

Summary
Faithful maintenance of immune homeostasis relies on the capacity of the cellular immune surveillance machinery to recognize ‘nonself’, such as the presence of pathogenic RNA. Several families of pattern-recognition receptors exist that detect immunostimulatory RNA and then induce cytokine-mediated antiviral and proinflammatory responses. Here we review the distinct features of bona fide RNA sensors – Toll-like receptors and RIG-I-like receptors in particular – with a focus on their functional specificity imposed by cell type-dependent expression, subcellular localization, and ligand preference. Furthermore, we highlight recent advances on the roles of NOD-like receptors and DEAD-box or DEAH-box RNA helicases in an orchestrated RNA-sensing network and also discuss the relevance of RNA sensor polymorphisms in human disease.
eTOC Blurb:
Liu and Gack summarize recent advances on the physiological role of classical and emerging RNA sensors and co-sensors in innate immunity, their ‘cross-talk’ to form an orchestrated RNA-sensing network, and the relevance of polymorphisms in RNA sensors for human disease.
eTOC Blurb:
Recent advances identify several RNA sensors and co-sensors of innate immunity which are critical for the detection of viral pathogens. Gack and colleague review the individual roles of these sensors, their ‘cross-talk’ to form an orchestrated RNA-sensing network, and the relevance of polymorphisms in RNA sensors for human disease.
Introduction
Loss of homeostatic control of the immune response is a key determinant of human disease pathogenesis. Exogenous insults such as microbial infection or danger signals from within the cell (e.g. during cell injury) are rapidly recognized by the innate immune system, which elicits host mechanisms to resolve the threats. Failures in tightly regulating these defense mechanisms can lead to detrimental consequences such as chronic infection or autoimmunity. Two major innate immune defense programs are the type I interferon (IFN) and IL-1-mediated proinflammatory responses. While type I IFNs signal through the IFNα/β (IFNAR) receptor to upregulate IFN-stimulated gene (ISG) products that have antiviral or immunomodulatory activities, IL-1β of the IL-1 cytokine family engages the cognate IL-1 receptor to induce inflammation during microbial infections. Accumulating evidence shows extensive ‘cross-talk’ between type I IFN and IL-1 responses in microbial infections and autoinflammatory disorders, wherein either cooperative or negatively-regulatory circuits have been reported (Mayer-Barber et al., 2014; Mayer-Barber and Yan, 2017).
The production of type I IFNs and IL-1β relies on the activation of a set of sensors of the innate immune system, collectively known as pattern-recognition receptors (PRRs). These PRRs survey different cellular compartments and have evolutionarily gained the capability to distinguish foreign or abnormal molecules from host-cell components. Viral RNA species represent a pivotal class of pathogen-associated molecular patterns that activate these sensors. Depending on their cellular localization, which is determined by the route of viral entry and site of virus replication, viral RNA-ligands are recognized by one or more specific PRRs which subsequently mount coordinated host responses. Recent advances have expanded the repertoire of immunostimulatory RNA to certain RNA species of host origin, in particular different types of cellular noncoding RNAs. These host RNAs become immunostimulatory when chronically-upregulated, mislocalized, and/or misprocessed during viral infection or in disease settings such as autoimmunity. Accordingly, the recognition of cellular immunostimulatory RNAs triggers innate immune responses to combat viral infection, or it can cause chronic inflammation. The vast majority of immunostimulatory RNAs – both pathogen-and host-derived RNAs – reside in the endosome and cytoplasm, where also most RNA sensors are localized. Intriguingly, recent studies show that PRR-mediated RNA sensing can also occur in the nucleus and mitochondrion (Cao et al., 2019a; Liu et al., 2018; Wang et al., 2019; Zhang et al., 2020), highlighting an orchestrated multi-compartmental RNA-sensing paradigm.
Here, we summarize our current knowledge on classical and emerging RNA sensors and co-sensors of the innate immune system, with an emphasis on their functional distinction imposed by cell type-dependent expression, subcellular localization, and ligand specificity. We also highlight their physiological roles in type I IFN and IL-1β responses as well as their interdependent regulation. Finally, we discuss the relevance of polymorphisms in genes encoding RNA sensors for human disease conditions.
Toll-like Receptors
Toll-like receptors (TLRs) represent well-characterized PRRs in mammals whose identification preceded the other PRR families. They are named after the Drosophila Toll proteins which are involved in Drosophila embryo development and antimicrobial peptide expression (O’Neill et al., 2013). To date, the mammalian TLR family contains ten (TLR1–10) and twelve (TLR1–9, TLR11–13) members in human and mouse, respectively, among which TLR3, TLR7, TLR8, and TLR13 sense RNA. TLRs primarily localize to the cell surface and/or endosomes, and share a common structural architecture consisting of a leucine-rich repeats (LRR)-containing domain at the amino (N)-terminus, a transmembrane domain, and a carboxyl (C)-terminal cytoplasmic Toll/IL-1 receptor (TIR) domain (Jin and Lee, 2008). Upon ligand binding to the LRR-containing ectodomain, TLRs predominantly form homodimers that are characterized by an “m”-shaped architecture. Unlike that of most TLRs, the apo form LRR domain of TLR8 has been crystallized as a pre-existing inactive homodimer, but not a monomer (Choe et al., 2005; Tanji et al., 2013; Zhang et al., 2016). In the dimer, the cytoplasmic TIR domains of TLRs are orientated in such a way that allows for the recruitment of downstream adaptor proteins for signaling transduction (Figure 1). The myeloid differentiation primary-response protein 88 (MyD88) is the key adaptor of all TLRs except for TLR3. It contains a TIR domain which mediates homotypic interactions with the TIR domain of TLRs. The recruitment of MyD88 triggers the assembly of the ‘Myddosome’, which is mediated by homotypic interactions between the death domains (DD) of MyD88 and also the DD–DD interactions between MyD88 and IL-1R-associated kinases (IRAK) such as IRAK1, IRAK2 and IRAK4 (Gay et al., 2011). This higher-order complex serves as a signaling platform for the recruitment of TNF receptor-associated factor 6 (TRAF6), which then activates TGFβ-activated kinase 1 (TAK1). TAK1 leads to the activation of mitogen-activated protein kinases (MAPKs), nuclear factor (NF)-κB, and IFN-regulatory factor 5 (IRF5) (Bergstrom et al., 2015; Kawai and Akira, 2010; Takaoka et al., 2005). TLR3 signals exclusively through the TIR-domain-containing adapter-inducing interferon-β (TRIF) (Hoebe et al., 2003; Oshiumi et al., 2003; Yamamoto et al., 2003). TRIF interacts with TRAF3 to trigger the TANK-binding kinase 1 (TBK1)–IκB kinase ε (IKKε) axis for IRF3 activation, or associates with TRAF6 and receptor-interacting protein kinase 1 (RIPK1) to activate NF-κB and MAPKs via TAK1 (Kawai and Akira, 2010). In plasmacytoid dendritic cells (pDCs), TLR7 activates IRF7 exclusively through a MyD88– IRAK1/4–TRAF6 axis (Honda et al., 2004; Kawai et al., 2004; Uematsu et al., 2005). The activation of NF-κB, activator protein 1 (AP1), and IRF3 or IRF7 ultimately leads to the expression of type I and III IFNs as well as pro-inflammatory cytokines (Figure 1).
Figure 1. TLR and RLR signaling pathways.
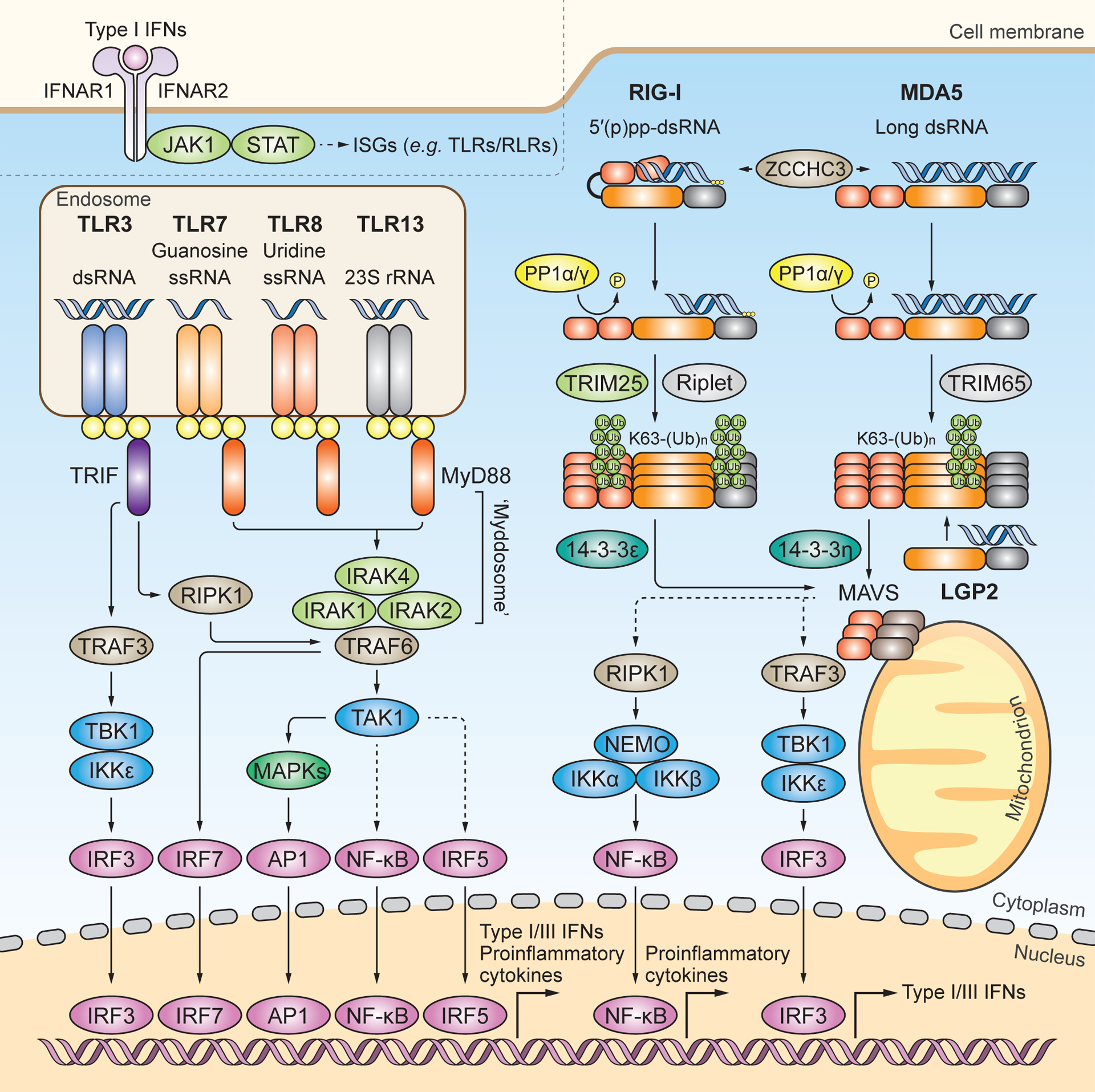
RNA-sensing TLRs predominately localize to the endosome and include TLR3, TLR7, TLR8 and TLR13. TLR3 recognizes dsRNA. TLR7 and TLR8 contain one binding site with specificity for guanosine and uridine, respectively, and the other binding site for ssRNA. TLR13 recognizes bacterial 23S rRNA. All RNA-sensing TLRs form homodimers upon activation. TLR7, TLR8 and TLR13 engage MyD88 as an adaptor protein to form the ‘Myddosome’, which is a multi-protein complex of MyD88, IRAK1, IRAK2 and IRAK4. The ‘Myddosome’ recruits TRAF6 and TAK1 to activate MAPKs, NF-κB, and IRF5. TLR3 signals through the adaptor protein TRIF to activate IRF3 via the TRAF3-TBK1–IKKε axis, or to activate MAPKs and NF-κB via the TRAF6-RIPK1-TAK1 axis. In plasmacytoid DCs, TLR7 activates IRF7 through TRAF6. It is noted that, for simplicity, this cell-type specific pathway is not illustrated separately. Nuclear translocation of activated transcription factors ultimately drives the expression of proinflammatory cytokines and type I and III IFNs. The RLR family members RIG-I and MDA5 localize to the cytoplasm and preferentially recognize 5’ di-or tri-phosphorylated short dsRNA and long dsRNA, respectively. ZCCHC3 facilitates RNA binding of RIG-I and MDA5. Structurally, RIG-I is held in an auto-repressed, closed conformation, while MDA5 likely adopts an open and flexible conformation. Cognate RNA-ligand binding induces conformational changes in RIG-I and MDA5 that expose their N-terminal CARDs, which are then subjected to posttranslational modifications (PTMs). PP1α or PP1γ-mediated dephosphorylation and TRIM25-mediated K63-linked ubiquitination at the CARDs induce further activation of RIG-I, which subsequently engages the chaperon protein 14–3-3ε for cytosol-to-mitochondrial translocation. Riplet modifies the C-terminal domain (CTD) of RIG-I with K63-linked-polyubiquitin chains. TRIM65 mediates K63-linked ubiquitination of MDA5 in its helicase domain, which induces MDA5 activation. The cytosol-to-mitochondria translocation of MDA5 is facilitated by 14–3-3η. Both RIG-I and MDA5 interact with and activate MAVS on mitochondria (and also other organelles), which subsequently activates IRF3 via TBK1 or IKKε and NF-κB via the IκB kinase (IKK) complex. LGP2 has been shown to facilitate MDA5 activation, whereas it inhibits antiviral signaling by RIG-I. Type I IFNs induced by TLR or RLR signaling bind to the type I IFN receptor (consisting of IFNAR1 and IFNAR2) and activate the Janus kinase (JAK)–signal transducer of activators of transcription (STAT) pathway to transcriptionally upregulate many ISGs including TLRs and RLRs themselves.
