

Abstract
Mutations in approximately 80 genes have been implicated as the cause of various genetic kidney diseases. However, gene delivery to kidney cells from the blood is inefficient due to the natural filtering functions of the glomerulus, and research and development of gene therapy directed toward kidney disease has lagged behind as compared to hepatic, neuromuscular, and ocular gene therapy. This lack of progress is in spite of numerous genetic mouse models of human disease available to the research community and many vectors in existence that can theoretically deliver genes to kidney cells with high efficiency. In the past decade, several groups have begun to develop novel injection techniques in mice such as retrograde ureter, renal vein, and direct subcapsular injections to help resolve the issue of gene delivery to the kidney through the blood. In addition, the ability to retarget vectors specifically towards kidney cells has been underutilized but shows promise. This review discusses how recent advances in gene delivery to the kidney and the field of gene therapy can leverage the wealth of knowledge of kidney genetics to work towards developing gene therapy products for patients suffering from kidney disease.
Introduction: The Impact of Kidney Diseases
Kidney diseases affect up to 13% of humans worldwide [1, 2]. Chronic kidney diseases due to genetic mutations can occur as frequently as 1 in 1,000 live births. Multiple genes have been implicated as the causes of genetic kidney diseases (reviewed in [2]). These genetic diseases can affect nearly all structures in the kidney, implying that any one of a multitude of cell types within the kidney may need to be targeted in order to treat a particular disease. Congenital kidney diseases can be grouped as cystic, glomerular basement membrane disorders, and tubulopathies, depending on how the disease affects normal physiology (Table 1 and [3]).
Table 1.
Some genetically characterized kidney diseases, the genes underlying their pathogeneses, and potential gene therapy strategies.
| Disease | Underlying Genes | cDNA Size (bp) | Gene Therapy Strategy |
|---|---|---|---|
| ADPKD | PKD1 | 14,138 | Gene addition |
| PKD2 | 5056 | Gene addition | |
| GANAB | 3906 | Gene addition | |
| ARPKD | PKHD1 | 16,282 | Gene addition |
| Alport syndrome | COL4A3 | 8097 | Gene addition |
| COL4A4 | 9895 | Gene addition | |
| COL4A5 | 6483 | Gene addition | |
| ADTKD/Medullary cystic kidney disease | MUC1 (type I) | Variable | Gene addition |
| UMOD | 2477 | Gene knockdown | |
| REN | 1462 | Gene knockdown | |
| HNF1B | 2790 | Gene addition | |
| SEC61A1 | 1871 | Gene addition | |
| Nephronophthisis | NPHP1 | 3756 | Gene addition |
| Bartter Syndrome | SLC12A1 | 4707 | Gene addition |
| KCNJ1 (type II) | 4074 | Gene addition | |
| CLCNKB (type III and IV) | 1544 | Gene addition | |
| BSND (type IV) | 3472 | Gene addition | |
| CLCNKA (type IV) | 2581 | Gene addition | |
| MAGED2 (type V) | 2066 | Gene addition | |
| Von Hippel-Lindau syndrome | VHL | 3737 | Gene addition |
| Gitelman syndrome | SLC12A3 | 3119 | Gene addition |
| CLCNKB | 1544 | Gene addition | |
| Congenital nephrotic syndrome | NPHS1 | 4276 | Gene addition |
| NPHS2 | 1855 | Gene addition | |
| Primary hyperoxaluria | AGXT (type I) | 1865 | Gene addition |
| GRHPR (type II) | 1280 | Gene addition | |
| HOGA1 (type III) | 2488 | Gene addition | |
| Dent disease | CLCN5 (type I) | 10,108 | Gene addition |
| OCRL (type II) | 5138 | Gene addition | |
| Thin Basement Membrane Nephropathy | COL4A3 | 8097 | Gene addition |
| COL4A4 | 9895 | Gene addition | |
| COL4A5 | 6483 | Gene addition | |
| Cystinuria | SLC3A1 | 1737 | Gene addition |
| SLC7A9 | 1752 | Gene addition | |
| Liddle syndrome | SCNN1A | 3481 | Gene knockdown |
| SCNN1B | 2597 | Gene knockdown | |
| SCNN1G | 3507 | Gene knockdown | |
| Papillorenal syndrome | PAX2 | 4057 | Gene addition |
| Cystinosis | CTNS | 2866 | Gene addition |
cDNA sizes were determined by analyzing exons of RefSeq sequences on UCSC Genome Browser. The designation of “Variable” indicates that the gene has multiple splice isoforms. ADPKD = Autosomal Dominant Polycystic Kidney Disease. ARPKD = Autosomal Recessive Polycystic Kidney Disease. ADTKD = Autosomal Dominant Tubulointerstitial Kidney Disease.
The Need for Gene Therapy for Kidney Diseases
Some diseases are amenable to ex vivo genetic modification. These include tissues like the bone marrow and immune system in which cells can be removed from the body, modified, and then be re-injected where they can reconstitute the tissue. Given the complex structure and functions of the kidney, most ex vivo strategies will likely be difficult. This necessitates the use of a gene or an RNA therapy in vivo. The gene therapy field has markedly improved the ability to treat genetic diseases in vivo for many organs. However, relatively little progress has been made toward enabling these therapies for kidney diseases. One reason for the low level of progress is not only the complex structure of the kidney, but also the plethora of distinct cell types: there are over 20 different cell types in the kidney. Genetic kidney diseases can arise from mutations that affect various subtypes of cells such as podocytes, proximal tubules, distal tubules, and collecting duct tubules. This means that in general, in can be difficult to optimize targeting and transgene expression for a given cell type.
The Kidney as a Difficult Target for Intravenous Molecular Therapies
The liver, kidney, and lungs eliminate most waste from the body. The kidney is tasked primarily with removing water-soluble small molecules. The lungs can eliminate volatile waste products by exhalation. The liver eliminates hydrophobic compounds into the bile or modifies them to make them water soluble for elimination by the kidney.
The intravenous route is favored for delivery of most molecular therapies, since administration is easy and it allows the vectors to distribute throughout the body by being carried in the blood (Figure 1). This route is particularly favored for liver-directed therapy, since the liver has fenestrations or “windows” in its vasculature that allow the passage of large particulates like molecular therapy vectors. Unlike the liver, the structure of the kidney impedes rather than facilitates delivery of these vectors from the blood (Figure 2)
Figure 1. Expected target tissues for transduction after intravenous administration of typical viral vectors.
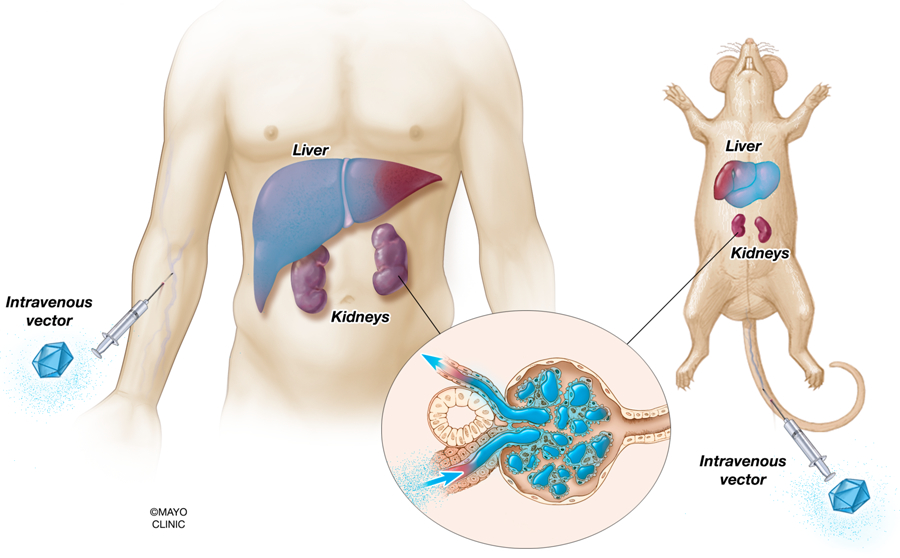
Liver-tropic vectors, such as AAV8 and Ad5, will highly transduce hepatocytes when administered systemically in large doses. Meanwhile, the vectors may physically reach the kidney from the blood, but will be limited to transducing cells in the glomerulus. Other tissues capable of being significantly transduced from systemic administration are the heart when AAV9 is used. Transduction is depicted as tissues and cells shaded in blue.
