

Abstract
The adoptive transfer of genetically engineered Chimeric Antigen Receptor (CAR) T-cells has opened a new frontier in cancer therapy. Unlike the paradigm of targeted therapies, the efficacy of CAR T-cell therapy depends not only on the choice of target, but also on a complex interplay of tumor, immune, and stromal cell communication. This presents both challenges and opportunities from a discovery standpoint. Whereas cancer consortia have traditionally focused on the genomic, transcriptomic, epigenomic, and proteomic landscape of cancer cells, there is an increasing need to expand studies to analyze the interactions between tumor, immune, and stromal cell populations in their relevant anatomical and functional compartments. Here, we focus on the promising application of systems biology to address key challenges in CAR T-cell therapy, from understanding the mechanisms of therapeutic resistance in hematologic and solid tumors to addressing important clinical challenges in biomarker discovery and therapeutic toxicity. We propose a systems biology view of key clinical objectives in CAR T-cell therapy, and suggest a path forward for a biomedical discovery process that leverages modern technological approaches in systems biology.
Introduction
CAR T-cell therapy has seen exceptional success in several hematologic malignancies (1–5), marked by the first round of FDA approvals and Phase II trials (4,6). Yet these studies have also highlighted key clinical challenges including therapeutic resistance in a subset of patients, challenges in translation to solid tumors, and therapeutic toxicity (7–9). As clinical knowledge increases, there is a promising opportunity to apply modern molecular technologies under a systems immunology approach to accelerate progress. Furthermore, the limited availability of clinical samples from early clinical trials is a major motivating factor for using advanced technologies to gain the most knowledge from the samples that exist. Broadly speaking, systems immunology encompasses the tools of systems biology framework to the unique biological characteristics of innate and adaptive immunity (10,11). The modern roots of systems biology date to the turn of the 21st century, as genome sequencing technologies began to rapidly accelerate (12,13). Under a systems biology paradigm, biological entities are modeled as a network of interacting units, typically requiring high-throughput data generation paired with integrative computational and statistical models. Each foundational unit may be defined as an intracellular entity such as a gene or metabolite, or a cellular entity such as an individual cell or cell type (10). Technological advances have made possible many approaches that were previously untenable except under theoretical circumstances. Massively parallel molecular assays can quantify not only the levels of various molecular analytes such as RNA, protein, and metabolites, but also interrogate their interactions at increasingly higher throughput. These molecular assays can also be applied to measure the cellular states and interactions after either genetic or chemical perturbations in a high-throughput fashion (14–16). The advent of single-cell technologies in particular has led to an influx of data, and ongoing consortia such as the Human Cell Atlas (17), the Human BioMolecular Atlas Program (18), and the Human Tumor Atlas Network will provide key resources to characterize the underlying basis of normal and malignant cellular phenotypes and tumor microenvironment. The immune system has been an important focus in multiple consortia, leading to opportunities to integrate knowledge into the field of CAR T-cell therapy and other immunotherapies.
In parallel to experimental technology development, a vast array of computational and statistical approaches have been developed to advance systems biology. Mathematical models of tumor-immune interactions have been developed using principles of optimal control theory, treating cellular populations as interacting units within a dynamic system (19,20). Another key class of systems biology algorithms model biological processes as molecular interaction networks, and both computational and technological advancements have allowed for construction and analysis of these networks at a tissue- or cell type- specific resolution (21,22). The relatively high sample sizes produced by single-cell experiments have been increasingly capable of powering for machine learning methods, such as probabilistic graphical models and deep learning, to interpret the data and identify regulatory mechanisms (23–25). A major ongoing challenge in the field is the integration of data across experimental protocols and molecular assays, and approaches such as factor analysis and transfer learning have been developed to address this challenge (26,27).
The mechanisms of CAR T-cell treatment success and failure are likely multimodal and involve tumor, T-cell, and microenvironmental factors (9). From a systems biology perspective, tumor-immune interactions can be viewed as a complex adaptive system that underlies the major clinical challenges, from treatment failures to therapeutic toxicities (Figure 1). In hematological malignancies such as B-cell acute lymphoblastic leukemia (B-ALL), cancer cells may be present in large numbers in peripheral circulation as well as the bone marrow, allowing T-cells to have relatively free access to the tumor throughout the body and in the marrow microenvironment. In contrast, CAR T-cell therapy for solid tumors faces multiple additional barriers including T-cell exclusion, vascular and hypoxic constraints, and immune suppression mediated by additional cell types such as cancer associated fibroblasts, tumor-associated macrophages, and myeloid-derived suppressor cells (28,29). Understanding these factors will be a crucial component of improving existing CAR T-cell therapies and developing new therapies for hematological and solid tumors. As the number of clinical and preclinical trials of CAR T-cell therapies expand, a systems biology approach will become a powerful complement to advance biological discovery (Table 1). Paramount to a systems biology approach is an understanding of the strengths and limitations of each technology, including sources of technical variability and the fundamental role of each molecular analyte in a biological system.
Figure 1.