The RNA-sensing TLRs are known to recognize their ligands with defined specificity, which has also been supported by structural studies (Table 1). TLR3 contains two RNA-binding sites that recognize double-stranded (ds)RNA in a sequence-independent manner (Alexopoulou et al., 2001). The minimal dsRNA length required for TLR3 activation in vitro has been shown to be 39–48 bp, which conforms to the length of dsRNA co-crystallized with the TLR3 ectodomains (Leonard et al., 2008; Liu et al., 2008). In cells engineered to express intracellular (but not cell surface-bound) TLR3, a minimal length of 90 bp is required for activation (Leonard et al., 2008). TLR3 has also been shown to be activated by small-interfering RNAs (siRNA) that are ~20 bp in length (Kariko et al., 2004a; Kleinman et al., 2008), though the underlying mechanism of siRNA recognition by TLR3 remains unknown. TLR3 plays an important role in the innate immune response to viral pathogens such as West Nile virus (WNV), influenza A virus (IAV), and poliovirus (Abe et al., 2012; Daffis et al., 2008; Le Goffic et al., 2006; Wang et al., 2004); however, the physiological TLR3 ligands in these contexts remain uncharacterized, except for poliovirus-derived RNA harboring stem-loop core structures (Tatematsu et al., 2013). In myeloid DCs, TLR3 also responds to phagocytosed RNA of viral (and likely also host) origin (Kariko et al., 2004b; Tatematsu et al., 2013), and phagocytosed viral RNA enhances cross-priming of cytotoxic T cells (Schulz et al., 2005). TLR7 and TLR8 are highly homologous RNA sensors with an RNA ligand preference for single-stranded (ss)RNA (Diebold et al., 2004; Heil et al., 2004; Lund et al., 2004). In humans, TLR7 is primarily expressed in pDCs and B cells, while TLR8 is expressed in myeloid DCs, monocytes, and monocyte-derived DCs (Gorden et al., 2005). Both TLRs contain two RNA ligand-binding sites within their LRR domain, designated site 1 and site 2, both of which are indispensable for TLR7 and TLR8 activation. The site 1 is a highly conserved binding site for nucleosides, with TLR7 and TLR8 having a preference for guanosine (G) and uridine (U), respectively (Tanji et al., 2015; Zhang et al., 2016; Zhang et al., 2018b). The oligonucleotide-binding activity is conferred by site 2. While TLR8 binds the UG dinucleotide, TLR7 requires at least a 3-mer motif containing a U in the second position. Further analysis reveals that the site 2 ssRNA sequence preference is UU(U/C) > UU(G/A) > XUX (where X stands for non-U) (Tanji et al., 2015; Zhang et al., 2016; Zhang et al., 2018b). However, it remains unclear how exactly physiological ssRNAs activate TLR7 and TLR8 by coordinating both binding sites. To date, TLR7 has been shown to mediate type I IFN responses to several RNA viruses, such as IAV, vesicular stomatitis virus (VSV), and human immunodeficiency virus-1 (HIV-1) (Diebold et al., 2004; Heil et al., 2004; Lee et al., 2007; Lund et al., 2004). The detailed ligand characteristics of a few microbial and cellular TLR7 agonists have been identified, which include HIV-derived GU-rich ssRNA, microRNA let-7, and multiple siRNA sequences (Heil et al., 2004; Hornung et al., 2005; Lehmann et al., 2012). The orphan receptor TLR13 is a mouse-specific endosomal TLR that is predominantly expressed in DCs and macrophages. It recognizes bacterially and virally derived RNAs, leading to proinflammatory and type I IFN responses through NF-κB and IRF7, respectively (Hidmark et al., 2012; Shi et al., 2011). Furthermore, the bacterial 23S ribosomal RNA (rRNA) has been identified to be a physiological ligand of TLR13. This RNA encodes a highly conserved 13-nt sequence within the ribozyme catalytic domain that mediates TLR13 activation in vitro and in vivo (Li and Chen, 2012; Oldenburg et al., 2012). Structural analysis of the TLR13 ectodomain in complex with the 23S rRNA-derived 13-nt ssRNA reveals that a unique stem-loop structure is crucial for TLR13 binding in a sequence-and conformation-specific manner. A similar RNA motif activating TLR13 is found in the VSV genome (Shi et al., 2011; Song et al., 2015) (Table 1).
TABLE 1.
TLR and RLR ligands
| Sensors | Ligand characteristics | Synthetic RNA | Microbial RNA | Host RNA | Key references |
|---|---|---|---|---|---|
| TLR3 | dsRNA |
|
Viral and bacterial RNA (e.g. long stem-loop containing poliovirus- derived RNA) | Phagocytosed host RNA (context-dependent) | (Kariko et al., 2004a; Kariko et al., 2004b; Kleinman et al., 2008; Leonard et al., 2008; Liu et al., 2008; Tatematsu et al., 2013) |
| TLR7 | Site 1: guanosine |
|
Viral and bacterial RNA (e.g. HIV GU-rich RNA) | Let-7 miRNA | (Heil et al., 2004; Hornung et al., 2005; Lehmann et al., 2012; Tanji et al., 2015; Zhang et al., 2016; Zhang et al., 2018b) |
| Site 2: ssRNA | |||||
| TLR8 | Site 1: uridine | UG dinucleotide | Viral and bacterial RNA | N/A | |
| Site 2: ssRNA | |||||
| TLR13 | Stem-loop ssRNA | ≥ 13 nt |
|
N/A | (Li and Chen, 2012; Oldenburg et al., 2012; Song et al., 2015) |
| RIG-I | 5’-di- or triphosphate and blunt-ended dsRNA | Short dsRNA of ≥ 10–19 bp |
|
N/A | (Baum et al., 2010; Goubau et al., 2014; Hornung et al., 2006; Liu et al., 2015; Pichlmair et al., 2006; Schlee et al., 2009; Schmidt et al., 2009) |
| ‘Non-canonical’ |
|
|
|
(Ablasser et al., 2009; Chen et al., 2017; Chiang et al., 2018; Chiu et al., 2009; Eckard et al., 2014; Jiang et al., 2018; Kato et al., 2008; Malathi et al., 2007; Minamitani et al., 2011; Runge et al., 2014; Saito et al., 2008; Zhang et al., 2018a; Zhao et al., 2018) | |
| MDA5 | Long irregular dsRNA | Poly(I:C) of > 300 bp |
|
|
(Ahmad et al., 2018; Deddouche et al., 2014; Dhir et al., 2018; Kato et al., 2008; Luthra et al., 2011; Pichlmair et al., 2009; Runge et al., 2014) |
| LGP2 | dsRNA; no end specificity | dsRNA that is blunt-ended or has overhangs with or without 5’-triphosphate moiety | EMCV L antisense RNA | N/A | (Deddouche et al., 2014; Uchikawa et al., 2016) |
Abbreviations: HIV, human immunodeficiency virus; VSV, vesicular stomatitis virus; IAV, influenza A virus; HCV, hepatitis C virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; EBV, Epstein-Barr virus; IRE-1, inositol-requiring enzyme 1; EMCV, encephalomyocarditis virus; Alu, Arthrobacter luteus; N/A, not available.
The RNA-sensing TLRs are subjected to regulation by multiple mechanisms. Among those, regulation of TLR subcellular compartmentalization represents a key regulatory mechanism (Miyake et al., 2017; Pelka et al., 2016), and mislocalization of TLRs has been shown to lead to autoimmunity (Fukui et al., 2011; Kono et al., 2009). TLR trafficking from the endoplasmic reticulum (ER) to endolysosomes is primarily mediated by the multipass-transmembrane protein Unc-93 homolog B1 (UNC93B1), which directly interacts with TLRs (Kim et al., 2008; Lee et al., 2013; Tabeta et al., 2006). UNC93B1 can also direct TLR3 and TLR7 to the cell surface (Kanno et al., 2015; Pohar et al., 2013). Moreover, UNC93B1 has been shown to stabilize TLR3 and TLR7 prior to their endosomal trafficking in mouse DCs and macrophages (Pelka et al., 2018), and to limit TLR7 activation by recruiting syntenin-1 which facilitates TLR7 sorting into exosomes (Majer et al., 2019). Another major regulatory mechanism for TLR7 and TLR8 is proteolytic cleavage. Proteolytic processing by compartment-specific proteases, such as the furin-like propeptide convertases, ultimately ensures proper maturation of TLR7 and TLR8 in endosomes (Hipp et al., 2013; Ishii et al., 2014). The protease cleavage sites locate to the site 2 (within a loop structure called ‘Z-loop’) that is involved in ssRNA binding (Zhang et al., 2016). A failure in Z-loop cleavage inhibits TLR8 dimerization and renders it inactive (Hipp et al., 2013; Tanji et al., 2016; Tanji et al., 2013). Phosphorylation has also been shown to regulate the activation of RNA-sensing TLRs. Activation of TLR3 and TLR8 has been shown to require phosphorylation of several tyrosine residues located in their TIR domains, although the kinases responsible for phosphorylation remain controversial (Leifer and Medvedev, 2016). Moreover, TLR3 has been shown to undergo N-linked glycosylation, and chemical inhibition of glycosylation or mutational perturbation of the predicted glycosylation sites impairs TLR3-mediated NF-κB activation (Sun et al., 2006).
RIG-I-like Receptors
The family of retinoic-acid inducible gene-I (RIG-I)-like receptors (RLR) encompasses three members: RIG-I, melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) (Andrejeva et al., 2004; Cui et al., 2001; Yoneyama et al., 2004). They belong to the SF2 helicase superfamily whose members mostly act as NTP-dependent RNA helicases. RLRs predominately localize to the cytoplasm, with a small portion of RIG-I also localized to the nucleus (Liu et al., 2018). RIG-I and MDA5 are the two signaling-competent RLR members with a common domain architecture that consists of a central helicase flanked by an N-terminal tandem caspase activation and recruitment domain (CARDs) and a C-terminal domain (CTD). LGP2 contains the helicase domain and CTD but lacks the N-terminal CARDs (Saito et al., 2007), and thus mainly acts as a regulator of RIG-I and MDA5 due to a lack of signaling competency (Bruns and Horvath, 2015). In the absence of RNA ligands, RIG-I resides in an auto-inhibitory conformation in which the CARDs are sequestered through an interaction of CARD2 with the Hel-2i subdomain of the helicase. In contrast, MDA5 is believed to adopt an open, quite flexible conformation in the absence of stimulatory RNA. RNA binding to the CTD and helicase of RIG-I and MDA5 liberates the CARDs for downstream protein association (Kolakofsky et al., 2012) (Figure 1). The exposed CARDs interact with several regulatory enzymes (e.g. ubiquitin E3 ligases and phosphatases), which posttranslationally modify the CARDs, making them fully signaling-active. RLRs in their signaling-primed forms then interact with members of the 14–3-3 protein family, specifically 14–3-3ε and 14–3-3η which, respectively, mediate the translocation of RIG-I and MDA5, to their common adaptor protein MAVS (mitochondrial antiviral signaling protein). MAVS predominately localizes to the outer mitochondrial membrane and has also been found at mitochondria-associated membranes of the endoplasmic reticulum (MAMs) and peroxisomes (Chow et al., 2018a; Horner et al., 2011; Tan et al., 2018). MAVS contains a C-terminal transmembrane domain and an N-terminal CARD domain involved in the homotypic interaction with the CARDs of RIG-I and MDA5. Activated MAVS recruits TRAF2, TRAF5, TRAF6, and TRADD which – via the TRAF3–TANK–TBK1 axis – ultimately leads to IRF3 or IRF7 activation and the induction of type I and III IFNs. Additionally, MAVS signaling induces NF-κB activation for pro-inflammatory cytokine expression through the FADD–RIP1–IKK axis (Liu et al., 2013; Michallet et al., 2008) (Figure 1). RLR signaling also ‘cross-talks’ with other sensing pathways such as TLRs, NLRs, and the cGAS-STING pathway (Barber, 2015; Zevini et al., 2017). Conceptually, PRR crosstalk can occur at the transcriptional level whereby activation of an immediate sensor, via autocrine or paracrine IFNAR signaling, transcriptionally upregulates other PRRs, which usually are themselves ISGs. Furthermore, different sensors can collaborate at the level of downstream signaling molecules. For example, TLR7 and RLRs have been shown to pair respectively with IRF5 and IRF3 to induce partitioned gene expression and immune cytokine production in pDCs, leading to polarized immune response outcomes (Chow et al., 2018b). Moreover, RLRs have recently been shown to also suppress viral replication in a signaling-independent manner (Sato et al., 2015; Weber et al., 2015).
