

Abstract
VNLG-152 is a novel retinamide (NR) shown to suppress growth and progression of genetically diverse prostate cancer cells via inhibition of androgen receptor signaling and eukaryotic initiation factor 4E (eIF4E) translational machinery. Herein, we report therapeutic effects of VNLG-152 on castration-resistant prostate cancer (CRPC) growth and metastatic phenotype in a CRPC tumor xenograft model. Administration of VNLG-152 significantly and dose-dependently suppressed the growth of aggressive CWR22Rv1 tumors by 63.4% and 76.3% at 10 and 20 mg·kg−1 bw, respectively (P < 0.0001), vs. vehicle with no host toxicity. Strikingly, the expression of full-length androgen receptor (f-AR)/androgen receptor splice variant-7 (AR-V7), mitogen-activated protein kinase-interacting kinases 1 and 2 (MNK1/2), phosphorylated eIF4E and their associated target proteins, including prostate-specific antigen, cyclin D1 and Bcl-2, were strongly decreased in VNLG-152-treated tumors signifying inhibition of f-AR/AR-V7 and MNK–eIF4E signaling in VNLG-152-treated CWR22Rv1 tumors as observed in vitro. VNLG-152 also suppressed the epithelial to mesenchymal transition in CWR22Rv1 tumors as evidenced by repression of N-cadherin, β-catenin, claudin, Slug, Snail, Twist, vimentin and matrix metalloproteinases (MMP-2 and MMP-9) with upsurge in E-cadherin. These results highlight the promising use of VNLG-152 in CRPC therapy and justify its further development towards clinical trials.
Keywords: CRPC/mCRPC, f-AR/AR-V7, Mnk-eIF4E, VNLG-152
Introduction
Prostate cancer (PCa) is one of the most prevalent malignancies among men worldwide [1]. Androgen ablation therapy is typically used to treat advanced and metastatic prostate cancers. However, in most cases, the prostate cancer becomes ultimately unresponsive to androgen ablation and develops into castration-resistant prostate cancer (CRPC), and then progresses rapidly. Therapy for CRPC remains limited and a very few drugs have given modest survival in patients with metastatic (m) CRPC [2,3]. Thus, new effective therapeutic agents for mCRPC that have potential anti-tumor activity and low toxicity are urgently needed.
Epithelial to mesenchymal transition (EMT), a hallmark of cancer cell migration, invasion and metastasis, is a highly conserved process that allows polarized, immobile epithelial cells to transdifferentiate to those with motile mesenchymal phenotypes [4,5]. Accumulating evidence indicates that EMT plays a pivotal role in the development of mCRPC [6,7]. Full-length androgen receptor (f-AR) signaling and activation of the mitogen-activated protein kinase-interacting kinase 1/2 (MNK1/2)–eukaryotic initiation factor 4E (eIF4E) pathway are known to contribute to metastasis by facilitating EMT and promoting signaling interactions [8–10]. f-AR induces EMT through activation of the Snail transcription factor or via repression of E-cadherin [8]. Activation of Snail increases the expression of mesenchymal markers and proteins associated with invasion. In contrast to Snail’s activation, transcriptional repression of E-cadherin, a key mediator of intracellular adhesions at adherens junctions, results in collapse of cell–cell communication and onset of EMT [11,12].
Activation of eIF4E is also recognized to stimulate EMT via promoting the expression of pro-metastatic factors such as Snail and matrix metalloproteinases (MMPs) [10]. MMPs cleave several component proteins of the extracellular matrix (ECM) to promote tissue invasion and metastasis [13,14]. Recently, Robichaud et al. [10] demonstrated that besides endorsing tumor development, activation of eIF4E promotes metastatic progression via translation of several EMT-associated mRNAs such as those of Snail and MMP-3.
In an earlier study, we demonstrated that novel retinamides (NRs), including VNLG-152, simultaneously target both f-AR/androgen receptor splice variant-7 (AR-V7) signaling and MNK–eIF4E translation in several androgen-sensitive cells and CRPC cells by enhancing f-AR/AR-V7 and MNK degradation through the ubiquitin–proteasome pathway [15]. Herein, we investigated the role of VNLG-152 in inhibiting in vivo tumor growth using a hormone-refractory castration-resistant human PCa xenograft model, specifically in the context of f-AR/AR-V7 and MNK–eIF4E signaling inhibition. We demonstrate, for the first time, that administration of VNLG-152 dramatically suppresses tumor growth and expression of several EMT-associated markers (N-cadherin, MMPs, Snail and Slug) in a CRPC xenograft model. An in vitro invasion assay conducted with CWR22Rv1 (22Rv1) cells knocked down for AR and/or MNK1 indicated cell invasion to be significantly repressed in 22Rv1 cells harboring both AR and MNK1 gene knockdown. In parallel, 22Rv1 cells treated with VNLG-152 displayed almost equivalent inhibition of cell invasion compared with cells that harbored AR/MNK1 double knockdown. Besides, VNLG-152 treatment also caused substantial reduction of dihydrotestosterone (DHT) -induced increase in f-AR activity and formation of the eIF4F cap complex. Thus, inhibition of 22Rv1 xenograft tumor growth and down-regulation of EMT-associated markers are allied to suppression of f-AR and MNK–eIF4E signaling, the key inducers of tumor growth and EMT. The molecular alterations observed in vivo showed a translational relevance to our earlier in vitro findings in PCa cells with VNLG-152. A preliminary account of part of this study has recently been reported [16].
Results
VNLG-152 suppresses tumor growth of CWR22Rv1 cells in castrated mice
Since VNLG-152 exhibited potent anticancer effects in androgen-dependent and castration-resistant PCa cells, we investigated the efficacy of VNLG-152 in vivo, especially at the castration-resistant stage using CRPC AR-V7-positive 22Rv1 xenografts. We found that even with complete androgen deprivation therapy in mice through surgical castration, 22Rv1 cells inoculated on flanks of mice in vehicle-treated groups were still able to grow into tumors of significant size (Fig. 1A). In contrast, castrated mice bearing 22Rv1 tumors treated with VNLG-152 at two different doses (10 and 20 mg·kg−1 bw) had smaller tumors compared with those with vehicle injection. The tumor growth inhibition values in the groups administered with 10 and 20 mg·kg−1 bw VNLG-152 were 63.4% and 76.3% vs. vehicle, respectively (P < 0.0001). In addition, no host toxicity was observed as monitored by changes in body weight throughout the study (Fig. 1B). The hematoxylin and eosin staining of liver, lung and kidney in the treated groups did not show any gross organ abnormalities on histological examination (Fig. 1C).
