

Abstract
Purpose
Translocation renal cell carcinoma (tRCC) is a rare, aggressive RCC subtype. There is currently limited understanding on the role of molecular alterations in the pathogenesis and progression of these tumors. We investigated the association between somatic alterations and clinical outcomes in two independent cohorts profiled using DNA-sequencing.
Experimental Design
Twenty-two tRCCs underwent targeted sequencing (MSK-IMPACT); a subset was profiled using exome-sequencing and combined with exome data from The Cancer Genome Atlas (TCGA) for analysis. The prognostic value of specific somatic aberrations, tumor mutation burden (TMB) and fraction of copy-number altered genome (FCNAg) was explored. In TCGA cases, neoantigen prediction and immune cell deconvolution were performed using RNA-seq and exome data. Overall survival estimates were computed using the Kaplan-Meier method; time-on-treatment (TOT) was calculated for 14 MSK-IMPACT patients who underwent systemic therapy. Associations between molecular features and outcomes were evaluated using non-parametric testing
Results
Copy-number aberrant tRCCs were associated with poor overall survival (p=0.03). Pediatric patients had tumors with lower FCNAg (p=0.01). In one adult case with two chronologically distinct tumor samples sequenced, we confirmed that copy-number events occurred early during evolution. TERT promoter mutations were found exclusively in high-stage tumors. We found that tRCCs displayed distinct angiogenesis and PD-L1 gene expression profiles compared to other RCC subtypes.
Conclusions
Tumors molecularly defined by increased CNVs were associated with aggressive disease in tRCC. A higher burden of genomic events in adults compared to pediatric cases likely reflects a more aggressive clinical course. The unique immunophenotypic characteristics of tRCC merit further exploration.
Introduction
Microphthalmia transcription factor (MiTF)-family translocation renal cell carcinoma (tRCC) constitutes approximately 4% and 41.5% of renal cell carcinoma (RCC) cases in adults and children, respectively, with pediatric cases generally showing more favorable outcomes. The pathognomonic event in this disease is a fusion between transcription factors TFE3 or TFEB (on chromosomal loci Xp11.2 and 6p21, respectively) to a potentially more transcriptionally active locus. MiTF transcription factors are involved in the regulation of genes related to cell growth, metabolism, and lysosomal biogenesis, but the specific pathways and molecular mechanisms triggering oncogenesis and driving aggressiveness in tRCC remain unclear(1–4). There are currently no prognostic biomarkers, nor standard of care therapy for advanced disease, in this rare and aggressive RCC subtype.
Prior analyses of recurrent genomic alterations in tRCC have been limited to relatively small cohorts and predominantly focused on the analysis of copy number variations (CNVs). In a previous report of 16 primary tRCC specimens, distinct somatic CNVs were reported to be associated with poor prognosis. The most frequent CNVs in this cohort were 17q gain and 9p loss, the latter also representing a known prognostic feature of clear cell RCC (ccRCC)(5),(6). Additionally, only a small number of patients with advanced tRCC have been included in clinical trials of systemic therapy for RCC. Therefore, there is currently not enough information about the efficacy of different treatments and, as a result, no established standard of care (7–9). In a recent retrospective, multi-institutional study of 24 patients undergoing immune-checkpoint blockade (ICB) therapy, to date the largest report on outcomes to systemic therapy in tRCC, the authors reported a benefit in patients harboring mutations in bromodomain-containing genes. However, an evaluation of copy number status was not performed (10).
Here, we investigated the presence of recurrent genomic alterations (mutations and CNVs) and their association with prognosis in 22 patients prospectively sequenced at our institution using a combination of targeted and exome-wide sequencing. Additionally, we explored the frequency of these events in a subset of cases with response to systemic therapy. We evaluated the reproducibility of our findings in an independent cohort of 14 tRCC cases from the TCGA consortium. We further carried out neoantigen prediction of mutations and fusion-generated peptides and investigated the tumor microenvironment using RNA sequencing data. All of the underlying mutation and copy number data associated with our analysis is provided in the Supplementary Material as a public resource for research into this rare entity.
Patients and methods
Patient cohorts
The study was conducted in accordance with the Declaration of Helsinki and was approved by an Institutional Review Board. All subjects or subjects’ guardians gave written informed consent.
MSK-IMPACT cohort (discovery).
After obtaining Institutional Review Board approval, our internal database was queried for patients with a histological diagnosis of tRCC. Diagnoses were made by dedicated genitourinary pathologists and were based on positive immunohistochemistry (IHC) or the confirmation of an MiTF gene fusion by either fluorescence-in-situ hybridization (FISH), anchored multiplex polymerase chain reaction assay (ARCHER®) or next-generation sequencing (NGS). Since false-positive and false-negative results have been reported for IHC-based diagnoses, patients who did not have cytogenetic proof of a translocation were excluded from further analysis (Supplementary Figure 1) (11).
MSK-exome cohort.
A subset of the MSK-IMPACT cohort (n=10) underwent additional whole-exome sequencing (WES). Sequencing methods are detailed in the relevant section below. An additional tumor sample, derived from the same patient as TCGA sample BQ-5887, was also submitted for exome sequencing.
TCGA-exome cohort (validation).
Non-MSK patients with tRCC were identified from The Cancer Genome Atlas pan-kidney (TCGA-KIPAN) cohort, with available RNA sequencing (RNA-seq) and exome sequencing data. Clinical information for these cases was obtained from a publicly available dataset (www.firebrowse.org, TCGA data version 2016_01_28). MiTF gene fusion was confirmed using two independent datasets of transcript fusions across TCGA samples (12,13). Baseline characteristics of these patients and sample metadata are available in Supplementary Tables S1 and S2.
MSK-IMPACT Targeted Sequencing
Tumor and matched-normal specimens were sent for targeted next-generation sequencing (NGS) using our previously-validated sequencing panel (MSK-IMPACT®), which targets 341, 410, or 468 actionable genes based on the platform version(14). In brief, DNA from tumor and matched blood normal specimens from each patient were extracted and sheared to create barcoded DNA libraries. Using captured DNA, all coding exons of cancer genes and a subset of polymorphic loci (for copy number analysis) were sequenced. Deep sequencing was performed at an average of 500 to 1,000X coverage. After alignment to the reference human genome, somatic alterations (missense mutations, small insertions and deletions, structural rearrangements, and DNA copy number changes) were identified using a bioinformatics pipeline described in detail previously (14). To assess oncogenicity, mutations were annotated using the oncology knowledge database OncoKB® (15).
Whole-Exome Sequencing, Processing, and Mutation Analysis
Alignment of FASTQ-formatted raw sequencing data to the human reference genome (b37) was performed using the Burrows–Wheeler aligner (BWA v.0.7.10). The Genome Analysis Toolkit was used to perform local realignment (GATK v4.1.4.1(16)). Duplicate reads were excluded using Picard v.2.13. Four different variant calling tools were used for mutation calling: MuTect2 (part of GATKv4.1.4.1(16)), Strelka2 v2.9.10 (17), Varscan v2.4.3 (18) and Platypus (19). To pass filtering, mutations were required to be detected by at least two caller algorithms. To obtain highly accurate variant calls, ancillary filters were used. These filters included a base coverage of a minimum of 10 reads in the tumor, at least 5 reads supporting the mutation and an allelic frequency below 2% in the matched-blood sample. Furthermore, single-nucleotide variants (SNVs) identified at a frequency higher than 1% in dbSNP (20) or 1000Genomes project (21) were excluded. Mutation calls from both MSK-IMPACT and exome sequencing have been provided (Supplementary Table S3).