Figure 2. Intravenous and direct kidney vector administration in the context of the renal corpuscle.
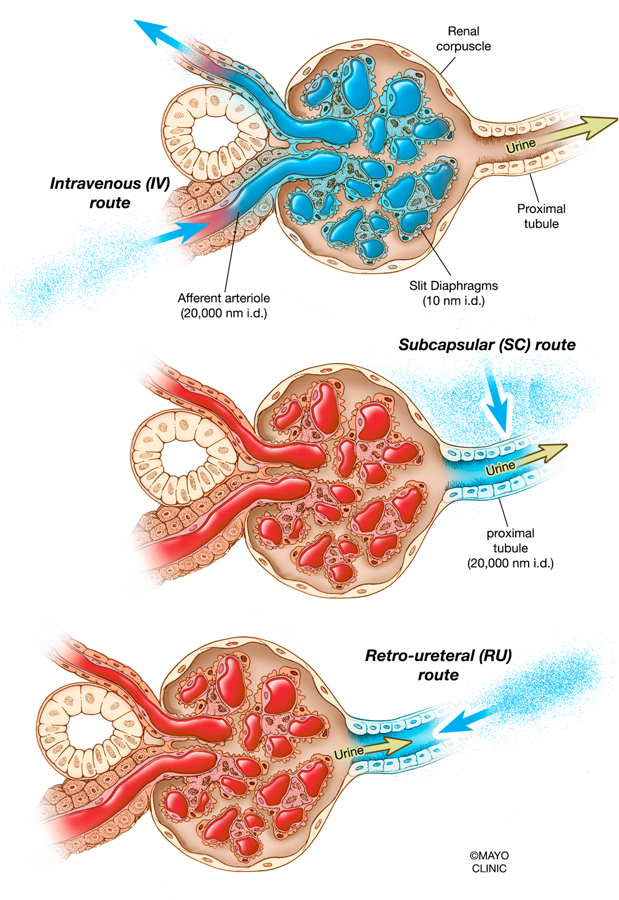
Injecting vectors by the intravenous route would result in most vectors being stuck in the glomeruli of the kidneys or recirculated into the bloodstream, since slit diaphragms are approximately 10 nm in diameter (upper). Injecting vectors by the subcapsular route would bypass the glomeruli and potentially give vectors access to the apical and basolateral sides of some tubules (middle). Injecting vectors by the retro-ureteral route would theoretically grant vectors access to the apical side of tubules unless they escape from within the tubule epithelium, since proximal tubules are up to 20,000 nm in diameter (lower). nm, nanometer; i.d., in diameter.
Mammalian kidneys have evolved to filter out large compounds and only pass small molecules. This filtration is performed in the glomerulus of each nephron. The glomerulus actively excludes proteins above 50 kiloDaltons (kDa) in size [4]. In addition, podocytes within the glomerulus create slit diaphragms with diameters of only 10 nm (Figure 2). In contrast, many viral and non-viral gene therapy vectors are larger than 10 nm in diameter and many have masses on the order of hundreds of megaDaltons (Figure 3). For example, adeno-associated virus is a relatively small viral vector, with a capsid diameter of 25 nm and a mass of 5 to 6 megaDaltons. Even this vector does not pass the stringent criteria of the glomerular filter. Other viral vectors, such as adenovirus and lentivirus, are significantly larger.
Figure 3. Molecular therapy vectors and their respective sizes.
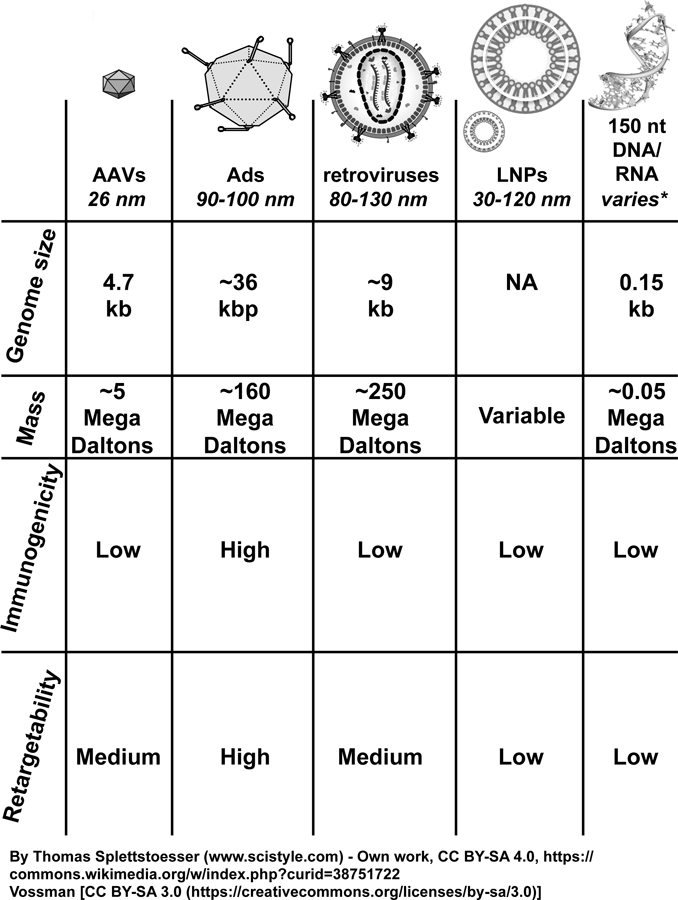
None of the molecular therapy vectors shown here, with the exception of 150 nt or smaller nucleic acids, would be expected to penetrate past the glomerular barrier and into the epithelium of the kidney, based on their respective masses and diameters. AAVs, adeno-associated viruses; Ads, adenoviruses; LNPs, lipid nanoparticles; nt, nucleotide; kb, kilobase; kbp, kilobasepair; kDa, kiloDalton.
The permselectivity of the glomerulus will prevent entry of most molecular therapies from the blood into the kidney. Therefore, establishment of alternate entry routes for vectors is key to accessing kidney cells downstream of the glomerulus. One potential special path is entry through lymphatics. The lymphatic system of the kidney began to be elucidated in the 1940’s and 1950’s. By injecting dog and sheep kidneys with traceable fluids, researchers found that lymphatics run beside or between the renal artery and renal vein to the renal lymph node, located proximal to the intersection of the aorta and renal artery [5, 6]. This route has not necessarily ever been explored as a means for gene delivery.
Other special paths include efforts to use hydrodynamic pressure to increase vector penetration into the kidney from the blood. Others include retrograde injection of vectors up the ureter or direct injection into the parenchyma of the organ through the kidney capsule (Figure 4). These alternate strategies as they have been applied for gene delivery will be discussed below.
Figure 4. Various modalities of vector administration in the context of the whole kidney.