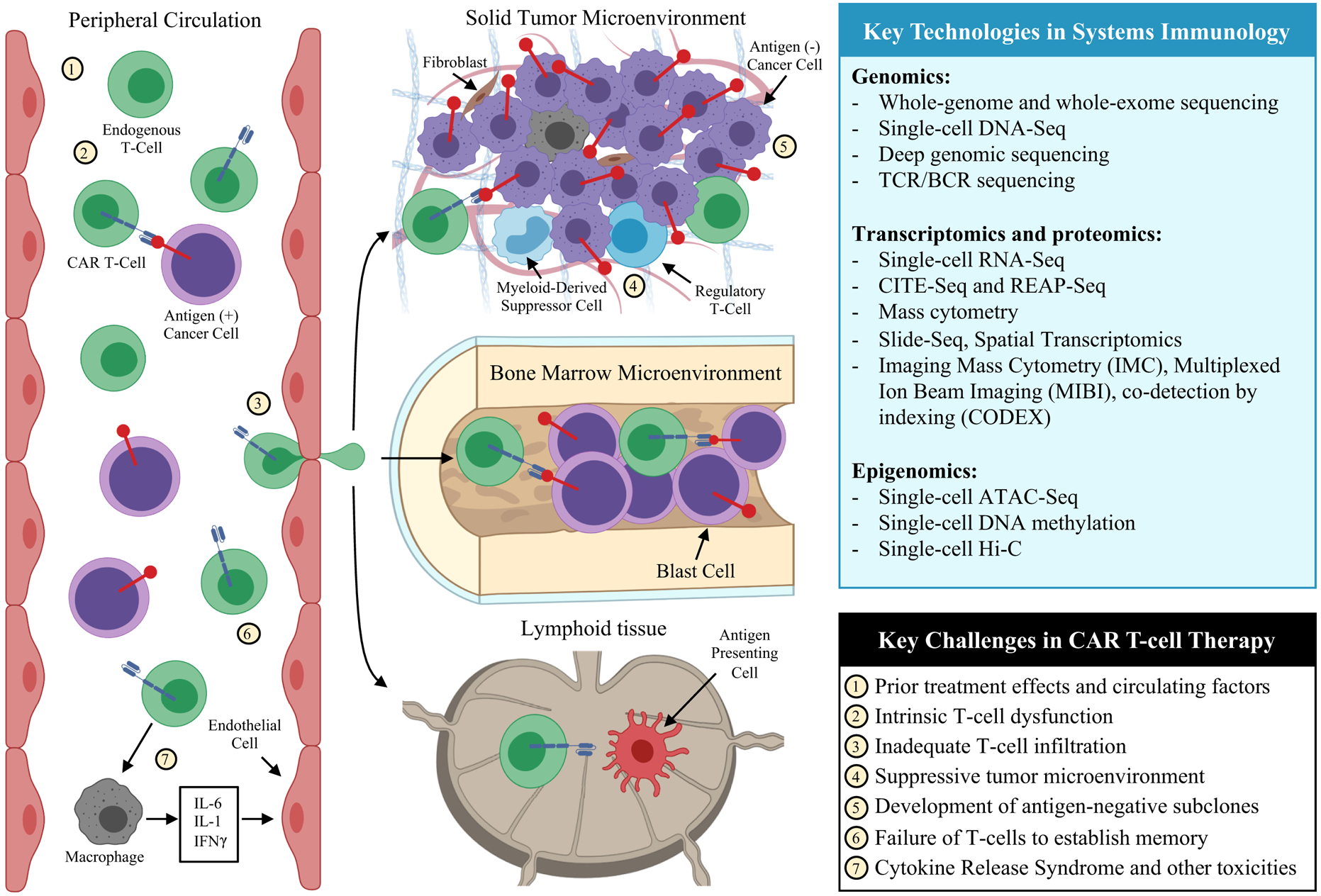
A systems immunology view of biomedical discovery for CAR T-cell therapy. Immunotherapy requires cellular interactions between tumor and immune populations in multiple physiological compartments including the peripheral circulation, tumor microenvironment, and lymphoid tissues. Addressing the key challenges in CAR T-cell therapy will require an understanding of these interactions among cellular phenotypes and microenvironments. Experimental and computational advances in systems immunology provide the capacity to simultaneously profile multiple cellular populations within each anatomical component, making it possible to investigate the tumor-immune interactions that underlie the major challenges in CAR T-cell therapy. Recent technological advances enable investigators to query genomic, transcriptomic, proteomic, and epigenomic features at a single-cell level. A number of methods for joint profiling of multiple molecular features have been developed, and imaging-based approaches have been developed to resolve spatial context of transcriptomic and proteomic data.
Table 1.
A framework for addressing the major clinical challenges of CAR T-cell therapy using systems immunology approaches*
| Clinical challenge | Systems immunology approach |
|---|---|
| Biomarker discovery |
|
| Pre-treatment effects and identifying combination therapies |
|
| Understanding T-cell aspects to therapy failure |
|
| Expanding CAR T-cell therapy to solid tumors |
|
| Antigen escape and novel target identification |
|
| Addressing major toxicities |
|
Completed and ongoing clinical trials have brought to light a number of major clinical challenges for CAR T-cell therapy moving forward: Biomarker discovery, pre-treatment effects and identifying combination therapies, understanding T-cell aspects to therapeutic failure, expanding CAR T-cell therapy to solid tumors, antigen escape and novel target identification, and addressing major toxicities. For each of these major clinical challenges, the tools of systems immunology provide a strategy to better understand the complex interactions between tumor and immune cell populations, providing a path forward for the next generation of cellular therapeutics.
