

Abstract
Purpose of review:
In recent years, we have witnessed a remarkable surge in the clinical development of effective biological and cellular therapies for the treatment of neoplastic and autoimmune disorders. This review summarizes our understanding of the pathogen-specific infection risk associated with the use of such therapies.
Recent findings:
A variety of biologics, in the form of either monoclonal antibodies (Mabs) or small molecule kinase inhibitors (Nibs), are continuously introduced in the clinic for the management of autoimmune and malignant diseases. In addition, cellular therapies such as the infusion of chimeric antigen receptor (CAR) T-cells are becoming increasingly available for patients with treatment-refractory lymphoid malignancies. Some of these biological and cellular interventions exert direct or indirect adverse effects on the induction of protective immune responses against various pathogens, resulting in heightened infection susceptibility.
Summary:
The introduction of biological and cellular therapies for the treatment of malignant and autoimmune diseases has been associated with increased infection susceptiblity, which varies greatly depending on the specific immunomodulatory therapy, the infecting pathogen and the recipient patient population. A high index of clinical suspicion and efforts aiming at early diagnosis, targeted vaccination or prophylaxis, and prompt initiation of antimicrobial treatment should help improve infection outcomes.
Keywords: biologics, monoclonal antibodies, small molecule kinase inhibitors, infection, CAR T-cells
Introduction
In recent decades, the advent of chemotherapeutic and immunomodulatory therapies and of hematopoietic stem cell transplantation (HSCT) and solid organ transplantation (SOT) has revolutionized the management of patients with malignancies, autoimmunity, and end-organ failure. Collectively, these interventions have caused an expansion of human populations at-risk for developing certain infections.
Since the early 2000s, when the BCR-ABL tyrosine kinase inhibitor imatinib proved effective and safe for treating chronic myelogenous leukemia and gastrointestinal stromal tumors (1, 2), an exponential surge in the clinical use of novel mechanism-based biological therapies [i.e., monoclonal antibodies (Mabs), small molecule kinase inhibitors (Nibs)] has further transformed the management of malignant and autoimmune diseases. Moreover, the increasingly broader use of novel cellular therapies such as chimeric antigen receptor (CAR) T-cells shows promise for the successful management of patients with treatment-refractory malignancies (3).
Some biological and cellular therapies have been associated with the emergence of certain infections, which are accounted for by inhibitory effects on immune-related molecules and pathways that are critical for mounting protective innate and adaptive immune responses in humans (4–6). Herein, we briefly outline key observations pertaining to heightened infection susceptibility in the setting of certain biological and cellular therapies; an exhaustive survey of all such infections and therapies is beyond the scope of this mini-review.
Infectious complications associated with Mabs
A few important Mab-associated infectious complications are worth briefly discussing (Table 1). In 2001, the first association between a Mab and infection susceptibility was revealed with the reporting of tuberculosis -often extrapulmonary (including disseminated)- in recipients of the TNF-α inhibitor infliximab (7). TNF-α inhibitors are now well-recognized to predispose to infections by mycobacteria and, less often, endemic dimorphic fungi (e.g., histoplasmosis), consonant with the critical contribution of TNF-α in priming intra-macrophage pathogen clearance (5, 8, 9); infection risk is greater with the Mabs infliximab and adalimumab relative to the soluble TNF-α receptor etanercept, and screening for latent tuberculosis is recommended prior to initiation of anti-TNF-α therapy.
Table 1.
Monoclonal antibodies presented in the current review and their associated infection complications.
| Mab | Target | Main FDA-approved indications | Relative risk for infection | Predominant infection susceptibility |
|---|---|---|---|---|
| Infliximab Adalimumab Etanercept* |
TNF-α | Psoriasis, rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease | High | Mycobacterial infections (including disseminated TB) > endemic fungal infections |
| Tocilizumab | IL-6 receptor | Rheumatoid arthritis, CRS during CAR T-cell therapy | Low (short-term exposures) Intermediate (prolonged administration) |
Bacterial infections |
| Brodalumab Ixekizumab Secukinumab Bimekizumab Ustekinumab Risankizumab Tildrakizumab Guselkumab |
IL-17RA IL-17A IL-17A IL-17A/F IL-12p40 IL-23p19 IL-23p19 IL-23p19 |
Psoriasis, inflammatory bowel disease | Low | Mucosal candidiasis (2–4%) |
| Emapalumab | IFN-γ | Hemophagocytic lymphohistiocytosis |
High | Viral > bacterial > fungal infections (overall frequency, 32%) |
| Alemtuzumab | CD52 | Chronic lymphocytic leukemia, multiple sclerosis | High | Herpetic infections, bacterial pneumonia, CMV reactivation in SOT recipients (common) > mucosal candidiasis and other AIDS-defining opportunistic infections |
| Basiliximab | CD20 | Prophylaxis against organ rejection during kidney transplantation | Intermediate | Bacterial infections, CMV reactivation |
| Rituximab | CD20 | Chronic lymphocytic leukemia, lymphomas, rheumatoid arthritis | High | Bacterial and viral infections (common); hepatitis B reactivation (common in HBsAg positive or HBsAg negative/anti-HBc positive individuals); PML (~1:40,000) |
| Daratumumab | CD38 | Multiple myeloma | High | Bacterial and viral infections |
| Natalizumab | α4 integrin | Multiple sclerosis, inflammatory bowel disease | Intermediate | PML (~1:250) |
| Eculizumab | C5a | Hemolytic syndromes | HIgh | Meningococcal infections, pneumococcal infections; Haemophilus infections (common); invasive fungal disease (in patients with additional risk factors) |
| Pembrolizumab | PD-1 | Several malignancies | Low | TB |
etanercept is a soluble TNF-α receptor, listed here together with the TNF- α targeting Mabs.