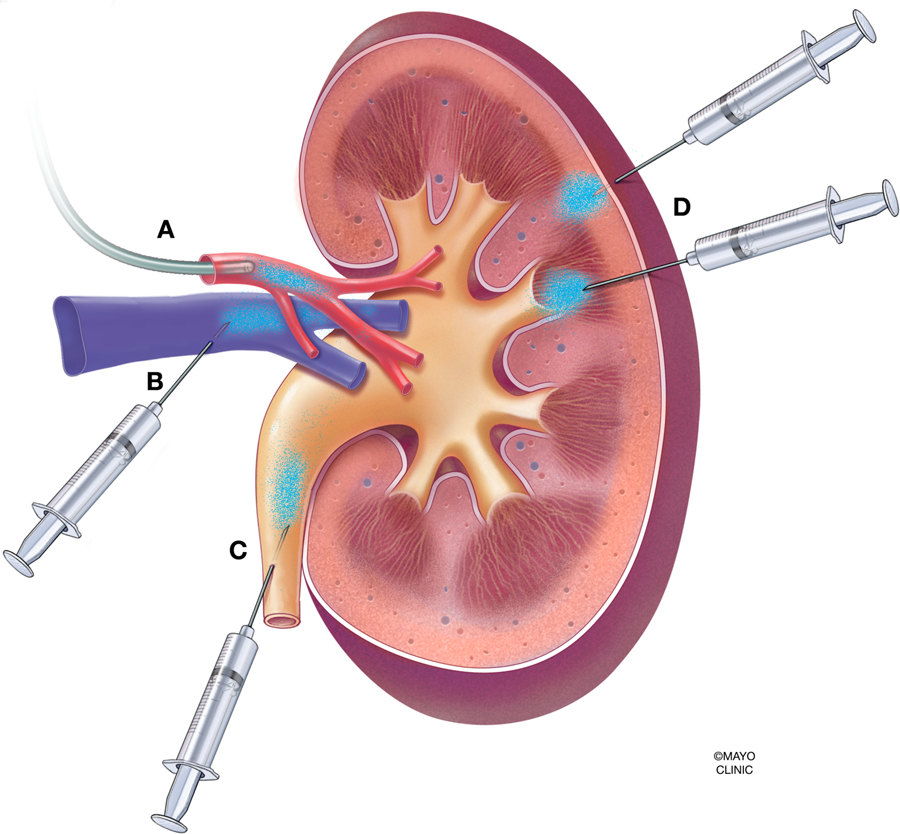
(A) Infusion into the renal artery, via catheter or injection. (B) Retrograde infusion into the renal vein. (C) Retrograde infusion into the ureter. (D) Subcapsular injection into the parenchyma of the kidney
Vectors for Molecular Therapy
Molecular therapy strategies vary based on the genetics of the disease. A recessive genetic disease may only require delivery of a “good” copy of the gene into cells to replace the damaged function. In contrast, a dominant genetic disease may require that one not only deliver a repair gene or mRNA, but also knock-down, knock-out, or edit out the damaged gene to remove its malign effects on the cell.
To do this, one usually needs some vector to carry the nucleic acid into the cell. Certain cells like skeletal muscle can import naked DNA or RNA, but this is usually too inefficient to mediate substantial effects on cell biology. Delivery of naked small RNA therapies can be achieved in some cases, but this usually achieved by mass action and requires the use of massive amounts of the agent.
Many different types of viral and non-viral vectors can be used to increase nucleic acid delivery into cells and to protect them from destruction by extracellular and intracellular nucleases. In general, viral vectors are most potent for in vivo delivery, but newer lipid nanoparticle (LNP) and other non-viral vector platforms are catching up. Of note, the recent FDA approval of patisiran (discussed further below) brings to the market a short interfering RNA (siRNA) and LNP formulation that effectively treats transthyretin amyloidosis. Two of the most robust in vivo viral vector gene delivery platforms are adeno-associated virus (AAV) and adenovirus (Ad) (Figure 3). Both of these viral vector systems exist in diverse genetically varied serotypes that are immunologically distinct and that can target different cells in the body. Other vectors like retroviral and lentiviral vectors have had most of their use for ex vivo gene therapy. However, these may also have utility for in vivo kidney therapy. These vector platforms are discussed in detail below.
Adenovirus Vectors
Adenoviral vectors (Ad) are one of the oldest platforms for in vivo gene therapy. Because of its relatively early adoption several decades ago, some of the earliest research in gene transfer to the kidney was performed using adenovirus for gene delivery. Tragically, a second generation Ad vector led to the death of Jesse Gelsinger in a clinical gene therapy trial to treat ornithine transcarbamylase (OTC) deficiency in 1999[7]. This event damaged the perception of Ad as a gene therapy vector. The aftermath of this incident is clear: as of the publication of this review, there are no clinical trials in progress to treat hereditary disease with adenovirus-mediated gene therapy. (There are, however, two adenovirus clinical trials for delivery of vascular endothelial growth factor [VEGF] to treat refractory angina pectoris: ClinicalTrials.gov Identifiers are NCT04125732 and NCT03039751.) While this was a public relations disaster for this vector system, the lessons learned from Mr. Gelsinger’s death and vector improvements now argue that Ad can be used safely for human gene therapy.
Kidney researchers continued to use Ad to transfer genes to kidney cells. In most cases, this work has involved the use of one serotype of Ad called Ad5. Most studies use replication-defective first generation Ad5 (FG-Ad5) with its E1 and E3 genes deleted. These vectors still contain most Ad open reading frames (ORFs) and are strongly targeted by antiviral T cells within 2 weeks of in vivo administration [8]. Ad5 targets the coxsackie and adenovirus receptor (CAR) and αv integrins (reviewed in [9, 10]). Other Ad serotypes target different receptors, but have largely not been tested in the kidney. When Ad5 is administered systemically (i.e. intravenously (IV)), about 98% of the vector is sequestered by Kupffer cells in the liver [11, 12]. As a result, very little of the virus lands in the kidney. Mammalian kidneys have evolved to allow only small molecules and proteins to pass the glomerulus. Kidneys actively exclude proteins above 50 kDa in size[4]. In addition, podocytes within the glomerulus create slit diaphragms that are only 10 nm in diameter, which are well below the diameter of gene therapy vectors including popular AAV vectors. In 1997, Zhu et al. demonstrated this theory elegantly by showing that after either intravenous or renal artery infusions of replication-defective Ad5 encoding β-galactosidase (Ad5-β-gal), the activity of β-gal in rat glomeruli was only detectable after transduced hepatocytes had undergone immune-mediated destruction and the β-gal protein product was deposited in the glomerulus from the bloodstream [13]. In addition, in 2000, Ye et al. showed that liver bypass via clamping the portal vein and hepatic artery prior to systemic administration of Ad5-β-gal resulted in increased transduction in the lung, intestinal wall, and renal glomeruli, with the liver being transduced only after the clamps were released [14].