Biomarker discovery: leveraging prior immunological knowledge
The identification of prognostic biomarkers of response to CAR T-cell therapy is of great clinical interest due to the potential to aid in risk stratification and identification of patients most likely to benefit from therapy. There is an urgent need to define T-cell characteristics in the CAR T-cell product that predict key attributes such as proliferation and persistence of CAR T-cells after infusion. A number of cellular and molecular biomarkers have been identified in immune checkpoint blockade: circulating factors such as lactate dehydrogenase (LDH) (30,31); immunological markers such as immune cell composition, T-cell activation pathways, lymphocytic infiltration, and T-cell repertoire (32–35); and tumor markers such as PD-L1 signaling (36) and mutational load (36–38). Some, but not all, of these biomarkers have the potential to generalize to CAR T-cell therapy. Transcriptomic phenotypes of lymphocytic activation and migration may be shared between immunotherapy modalities. On the other hand, biomarkers that assess the diversity in antigen recognition, such as T-cell repertoire and mutational load, may be of lesser significance in CAR T-cell therapy compared to immunotherapies that depend primarily on cytotoxicity through endogenous T-cell receptor signaling. The utility of these biomarkers in combined checkpoint blockade and CAR T-cell therapy, or in future CAR T-cell therapies that may require general T-cell activation and epitope spreading for efficacy, has yet to be assessed in clinical trials.
Circulating factors may play a general role in suppression or activation of adaptive immune responses. Lactic acid has been shown to blunt antitumor responses by T-cell and NK-cells (39), and serum lactate dehydrogenase (LDH) has been shown to correlate with clinical outcomes in immune checkpoint blockade. In CAR T-cell therapy, post-infusion serum cytokine markers have been shown to be predictive of cytokine release syndrome (CRS) (40), the most significant characteristic adverse effect of the therapy; but biomarkers predicting or modulating efficacy in either the product or the patient have not been clearly identified. Transcriptional pathways in T-cell populations, such as T-cell exhaustion, have been associated with resistance to both immune checkpoint blockade and CAR T-cell therapy (41,42). Insights related to tumor evasion of adaptive immunity may be shared between CAR T-cell therapy and other therapeutic modalities. Among the best-known example is expression of the programmed death-ligand 1 (PD-L1) on tumor cells, which interacts with PD-1 on T-cells to suppress T-cell activity. In an in vivo mouse model of pleural mesothelioma, tumor-cell PD-L1 expression inhibited CAR T-cell effector functions (43).
The transfer of knowledge from prior studies of immunology and cancer immunotherapy to CAR T-cell therapy will be key to the systems biology approach. The Immunological Genome Project (44) and more recently, the Human Cell Atlas (17), the Human Tumor Atlas Network, and the Immuno-Oncology Translational Network will provide a wealth of reference data on the molecular physiology of multiple immune populations in normal and malignant environments. In clinical and pre-clinical studies of cancer immunotherapy, particularly in immune checkpoint blockade, there has been an expansion in the use of high-throughput technologies to generate large datasets and computational models of tumor and immune populations. For example, Jiang et al. used publicly available bulk RNA-Seq profiles to derive a predictive signature of T-cell exclusion and dysfunction in anti-PD1 and anti-CTLA4 therapy (45); and Krieg et al. used single-cell mass cytometry to assess cell surface protein phenotypes of peripheral blood mononuclear cells at multiple therapeutic time points (46).
The computational framework for biomarker discovery for CAR T-cell therapy from prior immunological data can be formulated under the principles of transfer learning, which involves the exchange of knowledge from a source domain and task to a target domain and task (47). Under this framework, the source domain consists of data generated from a previous immunological study, and target domain consists of data generated from one or more CAR T-cell studies (Figure 2). Knowledge transfer can occur in the form of model parameters, feature representations, or instances in an integrative model. In the case of immunological data, the most direct methods of knowledge transfer can involve gene signatures, pathway signatures, gene network topology and edge weights, or model coefficients to be used as a statistical prior. Transfer learning has been successfully used for prediction of MHC class I protein binding, clinical and histopathological image analysis, integration of molecular data from genomic and RNAi screens, and integration of single-cell RNA-Seq data (48–52). Immune cell populations are among the most common cells to be studied by high-throughput technologies, and the wealth of publicly available immunological data is an opportunity for the development and deployment of integrative computational methods to be used in studies of CAR T-cell therapy.
Figure 2.

The challenge of integrating data from heterogeneous molecular assays and experimental sources. Prior and ongoing consortia have provided a wealth of data for multiple immune cell populations in isolation and in interaction with each other and tumor cells. Integrative data analysis is a major challenge in the field, and leveraging publicly available immunological data may be a valuable approach for systems immunology model development. The use of unlabeled immune cell data may be framed under an unsupervised transfer learning framework, in which the target task may be dimensionality reduction or clustering of immune cell populations. The use of labeled source data - for example, transcriptomic profiles paired with clinical outcome of another immunotherapy modality – may follow a framework of inductive transfer learning, in which the target task is the prediction of CAR T-cell clinical outcome.
