populations are boxed for easy visualization.
In addition to SkM and neural cells, we also found multiple other cell types dynamically present in live-sorted populations during the course of differentiation. At week 3 of differentiation, we saw a large portion of chondrogenic cells (SOX9+/COL2A1+) and SMCs (MYLK+) dominating the cultures (Figure 5A), and these populations decreased over time and were absent at 6–8 week time points (Figures 5D and 5E). Meanwhile, a mesenchymal population expressing high levels of PDGFRA and THY1 but not the chondrogenic markers SOX9 or COL2A1, arose at week 4 and increased in proportion towards later time points of differentiation (Figures 5B–5E). Another small but persistent cell type seen during the course of directed differentiation (except week 5) was the Schwann cell population (CDH19+) (Figures 5A, 5B, 5D and 5E).
Using our scRNA-seq strategy, we also examined the directed myogenic differentiation cultures from two additional protocols widely used by our lab and others published by Chal et al and Shelton et al (here termed JC and MS protocol, respectively) (Chal et al., 2015; Shelton et al., 2014). At week 5 of differentiation by JC protocol, we observed both myogenic and non-myogenic populations present in cultures (Figure S5A). The latter included NPCs, neurons, Schwann cells as well as a mesenchymal population expressing high levels of PDGFRA, THY1 and DCN which is likely composed of subpopulations indicated by varying degrees of expression of additional markers (e.g., ALCAM, LUM and COL11A1). The cellular composition of the differentiation culture at week 6–7 using MS protocol were found to be quite different from that obtained from HX and JC protocols (Figure S5B). In addition to SkM cells, we observed a robust population highly expressing genes encoding cytokeratins (e.g., KRT19) or those pertaining to keratinization (e.g., PERP), and therefore is likely involved in epithelium development. There was another major population enriched for genes involved in skeletal development (e.g., COL1A1 and OGN) but lacking strong expression of the canonical commitment markers for the osteogenic, chondrogenic or tenogenic lineages. We also found a small subset of cells enriched for genes participating in cholesterol biosynthesis (CRABP1 and CRABP2) but the accurate identity of this population is yet to be determined.
In conclusion, our scRNA-seq approach identified dynamic cellular compositions, both myogenic and non-myogenic, during the course of hPSC SkM directed differentiation across multiple protocols. This provides a unique resource to not only further explore hPSC-derived myogenic cells, but also other cell types present in the differentiation cultures and their potential influences on in vitro hPSC myogenesis.
Skeletal muscle cells derived in vitro from hPSCs harbor multiple myogenic subpopulations during the course of directed differentiation
Similar to our approach on studying in vivo human myogenesis, we bioinformatically isolated the SkM cells from cultures examined during 4–8 weeks of in vitro hPSC directed differentiation using HX protocol. Consistent with our in vivo findings, we also found subpopulations representing different myogenic commitment status, i.e., MP, MB and MC cells at all time points of directed differentiation, and the relative distribution of these three subpopulations largely stayed constant regardless of differentiation timing or enrichment strategies (Figures 6A–6D). Of note, we detected MBs and MCs within the SkM populations even from PAX7-GFP+-sorted fractions. This is likely due to the low expression of PAX7 in early committed MBs (Figures 6A–6D, middle panels) and the high stability of the GFP proteins (Li et al., 1998) retained in committed cells that have previously expressed PAX7. Interestingly, MPs at 4 weeks of directed differentiation from live-sorted populations could be further subdivided into two subsets, enriching for PAX3 and PAX7, respectively. As expected, MPs at this differentiation time point from PAX7-GFP+-sorted SkM cells were mainly comprised of PAX7+ with only barely detectable PAX3+ progenitors (Figure 6A). However, at later time points MPs from either live- or PAX7-GFP+-sorted fractions did not show obvious expression of PAX3 and only expressed PAX7 (Figures 6B–6D). This is similar to the PAX3 to PAX7 transition that we observed at early in vivo human limb myogenesis (Figures 2A–2H, 4L and S3B-S3E). Reminiscent of the SkM.Mesen subpopulation found during week 7–18 of prenatal development (Figures 2C–2F), we observed a small but consistent “side” population in all examined directed differentiation time points (we also termed these cells “SkM.Mesen” but in an in vitro context). This subset of cells showed slightly higher expression of myogenic activation and commitment markers MYOD1, MYOG and MYH3 than MPs, but much lower than MBs and MCs, suggesting they are not fully committed terminally differentiated muscle cells. Meanwhile, they showed appreciably lower expression of the stem/progenitor marker PAX7 than MPs, and indeed this subpopulation was only detectable in live-sorted but not PAX7-GFP+-enriched cell fractions (Figures 6A–6D).
Figure 6. scRNA-seq identifies myogenic subpopulations during hPSC myogenic differentiation. See also Figures S4 and S5.
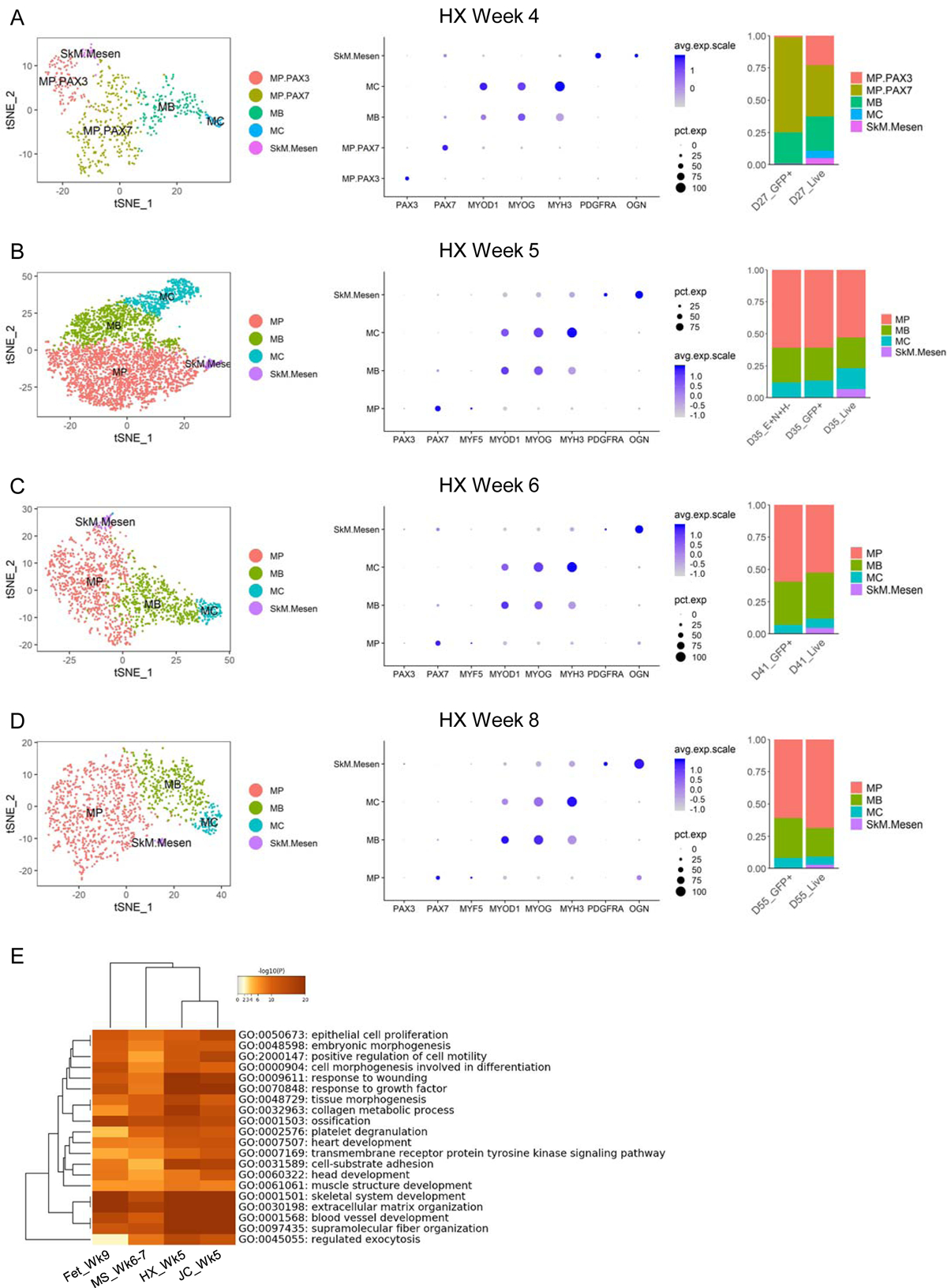
(A-D) Left panels: single cells classified as “SkM” derived using HX protocol at similar time points on tSNE plots and colored by myogenic subtype. Middle panels: dot plots of selected subtype markers. Right panels: bar plots of subtype distribution in enriched or unenriched samples at similar differentiation time points. (E) DEGs upregulated in SkM.Mesen vs. the main SkM subpopulations (MP, MB and MC) from three hPSC differentiation protocols as well as human fetal week 9 samples were subjected to GO enrichment analysis. Heatmap clustering of the top 20 shared GO groups based on enrichment p values.
When examining the SkM subpopulations from JC and MS protocols, we found similar MP, MB and MC subsets, though their relative proportions varied across different protocols (Figures S5D and S5E). Again, we observed the SkM.Mesen subpopulations from both protocols that share many of the enriched genes and biological processes with similar populations from HX protocol as well as in vivo week 9 fetal samples (Figures 6E and S5F and Table S2).
Here, we consistently identified, across multiple hPSC myogenic differentiation protocols, major and rare subpopulations within the SkM cells. This allows us to better understand the dynamics of myogenic lineage development modeled in vitro by hPSCs.
hPSC-SMPCs generated from multiple protocols align to a developmental stage of late embryonic to early fetal transition
To determine the molecular identity of hPSC-derived SMPCs, we mapped the MP subpopulations from all differentiation time points generated from HX protocol along with the in vivo progenitor and stem cells on DM space. The in vivo cells largely retain their developmental trajectory from stage 1 to 5 as previously analyzed (Figure 4A), with minor changes possibly due to variations introduced by adding in the in vitro cells. SMPCs derived from hPSCs aligned to the stage 2–3 in vivo SMPCs along the DM1 component and diverged along DM2 which likely results from culture-related effects (Figures 7A and 7B). To more quantitatively assess the developmental timing of the cells, we developed a more linear method to calculate each cell’s developmental score (“Dev.Score”), where we took into account the expression levels of postnatal vs. embryonic enriched genes in individual cells (see STAR Methods). Using this independent method, we again found in vitro hPSC-derived SMPCs aligned to in vivo SMPCs of stage 2 to 3, which corresponds to the embryonic week 7 to fetal week 12 transition period (Figure 7C). Furthermore, we included additional SMPCs generated from JC and MS protocols in our analysis pipeline and found that hPSC-SMPCs derived from all protocols mapped to a similar late embryonic to early fetal transition stage of human myogenesis (Figures S6A–S6D).
Figure 7. In vitro hPSC-SMPCs align to an embryonic-to-fetal transition stage of in vivo human myogenesis. See also Figures S6 and S7.