Allele-specific copy number analysis
Allele-specific copy number analysis (ASCN) was performed using our institutional bioinformatics algorithm FACETS (22). After genome segmentation, each segment was categorized as being diploid or non-diploid according to its CN integer values (i.e. segments with a total copy number ≠ 2 or minor copy number ≠ 1, according to the FACETS outputs). In cases where FACETS failed to produce any estimates, the values from the closest adjacent segment (on the same chromosomal arm) were used instead. In order to compute the fraction of copy number-altered genome (FCNAg), the length of each autosomal genomic segment (in base pairs) was determined. FCNAg was then calculated by adding the non-diploid segments and comparing their total length to the sum of all segments. Copy-neutral segments showing allelic imbalance were also considered to be aberrant (i.e. total CN = 2 and lower CN = 0). Gains were called if the total copy number of a segment was greater than 2 (diploid). Losses were called if the minor copy number of an arm segment was equal to 0 (corresponding to loss-of-heterozygosity [LOH]), regardless of the total number of copies. No arm-level CNVs were found to have both LOH and a total copy number greater than 2 (Supplementary Table 4). Arm-level events were defined as any gain or loss occurring in an autosome that involved at least 10% of the arm. We used this relatively low arm fraction threshold both to remove potential artifacts from the assay, such as drops in coverage around centromeric or telomeric regions (which could be captured as losses), and at the same time capture any somatic copy number events regardless of their breadth. In general, very few focal CN events were observed, except for some focal losses in 9p21.3 involving CDKN2A/2B loci (which were considered 9p losses in the analysis). Copy number-aberrant tumors were classified as such when they satisfied at least one of the following criteria: (1) 9p loss/focal loss of CDKN2A/2B, (2) 17q gain, (3) FCNAg greater than the median in the relevant cohort. All 9p and 17q calls were manually reviewed using the raw FACETS output, and in cases where it was clear that FACETS had misclassified a subclonal gain/loss at these loci, this was corrected and recorded. All copy number calls, including notation of manual review, are available in Supplementary Table S4.
Calculation of TMB
All non-silent exonic mutations were considered for computation of TMB and this number was corrected for the total genomic length covered in the hybridization assay (1 Mb and 1.1 Mb for cases of MSK-IMPACT® versions 5 and 6). Since the MSK and TCGA whole-exome assays covered different lengths of the genome, appropriate corrections were made (38.9 Mb for MSK-exome and 36.8 Mb for TCGA-exome. Mutation calling was not completed on 2 TCGA samples sequenced with the SOLiD sequencing platform, and therefore tumor mutation burden for these samples was determined using public TCGA mutation calls.
Cancer cell fraction analysis
Mutations with a cancer cell fraction (CCF) of ≥ 0.9 were defined as clonal, and all other mutations were considered subclonal. Using these definitions, an intratumoral heterogeneity (ITH) index (range 0, +∞) was computed by dividing the number of subclonal and clonal mutations (#subclonal / #clonal).
Neoantigen prediction and immune cell deconvolution
We used INTEGRATE, as described by Zhang et al(23), for identifying gene fusions from RNA-seq data. HLA alleles were identified using POLYSOLVER(24). For the neoantigen prediction of the fusion proteins called by INTEGRATE, we used the INTEGRATE-neo pipeline(23) which reports any fusion-derived peptides found with binding affinity less than 500 nM (default setting) for the given HLA allele through NetMHC v4.0(25).
We further assessed differences in the expression of signatures associated with angiogenesis, PD1, PD-L1 and CTLA4 between tRCC cases and other RCC histologies. To do this, we performed Gene Set Enrichment Analysis (ssGSEA) using previously described gene signatures in single-sample RNA-seq data (26)(27)(28). We used the ssGSEA algorithm implementation through the R package ‘gsva’(29) to calculate the relative overexpression of gene sets of interest in each of the samples (30). Expression data was downloaded from the TCGA firebrowse website.
Statistical analysis
Cases with complete genomic information were included in the analysis (n=22). Patient age was recorded at the time of diagnosis and pediatric cases were defined as patients diagnosed before 18 years of age. The association between FCNAg and clinicopathological variables at baseline (i.e. clinical stage and age) was tested using the Mann-Whitney U test. The same approach was used to perform pairwise TMB comparisons between the tRCC cohorts and other RCC subtypes in the TCGA (KIRC, KIRP, KICH), as well as when assessing differences in FCNAg between two groups. Differences in PD-L1 gene expression were tested between tRCCs and other tumors within the corresponding TCGA subgroup (KIRC, KIRP).
The primary outcome was overall survival (OS), which was calculated from the time of pathologic diagnosis to the date of death or last follow-up. Survival estimates were computed using the Kaplan-Meier method and estimates were compared using the log-rank test. Two TCGA samples sequenced with SOLiD sequencing were excluded from survival analysis due to absence of copy number data (Supplementary Table 2). In order to assess the response to systemic therapy, we measured time-to-treatment (TTT) and time-on-treatment (TOT). TTT was defined as the time interval from diagnosis to systemic therapy start; TOT was specified as the time from treatment start to discontinuation, either due to therapy-related toxicity or disease progression. A TOT > 12 months was used as a surrogate for response. All hypothesis tests were two-sided and statistical significance was defined as p<0.05. All analyses were performed using the R platform v3.5.3.
Results
Patient characteristics
We identified 37 patients treated for tRCC at our center between 1997 and 2019, with the diagnosis based on immunohistochemistry, FISH analysis, anchored multiplex polymerase chain reaction or NGS. We excluded 15 samples from further analysis due to lack of consent to sequencing or negative cytogenetic proof (i.e. FISH, ARCHER® or NGS) of gene rearrangement (Supplementary Figure 1). An overview of the clinicopathological and molecular features of the Memorial Sloan Kettering Cancer Center (MSK) cohort who underwent NGS (n=22) is provided in Supplementary Tables 1 and 2. A summary of the clinical and genomic features of the MSK-IMPACT cohort (n=22) is also shown (Figure 1). Gene fusions were found in both TFE3 (n=20) and TFEB (n=2). Additionally, we identified an independent cohort from the pan-kidney (KIPAN) TCGA cohort of 14 primary tumors with a confirmed MiTF gene fusion and WES data (Supplementary Table 2), TFE3 and TFEB rearrangements were identified in 13 and 1 cases, respectively. Only 12 of the TCGA samples were included in most analyses, as two of them could not be processed due to our pipeline not being compatible with SOLiD sequencing (see Methods). The median follow-up time in the MSK-IMPACT and TCGA cohorts was 27.1 and 41.4 months, respectively.