Pre-treatment effects and the search for combination therapy
Patients on clinical trials of CAR T-cell therapy to date have received CAR T-cell therapy subsequent to other treatments, often including multiple rounds of cytotoxic chemotherapy. These prior treatment effects may have a profound impact on the immune cells, tumor cells, and tumor microenvironment involved in CAR T-cell therapy. In studies of other immunotherapies, chemotherapeutic agents have been shown to increase immunogenicity of tumor cells through increased antigenicity and depletion of suppressive immune cell populations (53,54) and increase tumor infiltration by T-cells in solid tumors (55). Combination therapy between certain chemotherapeutic agents and immune checkpoint blockade is the standard of care in some cancers (56). It is unclear to what extent these factors apply to CAR T-cell therapy. Lymphodepleting chemotherapy is routinely given to patients prior to CAR T-cell infusion, and has been associated with improved in vivo expansion and persistence (57), owing to cytokine upregulation as well as suppression of regulatory T-cells and myeloid-derived suppressor cells that has been observed in earlier adoptive cell transfer therapies (58). Clinical and preclinical studies comparing the impact of prior therapy on CAR T-cells themselves are scarce, and such studies are limited by the heterogeneity of prior treatment regimens among patients enrolled in clinical trials. It is possible that certain chemotherapies could have a deleterious impact on CAR T-cell therapy due to their impact on T-cells and activation of pathways hindering expansion in vitro or post-infusion (59).
With over a hundred approved anticancer drugs and over a thousand in development (60), the identification of agents that sensitize cancer cells or augment immunological function in CAR T-cell therapy is a promising objective. Some drug combinations, such as combining immune checkpoint blockade and CAR T-cell therapy, have initial clinical evidence of benefit (61). A highly active area of systems biology has been the development of technologies and computational methods to identify synergistic and antagonistic drug combinations in cancer therapy. High-throughput drug sensitivity screens on cell lines, paired with genomic and transcriptomic data, have provided a resource for modelling individual drug perturbations in cancer cells (62,63), and CRISPR screens of gene pairs provide a functional assay to assess for genetic interactions among known drug targets in cancer cells (64). A number of computational methods have been developed to model the gene regulatory networks of cancer cells, with the objective of identifying synergistic drug combinations or pairs of potentially targetable genes that exhibit synthetic lethality or synthetic dose lethality (65–68). These approaches, however, are challenging to generalize to immunotherapy. Immunotherapy fundamentally differs from conventional pharmacotherapy, in that the mechanism of tumor cell killing is dependent on complex interactions between immune cells and tumor cells, which are not adequately modeled in assays of cancer cell lines. One approach has been to identify differentially expressed genes and gene modules between immunotherapy responders and non-responders, and consider regulators of these pathways as candidate targets. For example, Lesterhuis et al. performed network analysis of immunotherapy responsive and resistant tumor transcriptomes to identify candidate target genes to augment checkpoint blockade (69), and Jerby-Arnon et al. performed single-cell RNA-Seq on 33 melanoma tumors to identify an immune evasion cellular program that suggested CDK4/6 inhibition in combination with immune checkpoint blockade (70). Intratumor targets, however, only represent a subset of drug targets among the potentially targetable cellular populations. For example, stimulation of innate immunity, modification of the tumor immunosuppressive environment, and modulation of T-cell function represent classes of potential drug targets directed toward non-tumoral cells (71). To address these broader opportunities of combination therapies, computational models must consider immune cell populations and tumor-immune interactions.
Use of single-cell technology to better understand the T-cell contribution to CAR T-cell therapy
The underlying T-cell populations used for generation of CAR T-cell products likely play a substantial role in determining the ability of the CAR T-cells to proliferate and kill tumor cells in vivo. Until recently, most protocols for CAR T-cell generation have not selected for T-cell subtypes for engineering, and thus contain a heterogeneous mix of CD4+ and CD8+ T-cells along the full spectrum of naive to effector phenotypes. A recent exception to this is the JCAR017 approach, using defined populations of CD4 and CD8 cells, with impacts on efficacy and persistence that are still being studied (72). A number of studies have assessed T-cell predictors of therapeutic response in chronic lymphocytic leukemia (42), non-Hodgkin lymphoma (73), and multiple myeloma (74). Fraietta et al. found that sustained response to CAR T-cell therapy in chronic lymphocytic leukemia was associated with increased prevalence of CD27+CD45RO−CD8+ memory-like T-cells prior to CAR T-cell generation, and the presence of CD27+PD-1−CD8+ CAR T-cells expressing high levels of the IL-6 receptor-β chain was predictive of therapeutic response (42). In a study of non-Hodgkin lymphoma, Rossi et al. applied a single-cell secreted cytokine assay on pre-infusion CAR T-cells, and developed a predictive score based on the proportion of cells secreting multiple cytokines and the mean fluorescence intensity of secreted proteins per cell; they reported that their score was significantly associated with treatment response (73). In a study of multiple myeloma, Cohen et al. found that a higher CD4:CD8 ratio in the initial leukapheresis product was associated with greater CAR T-cell expansion in vivo (74). An important area of study has been understanding of the role of early memory phenotypes (75), such as naive T-cells, in establishing proliferative populations and long-term memory. Selection or enrichment of T-cell subtypes has been proposed as a major area of interest for improving CAR T-cell therapies (76,77), which may be aided by the recent proliferation of single-cell transcriptomic, epigenomic, and proteomic technologies.
