

Abstract
Rationale:
Mitochondrial Ca2+ loading augments oxidative metabolism to match functional demands during times of increased work or injury. However, mitochondrial Ca2+ overload also directly causes mitochondrial rupture and cardiomyocyte death during ischemia-reperfusion injury by inducing mitochondrial permeability transition pore opening. The mitochondrial Ca2+ uniporter (MCU) mediates mitochondrial Ca2+ influx, and its activity is modulated by partner proteins in its molecular complex, including the MCUb subunit.
Objective:
Here we sought to examine the function of the MCUb subunit of the MCU-complex in regulating mitochondria Ca2+ influx dynamics, acute cardiac injury and long-term adaptation after ischemic injury.
Methods and Results:
Cardiomyocyte-specific MCUb overexpressing transgenic mice and Mcub gene-deleted (Mcub−/−) mice were generated to dissect the molecular function of this protein in the heart. We observed that MCUb protein is undetectable in the adult mouse heart at baseline, but mRNA and protein are induced after ischemia-reperfusion injury. MCUb overexpressing mice demonstrated inhibited mitochondrial Ca2+ uptake in cardiomyocytes and partial protection from ischemia-reperfusion injury by reducing mitochondrial permeability transition pore opening. Antithetically, deletion of the Mcub gene exacerbated pathologic cardiac remodeling and infarct expansion after ischemic injury in association with greater mitochondrial Ca2+ uptake. Furthermore, hindlimb remote ischemic pre-conditioning induced MCUb expression in the heart, which was associated with decreased mitochondrial Ca2+ uptake, collectively suggesting that induction of MCUb protein in the heart is protective. Similarly, mouse embryonic fibroblasts from Mcub−/− mice were more sensitive to Ca2+ overload.
Conclusions:
Our studies suggest that Mcub is a protective cardiac inducible gene that reduces mitochondrial Ca2+ influx and permeability transition pore opening after ischemic injury to reduce ongoing pathological remodeling.
Subject Terms: Animal Models of Human Disease, Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling, Heart Failure, Ischemia
Keywords: Mitochondrial calcium, mitochondria, Ca2+ handling, heart, ischemic injury, reperfusion injury, calcium regulation, infarction
Graphical Abstract
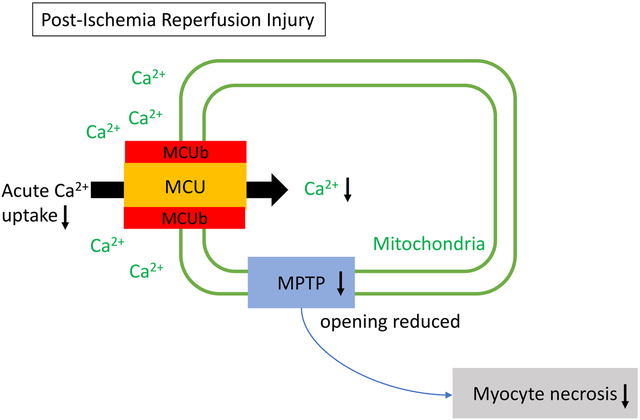
Our study uniquely demonstrates the physiological relevance of MCUb in the heart. We utilized both MCUb-overexpressing and Mcub−/− mice. Importantly, this is the first report of the phenotype of genetic loss of Mcub in the heart. We show that MCUb is uniquely induced in the heart within 2–3 days after ischemic injury, where it then decreases mitochondrial Ca2+ uptake via the mitochondrial Ca2+ uniporter, which reduces the extent of cardiac damage in the proceeding days. We also observed that MCUb expression is induced in the heart by remote ischemia preconditioning of the hindlimb, which similarly reduces mitochondrial Ca2+ influx. Taken together, our data show that MCUb induction is an endogenous protective compensatory measure that reduces mitochondrial Ca2+ overload-induced injury and ongoing borderzone expansion through cardiomyocyte death.
INTRODUCTION
Ischemic heart disease is one of the leading causes of death worldwide. During ischemia, obstruction of the coronary arteries restricts blood flow causing a shortage of oxygen and nutrients in the affected myocardium, which can directly kill cardiomyocytes.1, 2 In some settings vascular flow can be reestablished by surgical intervention that often reduces the total level of myocardial wall necrosis, although the reperfusion phase itself can also kill cardiomyocytes.3 In animal models ischemia-reperfusion (I/R) injury causes cardiomyocyte death by exposing these cells to high levels of free cytosolic Ca2+, which then causes mitochondrial permeability transition pore (mPTP) opening.1, 3 The mPTP is a pore-forming protein complex spanning the mitochondrial inner and outer membranes opens in response to high intracellular Ca2+ and reactive oxygen species, where it leads to dissipation of mitochondrial membrane potential, organelle swelling and rupture, ultimately leading to necrotic cell death.4, 5 Cyclophilin D is an important regulator of mPTP opening and use of cyclophilin inhibitory drugs such as cyclosporine A (CsA) desensitize pore opening, resulting in less cardiomyocyte death after I/R injury.1, 6 Moreover, reducing mitochondrial Ca2+ uptake in the heart during I/R injury can also reduce mPTP opening and subsequent myocyte necrosis.7
Transport of Ca2+ into the mitochondrial matrix is mediated by the mitochondrial Ca2+ uniporter (MCU)-complex, which serves as the major Ca2+ influx pathway across the inner membrane, and this activity can be inhibited by Ru360 or ruthenium red.8, 9 Ca2+ efflux is due to the activity of the Na+/Ca2+/Li+ exchanger (NCLX), which is also in the mitochondrial inner membrane.10 The MCU-complex contains four MCU-subunits, produced by either the Mcu or Mcub gene.11 If the tetramer is comprised of the Mcu gene product (MCU) it forms a pore that readily transduces Ca2+; however, the presence of the Mcub gene product (MCUb) appears to antagonize Ca2+ influx by the MCU-complex.12 The MCU-complex also contains a number of regulatory components that alter the kinetics of Ca2+ uptake or even assembly of the complex in the inner membrane, such as mitochondrial Ca2+ uptake 1/2 proteins (MICU1/MICU2) and the essential MCU regulator, mitochondria (EMRE).13, 14 The minimal functional Ca2+ influx channel has recently been reported to consist of MCU and EMRE.15
MCUb is encoded by the gene originally annotated as ccdc109b, which is now officially known as Mcub. MCUb is structurally similar to MCU although the critical “DIME” motif present in MCU that acts as the Ca2+ selectivity region of the channel is slightly different in MCUb.12 As stated above, in vitro studies suggest that MCUb becomes part of the tetrameric core of the MCU-complex where it appears to inhibit Ca2+ influx.12 However, deletion of MCU from neurons of the mouse brain did not eliminate all Ca uptake (only 80% inhibited) and the observed residual activity was suggested to arise from MCUb.16 Cardiac-specific transgenic (TG) mice overexpressing a dominant negative (dn) MCU mutant were generated, which like MCUb, has alterations in the “DIME” motif.17 Surprisingly, dnMCU mice were not protected from I/R injury despite near complete inhibition of acute mitochondrial Ca2+ uptake.12 Consistent with this, Mcu−/− mice, which also show a complete lack in acute mitochondrial Ca2+ uptake, did not show cardioprotection after I/R injury.18, 19 In contrast, inducible deletion of the Mcu gene from the adult mouse heart did protect from I/R injury and reduce mPTP opening.7, 20 This inconsistency was attributed to an unknown compensatory effect that occurs during development in total somatic Mcu−/− mice, but which does not occur when the gene is deleted in the adult heart for the first time.18, 19 More recently, cardiac-specific MCUb TG mice were generated using a tamoxifen inducible, DNA recombination-dependent overexpression strategy.21 These inducible MCUb-TG mice showed extreme lethality (12/13) when I/R was performed 1 wk after acute tamoxifen-mediated activation of MCUb expression, although when performed 1 month after tamoxifen treatment this increased lethality was no longer observed and instead mice showed significantly reduced infarct sizes.21 Collectively, these results highlight the complexity and continuing uncertainty surrounding MCUb function in the heart, and no one has yet investigated mice lacking the Mcub gene in the heart.
Here we observed that MCUb is essential in regulating mitochondrial Ca2+ dynamics by limiting MCU-complex Ca2+ uptake. Transgene-mediated constitutive overexpression of MCUb in the heart lowered mitochondrial Ca2+ uptake rates and was protective during acute I/R injury by reducing cell death and mPTP opening frequency. With respect to physiologic significance, MCUb protein is normally not detectable in the adult heart but upon injury this gene product is induced where it serves a protective function. Indeed, Mcub−/− mice show greater mitochondrial Ca2+ uptake days after I/R injury that is associated with greater ventricular pathology and remodeling compared with wildtype (WT) controls. Moreover, we observed that remote ischemic pre-conditioning (RIPC) also induces MCUb expression in the heart and that this limits mitochondrial Ca2+ uptake. Our results are the first to show that the Mcub gene plays a necessary physiologic role in the heart where it’s induction after I/R injury limits ongoing pathological remodeling and infarct expansion.
METHODS
Detailed methods are available in the Online Data Supplement. All original data, additional detailed methods and materials used will be provided upon reasonable request to the corresponding author.
Animals.