Mab, monoclonal antibody; CAR, chimeric antigen receptor; CRS, cytokine release syndrome, CMV, cytomegalovirus; SOT, solid organ transplantation; TB, tuberculosis, PML, progressive multifocal leukoencephalopathy.
Since the advent of TNF-α inhibitors, several other cytokine-targeting Mabs have been introduced in clinical practice, associated with differential Mab-specific infection risk. For example, inhibition of IL-1–related signaling by IL-1β–targeting canakinumab or IL-1 receptor–targeting anakinra is overall well-tolerated from an infection standpoint in the absence of other risk factors. Inhibition of IL-6 receptor signaling by tocilizumab increases the risk for serious bacterial infections in patients with rheumatologic diseases who receive it recursively, whereas it is typically well-tolerated in patients with short-term exposures (i.e., 1–2 doses within 48 hours); sporadic opportunistic fungal infections have also been reported with long-term tocilizumab use, primarily in patients receiving additional immunomodulators such as corticosteroids (10–13).
The recent introduction of Mabs that target IL-17 receptor signaling at various levels (e.g., IL-12p40, IL-12p19, IL-17A, IL-17A/IL-17F, IL-17RA) for the management of psoriasis and inflammatory bowel disease has been associated with refractory mucosal candidiasis in ~2–4% of treated individuals, consistent with the known requirement for intact IL-17 signaling in promoting mucosal antifungal immunity (5, 14–16). Early clinical experience with the GM-CSF receptor-targeting mAb mavrilimumab is suggestive of a possible increased risk for pneumonia (17), whereas early experience with the IFN-γ–targeting mAb emapalumab points to an increased risk for serious infections by bacteria (including bacteremia, pneumonia, sepsis and necrotizing fasciitis), mycobacteria, endemic dimorphic fungi, Pneumocystis jirovecii (PJP) and viruses (particularly herpes zoster) (overall frequency, 32%; FDA package insert). Hence, screening for latent tuberculosis and antiviral and antifungal prophylaxis should be considered in emapalumab-treated patients.
In addition to cytokine-targeting Mabs, Mabs that target and/or deplete lymphocytes also enhance the risk of infection. For example, the CD52-targeting Mab alemtuzumab causes profound T-cell lymphocytopenia, which may last for up to 3–5 years (18). Accordingly, alemtuzumab-treated patients are at high-risk (frequency, >5–10%) for bacterial pneumonia and for reactivation of viral infections with herpes simplex and herpes zoster. As such, vaccination for pneumococcus and herpes zoster is advised prior to alemtuzumab initiation and valacyclovir prophylaxis should be strongly considered in alemtuzumab-treated individuals. Moreover, alemtuzumab given for rejection prevention predisposes to CMV reactivation in SOT recipients, typically within the first year post-transplantation (frequency, ~5–15% depending on the patient population). CMV and, less often, BK virus reactivation may also be observed in SOT recipients treated with basiliximab, a Mab that targets the α-chain (CD25) of the IL-2 receptor on activated T-cells (19); however, that risk appears lower with basiliximab relative to that conferred by alemtuzumab or anti-thymocyte globulin. Alemtuzumab-treated patients may also develop, yet with less frequency, other AIDS-associated opportunistic infections such as mucosal candidiasis PJP, progressive multifocal leukoencephalopathy (PML), cryptococcosis, and toxoplasmosis, while sporadic cases of listeriosis and nocardiosis have also been reported early on after alemtuzumab initiation (20). Due to the risk of tuberculosis and HPV reactivation in high-risk patients, HPV vaccination and screening for latent tuberculosis are also advised before initiating alemtuzumab. Last, besides valacyclovir prophylaxis, consideration can be given for prophylaxis with fluconazole and/or trimethoprim-sulfamethoxazole depending on the patient population and co-existent risk factors.
The CD20-targeting Mab rituximab causes prolonged B-cell depletion and hypogammaglobulinemia, thereby predisposing to severe bacterial and viral infections (21); similar infections occur with the CD38-targeting Mab daratumumab, which depletes plasma cells/plasmablasts and also causes hypogammaglobulinemia (22). A recent study showed a correlation between decreased IgG levels before or during rituximab therapy with the development of serious infections; therefore, monitoring of gamma globulin levels and immunoglobulin replacement therapy should be considered in rituximab-treated patients with inadequate vaccination responses (23). Importantly, rituximab is a major risk factor for hepatitis B reactivation, which can cause fulminant liver failure and death; therefore, prophylaxis is advised in high-risk individuals (i.e., HBsAg positive or HBsAg negative/anti-HBc positive) (24). PML may also occur with rituximab (frequency, ~1:40,000), and it is more often observed after repeated, prolonged use (25). Similarly, PML develops with the α4-integrin–targeting mAb natalizumab (frequency, ~1:250), more often also after prolonged (>2-year) administration, and in individuals with positive anti-JC antibodies and/or receiving additional immunosuppressive medications (26). Rituximab may also promote PJP susceptibility, while severe West Nile encephalitis and babesiosis have been reported in rituximab-treated patients (21).