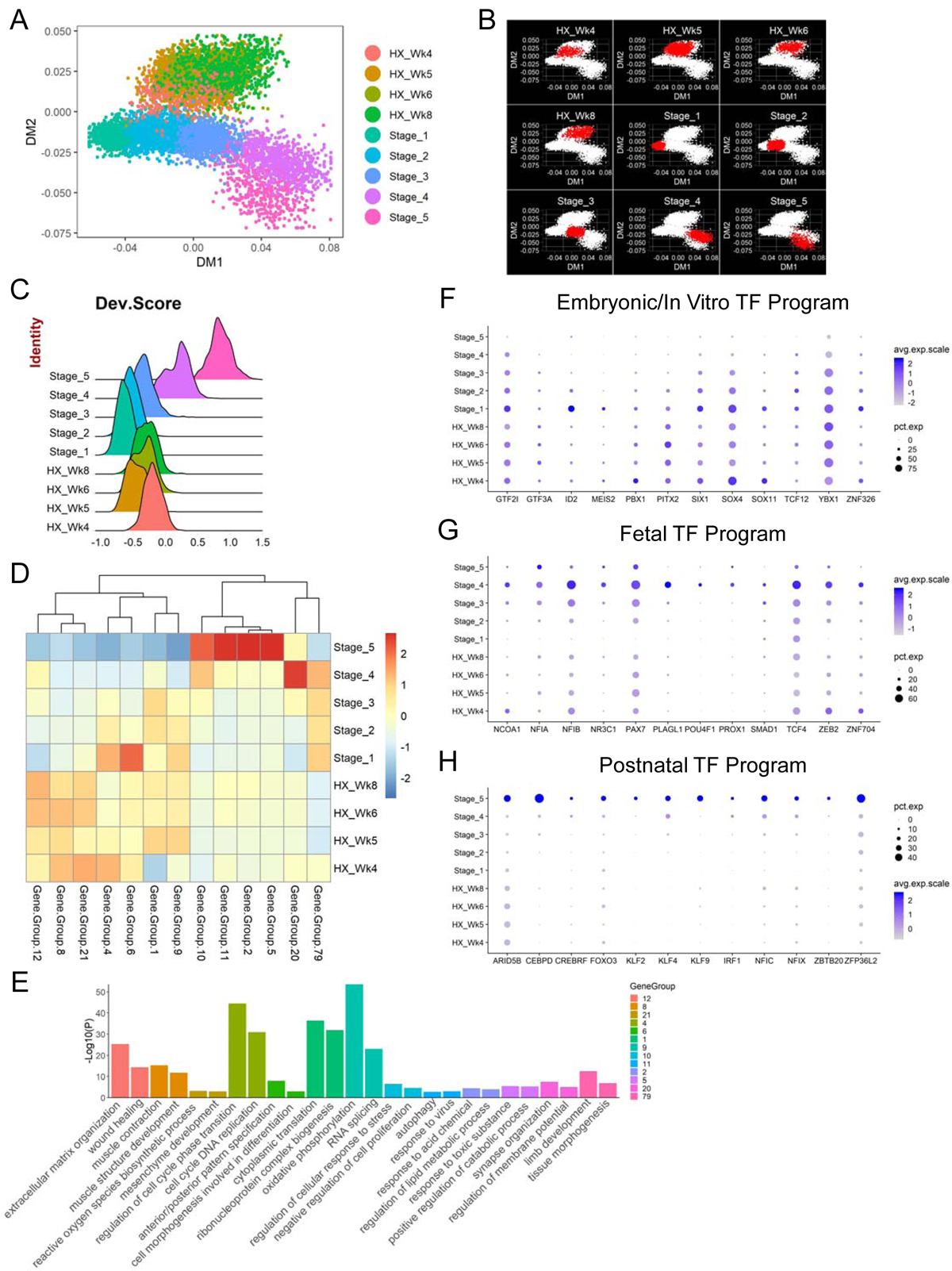
(A) DM plot of single cells of in vivo and in vitro (HX protocol) SMPCs and SCs. (B) DM plots highlighting cells (in red) from individual in vivo or in vitro (HX protocol) stages. (C) Ridge plot of developmental score (“Dev.Score”) distribution across in vivo or in vitro (HX protocol) stages. (D) Heatmap of selected co-regulated gene groups (gene number > 50) across averaged in vivo or in vitro (HX protocol) stages. (E) Two selected enriched GO terms from each gene group are plotted and color-coded. (F-H) Dot plots of selected TFs differentially enriched in embryonic/in vitro, fetal and postnatal stages.
To further explore the differences underlying the separation of in vivo and in vitro SMPCs, we compared the gene expression profiles of hPSC-derived myogenic progenitors from all three protocols to in vivo progenitors from stage 2 and 3, a developmental period that the hPSC-SMPCs most closely align to. Hierarchical clustering of these five groups of cells showed major segregation based on source of in vivo or in vitro derivation, and within the in vitro hPSC-SMPCs those generated from HX and JC protocols were closer to each other than those from MS protocol (Figure S6E). Next, we performed differential gene expression analysis between each of the three hPSC-SMPC populations compared to the stage 2 or 3 populations (Table S5) and found genes that are commonly enriched in either in vivo stage 2 or 3 cells (Figures S6F and S6G), and vice versa (Figures S6H and S6I). Subsequently, GO analysis of these genes revealed biological processes and signaling pathways consistently upregulated in SMPCs from in vivo stages compared to all of the three in vitro myogenic protocols. These include both positive (CCND1 and CDK6) and negative (SPRY1 and DUSP1) regulation of cell cycle indicating more orchestrated cell cycle progression, RNA splicing (RPS26 and RBM39), WNT signaling pathways (FRZB and TCF12) and SkM development (MYF5, MSTN and VGLL2) (Figures S6F, S6G and S6J). On the other hand, processes and pathways consistently enriched in in vitro derived cells from all three protocols include muscle contraction (MYL1, CKB and KLHL41), cell motility (NEFL and YBX3), lipid metabolism (FDFT1, NPC2 and TSPO) and ECM (DCN and MGP) (Figures S6H, S6I and S6K). These findings suggest that there are fundamental differences between SMPCs derived in vivo compared to in vitro, although they might represent a similar developmental stage.
To better understand the gene regulatory networks distinguishing the different myogenic stem and progenitor cells arising during in vivo human development and derived from hPSC directed differentiation, we performed gene co-regulation analysis on our scRNA-seq data (see STAR Methods). We found co-regulated gene groups differentially expressed at distinct stages of myogenesis (Figure 7D and Table S6) and performed GO analysis to explore the key biological processes/pathways enriched in these gene networks (Figure 7E). For example, gene groups 12, 8 and 21 were upregulated in the in vitro hPSC-SMPCs compared to the in vivo cells, and they were enriched for GO terms such as ECM, muscle contraction and reactive oxygen species. Cell cycle, translation, energy metabolism as well as morphogenesis and patterning were enriched in gene groups upregulated in early embryonic- as well as hPSC-SMPCs, such as groups 4, 1, 9 and 6. For gene groups upregulated in postnatal SCs (groups 10, 11, 2 and 5), enriched biological processes were in general involved in maintaining cellular homeostasis. Group 20 was found to be uniquely expressed at high levels in stage 4 SMPCs (fetal week 17–18) and was enriched for genes participating in neuromuscular junction establishment. Group 79 was expressed at a relatively stable level across prenatal development, but at low levels in hPSC-SMPCs or postnatal SCs, and this group enriched for processes such as limb morphogenesis. Next, we focused on the TFs within each of the gene groups, as they have been shown to be the master regulators in cell fate decisions in multiple systems (Oh and Jang, 2019). We found distinct TF programs that were differentially enriched in embryonic/in vitro, fetal and postnatal stages (Figures 7F–7H). These TFs included some canonical myogenic factors such as PITX2 and SIX1 that were enriched in SMPCs from early in vivo stages and derived from hPSCs (Figure 7F), which is consistent with our previous findings (Figure 4L). However, most of these TFs are not classic myogenic genes, which indicates that maturation of myogenic progenitor and stem cells involves processes beyond the regulation of myogenic identity. Furthermore, using RNAscope coupled with IHC, we confirmed the dynamic expression patterns of selected TFs (NFIX, NFIC, KLF9 and CEBPD) in PAX7+ SMPCs/SCs in limb tissues from different embryonic, fetal and adult stages (Figure S7). Overall, these analyses provide potential candidate pathways and TFs to manipulate the maturation status of SMPCs in the future.
Discussion
Myogenesis occurs from early embryonic to postnatal periods and involves myogenic as well as other supportive cell types. Yet myogenic development in human is poorly understood. Although recent work has profiled skeletal muscles using scRNA-seq (Barruet et al., 2020; De Micheli et al., 2020; Dell’Orso et al., 2019; Giordani et al., 2019; Rubenstein et al., 2020; Tabula Muris Consortium et al., 2018), the scope was limited to adult tissues. In this work, we provide a comprehensive roadmap of in vivo human limb myogenesis at the single cell level across development from as early as embryonic week 5 up to adulthood. We also interrogated in vitro hPSC myogenic differentiation from multiple published protocols. Through trajectory analysis, we showed that myogenic progenitor and stem cells from different developmental stages possess distinct gene expression profiles, and hPSC-derived SMPCs align to an in vivo stage of late embryonic to early fetal transition.
One interesting observation is the identification of a resident embryonic and fetal SkM subpopulation that expresses reduced canonical myogenic markers but increased levels of mesenchymal (e.g., PDGFRA, OGN, THY1 and DCN) and skeletal lineage genes (e.g., RUNX2, COL1A1, MGP and TNMD) (Figures 2, 3 and S2 and Table S2). When isolated and cultured in vitro, this SkM.Mesen subpopulation showed weaker myogenic fusion but stronger osteogenic differentiation capacities. These unique cells could represent a transient subset of myogenic cells existing during early myogenic development that have a higher propensity of osteogenic fate adoption. Indeed, it has been shown that human second trimester fetal SkM cells harbor myogenic and osteogenic bipotency when isolated and cultured in vitro (Castiglioni et al., 2014; Tanaka et al., 2012). A similar “side” population of SkM.Mesen was also detected from all three hPSC myogenic differentiation protocols (Figures 6 and S5 and Table S2). However, whether these in vitro SkM.Mesen cells are the same as those detected in vivo or a small subset drifting away from their myogenic identity due to culture conditions, needs to be further explored. It will also be interesting to fully characterize other cell types during the transition from prenatal to postnatal limb development. Deciphering and co-opting the roles of supportive cells in vivo could increase our ability to mature and improve the functional potential of SMPCs derived from hPSCs in vitro.
Our scRNA-seq pipeline enabled us to focus on the differences of the progenitor and stem cell subpopulations within the SkM lineage across development, avoiding potential influences such as commitment status from the other myogenic subpopulations. Accordingly, we were able to confidently map the developmental trajectory of SMPCs and SCs across development and identify gene expression differences that distinguishes each of them (Figures 4 and S3).
Striking differences in ECM components have been recently reported in fetal and postnatal mouse myogenic progenitor and stem cells, and they are critical for the differential regenerative capacities of cells from different developmental stages (Tierney et al., 2016). Here, we also found that ECM gene expression is one of the key features that significantly changed across human development (Figure 4F and Table S3), suggesting ECM remodeling as a critical process in response to both the intrinsic cues and extrinsic cell-cell/cell-matrix interactions during the SMPC-to-SC transition in human development.
Metabolism is becoming a key feature of cell fate regulation in model organisms, including somite specification and mouse SC states (Koopman et al., 2014; Oginuma et al., 2017; Pala et al., 2018; Ryall, 2013; Ryall et al., 2015; Yucel et al., 2019), but has not been carefully evaluated throughout embryonic and fetal to adult development. We found that multiple genes participating in central metabolism were expressed at higher levels in early embryonic SMPCs and gradually decreased as cells transition to postnatal SCs. Consistently, negative metabolic regulators such as TXNIP, an inhibitor of glucose uptake and glycolysis and PDK4, which downregulates pyruvate entry into the mitochondrial TCA cycle, were found to be upregulated in postnatal SCs (Figure 4G and Table S3). This gene expression pattern most likely reflects the changing metabolic demands as actively expanding SMPCs during prenatal muscle establishment transition to quiescent SCs in postnatal homeostasis, and suggests that metabolic wiring distinguishes SMPC and SC states.