All experimental use of mice was approved by the Institutional Animal Care and Use Committee (IACUC). Cardiac-specific MCUb overexpressing TG mice were generated using the cardiac-specific tetracycline (Tet)-off expression system as described previously.22 A tetracycline transactivator (tTA) TG mouse line with a cardiac-specific promoter (α-myosin heavy chain, α-MHC) was used along with another TG mouse line in which the mouse Mcub cDNA was under the control of the α-MHC-tetracycline operator sequences. Mcub-null mice were also generated from previously targeted ES cells using the “knockout-first allele” strategy that is targeted between the 1st and 2nd exon (Mcubtm1a(KOMP)Mbp, European Conditional Mouse Mutagenesis program, MGI ID: 1914065). Mouse embryonic fibroblasts (MEFs) were generated and cultured as described previously.23 Mouse embryonic fibroblasts (MEFs) were generated from Mcub-loxP (fl) and Mcufl/fl mice.7 These mice were also crossed together to generate Mcufl/flMcubfl/fl double-targeted MEFs.
Surgery.
I/R injury was performed as previously described.7, 24 Mice were closely monitored after surgery and received necessary pain relief treatment (Buprenorphine, 0.03mg/mL, subcutaneous injection) to minimize discomfort. For myocardial infarction (MI) injury, mice were challenged with permanent left coronary artery ligation and pain medication was given (Buprenorphine, 0.03mg/mL, subcutaneous injection).
Mitochondrial Isolation and analyses.
Heart or MEF mitochondria were isolated by differential centrifugation as described previously.7, 23
Ca2+ and mPTP measurement in cardiomyocytes.
Adult cardiac ventricular myocytes were isolated from mouse hearts using previously described methods.25
Ca2+ sparks and transient measurements.
Intact ventricular myocytes were loaded with Fluo-4 AM dye (5 μmol/L) for 30 minutes, transients and sparks were recorded as previously described.25, 26 Calcium transients were obtained by field stimulation at 1 Hz in normal Tyrode’s buffer with 1.8 mmol/L Ca2+. Sarcoplasmic reticulum (SR) Ca2+ load was evaluated by the Ca2+ transient amplitude upon rapid application of caffeine (10 mmol/L). Images were acquired with confocal microscopy (Nikon, ×40 objective) using line scan mode with excitation at 488 nm, emission at >505 nm. Images were analyzed using ImageJ software and Sparkmaster.27
RESULTS
MCUb expression is induced following I/R injury.
To investigate the function of the Mcub gene in regulating mitochondrial Ca2+ influx with ischemic injury we first carefully examined Mcub mRNA and MCUb protein expression, along with the other components of the MCU-complex, following a time-course of I/R injury (Fig. 1A–B). WT mice were challenged with I/R injury to their hearts (60 minutes of ischemia) and then harvested 1-, 3- or 7-days afterwards. The left ventricle of the heart was used for analysis (either whole tissue) to focus on the infarct and border-zone regions. We observed that both Mcub mRNA and MCUb protein expression were increased starting 3 days after I/R injury compared to sham controls, and this induction was even stronger 7 days after injury (Fig. 1A–B). Importantly, the specificity of the MCUb custom made antibody was verified in Mcub−/− hearts after I/R injury (and null MEFs), as all commercial antibodies that we surveyed failed and were either non-specific or did not detect MCUb. We believe that the increase in MCUb protein observed from purified heart mitochondria after I/R injury is dominantly due to cardiomyocyte expression since the contribution to the total mitochondrial content from immune cells, fibroblasts and endothelial cell fraction in the heart is negligible. mRNA for other MCU-complex components (Mcu, Micu1, Micu2 and Smdt1 (EMRE encoding gene)) also showed increased expression following I/R injury, but to a lower degree than Mcub (Fig.1A). Interestingly, we also observed that MCU and EMRE protein levels were increased in the heart 7 days post I/R injury (Fig 1B). To extend these results permanent myocardial infarction (MI) injury was performed on WT mice, which similarly showed induction of MCUb protein in the heart by 3 and 7 days after injury (Fig. 1C). However, it should be noted that we were unable to detect MCUb protein in the hearts of uninjured adult mice, suggesting that this protein might only have a functional role in the heart days after an ischemic injury event.
Figure 1. MCUb expression is induced following cardiac ischemic injury.
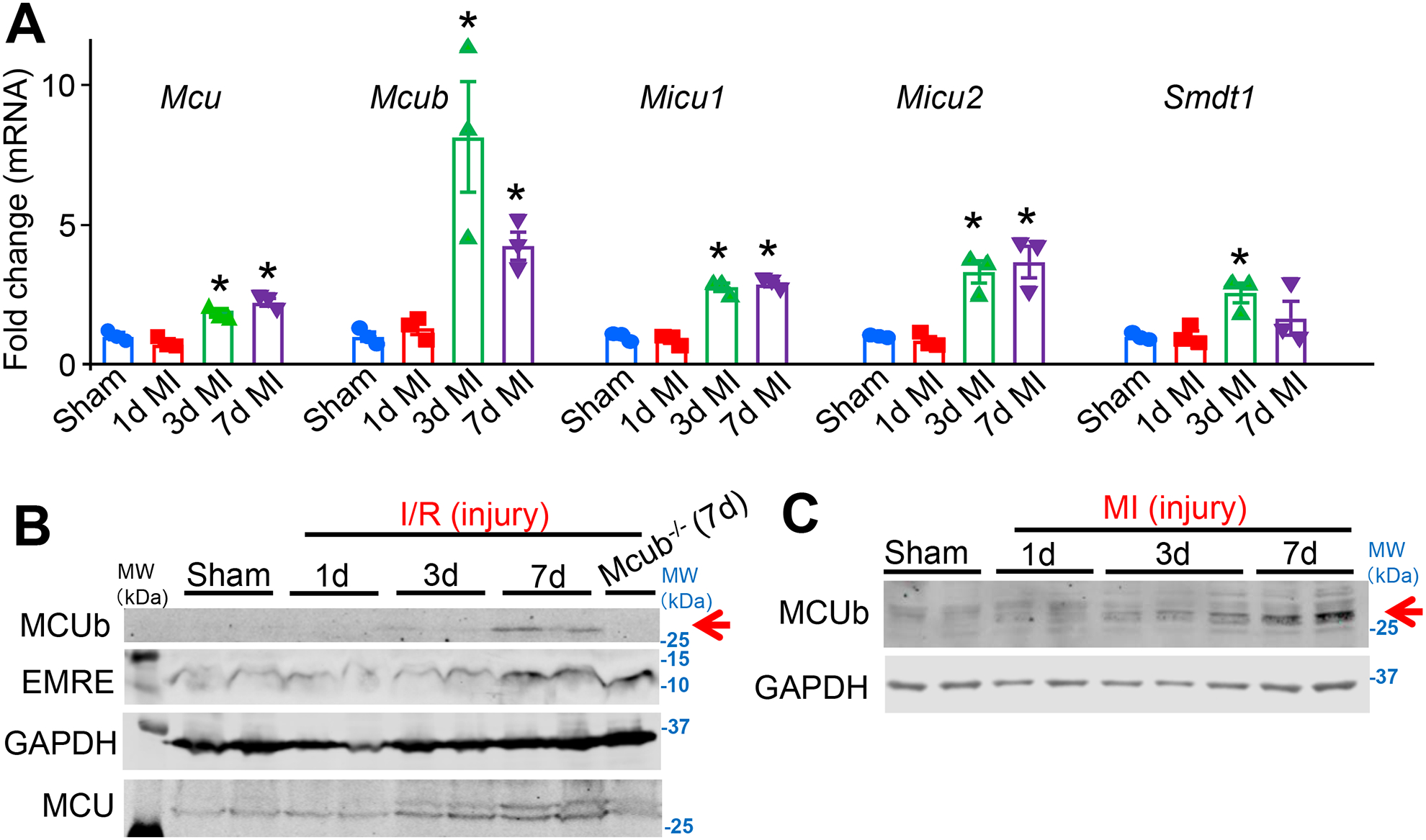
A) MCU-complex components Mcu, Mcub, Micu1, Micu2 and Smdt1 (EMRE) mRNA levels from cardiac left ventricle tissue and isolated myocytes following ischemia reperfusion (I/R) injury for 1-, 3- or 7-days versus sham. n=3 per group. Data presented as mean ± standard deviation of mean (SEM). One-way ANOVA and post hoc Bonferroni test was used for statistical analysis. *p<0.05 versus sham by post hoc Bonferroni’s multiple comparison test. B) Western blot of MCUb, MCU and EMRE from left ventricle of the WT mouse heart following a time-course of I/R injury as shown in days. GAPDH was used as the protein loading control. Mcub−/− heart tissues 7 days after I/R injury is used as an antibody negative control. Arrow shows position of MCUb protein. The molecular weight marker shown is based on the uncropped western blots in which standards can (Online Supplement, and applicable to the remaining western blots in the paper). C) Western blot of MCUb from the heart following a time-course of myocardial infarction (MI) injury as shown. GAPDH was used as the protein loading control.
Cardiomyocyte-specific MCUb overexpressing mice.