Because of this strong size exclusion by the glomerulus, some Ad kidney gene transfer studies delivered the vector by infusing it into the kidney using a variety of methods. In 1994, Moullier et al. used Ad5-β-gal to transduce rat kidney through both arterial infusion and catheter mediated retrograde infusion (Figure 4A). The former method resulted in transduction of proximal tubule cells and the latter resulted in transduction of tubular cells in the papilla and medulla [15]. In 1996, Heikkila et al. developed an ex vivo and in vivo perfusion system that was able to transduce approximately 85% of glomeruli in pigs with β-gal after a two hour perfusion [16]. In 1999, McDonald et al. infused Ad5-β-gal into the renal artery of rats to monitor transduction of the renal vascular endothelium [17]. Since Ad5 uses αv integrin as a receptor, and this integrin is present on the vascular endothelium, the authors reasoned that this vector could transduce these. They found that using Ad5 with a fiber modified with RGD peptide, which specifically binds to the integrin, did in fact transduce vasculature throughout the kidney. In 2001, Ye et al. showed that a slow, 0.1 mL/min infusion of Ad5-β-gal into the renal artery over the course of 15 minutes efficiently transduced glomerular endothelium, but no downstream tubule epithelium [18]. In 2001, Chetboul et al. injected Ad5-β-gal into two-month-old beagle puppies through both the intra-renal-ureteral and intra-renal-arterial routes to examine any vector-related toxicity in addition to transduction efficiency [19]. While no toxicity was observed, the arterial route transduced sporadically in the cortical interstitium and the ureteral route transduced distal tubular pyelic epithelial cells sporadically. In 2003, Choi et al. performed an intraparenchymal injection of FG-Ad5 expressing human interleukin 10 in FGS/Kist mice, a strain that naturally develops proteinuria and nephritis at 6 weeks of age [20]. This study noted sustained transgene expression for two weeks after injection and transgene delivery to the cortex and medulla, as well as attenuation of the glomerular sclerosis phenotype. In 2005, Fujishiro et al. used a different catherization technique in rats that mimicked human angiography [21]. By this approach, they noted stochastic transduction of kidney cells by Ad5. In 2001, Heikkila et al. used FG-Ad5 to deliver type IV collagen alpha chain gene to the glomerular basement membrane (GBM) by infusion into pig kidneys towards a therapy for Alport syndrome [22]. In 2013, Corridon et al. performed retrograde renal vein hydrodynamic injections of plasmid DNA, adenovirus, and a less commonly used vector, baculovirus, in rats (Figure 4B). In this study, the authors used intravital fluorescence two-photon microscopy to quantitate that plasmid and adenovirus injections granted a gene transfer efficiency of 50–90% while baculovirus granted 10–50%. The gene transfer efficiency throughout an entire plane of the rat kidney was determined by ascertaining the basal level of fluorescence in a control mouse (autofluorescence) and then counting only signals double the basal value as a true positive. This study is unique both because it quantifies fluorescence throughout a whole kidney in vivo and also because it reports such high efficiencies of gene transfer. However, to our knowledge, no subsequent study has followed up to replicate this high efficiency of gene transfer, or more importantly, deliver a therapeutic gene to a rat model of human disease. More recently, in 2019, Rubin et al. showed that injecting Ad5-Cre retro-ureterally (into the ureter retrograde toward the kidney) and subcapsularly (directly into the kidney capsule) mediated robust transduction of kidneys in loxP-STOP-loxP reporter mice, compared to intravenous injections (Figures 4C and 4D)[23].
The aforementioned studies involving gene delivery to the kidney were successful in the sense that they achieved transgene expression in kidney cells using Ad, however, transgene modification of cells was generally stochastic and non-specific. The exception to this was Watanabe et al., who was able to direct transgene expression to specific regions and cells of the kidney using a clever kidney specific promoter expression system[24]. In this study, they engineered Ads carrying transgenes that were regulated by the promoters from different kidney solute transporters. When these vectors were injected by the intra-arterial route, they demonstrated varied cell-specific gene delivery, albeit, only by immunohistochemical (IHC) staining of whole kidney sections. However, the question still remains of whether any of the Ads used for these studies are translational for human diseases, or whether an intra-arterial infusion method of delivery is feasible. Ad is a non-enveloped virus, and can be re-targeted by making genetic or chemical modifications to its various capsid proteins (reviewed in [9, 10]). Ad capsids are composed of a patterned array of 720 hexon proteins, 60 penton base proteins, and 36 fiber proteins [25]. This pattern is very efficient at attracting blood proteins that change virus tropism and influence efficacy and side effects. For example, Species C adenoviruses such as Ad5 and Ad6 bind to clotting factors FIX and FX in the blood, which protect the viruses from destruction by macrophages and also target them to the liver [26–28].
Due to the exposure of the hexon and fiber proteins during physical virus-cell interactions, these proteins have been of interest for modifications that target specific cells based on the presence of particular ligands or receptors. The C-termini of the fiber trimer forms a “knob” which binds to CAR or other receptors and initiates natural adenovirus infections. As such, the first attempts to genetically retarget adenovirus involved adding peptides into a region of the fiber protein C-terminus called the HI loop[29–34] (reviewed in [35]).
Modifications to the HI loop became particularly relevant to kidney gene delivery using peptides selected by phage display. Phage display technology involves exposing a library of up to 1010 phages displaying random peptides to cells in tissue culture, tissues in vivo, or even a chemically coated surface, to identify peptides that will bind with cellular receptors as ligands[36, 37] (reviewed in [38, 39]). This technology was used for kidney targeting by Denby et al. and Diaconu et al. by performing in vivo phage selection in rats by IV injection and selection of peptide libraries [40, 41]. By this approach, they identified three different peptides that were each individually inserted into the HI loop of the Ad5 fiber. Two of these peptides succeeded in increasing transduction of kidney cells after IV injection making this a remarkable engineering achievement. However, transduction of kidney cells was still stochastic.
Other approaches to modify Ad vectors include addition of a biotin acceptor peptide (BAP) and chemical modifications (reviewed in [42]). BAP has been genetically inserted into the hexon, pIX, and fiber proteins of Ad5 and Ad657 [43–45]. When these proteins are expressed in mammalian cells, they are metabolically biotinylated by endogenous biotin ligase. These covalently biotinylated capsomer proteins are inserted into the virion. This allows Ads to be purified on monomeric avidin columns, but more importantly, any biotinylated ligand, such as antibodies, peptides, etc, can be ligated to the Ad using tetrameric avidin[43, 44]. The retargeting potential of these biotinylated Ads is theoretically limited only to knowledge of what receptors or ligands a target cell or tissue express, and what antibodies or ligands will bind to those receptors.
Ad vectors can also be retargeted by conjugating cell targeting ligands to the virus with polymers like polyethylene glycol (PEG) [46, 47]. In these approaches, bifunctional PEG cross-linkers are generally first reacted with lysines on the capsid surface and then to cysteine-labeled peptides or proteins using a maleimide linker. Both peptides and proteins have been used to retarget Ad to new receptors in vitro and in vivo. Early work used phage-selected peptides to target lung epithelial cells [47] or chronic lymphocytic leukemia cells [48] and EGF or FGF to increase infection of cancer cells and skeletal muscle [46, 49]. In addition to its utility in adding ligands, PEG can also be used to protect Ad from neutralizing antibody responses, without attenuation to its ability to transduce cells in vivo[50, 51]. Later studies showed that the length of the PEG changed Ad tropism, specifically with reduced targeting to the liver[9, 52]. Overall, these studies indicate there is promise in genetic and chemical modifications made to Ad vector capsids for gene delivery to specific cell types, such as kidney cells.
Adeno-associated Virus Vectors
Adeno-associated virus (AAV) was initially discovered in the 1960’s as a contaminant in adenovirus culture[53]. AAV is of the family Parvoviridae and of the genus Dependoparvovirus, I.E., it depends on several adenoviral proteins and one RNA to enable it to replicate (reviewed in [54]). AAV’s replication-defective nature, non-pathogenicity in humans, and relative non-immunogenicity have led to decades of widespread effort to vectorize the virus and ultimately AAV has become one of the most popular platforms for gene therapy. Notably, AAV has not attained the same level of widespread use in the kidney as compared to other organ systems. This is generally due to the issue of gene transfer efficiency. AAV is most commonly delivered via the systemic route, which mediates minimal transduction of kidney cells, and those that are transduced are typically glomerular endothelium (Figure 5A)[55–57]. While various novel direct kidney injection methods have been tested with AAV, the efficiency of gene delivery may still be too small to effectively treat disease. These studies are discussed in more detail below.
Figure 5. Routes of vector entry in the context of the nephron.
