

Abstract
The sequestration of the essential nutrient iron (Fe) from bacterial invaders that colonize the vertebrate host is a central feature of nutritional immunity and the “fight over transition metals” at the host-pathogen interface. The Fe quota for many bacterial pathogens is large, as Fe enzymes often comprise a significant share of the metalloproteome. Fe enzymes play critical roles in respiration, energy metabolism and other cellular processes by catalyzing a wide range of oxidation-reduction, electron transfer, and oxygen activation reactions. In this Concepts report, we discuss recent insights into a diversity of ways that bacterial pathogens acquire this essential nutrient beyond well-characterized tris-catecholate FeIII complexes, in competition and cooperation with significant host efforts to cripple these processes. We also discuss pathogen strategies to adapt their metabolism to less-than-optimal Fe, and briefly speculate on what may be an integrated adaptive response to concurrent limitation of both Fe and Zn in the infected host.
Keywords: Iron; iron uptake, host-pathogen interface; metal nutrient acquisition; siderophore; catechols
Graphical Abstract
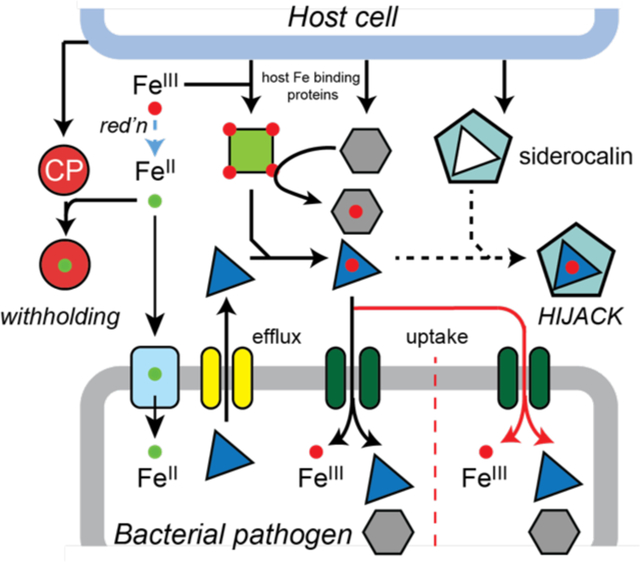
Bacterial pathogens employ myriad strategies to acquire the essential nutrient iron. Here we highlight newly emerging features of bacterial Fe acquisition beyond classical FeIII-siderophores, including the use of coordinatively unsaturated FeIII-catechol complexes and FeII uptake. The bacterial adaptive response to host efforts to restrict Fe availability is also discussed.
1. Introduction
1.1. Iron in bacterial cellular metabolism
Iron (Fe) is among the most Earth-abundant transition elements and is extensively used by nearly all living organisms to perform an impressive array of chemical transformations. Fe-dependent catalysis impacts many aspects of metabolism, including energy metabolism, respiration, lipid metabolism, amino acid and cofactor biogenesis and DNA metabolism.[1] Fe enzymes play important roles in electron transfer processes and as oxidoreductases, and both functions derive from the accessibility of ferrous (FeII) and ferric (FeIII) oxidation states. Fe-dependent mono- and dioxygenases often cycle to higher oxidation states, e.g., FeIV, which is a potent oxidant. Fe speciation in proteins involves ionic mononuclear Fe and multi-metal (Fe-Fe, or mixed metal, e.g., Ni-Fe) coordination complexes, Fe-heme or iron-sulfur (Fe-S) clusters, the latter of which are inorganic complexes of Fe with sulfide (S2−), as [2Fe-2S] or [4Fe-4S] cubane structures. In a typical Fe-centric, facultative anaerobic, Gram-negative bacterium, e.g., Escherichia coli, characterized by an outer membrane (OM) and an inner cytoplasmic membrane (CM) that collectively encompass the periplasm, Fe-containing proteins are estimated to comprise upwards of 85% of the metalloproteome,[2] which itself accounts for ≈30% of the entire proteome.[3] This metal quota is significant, with the next largest contributor to the metalloproteome zinc (Zn) metalloenzymes, which comprise the majority of what remains (≈15%; or 5–6% of a typical bacterial proteome).[4]
1.2. Fe bioavailability and the microenvironmental niche
Since the footprint of Fe-dependent metabolic activity is large, Fe withholding from a bacterial pathogen that colonizes the vertebrate host is foundational to the concept of nutritional immunity (see below).[5] Fe sparing[6] has emerged in response as a broadly employed strategy that bacterial cells use to lower their cellular demand during extreme Fe restriction during infection.[7] In addition, the successful pathogen employs a myriad of strategies to secure Fe from the surrounding microenvironment The latter helps to avoid the collateral damage of weakly complexed “free” FeII in an aerobic environment, which becomes a potent autocatalytic producer of oxidative stress, as highly reactive hydroxyl radicals (OH•) formed by the Fenton reaction. FeII will predominate in the reducing environment of the cytoplasm, and is considered a weakly competitive transition metal, which generally binds to protein sites with only modest affinity, only greater than that of MnII.[8] Some bacteria have evolved FeII efflux transporters, likely as a means to mitigate the impact of FeII excess,[6a, 9] particularly under conditions of host-derived oxidative stress.[9–10] Maintaining bioavailable Fe concentrations and optimal Fe speciation that is compatible with cellular viability during infection is controlled by metalloregulatory proteins that sense either FeII, e.g., Fur (ferric uptake regulator),[11] Fe-S loads,[12] or in some cases, heme-Fe.[13] Fur, for example, represses the expression of the Fur regulon when Fe is replete.[8] As Fe becomes increasingly scarce, FeII dissociates from Fur, and Fur, in turn, sequentially releases from individual DNA operator sites in “waves”, driving a graded transcriptional response to cellular Fe deprivation.[6a] FeII sensing in many organisms is intertwined with oxidative stress resistance[14] and MnII acquisition[15] since MnII can substitute for FeII in some non-heme Fe enzymes, thus protecting these enzymes against Fe-catalyzed oxidative damage.[16]
Since Fe is clearly a precious commodity, bacteria have evolved a remarkable array of mechanisms to scavenge this nutrient from the environment. In this Concepts paper, we review recent progress in our understanding of Fe uptake by bacterial pathogens that goes beyond the well-studied FeIII-tris-catecholate systems, exemplified by the FeIII-enterobactin (enterochelin) extensively characterized in E. coli (Figure 1).[17] The work reviewed here is not meant to be comprehensive, but instead highlights newly emerging features of Fe acquisition and the bacterial adaptive response to host efforts to restrict the availability of this essential metal.
Figure 1.
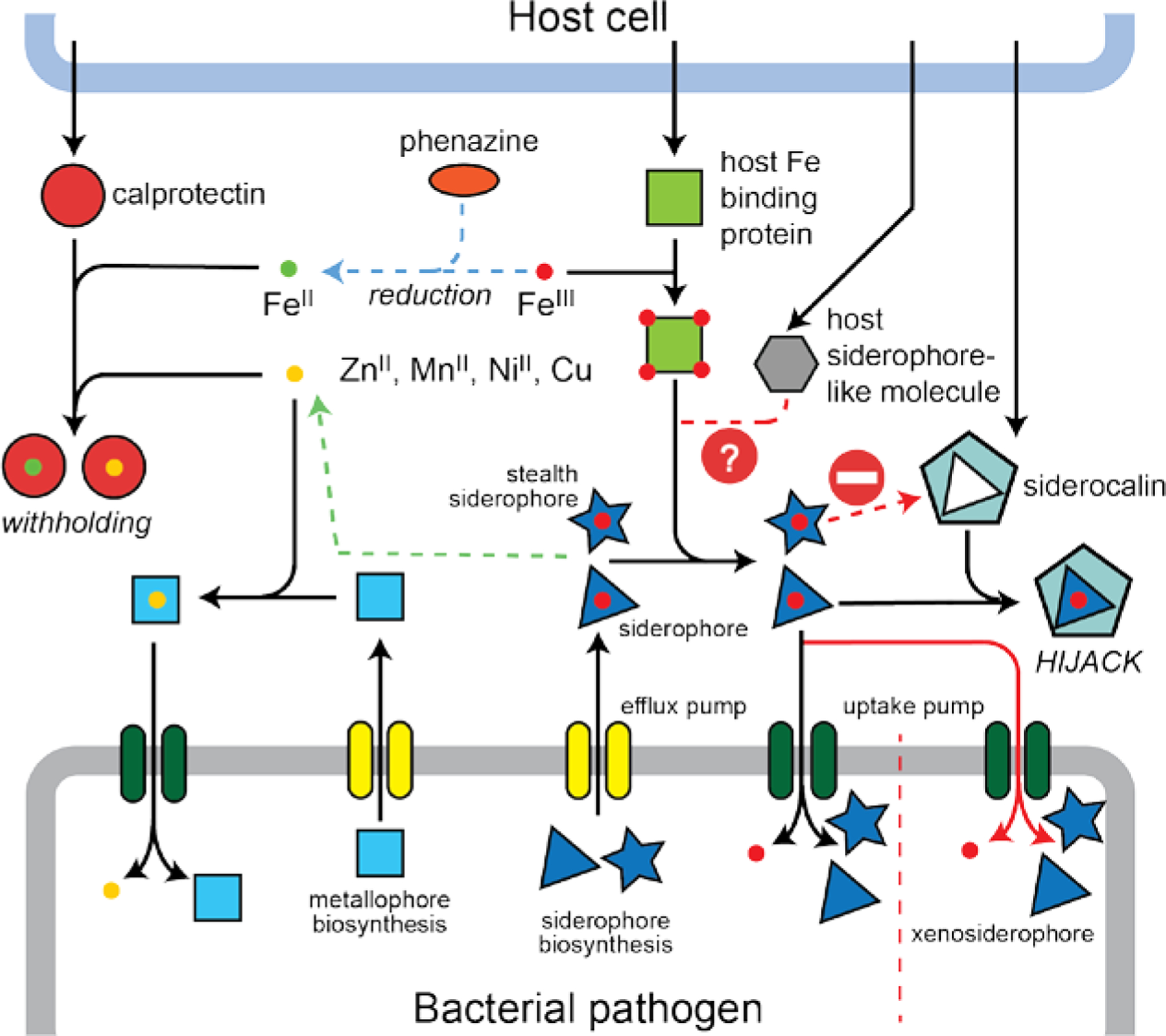
A schematic overview of the fight over nutrient Fe at the host (top)-bacterial pathogen (bottom) interface during infections. The green arrow is meant to indicate that a previously characterized FeIII-siderophore, e.g., yersiniobactin, from Yersinia spp. and uropathogenic E. coli, is capable of binding other transition metals (yellow sphere) in infected animals.[116a, 116c] A host siderophore-like molecule is the simple catecholamine, norepinephrine (NE) (Figure 2A) that is proposed to strip FeIII from transferrin in redox-dependent manner.[59]
2. The tris-catecholate complexes
2.1. General features and regulation
Siderophores are small molecule, high affinity FeIII chelators that harbor up to six coordinating donor atoms and typically form coordinatively saturated complexes with the metal.[18] Four structural classes of siderophores have thus far been identified, and these include the catecholate, hydroxamate, carboxylate and mixed-type siderophores distinguished by their distinct combinations of FeIII coordinating donors (Figure 2A).[19] The archetypal siderophore is enterobactin (Ent), a tris-catechol derivative of a cyclic triserine lactone. The metal coordinating motif is an ortho 2,3-dihydroxybenzoate (DHB) substituent coupled to the α-amino group of L-serine via an amide linkage. The cyclic trilactone core contains three ester linkages with three dihydroxybenzylserine (DHBS) metal coordinating groups that completely surround the FeIII atom (Figure 2A). FeIII binds with a stability constant of log KFe of ≈36 at pH 7.0 with a significant contribution of the chelate effect, and thus competes well with host mononuclear FeIII complexes of the major serum Fe transport protein, e.g., transferrin (log KFe of 22[20]) or lactoferrin. These ester linkages are susceptible to slow hydrolysis in aqueous solution [21] or in a reaction catalyzed by esterases (Figure 2A, top).