Àctivation of RIG-I and MDA5 requires multiple steps which include, besides RNA binding, the multimerization and posttranslational modifications (PTMs) of the sensors. Both receptors form ATP-mediated filaments on dsRNA, however with distinct mechanisms [(described in detail elsewhere) (Reikine et al., 2014)]. Upon filament formation, the ‘head-to-tail’ stacking of monomeric RIG-I or MDA5 brings the nearby CARDs into proximity to oligomerize into a ‘lock-washer’-like helical assembly. This helical oligomer then induces the formation of prion-like MAVS filaments (Hou et al., 2011; Peisley et al., 2014; Wu et al., 2014). Multiple PTMs that regulate either RNA binding or CARD signaling by RLRs have been identified (Chiang and Gack, 2017). Acetylation of RIG-I at K909, which is located within its CTD, inhibits RNA binding and RNA-dependent RIG-I oligomerization. As such, deacetylation of the RIG-I CTD by histone deacetylase 6 (HDAC6) is necessary for RIG-I activation (Choi et al., 2016; Liu et al., 2016). Serine or threonine phosphorylation of the CTD and CARDs prevents aberrant activation of RIG-I and MDA5 in uninfected cells (Gack et al., 2010; Maharaj et al., 2012; Sun et al., 2011; Takashima et al., 2015; Wies et al., 2013). Dephosphorylation by cellular phosphatases PP1α or PP1γ upon RNA stimulation is required for RIG-I and MDA5 activation (Wies et al., 2013). In the case of RIG-I, dephosphorylation of the CARDs by PP1 induces K63-linked polyubiquitination, which is required for RIG-I activation by promoting its oligomerization (Gack et al., 2007; Jiang et al., 2012). RIG-I undergoes covalent K63-linked ubiquitination not only in the CARDs but also CTD (Gack et al., 2007; Oshiumi et al., 2009; Oshiumi et al., 2013). In addition, noncovalently-bound K63-polyubiquitin at the CARDs has been shown to activate RIG-I in vitro (Zeng et al., 2010); however, the relevance of noncovalent K63-polyubiquitin in RIG-I activation in infected cells is less well established. Over the past years, several E3 ubiquitin ligases have been proposed to be involved in RIG-I K63-ubiquitination; however, only tripartite motif-containing protein 25 (TRIM25) and ring finger protein 135 (RNF135, also known as Riplet) have been demonstrated to directly catalyze RIG-I K63-ubiquitination and to mediate antiviral innate immune responses (Gack et al., 2007; Oshiumi et al., 2009; Rehwinkel and Gack, 2020). For TRIM25 in particular, multiple cellular interacting proteins (e.g. NLRP12, caspase 12, and NDR2) and host noncoding RNAs have been shown to modulate the TRIM25-mediated RIG-I CARD ubiquitination (Chen et al., 2019; Lin et al., 2019; Liu et al., 2019; Sanchez et al., 2018; Wang et al., 2010). TRIM25 has also been shown to be a major target of viral evasion strategies (Chan and Gack, 2016). In the context of full-length RIG-I, the current model is that K63-linked ubiquitination of the RIG-I CTD at K788 by Riplet relieves the auto-repression of RIG-I, which serves as a prerequisite for subsequent K63-linked ubiquitination at K172 within the CARD by TRIM25 (Gack et al., 2007; Oshiumi et al., 2013). Of note, a recent study claims that Riplet is the only required E3 ligase for the K63-linked ubiquitination and activation of RIG-I (Cadena et al., 2019). However, the data from this study contradict an abundance of work by several independent groups which demonstrates that TRIM25 directly ubiquitinates RIG-I (both in cells and in vitro) and is necessary for efficient RIG-I signaling in response to a variety of viruses [(reviewed in detail elsewhere) (Rehwinkel and Gack, 2020)]. The requirement of TRIM25 and/or Riplet in RIG-I signaling may be cell-type-or virus-specific, or temporally-distinct. Of note, Riplet has also been recently shown to regulate type I IFN responses independently of RIG-I by acting at the step of IRF3 activation (Vazquez et al., 2019). Whether K63-linked ubiquitination is necessary for MDA5 activation is less well understood. Unanchored K63-polyubiquitin chains have been reported to promote MDA5 CARD oligomerization (Jiang et al., 2012), presumably through a mechanism similar to that for RIG-I. TRIM65 has been identified as an E3 ubiquitin ligase that catalyzes covalent K63-linked ubiquitination of the MDA5 helicase (at K743), thereby triggering MDA5 activation during EMCV infection (Lang et al., 2017). Recently, ZCCHC3 has been identified as a coreceptor for both RIG-I and MDA5, which facilitates dsRNA binding and TRIM25-mediated K63-linked ubiquitination of both RLRs (Lian et al., 2018). The activities of many signaling proteins downstream of RLRs are also regulated by PTMs such as phosphorylation and different types of polyubiquitination, and E3 ligases of the TRIM protein family have emerged as key regulators of RLR signal transduction (van Gent et al., 2018). Alternative splicing represents another mechanism of RLR regulation. A splice variant of RIG-I which carries a short deletion within the first CARD has been shown to lose TRIM25 binding and K63-linked ubiquitination, thereby acting as a dominant-negative inhibitor of RIG-I signaling (Gack et al., 2008). Similarly, an N-terminally truncated form of MAVS produced from leaky ribosomal scanning dampens RLR signaling (Brubaker et al., 2014).
LGP2 is a major regulator of RLR signaling where it can act both positively and negatively. Several mechanisms for inhibition of RIG-I by LGP2 have been proposed, including competitive RNA binding (Murali et al., 2008; Rothenfusser et al., 2005; Yoneyama et al., 2005), competition with IKKε for MAVS interaction (Komuro and Horvath, 2006), blockage of RIG-I multimerization (Saito et al., 2007), and inhibition of the TRIM25-mediated K63-linked ubiquitination of RIG-I (Quicke et al., 2019). Studies in Lgp2−/− mice reveal a role of LGP2 in sensitizing MDA5-dependent antiviral responses to encephalomyocarditis virus (EMCV) infection (Satoh et al., 2010; Venkataraman et al., 2007), which requires the ATPase activity of LGP2 (Bruns et al., 2013; Satoh et al., 2010). The intimate cooperation of LGP2 with MDA5 during EMCV infection has further been substantiated by the fact that LGP2 associates with MDA5-stimulatory RNA (Deddouche et al., 2014). LGP2 facilitates the interaction of MDA5 with dsRNA as well as the formation of shorter MDA5 filaments (which also contain LGP2), which have been shown to be more immunostimulatory than long MDA5-only filaments (Bruns et al., 2014). Structurally, LGP2 preferentially interacts with dsRNA in an ‘end-capping’ mode. This is reminiscent of RIG-I binding but with relaxed ligand specificity. LGP2 can also form filaments on dsRNA in a less cooperative way than does MDA5. At a limiting concentration, LGP2 potentiates MDA5 signaling in response to poly(I:C), which is likely due to promoting MDA5 nucleation and oligomerization on dsRNA (Bruns et al., 2014; Uchikawa et al., 2016).
RLR ligands and their features
RIG-I and MDA5 play critical roles in sensing RNA derived from RNA viruses, leading to the induction of type I and III IFN responses. Accumulating evidence demonstrates that RIG-I and MDA5 can act distinctly or cooperatively to detect certain viruses. Negative-strand RNA viruses of the Paramyxoviridae, Orthomyxoviridae, Rhabdoviridae, Bunyaviridae, and Filoviridae families are mainly sensed by RIG-I, whereas positive-strand RNA viruses of the Picornaviridae family are sensed by MDA5 (Kato et al., 2008; Kato et al., 2006; Loo et al., 2008). The sensing of Reoviridae and Flaviviridae family members is mediated by both RIG-I and MDA5, highlighting the collaboration of the two receptors in antiviral immunity (Errett et al., 2013; Loo et al., 2008). Over the past decade, the RNA-ligand specificity of RIG-I has been extensively studied. Two canonical RNA characteristics are generally required for optimal RIG-I activation (Table 1). First, the RIG-I ligand requires a relatively short (~10 to 19 bp in length) dsRNA region that is usually blunt-ended (Schlee and Hartmann, 2016). RIG-I ligands also tolerate wobble base pairs, mismatches, and bulge elements as those contained in the panhandle structures of the RNA genomes of negative-strand RNA viruses (Liu et al., 2015; Schlee et al., 2009). While RNA recognition by RIG-I is generally believed to be sequence-independent, certain motifs in RNA ligands have also been proposed to confer, or facilitate, RIG-I binding. These include the poly(U/UC) tract in the 3’ UTR of the hepatitis C virus (HCV) genomic RNA (Saito et al., 2008), the GA-rich motifs embedded in the multi-branch loop structures of the cellular long noncoding RNA lnc-Lsm3b (Jiang et al., 2018), as well as AU-rich motifs found in the L gene mRNA of measles virus (Runge et al., 2014), Sendai virus (SeV) defective-interfering genome (Xu et al., 2015) and Kaposi’s sarcoma-associated herpesvirus (KSHV)-derived RNA transcripts (Zhang et al., 2018a). Second, typical RIG-I agonists harbor a 5’ di-or tri-phosphate group (Goubau et al., 2014; Hornung et al., 2006; Pichlmair et al., 2006; Schmidt et al., 2009), which is commonly present in the (sub)genomes and replication intermediates of negative-strand RNA viruses but is absent in most cellular RNAs. RIG-I ligands that lack a 5’ di-/triphosphate moiety have also been reported, such as RNase L-cleaved cellular RNA bearing 5’-OH and 3’-monophosphates (Malathi et al., 2007), RNase III-digested poly(I:C) (Kato et al., 2008), cellular RNA cleaved by inositol-requiring enzyme 1 (IRE-1) (Eckard et al., 2014), and exogenous circular RNA (Chen et al., 2017). The structural basis for RIG-I activation by these ‘non-canonical’ ligands warrants further investigation.
Accumulating evidence highlights the importance of RIG-I in sensing DNA viruses, such as herpes simplex virus type 1 (HSV-1), Epstein–Barr virus (EBV), KSHV, and adenovirus. RNAs detected by RIG-I during DNA virus infection can be from either viral or host origin and, further, are often RNA polymerase (Pol) III transcripts, which have been shown to activate RIG-I (Ablasser et al., 2009; Chiu et al., 2009). The 5S ribosomal RNA pseudogene transcript 141 (RNA5SP141) has been shown to activate RIG-I during HSV-1, EBV and also IAV infection. Viral infection induces the nucleus-to-cytoplasm relocalization of RNA5SP141, and viral host shutoff mechanisms downregulate specific proteins that normally associate with RNA5SP141. Consequently, RNA5SP141 accumulates in the cytoplasm of infected cells and becomes liberated for RIG-I binding (Chiang et al., 2018). During KSHV infection, viral downregulation of the cellular triphosphatase DUSP11 (Dual specificity phosphatase 11) leads to the accumulation of 5’-triphosphorylated vault RNAs, which then activate RIG-I (Zhao et al., 2018). Furthermore, Pol III-driven small RNA transcripts derived from EBV (EBER RNAs) and adenovirus (VA RNAs) induce type I IFN responses in a RIG-I-dependent manner (Ablasser et al., 2009; Minamitani et al., 2011) (Table 1).
The characteristics of MDA5 ligands are much less well-understood than those of RIG-I agonists. MDA5 is preferentially activated by long irregular dsRNA which presumably exists in a complex higher-order configuration (Kato et al., 2008; Pichlmair et al., 2009). Partial digestion of synthetic poly(I:C) by RNase III shows that dsRNA longer than 300 bp activates MDA5 (Kato et al., 2008). The replication of DNA viruses, positive-strand RNA viruses, and dsRNA viruses is generally believed to produce long dsRNAs (Weber et al., 2006), which are potential sources of MDA5 ligands. Furthermore, parainfluenza virus 5 (PIV5) and measles virus (which harbor negative-strand RNA genomes) also elicit MDA5-mediated type I IFN responses, which has been shown to be mediated by an association of MDA5 with the mRNA of the viral L gene (Luthra et al., 2011; Runge et al., 2014).