Figure 1:
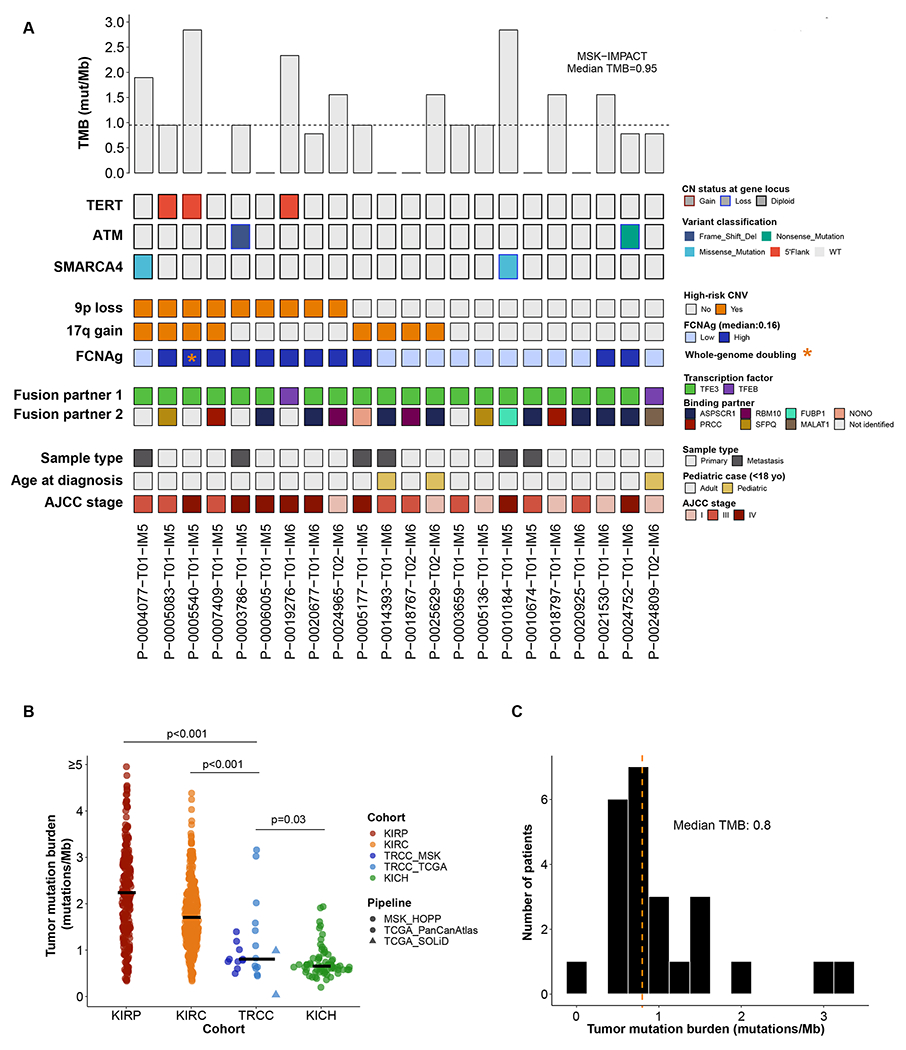
(A) Oncoprint displaying recurrent somatic mutations (genes mutated in >1 sample) and frequent copy number variants (CNVs) in the MSK-IMPACT cohort. Copy number variants (CNVs) CNVs are mostly found in cases with a high AJCC stage. The most frequent MiTF fusion gene was TFE3. (B) TMB in merged MSK-TCGA exomes compared to other TCGA-RCC cohorts. (C) Tumor mutation burden of tRCC is centered around a median value of 0.8 non-synonymous mutations per megabase, with two outlier cases. Mann-Whitney U tests were used for pairwise TMB comparisons between RCC subtypes.
Somatic driver mutations, tumor mutation burden, intratumoral heterogeneity and immunogenicity of fusion peptides in tRCC
Previous analyses of tRCC tumors were limited to comparatively small cohorts, and therefore were limited in their power to detect recurrent somatic mutations. We therefore first focused on the analysis of somatic mutations in the MSK-IMPACT cohort. After excluding TFE fusions and copy number alterations, 15/22 (68.2%) patients demonstrated somatic single nucleotide variations or indels, with putatively oncogenic driver mutations in SWI/SNF and DNA damage repair (DDR) pathways in 5/22 (22.7%) samples. We identified activating TERT promoter mutations (C228T) in three patients, all of which occurred in high AJCC stages, suggesting that activation of TERT may be a novel marker of aggressive tRCC. Where possible, we quantitatively determined the clonality of putatively oncogenic mutations using allele-specific copy number analysis. We determined that 1/2 ATM mutations and 1/1 of the SMARCA4 mutations for which allele-specific copy number data was available were clonal (Supplementary Table 3). Of these, only the clonal ATM mutation was accompanied by loss-of-heterozygosity of the complementary allele, suggesting that this lesion may be a truncal driving event in the pathogenesis of the tumor.
High tumor mutational burden (TMB) has been used as a biomarker of favorable response to ICB in many solid cancers(31),(32). Because accurate quantification of TMB in low mutation burden tumors is difficult using targeted NGS panels, we assessed TMB from exome sequencing of 10 patients in the MSK-exome and 12 patients from the TCGA-exome cohort, for which single nucleotide variations were available from the public repository (see Methods). We computed a median TMB of 0.80 mut/Mb for tRCCs, different from all other TCGA-RCC cohorts (Figure 1). TMB was consistent between the TCGA and MSK-exome cohorts (net difference −0.12, Mann-Whitney U, p=0.47). Furthermore, the TMB of tRCC was characteristically different than that of other RCC subtypes: lower compared to clear cell (TCGA KIRC, −0.76 [−0.49, −1.02], Mann-Whitney U, p<0.001) and papillary (KIRP, −1.17 [−0.77, −1.54], Mann-Whitney U, p<0.001), but higher compared to chromophobe RCC (KICH, 0.16 [0.02, 0.34], Mann-Whitney U, p=0.03). No putatively explanatory mutations were identified in the outlier tRCC cases of exceptionally high TMB (Figure 1). We summarized the clonality of somatic mutations using a metric for overall intratumoral heterogeneity (ITH-index, see Methods). The median ITH-index across all tRCC exome samples considering all mutations was 0.82 (IQR 0.55-1.36), indicating a higher rate of clonal events relative to subclonal events. This is consistent with a model for the evolution of tRCC whereby early somatic mutations occur which are succeeded later by a small number of subclonal mutations. Taken together, these results indicate that, from a mutational perspective, tRCC tumors exhibit (1) relatively low mutation burden, (2) relatively few canonically oncogenic mutational events aside from MiTF translocation, and (3) in cases with loss-of-function mutations to tumor suppressors, the wild-type allele infrequently undergoes concomitant loss-of-heterozygosity (LOH). Mutation annotation files for the MSK-IMPACT and TCGA-exome cohorts are provided in Supplementary Table 3.
Given the recent report of immunogenic fusion-derived neoantigens in metastatic head and neck cancer (33) and the concept of immunogenicity in tumors with relatively low mutational burden, we assessed immunogenicity and the composition of the tumor microenvironment in TCGA-tRCC samples for which exome and RNA-seq data was available. We identified a significant HLA-epitope binding affinity of fusion-derived peptides in 8/14 (57.1%) cases, with a median affinity of 98.5 nM (range 58.6-496.8). The relevant data, including the HLA type as well as the epitope peptide sequence, are provided in Supplementary Table 2. We also used immune deconvolution techniques to test for differences in gene signatures for angiogenesis and three immunotherapeutic targets in ccRCC: PD1, PD-L1, and CTLA4. The angiogenesis gene expression signature was higher in tRCCs compared to papillary RCC (Mann-Whitney U, p=0.007) and lower but almost equivalent with respect to clear cell tumors (Mann-Whitney U, p=0.053). tRCCs also showed a higher PD-L1 expression signature compared to the KIRC (p=0.01) and KIRP cohorts (p=0.002). However, we found no significant differences in CTLA4 (p=0.2 and p=0.71, respectively) or PD1 signatures (p=0.74 and p=0.7, respectively, Supplementary Figure 2).
Prognostic implications of copy number alterations
In prior analyses of tRCC, loss of chromosome arm 9p and gain of arm 17q were identified as common and, in the case of 17q gain, potential prognostic biomarkers(5). In contrast to earlier studies, our approach used allele-specific copy number analysis that could more sensitively detect small gains and copy number-neutral loss-of-heterozygosity events (see Methods). We therefore set out to evaluate the association between gross copy number events and both clinical features and OS in tRCC. We began by calculating the fraction of the genome exhibiting any copy number alteration (FCNAg). The median FCNAg was 0.16 (IQR: 0.09-0.32) in the MSK-IMPACT cohort and 0.09 (IQR: 0.04-0.33) in the TCGA cohort. High AJCC stage (III/IV) was found to be associated with higher FCNAg in the TCGA cohort (Mann-Whitney U, p=0.006) but this finding did not reach statistical significance in the MSK-IMPACT cohort (p=0.09, Figure 2).
Figure 2.