Figure 2.
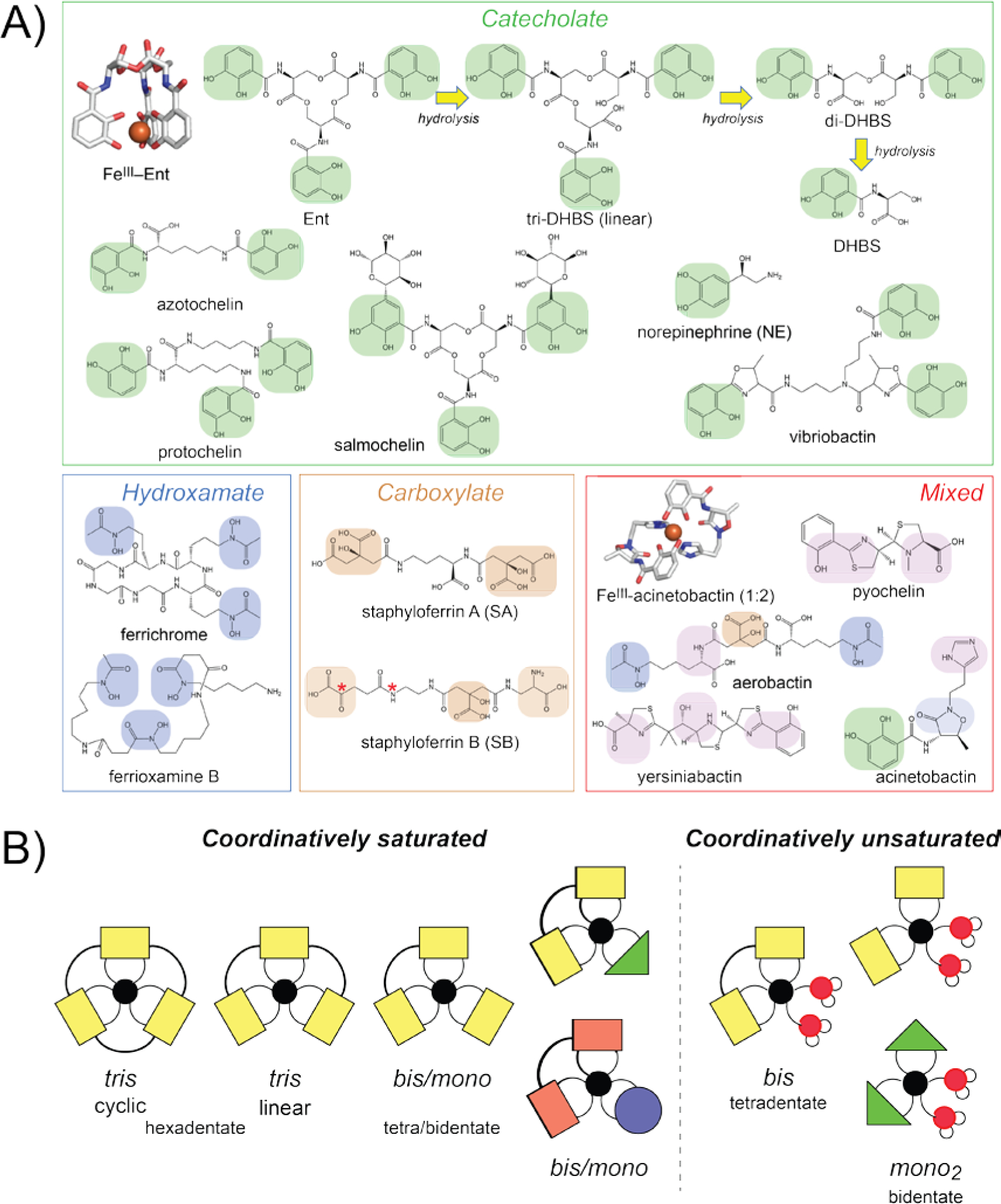
A) Chemical structures of the four major classes of FeIII siderophores, including the Ent hydrolysis products (top). This table is not meant to be exhaustive, but instead provides a snapshot of the structural diversity of the FeIII-chelating small molecules discussed in the text. The three-dimensional structures of the FeIII-Ent[26a] (pdb 6q5e; P. aeruginosa PfeA, see Figure 3B) and FeIII-acinetobactin[38] (pdb 6mfl; A. baumannii BauB, the SBP) are shown, derived from protein-ligand complexes. The α-hydroxy carboxylate and α-amino carboxylate bidentate chelating moieties of the citrate-derived polycarboxylate siderophores SA and SB are highlighted in brown. Red asterisks, although the linear structure of the SB is shown, these two atoms are known to cyclize to form a hemiaminal and resulting α-hydroxy carboxylate moiety in the active molecule.[31c] B) Cartoon illustrations of coordinatively saturated and coordinatively unsaturated FeIII-siderophore complexes. The different shapes and colors represent distinct bidentate FeIII-chelating (e.g., catechol, hydroxamate, carboxylate) moieties. Thick lines, covalent connection of the component bidentate coordinating units; thin lines, coordination bonds to the FeIII atom (black filled circle). Red filled circle symbol, solvent water molecule.
A typical Gram-negative bacterium like E. coli or Salmonella enterica sevovar Typhimurium harbors the biosynthetic machinery to synthesize Ent. The biosynthesis is typically controlled by Fur, which in E. coli regulates the expression of three Ent-related gene clusters, fepDGC-entS, fepB-entC, and fepA-fes.[22] The fepDGC genes encode an inner (cytoplasmic) membrane localized ATP-binding cassette (ABC) transporter to which FepB, the periplasmic solute binding protein (SBP), binds when bound to cargo FeIII-Ent, and is transported across this membrane using the energy of ATP hydrolysis (Figure 3A). entS encodes the apo-Ent effluxer that pumps Ent into the periplasm and ultimately into the extracellular milieu where it captures FeIII. FeIII-Ent then binds to an outer membrane (OM)-integrated 22-stranded β-barrel receptor (FepA), which via a TonB-ExbB-ExbD-dependent transport process, is brought into the periplasm (Figure 3A).[23] Once across the cytoplasmic membrane via FepBCDG, FeIII-Ent is subjected to esterase-catalyzed cleavage by Fes and/or reduction to FeII in the cytoplasm by resident ferric siderophore reductase(s); this makes FeII bioavailable to client biomolecules.[24] Other bacteria, including Acinetobacter baumannii and Pseudomonas aeruginosa, lack the machinery to biosynthesize Ent, but express an OM receptor that specifically brings FeIII-Ent into the periplasm (P. aeruginosa PfeA; Figure 3B).[25] Thus, Ent is a xenosiderophore for these organisms (Figure 1). This strategy of iron piracy is ubiquitous in polymicrobial niches, and highlights the importance to the bacterium of tapping any and all bioavailable sources of Fe in that niche.
Figure 3.
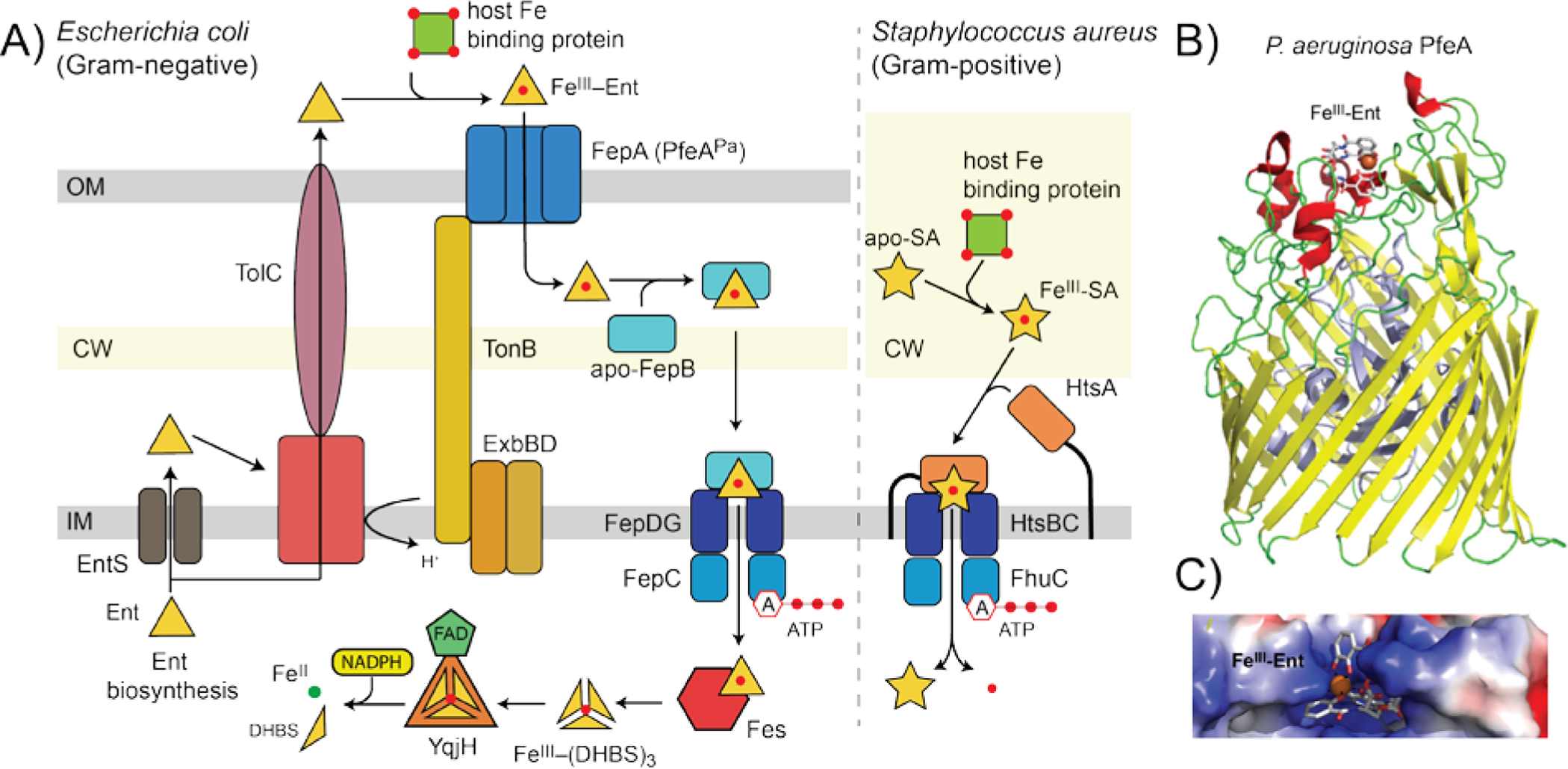
A) An expanded view of FeIII-Ent uptake (left) and FeIII-staphyloferrin A (SA)[30] (right) as representative of steps involved in FeIII-tris-catecholate uptake by Gram-negative bacteria and FeIII-polycarboxylate siderophore uptake in Gram-positive pathogens, respectively. Siderophore biosynthesis is not shown. Although not discussed here, TolC is a periplasm-spanning protein required to pump apo-Ent and presumably other apo-siderophores into the extracellular space.[117] B) Ribbon representation of the structure of the OM β-barrel receptor P. aeruginosa PfeA bound to FeIII-Ent (pdb 6q5c).[26a] C) Expanded view of the favorable electrostatics in the binding pocket for FeIII-Ent.