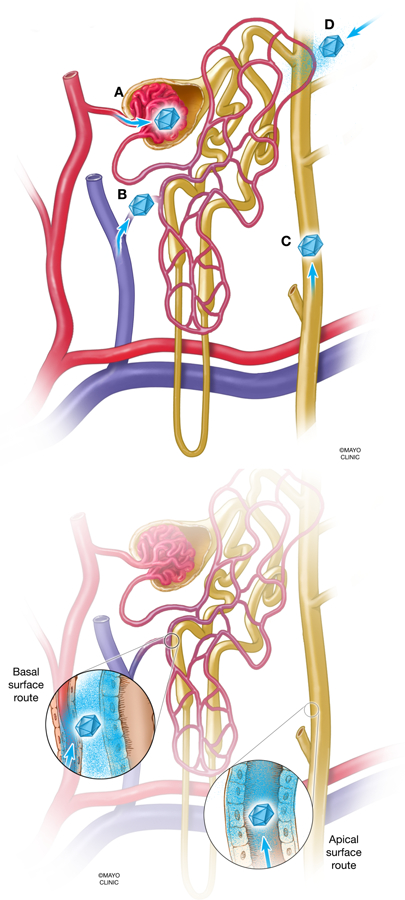
(A) Blood enters the glomerulus of the nephron through the afferent arteriole, which has an approximate diameter of 20,000 nm. However, filtrate that penetrates further into the nephron is limited by the diameter of the slit diaphragms in the glomerular basement membrane, which are approximately 10 nm in diameter. The small diameter of the slit diaphragms excludes molecular therapy vectors from entering the kidney from the bloodstream, while the much broader diameter of the tubule epithelium (20,000 nm or greater) may allow entrance of vectors from the ureteral route. A high pressure delivery, I.E., a hydrodynamic injection, may change the principles of which vectors can pass through the glomerular barrier. (B) Vector access to the nephron after injection into the renal vein. Vectors may escape from the vasculature endothelium and access the basolateral surface of tubules (inset). (C) Vector access to the nephron after retrograde ureteral infusion. Vectors theoretically have exclusive access to the apical side of tubules, from the collecting duct to the proximal tubule (inset). (D) Vector access to the nephron after subcapsular injection. Vectors come into contact with tubules in a more undirected fashion, which theoretically is from “extra-tubular” space of kidney. Vectors may have access to basolateral or apical sides of tubules.
By the late 1990’s, researchers had begun to use AAV for in vitro and in vivo kidney gene delivery studies. In 1999, Lipkowitz et al. transduced mouse kidney cells in vitro as well as in vivo by intraparenchymal injection of AAV5 expressing the green fluorescent protein (GFP)[58]. In 2003, Chen et al. infused AAV2-GFP into the rat renal arteries and showed that GFP could be detected in the S3 segment of proximal tubules[59]. In 2004, Takeda et al. showed that AAV serotype 2 could deliver the β-galactosidase gene to kidney cells by a catheterization technique, while serotypes 1, 3, 4, and 5 could not. Briefly, Takeda et al. inserted catheters in mice and rats via the left iliac artery and the abdominal aorta, clamped the aorta, injected saline to flush out the blood, injected AAV vector solution, and then clamped the renal vein for 10 minutes. Although this method seems technically robust, it resulted in only small amount of transduced cells, demonstrating AAV serotypes 1 through 5 to be ineffective[60]. In 2008, Ito et al. showed that AAV-GFP (serotype not documented) could deliver GFP to both the kidney cortex and medulla using an intra-renal injection after ureteral unilateral obstruction (UUO)[61]. In 2012, Qi et al. used a microinjection technique to deliver GFP to tubular epithelial cells in rats with mutated AAV2, AAV8, and AAV9 [62].
While these early studies were good proof of principle for AAV’s ability to transduce kidney cells in vitro and in vivo, the actual level of in vivo transduction was largely inconsistent. This may have been in part due to the delivery method used for getting AAV to the kidney and in part due to the inability of these serotypes to transduce kidney cells with high frequency. In several more recent studies, advances have been made both in terms of optimized delivery method of AAV and more robust serotypes being use for gene delivery. Specifically, the innovation of the retrograde ureteral injection by Chung et al. in 2011 was a significant advance for the field [63]. By this point in time, it was already understood that AAV-mediated gene delivery to the epithelium of the kidney after intravenous injection is highly inefficient due to restriction by the glomerular filter. Chung et al. sought to overcome this obstacle by injecting AAV into the ureter in the direction opposite of natural fluid flow, allowing AAV to access kidney epithelium through the apical side (lumen), or perhaps even diffuse through the epithelium and access the basolateral side of cells (Figure 4). This initial study had some success in transducing mouse kidney epithelium in vivo using AAV8 and AAV9. While the transduction of tubule epithelial cells reported was higher than that of previous studies, immunofluorescent sections still only showed relatively small numbers of tubule cells transduced. In 2016, Konkalmatt et al. used a synonymous injection method to deliver AAV9 reporter vectors to the kidney. This study showed high transduction of proximal tubule cells and some transduction of collecting duct cells with AAV9 expressing enhanced green fluorescent protein (EGFP). In 2018, Asico et al. built upon the aforementioned study by using kidney-specific promoters in tandem with retrograde ureteral injections to show that reporter gene expression could be limited to specific cell types in the kidney. For example, the 1.1 kb sodium-glucose cotransporter 2 (SGLT2) promoter limited expression to proximal tubule cells in vivo. Although the latter two studies demonstrated more efficient gene delivery, one drawback is high level of known autofluorescence that occurs while examining GFP fluorescence in kidney tissue potentially making results difficult to interpret. Also in 2018, Shen et al. developed a transparenchymal renal pelvis injection technique in mice in which a 30 gauge needle is pierced through the mid pole of the left kidney towards the renal pelvis, and an injection is made[64]. The described technique is reminiscent of a percutaneous nephrolithotomy, a technique used in humans to break up kidney stones. Using AAV9-GFP and AAV9-luciferase as vectors, reporter gene expression was seen in the injected kidney. Notably, these four studies employed the use of clips on the renal artery, renal vein, and ureter to enhance the physical exposure time that the AAV in the injection fluid had in contact to cells in the kidney. (Chung et al. and Shen et al. did not use renal artery and vein clamps, only ureteral clamps.) As discussed in the previous section, another recent innovation with respect to injection methods is the renal vein injections. In 2014, Rocca et al. performed renal vein injections of AAV5, 6, 8, and 9 in mice in tandem with clamping the vein and showed that this method markedly increased kidney transduction as compared to intravenous injections (Figure 5B)[65]. This study also showed that AAV9 was the only serotype tested that allowed substantial transduction of both the cortex and medulla via the renal vein injection route. However, one limitation of this study is that it did not histologically counterstain for many epithelial cell types in the kidney to determine the cell types being transduced, which is important for treating genetic diseases. In addition, it is unclear how the transduction efficiency compares to the 50–90% gene transfer efficiency claimed by Corridon et al. when injecting plasmid and adenovirus using this route in rats [66]. In 2019, Saito et al. tested AAV1, 2, 5, 6, 7, 8, and 9 head to head. Surprisingly, the study found that AAV6 was superior to the other serotypes tested for transducing rat kidney fibroblasts in vitro, human kidney proximal tubule epithelial cells in vitro, and mouse kidney tubule cells in vivo via renal pelvis injection (Figure 5C)[67]. In somewhat of a contrast to other studies mentioned here a renal pelvis injection of 5e9 genome copies (GC) of AAV6-GFP in mice resulted in wide fields of transduced tubule cells, which is even more pronounced when considering this dose in comparison to the more typical 1e11 GC or 1e12 GC administered to mice. While this study was lacking a quantification of number of cells transduced in vivo, the authors were able to ameliorate fibrosis after UUO injury by delivering AAV6 encoding for mIR-29b, an endogenous inhibitor of renal fibrosis, via renal pelvis injection, showing that their method had a physiological effect. In 2019, we showed that retrograde ureteral injections and subcapsular injections, without the use of clamps, lead to massive leakage and transduction of the liver, indicating that clamps may be a good physical way to restrict transduction to the kidney (Figure 5D)[23]. Leakage from the target organ and site of injection, the kidney, to off-target tissues, such as the liver, is an important safety issue, especially given that recent studies have shown deleterious effects of high-dose intravenous AAV administration [68–70].
Since these earlier studies, several techniques have been developed to either engineer or select for novel AAV capsids that have the capacity to target a particular cell type. Directed evolution of AAV can be employed by performing error-prone PCR on specific regions of the AAV capsid, such as VP1, creating a library of capsid mutants, then screening the mutants in vitro or in vivo, as in [71]. “Capsid shuffling” can be performed by fragmenting the Cap genes of various AAV serotypes then recombining them via PCR. Li et al. used this technique to develop a novel AAV1/2/8/9 capsid that had increased ability to transduce melanoma cells [72].