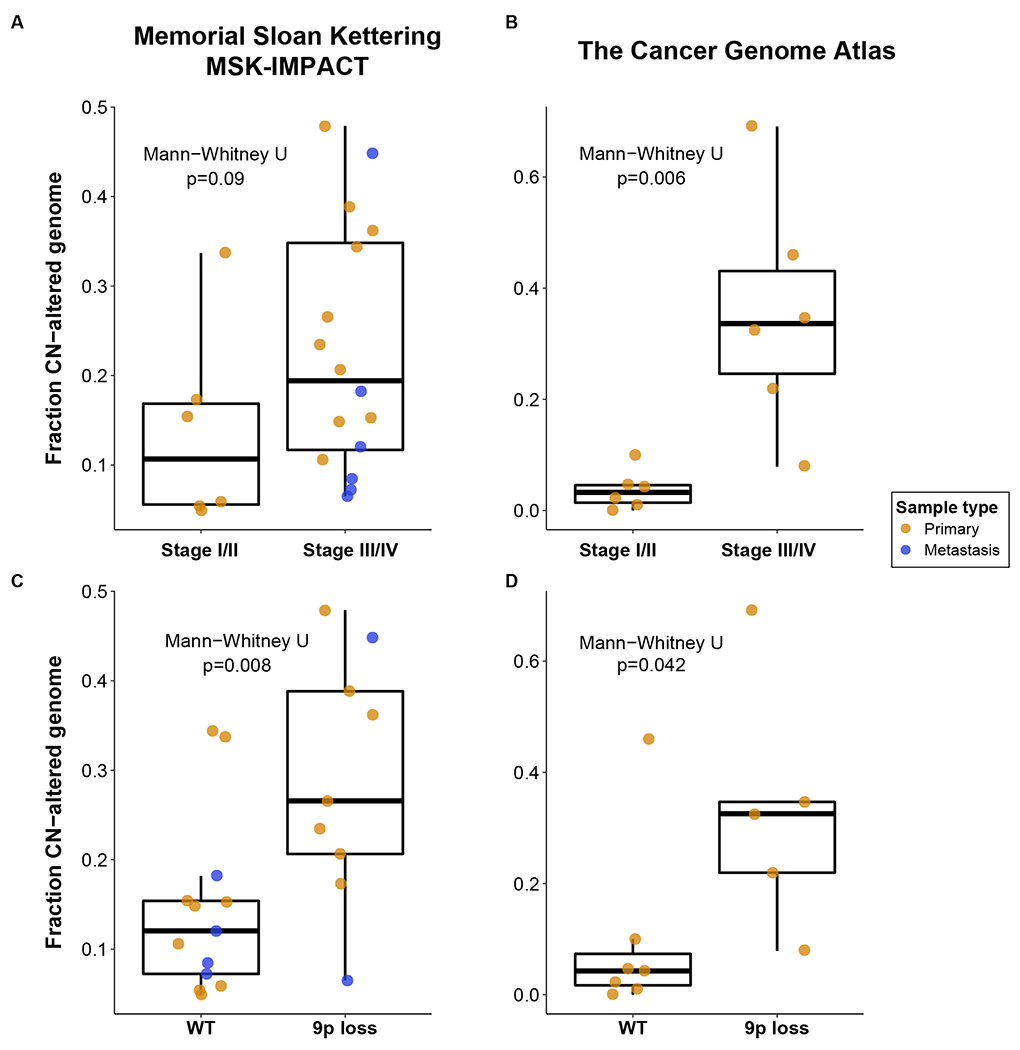
Association of FCNAg with AJCC stage and 9p loss. The broad line of the boxplots illustrate the median FCNAg, upper and lower hinges showing the interquartile ranges (IQR, 25% and 75% of values) and the whiskers illustrating values within 1.5x the IQR in the MSK (A) and TCGA (B) cohorts. Loss of chromosome 9p was found to be associated with higher FCNAg in the MSK (C) and TCGA cohorts (D).
We next examined copy number alterations in the MSK-IMPACT cohort at a more granular level. The most common arm-level copy number alterations observed in the MSK-IMPACT cohort were loss of 9p in 9/22 (41%) patients, gain of 17q in 8/22 (36%) patients, and gain of 6p in 8/22 (36%) patients. No single copy number event was significantly associated with overall survival in the MSK-IMPACT cohort (Supplementary Table 5). However, we did note that 3/3 TERT mutations were identified in tumors harboring 9p loss, and that the presence of 9p loss was significantly associated with higher FCNAg in both cohorts (Figure 3, Mann-Whitney U, p=0.008 and 0.042 in the MSK and TCGA cohorts, respectively).
Figure 3:
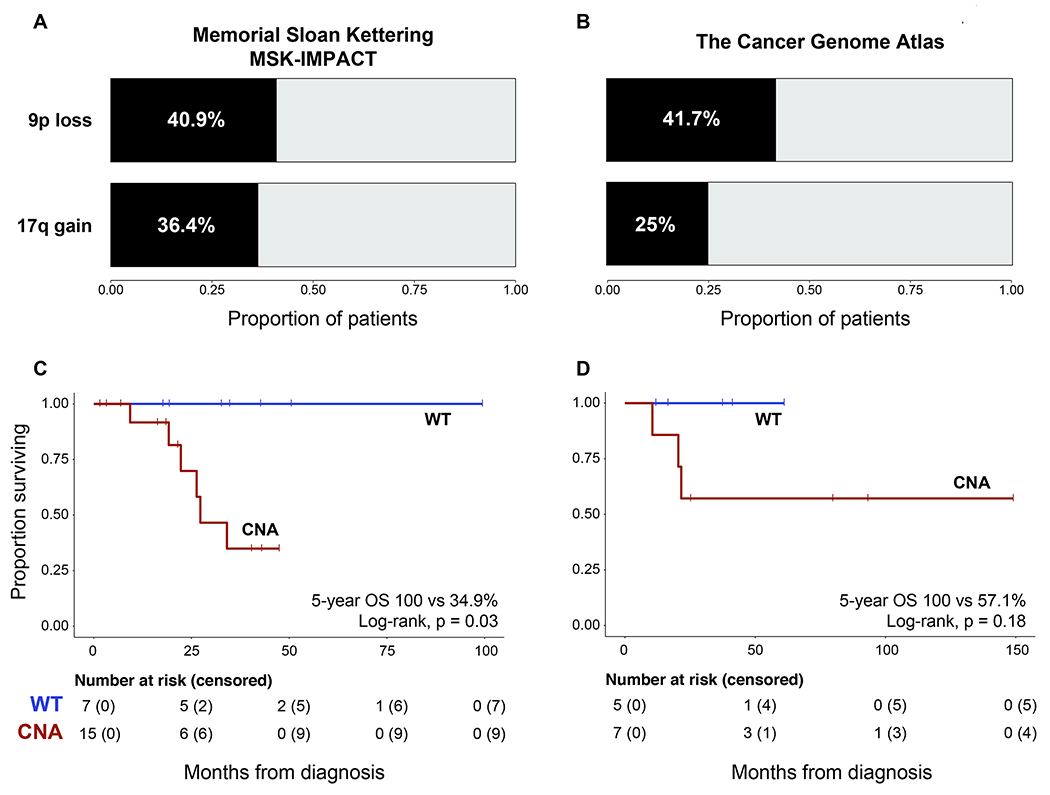
Proportion of patients with 9p loss and 17q gain in the MSK (A) and TCGA cohorts (B). Kaplan-Meier analysis of overall survival for patients with copy number-aberrant tumors (i.e. 9p loss, 17q gain, or high FCNAg [>within-cohort median]) in the MSK-IMPACT (C) and TCGA cohorts (D). Log-rank test results from both cohorts were combined in a meta-analysis using Fisher’s method (Chi-squared [4d.f]=10.52, p=0.03).