2.2. Siderophore-dependent FeIII acquisition across membranes
The ligand specificity of the FeIII-siderophore uptake appears to lie largely at the level of the OM receptor in Gram-negative organisms (Figure 3A). Although the structure of the ligand-free OM FeIII-Ent receptor E. coli FepA has been known for over twenty years,[23c] the structure of the FepA homolog from P. aeruginosa, PfeA, bound to FeIII-Ent provides new insights into the transport mechanism.[23c] This structure reveals a series of large extracellular loops that capture FeIII-Ent, coupled to an N-terminal globular plug domain that is inserted into the pore of the barrel (Figure 3B, C). FeIII-Ent binds to these extracellular loops that extend above the barrel itself, which function as “fingers on a hand” to grab the ligand. FeIII-Ent binding triggers a conformational change in the N-terminal plug domain that appears to push the FeIII-Ent complex to a second, intramembrane binding site. This allows the “Ton-box” on the plug domain to physically engage TonB which in turn triggers a relocation of plug domain to open a periplasmic gate to allow FeIII-Ent uptake into the periplasm.[26] Although both FepA and PfeA transport FeIII-Ent, PfeA also binds and transports other tris-catechol xenosiderophores, e.g., protochelin, as well as a bis-catechol, FeIII-azotochelin (Figure 2), which form complexes that are nearly isostructural with that of FeIII-Ent [26a] and thus represents a further example of Fe piracy (Figure 1). Other OM receptors reported as catecholate-type siderophore uptake systems in E. coli are CirA and Fiu, the latter of which is a candidate receptor of FeIII-DHBSn complexes (see Section 3).[27] The structure of Fiu reveals a selectively-gated cavity in the transporter that binds the Fiu substrate and appears to function in a P. aeruginosa PfeA-like two-step mechanism.[28]
In Gram-negative organisms, the FeIII-siderophore complex enters the periplasm after crossing OM where it is captured by an SBP and then transported into the cytoplasm via an ABC-type active transporter (Figure 3A, left).[29] In Gram-positive organisms, SBPs are anchored to the cell membrane coupled with an ABC transporter and are termed lipoproteins (Figure 3A, right).[29] In these organisms, the lipoprotein SBP actively discriminates among various FeIII-chelates, to form specific, high affinity, yet transport-competent complexes with their cargo.[30] For example, the major human pathogen Staphylococcus aureus synthesizes two citrate-derived polycarboxylate-type siderophores, staphyloferrin A (SA) and staphyloferrin B (SB) (Figure 2A)[31] The lipoprotein SBPs HtsA[32] and SirA[31a] bind with high affinity and specificity to only their cognate FeIII complexes formed by SA and SB, respectively (Figure 3A). All metal-transporting SBPs and lipoproteins, including those that transport FeIII-siderophore complexes, are members of the class III (cluster A) clade in which the two internally duplicated “lobes” are connected by a long α-helix or “brace” helix, with the cavity between the lobes defining the ligand binding pocket (see Figure 4B, below).[29] Ligand binding to class III SBPs results in little or no overall conformational change, unlike other SBP classes.[33] E. coli FepB, like FepA, has high specificity for FeIII-Ent[34] and even a linear tris-catecholate FeIII-siderophore such as agrobactin cannot be transported by FepB.[35]
Figure 4.
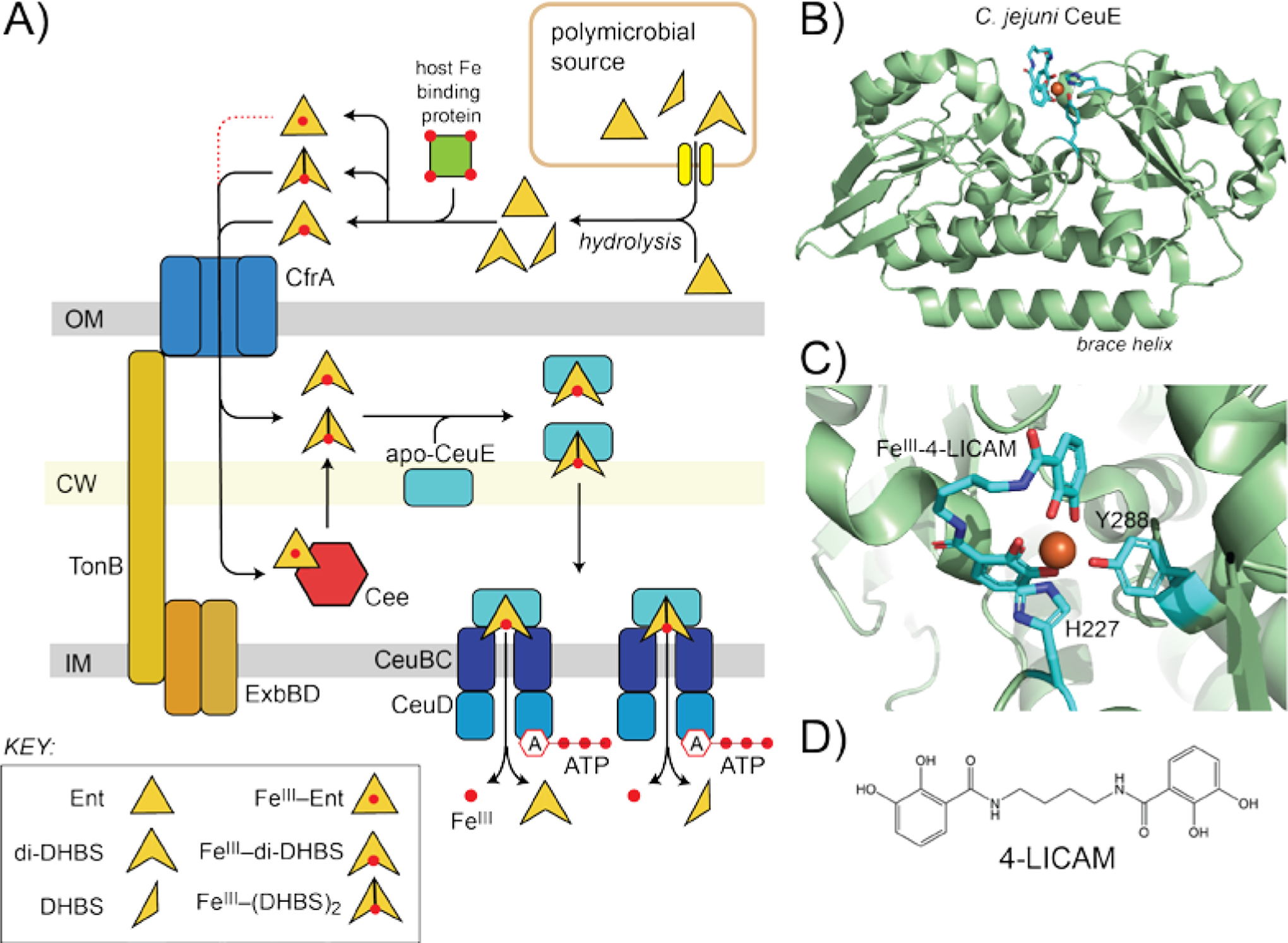
A pictorial representation of the uptake of enterobactin (Ent) hydrolysis products by Ceu system in C. jejuni. A) Overall process. B) Ribbon representation of the structure of the FeIII-4-LICAM complexed to the SPB CeuE (pdb 5a1j).[65] C) Close-up of the first coordination sphere around the FeIII, with CeuE-derived ligands H227 and Y288 shown coordinating the metal. D) Chemical structure of the synthetic tetradentate bis-catechol, 4-LICAM.[65]
Analogous transporters have been described for other siderophore classes in many human pathogens (Figure 2A). These include the E. coli ferrichrome (hydroxamate) uptake system, composed of the OM receptor FhuA and the FeIII-ferrichrome binding protein FhuD.[36] In the Gram-negative pathogen, A. baumannii, a mixed type siderophore FeIII-acinetobactin is first transported across the OM as 1:1 or 1:2 FeIII-(pre)acinetobactin complexes through BauA,[37] which is then bound by the SBP, BauB, as a 1:2 FeIII-acinetobactin complex (Figure 2).[38] There is evidence in some organisms that periplasm-resident esterases might process (hydrolyze) cyclic siderophores once brought across the OM, as shown for the tris-lactone esterase IroE which required for linear salmochelin uptake in uropathogenic E. coli.[24a] The extent to which this is generally true is not known, and competing models for IroE function exist, including hydrolysis of the metal-free tris-catecholate prior to export.[39]
2.3. The intracellular fate of FeIII-siderophore complexes
FeIII release once inside the cytoplasm is generally accomplished by two general strategies. These are covalent modification or hydrolysis of, for example, the tris-lactone siderophore, and/or reduction of the bound FeIII to FeII. Both lower metal-chelate stability, and in the case of the reduction to FeII, greatly enhances Fe aqueous solubility (Figure 3A).[40] In E. coli for example, the Fur-regulated cytoplasmic FeIII-Ent esterase Fes hydrolyzes the tris-lactone ester bonds to generate DHBS monomers as the final product (Figure 2A, top).[39] Although each DHBS harbors the bidentate catechol motif, the complex is less thermodynamically stable than FeIII-Ent. YqjH is a widespread NADPH, FAD-dependent FeIII reductase in E. coli capable of promoting FeII release from a variety of tris-catecholates including Ent, vibriobactin and aerobactin;[41] others have been characterized in S. aureus.[40] The reduction efficiency for FeIII bound to bidentate or simple organic ligands, such as DHBS and dicitrate, is high thus increasing the bioavailability of FeII in the cytoplasm.[41] These iron release mechanisms mentioned above are generally transcriptionally controlled by the global iron response regulator, Fur. YqjH is regulated by both Fur and YqjI, a transcriptional regulator that senses nickel toxicity in E. coli, as expected since nickel toxicity is known to disrupt Fe homeostasis.[42]
A cytoplasmic membrane associated [2Fe-2S] cluster protein, E. coli FhuF, is capable of reducing FeIII-hydroxamate siderophore complexes, including those formed with ferrichrome and ferrioxamine B (Figure 2).[43] In addition, there is evidence to suggest that one of the N-hydroxyl groups of ferrichrome is subject to acetylation after FeIII reduction so as not to negatively impact Fe speciation in the cytoplasm; the acetylated ferrichrome is then effluxed into the extracellular space where it is slowly hydrolyzed.[44] In this way, ferrichrome is recycled so as to maximize the benefit of energy-intensive de novo siderophore biosynthesis.