Shuffling may indeed identify AAV vectors with improved kidney tropism. However, it is unclear how many “new” cell binding ligands can be found by this process or whether cryptic binding motifs are simply being revealed. If you shuffle a deck of 52 cards and get a new card, you will probably get thrown out of the game or have a really great story to tell.
Lastly, rational design of AAV capsid involves tailoring mutations in the capsid proteins toward a specific end. Though this technique is somewhat limited by basic science knowledge of AAV cell entry, it has also had breakthroughs. In 2015, Zinn et al. used this technique to generate a capsid based on AAV1, 2, 8, and 9 sequences that is the “predicted ancestor” of these common AAV vector capsids [73]. In 2018, Ikeda et al. used the aforementioned capsid, Anc80, to successfully deliver genes to kidney mesenchymal cells in vitro and in vivo [74]. Notably, this application has potential implications for treating kidney fibrosis with AAV-based gene therapy, since Anc80 was used to deliver Cre recombinase to a floxed mouse model, rather than a therapeutic transgene itself. This approach also brings enthusiasm to the idea that AAV can be re-engineered to target other cell types in the kidney. Certain positions in the AAV capsid are amenable to peptide inserts, such as those discovered using phage display libraries (reviewed in [75]). However, the very small 4.7 kilobase (kb) genome capacity of AAV vectors limits their use for large genes involved in certain kidney diseases (Table 1).
There are currently two FDA approved AAV gene therapies. The first, LUXTURNA®, was approved in December 2017 and delivers a functional copy of the RPE65 gene to retinal pigment epithelium to treat Leber’s congenital amaurosis, a form of retinitis pigmentosa. The second, ZOLGENSMA®, was approved in May 2019 and delivers a functional copy of the SMN1 gene to spinal motor neurons to treat spinal muscular atrophy. The former uses an AAV2 capsid while the latter uses an AAV9 capsid. These two recent FDA approvals for AAV gene therapy are in contrast to Ad and lentiviruses, neither of which is currently approved for in vivo gene therapy. (Notably, lentiviruses are FDA approved for ex vivo cell therapy in the context of creating chimeric antigen receptor (CAR) T therapies. The two therapies are KYMRIAH®, approved in August 2017, and YESCARTA®, approved in October 2017. However, the relative success of AAV does not come without its cautions. AAV has been shown to induce hepatocellular carcinoma (HCC) experimentally in mice and has also been implicated in insertional mutagenesis causing human HCC[68, 69, 76–78]. In addition, several recent studies demonstrated severe toxicity requiring euthanasia when delivering AAV9 capsid variants in high doses to nonhuman primates intravenously [70, 79]. While AAV remains a non-pathogenic and relatively non-immunogenic vector, it still has safety concerns. AAV gene therapy with respect to the kidney via direct injection methods holds certain potential advantages, such as limiting vector interactions to only the kidney and restricting systemic effects such as hepatic toxicity.
Lentivirus and Retrovirus Vectors
Ad and AAV are DNA viruses. In most cases, Ad and AAV deliver their genes and persist in non-dividing cells as episomes. If the cell divides, Ad and AAV transgenes are lost unless they become integrated into chromosomes by random events. Ad typically provides rapid, high transgene expression and AAV typically provides slower onset, long-term gene expression. Cells transduced by Ad can also be readily eliminated by the immune system due to the immunogenicity of its proteins. In contrast, retroviruses and lentiviruses have RNA genomes. In nature, retroviruses infect cells, reverse transcribe their RNA genomes to DNA, then integrate that pro-viral DNA into the host cell genome. Lentiviruses have the same modus operandi. When these two viruses are modified to deliver transgenes, both have the unique advantage of inserting genes into the host cell’s chromosomes. Therefore, if this cell divides, the transgene is carried on by all daughter cells, in contrast to episomal vectors such as Ad and AAV.
One primary disadvantage of retroviral vectors is that they only have the ability to transduce dividing cells – the intact nucleus of a non-dividing cell is not permissive to infection or transduction. Lentiviral vectors are built upon the backbone of the human immunodeficiency virus type 1 (HIV1) genome and are able to transduce both dividing and non-dividing cells, overcoming this limitation of retroviral vectors (reviewed in [80]).
Lentiviral vectors have become popular in treating genetic disorders of blood cells, primarily because patient bone marrow or hematopoietic cells can be extracted, transduced ex vivo, re-implanted, and yield permanent transgene expression for all daughter blood cells of the genetically corrected cells. Despite the concern of introducing a replication-competent lentivirus into a patient, there is considerable potential in using lentivirus to treat genetic disorders of other tissue types. Retargeting of lentiviral vectors can be achieved by pseudotyping the vectors with various envelope or glycoproteins. For example, retroviral and lentiviral vectors are often pseudotyped with vesicular stomatitis virus G-protein (VSV-G), which grants the vector extremely broad tropism due to VSV-G’s use of low-density lipoprotein receptor (LDLR) to enter cells [81, 82]. While pseudotyping lentiviral vectors with VSV-G eliminates the problem of poor transduction, it also increases the problem of off-target effects when administrating the vector.
In 2005, Morizono et al. showed that lentiviral vectors pseudotyped with an engineered Sindbis virus envelope could then be conjugated with a monoclonal antibody to retarget the lentivectors to melanoma cells expressing P-glycoprotein [83]. In 2009, Gennari et al. pseudotyped lentivectors with murine leukemia virus envelope fused to two single chain monoclonal antibodies (scFv’s) to increase transduction of bone-marrow derived dendritic cells by 5–10 fold, an important target for vaccination[84]. The most flexible method of retargeting lentiviruses that has emerged is pseudotyping the lentiviral envelope with measles virus (MV) hemagglutinin (H) and fusion (F) glycoproteins, the glycoproteins responsible for MV cell receptor binding and entry during infection (reviewed in [85]). Kneissel et al. fused scFv’s to the H glycoprotein, which allowed for specific targeting and ablated MV’s interaction with its natural receptors, CD46 and SLAM[86]. Even more flexible is Munch et al.’s addition of designed ankyrin repeat proteins (DARPins) to H glycoprotein, which allows for precise targeting using ribosomal display and phage display selected ligands [87]. Lentivectors retargeted by DARPins were eventually used by Zhou et al. to specifically target CD4+ T cells, an important goal for immunotherapy and anti-HIV gene therapy [88].