T-cell subset assessment has conventionally relied on the use of individual cell-surface protein markers, but recent technologies have made it possible to interrogate large numbers of markers at a single-cell level in combination with other molecular and imaging assays. Single-cell mass cytometry uses flow cytometry and mass spectrometry with discrete isotopes to characterize large numbers of cell surface protein markers simultaneously at a single-cell level (78). Simultaneous profiling of single-cell RNA transcriptomes with proteomics has been made possible with CITE-Seq and REAP-Seq, methods that combine single-cell RNA-Seq with oligonucleotide-labeled antibodies that bind to cell surface proteins (79,80). These methods may be used to distinguish between CAR T-cells and non-CAR T-cells in single-cell transcriptomic studies, allowing for the capacity to identify gene expression pathways specific to the endogenous and engineered cell populations.
Single-cell RNA-Seq has been a breakthrough technology for characterizing whole-transcriptome gene expression at a single-cell resolution. Single-cell RNA-Seq has been used to profile infiltrating T-cells in several solid tumor types (81,82), demonstrating the ability to investigate T-cell populations within the tumor microenvironment. Tikhonova et al. and Baryawno et al. recently characterized the mouse bone marrow microenvironment using single-cell RNA-Seq, providing key insights into cellular phenotypes and signaling that underlies the bone marrow niche (83,84). Applying single-cell transcriptomics to complex populations of peripheral and infiltrating T-cells in CAR T-cell therapy may aid in identifying subpopulations and cellular pathways that mediate proliferation, persistence, and memory formation. Characterization of suppressive cell types in the tumor microenvironment, such as tumor-associated macrophages, cancer-associated fibroblasts, myeloid-derived suppressor cells, and regulatory T-cells, may aid in understanding the contribution of these cells in the efficacy of CAR T-cell therapy. Without doubt, current single-cell technologies still have a number of limitations, such as coverage and throughput. There has been significant work to address these and other technical challenges in single-cell RNA-Seq, including variability in sequencing depth and cell size, drop out, and batch effect correction (85).
Improving the engineering of CAR T-cells with the goal of increasing efficacy, speeding manufacturing, and decreasing toxicity is a central challenge in the field, with the potential to enhance existing therapies and accelerate the adoption of cellular therapeutics to other cancer types. Beyond cancer therapy, CAR T-cells have been designed for other therapeutic purposes, including autoimmune disease (86), prevention of allogeneic transplant rejection (87), and against infectious disease (88,89). Modeling of intracellular and extracellular interactions may play an important role in the design of novel engineering of CAR components. The intracellular pathway mediating cytotoxic response in CAR T-cells is incompletely understood, and recent studies have applied computational modeling to understand phosphorylation kinetics of certain CAR constructs (90). A number of synthetic biology approaches have been designed to augment and enhance CAR construction, including T-cells engineered with kill switches, growth switches, chemokine receptors, multiple targets, and avoidance of allogeneic rejection (91,92).
Addressing challenges in solid tumors
CAR T-cell therapies have seen their greatest success in hematological B-cell malignancies, and the design of an effective CAR T-cell therapy for solid tumors is a key objective in pediatric and adult clinical oncology. In contrast to hematological cancers, solid tumors pose particular challenges including the need for CAR T-cell trafficking and infiltration, overcoming a hypoxic and metabolically constrained tumor microenvironment, and the presence of suppressive cell populations (29). These factors have been hypothesized to be major barriers to the effective implementation of CAR T-cell therapy in solid tumors.
In CAR T-cell therapy, T-cells are typically extracted from and re-infused into the peripheral circulation, making it necessary for CAR T-cells to enter the tumor microenvironment. To address this challenge, some groups have explored intracavitary or intratumoral injection of CAR T-cells (93,94). This strategy may be technically challenging, and even more importantly, it is unclear to what extent regionally delivered CAR T-cells can migrate to other tumor sites in highly disseminated disease. The level of T-cell infiltration into solid tumors has been correlated with chemokine expression in the tumor microenvironment, and variability in chemokine expression may explain the low level of T-cell infiltration in some tumors (95). Some groups have focused on engineering approaches to localize CAR T-cells, such as the manufacturing of T-cells with receptors for chemokine CCR2 to increase cellular migration (96) or injection of amphiphile CAR T-cell ligands that traffic with endogenous albumin to lymph nodes and enhance the interaction between CAR T-cells and antigen-presenting cells (97).
Recent technological advances in proteomics and single-cell RNA-sequencing has improved our understanding of the composition and cellular interactions within the tumor-immune microenvironment (98). For example, Spitzer et al. performed mass cytometry of immune cells in multiple anatomical compartments of a mouse model of cancer immunotherapy, and demonstrated that immune activation occurs systemically in initial therapy but only in the peripheral compartment during tumor rejection (99). In particular, the role of regulatory T-cells, M2 macrophages, and myeloid-derived suppressor cells in immune evasion has been demonstrated in clinical and pre-clinical studies of other immunotherapies in solid tumors (100); however, limited sample sizes and technical challenges in raising immunocompetent mouse models have made it challenging to directly study these interactions of CAR T-cells with these cellular populations (28). Ongoing consortia, such as the Human Tumor Atlas Network and the Adult Immunotherapy Network, have dedicated significant resources into generating publicly accessible data that will be key to unravelling the basis of immune suppression in the tumor-immune microenvironment. An exciting advance in the field has been the integration of cellular imaging with single-cell molecular profiling. Technologies such as Spatial Transcriptomics and Slide-Seq have been recently developed to integrate single-cell RNA-Seq with cellular imaging (101,102), providing an opportunity to understand the spatial context of cellular populations in the tumor-immune microenvironment. Highly multiplexed analysis of proteins in the context of tissue imaging has been made possible with several imaging technologies, including Imaging Mass Cytometry (IMC) (103), Multiplexed Ion Beam Imaging (MIBI) (104), and co-detection by indexing (CODEX) (105). These resources will provide rich datasets for modeling the signal transduction pathways within the tumor microenvironment in CAR T-cell therapy.