3. Coordinatively unsaturated bis- and mono-catecholate FeIII complexes as Fe sources
3.1. General considerations
In order to form high affinity complexes with FeIII, a typical siderophore forms coordinate-covalent bonds with all six donor atoms derived from a single molecule, like those described for the cyclic tris-catecholate Ent (Figure 2A,B). However, atypical FeIII complexes can also be used by bacterial pathogens to acquire Fe. These can be operationally grouped into two groups: coordinatively saturated and coordinatively unsaturated FeIII complexes (Figure 2B). Coordinatively saturated FeIII complexes incorporate six donor atoms derived from one, two or three liganding molecules and thus might have mixed ligand donor sets (Figure 2B, left), an early example of which is the FeIII complex formed by pyochelin (tetradentate) and cepabactin (a bidentate, monomeric hydroxamate ligand) in Pseudomonas cepacia.[45] Coordinatively unsaturated complexes, in contrast, are formed by one tetradentate dimer molecule or by two bidentate monomers with the remaining two coordination sites occupied by solvent (Figure 2B, right). Even though these coordinatively unsaturated FeIII complexes are characterized by lower overall FeIII stability constants, they still function as biologically important FeIII scavengers. This structural diversity may offer significant growth advantages in specific tissue niches, particularly for those bacteria that do not synthesize their own siderophores. These bacteria include the obligate human pathogens Neisseria meningitidis and Neisseria gonorrhoeae,[46] the intestinal pathogen Campylobacter jejuni[47] and two respiratory pathogens capable of colonizing the human nasopharynx, Streptococcus pneumoniae and Haemophilus influenzae.[48]
3.2. Coordinatively unsaturated tetra- and bidentate catecholate siderophores
The most well-studied coordinately unsaturated FeIII-siderophores are the tetradentate and the bis-catecholate-type siderophores (Figure 2A). These siderophores include, but are not limited to, the endogenously synthesized bis-catecholates azotochelin and azotobactin d from Azotobacter vinelandii,[49] pyochelin from Pseudomonas ssp.[50] and amonabactin from Aeromonas spp.[51] The hydrolysis products of Ent including linear tri-DHBS, di-DHBS and DHBS monomers (Figure 2A) are also present, and although their origin within a complex milieu of FeIII-siderophores at infection sites[52] is often not known, they allow bacteria to grow under iron-limited conditions.[27b, 53] In addition to these siderophore hydrolysis products of likely bacterial origin, other host-derived candidate FeIII chelators are the monomeric catechols, e.g., the catecholamines, human stress hormones that localize to sites of infection[54] at concentrations approaching 10 μM.[55] The catecholamine norepinephrine (NE) (Figure 2A) is known to enhance the proliferation of a number of bacterial pathogens, including S. pneumoniae, under Fe-limited growth conditions,[47b, 56] particularly in tissue niches where transferrin is the primary available iron source.[57] Although some organisms, including Neisseria spp. and H. influenzae can extract FeIII from transferrin directly via an OM receptor,[46, 58] others cannot. It is thought that the auto-oxidation of NE reduces FeIII to FeII, thus triggering iron release from transferrin;[59] the released Fe is then re-oxidized to FeIII upon capture by NE. Consistent with this, it is known that catecholamines form 2:1 or 3:1 (Figure 2B) FeIII-catecholamine complexes at physiological pH.[60] The generality of these findings and their relevance during infections is not yet known. To explore this further, the metabolic fate of these catecholates will have to be elucidated as described in the context of FeIII-bacillibactin metabolism in Bacillus subtilis.[61]
3.3. Iron acquisition via coordinately unsaturated tetra- and bidentate FeIII complexes
How do these simple, coordinately unsaturated FeIII complexes get into cells? Recent studies confirm that catecholate siderophore FeIII complexes cross the OM via a TonB dependent OM receptor CfrA in Campylobacter ssp.; as expected, inactivation of CfrA strongly impairs norepinephrine-dependent growth (Figure 4A).[47b, 56a, 56b, 62] A periplasmic esterase Cee in C. jejuni, capable of hydrolyzing Ent to DHBS monomers analogous to that carried out by the cytoplasmic Ent esterase Fes in E. coli, [63] becomes essential when FeIII-Ent is the sole iron source in culture, consistent with the hypothesis that periplasmic hydrolysis of Ent is required for FeIII uptake in this organism.[63] This finding predicts the presence of a periplasmic SBP that is specific for coordinately unsaturated FeIII-NE and FeIII-Ent hydrolysis products localized to the cytoplasmic membrane (see Figure 2B, right); this is, in fact, CeuE, from the ABC transporter, Ceu.[64] Although the structure of CeuE (Figure 4B) reveals a positively charged, ligand binding pocket reminiscent of the FeIII-Ent binding pocket of E. coli FepB or P. aeruginosa PfeA,[56c] CeuE binds FeIII-Ent relatively weakly, while coordinatively unsaturated FeIII-di-DHBS or FeIII-[DHBS]2 complexes bind with far higher affinity.[56c] The crystal structures of CeuE bound to a series of 1:1 FeIII complexes of an artificial tetradentate, bis-catecholate complex, 4-LICAM, establish why: the two open FeIII coordination sites on one side of the bis-catecholate chelate are occupied by a conserved tyrosine (Y288), buried in the binding pocket, and a conserved histidine (H227) derived from a flexible loop that covers the binding pocket (Figure 4C,D).[56c, 65]
This FeIII acquisition system in C. jejuni establishes the physiological importance of coordinatively unsaturated FeIII-siderophore complexes as nutritional iron sources. In fact, CeuE-type SBPs that conserve both Tyr and His ligands are found in Gram-negative and Gram-positive pathogens.[56c] This includes Vibrio cholerae VctP[66] which is known to bind the FeIII complex of a tetradentate bis-catecholate, salmochelin S1, preferentially over hexadentate siderophores. A similar CueE-type SBP, SstD, is found in Staphylococcus aureus and has been shown to be required for Ent-dependent FeIII acquisition and catecholamine-stimulated growth under severely iron-restricted conditions.[31b] This occurs despite the fact that both V. cholerae and S. aureus biosynthesize their own siderophores (Figure 2). The capacity to utilize hydrolysis products of tris-catecholate siderophores in polymicrobial niches is likely important, since intact siderophores may be limiting due to scavenging by other bacteria or sequestration by host siderocalin (Section 5).[56c, 66]
4. FeII uptake
4.1. FeII bioavailability at the host-pathogen interface
Although FeIII has long been considered the nutritionally important oxidation state of Fe at the host-pathogen interface, FeII can also be bioavailable particularly in anaerobic and/or acidic niches, both of which would enhance the solubility of FeII.[67][68] However, FeII can be accessible in other microenvironments as well. For example, extracellular FeII bioavailability may be increased by redox active metabolites like phenazines (see Figure 1) that are secreted by pathogens like P. aeruginosa and are capable of reducing FeIII to FeII.[68a, 69] Weakly complexed FeII can diffuse freely into the periplasm through outer membrane porins in Gram-negative organisms; alternatively, a ferric reductase anchored to the cytoplasmic membrane functions to reduce periplasmic FeIII to FeII,[70] which is then transported into the cytoplasm by the Feo system (Section 4.2). The source of electrons for FeIII reduction by these ferrireductases could be heme,[70b] NADH[70a] or flavins.[71] In another variation on FeII uptake, the obligate intravacuolar pathogen Legionella pneumophila directly accesses the labile cytoplasmic FeII pool of the host cell by inserting an FeII selective transmembrane importer into the vacuolar membrane; once enriched inside the vacuole, FeII is then likely imported by the Feo transporter (Section 4.2).[72]
4.2. Feo system for FeII uptake
The major bacterial system that transports FeII across the cytoplasmic membrane is the Feo system (Figure 5A).[73] The canonical feo operon is comprised of three genes, named feoABC and is Fur regulated; however, the core transporter is FeoAB since FeoC is not encoded by the vast majority of feo operons.[74] FeoA is a small, cytoplasmic β-rich protein, while FeoB is a membrane protein typically comprised of three domains: an N-terminal cytoplasmic G protein domain, a GDP dissociation inhibitor domain (essentially a linker domain that connects the G domain to the transmembrane domain) and the transmembrane domain itself, thought to form the FeII transport channel.[73] FeoB is anticipated to couple FeII transport with GTP hydrolysis with FeoA and FeoC functioning as accessory proteins in this process.[73–75]
Figure 5.
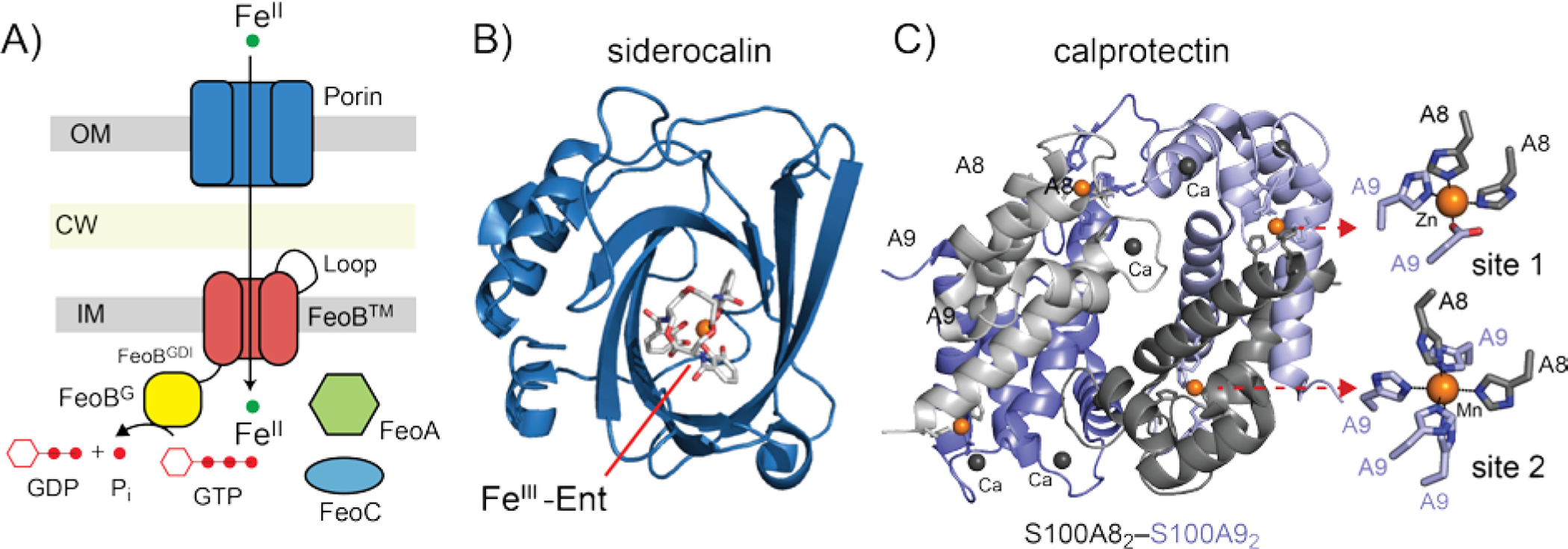
A) Schematic illustration of FeII FeoB uptake system, with the three domains of FeoB indicated. See text for details. B, C) Host-derived weapons that limit the bioavailability of FeIII and FeII, respectively, at infection sites. B) K125A derivative of human siderocalin bound to FeIII-Ent (pdb 3cmp).[118] FeIII-Ent binds to positively charged calyx in the center of the β-barrel where favorable electrostatics govern the affinity of the complex.[119] C) Calprotectin. Ribbon representation of the structure of the S100A82–S100A92 heterotetramer is shown (pdb 4ggf), with an expanded view of the site 1 and site 2 transition metal coordination sites, bound to ZnII and MnII respectively, in this structure (right).[89] The locations of the four CaII binding sites are also shown.