Based on the relatively low TMB of tRCC and the comparatively high frequency of copy number alterations, we sought to evaluate the association between clinical outcomes and a summary measure of copy number changes. We defined a copy number-aberrant cohort of patients which harbored either 9p loss, 17q gain, or an FCNAg higher than the within-cohort median. In the MSK-IMPACT cohort, the 15 patients with copy number-aberrant (CNA) tumors demonstrated worse overall survival (5-year OS 34.9% vs 100%, log-rank p=0.03). In the TCGA cohort, this analysis was not statistically significant (log-rank, 0.18), however, the point survival estimates were very similar (5-year OS 57.1% vs 100%) suggesting that the analysis in the TCGA data may have been underpowered (with only n=10 samples). Two tumors in the MSK-IMPACT cohort and two tumors in the TCGA cohort exhibited bi-allelic loss of CDKN2A/B; treating these tumors as a separate group for the purposes of survival analysis did not meaningfully affect our findings, suggesting that bi-allelic loss of this gene is not the sole driver of poor prognosis in CNA tumors. Therefore, we performed a meta-analysis on the results with the two cohorts which demonstrated a statistically significant association between copy number-aberrant tumor status and poor survival (p=0.03, Fisher’s combined probability test).
Temporal Evolution of a metastatic tRCC tumor
We retrospectively identified an additional tRCC tumor derived from a patient treated at our institution who had an earlier tRCC tumor sample sequenced as part of the TCGA consortium (only a single such overlapping patient was identified). This patient initially underwent a partial nephrectomy (at our institution, sample unavailable) approximately 20 years ago for a 4.5 cm renal mass that was diagnosed as a high-grade papillary RCC. After 8 years, a local recurrence developed in the same kidney where the original tumor was removed, leading the patient to undergo a completion radical nephrectomy with retroperitoneal lymph node dissection (TCGA sample BQ-5887). To note, all the lymph nodes obtained during this procedure were found to be free of tumor. This local kidney recurrence was found to have concordant morphologic features when compared to the previous specimen, as well as a TFE3 gene rearrangement; prompting a change in the original diagnosis to MiTF-family tRCC. The patient remained disease-free for about 8 more years, after which he developed severe abdominal and back pain due to extensive disease spread in the upper abdomen and retroperitoneum. One of the retroperitoneal nodes was biopsied and also found to bear a PRCC-TFE3 fusion (MSK-RP-5887 sample). At this point the patient was diagnosed with metastatic disease and started on systemic therapy (Figure 4). After an initial period of partial response to everolimus/bevacizumab, the abdominal lesions became stable and subsequently grew again (with a new disease site also evidenced in the thorax). At this point, the treatment was stopped and the patient was administered two additional lines of therapy, which resulted in mixed responses (i.e. shrinkage of some lesions but progression in others). After progression on the third-line of therapy (plus significant TKI-related toxicity), the patient refused additional treatments and decided to undergo hospice care instead.
Figure 4:
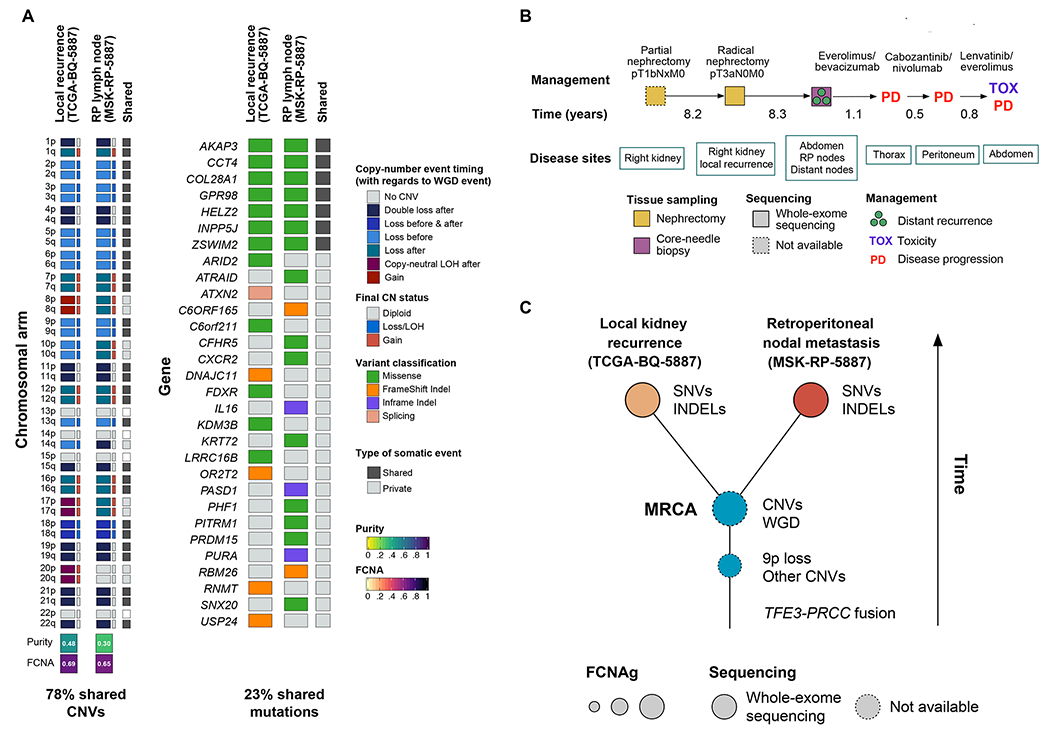
Temporal evolution of tRCC in a single patient. (A) Private and shared copy number alterations and somatic mutations in two tRCCs tumors from a single patient. (B) Clinical course of tRCC patient from which tumor samples were derived. (C) Putative model for tRCC evolution in this case. Early loss of 9p and other copy number alterations was followed by large-scale whole genome duplication. The majority of somatic mutations were acquired in the period following the appearance of the most recent common ancestor (MRCA), which bore a high-burden of CNVs.
The newly identified tumor was a pathologically confirmed lymph node metastasis with evidence of a PRCC-TFE3 fusion (confirmed by IHC and FISH, as well as MSK-IMPACT sequencing). To understand the clonal relatedness of these tumors, we completed exome sequencing on the nodal metastasis, and compared copy number aberrations and mutations between MSK-RP-5887 and TCGA-BQ-5887. Interestingly, we noted that both samples exhibited whole-genome doubling (WGD) and extensive copy number alterations across the entire genome and therefore an aberrantly high FCNAg compared to other tRCC tumors. In fact, the vast majority of arm-level events (78%) were shared between the two tumors. Notably, loss of chromosome 9p was evident in the two samples and, in both cases, it preceded the WGD event. These findings are consistent with CNVs being an early event in the development of the tumor. In contrast, only 23% of somatic mutations were shared between the two tumors (ITH-index, MSK-RP-5887: 5.3, TCGA-BQ-5887: 16), indicating that extensive mutational diversification occurred after divergence of the most recent common ancestor (MRCA). Together, this suggests that in this particular tRCC tumor, copy number alterations occurred early during tumor evolution, whereas the acquisition of the bulk of the somatic mutations occurred later (Figure 4). This result is not in agreement with the median ITH-index result reported above (of 0.82), and may reflect different evolutionary patterns in metastatic vs. primary tRCC.
Response of tRCC patients to systemic therapies in the MSK-IMPACT cohort
We assessed treatment response in 14 patients of the MSK-IMPACT cohort who underwent, in their majority, multiple lines of systemic treatment. Five out of fourteen (35.7%) patients showed a prolonged TOT (>12 months) in the first- or second-line of therapy. We observed no difference in the median FCNAg when comparing responders to non-responders (Mann-Whitney U, p=0.55). We did not find any association between response to any therapy and genomic features. Notably, a patient with a prolonged response to ICB therapy harbored missense mutations in two genes of the PBAF chromatin-remodeling complex, SMARCA4 and PBRM1; variants in both genes have recently been linked to immunogenic features (34),(35). The treatment course and genomic characteristics of tRCC patients who showed prolonged responses to first- or second-line systemic therapy are shown in Figure 5.