The complement C5a-targeting Mab eculizumab causes a ~1000–2000–fold increased risk for life-threatening meningococcal disease (27) and predisposes to invasive infections by pneumococcus, gonorrhea, and Haemophilus, in keeping with similar susceptibility of patients with inherited terminal complement deficiencies (28). Thus, vaccination against these pathogens before eculizumab initiation is critically important. In 2018, the FDA updated eculizumab’s package insert to warn for the risk for aspergillosis, especially in patients with additional predisposing factors; concordantly, a small report of eculizumab-treated HSCT recipients with thrombotic microangiopathy showed that 20% developed aspergillosis despite receiving mold-active antifungal prophylaxis (29). More studies are needed to further clarify the risk of fungal disease in eculizumab-treated patients.
The checkpoint molecule (PD-1, CTLA-4)-targeting Mabs have transformed the management of several neoplasms. Except for recent reports that indicate that checkpoint inhibitors (CPIs) may predispose or unmask tuberculosis (30), these molecules do not appear to drive infection susceptibility (31); in fact, early clinical reports show promise for the potential use of CPIs for the treatment of refractory infections (e.g., PML, mucormycosis) (32, 33). Nonetheless, CPI-associated immune-related adverse events occur frequently, and their management with immunomodulators such as corticosteroids or TNF-α inhibitors can predispose to opportunistic infections (34).
Infectious complications associated with Nibs
A few important Nib-associated infectious complications are worth highlighting herein (Table 2). The Janus kinase (JAK) inhibitors (ruxolitinib, tofacitinib, baricitinib, upadacitinib) are increasingly used in clinical practice with >300 ongoing clinical trials aimed at evaluating their efficacy in numerous autoimmune, inflammatory, and malignant conditions (clinicaltrials.gov). These drugs potently inhibit the JAK/STAT pathway and impair signaling downstream of several inflammatory mediators including common γ-chain cytokines, type-I, type-II and type-III interferons, GM-CSF, IL-6, IL-12, IL-22 and IL-23 (35–37). Not surprisingly, JAK inhibitor-treated patients are at-risk for bacterial pneumonia and severe viral infections including herpes simplex, herpes zoster and, less often, CMV disease. Thus, valacyclovir prophylaxis should be strongly considered in patients receiving JAK inhibitors. Furthermore, in keeping with the crucial role of the cross-talk between IFN-γ–producing lymphocytes and macrophages for clearance of intracellular pathogens, JAK inhibitors are associated with developing infections by mycobacteria (tuberculosis and non-tuberculous), endemic dimorphic fungi, Cryptococcus, and PJP (38, 39). As such, PJP prophylaxis should be considered in JAK inhibitor-treated patients when additional risk factors co-exist. In addition, awareness is warranted in JAK inhibitor-treated patients for development of aspergillosis, especially when other immunomodulators are co-administered; indeed, sporadic reports of aspergillosis are now emerging (40, 41), consistent with mouse experimental evidence that IFN-λ/IFNLR1/JAK/STAT signaling promotes neutrophil oxidative burst and Aspergillus killing (42).
Table 2.
Small molecule kinase inhibitors presented in the current review and their associated infection complications.