Metal specificity of the Feo system.
FeoB was originally discovered by characterization of iron transport mutants that arose following the addition of antibiotics, which often generate intracellular ROS in the presence of Fe.[73, 76] Mutants that had low internal Fe and thus survived the treatment were isolated and feo locus was identified.[76] The specificity for FeII has generally been established by culturing siderophore biosynthesis deficient mutants under conditions where FeII speciation is favored. For example, in studies of V. cholerae, ascorbate added to the growth medium was found to stimulate 55Fe uptake in a Feo-dependent manner, and thus it was concluded that Feo was specific for FeII.[77] However, FeoB homologs in Porphyromonas gingivalis and Clostridium perfingens impact MnII uptake as well, but the extent to which this characterizes other bacterial systems is not known.[78] Clearly, Fe-specific FeoB proteins from E. coli and Klebsiella pneumoniae are more closely related than are Fe-only vs. Fe-Mn FeoB2 transporters (88 vs. 31% sequence identity), but the origin for this apparent relaxed metal specificity in the latter is unknown. The metal specificity of Feo transporters, particularly those present as second copies in genomes, clearly warrants further investigation.
The Feo system and virulence.
FeoB is known to be crucial for the virulence of several microbial pathogens. C. jejuni and Helicobacter pylori are expected to encounter significant FeII in the relatively acidic and oxygen-limited conditions of the upper gastrointestinal tract,[74] and feoB mutants in C. jejuni and H. pylori have lower viability and decreased colonization efficiencies in the piglet intestine and the mouse stomach, respectively.[79] The same is true of Salmonella in infected macrophages.[80] The feoB transcript has been detected in P. aeruginosa extracted from the lungs of patients suffering from cystic fibrosis.[68a] FeoB may act in concert with other Fe uptake systems in some bacterial species such as Yersinia pestis and Francisella tularensis which demonstrate reduced viability only when a feoB deletion is accompanied by deletion of other FeIII uptake systems.[81] Although most studies that investigate the impact of Feo system on bacterial virulence have focused on FeoB, a number of reports reveal that a feoA deletion can result in a significant reduction of Fe uptake and bacterial growth in several pathogens including V. cholerae, Salmonella enterica and A. baumannii, although the precise function of FeoA remains to be clarified.[82] For example, in S. enterica, both feoA and feoB deletion strains result in a ≈3-fold decrease in ferrous uptake, but only when other known FeII uptake systems are deleted. A feoA-feoB double mutant could be rescued from the defect in FeII uptake only by complementation of both genes, and FeoA interacts with FeoB in vivo, consistent with a functionally important interaction.[82b]
5. Mechanisms of Fe sequestration by the host
5.1. FeIII sequestration by the host
The host deploys siderocalin (lipocalin 2) at sites of infection to sequester FeIII-Ent and some other carboxymycobactin siderophores[83] thereby withholding FeIII from the pathogen. Abundant host proteins lactoferrin and transferrin also play a role in sequestering extracellular FeIII. Siderocalin belongs to lipocalin family of proteins characterized by eight-stranded antiparallel β-barrel harboring a calyx where the FeIII-siderophore complex binds (Figure 5B). Three positively charged side chains form a cation-π interaction with each of the three catechol rings of the tris-catecholate siderophore and there are additional hydrogen bonding interactions involving a highly conserved tyrosine residue (Figure 5B).[84] In the ongoing “arms race” for Fe in the infected host,[58, 85] some bacteria have adapted to siderocalin-mediated FeIII-Ent sequestration by synthesizing modified Ent molecules that cannot be bound by siderocalin, and are therefore designated stealth siderophores (Figure 1). These include salmochelin, which is essentially Ent derivatized with a bulky C-linked glucose substituent on the catechol moieties, and petrobactin, produced by Bacillus anthracis, which incorporates a 3,4 catechol instead of 2,3 catechol characteristic of all other catecholate family siderophores (see Figure 2A).[83]
5.2. FeII sequestration by the host
Although originally described as playing a role in manganese and zinc restriction at sites of infection,[86] the multi-metal withholding protein calprotectin (Figure 5C) is now believed to play an important role in FeII sequestration,[87] certainly in liquid culture-based assays,[4b, 88] and perhaps at the host-pathogen interface. Secreted by neutrophils and highly abundant at infection sites, calprotectin is a heterotetramer of two S100 family proteins (S100A8 and S100A9; α2β2) (Figure 5C).[89] Two distinct metal coordination sites present at its αβ heterodimer interface exhibit distinct metal specificities and are strongly activated to coordinate transition metals by CaII, which is abundant in the extracellular environment.[90] The His3Asp site 1 binds ZnII with picomolar affinity and is generally conserved in other S100-family metal-chelating proteins. The His6 site 2, on the other hand, is a “jack of all trades” and coordinates FeII, MnII, ZnII and NiII to form what are believed to be isostructural, His6 octahedral coordination complexes[91] of high thermodynamic and/or kinetic stability.[92] FeII binding by calprotectin shifts the redox speciation of Fe from FeIII to FeII in an aerobic environment in the absence of a extracellular reductant,[93] and is capable of withholding Fe from a number of Gram-positive and Gram-negative bacteria including P. aeruginosa, E. coli, S. enterica and K. pneumoniae.[88] [4b, 88, 92]
6. Physiological adaptation to host-mediated FeII or FeIII sequestration by the pathogen
A primary physiological response to severe Fe restriction is induction of the Fur regulon. Fur and other Fe-sensing transcriptional regulators are global regulators in many organisms, and thus goes well beyond the regulation of genes encoding Fe acquisition systems to a significant re-programming of cellular metabolism. An important mechanistic feature of this re-programming is the expression of one or more Fur-regulated small regulatory RNAs (sRNAs) that inhibit the synthesis of non-essential Fe requiring proteins and enzymes, which for P. aeruginosa is important in murine lung infections.[6b, 7, 94] This results in post-transcriptional downregulation of genes that encode for cell-abundant Fe enzymes, including succinate dehydrogenase and cytochromes, which optimizes use of scarce bioavailable iron to metallate essential enzymes, a prioritization mechanism termed Fe sparing.[95] In B. subtilis, sRNA-regulated Fe sparing is effectively an adaptation of “last resort” and is within the final “wave” of Fur-induced derepression observed upon increasingly severe degrees of Fe limitation.[6a] Some other effects include repression of chemotaxis and motility genes that has been observed during Fe limitation in A. baumannii.[96]
A major metabolic shift that may well represent a widespread, “frontline” adaptive response to Fe restriction is a shift toward flavin-anchored metabolic pathways, and away from a reliance on Fe-S-containing ferredoxins as cellular reductants. As ferredoxins and flavodoxins possess similar reduction potentials, flavodoxins are capable of substituting for ferredoxin as electron carriers (reductants) under conditions of Fe limitation, even for highly specific transformations including dehydration reactions in Acidaminococcus fermentans[97] and the desaturation of fatty acids in B. subtilis.[98] Indeed, genes encoding flavodoxins are highly transcriptionally induced under Fe limitation in Clostridium acetobutylicum[99] and in the strict anaerobe and intestinal pathogen, Clostridium difficile.[100] In many species of bacteria and algae, flavodoxin protein levels are detected only under conditions of Fe limitation [98c] and this is true for the flavodoxin WrbA in A. baumannii as well;[4b] indeed, the ratio of cellular flavodoxin to ferredoxin is often considered a benchmark for Fe limitation in marine phytoplankton.[101] In order to fully activate this metabolic switch, a concomitant prioritization of flavin biosynthesis must occur, which can result in an accumulation of riboflavin in the culture supernatant.[99, 102] The regulatory mechanisms that drive this switch, however, likely differ among organisms. In some bacteria, riboflavin biosynthesis is directly Fur-regulated, [95, 99, 102–103] while in others, this adaptive response to Fe limitation is not so straightforward.[104]
Recent work reveals that calprotectin induces an overlapping ZnII and Fe starvation response in A. baumannii, as determined by changes in both the transcriptome and the soluble proteome.[4b] These changes include transcriptional upregulation of the FeII importer FeoAB, several FeIII siderophore OM receptors, acinetobactin biosynthetic proteins and decreased cellular abundance of a number of Fe proteins, including the hemoprotein cytochrome b562 and the major ferredoxin FdxB, alongside induction of the entire Zur regulon.[4b, 105] More importantly, these studies uncover a strongly integrated coupling of extreme Fe/ZnII restriction and de novo riboflavin biosynthesis and perhaps other metabolic processes. To illustrate, the first enzyme in the convergent de novo flavin biosynthesis pathway is encoded by ribB which catalyzes the conversion of ribulose-5-phosphate to 3,4-dihydroxy-2-butanone 4-phosphate (DHBP) (DHBP synthase or RibB). In A. baumannii, a novel RibB fusion protein, RibBX, an active DHBPS, becomes detectable only under conditions of calprotectin stress; RibBX appears to enhance flavin biosynthesis by dramatically increasing the flavin toxicity “set-point” thus working around riboflavin-sensing riboswitch-mediated inhibition of authentic ribB expression.[4b] Furthermore, the Zur-inducible, candidate ZnII metallochaperone ZigA[106] becomes cell-abundant under calprotectin stress, and a calprotectin-treated ΔzigA A. baumannii strain grows poorly and fails to sustain cellular flavin levels. Riboflavin supplementation partially rescues this phenotype. How ZigA sustains flavin biosynthesis in A. baumannii is unknown, but it is known that this pathway harbors two obligate ZnII metalloenzymes, including the rate-determining enzyme, GTP cyclohydrolase II, encoded by ribA. It is interesting to note that the Zur-regulated ZigA homolog in Bacillus subtilis, ZagA, appears to sustain folate biosynthesis under conditions of zinc restriction, by interacting with an obligatory ZnII metalloenzyme GTP cyclohydrolase I, encoded by folE.[107]
Detailed studies like these raise the interesting possibility that in some microbial pathogens, the adaptive response to ZnII limitation may be more strongly interwoven with the Fe-limitation response than previously anticipated,[108] since both stresses may be present concurrently in the host, particularly for extracellular pathogens, and therefore potentially negatively impacts nearly the entire metalloproteome. For example, the biosynthesis of the broad-spectrum transition-metal binding metallophore, staphylopine, in Staphylococcus aureus, is under dual transcriptional control by both Fur and Zur.[108] In addition, low Zn or low Fe jointly regulate the expression of a “hybrid” ABC-family metal transporter in Corynebacterium diptheriae.[109] Detailed investigations of low Zn-low Fe crosstalk should yield new insights in a systems-level response to co-incident host-derived ZnII and Fe restriction during infections.