Fig. 1.

Therapeutic effect of VNLG-152 in vivo. (A) Representative tumors from three groups: vehicle and VNLG-152 at 10 and 20 mg·kg−1. Growth curves expressed as tumor volume (mm3) of primary tumors derived from injection of 22Rv1 cells. Effect of VNLG-152 at 10 and 20 mg·kg−1 was evaluated in castrated 22Rv1 xenograft-bearing mice. Mice (n = 5) were treated with VNLG-152 (10 and 20 mg·kg−1) twice daily, which was administered i.p., 5 days per week for 34 days. Tumors were measured twice a week. Tumor growth inhibition (TGI) values are indicated to the right of each growth curve, and the error bars are the SEM. (B) Mean body weight determination during VNLG-152 treatment. Body weights were measured in vehicle- or VNLG-152-injected mice once a week for the duration of the study. (C) Histological examination of liver, lung and kidney tissues in the various treatment groups after hematoxylin–eosin staining. Bars correspond to 20 μm. Results are represented as means ± SEM, *P < 0.0001 compared with control. Statistical significance was determined by Student’s t test and analysis of variance (ANOVA).
VNLG-152 reduces the expression of f-AR (full-length and splice variant), MNK1/2 and p-eIF4E and their downstream targets in CWR22Rv1 tumors
To validate the molecular mechanism(s) underlying the effect of VNLG-152, we evaluated the expression levels of f-AR, MNK1/2 and phosphorylated (p)-eIF4E and their downstream targets in 22Rv1 tumors. Immunoblotting with anti-f-AR, anti-MNK1, anti-MNK2 and anti-p-eIF4E antibodies using representative tumor samples revealed that VNLG-152 administration at both doses (10 and 20 mg·kg−1 bw) significantly reduced intensity and expression of f-AR, MNK1, MNK2 and p-eIF4E in treated samples (Fig. 2A,B). Western blot analysis also showed a significant reduction in the expression of both f-AR and AR-V7 in VNLG-152-administered xenograft tumors (Fig. 2C). A significant depletion of MNK1, MNK2 and p-eIF4E was also observed in VNLG-152-treated tumors (Fig. 2C). Densitometry analysis of f-AR, AR-V7, MNK1, MNK2 and p-eIF4E protein expression in two representative tumors in the VNLG-152- and vehicle-treated groups are shown in Fig. 2D.
Fig. 2.

Effect of VNLG-152 on f-AR, MNK1/2 and p-eIF4E. (A) Representative images of f-AR, MNK1, MNK2, p-eIF4E and PSA immunostaining. Immunostaining with MNK1, MNK2, p-eIF4E and PSA antibodies revealed reduced intensity and decrease in the number of positively stained cells in VNLG-152 (10 and 20 mg·kg−1)-treated groups compared with vehicle-treated controls. Bars correspond to 20 μm. (B) f-AR/MNKs/p-eIF4E-positive cells were counted in 12 fields in each group. The percentage of positive cells was calculated and presented as mean ± SD. (C) Xenograft tumor tissues were harvested to evaluate effect of VNLG-152 on f-AR, PSA, MNK and p-eIF4E by western blotting. Equal protein concentrations of tissue lysate were separated by SDS/PAGE and western blots probed with indicated antibodies. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (D) Quantitative analysis of the protein expression using a scanning densitometer. Results are represented as means ± SEM. *P < 0.01; †P < 0.05 compared with control. Statistical significance was determined by Student’s t test and ANOVA.
Depletion of f-AR, MNKs and p-eIF4E expression in VNLG-152-treated tumors was concomitant with a down-regulation in the expression of their downstream targets including prostate-specific antigen (PSA), cyclin D1 and B-cell lymphoma 2 (Bcl-2). A significant increase in the expression of proapoptotic Bax, caspase-3 and cleaved poly (ADP-ribose) polymerase (PARP) was also observed in VNLG-152-treated 22RV1 tumors (Fig. 3A–D).
Fig. 3.
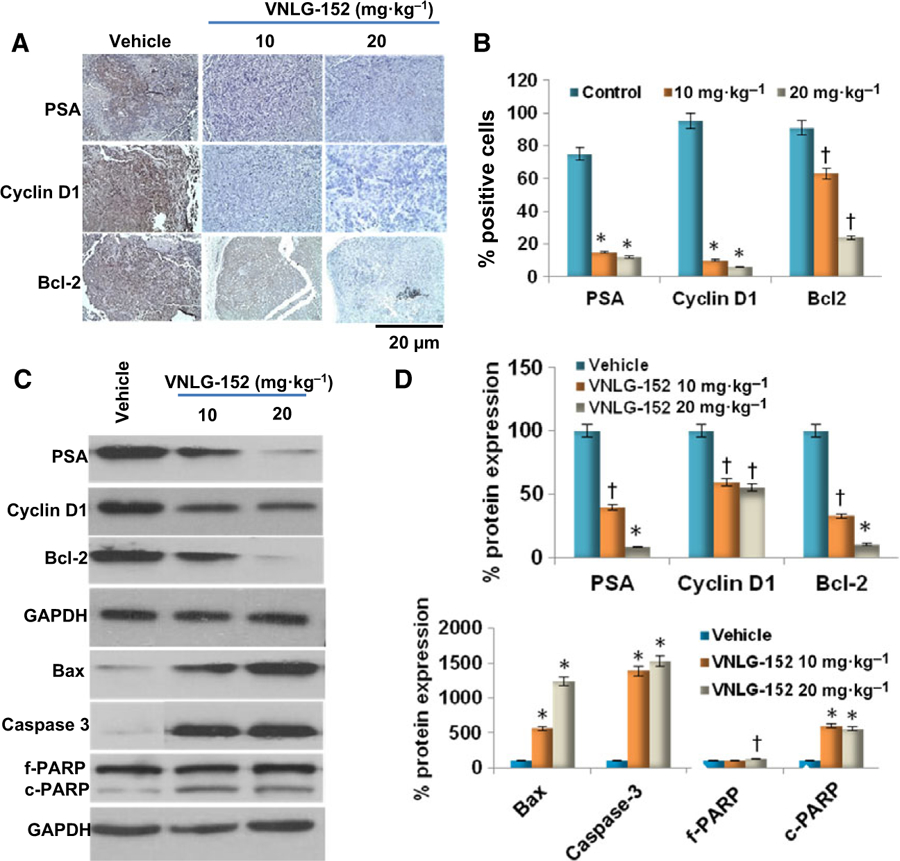
Effect of VNLG-152 on downstream targets of f-AR and MNK-peIF4E signaling. (A) Representative images of PSA, cyclin D1 and Bcl-2 immunostaining. Immunostaining with PSA, cyclin D1 and Bcl-2 antibodies revealed reduced intensity and decrease in the number of positively stained cells in VNLG-152 (10 and 20 mg·kg−1)-treated groups compared with vehicle-treated controls. Bars correspond to 20 μm. (B) Immunopositive cells were counted in 12 fields in each group. The percentage of positive cells was calculated and presented as mean ± SD. (C) Xenograft tumor tissues were harvested to evaluate effect of VNLG-152 on PSA, cyclin D1, Bcl-2, Bax, caspase-3 and PARP (full length (f) and cleaved (c)) by western blotting. Equal protein concentrations of tissue lysate were separated by SDS/PAGE and western blots probed with indicated antibodies. (D) Quantitative analysis of the protein expression using a scanning densitometer. Results are represented as means ± SEM. *P < 0.01; †P < 0.05 compared with control. Statistical significance was determined by Student’s t test and ANOVA.