To model the observed induction of MCUb in the heart after injury and to examine its potential function we developed cardiac-specific MCUb TG mice using the tet-off system.28 A tetracycline transactivator (tTA) TG mouse line with cardiac-specific expression (α-myosin heavy chain, α-MHC) was crossed with α-MHC tetracycline-operator responsive TG mice containing a MCUb cDNA (MCUb-TG) to generate experimental double TG (MCUb-DTG) mice (Fig.2A). Western blotting from purified mitochondria from the hearts of tTA-TG, single MCUb-TG and the MCUb-DTG mice was performed at 6 wks of age (Fig. 2B). The data show no expression of MCUb protein in tTA-TG hearts but substantial expression in the hearts of MCUb-DTG mice (Fig. 2B). We should also note that the single MCUb-TG line showed minimally detectable MCUb protein, due to slight leak of the single TG construct (Fig. 2B). MCU, MICU1, MICU2 and NCLX protein levels were not changed with MCUb overexpression in the heart, but interestingly, EMRE levels were reduced compared with the 2 controls (Fig. 2B). As recently proposed, EMRE is essential for MCU complex assembly such that MCUb overexpression might displace MCU from the tetramer and its association with EMRE.29 To examine this concept further we performed co-immunoprecipitation (Co-IP) for EMRE using MCU antibody, which showed that MCUb overexpression inhibited the presence of EMRE within the MCU complex (Fig. 2C). Previous studies showed that deleting Mcu in the heart decreased the expression level of NCLX to maintain overall mitochondrial Ca2+ flux.7 However, we did not observe a difference in NCLX protein levels with MCUb is overexpression in the heart (Fig. 2B), although NCLX activity was reduced in adult cardiomyocytes from these MCUb-DTG mice (Online Fig. I), presumably as a compensatory measure to again maintain total mitochondrial Ca2+ flux potential.
Figure 2. Generation of cardiomyocyte-specific MCUb overexpressing transgenic mice.
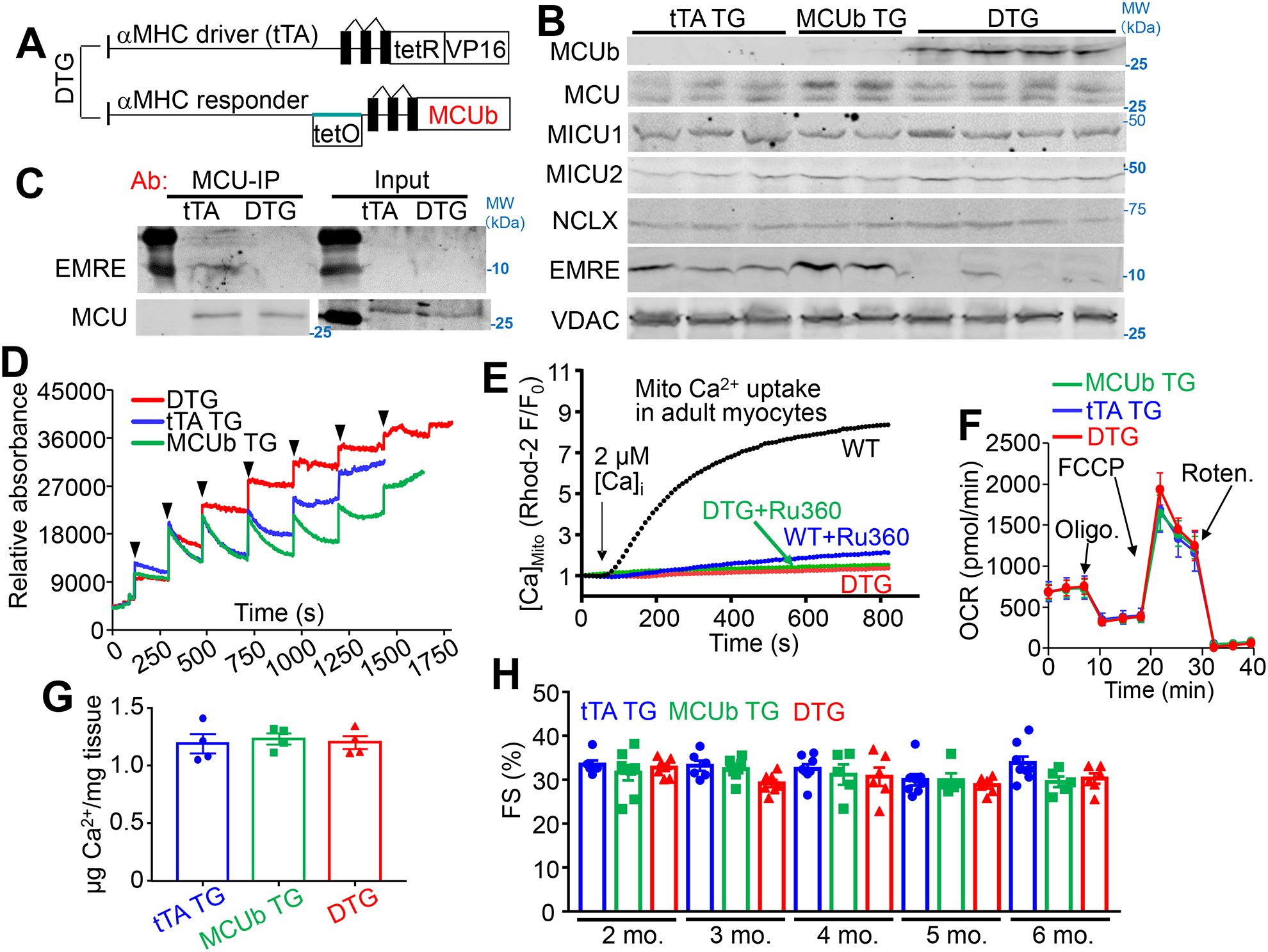
A) Schematic of cardiomyocyte-specific MCUb overexpressing mice using a double transgenic (DTG) tet-off system based on the α-myosin heavy chain (α-MHC) promoter. The driver line expresses the tetracycline transactivator cDNA (tTA), and the responder line contains the tet-operator and the MCUb cDNA. B) Western blot of MCUb, MCU, MICU1, MICU2, NCLX and EMRE in cardiac mitochondria from the indicated groups of mice. VDAC was used as a processing and loading control. C) Co-immunoprecipitation of EMRE with an MCU antibody from isolated heart mitochondria from the indicated two groups of mice. Western blotting for MCU from both immunoprecipitated samples as well as input samples is shown as controls. D) Mitochondrial Ca2+ uptake in isolated heart mitochondria from the indicated groups at 3 months of age. Calcium Green-5N was used as the Ca2+ indicator. The arrows indicate addition of 20 μM CaCl2 to the solution. E) Mitochondrial Ca2+ uptake in permeabilized adult cardiomyocytes from hearts of the indicated groups, with or without Ru360 under 2 μmol/L CaCl2 perfusion. F) Oxygen consumption rate (OCR) measurement in cardiac mitochondria from the indicated mouse groups of mice at 3 months of age. n=3 per group. Arrows show the position of the three different drugs given in temporal sequence. G) Quantification of baseline mitochondrial Ca2+ levels in isolated cardiac mitochondria from the indicated groups of mice at 6 months of age. n=4 per group. Data presented as mean ± SEM. One-way ANOVA was used for statistical analysis. H) Echocardiographic measurement of fractional shortening (FS, %) from indicated groups at the indicated ages in months. n=7 in tTA TG group, n=5 in MCUb TG group, and n=6 in DTG group. Data presented as mean ± SEM. Two-way ANOVA was used for statistical analysis, with no significant interactions or effects of variables.
Purified mitochondria from hearts of MCUb DTG mice showed inhibited Ca2+ uptake compared with the two control groups (Fig. 2D). We also examined mitochondrial Ca2+ uptake in living but permeabilized cardiomyocytes in culture using a Rhod-2-fluorescence assay, which again showed that MCUb overexpression blocked mitochondrial Ca2+ uptake compared to controls, and this block was similar in effect to treatment with the MCU-complex inhibitor Ru360 (Fig. 2E). Collectively these data suggest that MCUb functions as a type of dominant negative effector of the MCU-complex.
While acute mitochondrial Ca2+ uptake was inhibited by MCUb overexpression in both purified mitochondria and intact permeabilized adult cardiomyocytes, there was no effect on oxygen consumption rate (OCR) using a seahorse assay in purified mitochondria from MCUb-DTG hearts, nor was there a reduction in baseline mitochondrial Ca2+ content (Fig. 2F–G). We previously showed that mitochondria from conditional Mcu-deleted hearts also had no reduction in baseline mitochondrial Ca2+ levels nor oxygen consumption.7 Such results suggest that other Ca2+ influx pathways can partially compensate for the loss of MCU-complex activity to allow long-term control of Ca2+ levels in the mitochondrial matrix and associated metabolic dynamics.18 Indeed, overexpression of MCUb in the hearts of MCUb-DTG mice showed no pathological effect as these mice aged, such as no reduction in cardiac fractional shortening measured by echocardiography (Fig. 2H), nor a change in histopathological features, which would be expected if there was a metabolic deficiency (Online Fig. II). MCUb overexpression also did not alter cardiomyocyte intracellular Ca2+ handling, such as the amplitude and kinetics of the Ca2+ transient, SR Ca2+ content, or Ca2+ spark frequency (Online Fig. II). In addition, overexpressing MCUb in the heart did not change mitochondrial ultrastructure or show other pathological features by transmission electron microscopy (Online Fig. III). Collectively these findings indicate that MCUb overexpression is not deleterious to cardiac structure-function.