7. Concluding Remarks
In this Concepts report, we highlight new “variations on a theme” of pathogen Fe acquisition that diverge from the well-studied E. coli FeIII-Ent model. We place these new studies in the context of recent observations that begin to challenge the FeIII-centric view of Fe acquisition at even aerobic or oxic/anoxic interfacial sites of infection, that collectively suggest an important role for FeII acquisition by the pathogen in these niches. In these sites, calprotectin and perhaps other small molecule divalent metal-specific chelators yet to be discovered, e.g., those analogous to the broad-spectrum nicotianamine-like metallophores, staphylopine and pseudopaline from S. aureus and P. aeruginosa, respectively,[110] may well function here. Staphylopine biosynthesis, efflux, metal capture, and holo metallophore uptake precisely parallel the FeIII-siderophore systems described here (Figures 3–4).[111] Staphylopine, whose biosynthesis is strongly induced by calprotectin stress,[112] does in fact bind FeII but with a stability constant (log KFe 12.1) nearly 1000-fold weaker than ZnII (log KZn 15.0).[110a] Consistent with this, staphylopine outcompetes calprotectin for nutrient ZnII in S. aureus during infections.[111a] and the closely related pseudopaline functions to capture ZnII in P. aeruginosa cultures.[110b] It is interesting to note, in the context of a continuously evolving host-pathogen “fight over metals”,[58] that an OM-localized calprotectin receptor in N. gonorrhoeae is capable of hijacking ZnII from calprotectin for use as a nutritional source.[113] Although characterized in ZnII uptake, FeII-bound calprotectin might function in this way as well.
Multi-modal mass spectrometry-based imaging approaches will continue to play an important role in future efforts to detect and quantify the chemical constituents linked to transition metal (Fe, Zn) speciation at tissue sites of infection, particularly important in the context of polymicrobial communities.[114] Such an approach was recently used to map the tissue distribution of siderophores in animals infected with S. aureus as well as for the detection of secreted proteins, e.g., calprotectin,[86a, 115] at the host-pathogen interface.[52] Detailed mass spectrometry analysis of a pathogen secretome ex vivo or from infected animals may also be leveraged for the discovery of new metallophore-like small molecules bound to their biologically relevant metals, e.g., as previously illustrated for the virulence-associated metallophore yersiniabactin in uropathogenic E. coli infections of the urinary tract,[116] or extracellular ferric reductases that could be involved in enhancing FeII bioavailability at sites of infection. The “arms race” for Fe[58] in the context of broad host-mediated transition metal restriction continues unabated and new discoveries await.
Acknowledgements
We gratefully acknowledge financial support from the US National Institutes of Health (NIH grants R35 GM118157 and R01 AI101171) and from Indiana University.
Biographical Sketches

Prof. David Giedroc obtained his Ph.D. under the supervision of J. David Puett at Vanderbilt University. He then carried out postdoctoral research with the late Joseph E. Colman of Yale University, a pioneer in the field of bioinorganic chemistry and enzymology of zinc metalloenzymes. He then established his own research group, first at Texas A&M University (1988–2007), and then at Indiana University in Bloomington, where he is the Lilly Chemistry Alumni Distinguished Professor in the Department of Chemistry. His group takes a problem-oriented, multidisciplinary approach to understanding the physical and chemical biology of transition metal homeostasis and more recently, reactive sulfur species and hydrogen sulfide homeostasis, in bacterial cells. He is Chair of the Editorial Board of Metallomics, a publication of the Royal Society of Chemistry (UK), and a Fellow of the American Association for the Advancement of Science (AAAS), the Royal Society of Chemistry and the American Academy of Microbiology.

Yifan Zhang is a native of Shijiazhuang, China and earned his undergraduate degree in bioengineering from the Beijing Institute of Technology and an M.S. in physical biochemistry from the Illinois institute of Technology with Prof. Andrew Howard. He is currently a Ph.D. student in Biochemistry at Indiana University in the Giedroc group, where he studies iron homeostasis in Streptococcus pneumoniae.

Dr. Sambuddha Sen is a native of Kharagpur, India. He earned his B.Sc. in Chemistry from Jadavpur University, Kolkata, India, and his M.Sc. in Chemistry from the Indian Institute of Technology, Kanpur, India in materials chemistry with Dr. Debabrata Goswami. He took his Ph.D. in Chemistry from The Ohio State University under the supervision of Prof. James Cowan where he studied Fe-S cluster maturation and trafficking mechanisms in eukaryotic cells. Dr. Sen is a postdoctoral research associate in the Giedroc group interested in transition metal restriction in Acinetobacter baumannii.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Andreini C, Putignano V, Rosato A, Banci L, Metallomics 2018, 10, 1223–1231. [DOI] [PubMed] [Google Scholar]
- [2].Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL 2nd, Jenney FE Jr., Lancaster WA, Praissman JL, Shanmukh S, Vaccaro BJ, Trauger SA, Kalisiak E, Apon JV, Siuzdak G, Yannone SM, Tainer JA, Adams MW, Nature 2010, 466, 779–782. [DOI] [PubMed] [Google Scholar]
- [3].Waldron KJ, Rutherford JC, Ford D, and Robinson NJ, Nature 2009, 460, 823–830. [DOI] [PubMed] [Google Scholar]
- [4].a) Andreini C, Banci L, Bertini I, Rosato A, J Proteome Res 2006, 5, 3173–3178; [DOI] [PubMed] [Google Scholar]; b) Wang J, Lonergan ZR, Gonzalez-Gutierrez G, Nairn BL, Maxwell CN, Zhang Y, Andreini C, Karty JA, Chazin WJ, Trinidad JC, Skaar EP, Giedroc DP, Cell Chem Biol 2019, 26, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weinberg ED, JAMA 1975, 231, 39–41. [DOI] [PubMed] [Google Scholar]
- [6].a) Pi H, Helmann JD, Proc Natl Acad Sci U S A 2017, 114, 12785–12790; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Masse E, Gottesman S, Proc Natl Acad Sci U S A 2002, 99, 4620–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Reinhart AA, Nguyen AT, Brewer LK, Bevere J, Jones JW, Kane MA, Damron FH, Barbier M, Oglesby-Sherrouse AG, Infect Immun 2017, 85, e00764–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Osman D, Martini MA, Foster AW, Chen J, Scott AJP, Morton RJ, Steed JW, Lurie-Luke E, Huggins TG, Lawrence AD, Deery E, Warren MJ, Chivers PT, Robinson NJ, Nat Chem Biol 2019, 15, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guan G, Pinochet-Barros A, Gaballa A, Patel SJ, Arguello JM, Helmann JD, Mol Microbiol 2015, 98, 787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Grass G, Otto M, Fricke B, Haney CJ, Rensing C, Nies DH, Munkelt D, Arch Microbiol 2005, 183, 9–18; [DOI] [PubMed] [Google Scholar]; b) Sankari S, O’Brian MR, J Biol Chem 2014, 289, 16498–16507; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pi H, Helmann JD, Metallomics 2017, 9, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee JW, Helmann JD, Biometals 2007, 20, 485–499. [DOI] [PubMed] [Google Scholar]
- [12].Giel JL, Nesbit AD, Mettert EL, Fleischhacker AS, Wanta BT, Kiley PJ, Mol Microbiol 2013, 87, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Verstraete MM, Morales LD, Kobylarz MJ, Loutet SA, Laakso HA, Pinter TB, Stillman MJ, Heinrichs DE, Murphy MEP, J Biol Chem 2019, 294, 11622–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) VanderWal AR, Makthal N, Pinochet-Barros A, Helmann JD, Olsen RJ, Kumaraswami M, Infect Immun 2017, 85; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brenot A, Weston BF, Caparon MG, Mol Microbiol 2007, 63, 1185–1196. [DOI] [PubMed] [Google Scholar]
- [15].a) Turner AG, Djoko KY, Ong CY, Barnett TC, Walker MJ, McEwan AG, Biochem J 2019, 476, 595–611; [DOI] [PubMed] [Google Scholar]; b) Helmann JD, J Biol Chem 2014, 289, 28112–28120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Sobota JM, Imlay JA, Proc Natl Acad Sci U S A 2011, 108, 5402–5407; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anjem A, Imlay JA, J Biol Chem 2012, 287, 15544–15556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Johnstone TC, Nolan EM, J Am Chem Soc 2017, 139, 15245–15250; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Raymond KN, Dertz EA, Kim SS, Proc Natl Acad Sci U S A 2003, 100, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hider RC, Kong X, Nat Prod Rep 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- [19].Wilson BR, Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y, Trends Mol Med 2016, 22, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Martin RB, Savory J, Brown S, Bertholf RL, Wills MR, Clin Chem 1987, 33, 405–407. [PubMed] [Google Scholar]