Figure 5:
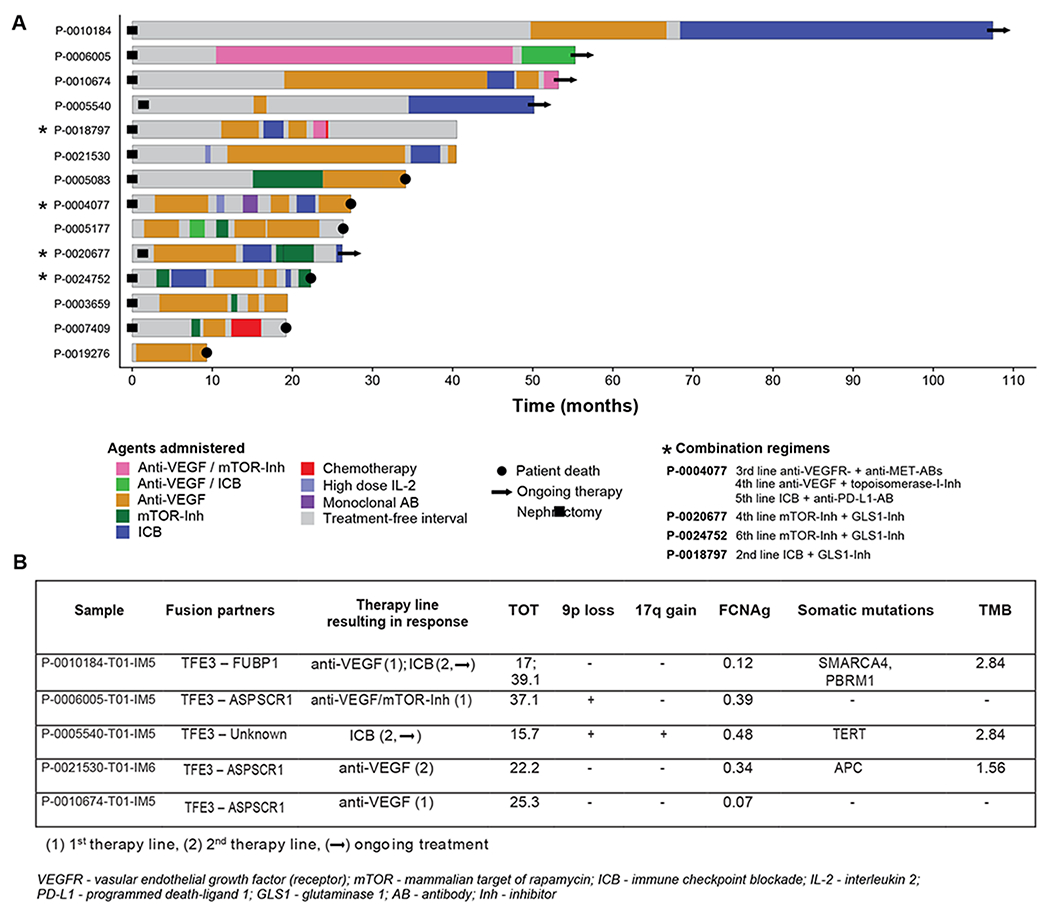
(A) Swimmer plot displaying a heterogeneous pattern of substances and response, a few cases showing an exceptionally prolonged time on treatment, (B) Molecular features identified in patients with TOT >12 months.
Pediatric tRCC cases in MSK-IMPACT cohort
Finally, we assessed the presence of molecular alterations in patients under 18 years of age (n=3). Their sites of disease and their therapeutic course are displayed in Figure 6. Two patients had undergone prior chemotherapy: one for retinoblastoma, who also had evidence of a germline RB1 mutation (P-0025629-T02-IM6, germline status identified from sequencing of matched normal blood per standard MSK-IMPACT sequencing), and the other for neuroblastoma (P-0024809-T02-IM6). None of the pediatric cases showed 9p loss or 17q gain and, consistently, they were also found to have a significantly lower FCNAg when compared to their older counterparts (Mann-Whitney U, p=0.02) (see Supplementary Table 3).
Figure 6:
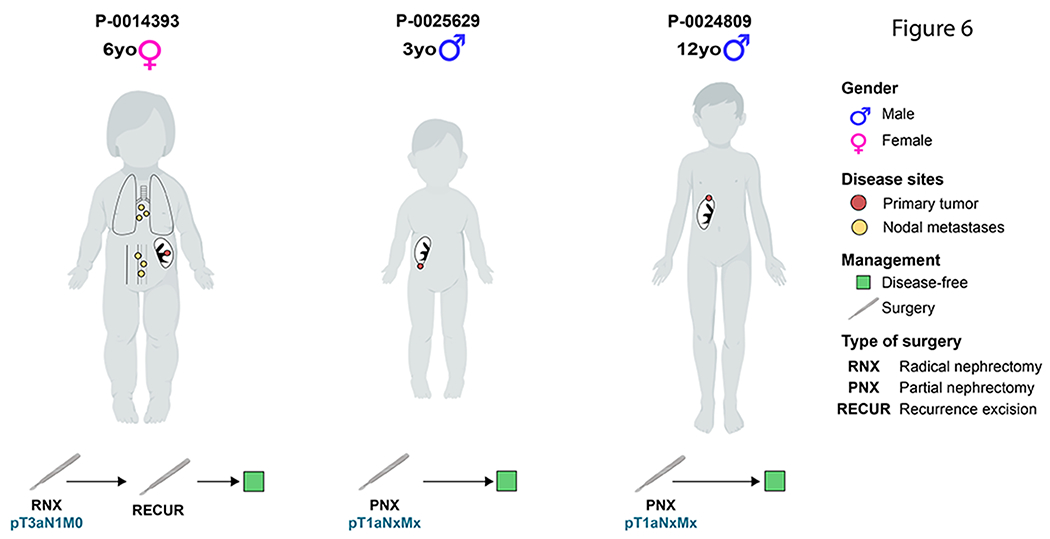
Three panels with pediatric cases in our MSK-IMPACT cohort displaying a favorable disease course after surgery as the only intervention (Created with BioRender.com).
Discussion
We present a comprehensive genomic analysis of 22 tRCC patients utilizing high-depth targeted sequencing and high-breadth exome sequencing, combining the results with additional exome sequencing data from 14 TCGA tRCC cases. To serve as a resource for future studies, we report the genomic and clinical features found in each tumor in both cohorts. Our results suggest that tRCCs are driven primarily by copy number aberrations, rather than by somatic mutations in tumor suppressors and oncogenes. We observed distinct RNA-seq deconvolution scores for angiogenesis and PD-L1 in TCGA-exome cases compared to clear cell and papillary tumors from the TCGA; as well as, a prediction of significant HLA-binding affinity in 57.1% of the fusion-derived neoantigens, suggesting that perhaps influencing the immune system to direct it against these clonal antigens may represent a viable treatment strategy for tRCC. The smaller proportion of non-diploid genome in pediatric tRCC cases, compared to adults, is in line with a less aggressive tumor biology. However, these results must be viewed with caution due to the small patient numbers.
Gene rearrangements are the key oncogenic event in tRCC, and prior work has associated certain fusion partners with worse outcomes (36). Additionally, recurrent somatic mutations and CNVs in tRCC have previously been explored. An earlier genomic study in 16 patients with tRCC found heterogeneous molecular profiles, with 17q gain and 9p loss identified as the most frequent CNVs. In that study, 17q gain was an essential feature of poor outcome, but presence of 9p loss was not significantly associated with poor OS(5). These results are partially consistent with our own findings, which, however, showed a significant association in the MSK-IMPACT cohort between poor outcome in a copy number-aberrant group of tumors defined by the presence of either 9p loss, 17q gain, or elevated FCNAg. 9p loss was previously associated with a worse outcome in ccRCC and papillary RCC(6,37),(38),(39). Although the processes underlying this association are unclear, it is notable that CDKN2A and CDKN2B, important cell-cycle regulators which have been shown to interact with HIF-1ɑ and cause VEGF upregulation (40), are located on 9p21 and were lost (sometimes focally) in a significant percentage of cases.