MCUb overexpression protects from I/R injury.
An area of controversy in the MCU cardiac literature is based on the observation that loss of MCU (Mcu−/− mice) in certain genetic backgrounds of mice does not cause lethality, and the viability is presumably due to genetic compensation by other Ca2+ influx mechanisms.19 Consistent with this idea, a study of mitoplasts isolated from cells where MCU expression was knocked-down with siRNA showed that MCU-independent Ca2+ currents were increased or induced when MCU levels are inhibited.30 Moreover, viable germline Mcu−/− mice were not protected from I/R injury, yet adult inducible and cardiomyocyte-specific Mcu deletion in a pure C57BL/6 genetic background did result in less I/R injury to the heart.7, 18 Inducible expression of MCUb in the adult mouse heart also produced protection21, hence we were uncertain how our mice with constitutive MCUb overexpression in the heart might impact acute injury responsiveness after I/R. Six-wk-old control and MCUb-DTG mice were subjected to 30 minutes of ischemia followed by 24 hours of reperfusion (Fig. 3A). Compared with controls, MCUb-DTG mice showed a significant reduction in ischemic injury with no difference in the area at risk (Fig. 3B–D). Mechanistically, we also observed that overexpression of MCUb decreased mPTP opening frequency to a similar extent as using the mPTP desensitizer CsA in tTA-TG control mitochondria (Fig. 3E). Collectively, these results suggest that chronic MCUb overexpression in the heart is not subject to compensation and that MCUb overexpression conveys acute cardioprotection following I/R injury.
Figure 3. MCUb overexpression generates protection from cardiac I/R injury.
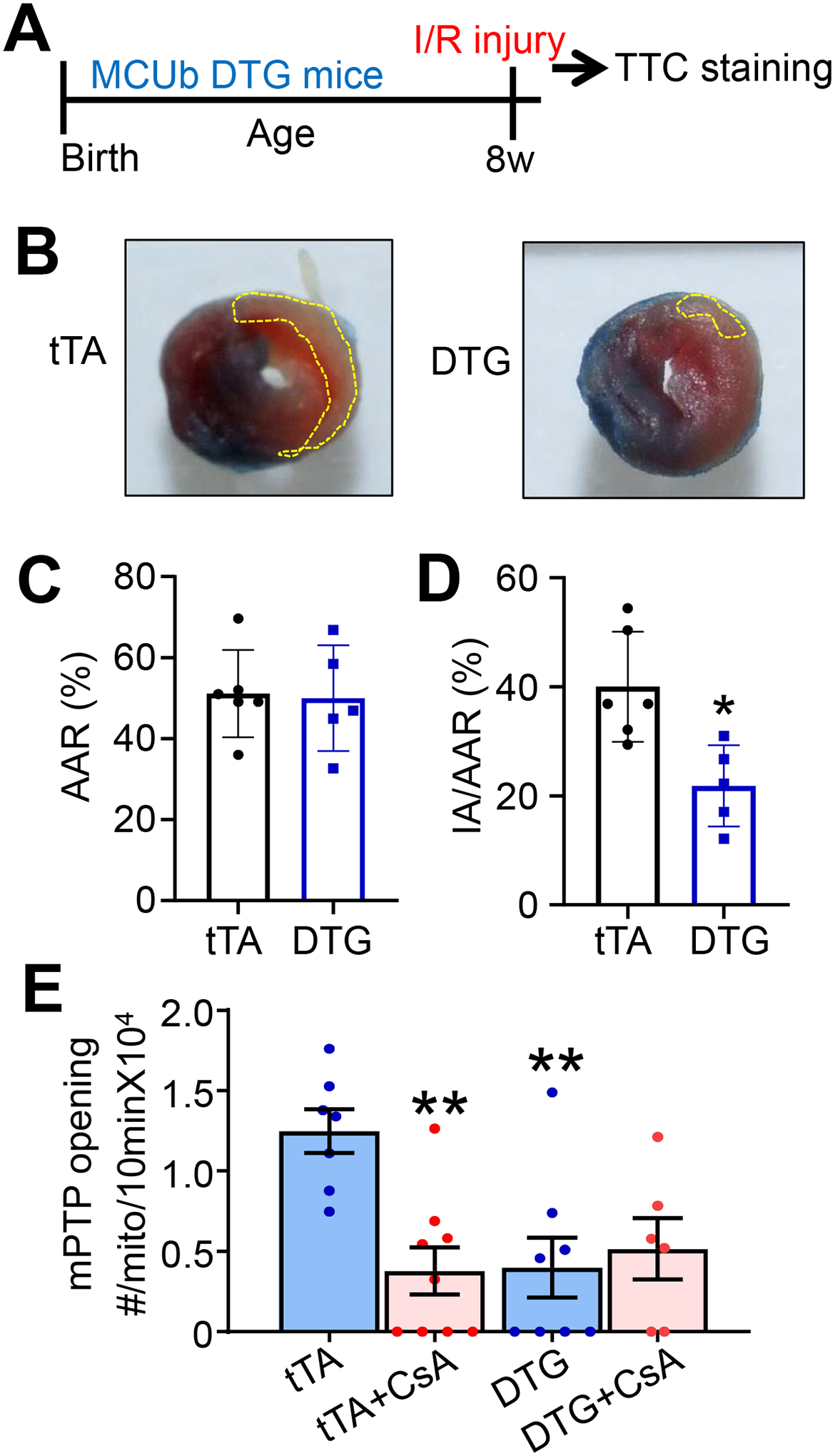
A) Temporal strategy of I/R injury in mice for panels “B”, “C” and “D”. Mice at 8 wks of age were challenged with 30 minutes of ischemia followed by 24 hours of reperfusion. B) Hearts were sacrificed for 2,3,5-triphenyltetrazolium chloride (TTC) staining. The yellow dotted area shows ischemic region. C,D) Average area at risk (AAR) and ischemic area (IA)/AAR of hearts from mice subjected to I/R injury from the indicated mouse groups. n=6 in tTA TG group, n=5 in DTG group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. *p<0.05 vs tTA. E) Mitochondrial permeability transition pore (mPTP) opening frequency measurements in permeabilized cardiomyocytes. mPTP inhibitor cyclosporine A (CsA) was used as a control. n=7 in tTA TG group, n=9 in tTA group with CsA treatment, n=8 in DTG group, n=6 in DTG group with CsA treatment. Data presented as mean ± SEM. Two-way ANOVA with a post-hoc Bonferroni’s multiple comparison test was performed. A significant interaction was found between treatment and genotype (p<0.05). **p<0.01 vs tTA.
Deletion of the Mcub gene does not affect the heart at baseline.
To understand the physiological role of MCUb, we generated and analyzed Mcub−/− mice in which this genetic locus was targeted with a “knock-out first” cDNA cassette containing β-galactosidase (LacZ) and neomycin (Neo). This allele construction disrupts protein expression without employing the conditional LoxP sites and a Cre-recombinase strategy that are built-in if needed (Fig. 4A). Since MCUb protein expression is not detectable by western blot in the heart under baseline conditions (Fig. 1B and 2B), we did not measure cardiac MCUb in our Mcub−/− groups at baseline. MCU, MICU1, MICU2 and NCLX expression were not altered in hearts of Mcub−/− mice compared to controls (Fig. 4B). Interestingly, an increase in EMRE protein expression was observed in Mcub−/− mice, which is the antithetic effect observed with MCUb overexpression whereby EMRE was reduced in the heart (Fig. 2B). No alterations were observed in Mcub−/− mouse body weights, activity, or gross anatomical features compared to controls.
Figure 4. Deletion of Mcub does not affect mitochondrial Ca2+ uptake or cardiac function at baseline.