- [21].Gomez-Bombarelli R, Calle E, Casado J, J Org Chem 2013, 78, 6868–6879. [DOI] [PubMed] [Google Scholar]
- [22].a) Lavrrar JL, Christoffersen CA, McIntosh MA, J Mol Biol 2002, 322, 983–995; [DOI] [PubMed] [Google Scholar]; b) Brickman TJ, Ozenberger BA, McIntosh MA, J Mol Biol 1990, 212, 669–682; [DOI] [PubMed] [Google Scholar]; c) Hunt MD, Pettis GS, McIntosh MA, J Bacteriol 1994, 176, 3944–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Schalk IJ, Mislin GL, Brillet K, Curr Top Membr 2012, 69, 37–66; [DOI] [PubMed] [Google Scholar]; b) Celia H, Noinaj N, Zakharov SD, Bordignon E, Botos I, Santamaria M, Barnard TJ, Cramer WA, Lloubes R, Buchanan SK, Nature 2016, 538, 60–65; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Buchanan SK, Smith BS, Venkatramani L, Xia D, Esser L, Palnitkar M, Chakraborty R, van der Helm D, Deisenhofer J, Nat Struct Biol 1999, 6, 56–63. [DOI] [PubMed] [Google Scholar]
- [24].a) Zhu M, Valdebenito M, Winkelmann G, Hantke K, Microbiology 2005, 151, 2363–2372; [DOI] [PubMed] [Google Scholar]; b) Neumann W, Sassone-Corsi M, Raffatellu M, Nolan EM, J Am Chem Soc 2018, 140, 5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Subashchandrabose S, Smith S, DeOrnellas V, Crepin S, Kole M, Zahdeh C, Mobley HL, mSphere 2016, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cornelis P, Dingemans J, Front Cell Infect Microbiol 2013, 3, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Moynie L, Milenkovic S, Mislin GLA, Gasser V, Malloci G, Baco E, McCaughan RP, Page MGP, Schalk IJ, Ceccarelli M, Naismith JH, Nat Commun 2019, 10, 3673; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zarate-Bonilla LJ, Del Portillo P, Saenz-Suarez H, Gonzales-Santos J, Barreto-Sampaio GE, Poutou-Pinales RA, Rey AF, Rey JG, Poult Sci 2014, 93, 221–230; [DOI] [PubMed] [Google Scholar]; c) Smallwood CR, Jordan L, Trinh V, Schuerch DW, Gala A, Hanson M, Shipelskiy Y, Majumdar A, Newton SM, Klebba PE, J Gen Physiol 2014, 144, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Braun V, Hantke K, Koster W, Met Ions Biol Syst 1998, 35, 67–145; [PubMed] [Google Scholar]; b) Hantke K, FEMS Microbiol Lett 1990, 55, 5–8. [DOI] [PubMed] [Google Scholar]
- [28].Grinter R, Lithgow T, J Biol Chem 2019, 294,19523–19534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Delepelaire P, Res Microbiol 2019, 170, 345–355. [DOI] [PubMed] [Google Scholar]
- [30].Cooper JD, Hannauer M, Marolda CL, Briere LA, Heinrichs DE, Biochemistry 2014, 53, 5060–5069. [DOI] [PubMed] [Google Scholar]
- [31].a) Grigg JC, Cheung J, Heinrichs DE, Murphy ME, J Biol Chem 2010, 285, 34579–34588; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Beasley FC, Marolda CL, Cheung J, Buac S, Heinrichs DE, Infect Immun 2011, 79, 2345–2355; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Madsen JL, Johnstone TC, Nolan EM, J Am Chem Soc 2015, 137, 9117–9127. [DOI] [PubMed] [Google Scholar]
- [32].Grigg JC, Cooper JD, Cheung J, Heinrichs DE, Murphy ME, J Biol Chem 2010, 285, 11162–11171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].de Boer M, Gouridis G, Vietrov R, Begg SL, Schuurman-Wolters GK, Husada F, Eleftheriadis N, Poolman B, McDevitt CA, Cordes T, Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stephens DL, Choe MD, Earhart CF, Microbiology 1995, 141, 1647–1654. [DOI] [PubMed] [Google Scholar]
- [35].Sprencel C, Cao Z, Qi Z, Scott DC, Montague MA, Ivanoff N, Xu J, Raymond KM, Newton SM, Klebba PE, J Bacteriol 2000, 182, 5359–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mademidis A, Koster W, Mol Gen Genet 1998, 258, 156–165. [DOI] [PubMed] [Google Scholar]
- [37].Moynie L, Serra I, Scorciapino MA, Oueis E, Page MG, Ceccarelli M, Naismith JH, Elife 2018, 7, e42270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bailey DC, Bohac TJ, Shapiro JA, Giblin DE, Wencewicz TA, Gulick AM, Biochemistry 2018, 57, 6653–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lin H, Fischbach MA, Liu DR, Walsh CT, J Am Chem Soc 2005, 127, 11075–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kobylarz MJ, Heieis GA, Loutet SA, Murphy MEP, ACS Chem Biol 2017, 12, 1778–1786. [DOI] [PubMed] [Google Scholar]
- [41].Miethke M, Hou J, Marahiel MA, Biochemistry 2011, 50, 10951–10964. [DOI] [PubMed] [Google Scholar]
- [42].Wang S, Wu Y, Outten FW, J Bacteriol 2011, 193, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Matzanke BF, Anemuller S, Schunemann V, Trautwein AX, Hantke K, Biochemistry 2004, 43, 1386–1392. [DOI] [PubMed] [Google Scholar]
- [44].Hartmann A, Braun V, J Bacteriol 1980, 143, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Meyer JM, Hohnadel D, Halle F, J Gen Microbiol 1989, 135, 1479–1487. [DOI] [PubMed] [Google Scholar]
- [46].Noinaj N, Easley NC, Oke M, Mizuno N, Gumbart J, Boura E, Steere AN, Zak O, Aisen P, Tajkhorshid E, Evans RW, Gorringe AR, Mason AB, Steven AC, Buchanan SK, Nature 2012, 483, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].a) Field LH, Headley VL, Payne SM, Berry LJ, Infect Immun 1986, 54, 126–132; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Naikare H, Butcher J, Flint A, Xu J, Raymond KN, Stintzi A, Metallomics 2013, 5, 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cheng W, Li Q, Jiang YL, Zhou CZ, Chen Y, PLoS One 2013, 8, e71451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].a) Knosp O, von Tigerstrom M, Page WJ, J Bacteriol 1984, 159, 341–347; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Palanche T, Blanc S, Hennard C, Abdallah MA, Albrecht-Gary AM, Inorg Chem 2004, 43, 1137–1152. [DOI] [PubMed] [Google Scholar]
- [50].Brandel J, Humbert N, Elhabiri M, Schalk IJ, Mislin GL, Albrecht-Gary AM, Dalton Trans 2012, 41, 2820–2834. [DOI] [PubMed] [Google Scholar]
- [51].a) Balado M, Souto A, Vences A, Careaga VP, Valderrama K, Segade Y, Rodriguez J, Osorio CR, Jimenez C, Lemos ML, ACS chemical biology 2015, 10, 2850–2860; [DOI] [PubMed] [Google Scholar]; b) Barghouthi S, Young R, Olson MO, Arceneaux JE, Clem LW, Byers BR, J Bacteriol 1989, 171, 1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Perry WJ, Spraggins JM, Sheldon JR, Grunenwald CM, Heinrichs DE, Cassat JE, Skaar EP, Caprioli RM, Proc Natl Acad Sci U S A 2019, 116, 21980–21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].a) Wyckoff EE, Allred BE, Raymond KN, Payne SM, J Bacteriol 2015, 197, 2840–2849; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wilkins TD, Lankford CE, J Infect Dis 1970, 121, 129–136; [DOI] [PubMed] [Google Scholar]; c) Wang CC, Newton A, J Bacteriol 1969, 98, 1142–1150; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rabsch W, Methner U, Voigt W, Tschape H, Reissbrodt R, Williams PH, Infect Immun 2003, 71, 6953–6961; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hancock RE, Hantke K, Braun V, Arch Microbiol 1977, 114, 231–239; [DOI] [PubMed] [Google Scholar]; f) Berner I, Greiner M, Metzger J, Jung G, Winkelmann G, Biol Met 1991, 4, 113–118. [DOI] [PubMed] [Google Scholar]
- [54].Freestone PPE, Sandrini SM, Haigh RD, Lyte M, Trends Microbiol 2008, 16, 55–64. [DOI] [PubMed] [Google Scholar]
- [55].a) Eisenhofer G, Kopin IJ, Goldstein DS, Pharmacol Rev 2004, 56, 331–349; [DOI] [PubMed] [Google Scholar]; b) Esler M, Jennings G, Korner P, Blombery P, Sacharias N, Leonard P, Am J Physiol 1984, 247, E21–28. [DOI] [PubMed] [Google Scholar]
- [56].a) Zeng X, Xu F, Lin J, Infect Immun 2009, 77, 5437–5448; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu F, Zeng X, Haigh RD, Ketley JM, Lin J, J Bacteriol 2010, 192, 4425–4435; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Raines DJ, Moroz OV, Blagova EV, Turkenburg JP, Wilson KS, Duhme-Klair AK, Proc Natl Acad Sci U S A 2016, 113, 5850–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].a) Neal CP, Freestone PP, Maggs AF, Haigh RD, Williams PH, Lyte M, FEMS Microbiol Lett 2001, 194, 163–169; [DOI] [PubMed] [Google Scholar]; b) Gonzales XF, Castillo-Rojas G, Castillo-Rodal AI, Tuomanen E, Lopez-Vidal Y, Microbiology 2013, 159, 2333–2341; [DOI] [PubMed] [Google Scholar]; c) Graziano TS, Closs P, Poppi T, Franco GC, Cortelli JR, Groppo FC, Cogo K, J Periodontal Res 2014, 49, 660–669; [DOI] [PubMed] [Google Scholar]; d) O’Donnell PM, Aviles H, Lyte M, Sonnenfeld G, Appl Environ Microbiol 2006, 72, 5097–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Barber MF, Elde NC, Science 2014, 346, 1362–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sandrini SM, Shergill R, Woodward J, Muralikuttan R, Haigh RD, Lyte M, Freestone PP, J Bacteriol 2010, 192, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jewett SL, Eggling S, Geller L, J Inorg Biochem 1997, 66, 165–173. [Google Scholar]
- [61].Pi H, Helmann JD, 2018.