With respect to gene delivery to the kidney, some progress has been made with retroviral and lentiviral vectors Since the early 1990’s. In 1993, Bosch et al. showed that retroviral vectors expressing β-gal could transduce epithelial cells in the kidney via direct injection, after inducing kidney injury to increase the mitotic index of the kidney cells[89]. Gusella et al. showed that some kidney cells are stochastically transduced 1 month after renal artery, renal vein, ureter, or renal parenchyma injection of a lentiviral vector[90]. Considering cells that are transduced by these vectors result in a genomic integration, off-target effects of vector administration are considered higher risk than Ad or AAV because the effects are permanent (or until transduced cells die). However, the impressive targeting flexibility that DARPins grant lentivectors warrants further investigation. The kidney in particular offers a unique array of targeting options due to its highly specific cell-to-cell physiology. Each segment of the nephron is responsible for transporting different types of solutes. Thus, the proximal tubule, distal tubule, loop of Henle, and collecting duct each express unique proteins that could act as targets for vectors. For example, sodium-dependent phosphate transporter type 2a (NPT2a) and sodium-hydrogen exchanger 3 (NHE3) are expressed only in the proximal tubule, Na+/glucose co-transporter (SGLT2) is expressed only in the S1 and S2 segments of the proximal tubule, sodium-potassium-2-chloride cotransporter (NKCC2) is expressed only in the thick ascending limb of Henle (TALH), and aquaporin 2 (AQP2) and E-cadherin (ECAD) are expressed only in the collecting duct [24, 91]. It is also feasible to target vector expression, including Ad, AAV, and LV using the promoters of these genes to become nephron segment-specific. In a striking recent study, Xu et al. demonstrated the ability of the deactivated Cas9 (dCas9) platform delivered in vivo by multiple parenchymal injections of lentiviral vectors to demethylate key promoters to attenuate kidney fibrosis [92]. (dCas9 demethylation platforms are engineered, applied, and reviewed in and [93–95].) In 2009, Kim et al. induced renal ischemia-reperfusion injury (IRI) in adenosine A1 receptor (A1AR) knockout mice and injected a lentivirus expressing human A1AR via parenchymal injection, showing that this gene delivery had a protective effect against IRI [96]. The same parenchymal delivery method of lentiviruses encoding A1AR and heat shock protein 27 was subsequently shown to have protective effects against liver ischemic injury and acute kidney injury, respectively [97–99]. More recently, in 2015, Espana-Agusti et al. used an ultrasound-guided approach to perform a non-invasive intrarenal parenchymal injection method of lentiviral vectors and showed that transgene expression persisted for over two months in proximal and distal tubules and a short hairpin RNA (shRNA) delivered by the vector knocked down the Tsc1 gene to detectable levels [100]. In a particularly striking example of the power of lentiviral vectors, in 2020, Yuzefovych et al. used ex vivo kidney perfusion to deliver 1.5e11 LV particles to a rat kidney destined for kidney transplantation over the course of two hours [101]. In this case, the LV encoded for shRNAs against rat beta2-microglobulin and rat class II transactivator in order to prevent expression of MHC class I and II, respectively, and create an “immunologically invisible” donor kidney. While the authors did not actually test the ability of the genetically modified rat kidney to resist allograft rejection, they did note that the modified kidney had roughly 25% expression of the two target genes compared to control. Theoretically, ex vivo kidney perfusion could be used on a donor kidney to transduce cells with LV, other viral vectors, or non-viral vectors, and could also permanently modify cells by delivering cargo like CRISPR-Cas9. The proof of principle for lentiviral vectors in kidney therapeutic gene delivery has been established and the field is open to new innovations.
Non-viral Vectors for Molecular Therapy
Transfection is the technical term for delivering naked DNA or RNA into cells without the use of a viral vector. Viral vector transduction has some clear advantages in efficiency over DNA transfection, but this also comes with a biohazard cost. Viral vectors are generally attenuated forms of infectious, lethal, wild type viruses. There is also a risk of recombination of viral vectors with wild type viruses during production or after in vivo delivery [102]. Other risks include oncogenesis and immune responses [103, 104][105][106][107]). In a more practical sense, constructing a recombinant DNA plasmid for gene delivery avoids many of the technical hardships and pitfalls of production of a viral vector, for example the transgene cloning and Cre-Lox system necessary to produce helper-dependent Ad (HDAd)[108].
DNA transfection methods have evolved and have experienced much iteration over the years. The simplest is “naked DNA” transfection, which is transfection of DNA without the use of any carrier molecule or vector other than solvent. In 1999, Liu et al. showed that administering mice tail vein injections of large amounts of plasmid DNA (5–25 μg) dissolved in saline resulted in the vast majority of the DNA targeting the liver [109]. In an attempt to increase transfection efficiency, both through increased cellular uptake and increase DNA stability, cationic lipoplexes and polyplexes became popular (reviewed in [110–116]). Lipoplexes are complexes of DNA and lipids while polyplexes are complexes of DNA and engineered molecules. Although these tools increased transfection efficiency as compared to naked DNA transfections, they still lack the ability to be robustly targeted to specific cell or tissue types. One parallel between systemic administration of DNA for transfection and systemic administration of Ad is that both are mostly taken up by the liver. One solution to this conundrum is to administer a direct injection to the organ of interest in order to “target” the transfection. In 1992, Tomita et al. used HVJ-liposomes, a liposome mixed with inactivated “hemagglutinating virus of Japan” (a synonym for Sendai virus), to show that some reporter gene deliver could be seen in glomeruli of rat kidneys by catheter delivery to the renal artery[117]. HVJ-liposomes were used because the Sendai virus membrane proteins can fuse cells efficiently, but the fact that this method utilizes inactivated virus brings another level of safety precautions (HVJ-liposome technology is reviewed in [118]). In 1993, Isaka et al. used the same HVJ-liposome and renal artery delivery method to transfect rat glomeruli with plasmids expressing either transforming growth factor-β (TGF-β) or platelet-derived growth factor (PDGF) and showed that the overexpression of these genes was sufficient to cause glomerulosclerosis [119]. In 1999, Isaka et al. developed a chimeric TGF-β type II receptor cDNA that when administered to nephritic rats systemically could suppress the extracellular matrix expansion around glomeruli by blocking TGF-β activity[120]. In 2005, Koike et al. demonstrated that injecting plasmid DNA into the left rat artery followed by application of an ultrasound transducer directly to the side of the kidney for 1–2 minutes mediated superior gene expression to the HVJ-liposome method, with 70–80% of glomeruli being transduced [121]. The theory behind using ultrasound is that it makes cell membranes more permeable and thus permissive to uptake of DNA lipoplexes [122–126]. Interestingly, ultrasound has also been used in tandem with adenovirus injection to enhance transduction by almost 2-fold in the context of an intra-tumoral injection for cancer gene therapy [127]. In 2009, Xing et al. used ultrasound in tandem with hydrodynamic injections of luciferase plasmid in rat renal vein to show that in increased luciferase activity by nearly 10-fold [128]. However, overexposure to ultrasound can result in tissue injury, creating an additional complication in the safety profile of this technique. In 2014, Kurosaki et al. used bubble lipopolyplexes composed of protamine sulfate (PS), distearoyl phosphatidylglycerol (DSPG), distearoyl phosphatidylcholine (DSPC), and methoxy-polyethyleneglycol 2000-distearoyl phosphatidylethanolamine (PEG-DSPE) as an ultrasound-responsive carrier for plasmid DNA[129]. Using this system, mice could be administered tail vein injection of 25 μg of DNA and luciferase reporter activity was primarily located in the kidney by ultrasound irradiating that particular tissue. In addition, the method did not cause kidney damage, at least compared to the cisplatin that was used as a positive control. In 2001, Tsujie et al. showed that electroporating plasmid DNA expressing luciferase into the left kidney of rats by clamping electrodes to each end of the kidney resulted in 75% of glomeruli being transfected and roughly ten-fold increased luciferase expression when compared to the HVJ-liposome method [130]. However, electroporation caused burns at high voltages (100V) and it is questionable how techniques like this would be translated to humans.
In 1998, Lai et al. performed what possibly the first rescuing of a genetic renal phenotype in mice by administering mice deficient in carbonic anhydrase II (CAII) a renal pelvis injection of a plasmid encoding human CAII mixed with Lipofectin [131]. The authors could detect CAII in the cortex and outer medulla tubule cells and the mice regained their ability to acidify urine after ammonium chloride administration. In 2002, Maruyama et al. showed that some kidney endothelial cells could be transduced by naked DNA after direct retrograde injection into rat renal vein [132, 133]. In 2004, Kameda et al. showed that the same methodology is applicable in mice [134]. More recently, in 2017, Woodard et al. showed that hydrodynamic injection of DNA solution into the renal pelvis of mice resulted in some transduction of proximal tubule, distal tubule, collecting duct, and interstitial cells, corroborating that direct delivery to the kidney is superior to systemic injections [135].