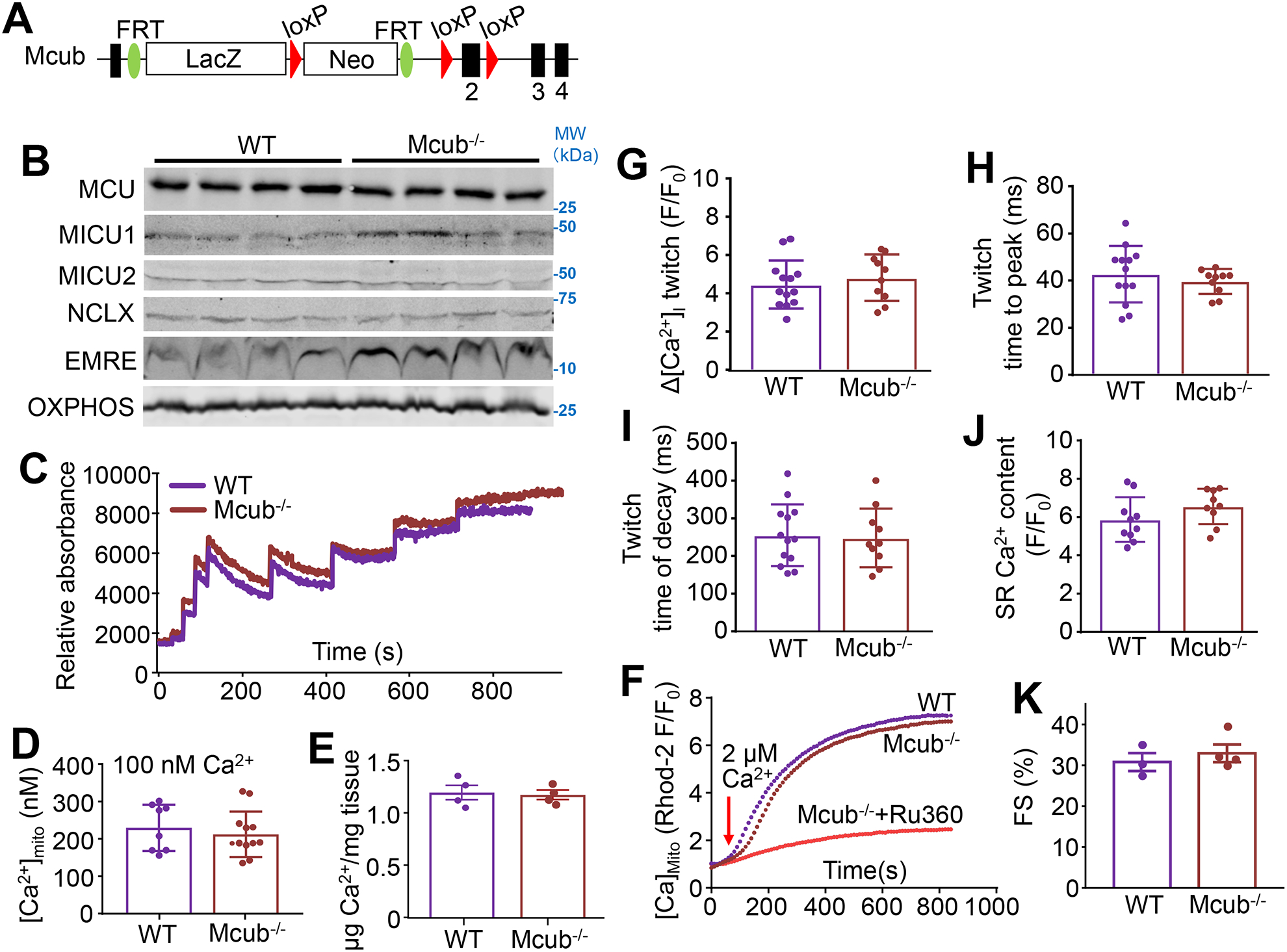
A) Strategy for generating Mcub-null mice with a “knock-out first” allele strategy in which a combined β-galactosidase/neomycin cDNA cassette was inserted between exon 1 and exon 2 of the Mcub gene. B) Western blot of MCU, MICU1, MICU2, NCLX and EMRE in cardiac mitochondria from the indicated groups of mice. OXPHOS antibody was used as a control. C) Mitochondrial Ca2+ uptake assay in isolated heart mitochondria from the indicated groups of mice at 4 months of age. D) Quantification of baseline mitochondrial Ca2+ content ([Ca2+]mito) in permeabilized adult cardiomyocytes from the indicated groups. n=8 in WT group, n=12 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. E) Quantification of baseline [Ca2+]mito levels in isolated cardiac mitochondria from the indicated groups of mice at 4 months of age. n=4 per group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. F) Measurement of mitochondrial Ca2+ uptake in permeabilized adult cardiomyocytes under 2 μmol/L CaCl2. Ru360 was used as a control. G-I) Measurements Ca2+ transient amplitude, time to peak and twitch decay (tau) from adult cardiomyocytes from hearts of the indicated groups of mice. n=13 in WT group, n=10 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. J) Measurements of sarcoplasmic reticulum (SR) Ca2+ content from adult cardiomyocytes from hearts of the indicated groups of mice. n=10 in WT group, n=9 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. K) Echocardiographic measurement of fractional shortening (FS, %) from the indicated groups at 4 months of age. n=3 in WT group, n=4 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis.
Mitochondrial Ca2+ uptake in isolated heart mitochondria from Mcub−/− mice was not altered versus control conditions, nor was baseline mitochondrial Ca2+ levels altered as measured by two different assays (Fig. 4C–E). Oxygen consumption rate was also not changed in isolated heart mitochondria from Mcub−/− mice compared to controls (Data not shown). Loss of MCUb caused no difference in the Rhod-2-fluorescence assay that measures mitochondrial Ca2+ uptake in permeabilized cardiomyocytes (Fig. 4F). Other quantitative aspects of the intracellular Ca2+ transient and Ca2+ handling kinetics were also unaltered in adult cardiomyocytes from Mcub−/− mice (Fig 4G–J). There was also no difference in Ca2+ spark frequency, Na+-dependent mitochondrial Ca2+ efflux rates, or in mPTP activity due to loss of Mcub−/− versus control animals (Online Figure IV). Cardiac function measured by echocardiography was also unaffected in Mcub−/− mice (Fig. 4K), nor was there discernible pathology in the heart by electron microscopy (Online Fig. III). Thus, loss of MCUb was of no discernable consequence to the heart at baseline.
MCUb deletion in mouse embryonic fibroblasts.
As presented earlier, adult cardiomyocytes lack detectable MCUb protein expression at baseline, which likely underlies our inability to identify a cardiac phenotype in Mcub−/− mice. However, mouse embryonic fibroblasts (MEFs) show MCUb expression at baseline (Online Fig. VA), so they were used as a more physiological model to examine the endogenous function of this gene. To generate Mcub targeted MEFs we used mice in which the LacZ-Neo cassette within the Mcub “knock-out first” allele was removed with a Flipase deletor mouse line, resulting in the generation of mice containing LoxP (fl) sites that could be used with Cre-recombinase for subsequent gene deletion. Immortalized MEFs were generated from these homozygous Mcubfl/fl mice and infection with an adenovirus expressing Cre recombinase (AdCre) in culture resulted in complete deletion and loss of MCUb protein (Online Fig. VA). We also generated Mcufl/fl MEFs as well as double-targeted Mcufl/fl/Mcubfl/fl-homozygous loxP MEFs for current analyses. AdCre infection deleted all MCU protein expression in the appropriate MEF lines (Online Fig. VA). We then assessed mitochondrial Ca2+ uptake dynamics from these MEFs. Deletion of Mcu (Mcufl/fl +AdCre) completely inhibited all Ca2+ uptake versus mitochondria from the same parent MEF cell line without adenovirus infection (Online Fig. VB), while double deletion of Mcu and Mcub (Mcufl/fl/Mcubfl/fl +AdCre) produced mitochondrial Ca2+ uptake dynamics identical to Mcu deletion, showing complete inhibition once again (Online Fig. VC). However, deletion of Mcub alone (Mcubfl/fl +AdCre) exhibited initial mitochondrial Ca2+ uptake dynamics similar to control (first 4 Ca2+ pulses) but then a much earlier transition to mPTP and release of all matrix Ca2+ was observed (i.e. reduced Ca2+ capacity and higher Ca2+ sensitivity of mPTP). This indicates that endogenous MCUb protein in MEFs normally limits Ca2+ uptake via the MCU-complex (Online Fig. VD).
Endogenous cardiac MCUb induction is cardioprotective.
To examine the physiologic function of MCUb protein induction following I/R injury we used 6-wk-old Mcub−/− mice subjected to 60 minutes of cardiac ischemia with a 24-hour reperfusion period, and then assessed injury (Fig. 5A). Importantly, there was no difference in acute injury to the heart as measured by ischemic area (IA) versus the area at risk (AAR) in Mcub−/− mice compared with control mice (Fig. 5B). Given that MCUb expression in the heart was observed 3 and 7 days after I/R injury, we next tested whether deleting MCUb alters post-injury infarct remodeling (Fig. 5C). Six-wk-old mice were challenged with 60 minutes of ischemia but were not harvested until 4 wks after surgery and assessed throughout for cardiac function by echocardiography (Fig. 5C). We observed that Mcub−/− mice developed significantly expanded left ventricle internal diameters, both end-diastolic and end-systolic, at 2 or 4 wks post injury (Fig. 5D–E, Online Fig. VI). However, Mcub−/− mice showed a trend towards a decrease in ventricular fractional shortening by 2 and 4 wks post injury as compared to controls (Fig. 5F). Gravimetric analysis showed a significantly greater increase in heart weight/body weight (HW/BW) and lung weight/body weight (LW/BW) in Mcub−/− mice 4 wks post I/R injury compared to control mice, suggesting greater pathology, most likely due to effects at the infarct borderzone (Fig. 5G–H). Indeed, direct measurements of scar area 4 wks after I/R injury showed a significant increase in Mcub−/− mice compared with control (Fig. 5I), even though initial infarct size 24 hours after I/R surgery was not different (Fig 5B). Collectively, these data indicate that MCUb induction normally plays a protective role in the heart by limiting Ca2+ influx in cardiomyocytes in the viable myocardium and borderzone, likely reducing ongoing cell death and scar expansion.
Figure 5. Cardiac MCUb protein induction protects the heart from damage post I/R injury.