TERT promoter mutations have also previously been associated with an aggressive disease course in more common variants of RCC (41), but the implications of these mutations in tRCC have not been described. In the MSK-IMPACT cohort, we detected clonal activating hotspot mutations in the TERT promoter of three tumors. All of these cases consisted of high-stage tRCC, and exhibited 9p loss as well as a high burden of somatic CNVs (Figure 1). This data collectively suggests that TERT mutations are associated with an aggressive phenotype in tRCC, however, it is impossible at this point to determine its role as a driver of aggressiveness.
In our cohort a first- or second-line treatment with a TKI led to extended TOT in three cases. The response to tyrosine kinase inhibitors (TKI) and mammalian target of rapamycin (mTOR) inhibitors was previously evaluated in a retrospective study which included 21 patients with tRCC. In that study, an objective response rate (ORR) of 33% could be observed, similar to ccRCC. Importantly, only 4/21 cases underwent cytogenetic confirmation of tRCC(9). The ORR for tRCC patients in a large nccRCC trial was comparable (29%)(42). Notably, one patient who underwent a combined mTOR-inhibitor/anti-VEGF regimen in our cohort showed a prolonged TOT of 37 months.
Our analysis here also provides an initial characterization of the tRCC tumor microenvironment, relative to clear cell and papillary RCC. We observed over-expression of PD-L1 in tRCC relative to KIRC and KIRP, which is consistent with findings from a large study of non-clear cell RCC in which 90% of tRCC cases showed PD-L1 overexpression in tumor-infiltrating mononuclear cells(43). Despite these apparent microenvironmental phenotypes, tRCC patients demonstrate poor response rates to TKI and immune checkpoint blockade, suggesting that emerging biomarkers for response to TKI and immunotherapy in ccRCC may not readily translate to tRCC. In this context, it is also notable that our analysis of RNA-seq data in the TCGA-exome cohort revealed fusion-derived neoantigens with high binding affinity to the HLA class-I molecules in 57% of the cases. In light of recent data describing the immunogenicity of fusion-derived neoantigens in head and neck cancers(33), this suggests that neoantigens produced by TFE3/TFEB translocation may be clinically meaningful. Taken together, these results argue for further exploration of the immunogenomic characteristics of tRCC, with special focus on T-cell response to ICB as well as potential neoantigen-directed vaccines (44). Furthermore, we observed a notable case of prolonged response to ICB harboring SMARCA4 and PBRM1 variants. An immunogenic effect of SMARCA4 mutations, via PD-L1 upregulation, has previously been postulated for small cell ovarian cancer(34). PBRM1 mutations were recently reported to have an increased clinical benefit in metastatic ccRCC patients undergoing ICB(35). We further investigated potential genomic biomarkers of response to ICB. Prior literature described an inverse association between aneuploidy and response to ICB in cohorts of patients with metastatic melanoma and non-small cell lung cancer (45,46). In our limited cohort, we did not find a statistically significant association between FCNAg and treatment response.
Pediatric tRCC cases commonly have better outcomes compared to adult variants, which was previously attributed to a smaller percentage of distant metastasis (47). In young patients, the disease is often manageable with surgery alone, which we could confirm for our three pediatric cases, all of whom were treated with surgery and were disease-free at the most recent follow-up. Furthermore, prior chemotherapy has been described as a risk factor for the development of tRCC in children(48). Two of our three pediatric patients had prior chemotherapy due to retinoblastoma and neuroblastoma. The occurrence of RCC as a secondary malignancy following neuroblastoma has been previously reported, with some of these tumors identified as tRCCs(49). RCC in neuroblastoma survivors is included as a provisional entity in the 2016 WHO classification of renal tumors(49,50). Notably, patient P-0025629-T02-IM6 is, to our knowledge, the first described case of an RCC following a diagnosis of retinoblastoma, however, the remaining RB1 copy was found to be unaffected in the tRCC sample. At the molecular level, fewer genomic events were previously reported in patients <18 years old (5). In our cohort, the favorable outcomes in pediatric cases were in line with a lower FCNAg and a lack of “high-risk” CN events.
Limitations of our study include the heterogeneous composition of sequenced specimens, originating from both primaries and metastatic sites (i.e. not paired). These samples may ultimately represent different snapshots of tumor evolution. Comparability with the TCGA cohort is limited as well, since TCGA samples were all obtained from untreated primary tumors. The rarity of tRCC imposes restrictions on the statistical power of our study; although we provided detailed clinical outcomes for all the patients included, another drawback is clearly the limited size of our cohort and of the TCGA cohort. However, our study only included cytogenetically confirmed tRCC and thus minimized contamination of the sample set with non-tRCC cases, which has not been routinely done in prior studies. Moreover, we acknowledge the fact that TFE3- and TFEB-associated tRCCs show distinct morphological features. However, due to our limited sample size, both variants were included in our tRCC cohorts. Furthermore, we did not have RNA-seq data available for our institutional cases, thus a validation of the neoantigen prediction in TCGA cases could not be performed. With respect to the systemic therapy data, we used TOT as a surrogate of treatment response which has potential interpretation issues. However, since RECIST scores were unavailable for these patients, the actual treatment courses were displayed graphically to provide an idea of the overall clinical effect.
Due to the low incidence of tRCC, future molecular studies should consider a multi-institutional pooling of cases to deepen our understanding of these tumors. Additionally, the collection of a larger number of patients who underwent systemic therapy could enable the evaluation of potential relationships between genomic aberrations and treatment response. Additional transcriptomic analyses could shed further light on potential immunogenic or angiogenic effects of the aforementioned copy number alterations. This is particularly interesting with regards to a recently opened multi-institutional prospective trial of an ICB/TKI combination therapy in tRCC patients (NCT03595124), a study of an ICB/anti-VEGF combination regimen including tRCC patients (NCT02724878) as well as potential trials involving neoantigen vaccines.
Conclusion
CNVs appear to play a key role in the prognosis of tRCC. Copy number aberrations are pervasive in tRCC, and appear to condition poor outcomes. The presence of TERT hotspot mutations in cases of advanced disease suggests an association with molecular progression in tRCC. An increased frequency of oncogenic events (including somatic mutations and CNVs) in adult patients could presumably be driving more aggressive disease phenotypes.
Supplementary Material
Translational relevance.
Translocation renal cell carcinoma (tRCC) is a rare, aggressive form of kidney cancer. Currently, there are no established biomarkers to stratify this disease by aggressiveness or to evaluate treatment response. Our study found that certain copy number variations (CNVs) were associated with disease aggressiveness in tRCC. By pooling our results and previously-published data, we could confirm that tumors bearing 9p loss, 17q gain or a generically high burden of CNVs was associated with poor survival in these patients. Additionally, three pediatric cases with an indolent disease course were found to have a lower copy number burden. The molecular features of patients showing exceptional responses to treatment are highlighted. We identified microenvironmental characteristics of tRCC tumors distinguishing them from common subtypes of kidney cancer. Finally, we explored the genetic evolution of tRCC in a patient with temporally distinct tumor samples and observed an early incidence of CNVs with later mutational diversification.
Acknowledgments:
This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Julian Marcon obtained financial support from the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, grant no. MA 8008/01 and /02).
Ed Reznik was supported by the Geoffrey Beene Cancer Research Center, the Department of Defense Kidney Cancer Research Program W81XWH-18-1-0318, and Kidney Cancer Association Young Investigator Award.
Conflicts of interest:
Robert J. Motzer reports serving in a consultancy or advisory role for Pfizer, Novartis, Genentech/Roche, Lilly Oncology, Eisai and Exelixis, and received research funding from Bristol-Myers Squibb, Pfizer, Eisai and Exelixis.
Victor E. Reuter is a non-compensated advisor for PAIGE.AI.