| Small molecule kinase inhibitor | Target | Main FDA-approved indications | Relative risk for infection | Predominant infection susceptibility |
|---|---|---|---|---|
| Ruxolitinib Tofacitinib Baricitinib |
JAK1 JAK2 JAK3 |
Myelofibrosis, polycythemia vera, rheumatoid arthritis, inflammatory bowel disease, graft-versus-host disease | HIgh | Herpetic infections (common) > CMV disease, other opportunistic infections (mycobacteria, endemic fungi, aspergillosis) |
| Ibrutinib | BTK | Chronic lymphocytic leukemia, lymphomas, Waldenstrom macroglobulinemia, graft-versus-host disease |
Intermediate | Bacterial infections (~5%); invasive fungal disease (~2–4% as monotherapy; 5–11% when combined with corticosteroids; >20% when combined with corticosteroids and chemotherapy); viral infections (~1%) |
| Dasatinib | BCR-ABL | Chronic myelogenous leukemia | High | Bacterial infections, CMV disease (HSCT), hepatitis B reactivation, Pneumocystis pneumonia |
| Idelalisib | PI3K (p100δ) | Chronic lymphocytic leukemia, lymphomas | High | CMV infection, Pneumocystis pneumonia |
| Fostamatinib | SYK | Immune thrombocytopenia | Unknown | Mucocutaneous fungal infections |
CMV, cytomegalovirus; HSCT, hematopoietic stem cell transplantation
The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib impairs B-cell development, survival and activation and has become a gamer-changing therapy for patients with chronic lymphocytic leukemia and other B-cell malignancies. Bacterial infections, primarily bacteremia and pneumonia, occur in ~6% of ibrutinib-treated patients; Staphylococcus aureus was the most common pathogen identified in a recent tertiary cancer center series (43). The emergence of invasive fungal disease post-ibrutinib was surprising given the low frequency of such infections in patients with X-linked agammaglobulinemia. Invasive mold infections (predominantly aspergillosis) are more common, followed by PJP, followed by cryptococcosis in ibrutinib-treated patients; in addition to inhibition of BTK in B-cells and myeloid cells, ibrutinib inhibits ITK in T-cells, which may contribute to the emergence of PJP and cryptococcosis that rely on T-cell–dependent immunity. Recent reports collectively suggest that ibrutinib monotherapy is associated with aspergillosis in ~2–4% of patients (43–45). Aspergillosis incidence is increased in patients receiving ibrutinib with corticosteroids (~5–11%) (46, 47), and is dramatically greater in those who receive ibrutinib with corticosteroids and chemotherapy (up to 39%) (48). Therefore, Aspergillus-active prophylaxis should be considered when ibrutinib and other immunosuppressive drugs are combined; isavuconazole exhibits lesser drug-drug interactions relative to other triazoles and may be favored as prophylaxis with ibrutinib co-administration (Lionakis, unpublished observations). Important clinical features of ibrutinib-associated invasive fungal disease include: a) the typical absence of conventional risk factors for such infections (e.g., neutropenia, lymphocytopenia), b) the development of most infections within the first 2–4 months post-ibrutinib initiation, but infrequently later on, c) and the dramatically increased frequency of central nervous system (CNS) aspergillosis (up to ~45% of infections) (44, 45, 48, 49). The emergence of invasive mold disease in ibrutinib-treated patients has uncovered the critical contributions of BTK in phagocyte activation and antifungal effector function (50). Last, severe viral infections, primarily respiratory viral pneumonia, has been reported in ~1% of ibrutinib-treated patients (43). It remains to be seen whether isolated BTK inhibition with second-generation inhibitors such as acalabrutinib will be associated with the same profile of infection susceptibilities as seen with combined BTK/ITK inhibition by ibrutinib.
Dasatinib, a second-generation multitargeted tyrosine kinase inhibitor has been associated with significant infection risk, ranging from severe bacterial infections (primarily sepsis, pneumonia and soft-tissue infections) to CMV disease in HSCT recipients, to PJP and hepatitis B reactivation (6). Severe CMV disease and PJP can also occur with the phosphatidylinositol-3-kinase (PI3K) inhibitor idelalisib; PJP prophylaxis is warranted in idelalisib-treated patients (6). Early experience with the spleen tyrosine kinase (SYK) inhibitor fostamatinib has not identified major infection susceptibility to-date. However, SYK is centrally located within the fungal sensing C-type lectin receptor/CARD9 signaling pathway, and patients with inherited CARD9 deficiency exhibit profound susceptibility to mucocutaneous and invasive fungal infections of the CNS, caused by impaired microglial-neutrophil responses leading to CNS neutropenia during infection (51–54). Therefore, clinical awareness is warranted for the possible development of fungal infections in fostamatinib-treated patients, as reports of mucosal candidiasis and skin fungal infection are emerging in such patients (55) (clinicaltrials.gov, NCT00798096).
Infectious complications following CAR T-cell therapy
CAR T-cells are an effective treatment option for patients with B-cell malignancies who are refractory to other therapies. CAR T-cells are genetically engineered to express a receptor recognizing a target protein on cancer cells that, when engaged, induces killing of the target-expressing cell, be it a tumor or normal cell (3). Current FDA-approved CAR T-cells target CD19 on B-cell malignancies (56, 57), but CAR T-cells targeting other antigens on B-cell malignancies and other cancers are currently in development.
While CAR T-cell therapy has proven successful in some patients, it is not effective in all and it is not without risks. The most well-described toxicities associated with CAR T-cell therapy are cytokine release syndrome (CRS) and Immune effector Cell-Associated Neurotoxicity Syndrome (ICANS), and B-cell aplasia, but additional toxicities are emerging, including prolonged cytopenias, which may be related to the underlying disease, prior treatment, or even CAR T-cell–related toxicities (4). CRS and ICANS are usually acute and are monitored closely following CAR T-cell treatment (Figure 1); both feature robust immune activation, leading to a pro-inflammatory cytokine storm that can have negative effects on multiple organs (58–60). These high cytokine levels, or the treatments used to manage them, may predispose to infections or prolonged cytopenias (11).
Figure 1. Schematic timeline of infection and inflammatory complications associated with CAR T-cell therapy.