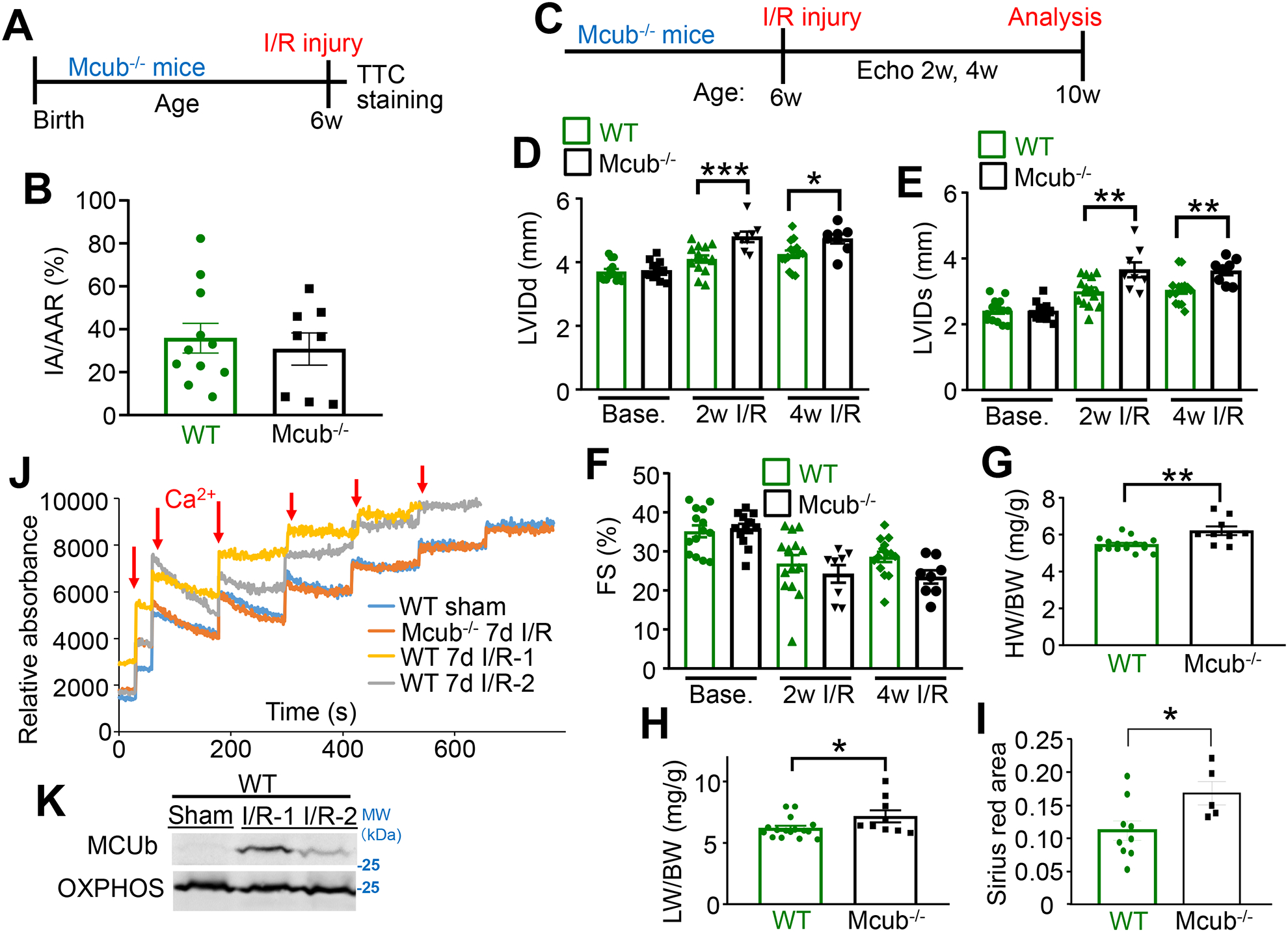
A) Temporal schematic of the I/R injury model in mice for panel “B”. Mice at 6 wks of age were challenged with 60 minutes of ischemia followed by 24 hours of reperfusion. Hearts were sacrificed for TTC staining. B) Both IA and AAR were analyzed and averaged for the indicated groups following I/R injury. n=11 in WT group, n=8 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. C) Temporal schematic of the strategy for assessing infarct expansion following I/R injury, for panels “D-H”. Mice at 6 wks of age were challenged with 60 minutes of ischemic injury. Echocardiographic measurements were performed before injury, then 2 and 4 wks of reperfusion. Mice were sacrificed at the 4 wk time point for additional analyses. D-F) Echocardiographic parameters in the indicated groups of mice at the indicated timepoints. LVIDd, end-diastolic left ventricle internal diameter; LVIDs, end-systolic left ventricle internal diameter; FS, fractional shortening. n=14 in WT group, n=12 in Mcub−/− group at baseline; n=14 in WT group, n=8 in Mcub−/− group post injury. Data presented as mean ± SEM. Two-way ANOVA with a post hoc Bonferroni’s multiple comparison test was performed. For “D&E”, there was a significant interaction (p<0.05) between time and genotype variables. For “F”, there was only a significant time effect (p<0.05) as the two-way ANOVA showed no significant interaction. Markings denote the following: *p<0.05, **p<0.01, ***p<0.001. G–H) Heart weight/body weight (HW/BW) ratio and lung weight/body weight (LW/BW) ratio in the indicated groups of mice 4 wks post I/R injury. n=14 in WT group, n=9 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. *p<0.05, **p<0.01. I) Quantification of Sirius Red staining of histological sections from hearts of mice 4 wks post I/R injury in the indicated groups. n=14 in WT group, n=9 in Mcub−/− group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. *p<0.05. J) Mitochondrial Ca2+ uptake in isolated mitochondria from the left ventricle of hearts in the indicated groups of mice. Mice were challenged with sham or I/R injury, and then collected 7 days post injury. K) Western blot of MCUb in the same isolated hearts used in “J”. OXPHOS antibody was used as the protein loading control.
We also examined the contribution of MCUb to mitochondrial Ca2+ uptake post I/R injury in control and the Mcub−/− mice (Fig. 5J–K). Western blotting from these same hearts again showed that I/R injury resulted in MCUb protein induction over 7 days (Fig. 5K). Isolated mitochondria from hearts of these WT mice 7-day post I/R injury generated a profile of inhibited mitochondrial Ca2+ uptake after injury compared to isolated mitochondria from hearts of sham controls (Fig. 5J). However, mitochondria isolated from hearts of Mcub−/− mice 7 days after I/R injury did not show this inhibited profile and instead showed normal Ca2+ uptake like that observed in uninjured sham hearts (Fig. 5J). Thus, the induction of MCUb protein expression in the heart in the first wk of I/R injury normally reduces mitochondrial Ca2+ influx to have a protective effect on the remaining myocardium. In contrast, Mcub−/− mice lack this protective mechanism and show unrestrained Ca2+ uptake after I/R injury leading to worsening of cardiac remodeling.
Remote ischemic pre-Conditioning induces MCUb expression.
We also examined if remote ischemic pre-conditioning (RIPC) induces cardiac MCUb expression prior to I/R injury. RIPC is a noninvasive ischemic procedure in rodents, whereby peripheral blood flow is interrupted to parts of the limb to activate the same molecular players in the heart that underlie protection against I/R injury.31, 32 The femoral artery of the mouse was occluded for 5-min followed by 5-min release, with a total of 4 cycles per day for seven consecutive days (Fig. 6A). The left ventricle of the heart was used for consistency with I/R groups in this paper. We found that both mRNA expression and protein levels of MCUb were induced in the heart following the RIPC procedure (Fig. 6B–C). Moreover, mitochondria isolated from the hearts of RIPC WT mice had reduced Ca2+ uptake rates compared to mitochondria isolated from sham-treated WT control mice (Fig. 6D). However, mitochondria isolated from pre-conditioned Mcub−/− mice showed intermediate Ca2+ uptake rates suggesting that MCUb was only part of the reason why RIPC affected cardiac mitochondrial Ca2+ uptake dynamics. Collectively, our data demonstrate that RIPC can induce MCUb expression in the heart, suggesting a collective mechanism whereby MCUb protein expression protects from later ischemic insults at the level of the mitochondrion.
Figure 6. MCUb is induced in the heart by post remote ischemic pre-conditioning from the hindlimb.