Recent studies indicate a role for MDA5 in sensing endogenous dsRNA. dsRNAs of ~300 bp in length formed through hybridization of inverted repeat transposable Arthrobacter luteus (Alu) elements have been identified as MDA5 ligands in Aicardi-Goutières syndrome (AGS)-associated type I IFN induction (Ahmad et al., 2018). Furthermore, mitochondrial dsRNA has been shown to activate MDA5 in cells deficient of mitochondrial RNA helicase SUV3 and polynucleotide phosphorylase (PNPase) (Dhir et al., 2018) (Table 1).
NLRs and Their Roles in Viral RNA Sensing
The nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are a group of intracellular proteins that mount immune responses to a variety of microorganisms and exogenous or cell-derived danger signals (Wen et al., 2013). They are characterized by the presence of a NOD domain which mediates dNTPase activity and NLR protein oligomerization. In addition, NLRs contain an N-terminal effector domain and a C-terminal LRR domain (Kanneganti et al., 2007). While the array of LRR motifs is responsible for ligand binding, the effector domain mediates binding to distinct downstream signaling proteins (Ting et al., 2008). Similar to RLRs, NLRs are in a monomeric state prior to activation in which an intramolecular interaction between the LRR and NOD domains prevents signaling. Ligand recognition stimulates conformational rearrangements that induce NLR oligomerization and the exposure of effector domains for protein recruitment (Lechtenberg et al., 2014). Multiple members of the 22 known human NLRs serve as scaffold proteins for the assembly of inflammasomes, which are large multiprotein complexes that catalyze caspase-1-mediated processing of pro-IL-1β and pro-IL-18 into their mature forms for secretion.
Other PRRs are known to ‘cross-talk’ with NLRs. For example, TLR signaling induces NF-κB-mediated transcriptional upregulation of NLRP3 and pro-IL-1β to prime inflammasome activation (Swanson et al., 2019). Recent studies show that some NLRs, in complex with specific DEAH-box RNA helicases, can sense viral dsRNA and then trigger type I IFN-and/or inflammasome-dependent antiviral responses. Furthermore, several NLR members function as regulators of other RNA-sensing pathways, in particular the RLR-MAVS pathway (Figure 2).
Figure 2. RNA sensing and regulatory roles of NLRs.
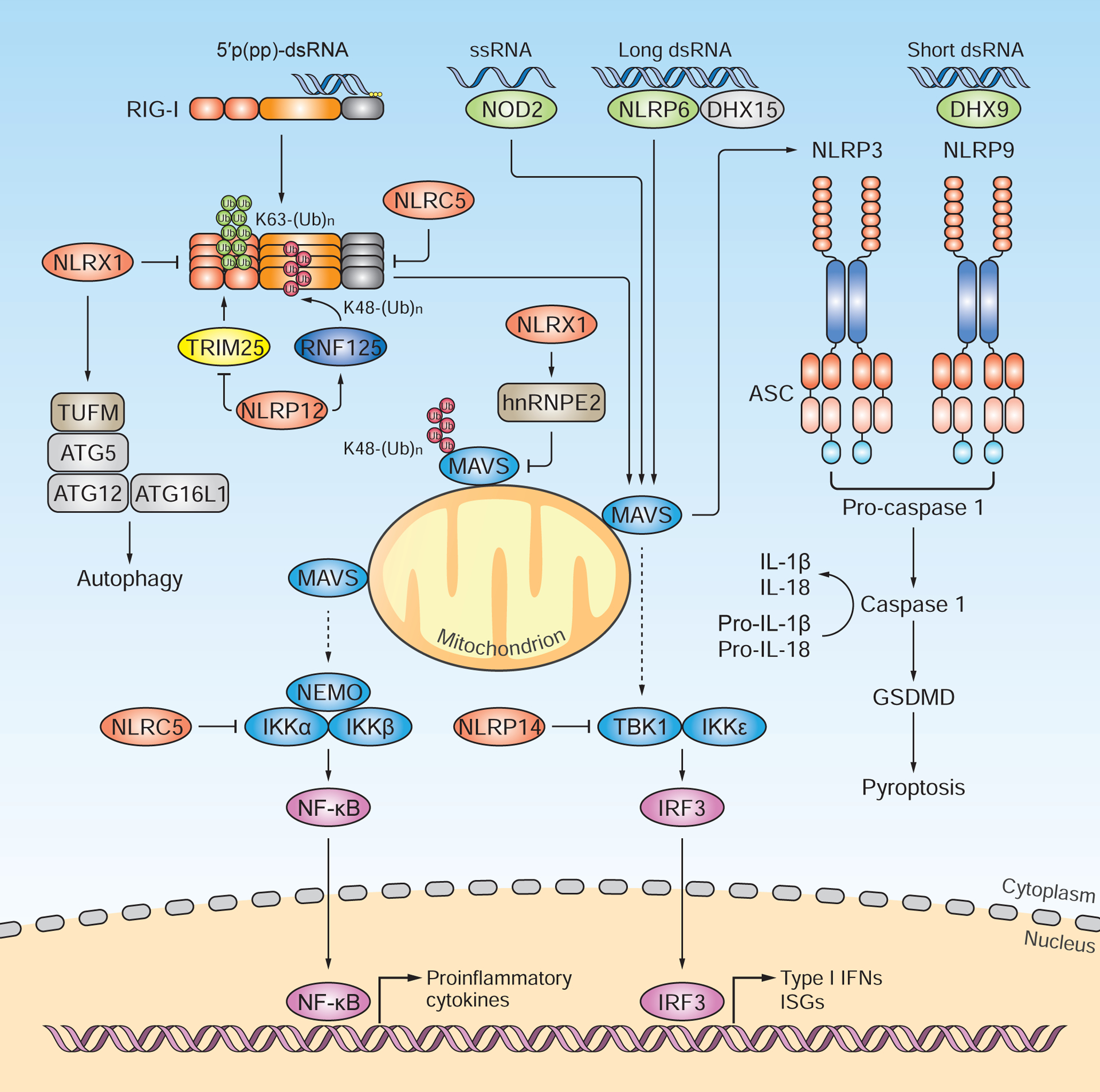
NLRs have been shown to sense viral RNA and trigger type I IFN-and/or inflammasome-dependent host responses, or they regulate other innate sensing pathways such as the RLR pathway. Of note, NLR proteins are expressed and/or function in a highly cell-type-specific or tissue-specific manner, which is not illustrated for simplicity. NOD2 recognizes the ssRNA genome of RSV and mounts MAVS-dependent antiviral responses in hematopoietic and non-hematopoietic cells. NLRP6, in complex with the DEAH-box RNA helicase DHX15, binds viral dsRNA from EMCV and induces MAVS-dependent type I IFN responses in intestinal epithelial cells. Another intestinally-expressed NLR member, NLRP9b, when complexed with DHX9, senses short dsRNA from rotavirus and triggers ASC-and caspase 1-mediated IL-18 production. Additionally, the NLRP9b-DHX9 complex activates caspase-1-dependent pyroptosis through gasdermin D (GSDMD). NLRP12 attenuates RIG-I signaling by counteracting the TRIM25-mediated K63-linked ubiquitination of RIG-I. Moreover, NLRP12 promotes RNF125-mediated K48-linked ubiquitination of RIG-I, which leads to its proteasomal degradation. NLRX1 prevents the RLR-MAVS interaction and also recruits hnRNPE2 to induce MAVS degradation. Furthermore, NLRX1 interacts with TUFM, which promotes autophagy via ATG5, ATG12 and ATG16L1, thereby dampening RLR signaling. NLRP3 interacts with MAVS, and the mitochondrial positioning of NLRP3 promotes inflammasome activation and IL-1β production. NLRC5 and NLRP14 negatively regulate RLR signaling by targeting IKK-mediated and TBK1-mediated innate immune responses, respectively. NLRC5 also binds directly to RLRs and thereby blocks IRF3-dependent cytokine induction. NLRs that promote cytokine responses are shown in green; those that dampen cytokine induction are indicated in red.
NOD2 (also known as NLRC2) has been shown to recognize viral genomic ssRNA during RSV infection and to mediate IRF3-dependent antiviral responses via MAVS in both hematopoietic and non-hematopoietic cells. Nod2−/− mice were impaired in type I IFN responses to RSV infection and exhibited more severe pathology (Sabbah et al., 2009). NLRP6, which is highly expressed in intestinal epithelial cells and exerts anti-bacterial defenses, also plays a central role in type I and III IFN-mediated antiviral immunity in the intestine (Wang et al., 2015). Nlrp6−/− mice that were orally infected with EMCV or murine norovirus 1 had higher viral titers and increased mortality compared to control mice. Mass spectrometry analysis of affinity-purified NLRP6 identifies the RNA helicase DHX15 as an interaction partner. The NLRP6-DHX15 complex interacts with long viral dsRNA in EMCV-infected cells and then is recruited to MAVS to transcriptionally induce type I and III IFNs as well as ISGs, which ultimately leads to virus restriction (Wang et al., 2015). Another intestinally-expressed NLR member, NLRP9b, is critical for the restriction of rotavirus infection in vivo (Zhu et al., 2017). NLRP9b together with the RNA helicase DHX9 senses short dsRNA and then triggers ASC-and caspase 1-mediated IL-18 production. In addition, the NLRP9b-DHX9-ASC axis induces pyroptosis through gasdermin D activation (Zhu et al., 2017) (Figure 2).
Several NLR proteins are key regulators of RLR signaling. NLRP12 functions as a critical checkpoint in RIG-I-mediated signaling by modulating nondegradative and degradative polyubiquitination events of the sensor (Chen et al., 2019). NLRP12 interacts with the E3 ligase TRIM25, and this interaction prevents K63-linked ubiquitination of the RIG-I CARDs and thereby binding to the adaptor MAVS. On the other hand, NLRP12 promotes RNF125-mediated K48-linked ubiquitination of RIG-I and thereby its degradation by the proteasome machinery. Both actions of NLRP12 attenuate RIG-I-mediated antiviral and proinflammatory cytokine responses. Myeloid-cell-specific Nlrp12−/− mice exhibited more robust immune signaling to RNA virus infection, in particular VSV, or upon stimulation with 5′ppp-dsRNA (Chen et al., 2019). Another NLR family member, NLRX1, regulates RLR signaling by acting more downstream in the pathway, at the step of MAVS activation (Allen et al., 2011; Moore et al., 2008). NLRX1 was found to prevent the association of MAVS and RLRs, thereby impeding type I IFN induction. During HCV infection, NLRX1 was also shown to recruit hnRNPE2 to induce MAVS protein degradation (Qin et al., 2017). NLRX1 together with TUFM (mitochondrial Tu translation elongation factor) and the classical autophagy proteins, ATG5, ATG12 and ATG16L1, increases the cellular autophagic response, which in turn further limits RLR signaling (Lei et al., 2012). NLRX1 was shown to have itself RNA-binding capacity in vitro (Hong et al., 2012); however, the impact of RNA binding on NLRX1’s role in the RLR pathway remains largely elusive. Another interconnection of the RLR and NLR pathways has been shown to be mediated by the NLRP3 inflammasome, which is important for in vivo host defenses against many pathogens including certain viral infections (e.g. IAV) (Swanson et al., 2019). MAVS was shown to function as a second critical adaptor of the NLRP3 inflammasome (besides the well-known adaptor ASC) by positioning NLRP3 at mitochondria, which ultimately promotes IL-1β production (Subramanian et al., 2013) (Figure 2).
At least two NLR proteins have been shown to regulate RNA-sensing pathways by targeting critical kinases downstream of innate immune sensors. NLRP14, which is specifically expressed in germ cells, interacts with TBK1, promoting its degradation (Abe et al., 2017). This action limits RNA sensing and DNA sensing by the RIG-I-MAVS and cGAS-STING axis, respectively, which, if aberrantly activated in germ cells, would have deleterious consequences for fertilization. NLRC5, which is the largest NLR protein, inhibits both NF-κB activation and RLR-mediated type I IFN induction (Cui et al., 2010). NLRC5 interacts with IKKα and IKKβ, inhibiting their kinase activity and ultimately suppressing NF-κB-dependent cellular responses. In addition, NLRC5 has been shown to bind to the sensors RIG-I and MDA5, blocking IRF3-dependent cytokine expression (Cui et al., 2010). Somewhat conflicting results have been reported for cytokine responses in Nlrc5−/− mice (Kumar et al., 2011; Tong et al., 2012), suggesting a pathogen-specific or temporal role for NLRC5 in type I IFN-driven innate immune responses (Figure 2).
RNA helicases, hnRNPs, and ZBP1: Emerging RNA Sensors and Co-Sensors
RNA helicases are critical orchestrators of RNA metabolism and also play emerging roles in innate immune sensing. The SF1 helicase ZNFX1 has been shown to localize to mitochondria where it binds viral RNA and mediates MAVS-dependent, but RLR-independent, type I IFN responses (Wang et al., 2019). Apart from RLRs, other SF2 superfamily members such as DEAD-box or DEAH-box helicase proteins, have been shown to sense RNA (Figure 3). Characteristic features of these RNA helicases are an Asp-Glu-Ala-Asp (DEAD) or Asp-Glu-Ala-His (DEAH) motif within their helicase domains, which also contain highly conserved motifs that confer NTP hydrolysis and RNA unwinding capabilities. Two principal ways of how DEAD-box or DEAH-box helicases participate in RNA sensing have been proposed: They either act as direct RNA sensors that function independently of other PRRs, or they serve as co-sensors of RLRs or NLRs potentiating their activation.