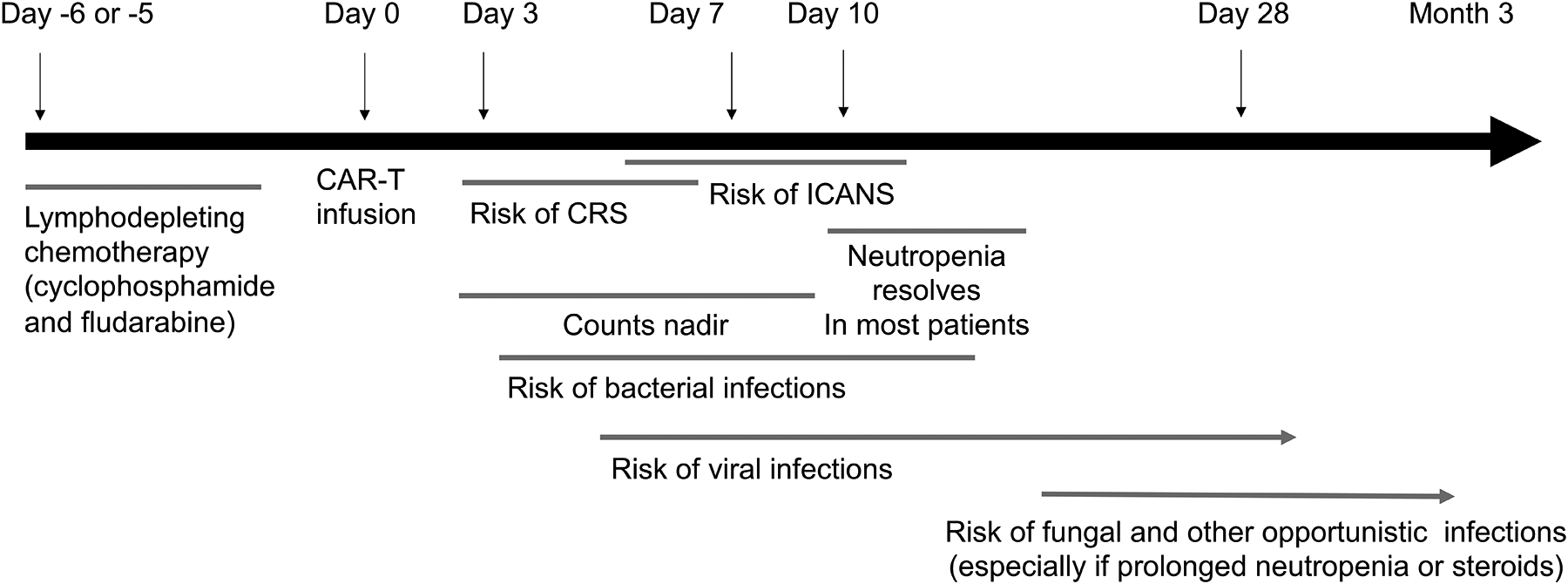
CAR T-cell therapy can be broken down into three phases: the week prior to infusion, when a preparative chemotherapy regimen is administered to achieve lymphodepletion prior to CAR T-cell infusion, which is known as Day 0. Not all protocols use chemotherapy, but many cancer protocols incorporate cyclophosphamide and fludarabine administered over 3–5 days, followed by a rest period of 1–2 days. During the first 2 weeks after CAR T-cell infusion, there is elevated risk of cytokine release syndrome (CRS) and immune-effector-cell associated neurotoxicity syndrome (ICANS), and this often coincides with the nadir of blood counts from the preparative regimen. Day 28 from CAR T-cell infusion usually marks a restaging evaluation and most (but not all) patients have achieved hematologic recovery. Host immunity is impaired during this period, and increased susceptibility to bacterial, viral, fungal and opportunistic infections can be see as a function of neutrophil decline (for bacteria and fungi) and T-cell and B-cell dysfunction, which can be multifactorial from prior therapies and the CAR T-cell target.
Infections in patients receiving CAR T-cell therapy may emerge as a direct result of the underlying malignancy and prior exposure to treatments for their hematologic malignancy. Most patients have undergone multiple prior therapies (61) that can contribute to infection risk, such as with the lymphodepleting drug fludarabine, which can cause prolonged lymphocyte dysfunction, or T-cell depleting antibodies like alemtuzumab, which are sometimes used during conditioning for allogeneic HSCT. Conditioning regimens (62–64) for CAR T-cell therapy typically include moderate doses of chemotherapy (cyclophosphamide and fludarabine), which cause lymphodepletion; normal cellular immunity may not recover for years, while myeloid recovery usually occurs within a week (Figure 1). However, some patients experience cytopenia that is unresolved even 28 days post-treatment. Growth-factor support is usually not initiated until the time window of greatest risk from CRS and ICANS has passed, or the patient recovers from CRS and ICANS (56, 65). Therefore, the first 28 days following treatment is when infections are most likely to occur (61). Most infections occur within the first six days following treatment, but infections can continue to occur even up to 21 months after treatment if the patient has prolonged cytopenias (Figure 1) (66). As with HSCT, early infections (developing within 28 days post-HSCT) tend to be bacterial, while late infections are typically caused by viruses (11, 61). The risk of fungal infection in CAR T-cell–treated patients has not been definitively assessed; guidelines in development suggest antifungal coverage for the first month post-infusion, which can be continued if cytopenias persist, as per standard-of-care.