Although there are multiple protocols reporting generation of SkM cells from hPSCs, the heterogeneity and dynamics of cell types present in culture and within the myogenic populations have not been adequately studied. Using scRNA-seq, we undoubtedly found myogenic as well as significant numbers of non-myogenic populations from all three representative protocols examined (Figures 5 and S5). Both HX and JC protocols employ a sequential specification through presomitic mesoderm, somite, dermomyotome and SkM, and they yielded similar cell types in the differentiation cultures. Both of these two protocols generated neural cell types including NPCs, neurons and Schwann cells. It is worth noting that the WNT activation and BMP and TGFβ inhibition approach used in these protocols have also been employed in strategies to differentiate hPSCs towards neural crest (NC) cells (Chambers et al., 2009), which are ancestors of multiple cell types including peripheral neurons, Schwann cells, SMCs and craniofacial cartilage and bone, among others (Cheung et al., 2019). Thus, it is conceivable that the neural cell types generated from these protocols might be derived from NC cells that were specified along with somite early on during differentiation. Moreover, in HX protocol, we observed SMCs and chondrogenic cells present at early time points (week 3–4) but with decreased proportions (week 5) and eventually undetectable (week 6–8) towards later time points. These populations could be derivatives from either NC or somite cells (Brent and Tabin, 2002), and the decrease of their presence might reflect the unsuitableness of the myogenic conditions to support them in long-term culture. The origin of the mesenchymal populations starting at week 4 will be interesting to explore further and might be derived from a rare population generated early on during differentiation, or from cells not well-supported in culture that drift away from their original identities. Future in vitro lineage tracing and depletion experiments will be required to delineate the origins of these non-myogenic populations and their influences on the myogenic specification efficiencies of the protocols.
This resource provides the ability for any lab performing hPSC differentiation to map the developmental identity of myogenic progenitor or stem cells. It is very striking that across all three different protocols, SMPCs derived from hPSCs align comparably to the in vivo embryonic-to-fetal transition stage and are not equivalent to the postnatal juvenile and adult SCs (Figure 7 and Figure S6). Prolonging the length of directed differentiation (HX protocol; up to 8 weeks) does not seem to drive hPSC-SMPCs beyond this transitioning stage. Of note, even compared to the in vivo SMPCs at embryonic-to-fetal transition, hPSC-SMPCs still show fundamental differences in a wide range of biological processes (Figure S6). These suggest that stringent evaluation is required to correctly determine cell identity, molecular property and functional potential of myogenic derivatives across differentiation strategies from hPSCs.
To better understand the regulatory network underlying myogenic development, we performed gene co-regulation analysis and identified developmental stage specific gene group signatures. Focusing on TFs within each group, we provide key TF programs that can serve as potential maturation factors for manipulating progenitor and stem cell states across development (Figure 7 and Table S6). We found canonical myogenic specification factors such as SIX1, PITX2 and PAX7. We also found other genes known to regulate SkM, such as ID2 and TCF12 (Zhao and Hoffman, 2004) that were enriched in the embryonic and hPSC-derived SMPCs, SMAD1 (BMP signaling) (Sartori and Sandri, 2015) and PROX1 (Kivela et al., 2016; Petchey et al., 2014) that were increased from early embryonic to late fetal stage and decreased postnatally, and FOXO3 (Sanchez et al., 2014) that was specifically expressed at high levels in postnatal SCs. Interestingly, we found all of the Nuclear Factor I family members (NFIA, NFIB, NFIC and NFIX) expressed at higher levels in late fetal or postnatal stages, suggesting this TF family might play an important role in myogenic maturation. In fact, NFIX has been reported to control the switch from embryonic-to-fetal myogenesis in both mouse and zebrafish (Messina et al., 2010; Pistocchi et al., 2013; Taglietti et al., 2018). Moreover, it is worth noting that the majority of the identified network genes are not typical myogenic TFs. For example, the Kruppel Like Factor family members KLF2, KLF4 and KLF9 were all enriched in postnatal SCs. This family of genes participates in the development and homeostasis of numerous tissues (McConnell and Yang, 2010), and KLF4 is well-known of its ability in induced pluripotency by acting as a pioneer factor that facilitates large scale chromatin remodeling (Schmidt and Plath, 2012; Takahashi and Yamanaka, 2016). Along this line, we also found other chromatin modifiers differentially expressed across development, including ARID5B, NCOA1 and NR3C1. These observations suggest a model where concerted efforts from canonical myogenic TFs as well as epigenetic and chromatin regulators are required to shape the gene regulatory landscapes and drive SMPC-to-SC transition during development. This intricate interplay will also likely be required to instruct hPSCs to gain a SC-like state and maintain their cell fate identity in culture.
In summary, this work serves as a resource for advancing our knowledge of human myogenesis. It also provides a tool for molecular identification of hPSC-derived SMPCs, and targets to guide the generation of the most regenerative cells for translational applications in SkM-based regenerative medicine.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, April D. Pyle (apyle@mednet.ucla.edu).
Materials availability
Plasmids generated in this study will be provided upon request.
Data and code availability
Both raw sequencing reads and processed digital gene expression (DGE) matrices of scRNA-seq datasets are deposited at NCBI GEO with accession number GSE147457. Interactive scRNA-seq data exploration can be accessed at skeletal-muscle.cells.ucsc.edu or aprilpylelab.com/datasets. General codes for computational analysis follow the instructions of the respective software and customized modifications will be available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tissues
Human tissues of 9 weeks of gestation or younger were obtained from electively aborted embryos and fetuses following informed consent and de-identification in accordance with institutional guidelines, which was approved by the local research ethics committee of the University of Tübingen (#312/2016BO1 and #634/2017BO1). Human tissues of 12–18 weeks of gestation were obtained from the University of California Los Angeles (UCLA) Center for AIDS Research (CFAR) Gene and Cellular Therapy Core using institutional review board (IRB)-approved de-identified and consented electively aborted human fetuses. Skeletal muscles from the 7 years old human juvenile subject were obtained from leftover tissues from surgical procedures approved by the UCLA institutional IRB, with patient consent and de-identification. Skeletal muscles from the 11 years old human juvenile subject and the two adult human subjects were obtained from donor autopsy provided by the National Disease Research Interchange (NDRI) with de-identification. Use of human tissues was IRB exempt by the UCLA Office of the Human Research Protection Program (IRB #15–000959).
Cell lines
The H9 human embryonic stem cells (WA09; WiCell Research Institute) are registered in the NIH Human Embryonic Stem Cell Registry with the Approval Number: NIH hESC-10–0062 (https://grants.nih.gov/stem_cells/registry/current.htm?id=414). The PAX7-GFP reporter cell lines are derived from the H9 cells.
METHOD DETAILS
Cell preparation for single cell RNA-sequencing
Embryonic week 7.25 and younger samples
Whole limbs were washed with wash buffer consisting of DMEM/F12, 10% fetal bovine serum (FBS), 1% Penicillin-Streptomycin (P/S) and 0.1% Amphotericin. Tissues were then mechanically chopped into small pieces at room temperature (RT) in digestion buffer consisting of wash buffer supplemented with 2 mg/ml of Collagenase IV and 1 mg/ml of Dispase II. Chopped tissues were further incubated in digestion buffer on a shaker at 37°C for 10–20 minutes with intermittent trituration. Digestion was stopped by adding surplus amount of Drop-seq buffer consisting of phosphate-buffered saline (PBS) supplemented with 0.01% bovine serum albumin (BSA). Digested tissues were filtered twice through 40 μm cell strainers, spun down and resuspended in small volumes of Drop-seq buffer. Cell number was counted and resuspended cells were kept on ice until subjected to the Drop-seq flow procedures.
Embryonic week 7.75 and fetal week 9 samples
Whole limbs excluding feet were washed with wash buffer and then mechanically chopped into small pieces at RT in digestion buffer consisting of wash buffer supplemented with 2 mg/ml of Collagenase II and 1 mg/ml of Dispase II. Chopped tissues were further incubated in digestion buffer on a shaker at 37°C for 20–25 minutes with intermittent trituration. Digestion was stopped by adding surplus amount of Drop-seq buffer. Digested tissues were filtered twice through 40 μm cell strainers, spun down and resuspended in small volumes of Drop-seq buffer. Cell number was counted and resuspended cells were kept on ice until subjected to the Drop-seq flow procedures.
Fetal week 12–18 samples
Skeletal muscles from whole limbs were separated from bones and skin. Muscles were washed with wash buffer and then mechanically chopped into small pieces at RT in digestion buffer consisting of wash buffer supplemented with 2 mg/ml of Collagenase II, 1 mg/ml of Dispase II and 50 μg/ml of DNase I. Chopped tissues were further incubated in digestion buffer on a shaker at 37°C for 20–25 minutes with intermittent trituration. Digestion was stopped by adding surplus amount of fluorescence-activated cell sorting (FACS) buffer consisting of PBS supplemented with 1% FBS and 1% P/S. Digested tissues were filtered through 100 μm cell strainers and spun down. Cell pellets were resuspended in FACS buffer, filtered through 70 μm cell strainers, spun down and resuspended again in small volumes of FACS buffer. Cells were then incubated on ice with antibodies against CD31, CD45 and CD235a. Stained cells were sorted on BD FACSAria sorters to collect the DAPI−/CD31−/CD45−/CD235a− fraction (live and depletion of the endothelial and hematopoietic lineages). Sorted cells were washed with Drop-seq buffer, spun down and resuspended in small volumes of Drop-seq buffer. Cell number was counted and resuspended cells were kept on ice until subjected to the Drop-seq flow procedures.
Postnatal juvenile and adult samples
Skeletal muscles from autopsy or surgical procedures were washed with wash buffer and then mechanically chopped into small pieces at RT in primary digestion buffer consisting of wash buffer supplemented with 2 mg/ml of Collagenase II. Chopped tissues were further incubated in primary digestion buffer on a shaker at 37°C for 10–20 minutes with intermittent trituration. Primary digestion was stopped by adding surplus amount of wash buffer and tissues spun down. Next, supernatant was removed and tissues were resuspended in secondary digestion buffer consisting of wash buffer supplemented with 7 mg/ml of Collagenase D, 1.5 mg/ml of Dispase II and 50 μg/ml of DNase I. Tissues were further digested on a shaker at 37°C for 15–20 minutes with intermittent trituration. Secondary digestion was stopped by adding surplus amount of FACS buffer. Digested tissues were filtered through 100 μm cell strainers and spun down. Cell pellets were resuspended in FACS buffer, filtered through 70 μm cell strainers, spun down and resuspended again in small volumes of FACS buffer. Cells were then incubated on ice with antibodies against CD31, CD45 and CD235a. Stained cells were sorted on BD FACSAria sorters to collect the DAPI−/CD31−/CD45−/CD235a− fraction (live and depletion of the endothelial and hematopoietic lineages). Sorted cells were washed with Drop-seq buffer, spun down and resuspended in small volumes of Drop-seq buffer. Cell number was counted and resuspended cells were kept on ice until subjected to the Drop-seq flow procedures.