VNLG-152 reverses EMT activity in CWR22Rv1 tumors
EMT is recognized as permitting polarized epithelial cells to undergo multiple changes that allow them to assume a mesenchymal phenotype, in which they display elevated resistance to apoptosis, amplified production of ECM components and increased capacity to migrate and invade [6]. During EMT, E-cadherin is replaced by N-cadherin and vimentin replaces cytokeratin. Other mesenchymal markers that are increased during EMT include Snail, Slug, Twist, and MMP-2, −3 and −9. Snail and Twist are transcription factors that act as repressors of E-cadherin [6,17]. We found that administration of VNLG-152 (10 and 20 mg·kg−1) to 22Rv1 tumors induced a significant re-expression of E-cadherin with concurrent reduction in the expression of N-cadherin, β-catenin, MMP-2, MMP-9, claudin, vimentin, Snail, Slug and Twist suggesting robust reversal in EMT activity (Fig. 4). This clearly suggests that VNLG-152 inhibition of tumor growth can also inhibit metastasis.
Fig. 4.
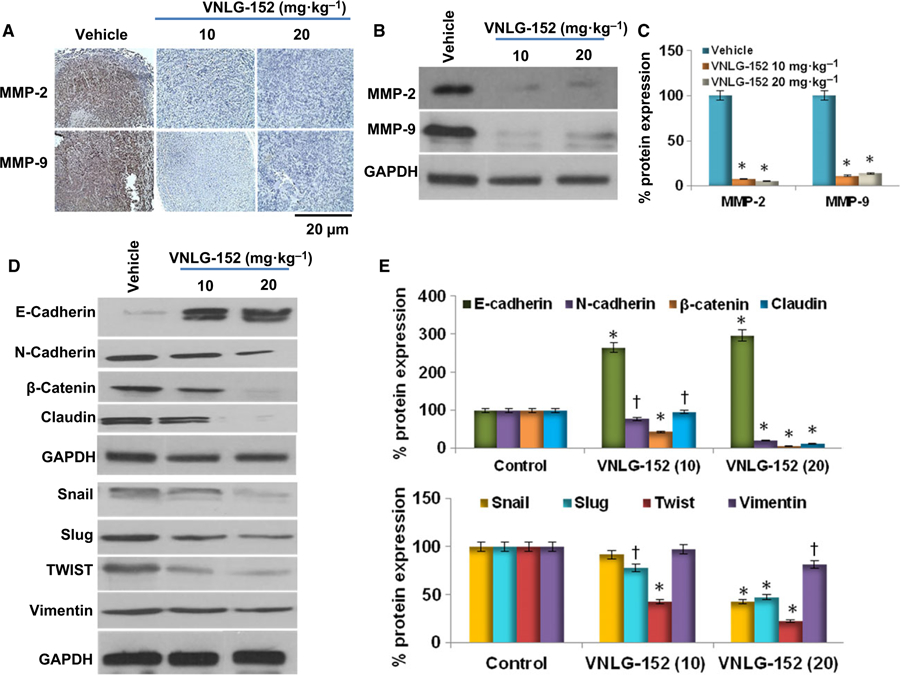
Effect of VNLG-152 on EMT-associated proteins. (A) Representative immunostaining images of MMP-2 and MMP-9. Immunostaining with MMP antibodies revealed reduced intensity and decrease in the number of positively stained cells in VNLG-152 (10 and 20 mg·kg−1)-treated groups compared with vehicle-treated controls. Bars correspond to 20 μm. (B) Xenograft tumor tissues were harvested to evaluate effect of VNLG-152 on MMP-2/9 by western blotting. Equal protein concentrations of tissue lysate were separated by SDS/PAGE and western blots probed with indicated antibodies. (C) Quantitative analysis of MMP-2/9 protein expression using a scanning densitometer. (D) Xenograft tumor tissues were harvested to evaluate effect of VNLG-152 on EMT-associated proteins by western blotting. (E) Quantitative analysis of the EMT-associated protein expression using a scanning densitometer. Results are represented as means ± SEM. *P < 0.01; †P < 0.05 compared with control. Statistical significance was determined by Student’s t test and ANOVA.
VNLG-152 inhibits cell invasion in vitro
To confirm the potential involvement of f-AR and MNK–eIF4E signaling in cell invasion and the impact of VNLG-152 on f-AR/MNK-induced cell invasion, we explored the invasive effects of 22Rv1 cells that were transiently knocked down for AR/MNK1, and 22Rv1 cells that were treated with VNLG-152 (5 μM) using a Boyden chamber invasion assay. 22Rv1 cells with both AR and MNK1 genes transiently knocked down exhibited reduced cell invasion across the Matrigel-coated membrane in contrast to cells that were transfected with either siAR or siMNK1 and scrambled siRNA. The invasion assay also demonstrated that control 22Rv1 cells exhibit enhanced ability to cross the basement membrane matrix, whereas treatment with VNLG-152 (5 μM) significantly inhibited invasion of 22Rv1 cells across the Matrigel-coated membrane (Fig. 5A,B). These results collectively suggest that f-AR and MNK1 expression/signaling is essential for 22Rv1 cell invasion, and that VNLG-152 inhibits cell invasion in 22Rv1 cells by impeding f-AR/MNK1 signaling.
Fig. 5.