Figure 3. RNA helicases, hnRNPs and ZBP1 as emerging RNA sensors and co-sensors.
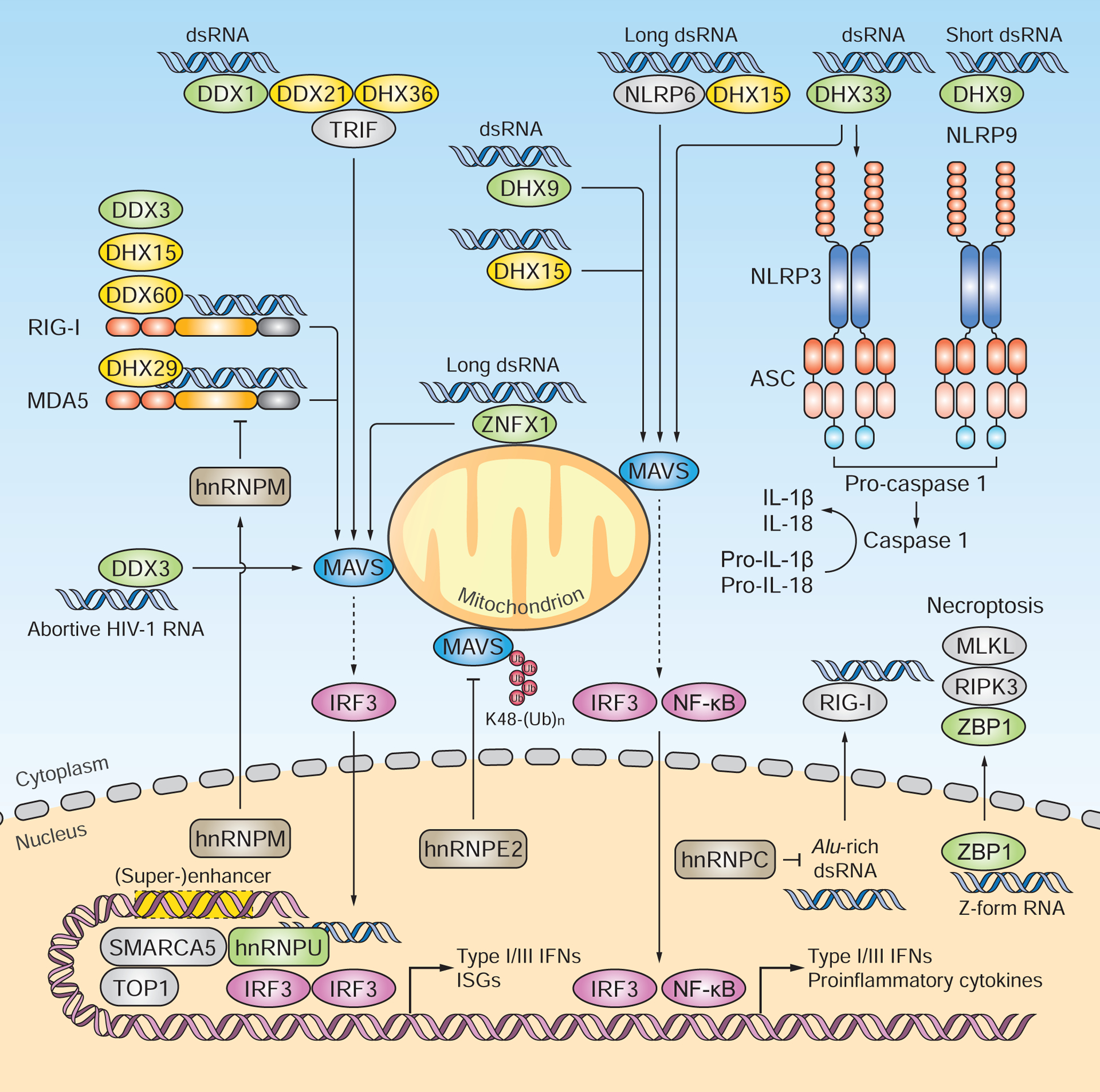
The DEAD-box or DEAH-box family proteins DDX1, DHX9 and DHX33 have been shown to sense RNA. DDX1, in complex with DDX21 and DHX36, recognizes poly(I:C) and induces type I IFN and proinflammatory responses via TRIF and MAVS. DHX9 associates with poly(I:C) and mediates MAVS-dependent cytokine responses in myeloid DCs. DHX9 also facilitates NLRP9b binding to short dsRNA [e.g. low-molecular-weight poly(I:C)] and thereby activates inflammasomes. DHX33 senses dsRNA [e.g. poly(I:C), reovirus genomic RNA, and RNase L cleavage products] and triggers NLRP3-mediated inflammasome activation and MAVS-dependent type I IFN induction. Multiple DEAD-box or DEAH-box helicases serve as RLR co-sensors. Most of these helicases (e.g. DDX3, DDX60, DHX15 and DHX29) interact with RLRs and potentiate their activation. DDX3 was found to also directly bind abortive HIV-1 RNA and to induce MAVS-dependent type I IFN responses. The hnRNP family members hnRNPE2, hnRNPM and hnRNPC negatively regulate RLR signaling. Upon virus-induced nucleus-to-cytoplasm translocation, HnRNPE2 interacts with MAVS and mediates its degradation. HnRNPM undergoes nucleus-to-cytoplasm redistribution and impairs the RNA-binding activity of RLRs. HnRNPC protects cells from accumulating RIG-I-stimulatory endogenous Alu-rich dsRNA. HnRNPU directly associates with HSV-1-derived dsRNA in the nucleus and, together with SMARCA5 and TOP1, activates the enhancers and super-enhancers that drive the transcription of antiviral and proinflammatory genes. ZBP1 senses both viral and host ‘Z-form’ dsRNA and then induces necroptosis through RIPK3 and MLKL. Green signifies proteins that are reported to function as direct RNA sensors; yellow indicates proteins that serve as RLR or NLR co-sensors.
The DEAD-box protein DDX1 has been identified as a poly(I:C)-binding protein in mouse myeloid DCs. It constitutively complexes with DHX36 through DDX21 to induce type I IFN and proinflammatory responses in a TRIF-dependent but TLR3-independent manner (Zhang et al., 2011a). Whereas the ablation of RLR expression leads to a major loss of type I IFN production in response to poly(I:C), silencing each component of the DDX1–DDX21–DHX36– TRIF complex further reduces type I IFN responses, suggesting an RLR-independent sensing pathway. DHX9 has been found to associate with poly(I:C) in vitro and to mediate cytokine responses to poly(I:C) or RNA virus infection in myeloid DCs in a RIG-I-independent but MAVS-dependent manner (Zhang et al., 2011b). DHX9 also interacts with specific NLR members to aid in viral RNA sensing and inflammasome activation (as described above) (Zhu et al., 2017). DHX33 has been shown to sense RNA and then trigger NLRP3 inflammasome activation and MAVS-dependent type I IFN induction in human macrophages and mouse myeloid DCs, respectively (Liu et al., 2014; Mitoma et al., 2013). DHX33 preferentially binds dsRNA, such as poly(I:C) or reovirus genomic RNA (Mitoma et al., 2013). DHX33 also associates with RNase L-cleaved RNAs bearing 2’,3’-cyclic phosphoryl termini in human macrophages, which triggers NLRP3-dependent IL-1β production during VSV and IAV infections (Chakrabarti et al., 2015). As with DHX9, DHX33-mediated type I IFN induction in myeloid DCs has been shown to require MAVS, but not RLRs.
Several helicase proteins have been identified to function as RLR co-sensors. DDX3 associates with MAVS and RLRs to strengthen type I IFN responses (Oshiumi et al., 2010), while DDX60 interacts only with RLRs, but not with MAVS, to facilitate the induction of type I IFNs and ISGs (Miyashita et al., 2011; Oshiumi et al., 2015). Both DDX3 and DDX60 bind to synthetic RNA in vitro; however, whether their RNA-binding ability is required for their potentiating effects on RLR signaling remains unclear. A recent study proposes that DDX3 can act as a direct RNA sensor that functions independently of RLRs. In human monocyte-derived DCs and macrophages, DDX3 associates with abortive HIV-1 RNA and endogenous cellular transcripts lacking 3’ poly(A) tails, and then triggers type I IFN responses in a MAVS-and TRAF3-dependent manner (Gringhuis et al., 2017). In response to poly(I:C), DHX15 interacts with MAVS, thereby activating IRF3, MAPKs and NF-κB (Lu et al., 2014; Mosallanejad et al., 2014). DHX15 has been shown to sense specifically RNA virus infection (i.e. SeV and reovirus), but does not detect poly(dG:dC) or DNA virus infection (i.e. HSV-1) (Lu et al., 2014). DHX15 directly binds poly(I:C) in an ATPase-independent manner, and the RNA-binding affinity of DHX15 is enhanced by DHX9 interaction (Lu et al., 2014). DHX15 also cooperates with specific NLR proteins to facilitate type I and III IFN responses during EMCV infection (Wang et al., 2015). In the context of RLR signaling, DHX15 has been shown to interact with the RIG-I CARDs, leading to RIG-I activation (Pattabhi et al., 2019). DHX29 is reported to bind poly(I:C) in vitro and to interact with RIG-I and MAVS. Individual silencing of DHX29 and RIG-I leads to a comparable reduction in type I IFN gene expression, suggesting a shared signaling axis (Sugimoto et al., 2014). A recent study proposes that DHX29 functions as a co-sensor for MDA5, but not for RIG-I. Upon interaction, DHX29 promotes MDA5 activation by enhancing its ability to bind RNA and to form filaments (Zhu et al., 2018).
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are nuclear or nucleocytoplasmic RNA-binding proteins that have recently been implicated in RNA sensing (Figure 3). Traditionally, hnRNPs are known to regulate RNA metabolism at either the transcriptional or translational level (Geuens et al., 2016). At least three hnRNP members have been identified to participate in the negative regulation of RLR signaling. Upon VSV infection, hnRNPE2 (also known as PCBP2) translocates from the nucleus to the cytoplasm, where it interacts with MAVS at the mitochondrial outer membrane to mediate MAVS degradation by the E3 ubiquitin ligase AIP4 (You et al., 2009). Such virus-induced nucleus-to-cytoplasm redistribution has also been observed for hnRNPM which, when present in the cytoplasm, interacts with RLRs to impair their RNA-binding activities (Cao et al., 2019b). Silencing of hnRNPC, a cancer biomarker, leads to the accumulation of endogenous dsRNA enriched in Alu elements. These RNAs induce RIG-I-mediated type I IFN responses, which, in turn, inhibit cancer cell proliferation and tumorigenesis (Wu et al., 2018). A recent study proposes hnRNPU (also known as SAFA) as a nuclear RNA sensor (Cao et al., 2019a). HnRNPU associates with HSV-1-derived dsRNA in the nucleus and mediates type I IFN induction. HnRNPU also interacts with SMARCA5 and TOP1, two key components of the SWI/SNF nucleosome remodeling complex, to activate enhancers and superenhancers that drive the transcription of antiviral and proinflammatory genes. hnRNPU deficiency reduces the residual type I IFN response in cells lacking MAVS, TBK1 or IRF3, indicating a nuclear signaling axis directly acting on type I IFN transcription.
ZBP1 (also known as DAI) contains two N-terminal Z-DNA-binding domains (Zα1 and Zα2) and was originally identified as a DNA sensor that mediates type I IFN responses to synthetic DNA or HSV-1 infection (Takaoka et al., 2007). More recently, ZBP1 has been shown to function as an RNA sensor activating cell death pathways in response to viral infection. During IAV infection in mice, ZBP1 associates with IAV (sub)genomic RNA through the Zα2 domain, and engages RIPK3 to recruit either the RIPK1–FADD–caspase-8 complex for the activation of apoptosis, or MLKL for necroptosis induction (Kuriakose et al., 2016; Nogusa et al., 2016; Thapa et al., 2016). Structural analyses support that the ZBP1 Zα2 domain can accommodate either dsDNA or dsRNA in a ‘Z’ conformation (Thapa et al., 2016), and IAV infection has recently been shown to generate Z-RNA (Zhang et al., 2020). Of note, Z-RNA ‘signature’ is also known to be present within the transcripts of endogenous retroelements (e.g. Alu), which engage the Zα domain of ADAR1-p150 (the IFN-inducible isoform of ADAR1) for RNA editing (Herbert, 2019). ZBP1-associated IAV RNAs show similarity to those associated with RIG-I during IAV infection (Baum et al., 2010; Thapa et al., 2016), suggesting a set of RNA-PAMPs that is collaboratively sensed by the two sensors. These two sensors likely act in a sequential mode since ZBP1-induced cell death has been shown to require RIG-I-mediated type I IFN induction (Kuriakose et al., 2016). Further supporting a role for ZBP1 in RNA recognition, ZBP1 has been found to associate with nascently synthesized viral (and likely also host) RNA, but not DNA, during murine cytomegalovirus (MCMV)-induced necroptosis (Maelfait et al., 2017; Sridharan et al., 2017). While ZBP1 localizes primarily to the cytoplasm of uninfected cells, MCMV and IAV infections induce nuclear accumulation of ZBP1 (Maelfait et al., 2017; Sridharan et al., 2017; Zhang et al., 2020). ZBP1 also directly binds IAV-derived Z-RNA in the nucleus, within which it initiates necroptosis (Zhang et al., 2020).