Identifying tumor-specific antigens is a challenge for the development of future CAR T-cell therapies for non-hematopoietic malignancies. One of the reasons for the success of CAR T-cell therapies for B-cell malignancies is that the ablation of normal B-cell populations is a manageable toxicity via immunoglobulin replacement. Cancer-associated antigens have to be chosen carefully to avoid off-tumor, on-target toxicity, which has been a challenge in some solid tumor CAR T-cell trials (14). Computational and functional screens have been designed to identify cell surface proteins that are ubiquitously expressed on tumor cells but not on normal cells (106,107). A major challenge to this approach is the lack of a comprehensive reference catalog of normal cell populations. Consortia such as GTEx have performed bulk RNA-Seq on multiple normal human tissues (108), but bulk assays may miss rare but important cell populations such as adult stem cells and non-parenchymal cells. Single-cell data sources have the potential to address this problem through their capacity to characterize rare and previously unknown cell populations (109). Designing CAR T-cell therapies to target multiple antigens may be an approach to circumvent antigen escape, as well as increase tumor specificity. Toward this goal, an integration of single-cell atlases with data from individual studies may provide the much-needed reference from which tumor-specific antigens and combinatorial antigen targets may be identified.
Therapy-induced toxicity
CRS is the most common and significant toxicity of CAR T-cell therapy, reported to occur in 54–91% of patients receiving anti-CD19 CAR T-cell therapy (110). This syndrome was not apparent in preclinical models, and only became manifest in early clinical trials (2). Neurotoxicity, referred to as immune effector cell-associated neurotoxicity syndrome, or ICANS (111), has been observed as an adverse event, with the most concerning trait being cerebral edema. While the underlying pathophysiology of these toxicities remain incompletely understood, clinical trials have implicated macrophages, IL-1, endothelial activation, and especially IL-6 in the pathogenesis of CRS. IL-6 blockage with tocilizumab is the only currently approved therapy for CRS (2,112). Novel murine models have been recently developed, and have suggested a role of myeloid lineages and endothelial activation in CRS (113). Giavirdis et al. (114) found that CAR T-cells recruit and activate macrophages, leading the myeloid cells to produce IL-6 and iNOS which contribute CRS. Norelli et al. (115) found that monocytes and macrophages were primarily responsible for increases in IL-1 and IL-6 in their mouse model of CRS, with IL-1 production preceding and possibly stimulating IL-6 production. In a clinical trial of CD19 CAR T-cell therapy in B-ALL, CLL, and NHL, investigators reported evidence of increased endothelial activation in patients with severe CRS, via elevated Von Willebrand Factor and increased Ang2:Ang1 ratio (110). Moreover, IL-6 has been shown to disrupt the blood-brain barrier in vitro, suggesting a possible mechanistic explanation for neurotoxicity that has been reported in some patients.
Toxicities of CAR T-cell therapy such as CRS and ICANS remain as important concerns that may or may not be class effects of the therapy, and prognostic and therapeutic strategies will require a deeper understanding of the underlying biological basis. While initial clinical studies have focused on tumor cells and T-cells, a systems immunology approach will involve investigation of cell-cell interactions with various other immune cell types in the peripheral and marrow microenvironments. Comprehensive analyses of myeloid and lymphoid compartments in clinical trials, as well as expansion of murine models, will be important paths forward for the study of these therapy-limiting toxicities.
Conclusions and future directions
CAR T-cell therapy has been a breakthrough in clinical oncology. Understanding the T-cell, tumor-cell, and tumor microenvironment factors mediating therapeutic efficacy will play a significant role in optimizing current therapies and applying CAR T-cells to other tumor types. Modern omics and systems biology technologies provide opportunities for biomarker discovery and therapeutic development. As clinical cohorts of patients treated with CAR T-cell therapy continue to grow, there will be increasing availability of clinical samples from which rich multi-omic profiling can advance the discovery process. Like many biomedical challenges in the 21st century, translational research will be best performed through a collaborative effort by interdisciplinary teams of clinicians, experimental and computational biologists.
Acknowledgements
This work was supported by National Institutes of Health of United States of America grants, GM108716, GM104369, CA226187, CA233285 (to K.T.), CA232361 (to D.B. and S.G.) and a Doris Duke Charitable Foundation Clinical Scientist Development Award (to D.B.).
Footnotes
Disclosure of potential conflicts of interest
The authors declare no competing interests.