Anti-invasive potential of VNLG-152 in 22Rv1 cells in vitro. (A) 22Rv1 cells were seeded on Matrigel coated Boyden chamber and treated with siAR and/or siMNK1 (A) and with/without VNLG-152 for 24 h. The images represent the extent of cell invasion in each of the treated cells. Bars correspond to 15 μm. (B) Western blot to confirm AR and MNK1 knockdown. (C) Quantification of invaded 22Rv1 cells was performed by counting cells in quadrants. Only invading cells at the bottom of inserts were counted. Results are represented as means ± SEM. †P < 0.05 compared with control. Statistical significance was determined by Student’s t test and ANOVA.
VNLG-152 inhibits DHT-induced f-AR activity and eIF4F complex formation in CWR22Rv1 cells
To validate the inhibitory effect of VNLG-152 on f-AR and eIF4E signaling in 22Rv1 cells, an f-AR–luciferase reporter gene expression assay and 7-methylguanosine 5′-triphosphate (m7GTP) pull-down assay were carried out, respectively. The f-AR–luciferase reporter gene expression assay determined the effect of VNLG-152 on DHT-induced f-AR activation. As shown in Fig. 6A, 22Rv1/f-AR cells treated with a combination of DHT and VNLG-152 (2.5 and 5 μM) displayed a strong inhibitory effect on the DHT-induced increase in f-AR activity in contrast to 22Rv1/f-AR cells treated with DHT alone.
Fig. 6.
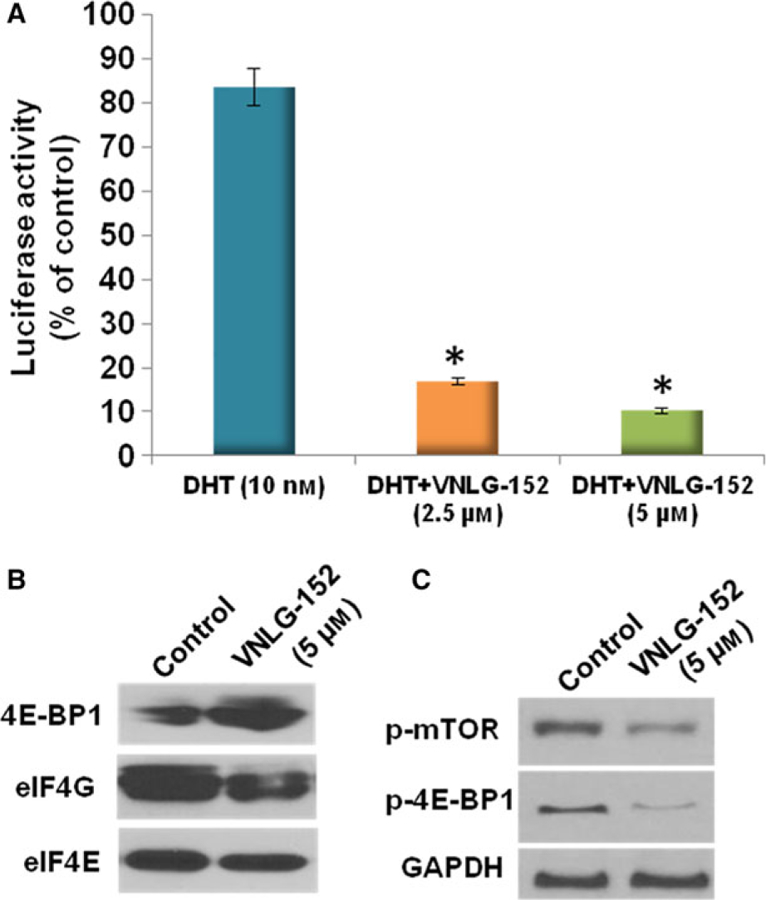
Effect of VNLG-152 on f-AR activity and eIF4F complex formation in 22Rv1 cells in vitro. (A) Effects of VNLG-152 on DHT-induced increase in f-AR reporter activity in 22Rv1/f-AR cells. 22Rv1/f-AR cells were seeded at a density of 0.1 × 105 cells·mL−1 of medium for 24 h. Then the medium was changed to RPMI 1640 without fetal bovine serum, and the cells were treated with vehicle (control) or DHT (10 nm) alone or in combination with VNLG-152 (5 and 10 μM) for 24 h. Luciferase activity was measured to etermine androgen receptor reporter activity in the 22Rv1/f-AR cells. Values are presented as the mean ± SEM from three separate experiments. *P < 0.01 compared with DHT-treated cells. Statistical significance was determined by Student’s t test and ANOVA. (B) Effect of VNLG-152 on eIF4E and eIF4G interaction in 22Rv1 cells. Western blot analysis of 4E-BP1, eIF4G and eIF4E from m7GTP purifications of vehicle- and VNLG-152 (5 μM, 24 h)-treated cell lysates. (C) 22Rv1 cells were treated with VNLG-152 (5 μM, 24 h) to evaluate the effect of p-mTOR and p-4E-BP1 by western blotting. Equal protein concentrations of cell lysate were separated by SDS/PAGE and western blots probed with indicated antibodies.
The potential effect of VNLG-152 on the interaction of eIF4E with the mRNA cap structure was investigated using the cap analog m7GTP immobilized to Sepharose in a pull-down assay. eIF4E assembly with the eukaryotic initiation factor 4G (eIF4G)–MNK subcomplex can be antagonized by eIF4E-binding proteins (4E-BPs) [18]. As summarized in Fig. 6B, VNLG-152 (5 μM, 24 h) treatment inhibited formation of the eIF4F cap complex by displacing eIF4G from eIF4E, and increasing eIF4E-binding protein (4E-BP1) expression. VNLG-152 treatment also reduced the expression of p-4E-BP1 and phosphorylated mechanistic target of rapamycin (p-mTOR) in 22Rv1 cells (Fig. 6C).
Discussion
The control and management of advanced PCa is a matter of concern arising from the first line of PCa treatment, which includes anti-androgen strategies. The hormone-refractory PCa also becomes resistant to chemotherapy and shows increased invasive and metastatic behavior [19]. In the present study administration of VNLG-152, at both the doses used, after the establishment of xenograft strongly inhibits the growth of f-AR/AR-V7-positive 22Rv1 xenografts in castrated SCID mice with no apparent host toxicity. The in vivo antitumor efficacy of VNLG-152 was accompanied by robust decreases in the expressions of f-AR, AR-V7, MNK1, MNK2 and p-eIF4E, the key players involved in f-AR and MNK–eIF4E signaling. Among the two VNLG-152-treated groups, the greater inhibitory effect observed in the 20 mg·kg−1 administered group might in part be due to greater drug availability. This finding is in line with our earlier reports in which C-4 heteroaryl retinamides were shown to be effective at inhibiting the growth of castration-resistant 22Rv1 human prostate tumor xenografts by depleting f-AR, AR-V7, MNK1/2 and p-eIF4E [20].