Martin H. Voss reports receiving commercial research support from Pfizer and honoraria from Pfizer, Exelixis, Eisai, Calithera Biosciences and Corvus Pharmaceuticals.
Timothy A. Chan is a cofounder of Gritstone Oncology and holds equity in An2H. Timothy A. Chan serves as an advisor for Bristol-Myers Squibb, Illumina and AstraZeneca. Timothy A. Chan receives research support from AstraZeneca and Illumina. Timothy A. Chan holds a patent for the use of TMB to predict immunotherapy response. This is licensed to PGDx and MSK and Timothy A. Chan are entitled to receive royalties.
Vladimir Makarov holds patent rights.
Julian Marcon, Renzo G. DiNatale, Alejandro Sanchez, Ritesh Kotecha, Sounak Gupta, Fengshen Kuo, Amar Sandhu, Roy Mano, Andrew W. Silagy, Kyle A. Blum, Daniel E. Nassau, Nicole E. Benfante, Michael V. Ortiz, Maria I. Carlo, Jonathan A. Coleman, Paul Russo, A. Ari Hakimi and Ed Reznik declare that they have no conflict of interest to report.
References:
- 1.Ross H, Argani P. Xp11 translocation renal cell carcinoma. Pathology. 2010;42:369–73. [DOI] [PubMed] [Google Scholar]
- 2.Cajaiba MM, Dyer LM, Geller JI, Jennings LJ, George D, Kirschmann D, et al. The classification of pediatric and young adult renal cell carcinomas registered on the children’s oncology group (COG) protocol AREN03B2 after focused genetic testing. Cancer. 2018;124:3381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magers MJ, Udager AM, Mehra R. MiT Family Translocation-Associated Renal Cell Carcinoma: A Contemporary Update With Emphasis on Morphologic, Immunophenotypic, and Molecular Mimics. Arch Pathol Lab Med. 2015;139:1224–33. [DOI] [PubMed] [Google Scholar]
- 4.Kauffman EC, Ricketts CJ, Rais-Bahrami S, Yang Y, Merino MJ, Bottaro DP, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014;11:465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malouf GG, Monzon FA, Couturier J, Molinié V, Escudier B, Camparo P, et al. Genomic heterogeneity of translocation renal cell carcinoma. Clin Cancer Res. 2013;19:4673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell. 2018;173:581–94.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKay RR, Bossé D, Xie W, Wankowicz SAM, Flaifel A, Brandao R, et al. The Clinical Activity of PD-1/PD-L1 Inhibitors in Metastatic Non-Clear Cell Renal Cell Carcinoma. Cancer Immunol Res. 2018;6:758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koshkin VS, Barata PC, Zhang T, George DJ, Atkins MB, Kelly WJ, et al. Clinical activity of nivolumab in patients with non-clear cell renal cell carcinoma. J Immunother Cancer. 2018;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malouf GG, Camparo P, Oudard S, Schleiermacher G, Theodore C, Rustine A, et al. Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol. 2010;21:1834–8. [DOI] [PubMed] [Google Scholar]
- 10.Boilève A, Carlo MI, Barthélémy P, Oudard S, Borchiellini D, Voss MH, et al. Immune checkpoint inhibitors in MITF family translocation renal cell carcinomas and genetic correlates of exceptional responders. J Immunother Cancer. 2018;6:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green WM, Yonescu R, Morsberger L, Morris K, Netto GJ, Epstein JI, et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am J Surg Pathol. 2013;37:1150–63. [DOI] [PubMed] [Google Scholar]
- 12.Gao Q, Liang W-W, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018;23:227–38.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, et al. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018;46:D1144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol [Internet]. 2017;2017 Available from: 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: Fast and accurate variant calling for clinical sequencing applications [Internet]. Available from: 10.1101/192872 [DOI] [PubMed] [Google Scholar]
- 18.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, WGS500 Consortium, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherry ST. dbSNP: the NCBI database of genetic variation [Internet]. Nucleic Acids Research. 2001. page 308–11. Available from: 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium T 1000 GP, The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing [Internet]. Nature. 2010. page 1061–73. Available from: 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016;26:108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–95. [DOI] [PubMed] [Google Scholar]
- 28.Şenbabaoğlu Y, Gejman RS. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA …. Genome [Internet]. genomebiology.biomedcentral.com; 2016; Available from: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-1092-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakimi AA, Voss MH, Kuo F, Sanchez A, Liu M, Nixon BG, et al. Transcriptomic Profiling of the Tumor Microenvironment Reveals Distinct Subgroups of Clear Cell Renal Cell Cancer: Data from a Randomized Phase III Trial. Cancer Discov. 2019;9:510–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 2017;16:2598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jelinic P, Ricca J, Van Oudenhove E, Olvera N, Merghoub T, Levine DA, et al. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. J Natl Cancer Inst. 2018;110:787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malouf GG, Camparo P, Molinié V, Dedet G, Oudard S, Schleiermacher G, et al. Transcription factor E3 and transcription factor EB renal cell carcinomas: clinical features, biological behavior and prognostic factors. J Urol. 2011;185:24–9. [DOI] [PubMed] [Google Scholar]
- 37.Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell. 2018;173:595–610.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunelli M, Eccher A, Gobbo S, Ficarra V, Novara G, Cossu-Rocca P, et al. Loss of chromosome 9p is an independent prognostic factor in patients with clear cell renal cell carcinoma. Mod Pathol. 2008;21:1–6. [DOI] [PubMed] [Google Scholar]
- 39.Klatte T, Pantuck AJ, Said JW, Seligson DB, Rao NP, LaRochelle JC, et al. Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma. Clin Cancer Res. 2009;15:1162–9. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Lu A, Li L, Yue J, Lu Y. p16 Modulates VEGF expression via its interaction with HIF-1alpha in breast cancer cells. Cancer Invest. 2010;28:588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casuscelli J, Becerra MF, Manley BJ, Zabor EC, Reznik E, Redzematovic A, et al. Characterization and Impact of TERT Promoter Region Mutations on Clinical Outcome in Renal Cell Carcinoma. Eur Urol Focus [Internet]. 2017; Available from: 10.1016/j.euf.2017.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez Chanzá N, Xie W, Asim Bilen M, Dzimitrowicz H, Burkart J, Geynisman DM, et al. Cabozantinib in advanced non-clear-cell renal cell carcinoma: a multicentre, retrospective, cohort study. Lancet Oncol. 2019;20:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choueiri TK, Fay AP, Gray KP, Callea M, Ho TH, Albiges L, et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol. 2014;25:2178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee C-H, Yelensky R, Jooss K, Chan TA. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018;39:536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J Clin Oncol. 2018;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science [Internet]. 2017;355 Available from: 10.1126/science.aaf8399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geller JI, Argani P, Adeniran A, Hampton E, De Marzo A, Hicks J, et al. Translocation renal cell carcinoma: lack of negative impact due to lymph node spread. Cancer. 2008;112:1607–16. [DOI] [PubMed] [Google Scholar]
- 48.Argani P, Laé M, Ballard ET, Amin M, Manivel C, Hutchinson B, et al. Translocation Carcinomas of the Kidney After Chemotherapy in Childhood [Internet]. Journal of Clinical Oncology. 2006. page 1529–34. Available from: 10.1200/jco.2005.04.4693 [DOI] [PubMed] [Google Scholar]
- 49.Falzarano SM, McKenney JK, Montironi R, Eble JN, Osunkoya AO, Guo J, et al. Renal Cell Carcinoma Occurring in Patients With Prior Neuroblastoma. Am J Surg Pathol. Wolters Kluwer; 2016;40:989–97. [DOI] [PubMed] [Google Scholar]
- 50.Udager AM, Mehra R. Morphologic, molecular, and taxonomic evolution of renal cell carcinoma: a conceptual perspective with emphasis on updates to the 2016 World Health Organization Classification. Arch Pathol Lab Med. the College of American Pathologists; 2016;140:1026–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.