References
- 1.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N Engl J Med. 2013;368:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139–303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol. 2019;19:73. [DOI] [PubMed] [Google Scholar]
- 8.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019;25:1341–55. [DOI] [PubMed] [Google Scholar]
- 9.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis MM, Tato CM, Furman D. Systems immunology: just getting started. Nat Immunol. 2017;18:725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villani A-C, Sarkizova S, Hacohen N. Systems Immunology: Learning the Rules of the Immune System. Annu Rev Immunol. 2018;36:813–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitano H. Computational systems biology. Nature. 2002;420:206. [DOI] [PubMed] [Google Scholar]
- 13.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–72. [DOI] [PubMed] [Google Scholar]
- 14.Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell. 2018;175:1958–1971.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, et al. Mapping the Genetic Landscape of Human Cells. Cell. 2018;174:953–967.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016;167:1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The Human Cell Atlas Gingeras TR, editor. eLife. 2017;6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder MP, Lin S, Posgai A, Atkinson M, Regev A, Rood J, et al. Mapping the Human Body at Cellular Resolution -- The NIH Common Fund Human BioMolecular Atlas Program. ArXiv190307231 Q-Bio [Internet]. 2019. [cited 2019 Aug 3]; Available from: http://arxiv.org/abs/1903.07231 [Google Scholar]
- 19.Castiglione F, Piccoli B. Cancer immunotherapy, mathematical modeling and optimal control. J Theor Biol. 2007;247:723–32. [DOI] [PubMed] [Google Scholar]
- 20.Mostolizadeh R, Afsharnezhad Z, Marciniak-Czochra A. Mathematical model of Chimeric Anti-gene Receptor (CAR) T cell therapy with presence of cytokine [Internet]. Numer. Algebra Control Optim Vol. 8 Pages 63–80. 2018. [cited 2019 Jul 31]. Available from: http://www.aimsciences.org.proxy.library.upenn.edu/article/doi/10.3934/naco.2018004 [Google Scholar]
- 21.Vidal M, Cusick ME, Barabási A-L. Interactome networks and human disease. Cell. 2011;144:986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitra K, Carvunis A-R, Ramesh SK, Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14:719–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iacono G, Massoni-Badosa R, Heyn H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019;20:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. Deep generative modeling for single-cell transcriptomics. Nat Methods. 2018;15:1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet. 2019;20:257–72. [DOI] [PubMed] [Google Scholar]
- 27.Finotello F, Rieder D, Hackl H, Trajanoski Z. Next-generation computational tools for interrogating cancer immunity. Nat Rev Genet. 2019;20:724–46. [DOI] [PubMed] [Google Scholar]
- 28.Newick K, O’Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Annu Rev Med. 2017;68:139–52. [DOI] [PubMed] [Google Scholar]
- 29.Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, et al. CAR T Cell Therapy of Non-hematopoietic Malignancies: Detours on the Road to Clinical Success. Front Immunol [Internet]. 2018. [cited 2019 Feb 15];9 Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02740/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res Off J Am Assoc Cancer Res. 2016;22:5487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017;18:e731–41. [DOI] [PubMed] [Google Scholar]
- 32.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res. 2010;16:2861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer [Internet]. 2015. [cited 2019 Apr 18];3 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4469400/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Caliò A, et al. Differential Activity of Nivolumab, Pembrolizumab and MPDL3280A according to the Tumor Expression of Programmed Death-Ligand-1 (PD-L1): Sensitivity Analysis of Trials in Melanoma, Lung and Genitourinary Cancers. PloS One. 2015;10:e0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016;24:657–71. [DOI] [PubMed] [Google Scholar]
- 40.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;CD-16–0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shay T, Kang J. Immunological Genome Project and systems immunology. Trends Immunol. 2013;34:602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24:144–53. [DOI] [PubMed] [Google Scholar]
- 47.Pan SJ, Yang Q. A Survey on Transfer Learning. IEEE Trans Knowl Data Eng. 2010;22:1345–59. [Google Scholar]
- 48.Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyö D, et al. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24:1559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM, et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature. 2017;542:115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharya R, Sivakumar A, Tokheim C, Guthrie VB, Anagnostou V, Velculescu VE, et al. Evaluation of machine learning methods to predict peptide binding to MHC Class I proteins. bioRxiv. 2017;154757. [Google Scholar]
- 51.Sanchez-Garcia F, Villagrasa P, Matsui J, Kotliar D, Castro V, Akavia U-D, et al. Integration of Genomic Data Enables Selective Discovery of Breast Cancer Drivers. Cell. 2014;159:1461–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stein-O’Brien GL, Clark BS, Sherman T, Zibetti C, Hu Q, Sealfon R, et al. Decomposing Cell Identity for Transfer Learning across Cellular Measurements, Platforms, Tissues, and Species. Cell Syst. 2019;8:395–411.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102:115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28:690–714. [DOI] [PubMed] [Google Scholar]
- 55.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44:343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84. [DOI] [PubMed] [Google Scholar]
- 57.Turtle CJ, Berger C, Sommermeyer D, Hanafi L-A, Pender B, Robinson EM, et al. Anti-CD19 Chimeric Antigen Receptor-Modified T Cell Therapy for B Cell Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Fludarabine and Cyclophosphamide Lymphodepletion Improves In Vivo Expansion and Persistence of CAR-T Cells and Clinical Outcomes. Blood. 2015;126:184–184. [Google Scholar]