Immunosuppression-related infections can also occur as a result of management strategies for CRS and ICANS. These syndromes are treated with high-dose corticosteroids and/or tocilizumab to suppress the amplified inflammation. The risks of corticosteroids are well-known to the clinical community, and rapid tapering is advised and instituted whenever possible. Although tocilizumab may effectively mask fevers, the incidence of severe infections related to tocilizumab use in CAR T-cell–treated patients has not been well-studied. In the rheumatologic literature, chronic tocilizumab use is associated with infection risk (10, 11). Although most CAR T-cell–treated patients who receive tocilizumab do so only once or twice in the 1–2 weeks following CAR T-cell therapy, the overall need to manage CRS is associated with increased infection risk. As in other patients, intensive care unit care that includes central venous and Foley catheters while managing CRS can further increase infection risk (58, 67).
In addition to targeting cancer cells, CD19 CAR T-cells can also deplete normal B-cells, resulting in varying degrees and durations of B-cell aplasia and hypogammaglobulinemia. This can also occur before CAR T-cell treatment, as a result of prior treatments, and can be exacerbated by CAR T-cell treatment. Due to expansion and persistence of CAR T-cells, hypogammaglobulinemia can persist for years. In one study, 67% of patients had hypogammaglobulinemia beyond 90 days post-treatment, although 43% had hypogammaglobulinemia before CAR T-cell infusion (66). These patients are likely susceptible to infections, particularly sino-pulmonary infections, as evident by infections occurring late in the course of treatment. In the same study, 61% of patients had at least one infection beyond 90 days after the first CAR T-cell treatment (66). Although hepatitis B reactivation has not been reported to occur clinically, this is likely because hepatitis serologies are routinely measured prior to CAR T-cell treatment and anti-viral prophylaxis is given with the same rationale as was developed for rituximab.
In summary, despite the risk of infection associated with CAR T-cell treatment, much of the risk is attributable to the history of hematologic malignancy and its prior treatments, and most infections are managed according to standard-of-care. In the first month after CAR T-cell infusion, the underlying malignancy, conditioning chemotherapy, and CRS and ICANS may all result in increased risk of serious infections, which are treated early and aggressively, even in the absence of confirmatory diagnosis (Figure 1). The long-term profile of CAR T-cell therapy suggests this is safe, though measurements of B-cell recovery and/or gamma globulins may aid in managing long-term infection risk with immunoglobulin replacement therapy. Replacement gamma globulin is routine in children with acute lymphoblastic leukemia and treated with CAR T-cell therapy, but may be reserved only for patients with recurrent sino-pulmonary infections in adults with lymphoma and ongoing hypogammaglobulinemia. The American Society of Transplantation and Cellular Therapy has established guidelines for grading toxicities (68), and management guidelines, including infection prophylaxis, are in development.
Conclusion
The use of novel biological and cellular therapies for neoplastic and autoimmune diseases results in complex iatrogenic immunodeficiency states and heightened risk for infections. Increased awareness, improved reporting and surveillance of infections in clinical trials, and research to understand the epidemiology and pathogenesis of infections associated with novel biological and cellular therapies should help clinicians develop better approaches for diagnosis, vaccination, prophylaxis and treatment for such infections.
Key points.
The introduction in clinical practice of novel biological (Mabs, Nibs) and cellular therapies (CAR T-cells) has transformed the management of patients with several malignant and autoimmune diseases.
Some of these therapies induce complex iatrogenic immunodeficiency states and heighten the risk for certain infections.
Awareness among clinicians and a better understanding of the epidemiology and pathogenesis of infections associated with novel biological and cellular therapies should help design improved strategies for vaccination, diagnosis, prophylaxis and treatment.
Financial support
This work was supported by the Division of Intramural Research of the National Institute of Allergy & Infectious Diseases, NIH.
Footnotes
Conflicts of interest
None
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–52. [DOI] [PubMed] [Google Scholar]
- 3.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379(1):64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4 *.Fishman JA, Hogan JI, Maus MV. Inflammatory and Infectious Syndromes Associated With Cancer Immunotherapies. Clin Infect Dis. 2019;69(6):909–20. [DOI] [PubMed] [Google Scholar]; This recent review summarizes the inflammatory and infection complications that develop following cancer immunotherapies.
- 5.Lionakis MS, Levitz SM. Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu Rev Immunol. 2018;36:157–91. [DOI] [PubMed] [Google Scholar]
- 6.Reinwald M, Silva JT, Mueller NJ, Fortun J, Garzoni C, de Fijter JW, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Intracellular signaling pathways: tyrosine kinase and mTOR inhibitors). Clin Microbiol Infect. 2018;24 Suppl 2:S53–S70. [DOI] [PubMed] [Google Scholar]
- 7.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345(15):1098–104. [DOI] [PubMed] [Google Scholar]
- 8.Vergidis P, Avery RK, Wheat LJ, Dotson JL, Assi MA, Antoun SA, et al. Histoplasmosis complicating tumor necrosis factor-alpha blocker therapy: a retrospective analysis of 98 cases. Clin Infect Dis. 2015;61(3):409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winthrop KL, Chang E, Yamashita S, Iademarco MF, LoBue PA. Nontuberculous mycobacteria infections and anti-tumor necrosis factor-alpha therapy. Emerg Infect Dis. 2009;15(10):1556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morel J, Constantin A, Baron G, Dernis E, Flipo RM, Rist S, et al. Risk factors of serious infections in patients with rheumatoid arthritis treated with tocilizumab in the French Registry REGATE. Rheumatology (Oxford). 2017;56(10):1746–54. [DOI] [PubMed] [Google Scholar]
- 11.Park JH, Romero FA, Taur Y, Sadelain M, Brentjens RJ, Hohl TM, et al. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin Infect Dis. 2018;67(4):533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12 *.Pawar A, Desai RJ, Solomon DH, Santiago Ortiz AJ, Gale S, Bao M, et al. Risk of serious infections in tocilizumab versus other biologic drugs in patients with rheumatoid arthritis: a multidatabase cohort study. Ann Rheum Dis. 2019;78(4):456–64. [DOI] [PubMed] [Google Scholar]; This study describes the risk of serious infections following treatment with the interleukin 6 receptor-targeted monoclonal antibody tocilizumab in patients with rheumatoid arthritis.