Human pluripotent stem cell-derived samples
At the end of directed differentiation, cells were dissociated by 2 mg/ml of Collagenase IV for about 5 min, followed by TrypLE Express for another 5–7 minutes. Dissociation was stopped by adding surplus amount of FACS buffer and dissociated cells were filtered sequentially through 100 and 70 μm cell strainers. Cells were spun down and resuspended in small volumes of FACS buffer. For some samples, cells were incubated on ice with antibodies against ERBB3, NGFR and HNK1. Cells were sorted on BD FACSAria sorters to collect the total live (DAPI−), DAPI−/ERBB3+/NGFR+/HNK1− or DAPI−/GFP+ fractions. Sorted cells were washed with Drop-seq buffer, spun down and resuspended in small volumes of Drop-seq buffer. Cell number was counted and resuspended cells were kept on ice until subjected to the Drop-seq flow procedures.
Cell capture and library construction for single cell RNA-sequencing
Prepared single cell solutions were subjected to single cell capture and droplet formation following instructions in the online Drop-seq protocol v.3.1 (http://mccarrolllab.org/download/905/) and those published in the original Drop-seq paper (Macosko et al., 2015). In brief, cells at 150,000 cells/ml, barcoded beads at 175,000 beads/ml and droplet generation oil were co-flowed at a rate of 4, 4, and 15 ml/hour, respectively, in a PDMS microfluidics chip to generate oil droplets containing beads and lysed cells. Post flow, droplets were breakdown and reverse transcription performed. Complementary DNA was PCR amplified, magnetically cleaned up and subjected to tagmentation and sequencing library construction. Prepared libraries were cleaned up and sequenced via Illumina HiSeq2500, HiSeq4000 or NovaSeq.
Human PSC maintenance
The parental H9 cells and engineered PAX7-GFP reporter cells were maintained on Matrigel-coated tissue culture plates in mTeSR1 medium. Cells were fed with fresh medium every day and passaged with 0.5 mM of EDTA every 4–6 days.
Human PSC skeletal myogenic directed differentiation
HX protocol
Differentiation was performed following procedures published by Xi, et al. (Xi et al., 2017) with minor modifications. Briefly, on day −1 hPSC colonies were dissociated into single cells with TrypLE Express and seeded on Matrigel-coated tissue culture plates at 12,500–25,000 cells/cm2 in mTeSR1 medium containing 10 μM of Y-27632. Differentiation was initiated the next day (day 0) when medium was switched to DMEM/F12 medium containing 1% ITS-G, 0.5% P/S and 3 μM of CHIR99021 (CHIR) for 2 days. On day 2, cells were switched to DMEM/F12 medium containing 1% ITS-G, 0.5% P/S, 200 nM of LDN193189 (LDN) and 10 μM of SB431542 (SB) for another 2 days. On day 4, LDN and SB from the previous medium were replaced with 10 μM of CHIR and 20 ng/ml of FGF2 for 2 days. On day 6, medium was switched to DMEM medium containing 0.5% P/S, 15% KSR (Knockout Serum Replacement), 10 ng/ml of HGF and 2 ng/ml of IGF1 until the end of differentiation. Cells were fed with fresh medium every day until day 6 and every other day thereafter.
JC protocol
Differentiation was performed following procedures published by Chal, et al. (Chal et al., 2015; Chal et al., 2016). Briefly, on day −1 hPSC colonies were dissociated into single cells with TrypLE Express and seeded on Matrigel-coated tissue culture plates at 15,000 cells/cm2 in mTeSR1 medium containing 10 μM of Y-27632. Differentiation was initiated on day 0 by switching to a medium containing DMEM/F12, 1% ITS-G, 1% nonessential amino acids (NEAA) and 0.5% P/S supplemented with 3 μM of CHIR and 0.5 μM of LDN. On day 3, 20 ng/ml of FGF2 was added to the differentiation medium for an additional 3 days. On day 6, medium was changed to a medium containing DMEM/F12, 15% KSR, 1% NEAA, 0.5% P/S and 0.1 mM of 2-mercaptoethanol supplemented with 10 ng/ml of HGF, 2 ng/ml of IGF1, 20 ng/ml of FGF2 and 0.5 μM of LDN for 2 days. On day 8, medium was changed to DMEM/F12 containing 15% KSR, 1% NEAA, 0.5% P/S and 0.1 mM of 2-mercaptoethanol supplemented with 2 ng/ml of IGF1. On day 12 until the end of differentiation, 10 ng/ml of HGF was added to the previous medium. Cells were fed with fresh medium every day until day 12 and every other day thereafter.
MS protocol
Differentiation was performed following procedures published by Shelton, et al. (Shelton et al., 2014) with minor modifications (Hicks et al., 2018). Briefly, on day −1 hPSC colonies were dissociated into single cells with TrypLE Express and seeded on Matrigel-coated tissue culture plates at 37,500 cells/cm2 in mTeSR1 medium containing 10 μM of Y-27632. On the next day (day 0), differentiation was initiated by switching to the E6 medium containing 0.5% P/S supplemented with 10 μM of CHIR for 2 days. On day 2, cells were switched to E6 medium containing 0.5% P/S for 10 days. On day 12, medium was changed to StemPro-34 medium supplemented with 0.5% P/S, 2 mM of L-glutamine, 0.45 mM of 1-thioglycerol, 11 μg/ml of human transferrin and 5 ng/ml of FGF2 for 6 to 8 days. On around day 20, medium was switched to E6 medium containing 0.5% P/S for about 10–15 days with the medium during the last 5–7 days of this period supplemented with 10 ng/ml of IGF1. From around day 30–35, medium was changed to DMEM/F12 containing 1.2% N2 supplement, 1% ITS-G, 0.5% P/S and 10 ng/ml of IGF1 for about 5 days. From then on cells were cultured in the same medium supplemented with 3 μM of SB until the end of differentiation. Cells were fed with fresh medium every day.
PAX7-GFP reporter cell construction
Candidate guide RNAs (gRNAs) targeting the 3’ untranslated region (UTR) of PAX7 transcript variant 3, which is conserved across species, were designed using the online tool at crispr.mit.edu. The targeting region was limited to the last 1600 bp of the 3’ UTR to exclude the potential human miR206/miR1–1/miR1–2 binding sites predicted by miRBase (http://www.mirbase.org/), as the mouse counterparts of these miRNAs have been shown to regulate Pax7 expression (Chen et al., 2010). Next, each of the candidate gRNAs in both the regular 20 bp form and short 17 bp form (which has been reported to increase specificity by (Fu et al., 2014)) was cloned into a gRNA cloning vector (Addgene, #41824; (Mali et al., 2013)) using the Gibson Assembly Cloning Kit following manufacturer’s instructions. The final gRNA used was selected based on the highest cleavage efficiencies in hPSCs when a hCas9 plasmid (Addgene, #41815; (Mali et al., 2013)) was co-expressed. The PAX7 targeting homology arms were then PCR amplified from the H9 cell genomic DNA based on the gRNA targeting region selected. For homologous recombination (HR) vector, the Oct4-IRES-eGFP-PGK-Neo plasmid (Addgene, #48681; (Yang et al., 2013)) was used and the Oct4 targeting homology arms were replaced by the ones targeting PAX7 using the Gibson Assembly Cloning Kit following manufacturer’s instructions. Plasmids encoding gRNA, hCas9 and the HR construct (2 μg each) were nucleofected together into 800,000 H9 cells following the Lonza Amaxa 4D guideline with program CA-137. Four days post nucleofection, neomycin/G418 selection at 50 μg/ml was applied for 5 days and then increased to 100 μg/ml afterwards. Individual resistant clones were expanded and genotyped to confirm correct insertion of the reporter cassette. One of the confirmed clones was incubated with recombinant TAT-Cre protein (a gift from Dr. William Pastor, McGill University) to remove the PGK-neomycin cassette between the LoxP sites. Single cell clones were selected, expanded and confirmed by genotyping and they regained sensitivity to neomycin/G418. Two of the final clones, #13 and #22 were used for downstream functional validation and clone #22 were used for directed differentiation for scRNA-seq experiments. Both clones were confirmed to express the pluripotency markers (NANOG, OCT4 and SOX2) by immunofluorescence staining. They were also examined and showed normal karyotypes.
PAX7-GFP reporter validation
Method of dCas9-VPR
Four gRNAs targeted to the PAX7 promoter region (Murmann et al., 2000) were designed using crispr.mit.edu. Each gRNA was cloned individually into the gRNA cloning vector (Addgene, #41824) similarly to previously described (Mali et al., 2013). In brief, 50 ng AflII-digested empty gRNA plasmid was mixed with 3.8 ng of the forward and reverse oligos and combined using the NEBuilder HiFi DNA Assembly Master Mix according to the manufacturer’s instructions. To activate endogenous PAX7 locus, plasmids encoding for all 4 gRNAs along with one for dCas9-VPR (Addgene, #63798; (Chavez et al., 2015)) were co-transfected using ViaFect according to manufacturer’s instructions. To limit nucleofection-related toxicity and increase transfection efficiency, H9 cells were dissociated into single cells with TrypLE Express and seeded on Matrigel-coated tissue culture plates at 25,000 cells/cm2 in mTeSR1 medium containing 10 μM of Y-27632. The next day medium was changed to DMEM/F12 medium containing 1% ITS-G, 0.5% P/S and 3 μM of CHIR for 2 days. One day before transfection, cells were dissociated into single cells and seeded on Matrigel-coated tissue culture plates at 75,000 cells/cm2 in DMEM medium supplemented with 20% FBS, 1% chicken embryo extract and 20 ng/ml of FGF2. One day after, cells were co-transfected in the same medium with 0.5 μg of each plasmids. Cells were grown for 3 more days with medium changing every day to express the vectors and activate the PAX7-GFP reporter cassette. Cells were then harvested and purified by FACS. Cells co-transfected with dCas9-VPR plasmid and the empty gRNA vector were used as controls. The GFP+ and GFP− cell fractions were collected and subjected to downstream analysis.
Method of directed differentiation
PAX7-GFP reporter cells were subjected to directed differentiation by the HX protocol as described above. Cells were harvested and purified by FACS. The H9 parental cells were differentiated alongside the reporter cells and used as controls. The GFP+ and GFP− cell fractions were collected and subjected to downstream analysis.
FACS cell sorting
Single cell solutions were filtered through 40 μm cell strainers and incubated with 1 μg/ml of DAPI as a live/dead cell indicator. When cell surface labelling was needed, cells were first blocked by Human TruStain FcX at RT for 5–10 minutes, followed by fluorophore-conjugated primary antibodies on ice for 20–30 minutes. For antigens requiring 2-step antibody staining, cells were stained on ice for 20–30 minutes with unconjugated primary antibodies followed by fluorophore-conjugated secondary antibodies on ice for another 20–30 minutes. Stained cells were washed with FACS buffer and processed as described above. Cells were sorted by BD FACSAria sorters with FACSDiva software. Standard gating strategies were applied to exclude the debris, doublets and dead cells. Marker specific gating was set up using fluorescence-minus-one stained controls. The parental H9 cells were used for GFP gating. Sorted cells were collected into buffers containing 10% FBS and kept cold until downstream processing. FACS plots were generated using FlowJo.