The maintenance of persistent f-AR activation in PCa cells is essential to CRPC growth and development. Several cellular and molecular alterations are related to this post-castration activation of the f-AR, including incomplete blockade of f-AR–ligand signaling, f-AR amplifications, f-AR mutations, aberrant f-AR co-regulator activities and f-AR splice-variant expression [21,22]. Intratumoral up-regulation of androgens even at low levels also drives continued expression of the f-AR transcriptional program and CRPC growth. A rise in serum PSA, which is partly related to f-AR transcriptional activity, serves as an index of CRPC growth and progression [23]. Interestingly in the present study, in vivo administration of VNLG-152 down-regulated the expression of both the f-AR and f-AR splice variants, with more potency towards the expression of an f-AR splice variant, AR-V7. Further, in vivo administration of VNLG-152 reduced the expression of PSA in 22Rv1 tumors. In a recent study, Zhou et al. [24] established that curcumin analogs inhibited 22Rv1 human prostate cancer cell growth by inhibiting testosterone-induced f-AR activity and PSA expression. Li et al. [25] established that kava components reduced growth in patient-derived prostate cancer xenografts in mice by down-regulating the expression of f-AR, f-AR splice variants and serum PSA levels. Galeterone and VNPT55 have demonstrated robust anti-tumor efficacy in CRPC xenografts via significant depletion of f-AR/AR-V7 expression [26]. Thus, observed down-regulation of f-AR-, f-AR-V7-, PSA- and DHT-induced f-AR activation upon administration of VNLG-152 is a reflection of its ability to inhibit f-AR activation and CRPC growth in 22Rv1 tumor xenografts.
Enhanced activation of eIF4E is another common feature in advanced prostate cancer [27–30]. In the nucleus, eIF4E associates with a subset of mRNAs containing ‘4E-sensitivity elements’ in their 3′-untranslated regions and promotes their export through nuclear pores. In the cytoplasm, eIF4E binds to the 5′-7-methylguanosine cap of mRNAs and recruits the eIF4F complex (eIF4G and eIF4A, an RNA helicase) to facilitate translation of mRNAs with complex 5′-untranslated regions [31,32]. Activation of eIF4E is mediated via serine-209 phosphorylation by MNK kinases (MNK1/2) that are dispensable for normal development and survival. mRNAs that are less well translated in the absence of MNK-mediated eIF4E phosphorylation include those that encode proteins involved in cellular growth and proliferation, apoptosis inhibition and ECM remodeling [28,33,34]. Bianchini et al. [35] established that eIF4E phosphorylation by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer. In a mouse model of prostate cancer, Furic et al. [28] validated that knock-in mice expressing a non-phosphorylatable form of eIF4E are resistant to tumorigenesis. Their findings also endorsed that eIF4E phosphorylation increases the translation efficiency of a subset of mRNAs encoding protumorigenic factors. Mice lacking MNKs are also reported to be resistant to cancer in the Lck-Pten mouse model [36,37]. MNK inhibition was also shown to impair xenograft growth in HCT116 tumor-bearing animals besides suppressing outgrowth of B16 melanoma lung metastases [37]. The present study identified a reduction in the expression of MNK1/2, p-eIF4E, cyclin D1 and Bcl-2 in VNLG-152-administered 22Rv1 tumors. An increase in the expression of Bax, caspase-3 and PARP proteins, whose mRNAs are translated in a cap-independent fashion, was also observed. Besides, a curbed formation of eIF4F complex in 22Rv1 cells through escalated expression of 4E-BP1 and diminished expression of eIF4G and eIF4E was also evident. 4E-BPs, the translational repressors of eIF4E, bind to eIF4E and prevent its association with eIF4G and further incorporation into the eIF4F complex [38]. Interaction of 4E-BP with eIF4E is, however, precluded when 4E-BP is phosphorylated by mTOR, a kinase that lies downstream of the growth factor-dependent phosphoinositide-3-kinase/AKT pathway [18]. The restricted formation of eIF4F together with reduced expression of MNK1/2, p-eIF4E, p-4E-BP1, p-mTOR and their downstream targets, cyclin D1 and Bax, observed upon VNLG-152 administration is indicative of the fact that VNLG-152 blocks the expression of MNKs, thereby inhibiting phosphorylation-mediated activation of eIF4E and subsequent eIF4F complex formation to inhibit 22Rv1 tumor growth and development. The marked inhibition of mTOR signaling by VNLG-152 treatment, as evident by strong depletion of p-mTOR and p-4E-BP1 (S65), corroborates previous studies by other investigators, albeit following MNK (kinase enzymatic activity) inhibition [39–42].
Multiple lines of evidence indicate that activation of f-AR and eIF4E commonly impinges upon metastatic phenotypes during advanced PCa [43–46]. Kong et al. [44] showed that AR-V7 is able to promote cell migration in a wound healing assay. Overexpression of AR-V7 in PCa cells also promotes prostasphere formation [44]. The expression of a number of EMT-associated genes including those for N-cadherin, vimentin, Snail, Twist, fibronectin and ZEB1 is also found increased in AR-V7-overexpressing PCa cells [45]. Phosphorylation of eIF4E was shown to promote invasion in transformed mouse embryonic fibroblasts, as well as transforming growth factor b-induced EMT in normal epithelial cells [10].
eIF4E phosphorylation was also reported to control the expression of key pro-invasive and pro-metastatic factors such as Snail and MMP-3/-9 [28]. MMPs cleave several component proteins of the extracellular matrix to promote migration, invasion and EMT [13,14]. Recently, Robichaud et al. [10] demonstrated mice harboring eIF4E mutations are resistant to lung metastases in a mammary tumor model. Cells isolated from these mice also exhibited impaired invasion. Their study also identified that a subset of mRNAs induced by canonical transforming growth factor β signaling is better translated when eIF4E becomes phosphorylated via MNK1. The impaired invasion observed in the present study upon transient knockdown of f-AR and MNK1 in 22Rv1 cells is an endorsement of the vital role of f-AR- and MNK-initiated eIF4E activation in cell migration and invasion. The reduced migratory and invasive phenotypes observed upon VNLG-152 treatment in 22Rv1 cells, coupled with diminished expression of MMPs, N-cadherin, b-catenin, claudin, Snail, Slug, Twist and vimentin, and increased E-cadherin expression observed in 22Rv1-treated tumors indicates that the inhibition of f-AR/AR-V7, MNK1/2 and p-eIF4E by VNLG-152 blocks further signals for cell invasion and EMT by f-AR/AR-V7 and/or MNK–eIF4E.