- [62].Zeng X, Xu F, Lin J, FEMS Microbiol Lett 2013, 347, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zeng X, Mo Y, Xu F, Lin J, Mol Microbiol 2013, 87, 594–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Palyada K, Threadgill D, Stintzi A, J Bacteriol 2004, 186, 4714–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wilde EJ, Hughes A, Blagova EV, Moroz OV, Thomas RP, Turkenburg JP, Raines DJ, Duhme-Klair AK, Wilson KS, Sci Rep 2017, 7, 45941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wilde EJ, Blagova EV, Sanderson TJ, Raines DJ, Thomas RP, Routledge A, Duhme-Klair AK, Wilson KS, J Inorg Biochem 2019, 190, 75–84. [DOI] [PubMed] [Google Scholar]
- [67].Aron AT, Heffern MC, Lonergan ZR, Vander Wal MN, Blank BR, Spangler B, Zhang Y, Park HM, Stahl A, Renslo AR, Skaar EP, Chang CJ, Proc Natl Acad Sci U S A 2017, 114, 12669–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].a) Hunter RC, Asfour F, Dingemans J, Osuna BL, Samad T, Malfroot A, Cornelis P, Newman DK, mBio 2013, 4, e00557–13; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blank BR, Talukder P, Muir RK, Green ER, Skaar EP, Renslo AR, ACS Infect Dis 2019, 5, 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cox CD, Infection and immunity 1986, 52, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].a) Peng ED, Payne SM, J Bacteriol 2017, 199, e00874–16; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Small SK, O’Brian MR, J Bacteriol 2011, 193, 4088–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Deneer HG, Healey V, Boychuk I, Microbiology 1995, 141, 1985–1992. [DOI] [PubMed] [Google Scholar]
- [72].Abeyrathna SS, Abeyrathna NS, Thai NK, Sarkar P, D’Arcy S, Meloni G, Biochemistry 2019, 58, 4337–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lau CK, Krewulak KD, Vogel HJ, FEMS Microbiol Rev 2016, 40, 273–298. [DOI] [PubMed] [Google Scholar]
- [74].Sestok AE, Linkous RO, Smith AT, Metallomics 2018, 10, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Smith AT, Linkous RO, Max NJ, Sestok AE, Szalai VA, Chacon KN, Biochemistry 2019, 58, 4935–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hantke K, FEMS <icrobiol Lett 1987, 44, 53–57. [Google Scholar]
- [77].Wyckoff EE, Mey AR, Leimbach A, Fisher CF, Payne SM, J Bacteriiol 2006, 188, 6515–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].a) Dashper SG, Butler CA, Lissel JP, Paolini RA, Hoffmann B, Veith PD, O’Brien-Simpson NM, Snelgrove SL, Tsiros JT, Reynolds EC, J Biol Chem 2005, 280, 28095–28102; [DOI] [PubMed] [Google Scholar]; b) Awad MM, Cheung JK, Tan JE, McEwan AG, Lyras D, Rood JI, Anaerobe 2016, 41, 10–17. [DOI] [PubMed] [Google Scholar]
- [79].a) Naikare H, Palyada K, Panciera R, Marlow D, Stintzi A, Infect Immun 2006, 74, 5433–5444; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Velayudhan J, Hughes NJ, McColm AA, Bagshaw J, Clayton CL, Andrews SC, Kelly DJ, Mol Microbiol 2000, 37, 274–286. [DOI] [PubMed] [Google Scholar]
- [80].Nagy TA, Moreland SM, Detweiler CS, Mol Microbiol 2014, 93, 1314–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].a) Perry RD, Mier I Jr., Fetherston JD, Biometals 2007, 20, 699–703; [DOI] [PubMed] [Google Scholar]; b) Perez N, Johnson R, Sen B, Ramakrishnan G, MicrobiologyOpen 2016, 5, 453–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].a) Weaver EA, Wyckoff EE, Mey AR, Morrison R, Payne SM, J Bacteriol 2013, 195, 4826–4835; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim H, Lee H, Shin D, Biochem Biophys Res Commun 2012, 423, 733–738; [DOI] [PubMed] [Google Scholar]; c) Alvarez-Fraga L, Vazquez-Ucha JC, Martinez-Guitian M, Vallejo JA, Bou G, Beceiro A, Poza M, Virulence 2018, 9, 496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dauner M, Skerra A, Chembiochem 2019, in the press.
- [84].a) Sia AK, Allred BE, Raymond KN, Curr Opin Chem Biol 2013, 17, 150–157; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang X, Slavkovic S, Song E, Botta A, Mehrazma B, Lento C, Johnson PE, Sweeney G, Wilson DJ, ACS Chem Biol 2019, in the press. [DOI] [PubMed]
- [85].Moura-Alves P, Puyskens A, Stinn A, Klemm M, Guhlich-Bornhof U, Dorhoi A, Furkert J, Kreuchwig A, Protze J, Lozza L, Pei G, Saikali P, Perdomo C, Mollenkopf HJ, Hurwitz R, Kirschhoefer F, Brenner-Weiss G, Weiner J 3rd, Oschkinat H, Kolbe M, Krause G, Kaufmann SHE, Science 2019, 366. [DOI] [PubMed] [Google Scholar]
- [86].a) Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP, Science 2008, 319, 962–965; [DOI] [PubMed] [Google Scholar]; b) Clohessy PA, Golden BE, Scand J Immunol 1995, 42, 551–556. [DOI] [PubMed] [Google Scholar]
- [87].Nakashige TG, Zhang B, Krebs C, Nolan EM, Nat Chem Biol 2015, 11, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zygiel EM, Nelson CE, Brewer LK, Oglesby-Sherrouse AG, Nolan EM, J Biol Chem 2019, 294, 3549–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, Zhang Y, Betz C, Hench L, Fritz G, Skaar EP, Chazin WJ, Proc Natl Acad Sci U S A 2013, 110, 3841–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].a) Brophy MB, Hayden JA, Nolan EM, J Am Chem Soc 2012, 134, 18089–18100; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hayden JA, Brophy MB, Cunden LS, Nolan EM, J Am Chem Soc 2013, 135, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Baker TM, Nakashige TG, Nolan EM, Neidig ML, Chem Sci 2017, 8, 1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zygiel EM, Nolan EM, Acc Chem Res 2019, 52, 2301–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nakashige TG, Nolan EM, Metallomics 2017, 9, 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Masse E, Salvail H, Desnoyers G, Arguin M, Cur Opin Microbiol 2007, 10, 140–145. [DOI] [PubMed] [Google Scholar]
- [95].da Silva Neto JF, Lourenco RF, Marques MV, BMC Genomics 2013, 14, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH, BMC Genomics 2011, 12, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Thamer W, Cirpus I, Hans M, Pierik AJ, Selmer T, Bill E, Linder D, Buckel W, Arch Microbiol 2003, 179, 197–204. [DOI] [PubMed] [Google Scholar]
- [98].a) Chazarreta-Cifre L, Martiarena L, de Mendoza D, Altabe SG, J Bacteriol 2011, 193, 4043–4048; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yoch DC, Valentine RC, Ann Rev Microbiol 1972, 26, 139–162; [DOI] [PubMed] [Google Scholar]; c) Sandmann G, Peleato ML, Fillat MF, Lazaro MC, Gomez-Moreno C, Photosynth Res 1990, 26, 119–125. [DOI] [PubMed] [Google Scholar]
- [99].Vasileva D, Janssen H, Honicke D, Ehrenreich A, Bahl H, Microbiology 2012, 158, 1918–1929. [DOI] [PubMed] [Google Scholar]
- [100].Hastie JL, Hanna PC, Carlson PE, Pathog Dis 2018, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Erdner DL, Price NM, Doucette GJ, Peleato ML, Anderson DM, Mar Ecol Progr Ser 1999, 184, 43–53. [Google Scholar]
- [102].Worst DJ, Gerrits MM, Vandenbroucke-Grauls CM, Kusters JG, J Bacteriol 1998, 180, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].a) Baichoo N, Wang T, Ye R, Helmann JD, Mol Microbiol 2002, 45, 1613–1629; [DOI] [PubMed] [Google Scholar]; b) Crossley RA, Gaskin DJ, Holmes K, Mulholland F, Wells JM, Kelly DJ, van Vliet AH, Walton NJ, Appl Environ Microbiol 2007, 73, 7819–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].a) Sepulveda Cisternas I, Salazar JC, Garcia-Angulo VA, Frontiers in microbiology 2018, 9, 1478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pajuelo D, Hernandez-Cabanyero C, Sanjuan E, Lee CT, Silva-Hernandez FX, Hor LI, MacKenzie S, Amaro C, Environ Microbiol 2016, 18, 4005–4022. [DOI] [PubMed] [Google Scholar]
- [105].Mortensen BL, Rathi S, Chazin WJ, Skaar EP, J Bacteriol 2014, 196, 2616–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].a) Nairn BL, Lonergan ZR, Wang J, Braymer JJ, Zhang Y, Calcutt MW, Lisher JP, Gilston BA, Chazin WJ, de Crecy-Lagard V, Giedroc DP, Skaar EP, Cell Host Microbe 2016, 19, 826–836; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jordan MR, Wang J, Weiss A, Skaar EP, Capdevila DA, Giedroc DP, Inorg Chem 2019, 58, 13661–13672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Chandrangsu P, Huang X, Gaballa A, Helmann JD, Mol Microbiol 2019. [DOI] [PMC free article] [PubMed]
- [108].Fojcik C, Arnoux P, Ouerdane L, Aigle M, Alfonsi L, Borezee-Durant E, Mol Microbiol 2018, 108, 159–177. [DOI] [PubMed] [Google Scholar]
- [109].Peng ED, Oram DM, Battistel MD, Lyman LR, Freedberg DI, Schmitt MP, J Bacteriol 2018, 200, e00051–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].a) Ghssein G, Brutesco C, Ouerdane L, Fojcik C, Izaute A, Wang S, Hajjar C, Lobinski R, Lemaire D, Richaud P, Voulhoux R, Espaillat A, Cava F, Pignol D, Borezee-Durant E, Arnoux P, Science 2016, 352, 1105–1109; [DOI] [PubMed] [Google Scholar]; b) Lhospice S, Gomez NO, Ouerdane L, Brutesco C, Ghssein G, Hajjar C, Liratni A, Wang S, Richaud P, Bleves S, Ball G, Borezee-Durant E, Lobinski R, Pignol D, Arnoux P, Voulhoux R, Sci Rep 2017, 7, 17132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].a) Grim KP, San Francisco B, Radin JN, Brazel EB, Kelliher JL, Parraga Solorzano PK, Kim PC, McDevitt CA, Kehl-Fie TE, MBio 2017, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Song L, Zhang Y, Chen W, Gu T, Zhang SY, Ji Q, Proc Natl Acad Sci U S A 2018, 115, 3942–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Peng H, Shen J, Edmonds KA, Luebke JL, Hickey AK, Palmer LD, Chang FJ, Bruce KA, Kehl-Fie TE, Skaar EP, Giedroc DP, mSphere 2017, 2, e00082–00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Stork M, Grijpstra J, Bos MP, Manas Torres C, Devos N, Poolman JT, Chazin WJ, Tommassen J, PLoS Pathog 2013, 9, e1003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].a) Wakeman CA, Moore JL, Noto MJ, Zhang Y, Singleton MD, Prentice BM, Gilston BA, Doster RS, Gaddy JA, Chazin WJ, Caprioli RM, Skaar EP, Nat Commun 2016, 7, 11951; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ohlemacher SI, Giblin DE, d’Avignon DA, Stapleton AE, Trautner BW, Henderson JP, J Clin Invest 2017, 127, 4018–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Moore JL, Becker KW, Nicklay JJ, Boyd KL, Skaar EP, Caprioli RM, Proteomics 2013, 14, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].a) Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, Henderson JP, Nat Chem Biol 2012, 8, 731–736; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robinson AE, Lowe JE, Koh EI, Henderson JP, J Biol Chem 2018, 293, 14953–14961; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Koh EI, Hung CS, Parker KS, Crowley JR, Giblin DE, Henderson JP, Metallomics 2015, 7, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bleuel C, Grosse C, Taudte N, Scherer J, Wesenberg D, Krauss GJ, Nies DH, Grass G, J Bacteriol 2005, 187, 6701–6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK, Mol Cell 2002, 10, 1033–1043. [DOI] [PubMed] [Google Scholar]
- [119].Hoette TM, Abergel RJ, Xu J, Strong RK, Raymond KN, J Am Chem Soc 2008, 130, 17584–17592. [DOI] [PMC free article] [PubMed] [Google Scholar]