- 58.Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother Hagerstown Md 1997. 2010;33:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das RK, Storm J, Barrett DM. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Cancer Res. 2018;78:1631–1631. [Google Scholar]
- 60.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–92. [DOI] [PubMed] [Google Scholar]
- 61.Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood. 2018;132:556–556. [Google Scholar]
- 62.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han K, Jeng EE, Hess GT, Morgens DW, Li A, Bassik MC. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat Biotechnol. 2017;35:463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jerby-Arnon L, Pfetzer N, Waldman YY, McGarry L, James D, Shanks E, et al. Predicting Cancer-Specific Vulnerability via Data-Driven Detection of Synthetic Lethality. Cell. 2014;158:1199–209. [DOI] [PubMed] [Google Scholar]
- 66.Hu Y, Chen C-H, Ding Y-Y, Wen X, Wang B, Gao L, et al. Optimal control nodes in disease-perturbed networks as targets for combination therapy. Nat Commun. 2019;10:2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JS, Das A, Jerby-Arnon L, Arafeh R, Auslander N, Davidson M, et al. Harnessing synthetic lethality to predict the response to cancer treatment. Nat Commun. 2018;9:2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sinha S, Thomas D, Chan S, Gao Y, Brunen D, Torabi D, et al. Systematic discovery of mutation-specific synthetic lethals by mining pan-cancer human primary tumor data. Nat Commun. 2017;8:15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lesterhuis WJ, Rinaldi C, Jones A, Rozali EN, Dick IM, Khong A, et al. Network analysis of immunotherapy-induced regressing tumours identifies novel synergistic drug combinations. Sci Rep. 2015;5:12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018;175:984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17:286–301. [DOI] [PubMed] [Google Scholar]
- 72.Abramson JS, Gordon LI, Palomba ML, Lunning MA, Arnason JE, Forero-Torres A, et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol. 2018;36:7505–7505. [Google Scholar]
- 73.Rossi J, Paczkowski P, Shen Y-W, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. 2018;132:804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cohen AD, Melenhorst JJ, Garfall AL, Lacey SF, Davis M, Vogl DT, et al. Predictors of T Cell Expansion and Clinical Responses Following B-Cell Maturation Antigen-Specific Chimeric Antigen Receptor T Cell Therapy (CART-BCMA) for Relapsed/Refractory Multiple Myeloma (MM). Blood. 2018;132:1974–1974.30089628 [Google Scholar]
- 75.Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8:320ra3–320ra3. [DOI] [PubMed] [Google Scholar]
- 76.Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qin H, Dong Z, Wang X, Cheng WA, Wen F, Xue W, et al. CAR T cells targeting BAFF-R can overcome CD19 antigen loss in B cell malignancies. Sci Transl Med. 2019;11:eaaw9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, et al. Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum. Science. 2011;332:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35:936–9. [DOI] [PubMed] [Google Scholar]
- 81.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. [DOI] [PubMed] [Google Scholar]
- 83.Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell. 2019;177:1915–1932.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet. 2015;16:133–45. [DOI] [PubMed] [Google Scholar]
- 86.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Noyan F, Zimmermann K, Hardtke‐Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am J Transplant. 2017;17:917–30. [DOI] [PubMed] [Google Scholar]
- 88.Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci. 2014;111:10660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scholler J, Brady TL, Binder-Scholl G, Hwang W-T, Plesa G, Hege KM, et al. Decade-Long Safety and Function of Retroviral-Modified Chimeric Antigen Receptor T Cells. Sci Transl Med. 2012;4:132ra53–132ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rohrs JA, Zheng D, Graham NA, Wang P, Finley SD. Computational Model of Chimeric Antigen Receptors Explains Site-Specific Phosphorylation Kinetics. Biophys J. 2018;115:1116–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chakravarti D, Wong WW. Synthetic biology in cell-based cancer immunotherapy. Trends Biotechnol. 2015;33:449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151–261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Schalkwyk MCI, Papa SE, Jeannon J-P, Urbano TG, Spicer JF, Maher J. Design of a Phase I Clinical Trial to Evaluate Intratumoral Delivery of ErbB-Targeted Chimeric Antigen Receptor T-Cells in Locally Advanced or Recurrent Head and Neck Cancer. Hum Gene Ther Clin Dev. 2013;24:134–42. [DOI] [PubMed] [Google Scholar]
- 95.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine Expression in Melanoma Metastases Associated with CD8+ T-Cell Recruitment. Cancer Res. 2009;69:3077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin Cancer Res. 2011;17:4719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487–502.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. [DOI] [PubMed] [Google Scholar]
- 102.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–22. [DOI] [PubMed] [Google Scholar]
- 104.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, et al. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell. 2018;174:968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li L-P, Lampert JC, Chen X, Leitao C, Popović J, Müller W, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med. 2010;16:1029–34. [DOI] [PubMed] [Google Scholar]
- 108.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. The Human Cell Atlas: from vision to reality. Nat News. 2017;550:451. [DOI] [PubMed] [Google Scholar]
- 110.Hay KA, Hanafi L-A, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood. 2017;130:2295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Clin Cancer Res. 2018;clincanres.2035.2018. [DOI] [PubMed] [Google Scholar]
- 113.van der Stegen SJC, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol Baltim Md 1950. 2013;191:4589–98. [DOI] [PubMed] [Google Scholar]
- 114.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739–48. [DOI] [PubMed] [Google Scholar]