In summary, we show for the first time that the novel retinamide VNLG-152 induces significant down-regulation of f-AR/AR-V7, MNK-1/2 and p-eIF4E to inhibit tumor growth and development mediated by f-AR- and MNK-induced eIF4E activation in a xenograft model of CRPC. We also provide mechanistic insight into the role of f-AR and MNK in PCa invasion and metastasis. We propose that combined suppression of f-AR/AR-V7 and MNK–eIF4E signaling by VNLG-152 inhibits the expression of key proteins chiefly implicated in CRPC development, progression and metastasis. The strong dose-dependent antitumor activity observed with VNLG-152 in castration-resistant PCa offers a novel and efficacious therapeutic approach for combatting advanced PCa and CRPC.
Materials and methods
Cell culture and reagents
CWR22Rv1 cells were acquired from the American Type Culture Collection (Rockville, MD, USA). RPMI 1640 tissue culture medium, penicillin–streptomycin, l-glutamine and fetal bovine serum (FBS) were from Gibco/Thermo Fisher Scientific (Waltham, MA, USA). The cells were maintained in RPMI 1640 culture medium, and the medium was supplemented with 10% FBS, penicillin (100 U·mL−1)–streptomycin (100 μg·mL−1) and l-glutamine (300 μg·mL−1). Cells were grown at 37 °C in a humidified atmosphere of 5% CO2 and were passaged two times per week.
f-AR luciferase reporter assay
f-AR transcriptional activity was measured by performing an f-AR luciferase reporter gene expression assay. 22Rv1 cells were dual transfected with ARR2–Luc and the Renilla luciferase reporting vector pRL-null with Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. After a 24 h incubation at 37 °C, the cells were incubated with fresh phenol-red-free serum-free RPMI 1640 medium and treated with DHT, ethanol vehicle and/or the specified compounds in triplicate. After a 24 h treatment period the cells were washed twice with ice-cold Dulbecco’s phosphate-buffered saline and assayed using the Dual Luciferase kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, cells were lysed with 100 μL of luciferase lysing buffer, collected in a microcentrifuge tube and pelleted by centrifugation. Supernatants (100 μL aliquots) were transferred to corresponding wells of opaque 96-well multiwell plates. Luciferin was added to each well, and the light produced during the luciferase reaction was measured in a Victor 1420 scanning multi-well spectrophotometer (Wallac, Inc., Gaithersburg, MD, USA). After measurement, Stop and Glo reagent (Promega) was added to quench the firefly luciferase signal and initiate the Renilla luciferase luminescence. Renilla luciferase luminescence was also measured in the Victor 1420. The results are presented as the fold induction, that is, the relative luciferase activity of the treated cells divided by that of the control, normalized to that of the Renilla [47].
7-Methyl GTP pull-down for analysis of eIF4F complex formation
eIF4F complex in cell extracts was detected using affinity chromatography m7GTP-Sepharose as described previously [48]. Briefly, 22Rv1 cells treated with 5 μm VNLG-152 for 24 h were lysed in buffer containing 20 mm Tris/HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1% Triton X-100, 2.5 mm sodium pyrophosphate and protease inhibitor cocktail, on ice for 15 min. After centrifugation at 12 000 g for 15 min at 4 °C, the supernatants were collected and incubated with m7GTP-Sepharose (GE Healthcare Bio-Sciences, Corp/Amersham, Piscataway, NJ, USA) at 4 °C for 2 h with constant shaking. Beads were washed three times with lysis buffer and three times with 1× PBS. The samples were then denatured, and the supernatants were loaded for SDS/PAGE followed by western blot analysis.
siRNA-mediated knockdown of AR/MNK1 gene expression
siRNA targeting the AR (siAR) and MNK1 (siMNK1) genes and corresponding scramble siRNA sequence (siControl) were purchased from Ambion (Foster City, CA, USA). 22Rv1 cells were seeded into 12-well cell culture plates (Corning, Tewksbury, MA, USA) and incubated for 24 h in Opti-MEM® reduced serum medium (Thermo Fisher Scientific) without FBS. Then, cells were subjected to transfection with 100 nm of siAR/siMNK1 or siControl using the RNAi Human/Mouse Starter Kit (Qiagen, Gaithersburg, MD, USA) following the manufacturer’s instructions. Briefly, 300 000 cells were mixed with siRNAs solution complexed with the transfection reagent, and the mixture was incubated at normal culture conditions for 48 h. Non-transfected cells treated in the same way were used as control [49]. Protein silencing was confirmed by immunoblot analysis.
Boyden chamber invasion assay
The invasion assay in 22Rv1 cells was performed using Matrigel (BD Biosciences, San Jose, CA, USA)-coated transwell cell culture chambers (8 μm pore size, EMD Millipore, Temecula, CA, USA) as described previously [15,50]. Briefly, untransfected and siRNA (AR/MNK1/scrambled)-transfected 22Rv1 cells (105 cells per well) were cultured in the upper chamber of the transwell insert for 24 h in serum-free RPMI 1640 medium. The cells were then treated with or without 5 μM of VNLG-152 for 24 h. RPMI 1640 medium containing 20% FBS was placed in the lower chamber. At the end of incubation, the top surface of the non-migrated cells was scraped gently with cotton swabs and the cells on the lower surface of the membrane (migrated cells) were fixed for 15 min with cold methanol and stained with crystal violet. Cells that had migrated to the bottom of the membrane were visualized and counted using an inverted microscope. For each replicate (n = 3), cells in three randomly selected fields were counted and averaged.
In vivo xenograft tumor growth
All animal studies were performed according to guidelines approved by the Animal Care Committee of the University of Maryland, School of Medicine, Baltimore. Male SCID mice, 5–6 weeks of age, were obtained from the National Cancer Institute (Fredrick, MD, USA). Surgically castrated mice were housed under complete aseptic conditions, and fed autoclaved pellets and sterile water ad libitum. Following a week of acclimatization, approximately 5 × 106 22Rv1 cells were inoculated into both flanks. Tumor-bearing mice (tumor volume around 50–70 mm3) were randomized into three groups (six mice in each group; compounds formulated in vehicle) and treated as follows: (a) vehicle control (40% β-cyclodextin in ddH2O, i.p.), (b) VNLG-152 (10 mg·kg−1, i.p., twice daily) and (c) VNLG-152 (20 mg·kg−1, i.p., twice daily) for 5 days per week. Tumors were measured twice weekly with calipers and tumor volume was calculated by the formula: length × width2 × 0.5 (mm3). Animals were also weighed weekly and monitored for general health status and signs of possible toxicity due to treatments. Mice were sacrificed after the indicated periods of treatment and tumors and organs excised. Tumors were divided and either flash-frozen in liquid nitrogen or placed in 10% buffered formalin for western blot analysis, immunocytochemistry and hematoxylin and eosin staining [20].