Immunofluorescence
Cells were fixed with 4% PFA for 10 minutes, followed by permeabilization with 0.3% Triton X-100 in PBS at RT for 10 minutes at RT. Samples were then blocked with 3% BSA, 10% goat serum and 0.3% Triton X-100 in PBS for 60 minutes at RT. Primary antibodies were applied for overnight at 4°C and fluorophore-conjugated secondary antibodies for 60 minutes at RT. Nuclei were counter stained with DAPI at 1 μg/ml. Images were captured using a Zeiss Axio Observer.Z1 microscope equipped with an AxioCamMR3 camera. Image processing and quantification were performed using Fiji/ImageJ (Schindelin et al., 2012) or Zeiss ZEN 3.1 (blue edition).
Cytospin
Sorted cells were spun down onto Superfrost Plus microscope slides using Shandon Double Cytofunnel in a Shandon Cytocentrifuge. Attached cells were processed for immunofluorescence (IF) staining and imaging as described above.
Immunohistochemistry with tyramide signal amplification
Human embryos and tissues were fixed with 4% PFA for one day at 4°C, washed and embedded in paraffin. To reduce tissue autofluorescence for samples of fetal week 9 and older, they were subjected to a dehydration-bleaching-rehydration process before embedding as described by The Collection of Immunolabeled Transparent Human Embryos and Fetuses project (https://transparent-human-embryo.com/?page_id=649) (Belle et al., 2014). Tissue blocks were then sectioned at a 4 μm interval onto Superfrost Plus microscope slides. For immunohistochemistry (IHC) staining, sections were deparaffinized with Xylene and rehydrated through EtOH/water gradient. Antigen retrieval was performed with a pressure cooker using 10 mM of sodium citrate buffer, followed by blocking with 3% BSA, 10% goat serum and 0.1% Tween-20 in PBS for 60 minutes at RT. Primary antibodies were applied for overnight at 4°C and HRP-conjugated secondary antibodies were applied for 45–60 minutes at RT. Tyramide signal amplification (TSA) was performed using the TSA Plus Fluorescence kits per the manufacturer’s instructions to amplify the fluorescent signals. Slides were mounted with DAPI nuclei counterstaining and proceeded to image capture and analysis as described above. Images showing whole limbs of early embryonic development were captured in a mosaic mode and stitched together using the Zeiss software.
RNAscope with Immunohistochemistry
Human tissues were processed similar to regular IHC procedures as described above, except that fixation was performed at RT instead of 4°C and the bleaching step was omitted according to manufacturer’s recommendations. Sections were hybridized with cataloged or custom-designed RNAscope probes and signal developed per manufacturer’s instructions using the RNAscope Multiplex Fluorescent Reagent Kit v2, with in-house protease treatment optimization (Protease Plus 15 minutes). Probe-hybridized sections were further subjected to IHC staining of PAX7 with TSA and imaged as described above. Quantification of RNAscope signals and PAX7 cells was performed using Zeiss ZEN 2.6 Pro (blue edition) software. RNAscope negative probes were applied on sections from different individual samples to set the threshold for positive signal counting.
Quantitative real time-PCR
Cells were harvested and RNA extracted using RNeasy Plus Mini or Micro Kit. Complementary DNA was synthesized using iScript Reverse Transcription Supermix and quantitative real time-PCR (qRT-PCR) was performed using SsoAdvanced Universal SYBR Green Supermix with technical triplicates on a Bio-Rad CFX384 Touch Real-Time PCR Detection System or a Thermo Fisher Scientific QuantStudio 6 Pro Real-Time PCR System. All primer pairs were selected from PrimerBank (Spandidos et al., 2010) or designed using Primer-BLAST (Ye et al., 2012) and tested in-house to ensure an amplification efficiency between 90–110%. Primer sequences for BGLAP, CKM, DCN, eGFP, IBSP, MYH8, OGN and RPL13A are listed in Methods S1. Other primer pairs are the same as previously reported (Xi et al., 2017).
In vitro myotube fusion assay
Sorted cells were resuspended in Lonza SkGM2 medium supplemented with 20 ng/ml of FGF2 and plated onto Matrigel-coated culture wells. Cells were cultured for 5–7 days until they reached >70–80% confluency. Then, medium was switched to DMEM/F12 medium containing 1% ITS-G, 0.5% P/S and 1% N2 supplement to induce fusion for 4–6 days. Medium was refreshed every other day during the culture, and cells at the end of fusion were subjected to IF staining and imaging as described above.
In vitro myogenic and osteogenic bipotential differentiation assays
Sorted cells were plated onto Matrigel-coated culture wells and expanded for 4–6 days in expansion medium (DMEM/F12 medium containing 20% FBS, 1% GlutaMAX, 1% NEAA, 1mM sodium pyruvate, 0.5% P/S and 20 ng/ml of FGF2). Cells were then split and cultured for another 2–3 days in expansion medium until they reached >70–80% confluency. For myogenic differentiation, medium was switched to fusion medium for 4–6 days as described above and cells were subjected to IF staining or harvested for qRT-PCR at the end of the fusion period. For osteogenic differentiation, medium was switched to Thermo Fisher Scientific StemPro Osteogenesis Differentiation medium for 2–3 weeks. At the end of the osteogenic period, cells were subjected to Alizarin Red S staining as previously reported (Xi et al., 2017) or harvested for qRT-PCR analysis. Medium was refreshed every other day during expansion and differentiation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Processing, read alignment and digital gene expression matrix generation
The raw sequencing reads were processed using the Drop-seq_tools-1.13 pipeline from the McCaroll lab (https://github.com/broadinstitute/Drop-seq/releases/tag/v1.13), following the general guidelines from the Drop-seq Alignment Cookbook v1.2 (https://github.com/broadinstitute/Drop-seq/files/2425535/Drop-seqAlignmentCookbookv1.2Jan2016.pdf) (Macosko et al., 2015). Briefly, reads were indexed and filtered by read quality. Sequencing adapter and polyA sequences were trimmed, and reads were further filtered to retain those of a length of at least 30 nucleotides. Processed reads were aligned to the human reference genome (hg19) using Bowtie2 (v2.2.9 with the ‘--very-sensitive’ mode) (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) (Langmead and Salzberg, 2012). Aligned reads were tagged to gene exons using Bedtools Intersect (v2.26.0) (https://github.com/arq5x/bedtools2) (Quinlan and Hall, 2010). Knee plots of cell-to-read fraction were generated to estimate the number of cell barcodes representing true cells. Digital gene expression matrices (DGEs) were then generated by counting gene transcripts for the number of cell barcodes selected based on the infliction points in the knee plots. To correct for any bead synthesis errors/read errors leading to false barcodes, transcript barcodes (unique molecular identifiers; UMIs) or cell barcodes were merged when they were within 1 Hamming or 2 Levenshtein distances, respectively. Barcodes containing < 2500 reads were excluded from the DGEs.
Computational analysis using Seurat
Data filtration, normalization and scaling
Downstream computational analyses of scRNA-seq data were mainly performed using the R package Seurat v2.3.3 (https://github.com/satijalab/seurat/releases/tag/v2.3.3) (Butler et al., 2018) by largely following the standard guidelines from the Satija lab (https://satijalab.org/seurat/). Seurat objects were generated with DGEs constructed as described above. Violin plots of number of expressed genes and unique transcripts (nGene and nUMI, respectively) of each cell were generated and outliers with too high or too low nGene and nUMI were removed to exclude potential cell doublets/aggregates or low quality cells/cell debris, respectively. As sequencing depth and cell type compositions vary across different samples, this filtration step was performed on a sample-to-sample basis. In general, prenatal and hPSC-derived samples were filtered with a minimum nGene of 500–1000. We consistently observed lower number of genes expressed from postnatal juvenile and adult samples, although in general they have the lowest unique read fraction levels (suggesting higher sequencing coverage) among all samples. Therefore, we set the nGene threshold of these samples to 250–400. After the cell filtration step, expression counts of each cell were normalized with the default Seurat setting using “NormalizeData”. To mitigate the cell cycle effects on potentially grouping different cell types based on their cell cycle states, we assigned “S.Score” and “G2M.Score” on each cell with the average normalized expression levels of core cell cycle genes using “CellCycleScoring” following the Seurat instructions (Tirosh et al., 2016). To reduce the effects of dissociation-related stress on gene expression analysis, we obtained the core stress genes identified from scRNA-seq studies on both mouse skeletal muscle and acinar (van den Brink et al., 2017) (Table S7), and assigned each cell a “Stress” score using the core stress gene list through the Seurat “AddModuleScore” function. Briefly, this function first assigned each of the genes to be analyzed into different bins based on the genes’ average expression across single cells. It then calculated a residual for each analyzed gene in each cell by subtracting the average expression of the control gene set from the expression level of the gene being analyzed, where the control genes were randomly selected from the bin that the analyzed gene was assigned to. This process was then reiterated through all the genes in the provided list, and the resulted aggregated expression was assigned as the score of the property the provided gene list represents. After this step, data scaling was performed using “ScaleData”, with “S.Score”, “G2M.Score” and “Stress” passed onto the “vars.to.regress” argument. At the same time, “nMUI” was also included in regression to control for the effects of cell size and/or sequencing depth.
Muscle.Score and Dev.Score
To readily detect skeletal muscle cells at various developmental or differentiation states, we assigned each cell a “Muscle.Score” using the above described “AddModuleScore” function using a list of conserved muscle cell genes (PAX3, PAX7, PITX2, MYF5, MYF6, MYOD1, MYOG, NEB and MYH3). To quantify the developmental status of myogenic progenitor and stem cells, we first used “AddModuleScore” to assign each cell a postnatal score (“Pst.Score”) using genes that were found to be upregulated in stage 5 SCs compared to stage 1 and 2 embryonic SMPCs (Figure 4 and Table S3). Similarly, we assigned each cell an embryonic score (“Emb.Score”) using genes upregulated in stage 1 and 2 SMPCs compared to stage 5 SCs. Finally, we calculated each cell’s myogenic developmental score (“Dev.Score”) by subtracting its “Emb.Score” from “Pst.Score”. Thus, a cell with a developmental “age” close to postnatal SCs would have a higher value of “Dev.Score”, and that similar to embryonic SMPCs a lower value.
Dimensional reduction and clustering
First, the most highly variable genes within each Seurat object were calculated and selected using “FindVariableGenes” (1500–2500 genes). Principle components (PCs) were calculated using the selected top variable genes by “RunPCA”, and the PCs were plotted using “PCElbowPlot”. Significant PCs were selected based on the elbow plot and used to further reduce data dimensionality using the T-distributed stochastic neighbor embedding (tSNE) method by “RunTSNE”. Cell clustering was performed by a shared nearest neighbor (SNN) modularity optimization based clustering algorithm using the Seurat function “FindClusters” with “reduction.type” set to “pca”. Identification of clusters/cell types were aided by known cell type specific markers as well as the distribution of cells on the tSNE space.
Differential gene expression analysis
Differentially expressed genes (DEGs) between one cell cluster versus all remaining cells or between individual clusters were identified by “FindAllMarkers” or “FindMarkers”, respectively. For both functions, “test.use” was set to “negbinom” to fit for the sparse data type generated from scRNA-seq, and “return.thresh” (p values) less than 0.01 (finding cluster markers) or 0.05 (comparing two clusters). We passed the same parameters as we did when scaling the data (“S.Score”, “G2M.Score”, “Stress” and “nUMI”) to the “latent.var” argument to regress out the effects of cell cycle, dissociation-related stress as well as cell size/sequencing depth on the identification of DEGs. In addition, DEGs must also meet the following default criteria in Seurat: 1) average expression difference exceeding 1.28-fold between the comparing group of cells (“logfc.threshold = 0.25”), and 2) detected in a minimum of 10% of cells in either of the comparing populations (“min.pct = 0.1”).