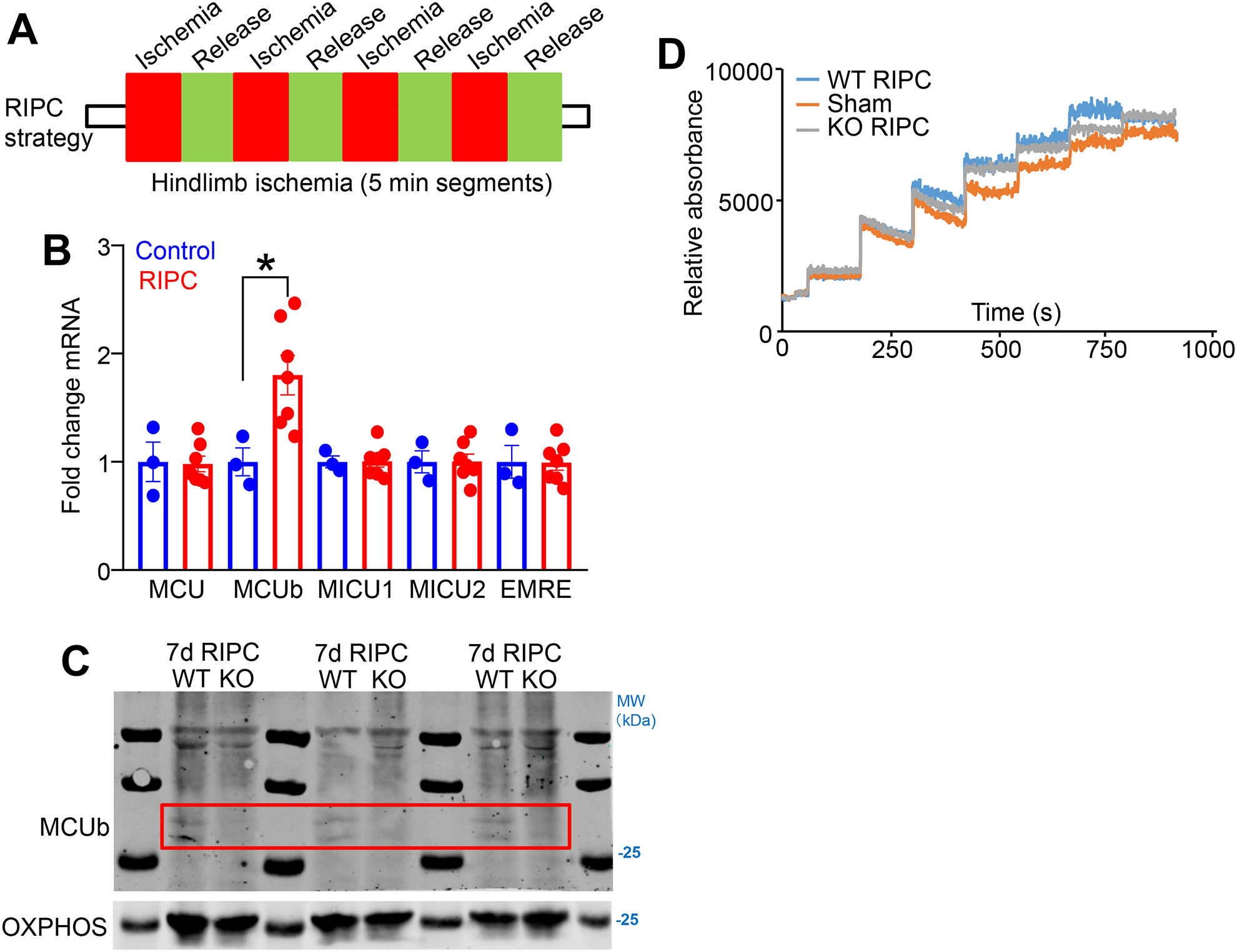
A) Temporal strategy of the remote ischemic pre-conditioning (RIPC) in mice for panels “B”, “C”, and “D”. Mice at 9–12 wks of age were challenged with hindlimb femoral artery occlusion for 5 minutes followed by 5 minutes of reperfusion, which was repeated with 4 cycles per day for 7 consecutive days. B) mRNA for MCU-complex components Mcu, Mcub, Micu1, Micu2, and Smdt1 (EMRE) from cardiac left ventricle tissue following the 7-day RIPC protocol, versus sham. n=3 in sham group, n=7 in RIPC group. Data presented as mean ± SEM. Student’s t-test was used for statistical analysis. *p<0.05. C) Western blot of MCUb in the same isolated hearts used in “D”. OXPHOS antibody was used as the protein loading control. The red box shows the area where MCUb protein is induced in the WT heart samples subject to RIPC but not in the Mcub−/− mice (KO). D) Mitochondrial Ca2+ uptake assay in isolated mitochondria from the left ventricle of hearts in the indicated groups of mice. Mice were challenged with sham or the 7-day RIPC protocol, and then hearts were collected and mitochondria were purified for analyses.
DISCUSSION
The precise biological role of MCUb remains controversial. Some studies have suggested that MCUb likely acts as a dominant negative regulator of MCU by inserting into the quaternary core of the MCU-complex and directly interfering with channel permeation of Ca2+.12 Other studies suggest that MCUb may function as a low conductance mitochondrial Ca2+ uptake channel.16, 33 However, most studies in cells with overexpressed proteins or in planar lipid bilayers with recombinant protein are consistent with mammalian MCUb functioning to inhibit acute Ca2+ uptake.8, 12, 15, 34 For example, Checchetto and Szabo showed that purified MCU protein generates a Ca2+ current in a planar lipid bilayer while purified MCUb could not, similar to results of Raffeallo et al.12, 35 However, the MCUb gene from trypanosomes was shown to conduct Ca2+ such that its absence caused reduced viability as well as reduced Ca2+ influx in isolated systems.33, 36 Although, it should be noted that the trypanosome MCU-complex is divergent from the mammalian form with additional subunit paralogs of unknown relevance.33, 36 Our studies support the idea that MCUb acts as a dominant negative regulator of the MCU-complex in mammalian systems, as least for acute mitochondrial Ca2+ uptake. In MEFs where MCUb protein is constitutively expressed, deletion of Mcub produced increased mitochondrial Ca2+ uptake sensitivity suggesting that it functions as a true inhibitor of both acute and slow Ca2+ influx.
Given the structural homology between MCUb and MCU it is believed that MCUb inserts into the quaternary core channel of the MCU-complex, and because MCUb has divergence in the “DIME” motif found in MCU, it no longer permits Ca2+ conduction through the pore.12 Alternately, MCUb may inhibit mitochondrial Ca2+ uptake by changing the composition of the MCU-complex such that it no longer contains the essential component EMRE, as our data suggest. Structural analysis of the MCU minimal conducting complex showed that EMRE must be present where it binds 1:1 with MCU in generating a proper pore conformation.15, 29 We observed that MCUb overexpression directly disrupted the interaction between MCU and EMRE by immunoprecipitation from mitochondrial protein extracts (Fig. 2B–C), while Mcub−/− hearts appear to show increased EMRE protein levels (Fig. 4B). However, future studies will be required to better define how MCUb inhibits mitochondrial Ca2+ influx.
We and others have shown that inhibition of mitochondrial Ca2+ influx by Mcu deletion in the adult mouse heart renders mitochondria insensitive to acute Ca2+ stimulated increases in mitochondrial energy production.7, 18 However, in the absence of acute fluxing of mitochondria Ca2+ a more dynamic compensation occurs that enhances long-term adaptation producing greater fatty acid utilization.37, 38 For example, skeletal muscle-specific deletion of Mcu in mice inhibited acute mitochondrial Ca2+ influx and Ca2+-stimulated mitochondrial respiration, resulting in impaired metabolism-contraction coupling and reduced acute exercise performance.39 However, loss of Mcu enhanced muscle performance under conditions of fatigue with a preferential shift towards fatty acid metabolism, resulting in reduced body fat with aging.39 In the current study we failed to observe an alteration in oxygen consumption in isolated mitochondria from MCUb-DTG or Mcub−/− hearts compared with controls when assayed in pyruvate/malate buffer. By comparison, recently generated adult inducible MCUb TG mice similarly failed to show an effect on basal oxygen consumption in isolated mitochondria, although a significant reduction in reserve and maximal respiratory activity was noted.21 dnMCU TG mice showed increased respiratory function in a working heart model but not at controlled Ca2+ levels in permeabilized myocardial fiber preparations, which the authors interpreted as resulting from greater energy production outside of the mitochondria.17 Thus, current evidence suggests that inhibition of MCU activity in the heart impacts respiratory function following acute stimulation with Ca2+, while baseline mitochondrial respiration is unaffected although during stress greater fatty acid utilization is observed.37, 38
A key finding here is that MCUb is an inducible cardioprotective gene in the context of I/R injury. MCUb protein is not detected in the adult mouse heart at baseline, but I/R injury results in a dramatic induction of expression within 3 days. MCUb induction has a delayed effect where it reduces the likelihood of infarct expansion into borderzone cardiomyocytes by reducing mitochondrial Ca2+ overload-induced necrosis. This would have a beneficial effect on remodeling after MI injury, supported by the observation that Mcub−/− mice succumb to greater injury in the proceeding days following an ischemic injury event. This is consistent with a mechanism of action referred to as delayed- or post-conditioning40, which we also observed in mice subjected to RIPC.31, 32 Indeed, we found that RIPC from the hindlimb also induced MCUb expression in the heart, suggesting that the MCUb gene is sensitive to and likely directly regulated by ischemia-dependent signaling and transcriptional pathways.
In conclusion, our study showed that MCUb is induced in the heart in response to ischemic stress, either by an I/R injury itself or by RIPC. This induction in MCUb in the heart is protective by limiting Ca2+ influx and Ca2+ overload in surviving cardiomyocytes at the level of the MCU-complex. We observed that while Mcub deletion did not affect initial I/R infarct size, it did limit infarct expansion and pathological remodeling compared with controls. These data suggest that therapeutically targeting the MCU-complex days after an infarction event could still provide benefit if an appropriate permeable and non-toxic MCU-complex inhibitory drug was identified.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Ischemia-reperfusion (I/R) injury causes cardiomyocyte death by exposing these cells to high levels of free cytosolic Ca2+, which then causes mitochondrial permeability transition pore (mPTP) opening.