Whereas these recent advances on the involvement of RNA helicases, hnRNPs, and ZBP1 in RNA sensing are exciting, additional studies are needed to confirm the physiological contribution of these proteins to innate immunity, and whether they primarily act by directly detecting RNA or by regulating RNA-dependent signaling through indirect mechanisms.
RNA Sensor Polymorphisms in Associated Autoinflammation and Infection
It has long been proposed that mutations that lead to aberrant nucleic sensing or constitutive activation of innate immune receptors may cause autoimmune or autoinflammatory diseases in humans. Indeed, common to several types of autoimmune diseases is an inborn excessive production of IFN-α/β, thus prompting the term ‘type I interferonopathies’. On the other hand, mutations that ablate the proper functioning of innate immune sensors are expected to decrease the host organism’s defense against pathogens. Recent research demonstrates that specific mutations in members of the RLR and TLR protein families are associated with several inherited autoimmune disorders, or an increased susceptibility to infections.
Gain-of-function mutations in the RLR members IFIH1 (encoding MDA5) and DDX58 (encoding RIG-I) have been shown to be causative of several rare, inherited autoimmune diseases: classical and atypical Singleton-Merten syndrome (SMS) as well as specific types of Aicardi-Goutières-syndrome (AGS) and Systemic Lupus Erythematosus (SLE). Common to these diseases are constantly elevated type I IFN amounts; yet, each disease is caused by a distinct RLR polymorphism and hence a specific underlying mechanism of RLR activation.
AGS is a multi-symptom autoimmune disease that most consistently affects the brain and the skin. AGS is caused by mutations – autosomal recessive or autosomal dominant ones – in one of several genes that are involved in RNA-or DNA-metabolizing processes: TREX1, ADAR1, SAMHD1 and RNASEH2 (subunit A, B or C). Additionally, multiple AGS-associated missense mutations in IFIH1 have been identified (p.Arg337Gly, p.Arg779Cys, p.Gly495Arg, p.Asp393Val, p.Arg720Gln, p.Arg779His, p.Ala452Thr, p.Leu372Phe), all of which are monoallelic and located within close proximity to the RNA-binding or ATP-binding sites in the MDA5 helicase domain (Oda et al., 2014; Rice et al., 2014). In vitro, these MDA5 mutations lead to heightened type I IFN induction as compared to wildtype MDA5, and several mechanisms (e.g. more effective RNA-binding ability, constitutive MDA5 activation without the need of RNA) have been proposed (Oda et al., 2014; Rice et al., 2014). Recent reports indicate a mechanistic interplay between MDA5 and ADAR1 in AGS. Whereas loss-of-function of ADAR (lack of A to I conversion) triggers aberrant activation of MDA5 by recognition of unmodified endogenous RNAs, such as Alu elements (Ahmad et al., 2018; Liddicoat et al., 2015; Pestal et al., 2015), MDA5 variants found in AGS individuals show reduced susceptibility to inhibition by RNA duplex structural irregularities as compared to wildtype MDA5; this ultimately leads to the formation of signaling-competent filaments by mutant MDA5 on Alu-dsRNAs (Ahmad et al., 2018).
Genetic mutations in several different genes that encode critical components of the type I IFN pathway have been associated with SLE, a chronic autoinflammatory disease with a multitude of symptoms including arthritis, skin rashes, nephritis, and neuroinflammation. At least two polymorphisms were found in the IFIH1 gene, p.Ala946Thr and p.Arg779His, the latter found by whole-exome sequencing in a patient with severe early-onset SLE (Cen et al., 2013; Gono et al., 2010; Molineros et al., 2013; Van Eyck et al., 2015). Both polymorphisms are proposed to lead to ‘hyper-active’ states of MDA5, which is consistent with the fact that SLE patients exhibit elevated amounts of type I IFNs.
Moreover, a missense mutations (p.Arg822Gln) in the gene encoding MDA5 has been found to be causative of SMS (Rutsch et al., 2015), which is an extremely rare, autosomal-dominant inherited disease that is characterized by dental and skeletal abnormalities and cardiac calcifications along with an enhanced type I IFN signature gene pattern. The Arg822Gln mutation is located in the HEL2 domain of MDA5 and is predicted to trigger conformational alterations that may increase the formation and/or stability of MDA5 filaments on dsRNA, resulting in constitutive activation of the sensor. Apart from this ‘classical’ type of SMS, a more recently discovered form of SMS called ‘atypical’ SMS has been found to be caused by monoallelic mutations in the DDX58 gene that encodes RIG-I (p.Cys268Phe and p.Glu373Ala), which are located in the ATPase motifs of the sensor (Jang et al., 2015). Functional characterization shows that these RIG-I mutations have deficient ATPase activity and increased binding to self RNA, which ultimately leads to aberrant innate immune signaling and transcriptional induction of IFN-β (Lassig et al., 2015).
Single nucleotide polymorphisms (SNPs) in the TLRs 3, 7 and 8 have been linked to an increased susceptibility to certain viral or bacterial infections. A rare variant of TLR3 (p.Pro554Ser), which is defective in activating IRF3 and NF-κB, has been identified in two patients that exhibited encephalitis caused by HSV-1 infection (Zhang et al., 2007). Two other TLR3 SNPs (p.Asn248Ile and p.Leu412Phe), with the latter being widely prevalent, locate in the LRR domain of TLR3 and were found to dampen TLR3 signaling activity in vitro by reducing its cell surface localization (Ranjith-Kumar et al., 2007). The p.Leu412Phe SNP was further proposed to increase cell survival responses in siRNA-based treatment of angiogenic disorders (Kleinman et al., 2008). Several SNPs in TLR7 – both coding changing and synonymous ones – were shown to affect chronic HCV infection or disease outcome (i.e., fibrosis, inflammation) (Askar et al., 2010; Schott et al., 2007). For example, p.Gln11Leu in TLR7 decreased IFN-λ expression in chronic HCV-infected individuals but had no direct effect on viral loads (Askar et al., 2010). HIV-infected individuals carrying the same TLR7 variant (p.Gln11Leu) showed an accelerated progression of HIV-induced disease and higher viral loads (Oh et al., 2009). Of note, the TLR7 Gln11Leu polymorphism has also been linked to a predisposition to SLE especially in males from East Asian origin (Shen et al., 2010). Furthermore, at least two variants of TLR8 have been linked to higher susceptibility to pulmonary tuberculosis and Crimean-Congo hemorrhagic fever (Davila et al., 2008; Engin et al., 2010).
Concluding Remarks
Molecular insight into the activation and regulation of RNA sensors in innate immunity is important for understanding their implications in microbial infection and sterile inflammation. With the continuous effort to identify additional RNA sensors, what remains less well understood is the physiological relevance and contribution of emerging sensors to antimicrobial host defenses and/or pathology in relation to the known bona fide sensors. For example, the physiologic role of DEAD-box or DEAH-box RNA helicases, whose contribution to the type I IFN response relies strictly on MAVS but appears to be independent of RLRs, is not very well understood. It is conceivable that the relative contribution of DEAD-box or DEAH-box RNA helicases to antiviral type I IFN induction is dictated by a distinct temporal expression pattern. Furthermore, for many RNA sensors, especially the recently identified ones, physiological ligands and their detailed characteristics remain to be determined.
Since many of the emerging RNA sensors also have important roles in cellular RNA metabolism or exert direct antiviral functions, it will be important to determine the contribution of their direct RNA-sensing capacity to antiviral immunity. Furthermore, the molecular details of how different RNA sensors ‘cross-talk’ have just begun to be elucidated. This information will, more broadly, enhance our understanding of how RNA sensing, RNA metabolism, and the direct activities of these proteins on viral nucleic acid cooperate in ‘nucleic acid immunity’.
Viral strategies to evade TLR and RLR signaling have been extensively investigated, which include (1) sequestration, masking, or modifying viral RNA, (2) antagonism of RNA sensors through direct binding or manipulation of their regulatory PTMs, and (3) inhibition of downstream signaling molecules through a variety of mechanisms (Chan and Gack, 2016). As such, exploration of how viruses evade detection by DEAD-box or DEAH-box RNA helicases and other emerging RNA sensors will be an important avenue for future research. Moreover, specific agonists of RLRs and TLRs showed promise in the treatment of cancers or infectious diseases (Barrat et al., 2016; Rehwinkel and Gack, 2020). Therefore, investigating the molecular features of RNA ligands may aid in developing therapeutic strategies to combat microbial infection, cancer, and autoinflammatory diseases.
Highlights:
RLRs and several TLRs sense microbial or cellular RNA in several cell compartments.
NLRs can directly sense RNA or they regulate other RNA-sensing pathways.
DEAD/DEAH-box helicases, hnRNPs and ZBP1 are emerging RNA sensors or co-sensors.
RNA sensor polymorphisms are implicated in autoinflammation and infection.