Trajectory analysis
For trajectory analysis, we reduced the dimensionality of the data by diffusion map (DM) (Haghverdi et al., 2015) using the top variable genes of the objects via the Seurat “RunDiffusion” function. For in vivo SMPC and SC only analysis, we further clustered the cells using “FindClusters” with “reduction.type = “dm” (using the first 2 DM dimensions) into distinct developmental stages. For analysis combining in vivo SMPCs and SCs as well as hPSC-SMPCs, “RunDiffusion” was performed using the top variable genes from the in vivo only dataset as a reference gene set. The developmental stage labels of the in vivo cells and the sample identities of the hPSC-derived cells were transferred and maintained from the original objects.
Analysis using Monocle3
Analysis in the Monocle3 R package (Cao et al., 2019) was performed according to Trapnell lab guidelines (https://cole-trapnell-lab.github.io/monocle3/monocle3_docs/). Gene expression data and cell metadata including cell type labels were carried over from the Seurat object. Parameters to regress were set similarly to analysis in Seurat by passing “nUMI”, “S.Score” “G2M.Score” and “Stress” to the “residual_model_formula_str” argument in the “preprocess_cds” function. Significant PCs were calculated and selected to further reduce the data dimensionality using uniform manifold approximation and projection (UMAP). Cells were plotted onto the UMAP space for visualization of their distribution and cell type identities.
Gene ontology enrichment analysis
Gene ontology (GO) enrichment was performed using Metascape (http://metascape.org/gp/index.html#/main/step1) (Zhou et al., 2019) against GO terms belonging to “Biological Processes”. Enriched GO terms with similar properties were further assigned to a common group, and the top 20 groups were retrieved. Select representative GO terms (members) from the consolidated groups were plotted against their negative Log10-transformed p values (no more than one member was selected from each group).
Gene set enrichment analysis
The gene set enrichment analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp) (Subramanian et al., 2005) was performed with the “GSEAPreranked” mode against the “Canonical Pathways” (c2.cp.v6.1) gene sets database. The “enrichment statistic” was set to “classic” and enriched gene sets containing more than 500 or less than 10 genes were excluded from the final enriched gene sets. The “normalization mode” was set to “meandiv” and permutations were performed 1000 times.
Co-regulated gene network analysis
To build the co-regulated gene network, the dataset containing all stages of in vivo SMPCs and SCs and in vitro hPSC-SMPCs derived using the HX protocol (Figure 7A), was used to compute a Pearson gene-to-gene correlation matrix and determine groups/networks including genes with correlation values greater than 0.125. Similar networks were condensed by segregation of cells into ample numbers of small cell clusters (roughly 50 cells per cluster), from which the expression of the primary networks was calculated and compared to each other again via a Pearson network-to-network correlation matrix, followed by merging similar networks with expression correlation of 0.7 or higher to generate the final networks. The expression level of a given gene group/network was calculated by averaging the normalized expression values of all genes in the group in a given cell. We manually inspected the gene groups to exclude those that were driven by an extremely high expression level of a few genes in random rare cells. To retrieve TFs from the gene groups, we intersected our identified genes with those annotated as transcription factors/regulators by the Animal Transcription Factor Database (bioinfo.life.hust.edu.cn/AnimalTFDB/). To mitigate the effects of tissue/cell dissociation-induced stress signatures, we compiled a common stress gene list (411 genes; Table S7) from published literature. Genes included in this list were chosen based on the following criteria: 1) included in the stress regression gene list as described above, or 2) significantly changed in the same direction (both induced or reduced) in response to dissociation-related stress as reported by van Velthoven et al and Machado et al (Machado et al., 2017; van Velthoven et al., 2017). Mouse genes were converted to their homologs in human and those mice only genes were removed from the final list. These common stress genes were intersected with the gene groups and TF sub-lists to exclude them from the final gene/TF lists for downstream analysis.
For gene group heatmaps, the average expression of selected groups was calculated for each developmental or directed differentiation stage/sample using the Seurat “AverageExpression” function. Only groups containing 50 or more genes were plotted. For GO analysis, all genes contained in a given group were used as input to Metascape for enrichment analysis against “GO Biological Processes”.
Hierarchical clustering
Average gene expression levels of single cells belonging to the same groups were calculated using the Seurat “AverageExpression” function. Spearman correlation coefficients were calculated between the averaged group of cells and visualized using the R package pheatmap. Hierarchical clustering was performed via the same package with default settings.
Gene list intersection and Venn diagram generation
Individual gene lists were supplied as input to the R package eulerr. All possible intersections of input gene lists were calculated and visualized in Venn diagram format with region areas proportional to the number of events in the regions.
Supplementary Material
Table S3. Related to Figure 4. DEG list of in vivo different stage SMPCs and SCs.
Table S5. Related to Figure 7. DEG list of in vivo stage 2 and 3 compared to in vitro hPSC-derived SMPCs.
Table S6. Related to Figure 7. Co-regulated gene groups and gene list from network analysis.
Table S7. Related to STAR Methods. Stress genes list.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-PAX3 | DSHB | Cat#: Pax3; RRID: AB_528426 |
| Anti-PAX7 | DSHB | Cat#: PAX7; RRID: AB_528428 |
| Anti-MyHC | DSHB | Cat#: MF 20; RRID: AB_2147781 |
| Anti-MET | Cell Signaling Technology | Cat# 8198; RRID: AB_10858224 |
| Anti-CD325 (CDH2) | BD Biosciences | Cat#: 561553; RRID: AB_10713831 |
| Anti-NANOG | Cell Signaling Technology | Cat# 3580; RRID: AB_2150399 |
| Anti-OCT-4A | Cell Signaling Technology | Cat# 2840; RRID: AB_2167691 |
| Anti-SOX2 | Cell Signaling Technology | Cat# 3579; RRID: AB_2195767 |
| Anti-PDGF Receptor alpha (PDGFRA) | Cell Signaling Technology | Cat# 3174; RRID: AB_2162345 |
| Anti-M-Cadherin/Cadherin-15 (CDH15) | R&D Systems | Cat#: AF4096; RRID: AB_10641849 |
| Anti-HGFR/c-MET (MET) - APC | R&D Systems | Cat#: FAB3582A; RRID: AB_1151927 |
| Anti-CD325 (CDH2) - PE | BD Biosciences | Cat#: 561554; RRID: AB_10714646 |
| Anti-CD31 - FITC | Thermo Fisher Scientific | Cat#: 11–0319-42; RRID: AB_2043835 |
| Anti-CD31 - PE | Thermo Fisher Scientific | Cat#: 12–0319-42; RRID: AB_10669160 |
| Anti-CD31 - APC/Cy7 | BioLegend | Cat#: 303120; RRID: AB_10640734 |
| Anti-CD45 - FITC | Thermo Fisher Scientific | Cat#: 11–0459-42; RRID: AB_10852703 |
| Anti-CD45 - PE | Thermo Fisher Scientific | Cat#: 12–0459-42; RRID: AB_1724079 |
| Anti-CD45 - APC/Cy7 | BioLegend | Cat#: 368516; RRID: AB_2566376 |
| Anti-CD235a - FITC | Thermo Fisher Scientific | Cat#: 11–9987-82; RRID: AB_465477 |
| Anti-CD235a - PE | Thermo Fisher Scientific | Cat#: 12–9987-82; RRID: AB_466300 |
| Anti- CD235a - APC/Cy7 | BioLegend | Cat#: 349116; RRID: AB_ 2650978 |
| Anti-ERBB3 - PE | BioLegend | Cat#: 324706; RRID: AB_2099569 |
| Anti-CD271 (NGFR) - PerCP/Cy5.5 | BioLegend | Cat#: 345111; RRID: AB_11204078 |
| Anti-CD57 (HNK-1) - PE/Cy7 | BioLegend | Cat#: 359624; RRID: AB_2632689 |
| Anti-CD140a (PDGFRA) - PE | BD Biosciences | Cat#: 556002; RRID: AB_396286 |
| Anti-CD140a (PDGFRA) - BV786 | BD Biosciences | Cat#: 742669; RRID: AB_2740957 |
| Anti-Rabbit IgG - Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat#: A32731; RRID: AB_2633280 |
| Anti-Mouse IgG2b - Alexa Fluor 568 | Thermo Fisher Scientific | Cat#: A21144; RRID: AB_ 2535780 |
| Anti-Sheep IgG - PerCP | Thermo Fisher Scientific | Cat#: A32731; RRID: AB_2633280 |
| Anti-Mouse IgG1 - HRP | Thermo Fisher Scientific | Cat#: A10551; RRID: AB_2534048 |
| Anti-Mouse IgG2a - HRP | Thermo Fisher Scientific | Cat#: A10685; RRID: AB_2534065 |
| Anti-Mouse IgG2b - HRP | Thermo Fisher Scientific | Cat#: M32507; RRID: AB_2536649 |
| Anti-Rabbit IgG - HRP | Promega | Cat#: W4011; RRID: AB_430833 |
| Anti-Sheep IgG - PerCP | R&D Systems | Cat#: F0128; RRID: AB_10892337 |
| Anti-Sheep IgG - NL557 | R&D Systems | Cat#: NL010; RRID: AB_884220 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Human embryos, fetuses and juvenile and adult skeletal muscle tissues | Table S1 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Y-27632 | Tocris | Cat#: 1254; CAS#: 129830–38-2 |
| CHIR99021 | Tocris | Cat#: 4423; CAS#: 252917–06-9 |
| LDN193189 | Tocris | Cat#: 6053; CAS#: 1435934–00-1 |
| SB431542 | Tocris | Cat#: 1614; CAS#: 301836–41-9 |
| FGF2 | Proteintech | Cat#: HZ-1285 |
| HGF | PeproTech | Cat#: 100–39H |
| IGF1 | Sigma-Aldrich | Cat#: I1271 |
| mTeSR1 | StemCell Technologies | Cat#: 85850 |
| DMEM/F-12, HEPES medium | Thermo Fisher Scientific | Cat#: 11330032 |
| DMEM, high glucose medium | Thermo Fisher Scientific | Cat#: 11965092 |
| E6 medium | Thermo Fisher Scientific | Cat#: A4238501 |
| StemPro-34 medium | Thermo Fisher Scientific | Cat#: 10639011 |
| SkGM-2 Skeletal Muscle Cell Growth Medium-2 BulletKit | Lonza | Cat#: CC-3245 |
| StemPro Osteogenesis Differentiation Kit | Thermo Fisher Scientific | Cat#: A1007201 |
| ITS -G supplement | Thermo Fisher Scientific | Cat#: 41400045 |
| N2 supplement | Thermo Fisher Scientific | Cat#: 17502048 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | Cat#: 16000044 |
| Knockout Serum Replacement (KSR) | Thermo Fisher Scientific | Cat#: 10828028 |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat#: 35050061 |
| Nonessential amino acids (NEAA) | Thermo Fisher Scientific | Cat#: 11140076 |
| Sodium Pyruvate | Thermo Fisher Scientific | Cat#: 11360070 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat#: 21985023; CAS#: 60–24-2 |
| 1-Thioglycerol | Sigma-Aldrich | Cat#: M6145; CAS#: 96–27-5 |
| Matrigel hESC-Qualified | Corning | Cat#: 354277 |
| TrypLE Express | Thermo Fisher Scientific | Cat#: 12605010 |
| Collagenase, Type 2 (Collagenase II) | Worthington-Biochem | Cat#: LS004177 |
| Collagenase IV | Thermo Fisher Scientific | Cat#: 17104019 |
| Dispase II | Thermo Fisher Scientific | Cat#: 17105041 |
| DNase I | Sigma-Aldrich | Cat#: D4513 |
| Critical Commercial Assays | ||
| PDMS chip - 26 Drop-seq generators on glass slide | FlowJEM | N/A |
| Barcoded beads for Drop-seq | ChemGenes | Cat#: MACOSKO-2011–1-0(V+) |
| Human TruStain FcX (Fc Receptor Blocking Solution) | BioLegend | Cat#: 422302 |
| Alizarin S, 2% Solution, pH 4.2 | Electron Microscopy Sciences | Cat#: 26206–01 |
| TSA Plus Fluorescein | PerkinElmer | Cat#: NEL741001KT |
| TSA Plus TMR | PerkinElmer | Cat#: NEL742001KT |
| TSA Plus Cyanine 5 | PerkinElmer | Cat#: NEL745001KT |
| RNAscope Multiplex Fluorescent Reagent Kit v2 | Advanced Cell Diagnostics | Cat#: 323100 |
| RNAscope 3-plex negative control probes | Advanced Cell Diagnostics | Cat#: 320871 |
| RNAscope Probe - Hs-NFIX | Advanced Cell Diagnostics | Cat#: 522341 |
| RNAscope Probe - Hs-KLF9-C2 | Advanced Cell Diagnostics | Cat#: 582551-C2 |
| RNAscope Probe - Hs-NFIC | Advanced Cell Diagnostics | Cat#: N/A; custom ordered new probe |