Immunohistochemical analysis
Tumor samples were fixed in 10% buffered formalin for 12 h and processed conventionally. The paraffin-embedded tumor sections (4 μm thick) were heat-immobilized and deparaffinized using xylene and rehydrated in a graded series of ethanol with a final wash in distilled water. Antigen retrieval was done in 10 mm citrate buffer (pH 6.0) in a microwave followed by quenching of endogenous peroxidase activity with 3.0% H2O2 in methanol (v/v). Sections were then incubated with specific primary antibodies and epitopes detected using the Ultra-sensitive ABC staining kit (Thermo Fisher Scientific). The images were captured with an EVOS® FL Auto Imaging System (Life Technologies, Carlsbad, CA, USA).
Tumor lysate preparation and western blot analysis
Western blotting of tumor tissue lysates was performed according to a standard protocol. Briefly, 60–80 μg protein per lysate was denatured with 2× sample buffer and resolved on 12 or 16% Tris–glycine gels by SDS/PAGE. Separated proteins were transferred onto a nitrocellulose membrane by western blotting, and the membrane was blocked for 1 h in blocking buffer and then incubated with specific primary antibodies followed by peroxidase-conjugated appropriate secondary antibody. Finally, proteins were visualized by enhanced chemiluminescence detection and exposure to X-ray film. To confirm equal protein loading, membranes were stripped and re-probed with mouse monoclonal anti-β-actin primary antibody (Sigma-Aldrich, St Louis, MO, USA).
Statistical analysis
All in vitro experiments were repeated at least three times and reported as means with standard error where applicable. Western blot on in vivo samples was repeated in at least two different tumor sections from different animals. Student’s t test and analysis of variance (ANOVA) were used to determine the significance of differences or lack thereof.
Acknowledgements
This work was supported in part by a grant from NIH and NCI (R21CA195694), start-up funds from University of Maryland School of Medicine and the Center for Biomolecular Therapeutics (CBT), Baltimore, USA to VCON. VPR was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Prostate Cancer Research Program under Award No. W81XWH-15-1-0586.
Abbreviations
- 22Rv1
CWR22Rv1
- 4E-BP
eIF4E-binding protein
- Bcl-2
B-cell lymphoma 2
- f-AR
full-length androgen receptor
- AR-V7
androgen receptor splice variant-7
- CRPC
castration-resistant prostate cancer
- DHT
dihydrotestosterone
- ECM
extracellular matrix
- eIF4E
eukaryotic initiation factor 4E
- eIF4G
eukaryotic initiation factor 4G
- EMT
epithelial–mesenchymal transition
- m7GTP
7-methylguanosine 5′-triphosphate
- MMP
matrix metalloproteinases
- MNK1/2
mitogen-activated protein kinase-interacting kinase 1/2
- mTOR
mechanistic target of rapamycin
- NR
novel retinamide
- PARP
poly (ADP-ribose) polymerase
- PCa
prostate cancer
- p-eIF4E
phosphorylated eIF4E
- PSA
prostate-specific antigen
Footnotes
Conflict of interest
VCON is the lead inventor of VNLG-152, and the patents and technologies thereof are owned by the University of Maryland, Baltimore. LKG is a co-inventor of VNLG-152. The other authors declare no potential conflicts of interest.
References
- 1.Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL et al. (2017) Annual Report to the Nation on the Status of Cancer, 1975–2014, featuring survival. J Natl Cancer Inst 109, djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saad F & Fizazi K (2015) Androgen deprivation therapy and secondary hormone therapy in the management of hormone-sensitive and castration-resistant prostate cancer. Urology 86, 852–861. [DOI] [PubMed] [Google Scholar]
- 3.Katzenwadel A & Wolf P (2015) Androgen deprivation of prostate cancer: leading to a therapeutic dead end. Cancer Lett 367, 12–17. [DOI] [PubMed] [Google Scholar]
- 4.Thompson EW, Newgreen DF & Tarin D (2005) Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res 65, 5991–5995; discussion 5995. [DOI] [PubMed] [Google Scholar]
- 5.Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2, 442–454. [DOI] [PubMed] [Google Scholar]
- 6.Grant CM & Kyprianou N (2013) Epithelial mesenchymal transition (EMT) in prostate growth and tumor progression. Transl Androl Urol 2, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montanari M, Rossetti S, Cavaliere C, D’Aniello C, Malzone MG, Vanacore D, Di Franco R, La Mantia E, Iovane G, Piscitelli R et al. (2017) Epithelial-mesenchymal transition in prostate cancer: an overview. Oncotarget 8, 35376–35389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu ML & Kyprianou N (2010) Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J 24, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matuszak EA & Kyprianou N (2011) Androgen regulation of epithelial-mesenchymal transition in prostate tumorigenesis. Expert Rev Endocrinol Metab 6, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L et al. (2015) Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 34, 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Shi J, Chai K, Ying X & Zhou BP (2013) The role of Snail in EMT and tumorigenesis. Curr Cancer Drug Targets 13, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen A, Beetham H, Black MA, Priya R, Telford BJ, Guest J, Wiggins GA, Godwin TD, Yap AS & Guilford PJ (2014) E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 14, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fink K & Boratynski J (2012) The role of metalloproteinases in modification of extracellular matrix in invasive tumor growth, metastasis and angiogenesis. Postepy Hig Med Dosw (Online) 66, 609–628. [DOI] [PubMed] [Google Scholar]
- 14.Brown GT & Murray GI (2015) Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J Pathol 237, 273–281. [DOI] [PubMed] [Google Scholar]
- 15.Ramamurthy VP, Ramalingam S, Gediya L, Kwegyir-Afful AK & Njar VC (2015) Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget 6, 3195–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramamurthy VR, Ramalingam S, Gediya LK & Njar VC (2016) Targeting eIF4E- the nexus of oncogenic circuitry with proprietary novel retinamides (NRs) for prostate cancer therapy. Paper presented at the AACR Precision Medicine Series: Targeting the Vulnerabilities of Cancer, Miami, FL. [Google Scholar]
- 17.De Craene B & Berx G (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13, 97–110. [DOI] [PubMed] [Google Scholar]