ACKNOWLEDGMENTS
We apologize to all whose important contributions could not be discussed and cited due to space constraints. Current research in Dr. Gack laboratory is supported by U.S. National Institutes of Health (NIH) grants (R01 AI087846, R01 AI127774 and R21 AI148082) and an award from the ClayCo Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abe T, Lee A, Sitharam R, Kesner J, Rabadan R, and Shapira SD (2017). Germ-Cell-Specific Inflammasome Component NLRP14 Negatively Regulates Cytosolic Nucleic Acid Sensing to Promote Fertilization. Immunity 46, 621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe Y, Fujii K, Nagata N, Takeuchi O, Akira S, Oshiumi H, Matsumoto M, Seya T, and Koike S (2012). The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J Virol 86, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, and Hornung V (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol 10, 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, and Hur S (2018). Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 172, 797–810 e713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, and Flavell RA (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]
- Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, et al. (2011). NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 34, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, and Randall RE (2004). The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 101, 17264–17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askar E, Ramadori G, and Mihm S (2010). Toll-like receptor 7 rs179008/Gln11Leu gene variants in chronic hepatitis C virus infection. Journal of medical virology 82, 1859–1868. [DOI] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat Rev Immunol 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Elkon KB, and Fitzgerald KA (2016). Importance of Nucleic Acid Recognition in Inflammation and Autoimmunity. Annu Rev Med 67, 323–336. [DOI] [PubMed] [Google Scholar]
- Baum A, Sachidanandam R, and Garcia-Sastre A (2010). Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 107, 16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, Flo TH, Mollnes TE, Espevik T, and Stenvik J (2015). TLR8 Senses Staphylococcus aureus RNA in Human Primary Monocytes and Macrophages and Induces IFN-beta Production via a TAK1-IKKbeta-IRF5 Signaling Pathway. J Immunol 195, 1100–1111. [DOI] [PubMed] [Google Scholar]
- Brubaker SW, Gauthier AE, Mills EW, Ingolia NT, and Kagan JC (2014). A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell 156, 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns AM, and Horvath CM (2015). LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine 74, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns AM, Leser GP, Lamb RA, and Horvath CM (2014). The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol Cell 55, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns AM, Pollpeter D, Hadizadeh N, Myong S, Marko JF, and Horvath CM (2013). ATP hydrolysis enhances RNA recognition and antiviral signal transduction by the innate immune sensor, laboratory of genetics and physiology 2 (LGP2). J Biol Chem 288, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena C, Ahmad S, Xavier A, Willemsen J, Park S, Park JW, Oh SW, Fujita T, Hou F, Binder M, and Hur S (2019). Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 177, 1187–1200 e1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Liu S, Li Y, Yang G, Luo Y, Li S, Du H, Zhao Y, Wang D, Chen J , et l. (2019a). The Nuclear Matrix Protein SAFA Surveils Viral RNA and Facilitates Immunity by Activating Antiviral Enhancers and Super-enhancers. Cell Host Microbe 26, 369–384 e368. [DOI] [PubMed] [Google Scholar]
- Cao P, Luo WW, Li C, Tong Z, Zheng ZQ, Zhou L, Xiong Y, and Li S (2019b). The heterogeneous nuclear ribonucleoprotein hnRNPM inhibits RNA virus-triggered innate immunity by antagonizing RNA sensing of RIG-I-like receptors. PLoS Pathog 15, e1007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen H, Leng RX, Wang W, Zhou M, Feng CC, Zhu Y, Yang XK, Yang M, Zhai Y, Li BZ, et al. (2013). Association study of IFIH1 rs1990760 polymorphism with systemic lupus erythematosus in a Chinese population. Inflammation 36, 444–448. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A, Banerjee S, Franchi L, Loo YM, Gale M Jr., Nunez G, and Silverman RH (2015). RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17, 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YK, and Gack MU (2016). Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14, 360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ST, Chen L, Lin DS, Chen SY, Tsao YP, Guo H, Li FJ, Tseng WT, Tam JW, Chao CW, et al. (2019). NLRP12 Regulates Anti-viral RIG-I Activation via Interaction with TRIM25. Cell Host Microbe 25, 602–616 e607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, and Chang HY (2017). Sensing Self and Foreign Circular RNAs by Intron Identity. Mol Cell 67, 228–238 e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, and Gack MU (2017). Post-translational Control of Intracellular Pathogen Sensing Pathways. Trends Immunol 38, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, Hopfner KP, and Gack MU (2018). Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol 19, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, and Chen ZJ (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe J, Kelker MS, and Wilson IA (2005). Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 309, 581–585. [DOI] [PubMed] [Google Scholar]
- Choi SJ, Lee HC, Kim JH, Park SY, Kim TH, Lee WK, Jang DJ, Yoon JE, Choi YI, Kim S, et al. (2016). HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J 35, 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KT, Gale M Jr., and Loo YM (2018a). RIG-I and Other RNA Sensors in Antiviral Immunity. Annu Rev Immunol 36, 667–694. [DOI] [PubMed] [Google Scholar]
- Chow KT, Wilkins C, Narita M, Green R, Knoll M, Loo YM, and Gale M Jr. (2018b). Differential and Overlapping Immune Programs Regulated by IRF3 and IRF5 in Plasmacytoid Dendritic Cells. J Immunol 201, 3036–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, and Wang RF (2010). NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 141, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Li M, Walton KD, Sun K, Hanover JA, Furth PA, and Hennighausen L (2001). The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics 78, 129–134. [DOI] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Suthar MS, Gale M Jr., and Diamond MS (2008). Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol 82, 10349–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila S, Hibberd ML, Hari Dass R, Wong HE, Sahiratmadja E, Bonnard C, Alisjahbana B, Szeszko JS, Balabanova Y, Drobniewski F, et al. (2008). Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS genetics 4, e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deddouche S, Goubau D, Rehwinkel J, Chakravarty P, Begum S, Maillard PV, Borg A, Matthews N, Feng Q, van Kuppeveld FJ, and Reis e Sousa C (2014). Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. Elife 3, e01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, Crow YJ, Rice GI, Duffy D, Tamby C, et al. (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, and Reis e Sousa C (2004). Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531. [DOI] [PubMed] [Google Scholar]
- Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, and Stetson DB (2014). The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol 15, 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin A, Arslan S, Kizildag S, Ozturk H, Elaldi N, Dokmetas I, and Bakir M (2010). Toll-like receptor 8 and 9 polymorphisms in Crimean-Congo hemorrhagic fever. Microbes and infection 12, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Errett JS, Suthar MS, McMillan A, Diamond MS, and Gale M Jr. (2013). The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol 87, 11416–11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, Onji M, Matsumoto M, Akira S, Yoshida N, and Miyake K (2011). Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 35, 69–81. [DOI] [PubMed] [Google Scholar]
- Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, and Jung JU (2008). Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A 105, 16743–16748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, and Jung JU (2010). Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol 84, 3220–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, and Jung JU (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. [DOI] [PubMed] [Google Scholar]
- Gay NJ, Gangloff M, and O’Neill LA (2011). What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol 32, 104–109. [DOI] [PubMed] [Google Scholar]
- Geuens T, Bouhy D, and Timmerman V (2016). The hnRNP family: insights into their role in health and disease. Hum Genet 135, 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gono T, Kawaguchi Y, Sugiura T, Furuya T, Kawamoto M, Hanaoka M, and Yamanaka H (2010). Interferon-induced helicase (IFIH1) polymorphism with systemic lupus erythematosus and dermatomyositis/polymyositis. Modern rheumatology 20, 466–470. [DOI] [PubMed] [Google Scholar]
- Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, and Vasilakos JP (2005). Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol 174, 1259–1268. [DOI] [PubMed] [Google Scholar]
- Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, et al. (2014). Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 514, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringhuis SI, Hertoghs N, Kaptein TM, Zijlstra-Willems EM, Sarrami-Forooshani R, Sprokholt JK, van Teijlingen NH, Kootstra NA, Booiman T, van Dort KA, et al. (2017). HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat Immunol 18, 225–235. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, and Bauer S (2004). Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529. [DOI] [PubMed] [Google Scholar]
- Herbert A (2019). Z-DNA and Z-RNA in human disease. Commun Biol 2, 7. Hidmark, A., von Saint Paul, A., and Dalpke, A.H. (2012). Cutting edge: TLR13 is a receptor for bacterial RNA. J Immunol 189, 2717–2721. [DOI] [PubMed] [Google Scholar]
- Hipp MM, Shepherd D, Gileadi U, Aichinger MC, Kessler BM, Edelmann MJ, Essalmani R, Seidah NG, Reis e Sousa C, and Cerundolo V (2013). Processing of human toll-like receptor 7 by furin-like proprotein convertases is required for its accumulation and activity in endosomes. Immunity 39, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. (2003). Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, and Taniguchi T (2004). Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A 101, 15416–15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Yoon SI, and Wilson IA (2012). Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 36, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner SM, Liu HM, Park HS, Briley J, and Gale M Jr. (2011). Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A 108, 14590–14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, et al. (2006). 5’-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997. [DOI] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, et al. (2005). Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11, 263–270. [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, and Chen ZJ (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Funami K, Tatematsu M, Seya T, and Matsumoto M (2014). Endosomal localization of TLR8 confers distinctive proteolytic processing on human myeloid cells. J Immunol 193, 5118–5128. [DOI] [PubMed] [Google Scholar]
- Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, Lee J, Jeong YM, Kim CH, Kim OH, et al. (2015). Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. American journal of human genetics 96, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zhang S, Yang Z, Lin H, Zhu J, Liu L, Wang W, Liu S, Liu W, Ma Y, et al. (2018). Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response. Cell 173, 906–919 e913. [DOI] [PubMed] [Google Scholar]
- Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, and Chen ZJ (2012). Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MS, and Lee JO (2008). Structures of the toll-like receptor family and its ligand complexes. Immunity 29, 182–191. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, and Nunez G (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559. [DOI] [PubMed] [Google Scholar]
- Kanno A, Tanimura N, Ishizaki M, Ohko K, Motoi Y, Onji M, Fukui R, Shimozato T, Yamamoto K, Shibata T, et al. (2015). Targeting cell surface TLR7 for therapeutic intervention in autoimmune diseases. Nat Commun 6, 6119. [DOI] [PubMed] [Google Scholar]
- Kariko K, Bhuyan P, Capodici J, and Weissman D (2004a). Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J Immunol 172, 6545–6549. [DOI] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, and Weissman D (2004b). mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 279, 12542–12550. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, and Akira S (2008). Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205, 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, et al. (2004). Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5, 1061–1068. [DOI] [PubMed] [Google Scholar]
- Kim YM, Brinkmann MM, Paquet ME, and Ploegh HL (2008). UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452, 234–238. [DOI] [PubMed] [Google Scholar]
- Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, et al. (2008). Sequence-and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452, 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakofsky D, Kowalinski E, and Cusack S (2012). A structure-based model of RIG-I activation. RNA 18, 2118–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro A, and Horvath CM (2006). RNA-and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol 80, 12332–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, and Theofilopoulos AN (2009). Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A 106, 12061–12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, and Kawai T (2011). NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. Journal of immunology 186, 994–1000. [DOI] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti TD (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang X, Tang T, Jin T, Ding C, Zhou R, and Jiang W (2017). TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J Exp Med 214, 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassig C, Matheisl S, Sparrer KM, de Oliveira Mann CC, Moldt M, Patel JR, Goldeck M, Hartmann G, Garcia-Sastre A, Hornung V, et al. (2015). ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, and Si-Tahar M (2006). Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtenberg BC, Mace PD, and Riedl SJ (2014). Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol 29, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, and Barton GM (2013). UNC93B1 mediates differential trafficking of endosomal TLRs. Elife 2, e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, and Iwasaki A (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401. [DOI] [PubMed] [Google Scholar]
- Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, et al. (2012). An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 15, 827–835. [DOI] [PubMed] [Google Scholar]
- Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, Swanson KV, Wen KW, Damania B, Moore CB, et al. (2012). The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36, 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifer CA, and Medvedev AE (2016). Molecular mechanisms of regulation of Toll-like receptor signaling. J Leukoc Biol 100, 927–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, and Segal DM (2008). The TLR3 signaling complex forms by cooperative receptor dimerization. Proc Natl Acad Sci U S A 105, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, and Chen ZJ (2012). Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. Elife 1, e00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Zang R, Wei J, Ye W, Hu MM, Chen YD, Zhang XN, Guo Y, Lei CQ, Yang Q, et al. (2018). The Zinc-Finger Protein ZCCHC3 Binds RNA and Facilitates Viral RNA Sensing and Activation of the RIG-I-like Receptors. Immunity 49, 438–448 e435. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Jiang M, Liu L, Yang Z, Ma Z, Liu S, Ma Y, Zhang L, and Cao X (2019). The long noncoding RNA Lnczc3h7a promotes a TRIM25-mediated RIG-I antiviral innate immune response. Nat Immunol 20, 812–823. [DOI] [PubMed] [Google Scholar]
- Liu G, Lu Y, Thulasi Raman SN, Xu F, Wu Q, Li Z, Brownlie R, Liu Q, and Zhou Y (2018). Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat Commun 9, 3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Park HS, Pyo HM, Liu Q, and Zhou Y (2015). Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J Virol 89, 6067–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, Jiang F, Loo YM, Hsu S, Hsiang TY, Marcotrigiano J, and Gale M Jr. (2016). Regulation of Retinoic Acid Inducible Gene-I (RIG-I) Activation by the Histone Deacetylase 6. EBioMedicine 9, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, and Davies DR (2008). Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, and Chen ZJ (2013). MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2, e00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu N, Yuan B, Weng L, Wang F, Liu YJ, and Zhang Z (2014). The interaction between the helicase DHX33 and IPS-1 as a novel pathway to sense double-stranded RNA and RNA viruses in myeloid dendritic cells. Cell Mol Immunol 11, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wu C, Pan Y, Liu H, Wang X, Yang Y, Gu M, Zhang Y, and Wang X (2019). NDR2 promotes the antiviral immune response via facilitating TRIM25-mediated RIG-I activation in macrophages. Sci Adv 5, eaav0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, and Gale M Jr. (2008). Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Lu N, Weng L, Yuan B, Liu YJ, and Zhang Z (2014). DHX15 senses double-stranded RNA in myeloid dendritic cells. J Immunol 193, 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, and Flavell RA (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 101, 5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Sun D, Silverman RH, and He B (2011). Activation of IFN-β expression by a viral mRNA through RNase L and MDA5. Proc Natl Acad Sci U S A 108, 2118–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, and Rehwinkel J (2017). Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J 36, 2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj NP, Wies E, Stoll A, and Gack MU (2012). Conventional protein kinase C-alpha (PKC-alpha) and PKC-beta negatively regulate RIG-I antiviral signal transduction. J Virol 86, 1358–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer O, Liu B, Kreuk LSM, Krogan N, and Barton GM (2019). UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature 575, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M Jr., and Silverman RH (2007). Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, et al. (2014). Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, and Yan B (2017). Clash of the Cytokine Titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol 14, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, Poeck H, Bscheider M, Hartmann G, Konig M, et al. (2008). TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 28, 651–661. [DOI] [PubMed] [Google Scholar]