| RNAscope Probe - Hs-CEBPD | Advanced Cell Diagnostics | Cat#: N/A; custom ordered new probe |
| RNeasy Plus Mini Kit | QIAGEN | Cat#: 74134 |
| RNeasy Plus Micro Kit | QIAGEN | Cat#: 74034 |
| iScript Reverse Transcription Supermix for RT-qPCR | Bio-Rad | Cat#: 1708841 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat#: 1725274 |
| Gibson Assembly Cloning Kit | New England BioLabs | Cat#: E5510S |
| NEBuilder HiFi DNA Assembly Master Mix | New England BioLabs | Cat#: E2621S |
| P3 Primary Cell 4D-Nucleofector X Kit L | Lonza | Cat#: V4XP-3024 |
| ViaFect Transfection Reagent | Promega | Cat#: E4981 |
| Deposited Data | ||
| Raw sequencing reads and processed DGE matrices | This paper | NCBI GEO accession#: GSE147457 |
| Interactive scRNA-seq exploration tools | This paper | skeletal-muscle.cells.ucsc.edu or aprilpylelab.com/datasets |
| Experimental Models: Cell Lines | ||
| Human: H9 (WA09) hESC line | WiCell | RRID: CVCL_9773 |
| Human: PAX7-GFP reporter lines derived from H9 cells | This Paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Forward sequence for cloning gRNA for homologous recombination of reporter cassette to the PAX7 locus: GTGAGTGGGTGTACGTGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | This paper | N/A |
| Reverse sequence for cloning gRNA for homologous recombination of reporter cassette to the PAX7 locus: CACGTACACCCACTCACGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAA | This paper | N/A |
| Forward sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C3): GTCAAACGCGTCCAGAAGCTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | This paper | N/A |
| Reverse sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C3): AGCTTCTGGACGCGTTTGACGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAA | This paper | N/A |
| Forward sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C4): GGGGCCAAAGTTTCCGAGCCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | This paper | N/A |
| Reverse sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C4): GGCTCGGAAACTTTGGCCCCGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAA | This paper | N/A |
| Forward sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C5): GGGTCCGGAGAAAGAAGGCGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | This paper | N/A |
| Reverse sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C5): CGCCTTCTTTCTCCGGACCCGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAA | This paper | N/A |
| Forward sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C6): GCCCCGGCTCGACCTCGTTTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTC | This paper | N/A |
| Reverse sequence for cloning gRNA targeting PAX7 promoter region for gene activation (Pax7C6): AAACGAGGTCGAGCCGGGGCGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAA | This paper | N/A |
| Primer pairs for qRT-PCR |
Xi et al., 2017 Methods S1 |
N/A |
| Recombinant DNA | ||
| gRNA_Cloning Vector | Mali et al., 2013 | RRID: Addgene_41824 |
| hCas9 | Mali et al., 2013 | RRID: Addgene_41815 |
| Oct4-IRES-eGFP-PGK-Neo | Yang et al., 2013 | RRID: Addgene_48681 |
| SP-dCas9-VPR | Chavez et al., 2015 | RRID: Addgene_63798 |
| Software and Algorithms | ||
| Drop-seq_tools-1.13 | Macosko et al., 2015 | https://github.com/broadinstitute/Drop-seq/releases/tag/v1.13 |
| Bowtie2 v2.2.9 | Langmead et al., 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Bedtools v2.26.0 | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| Seurat v2.3.3 | Butler et al., 2018 | https://github.com/satijalab/seurat/releases/tag/v2.3.3 |
| Monocle3 | Cao et al., 2019 | https://cole-trapnell-lab.github.io/monocle3/monocle3_docs/ |
| Pheatmap v1.0.12 | The Comprehensive R Archive Network | https://CRAN.R-project.org/package=pheatmap |
| Eulerr v6.0.0 | The Comprehensive R Archive Network | https://CRAN.R-project.org/package=eulerr |
| Metascape | Zhou et al., 2019 | http://metascape.org/gp/index.html#/main/step1 |
| GSEA | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| Gene network analysis | This paper | In STAR Methods; Will be provided upon request |
| Fiji/ImageJ | Schindelin et al., 2012 | http://fiji.sc/ |
| ZEN 3.1 (blue edition) and ZEN 2.6 Pro (blue edition) | ZEISS | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html |
| PrimerBank | Spandidos et al., 2010 | https://pga.mgh.harvard.edu/primerbank/ |
| Primer-BLAST | Ye et al., 2012 | https://www.ncbi.nlm.nih.gov/tools/primer-blast/ |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| Other | ||
Highlights.
Human atlas of limb skeletal muscle in embryonic, fetal and adult tissues
Human limb skeletal muscle populations and supportive cells vary across development
PAX7 muscle progenitor and stem cells are not identical across developmental states
hPSC-PAX7 cells align to the embryonic-to-fetal transition in human development
Acknowledgments
We thank the Flow Cytometry, High Throughput Sequencing and Stem Cell Bank cores from the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research (BSCRC) at UCLA. We acknowledge the Technology Center for Genomics & Bioinformatics, Translational Pathology and Janis V. Giorgi Flow Cytometry cores at UCLA Jonsson Comprehensive Cancer Center (JCCC). We also thank the UCLA Center for AIDS Research (NIH P30 CA016042 and 5P30 AI028697). We greatly appreciate Matthew Speir and Maximillian Haeussler for incorporating scRNA-seq data to the UCSC Cell Browser for interactive exploration. We thank KS-L for Deutsche Forschungsgemeinschaft (SCHE701/14-1) and the Ministry of Science, Research and Arts of Baden Württemberg (Az.: 33-729.55-3/214-8). ADP is funded by CIRM Quest (DISC2-10696), NIAMS (R01 AR064327), UCLA BSCRC, the Rose Hills Foundation and the Ablon Scholars Award. KP is funded by NIH (P01 GM099134), UCLA BSCRC, JCCC and David Geffen School of Medicine, as well as a Faculty Scholar grant from the Howard Hughes Medical Institute. JL is supported by the UCLA Tumor Cell Biology Training Program (USHHS Ruth L. Kirschstein Institutional NRSA T32 CA009056). SS is supported by the Rose Hills Foundation award from BSCRC at UCLA and is a UCLA graduate dissertation year fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
CSY, MS and ADP are co-founders of and have financial interests in Myogene Bio. The Regents of the University of California have licensed intellectual property invented by CSY, MS and ADP to Myogene Bio. MS and ADP serve on the scientific advisory board of Myogene Bio. CSY is currently CEO of Myogene Bio. The other authors declare no competing interests.
References
- Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, and de Crombrugghe B (2005). Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. U. S. A 102, 14665–14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MS, Rozkalne A, Colletta A, Spinazzola JM, Johnson S, Rahimov F, Meng H, Lawlor MW, Estrella E, Kunkel LM, and Gussoni E (2016). CD82 Is a Marker for Prospective Isolation of Human Muscle Satellite Cells and Is Linked to Muscular Dystrophies. Cell. Stem Cell 19, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applebaum M, and Kalcheim C (2015). Mechanisms of myogenic specification and patterning. Results Probl. Cell Differ 56, 77–98. [DOI] [PubMed] [Google Scholar]
- Bareja A, Holt JA, Luo G, Chang C, Lin J, Hinken AC, Freudenberg JM, Kraus WE, Evans WJ, and Billin AN (2014). Human and mouse skeletal muscle stem cells: convergent and divergent mechanisms of myogenesis. PLoS One 9, e90398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M, and Niswander L (2007). Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev. Cell 12, 931–941. [DOI] [PubMed] [Google Scholar]
- Barruet E, Garcia SM, Striedinger K, Wu J, Lee S, Byrnes L, Wong A, Xuefeng S, Tamaki S, Brack AS, and Pomerantz JH (2020). Functionally heterogeneous human satellite cells identified by single cell RNA sequencing. Elife 9, 10.7554/eLife.51576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle M, Godefroy D, Dominici C, Heitz-Marchaland C, Zelina P, Hellal F, Bradke F, and Chedotal A (2014). A simple method for 3D analysis of immunolabeled axonal tracts in a transparent nervous system. Cell. Rep 9, 1191–1201. [DOI] [PubMed] [Google Scholar]
- Borchin B, Chen J, and Barberi T (2013). Derivation and FACS-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent stem cells. Stem Cell. Reports 1, 620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent AE, and Tabin CJ (2002). Developmental regulation of somite derivatives: muscle, cartilage and tendon. Curr. Opin. Genet. Dev 12, 548–557. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C, and Shendure J (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni A, Hettmer S, Lynes MD, Rao TN, Tchessalova D, Sinha I, Lee BT, Tseng YH, and Wagers AJ (2014). Isolation of progenitors that exhibit myogenic/osteogenic bipotency in vitro by fluorescence-activated cell sorting from human fetal muscle. Stem Cell. Reports 2, 92–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerletti M, Shadrach JL, Jurga S, Sherwood R, and Wagers AJ (2008). Regulation and function of skeletal muscle stem cells. Cold Spring Harb. Symp. Quant. Biol 73, 317–322. [DOI] [PubMed] [Google Scholar]
- Chal J, Al Tanoury Z, Hestin M, Gobert B, Aivio S, Hick A, Cherrier T, Nesmith AP, Parker KK, and Pourquie O (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc 11, 1833–1850. [DOI] [PubMed] [Google Scholar]
- Chal J, Oginuma M, Al Tanoury Z, Gobert B, Sumara O, Hick A, Bousson F, Zidouni Y, Mursch C, Moncuquet P, et al. (2015). Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol 33, 962–969. [DOI] [PubMed] [Google Scholar]
- Chal J, and Pourquie O (2017). Making muscle: skeletal myogenesis in vivo and in vitro. Development 144, 2104–2122. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, and Wang DZ (2010). microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol 190, 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, Tai A, Lu PJ, and Cheah KS (2019). Acquisition of multipotent and migratory neural crest cells in vertebrate evolution. Curr. Opin. Genet. Dev 57, 84–90. [DOI] [PubMed] [Google Scholar]
- De Micheli AJ, Laurilliard EJ, Heinke CL, Ravichandran H, Fraczek P, Soueid-Baumgarten S, De Vlaminck I, Elemento O, and Cosgrove BD (2020). Single-Cell Analysis of the Muscle Stem Cell Hierarchy Identifies Heterotypic Communication Signals Involved in Skeletal Muscle Regeneration. Cell. Rep 30, 3583–3595.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Orso S, Juan AH, Ko KD, Naz F, Perovanovic J, Gutierrez-Cruz G, Feng X, and Sartorelli V (2019). Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 146, 10.1242/dev.174177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamini V, Ghadiali RS, Antczak P, Rothwell A, Turnbull JE, and Pisconti A (2018). The Satellite Cell Niche Regulates the Balance between Myoblast Differentiation and Self-Renewal via p53. Stem Cell. Reports 10, 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, and Joung JK (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol 32, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordani L, He GJ, Negroni E, Sakai H, Law JYC, Siu MM, Wan R, Corneau A, Tajbakhsh S, Cheung TH, and Le Grand F (2019). High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol. Cell 74, 609–621.e6. [DOI] [PubMed] [Google Scholar]
- Gopinath SD, Webb AE, Brunet A, and Rando TA (2014). FOXO3 promotes quiescence in adult muscle stem cells during the process of self-renewal. Stem Cell. Reports 2, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros J, and Tabin CJ (2014). Vertebrate limb bud formation is initiated by localized epithelial-to-mesenchymal transition. Science 343, 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Buettner F, and Theis FJ (2015). Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998. [DOI] [PubMed] [Google Scholar]