- 18.Muller D, Lasfargues C, El Khawand S, Alard A, Schneider RJ, Bousquet C, Pyronnet S & Martineau Y (2013) 4E-BP restrains eIF4E phosphorylation. Translation (Austin) 1, e25819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semenas J, Allegrucci C, Boorjian SA, Mongan NP & Persson JL (2012) Overcoming drug resistance and treating advanced prostate cancer. Curr Drug Targets 13, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mbatia HW, Ramalingam S, Ramamurthy VP, Martin MS, Kwegyir-Afful AK & Njar VC (2015) Novel C-4 heteroaryl 13-cis-retinamide Mnk/AR degrading agents inhibit cell proliferation and migration and induce apoptosis in human breast and prostate cancer cells and suppress growth of MDA-MB-231 human breast and CWR22Rv1 human prostate tumor xenografts in mice. J Med Chem 58, 1900–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karantanos T, Corn PG & Thompson TC (2013) Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 32, 5501–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramalingam S, Ramamurthy VP & Njar VC (2017) Dissecting major signaling pathways in prostate cancer development and progression: mechanisms and novel therapeutic targets. J Steroid Biochem Mol Biol 166, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perner S, Cronauer MV, Schrader AJ, Klocker H, Culig Z & Baniahmad A (2015) Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 6, 35542–35555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou DY, Zhao SQ, Du ZY, Zheng XI & Zhang K (2016) Pyridine analogues of curcumin exhibit high activity for inhibiting CWR-22Rv1 human prostate cancer cell growth and androgen receptor activation. Oncol Lett 11, 4160–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Liu Z, Xu X, Blair CA, Sun Z, Xie J, Lilly MB & Zi X (2012) Kava components down-regulate expression of AR and AR splice variants and reduce growth in patient-derived prostate cancer xenografts in mice. PLoS One 7, e31213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP & Njar VC (2015) Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget 6, 27440–27460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwegyir-Afful AK, Bruno RD, Purushottamachar P, Murigi FN & Njar VC (2016) Galeterone and VNPT55 disrupt Mnk-eIF4E to inhibit prostate cancer cell migration and invasion. FEBS J 283, 3898–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA et al. (2010) eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A 107, 14134–14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atala A (2011) Re: eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. J Urol 185, 1533. [DOI] [PubMed] [Google Scholar]
- 30.Ramamurthy VP, Ramalingam S, Kwegyir-Afful AK, Hussain A & Njar VC (2017) Targeting of protein translation as a new treatment paradigm for prostate cancer. Curr Opin Oncol 29, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silva RL & Wendel HG (2008) MNK, EIF4E and targeting translation for therapy. Cell Cycle 7, 553–555. [DOI] [PubMed] [Google Scholar]
- 32.Graff JR, Konicek BW, Carter JH & Marcusson EG (2008) Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res 68, 631–634. [DOI] [PubMed] [Google Scholar]
- 33.Hay N (2010) Mnk earmarks eIF4E for cancer therapy. Proc Natl Acad Sci U S A 107, 13975–13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S & Fukunaga R (2004) Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol 24, 6539–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bianchini A, Loiarro M, Bielli P, Busa R, Paronetto MP, Loreni F, Geremia R & Sette C (2008) Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. Carcinogenesis 29, 2279–2288. [DOI] [PubMed] [Google Scholar]
- 36.Ueda T, Sasaki M, Elia AJ, Chio II, Hamada K, Fukunaga R & Mak TW (2010) Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci U S A 107, 13984–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE et al. (2011) Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res 71, 1849–1857. [DOI] [PubMed] [Google Scholar]
- 38.Musa J, Orth MF, Dallmayer M, Baldauf M, Pardo C, Rotblat B, Kirchner T, Leprivier G & Grunewald TG (2016) Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis. Oncogene 35, 4675–4688. [DOI] [PubMed] [Google Scholar]
- 39.Brown MC & Gromeier M (2017) MNK controls mTORC1: substrate association through regulation of TELO2 binding with mTORC1. Cell Rep 18, 1444–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown MC, Dobrikov MI & Gromeier M (2014) Mitogen-activated protein kinase-interacting kinase regulates mTOR/AKT signaling and controls the serine/arginine-rich protein kinase-responsive type 1 internal ribosome entry site-mediated translation and viral oncolysis. J Virol 88, 13149–13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grzmil M, Huber RM, Hess D, Frank S, Hynx D, Moncayo G, Klein D, Merlo A & Hemmings BA (2014) MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J Clin Invest 124, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teo T, Yu M, Yang Y, Gillam T, Lam F, Sykes MJ & Wang S (2015) Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis-chronic myeloid leukemia cells. Cancer Lett 357, 612–623. [DOI] [PubMed] [Google Scholar]
- 43.Xu J & Qiu Y (2016) Role of androgen receptor splice variants in prostate cancer metastasis. Asian J Urol 3, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E & Sarkar FH (2015) Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate 75, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao L, Yang L, Li R, Rodrigues DN, Crespo M, Hsieh JT, Tilley WD, de Bono J, Selth LA & Raj GV (2017) Disrupting androgen receptor signaling induces Snail-mediated epithelial-mesenchymal plasticity in prostate cancer. Cancer Res 77, 3101–3112. [DOI] [PubMed] [Google Scholar]
- 46.De Benedetti A & Graff JR (2004) eIF-4E expression and its role in malignancies and metastases. Oncogene 23, 3189–3199. [DOI] [PubMed] [Google Scholar]
- 47.Purushottamachar P, Godbole AM, Gediya LK, Martin MS, Vasaitis TS, Kwegyir-Afful AK, Ramalingam S, Ates-Alagoz Z & Njar VC (2013) Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer. J Med Chem 56, 4880–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan S, Li Y, Yue P, Khuri FR & Sun SY (2010) The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia 12, 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebollo J, Geliebter J & Reyes N (2017) ESM-1 siRNA knockdown decreased migration and expression of CXCL3 in prostate cancer cells. Int J Biomed Sci 13, 35–42. [PMC free article] [PubMed] [Google Scholar]
- 50.Lin SJ, Lin CY, Yang DR, Izumi K, Yan E, Niu X, Chang HC, Miyamoto H, Wang N, Li G et al. (2015) The differential effects of anti-diabetic thiazolidinedione on prostate cancer progression are linked to the tr4 nuclear receptor expression status. Neoplasia 17, 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]