- Minamitani T, Iwakiri D, and Takada K (2011). Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J Virol 85, 4035–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, and Liu YJ (2013). The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, Shibata T, Ohto U, and Shimizu T (2017). Emerging roles of the processing of nucleic acids and Toll-like receptors in innate immune responses to nucleic acids. J Leukoc Biol 101, 135–142. [DOI] [PubMed] [Google Scholar]
- Miyashita M, Oshiumi H, Matsumoto M, and Seya T (2011). DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol 31, 3802–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineros JE, Maiti AK, Sun C, Looger LL, Han S, Kim-Howard X, Glenn S, Adler A, Kelly JA, Niewold TB, et al. (2013). Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS genetics 9, e1003222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, et al. (2008). NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451, 573–577. [DOI] [PubMed] [Google Scholar]
- Mosallanejad K, Sekine Y, Ishikura-Kinoshita S, Kumagai K, Nagano T, Matsuzawa A, Takeda K, Naguro I, and Ichijo H (2014). The DEAH-box RNA helicase DHX15 activates NF-kappaB and MAPK signaling downstream of MAVS during antiviral responses. Sci Signal 7, ra40. [DOI] [PubMed] [Google Scholar]
- Murali A, Li X, Ranjith-Kumar CT, Bhardwaj K, Holzenburg A, Li P, and Kao CC (2008). Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J Biol Chem 283, 15825–15833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. (2016). RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Golenbock D, and Bowie AG (2013). The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol 13, 453–460. [DOI] [PubMed] [Google Scholar]
- Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, et al. (2014). Aicardi-Goutieres syndrome is caused by IFIH1 mutations. American journal of human genetics 95, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Baumann K, Hamouda O, Eckert JK, Neumann K, Kucherer C, Bartmeyer B, Poggensee G, Oh N, Pruss A, et al. (2009). A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. Aids 23, 297–307. [DOI] [PubMed] [Google Scholar]
- Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, et al. (2012). TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337, 1111–1115. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Matsumoto M, Funami K, Akazawa T, and Seya T (2003). TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol 4, 161–167. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Matsumoto M, Hatakeyama S, and Seya T (2009). Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem 284, 807–817. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Miyashita M, Matsumoto M, and Seya T (2013). A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog 9, e1003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiumi H, Miyashita M, Okamoto M, Morioka Y, Okabe M, Matsumoto M, and Seya T (2015). DDX60 Is Involved in RIG-I-Dependent and Independent Antiviral Responses, and Its Function Is Attenuated by Virus-Induced EGFR Activation. Cell Rep 11, 1193–1207. [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Sakai K, Matsumoto M, and Seya T (2010). DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur J Immunol 40, 940–948. [DOI] [PubMed] [Google Scholar]
- Pattabhi S, Knoll ML, Gale M Jr., and Loo YM (2019). DHX15 Is a Coreceptor for RLR Signaling That Promotes Antiviral Defense Against RNA Virus Infection. J Interferon Cytokine Res 39, 331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Wu B, Xu H, Chen ZJ, and Hur S (2014). Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, Stahl R, Watson N, Miyake K, Hacohen N, et al. (2018). The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity 48, 911–922 e917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelka K, Shibata T, Miyake K, and Latz E (2016). Nucleic acid-sensing TLRs and autoimmunity: novel insights from structural and cell biology. Immunol Rev 269, 60–75. [DOI] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, and Stetson DB (2015). Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, and Reis e Sousa C (2006). RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314, 997–1001. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, and Reis e Sousa C (2009). Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83, 10761–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohar J, Pirher N, Bencina M, Mancek-Keber M, and Jerala R (2013). The role of UNC93B1 protein in surface localization of TLR3 receptor and in cell priming to nucleic acid agonists. J Biol Chem 288, 442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Xue B, Liu C, Wang X, Tian R, Xie Q, Guo M, Li G, Yang D, and Zhu H (2017). NLRX1 Mediates MAVS Degradation To Attenuate the Hepatitis C Virus-Induced Innate Immune Response through PCBP2. Journal of virology 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke KM, Kim KY, Horvath CM, and Suthar MS (2019). RNA Helicase LGP2 Negatively Regulates RIG-I Signaling by Preventing TRIM25-Mediated Caspase Activation and Recruitment Domain Ubiquitination. J Interferon Cytokine Res 39, 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjith-Kumar CT, Miller W, Sun J, Xiong J, Santos J, Yarbrough I, Lamb RJ, Mills J, Duffy KE, Hoose S, et al. (2007). Effects of single nucleotide polymorphisms on Toll-like receptor 3 activity and expression in cultured cells. J Biol Chem 282, 17696–17705. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, and Gack MU (2020). RIG-I-like receptors: their regulation and roles in RNA sensing. Nature Reviews Immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikine S, Nguyen JB, and Modis Y (2014). Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front Immunol 5, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. (2014). Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nature genetics 46, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, and Fitzgerald KA (2005). The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol 175, 5260–5268. [DOI] [PubMed] [Google Scholar]
- Runge S, Sparrer KM, Lassig C, Hembach K, Baum A, Garcia-Sastre A, Soding J, Conzelmann KK, and Hopfner KP (2014). In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog 10, e1004081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, et al. (2015). A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. American journal of human genetics 96, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, and Bose S (2009). Activation of innate immune antiviral responses by Nod2. Nature immunology 10, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, and Gale M Jr. (2007). Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A 104, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Owen DM, Jiang F, Marcotrigiano J, and Gale M Jr. (2008). Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JG, Sparrer KMJ, Chiang C, Reis RA, Chiang JJ, Zurenski MA, Wan Y, Gack MU, and Pornillos O (2018). TRIM25 Binds RNA to Modulate Cellular Anti-viral Defense. J Mol Biol 430, 5280–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, Watanabe T, Iijima S, Sakurai Y, Watashi K, et al. (2015). The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132. [DOI] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, and Takeuchi O (2010). LGP2 is a positive regulator of RIG-I-and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A 107, 1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, and Hartmann G (2016). Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 16, 566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, et al. (2009). Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP, et al. (2009). 5’-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A 106, 12067–12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott E, Witt H, Neumann K, Taube S, Oh DY, Schreier E, Vierich S, Puhl G, Bergk A, Halangk J, et al. (2007). A Toll-like receptor 7 single nucleotide polymorphism protects from advanced inflammation and fibrosis in male patients with chronic HCV-infection. Journal of hepatology 47, 203–211. [DOI] [PubMed] [Google Scholar]
- Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, and Reis e Sousa C (2005). Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433, 887–892. [DOI] [PubMed] [Google Scholar]
- Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, Wu YL, Yu CY, Tang Y, Chen JY, et al. (2010). Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America 107, 15838–15843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Cai Z, Sanchez A, Zhang T, Wen S, Wang J, Yang J, Fu S, and Zhang D (2011). A novel Toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem 286, 4517–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Wang J, Han Z, Zhang Y, Zhang H, Wang W, Chang J, Xia B, Fan S, Zhang D, et al. (2015). Structural basis for specific recognition of single-stranded RNA by Toll-like receptor 13. Nat Struct Mol Biol 22, 782–787. [DOI] [PubMed] [Google Scholar]
- Sridharan H, Ragan KB, Guo H, Gilley RP, Landsteiner VJ, Kaiser WJ, and Upton JW (2017). Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep 18, 1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, and Germain RN (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto N, Mitoma H, Kim T, Hanabuchi S, and Liu YJ (2014). Helicase proteins DHX29 and RIG-I cosense cytosolic nucleic acids in the human airway system. Proc Natl Acad Sci U S A 111, 7747–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Duffy KE, Ranjith-Kumar CT, Xiong J, Lamb RJ, Santos J, Masarapu H, Cunningham M, Holzenburg A, Sarisky RT, et al. (2006). Structural and functional analyses of the human Toll-like receptor 3. Role of glycosylation. J Biol Chem 281, 11144–11151. [DOI] [PubMed] [Google Scholar]
- Sun Z, Ren H, Liu Y, Teeling JL, and Gu J (2011). Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J Virol 85, 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, and Ting JP (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. (2006). The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 7, 156–164. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. (2007). DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, and Taniguchi T (2005). Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249. [DOI] [PubMed] [Google Scholar]
- Takashima K, Oshiumi H, Takaki H, Matsumoto M, and Seya T (2015). RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep 11, 192–200. [DOI] [PubMed] [Google Scholar]
- Tan X, Sun L, Chen J, and Chen ZJ (2018). Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu Rev Microbiol 72, 447–478. [DOI] [PubMed] [Google Scholar]
- Tanji H, Ohto U, Motoi Y, Shibata T, Miyake K, and Shimizu T (2016). Autoinhibition and relief mechanism by the proteolytic processing of Toll-like receptor 8. Proc Natl Acad Sci U S A 113, 3012–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji H, Ohto U, Shibata T, Miyake K, and Shimizu T (2013). Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339, 1426–1429. [DOI] [PubMed] [Google Scholar]
- Tanji H, Ohto U, Shibata T, Taoka M, Yamauchi Y, Isobe T, Miyake K, and Shimizu T (2015). Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat Struct Mol Biol 22, 109–115. [DOI] [PubMed] [Google Scholar]
- Tatematsu M, Nishikawa F, Seya T, and Matsumoto M (2013). Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat Commun 4, 1833. [DOI] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al. (2016). DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 20, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. (2008). The NLR gene family: a standard nomenclature. Immunity 28, 285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Cui J, Li Q, Zou J, Wang HY, and Wang RF (2012). Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell research 22, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikawa E, Lethier M, Malet H, Brunel J, Gerlier D, and Cusack S (2016). Structural Analysis of dsRNA Binding to Anti-viral Pattern Recognition Receptors LGP2 and MDA5. Mol Cell 62, 586–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, et al. (2005). Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7-and TLR9-mediated interferon-{alpha} induction. J Exp Med 201, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyck L, De Somer L, Pombal D, Bornschein S, Frans G, Humblet-Baron S, Moens L, de Zegher F, Bossuyt X, Wouters C, and Liston A (2015). Brief Report: IFIH1 Mutation Causes Systemic Lupus Erythematosus With Selective IgA Deficiency. Arthritis & rheumatology 67, 1592–1597. [DOI] [PubMed] [Google Scholar]
- van Gent M, Sparrer KMJ, and Gack MU (2018). TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu Rev Virol 5, 385–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez C, Tan CY, and Horner SM (2019). Hepatitis C Virus Infection Is Inhibited by a Noncanonical Antiviral Signaling Pathway Targeted by NS3-NS4 A. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, and Barber GN (2007). Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol 178, 6444–6455. [DOI] [PubMed] [Google Scholar]
- Wang P, Arjona A, Zhang Y, Sultana H, Dai J, Yang L, LeBlanc PM, Doiron K, Saleh M, and Fikrig E (2010). Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat Immunol 11, 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, Zheng Y, Rongvaux A, Sun Q, Yang G, et al. (2015). Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, and Flavell RA (2004). Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10, 1366–1373. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yuan S, Jia X, Ge Y, Ling T, Nie M, Lan X, Chen S, and Xu A (2019). Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS. Nat Cell Biol 21, 1346–1356. [DOI] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, and Paludan SR (2006). Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80, 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Sediri H, Felgenhauer U, Binzen I, Banfer S, Jacob R, Brunotte L, Garcia-Sastre A, Schmid-Burgk JL, Schmidt T, et al. (2015). Influenza virus adaptation PB2–627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Miao EA, and Ting JP (2013). Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39, 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, and Gack MU (2013). Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, Magor KE, Walz T, Penczek PA, and Hur S (2014). Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol Cell 55, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhao W, Liu Y, Tan X, Li X, Zou Q, Xiao Z, Xu H, Wang Y, and Yang X (2018). Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mercado-Lopez X, Grier JT, Kim WK, Chun LF, Irvine EB, Del Toro Duany Y, Kell A, Hur S, Gale M Jr., et al. (2015). Identification of a Natural Viral RNA Motif That Optimizes Sensing of Viral RNA by RIG-I. MBio 6, e01265–01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, and Akira S (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr., Akira S, et al. (2005). Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175, 2851–2858. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, and Fujita T (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5, 730–737. [DOI] [PubMed] [Google Scholar]
- You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, and Jiang Z (2009). PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol 10, 1300–1308. [DOI] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, and Chen ZJ (2010). Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141, 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zevini A, Olagnier D, and Hiscott J (2017). Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol 38, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527. [DOI] [PubMed] [Google Scholar]
- Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, Ragan KB, Ishizuka T, Crawford JC, Tummers B, et al. (2020). Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 180, 1115–1129.e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dittmer DP, Mieczkowski PA, Host KM, Fusco WG, Duncan JA, and Damania B (2018a). RIG-I Detects Kaposi’s Sarcoma-Associated Herpesvirus Transcripts in a RNA Polymerase III-Independent Manner. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, and Liu YJ (2011a). DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34, 866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ohto U, Shibata T, Krayukhina E, Taoka M, Yamauchi Y, Tanji H, Isobe T, Uchiyama S, Miyake K, and Shimizu T (2016). Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 45, 737–748. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Ohto U, Shibata T, Taoka M, Yamauchi Y, Sato R, Shukla NM, David SA, Isobe T, Miyake K, and Shimizu T (2018b). Structural Analyses of Toll-like Receptor 7 Reveal Detailed RNA Sequence Specificity and Recognition Mechanism of Agonistic Ligands. Cell Rep 25, 3371–3381 e3375. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Lu N, Facchinetti V, and Liu YJ (2011b). DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J Immunol 187, 4501–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ye X, Dunker W, Song Y, and Karijolich J (2018). RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat Commun 9, 4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Tan P, Li Y, Lin M, Li C, Mao J, Cui J, Zhao W, Wang HY, and Wang RF (2018). DHX29 functions as an RNA co-sensor for MDA5-mediated EMCV-specific antiviral immunity. PLoS Pathog 14, e1006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, Yang Y, Yu H, Li HB, Wang G, et al. (2017). Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]