- Hayashi K, and Ozawa E (1995). Myogenic cell migration from somites is induced by tissue contact with medial region of the presumptive limb mesoderm in chick embryos. Development 121, 661–669. [DOI] [PubMed] [Google Scholar]
- Hicks MR, Hiserodt J, Paras K, Fujiwara W, Eskin A, Jan M, Xi H, Young CS, Evseenko D, Nelson SF, et al. (2018). ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell Biol 20, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, and Rossi FM (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol 12, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Magli A, Chan SSK, Oliveira VKP, Wu J, Darabi R, Kyba M, and Perlingeiro RCR (2017). Expansion and Purification Are Critical for the Therapeutic Application of Pluripotent Stem Cell-Derived Myogenic Progenitors. Stem Cell. Reports 9, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivela R, Salmela I, Nguyen YH, Petrova TV, Koistinen HA, Wiener Z, and Alitalo K (2016). The transcription factor Prox1 is essential for satellite cell differentiation and muscle fibre-type regulation. Nat. Commun 7, 13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman R, Ly CH, and Ryall JG (2014). A metabolic link to skeletal muscle wasting and regeneration. Front. Physiol 5, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, and Kain SR (1998). Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem 273, 34970–34975. [DOI] [PubMed] [Google Scholar]
- Machado L, Esteves de Lima J, Fabre O, Proux C, Legendre R, Szegedi A, Varet H, Ingerslev LR, Barres R, Relaix F, and Mourikis P (2017). In Situ Fixation Redefines Quiescence and Early Activation of Skeletal Muscle Stem Cells. Cell. Rep 21, 1982–1993. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magli A, and Perlingeiro RRC (2017). Myogenic progenitor specification from pluripotent stem cells. Semin. Cell Dev. Biol 72, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell BB, and Yang VW (2010). Mammalian Kruppel-like factors in health and diseases. Physiol. Rev 90, 1337–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina G, Biressi S, Monteverde S, Magli A, Cassano M, Perani L, Roncaglia E, Tagliafico E, Starnes L, Campbell CE, et al. (2010). Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 140, 554–566. [DOI] [PubMed] [Google Scholar]
- Murmann OV, Niggli F, and Schafer BW (2000). Cloning and characterization of the human PAX7 promoter. Biol. Chem 381, 331–335. [DOI] [PubMed] [Google Scholar]
- Neufeld SJ, Wang F, and Cobb J (2014). Genetic interactions between Shox2 and Hox genes during the regional growth and development of the mouse limb. Genetics 198, 1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oginuma M, Moncuquet P, Xiong F, Karoly E, Chal J, Guevorkian K, and Pourquie O (2017). A Gradient of Glycolytic Activity Coordinates FGF and Wnt Signaling during Elongation of the Body Axis in Amniote Embryos. Dev. Cell 40, 342–353.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, and Jang J (2019). Directed Differentiation of Pluripotent Stem Cells by Transcription Factors. Mol. Cells 42, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pala F, Di Girolamo D, Mella S, Yennek S, Chatre L, Ricchetti M, and Tajbakhsh S (2018). Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J. Cell. Sci 131, 10.1242/jcs.212977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petchey LK, Risebro CA, Vieira JM, Roberts T, Bryson JB, Greensmith L, Lythgoe MF, and Riley PR (2014). Loss of Prox1 in striated muscle causes slow to fast skeletal muscle fiber conversion and dilated cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A 111, 9515–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistocchi A, Gaudenzi G, Foglia E, Monteverde S, Moreno-Fortuny A, Pianca A, Cossu G, Cotelli F, and Messina G (2013). Conserved and divergent functions of Nfix in skeletal muscle development during vertebrate evolution. Development 140, 1528–1536. [DOI] [PubMed] [Google Scholar]
- Price FD, von Maltzahn J, Bentzinger CF, Dumont NA, Yin H, Chang NC, Wilson DH, Frenette J, and Rudnicki MA (2014). Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med 20, 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt R, Gullotta F, Nusspaumer G, Unal E, Ivanek R, Zuniga A, and Zeller R (2019). Molecular signatures identify immature mesenchymal progenitors in early mouse limb buds that respond differentially to morphogen signaling. Development 146, 10.1242/dev.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein AB, Smith GR, Raue U, Begue G, Minchev K, Ruf-Zamojski F, Nair VD, Wang X, Zhou L, Zaslavsky E, et al. (2020). Single-cell transcriptional profiles in human skeletal muscle. Sci. Rep 10,229-019-57110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG (2013). Metabolic reprogramming as a novel regulator of skeletal muscle development and regeneration. Febs j. 280, 4004–4013. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M, and Sartorelli V (2015). The NAD(+) -dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell. Stem Cell 16, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A, Doyonnas R, Kraft P, Vitorovic S, and Blau HM (2008). Self-renewal and expansion of single transplanted muscle stem cells. Nature 456, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R, and Tajbakhsh S (2007). Skeletal muscle stem cell birth and properties. Semin. Cell Dev. Biol 18, 870–882. [DOI] [PubMed] [Google Scholar]
- Sanchez AM, Candau RB, and Bernardi H (2014). FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol. Life Sci 71, 1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, and Sandri M (2015). Bone and morphogenetic protein signalling and muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 18, 215–220. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, and Reggiani C (2015). Developmental myosins: expression patterns and functional significance. Skelet Muscle 5, 22–015–0046–6. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, and Plath K (2012). The roles of the reprogramming factors Oct4, Sox2 and Klf4 in resetting the somatic cell epigenome during induced pluripotent stem cell generation. Genome Biol. 13, 251–2012-13–10-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea KL, Xiang W, LaPorta VS, Licht JD, Keller C, Basson MA, and Brack AS (2010). Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell. Stem Cell 6, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton M, Metz J, Liu J, Carpenedo RL, Demers SP, Stanford WL, and Skerjanc IS (2014). Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell. Reports 3, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, and Seed B (2010). PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 38, D792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, and Principal investigators. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglietti V, Angelini G, Mura G, Bonfanti C, Caruso E, Monteverde S, Le Carrou G, Tajbakhsh S, Relaix F, and Messina G (2018). RhoA and ERK signalling regulate the expression of the transcription factor Nfix in myogenic cells. Development 145, 10.1242/dev.163956. [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2016). A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol 17, 183–193. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Matsumoto E, Higashimaki Y, Katagiri T, Sugimoto T, Seino S, and Kaji H (2012). Role of osteoglycin in the linkage between muscle and bone. J. Biol. Chem 287, 11616–11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, and Sacco A (2014). STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med 20, 1182–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney MT, Gromova A, Sesillo FB, Sala D, Spenle C, Orend G, and Sacco A (2016). Autonomous Extracellular Matrix Remodeling Controls a Progressive Adaptation in Muscle Stem Cell Regenerative Capacity during Development. Cell. Rep 14, 1940–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney MT, and Sacco A (2016). Satellite Cell Heterogeneity in Skeletal Muscle Homeostasis. Trends Cell Biol. 26, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, and Tsuchida K (2010). Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol 12, 143–152. [DOI] [PubMed] [Google Scholar]
- Uezumi A, Nakatani M, Ikemoto-Uezumi M, Yamamoto N, Morita M, Yamaguchi A, Yamada H, Kasai T, Masuda S, Narita A, et al. (2016). Cell-Surface Protein Profiling Identifies Distinctive Markers of Progenitor Cells in Human Skeletal Muscle. Stem Cell. Reports 7, 263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vertesy A, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936. [DOI] [PubMed] [Google Scholar]
- van Velthoven CTJ, de Morree A, Egner IM, Brett JO, and Rando TA (2017). Transcriptional Profiling of Quiescent Muscle Stem Cells In Vivo. Cell. Rep 21, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Fujiwara W, Gonzalez K, Jan M, Liebscher S, Van Handel B, Schenke-Layland K, and Pyle AD (2017). In Vivo Human Somitogenesis Guides Somite Development from hPSCs. Cell. Rep 18, 1573–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Tabebordbar M, Iovino S, Ciarlo C, Liu J, Castiglioni A, Price E, Liu M, Barton ER, Kahn CR, Wagers AJ, and Zon LI (2013). A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species. Cell 155, 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wilschut KJ, Kouklis G, Tian H, Hesse R, Garland C, Sbitany H, Hansen S, Seth R, Knott PD, Hoffman WY, and Pomerantz JH (2015). Human Satellite Cell Transplantation and Regeneration from Diverse Skeletal Muscles. Stem Cell. Reports 5, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima H, Yoneitamura S, Watanabe N, Tamura K, and Ide H (1999). Role of N-cadherin in the sorting-out of mesenchymal cells and in the positional identity along the proximodistal axis of the chick limb bud. Dev. Dyn 216, 274–284. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, and Jaenisch R (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, and Madden TL (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134–2105–13–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Price F, and Rudnicki MA (2013). Satellite cells and the muscle stem cell niche. Physiol. Rev 93, 23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yucel N, Wang YX, Mai T, Porpiglia E, Lund PJ, Markov G, Garcia BA, Bendall SC, Angelo M, and Blau HM (2019). Glucose Metabolism Drives Histone Acetylation Landscape Transitions that Dictate Muscle Stem Cell Function. Cell. Rep 27, 3939–3955.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, and Hoffman EP (2004). Embryonic myogenesis pathways in muscle regeneration. Dev. Dyn 229, 380–392. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523–019–09234–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Related to Figure 4. DEG list of in vivo different stage SMPCs and SCs.
Table S5. Related to Figure 7. DEG list of in vivo stage 2 and 3 compared to in vitro hPSC-derived SMPCs.
Table S6. Related to Figure 7. Co-regulated gene groups and gene list from network analysis.
Table S7. Related to STAR Methods. Stress genes list.
Data Availability Statement
Both raw sequencing reads and processed digital gene expression (DGE) matrices of scRNA-seq datasets are deposited at NCBI GEO with accession number GSE147457. Interactive scRNA-seq data exploration can be accessed at skeletal-muscle.cells.ucsc.edu or aprilpylelab.com/datasets. General codes for computational analysis follow the instructions of the respective software and customized modifications will be available upon request.