Mcub is an inhibitory subunit of the mitochondrial Ca2+ uniporter (MCU) that reduces mitochondrial Ca2+ influx.
Acute overexpression of Mcub protects the heart from I/R injury.
What New Information Does This Article Contribute?
Mcub expression is induced in the heart after ischemia-reperfusion injury or remote ischemic pre-conditioning.
Genetic deletion of Mcub in the heart exacerbated damage after ischemia-reperfusion injury.
Mcub induction in the heart is a protective mechanism that reduces mitochondrial Ca2+ overload and borerzone cardiomyocyte necrosis.
SOURCES OF FUNDING
This work was supported by grants from the National Heart Lung and Blood Institute of the National Institutes of Health (R01-HL132831 to JDM and DMB, R01-HL132831 JDM, R01-HL030077 to DMB), the Howard Hughes Medical Institute (JDM), and by a grant from the Fondation Leducq (JDM). J.H., S.L., J.Q.K and M.J.B were supported by grants from American Heart Association. K.M.G. was funded by a National Institutes of Health grant (F32HL138747).
Nonstandard Abbreviations and Acronyms:
- α-MHC
α-myosin heavy chain
- AAR
area at risk
- [Ca2+]mito
mitochondrial calcium concentration
- BW
body weight
- Co-IP
co-immunoprecipitation
- CsA
cyclosporine A
- dnMCU
dominant negative MCU mutant
- DTG
MCUb overexpressing mice with 2 transgenes (MCUb + tTA)
- EMRE
essential MCU regulator, mitochondrial (Smdt1 gene product)
- FCCP
carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone
- FS
fractional shortening
- HW
heart weight
- IA
ischemic area
- I/R
Ischemia-reperfusion
- LacZ
β-galactosidase
- LW
lung weight
- MCU-complex
mitochondrial calcium uniporter complex
- MCU
mitochondrial calcium uniporter pore-forming subunit
- Mcu−/−
null mice lacking gene encoding MCU protein
- MCUb
mitochondrial calcium uniporter b subunit
- Mcub−/−
null mice lacking gene encoding MCUb protein
- MCUb TG
α-MHC tetracycline-operator mouse with MCUb cDNA
- MEF
mouse embryo fibroblast
- MICU1
mitochondrial calcium uptake 1
- MICU2
mitochondrial calcium uptake 2
- MI
myocardial infarction
- mPTP
mitochondrial permeability transition pore
- Neo
neomycin
- NCLX
Na+/Ca2+/Li+ exchanger
- OCR
oxygen consumption rate
- OXPHOS
mitochondrial oxidative phosphorylation complex
- RIPC
remote ischemic pre-conditioning
- SR
sarcoplasmic reticulum
- tTA TG
tetracycline transactivator cardiac-specific transgenic mouse
- VDAC
Voltage-dependent anion channel
- wk
week
- WT
wildtype
- ΔΨm
mitochondrial membrane potential
Footnotes
DISCLOSURES
All authors confirm no conflict of interest.
SUPPLEMENTAL MATERIALS
1) Detailed Online Methods
2) Online Figures I-VI with legends
3) References 41–45
REFERENCES
- 1.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17:1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J Clin Invest. 2013;123:92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–385 [DOI] [PubMed] [Google Scholar]
- 5.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831 [DOI] [PubMed] [Google Scholar]
- 6.Halestrap AP. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem Soc Trans. 2006;34:232–237 [DOI] [PubMed] [Google Scholar]
- 7.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies mcu as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–U104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. Nclx is an essential component of mitochondrial na+/ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Stefani D, Patron M, Rizzuto R. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta. 2015;1853:2006–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. Emre is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R. Micu1 and micu2 finely tune the mitochondrial ca2+ uniporter by exerting opposite effects on mcu activity. Mol Cell. 2014;53:726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai XC, Jiang Y. Structural mechanism of emre-dependent gating of the human mitochondrial calcium uniporter. Cell. 2019;177:1252–1261 e1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, Brustovetsky N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J Biol Chem. 2018;293:15652–15663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME. Inhibition of mcu forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A. 2015;112:9129–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrington JL, Murphy E. The mitochondrial calcium uniporter: Mice can live and die without it. J Mol Cell Cardiol. 2015;78:46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW. Mcub regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616 [DOI] [PubMed] [Google Scholar]
- 23.Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD. Inhibition of mitochondrial permeability transition by deletion of the ant family and cypd. Sci Adv. 2019;5:eaaw4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pendergrass KD, Varghese ST, Maiellaro-Rafferty K, Brown ME, Taylor WR, Davis ME. Temporal effects of catalase overexpression on healing after myocardial infarction. Circ Heart Fail. 2011;4:98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates camkii and arrhythmias by o-linked glycosylation. Nature. 2013;502:372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase ii promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picht E, Zima AV, Blatter LA, Bers DM. Sparkmaster: Automated calcium spark analysis with imagej. Am J Physiol Cell Physiol. 2007;293:C1073–1081 [DOI] [PubMed] [Google Scholar]
- 28.Davis J, Maillet M, Miano JM, Molkentin JD. Lost in transgenesis: A user’s guide for genetically manipulating the mouse in cardiac research. Circ Res. 2012;111:761–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovacs-Bogdan E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD, Mootha VK. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci U S A. 2014;111:8985–8990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial ca(2+) uniporter (mcu)-dependent and mcu-independent ca(2+) channels coexist in the inner mitochondrial membrane. Pflugers Arch. 2014;466:1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honda T, He Q, Wang F, Redington AN. Acute and chronic remote ischemic conditioning attenuate septic cardiomyopathy, improve cardiac output, protect systemic organs, and improve mortality in a lipopolysaccharide-induced sepsis model. Basic Res Cardiol. 2019;114:15. [DOI] [PubMed] [Google Scholar]
- 32.Gertz ZM, Cain C, Kraskauskas D, Devarakonda T, Mauro AG, Thompson J, Samidurai A, Chen Q, Gordon SW, Lesnefsky EJ, Das A, Salloum FN. Remote ischemic pre-conditioning attenuates adverse cardiac remodeling and mortality following doxorubicin administration in mice. JACC CardioOncology. 2019;1:235–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiurillo MA, Lander N, Bertolini MS, Storey M, Vercesi AE, Docampo R. Different roles of mitochondrial calcium uniporter complex subunits in growth and infectivity of trypanosoma cruzi. MBio. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen NX, Armache JP, Lee C, Yang Y, Zeng W, Mootha VK, Cheng Y, Bai XC, Jiang Y. Cryo-em structure of a fungal mitochondrial calcium uniporter. Nature. 2018;559:570–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Checchetto V, Szabo I. Mcu regulation in lipid bilayer and electrophysiological recording. Methods Mol Biol. 2019;1925:59–63 [DOI] [PubMed] [Google Scholar]
- 36.Chiurillo MA, Lander N, Bertolini MS, Vercesi AE, Docampo R. Functional analysis and importance for host cell infection of the ca2+-conducting subunits of the mitochondrial calcium uniporter of trypanosoma cruzi. Mol Biol Cell. 2019;30:1676–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altamimi TR, Karwi QG, Uddin GM, Fukushima A, Kwong JQ, Molkentin JD, Lopaschuk GD. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J Mol Cell Cardiol. 2019;127:223–231 [DOI] [PubMed] [Google Scholar]
- 38.Jouaville LS, Pjnton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial atp synthesis by calcium: Evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96:13807–13812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwong JQ, Huo JZ, Bround MJ, Boyer JG, Schwanekamp JA, Ghazal N, Maxwell JT, Jang YC, Khuchua Z, Shi K, Bers DM, Davis J, Molkentin JD. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight. 2018;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ong SB, Dongworth RK, Cabrera-Fuentes HA, Hausenloy DJ. Role of the mptp in conditioning the heart - translatability and mechanism. Br J Pharmacol. 2015;172:2074–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gertz ZM, Cain C, Kraskauskas D, Devarakonda T, Mauro AG, Thompson J, Samidurai A, Chen Q, Gordon SW, Lesnefsky EJ, Das A, Salloum FN. Remote ischemic pre-conditioning attenuates adverse cardiac remodeling and mortality following doxorubicin administration in mice. JACC CardioOncology. 2019;1:235–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Honda T, He Q, Wang F, Redington AN. Acute and chronic remote ischemic conditioning attenuate septic cardiomyopathy, improve cardiac output, protect systemic organs, and improve mortality in a lipopolysaccharide-induced sepsis model. Basic Res Cardiol. 2019;114:15. [DOI] [PubMed] [Google Scholar]
- 44.Fewell JG, Osinska H, Klevitsky R, Ng W, Sfyris G, Bahrehmand F, Robbins J. A treadmill exercise regimen for identifying cardiovascular phenotypes in transgenic mice. Am J Physiol. 1997;273:H1595–1605 [DOI] [PubMed] [Google Scholar]
- 45.Kwong JQ, Huo JZ, Bround MJ, Boyer JG, Schwanekamp JA, Ghazal N, Maxwell JT, Jang YC, Khuchua Z, Shi K, Bers DM, Davis J, Molkentin JD. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight. 2018;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.