- 13.Lionakis MS, Kontoyiannis DP. Glucocorticoids and invasive fungal infections. Lancet. 2003;362(9398):1828–38. [DOI] [PubMed] [Google Scholar]
- 14.Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol. 2012;12(6):616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saunte DM, Mrowietz U, Puig L, Zachariae C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177(1):47–62. [DOI] [PubMed] [Google Scholar]
- 16.Krueger JG, Wharton KA Jr., Schlitt T, Suprun M, Torene RI, Jiang X, et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J Allergy Clin Immunol. 2019;144(3):750–63. [DOI] [PubMed] [Google Scholar]
- 17.Burmester GR, McInnes IB, Kremer J, Miranda P, Korkosz M, Vencovsky J, et al. A randomised phase IIb study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1020–30. [DOI] [PubMed] [Google Scholar]
- 18.Cox AL, Thompson SA, Jones JL, Robertson VH, Hale G, Waldmann H, et al. Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur J Immunol. 2005;35(11):3332–42. [DOI] [PubMed] [Google Scholar]
- 19.Group CSC, Haynes R, Harden P, Judge P, Blackwell L, Emberson J, et al. Alemtuzumab-based induction treatment versus basiliximab-based induction treatment in kidney transplantation (the 3C Study): a randomised trial. Lancet. 2014;384(9955):1684–90. [DOI] [PubMed] [Google Scholar]
- 20.Buonomo AR, Zappulo E, Viceconte G, Scotto R, Borgia G, Gentile I. Risk of opportunistic infections in patients treated with alemtuzumab for multiple sclerosis. Expert Opin Drug Saf. 2018;17(7):709–17. [DOI] [PubMed] [Google Scholar]
- 21.Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47(2):187–98. [DOI] [PubMed] [Google Scholar]
- 22.Johnsrud AJ, Johnsrud JJ, Susanibar SA, Kamimoto JJ, Kothari A, Burgess M, et al. Infectious and immunological sequelae of daratumumab in multiple myeloma. Br J Haematol. 2019;185(1):187–9. [DOI] [PubMed] [Google Scholar]
- 23 *.Md Yusof MY, Vital EM, McElvenny DM, Hensor EMA, Das S, Dass S, et al. Predicting Severe Infection and Effects of Hypogammaglobulinemia During Therapy With Rituximab in Rheumatic and Musculoskeletal Diseases. Arthritis Rheumatol. 2019;71(11):1812–23. [DOI] [PubMed] [Google Scholar]; This study describes the association between immunoglobulin levels and the risk of infection in rituximab-treated patients with rheumatoid arthritis.
- 24.Pattullo V Hepatitis B reactivation in the setting of chemotherapy and immunosuppression - prevention is better than cure. World J Hepatol. 2015;7(7):954–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berger JR, Malik V, Lacey S, Brunetta P, Lehane PB. Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event. J Neurovirol. 2018;24(3):323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho PR, Koendgen H, Campbell N, Haddock B, Richman S, Chang I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 2017;16(11):925–33. [DOI] [PubMed] [Google Scholar]
- 27.McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High Risk for Invasive Meningococcal Disease Among Patients Receiving Eculizumab (Soliris) Despite Receipt of Meningococcal Vaccine. MMWR Morb Mortal Wkly Rep. 2017;66(27):734–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine (Baltimore). 1984;63(5):243–73. [PubMed] [Google Scholar]
- 29.Bohl SR, Kuchenbauer F, von Harsdorf S, Kloevekorn N, Schonsteiner SS, Rouhi A, et al. Thrombotic Microangiopathy after Allogeneic Stem Cell Transplantation: A Comparison of Eculizumab Therapy and Conventional Therapy. Biol Blood Marrow Transplant. 2017;23(12):2172–7. [DOI] [PubMed] [Google Scholar]
- 30 **.Barber DL, Sakai S, Kudchadkar RR, Fling SP, Day TA, Vergara JA, et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci Transl Med. 2019;11(475). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the development of tuberculosis following PD-1 blockade, corroborating prior preclinical work that implicates PD-1 in effective host defense against mycobacterial infection.
- 31.Abers MS, Lionakis MS, Kontoyiannis DP. Checkpoint Inhibition and Infectious Diseases: A Good Thing? Trends Mol Med. 2019;25(12):1080–93. [DOI] [PubMed] [Google Scholar]
- 32 **.Cortese I, Muranski P, Enose-Akahata Y, Ha SK, Smith B, Monaco M, et al. Pembrolizumab Treatment for Progressive Multifocal Leukoencephalopathy. N Engl J Med. 2019;380(17):1597–605. [DOI] [PubMed] [Google Scholar]; This study describes the potential role of pembrolizumab as an immune-based treatment in patients with progressive multifocal leukoencephalopathy.
- 33.Grimaldi D, Pradier O, Hotchkiss RS, Vincent JL. Nivolumab plus interferon-gamma in the treatment of intractable mucormycosis. Lancet Infect Dis. 2017;17(1):18. [DOI] [PubMed] [Google Scholar]
- 34.Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med. 2018;378(2):158–68. [DOI] [PubMed] [Google Scholar]
- 35.Bechman K, Yates M, Galloway JB. The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol Res. 2019;147:104392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci U S A. 2016;113(35):9852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38 *.Bechman K, Subesinghe S, Norton S, Atzeni F, Galli M, Cope AP, et al. A systematic review and meta-analysis of infection risk with small molecule JAK inhibitors in rheumatoid arthritis. Rheumatology (Oxford). 2019;58(10):1755–66. [DOI] [PubMed] [Google Scholar]; This article summarizes the risk of infection with JAK inhibition in patients with rheumatoid arthritis.
- 39.Dioverti MV, Abu Saleh OM, Tande AJ. Infectious complications in patients on treatment with Ruxolitinib: case report and review of the literature. Infect Dis (Lond). 2018;50(5):381–7. [DOI] [PubMed] [Google Scholar]
- 40.Moruno-Rodriguez A, Sanchez-Vicente JL, Rueda-Rueda T, Lechon-Caballero B, Munoz-Morales A, Lopez-Herrero F. Invasive aspergillosis manifesting as retinal necrosis in a patient treated with ruxolitinib. Arch Soc Esp Oftalmol. 2019;94(5):237–41. [DOI] [PubMed] [Google Scholar]
- 41.Polverelli N, Breccia M, Benevolo G, Martino B, Tieghi A, Latagliata R, et al. Risk factors for infections in myelofibrosis: role of disease status and treatment. A multicenter study of 507 patients. Am J Hematol. 2017;92(1):37–41. [DOI] [PubMed] [Google Scholar]
- 42.Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol. 2017;2(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varughese T, Taur Y, Cohen N, Palomba ML, Seo SK, Hohl TM, et al. Serious Infections in Patients Receiving Ibrutinib for Treatment of Lymphoid Cancer. Clin Infect Dis. 2018;67(5):687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44 *.Rogers KA, Mousa L, Zhao Q, Bhat SA, Byrd JC, El Boghdadly Z, et al. Incidence of opportunistic infections during ibrutinib treatment for B-cell malignancies. Leukemia. 2019;33(10):2527–30. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the incidence of opportunistic infections during BTK inhibition with ibrutinib in patients with B cell malignancies.
- 45 *.Ruchlemer R, Ben-Ami R, Bar-Meir M, Brown JR, Malphettes M, Mous R, et al. Ibrutinib-associated invasive fungal diseases in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: An observational study. Mycoses. 2019;62(12):1140–7. [DOI] [PubMed] [Google Scholar]; This paper summarizes clinical and incidence daata on the development of opportunistic invasive fungal infections following treatment with the BTK inhibitor ibrutinib.
- 46.Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 2017;7(9):1018–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lakshmanan A, Byrd JC. Spotlight on Ibrutinib in PCNSL: Adding Another Feather to Its Cap. Cancer Discov. 2017;7(9):940–2. [DOI] [PubMed] [Google Scholar]
- 48.Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghez D, Calleja A, Protin C, Baron M, Ledoux MP, Damaj G, et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood. 2018;131(17):1955–9. [DOI] [PubMed] [Google Scholar]
- 50.Bercusson A, Colley T, Shah A, Warris A, Armstrong-James D. Ibrutinib blocks Btk-dependent NF-kB and NFAT responses in human macrophages during Aspergillus fumigatus phagocytosis. Blood. 2018;132(18):1985–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51 *.Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. 2019;20(5):559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uncovered the mechanisms that underlie local neutropenia in the fungal-infected central nervous system in the setting of CARD9 deficiency that has implications in the potential risk of patients treated with inhibitors of the SYK kinase, which is upstream of CARD9.
- 52.Lanternier F, Mahdaviani SA, Barbati E, Chaussade H, Koumar Y, Levy R, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J Allergy Clin Immunol. 2015;135(6):1558–68 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. 2013;369(18):1704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rieber N, Gazendam RP, Freeman AF, Hsu AP, Collar AL, Sugui JA, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight. 2016;1(17):e89890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New England Journal of Medicine. 2017;377(26):2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7(12):1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018;131(1):121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther. 2017;25(1):285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66 *.Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol Blood Marrow Transplant. 2020;26(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper outlines the complications that occur late after treatment with CAR T cells.
- 67.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68 *.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25(4):625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides a consensus grading of cytokine release syndrome and neurologic toxicity associated with CAR T cell therapy.