One particle challenge for non-viral vectors is their potential for retargeting. While Ad and AAV can be genetically modified to express peptides, and lentiviral vectors can contain an engineered scFv, non-viral vectors would theoretically need to be complexed with a lipid nanoparticle (LNP) that has specifically been designed to target a specific cell type, tissue type, or receptor. In 2002, Tomita et al. complexed HVJ-liposomes with anti-Thy1.1 antibody in order to target the OX-7 antigen present on rat mesangial cells and deliver an NF-κB decoy oligodeoxynucleotides (ODN’s) in order to squelch activity of the pro-inflammatory NF-κB promoter. While the FITC-labeled ODN’s did transfect kidney tissue and reduce IL-1β and ICAM-1 expression in glomeruli after systemic administration, the authors did not necessarily show that this method detargeted other tissues, such as the liver [136]. There is also evidence that particular non-viral vectors may naturally target the kidney. In 2019, Uchida et al. studied the in vivo effects of DNA-binding protein from starved cells (Dps) nanocages, a self-assembling tetrahedral dodecamer from Sulfolobus solfataricus, by injecting the nanocages intravenously in mice [137]. The authors found that the nanocages localize in the S1 segment of the proximal tubule, which is a unique property for any vector. These nanocages are approximately 9 nanometers in diameter, and therefore are actually small enough to clear the slit diaphgrams in the glomerulus. Although the authors did not attempt to deliver nucleic in the Dps nanocages, which is a future possibility, they did show that conjugating manganese to the nanocage provided an antioxidant effect and somewhat protected mice from LPS induced kidney injury. Retargeting non-viral vectors remains relatively difficult, however, LNPs have already proven their general efficacy to the public: in 2018, the FDA approved patisiran (ONPATTRO®), a small interfering RNA (siRNA) therapeutic directed at the liver to treat hereditary transthyretin amyloidosis (hATTR)[138, 139]. Thus, non-viral vectors definitely have the ability to treat human disease, and should not be ruled out for kidney disease.
RNA Vectors
Thus far, this review has covered vectors and delivery routes that have been more frequently used and made an overall larger impact on the field. However, there are other notable, albeit less studied, vectors and modalities of delivery that have shown to affect the kidney in vivo. In 2008, Suga et al. used a Sendai virus vector to deliver the human aquaporin 2 (AQP2) to rat medullary collecting duct cells by the retrograde ureter route, a therapy that could potentially treat nephrogenic diabetes insipidus in patients with mutations in vasopressin V2 receptor (AVPR2) or AQP2 [140]. Sendai virus is an enveloped, negative sense, single-stranded RNA genome virus of the family Paramyxoviridae. Sendai virus vectors are produced at intrinsically lower titers than DNA virus vectors such as Ad and AAV, which is one reason they have not attained the same widespread use[141]. Nevertheless, this study has laid the groundwork for use of therapeutic, non-replicating RNA viral vectors in kidney gene delivery.
RNAi
In addition to less commonly used vectors, important work has been over the last several decades utilizing antisense oligonucleotides (ASOs), also known as antisense oligodeoxynucleotides (ODNs). ASOs are typically twenty nucleotides in length and are designed to complement a target mRNA to induce steric blocking of the ribosome, degradation by RNase H, or even exon skipping (reviewed in [142]). Importantly, ASOs are often chemically modified (I.E., phosphorothioated oligonucleotides) for enhanced stability. Several studies in the early 1990’s showed that systemic delivery of ASOs in rodents resulted in accumulation of the ASOs in the liver and kidney [143–145]. In 1995, Oberbauer et al. determined that 18-mer ASOs remain their full length for up to four days after intravenous injection in rats, whereas they have been degraded in length when excreted in the urine [146]. In the same year, Rappaport et al. showed that P32-labeled ASOs infiltrated into the proximal tubules in mice after intravenous injections, indicating that ASOs do not run into the same problem of being sequestered at the glomerulus as viral vectors[147]. Indeed, in 1996, Noiri et al. designed an ASO targeting an exon in inducible nitric oxide synthase (iNOS) and showed that by delivering it intracardially immediately prior to induction of ischemia, ischemic injury was attenuated[148]. At this point in time, such an experiment was not possible at the level of protein inhibition since iNOS-specific antagonists did not exist, and antagonizing the other two isoforms in the kidney, endothelial constitutive NOS, and neuronal NOS aggravate kidney dysfunction during ischemia. Also in 1996, Akagi et al. showed that targeting TGF-β1 mRNA in a rat model of anti-Thy1.1 mesangial proliferative glomerulonephritis reduced TGF-β1 protein levels and extracellular matrix expansion [149]. Likewise, in 2003, Daniel et al. perfused phosphorothioate ASOs against thrombospondin-1 (TSP1), an activator of latent TGF-β, into kidneys of rats through the renal artery two days after induction of mesangial proliferative glomerulonephritis and showed that not only was expression TSP1 decreased, but TGF-β activation and extracellular matrix accumulation were both lowered [150]. As an alternative strategy, some studies involve implementing double-stranded ODNs as transcription factor-binding “decoys” that sequester transcription factors and thus modulate gene expression in renal diseases (reviewed in [151]). Close cousins of the ASO are small interfering RNAs (siRNAs), which are RNA oligonucleotides approximately twenty bases long that form secondary structure and cause an endogenous target RNA to be taken up by the RNA-induced silencing complex (RISC) and ultimately downregulated (reviewed in [142]). In 2009, Molitoris et al. showed that intravenously delivering an siRNA targeting p53 in a cisplatin-induced AKI model attenuated the kidney injury, potentially as an effect of reduced p53-mediated apoptosis in renal proximal tubule cells[152]. In 2014, Morishita et al. showed that intravenously delivering an siRNA targeting Smad4 in a folic acid-induced renal fibrosis model attenuated the level of renal fibrosis, possibly by downregulating downstream pro-fibrotic genes[153]. In 2016, Alidori et al. showed that siRNA complexed with ammonium-functionalized carbon nanotubes (fCNTs) increased biodistribution in the kidney from approximately 2% to 22%, then used this technology to prophylactically protect mice from cisplatin-induced injury by administering siRNAs against Trp53 and Mep1b, which are overexpressed in kidney injury [154]. Notably, the fCNT/siRNA accumulated preferentially in proximal tubule cells. These aforementioned studies show that ASOs and siRNAs still hold potential for treating AKI, CKD, or genetic kidney disorders through the systemic route of delivery.
“Kidney Effectors”
Much of this review has focused on the necessity of nucleic acid delivery directly to kidney cells to create the physiological change necessary to attenuate a disease phenotype. However, research has also been done on what we might call “kidney effectors”, or nucleic acid delivery strategies that affect kidney physiology without necessitating direct delivery to that tissue. In 2010, Schievenbusch et al. intravenously delivered AAV2, AAV8, and AAV9 encoding for human hepatocyte growth factor (HGF) to Col4a3 knockout mice, a model of chronic progressive renal disease and tubulointerstitial fibrosis (mutations in this gene also cause Alport Syndrome). This study found that AAV9 yielded superior higher transduction efficiency and transgene expression of a CMV-GFP cassette than AAV2 and AAV8 in both the liver and kidneys. However, when normalized to mouse Crp gene, AAV9 vector genomes had a median of approximately 80 relative units (RU) in the liver and approximately 1 RU in the kidneys (AAV2 and AAV8 were even less at approximately 0.4 and 0.6 RU, respectively)[155]. Subsequent IHC staining did show a small number of proximal and distal tubule cells expressing GFP. Notably, the authors were then able to ablate transduction of the spleen, lung, and heart, and somewhat enhance transduction in the liver and kidney by using a CMV enhancer fused to Ksp-cadherin promoter rather than the CMV promoter itself. When AAV9-CMV-Ksp-hHGF was delivered intravenously to 4-week old Col4a3 knockout mice (before fibrosis is histologically detectable), three markers of fibrosis (collagen 1α1 [Col1A1], α-smooth muscle actin [SMA], and platelet-derived growth factor receptor-β [PDGFR-β]) each decreased by approximately 50%, and ECM accumulation in the kidneys was improved in the treated mice. Although the authors argue that the antifibrogenic effect of the hHGF gene therapy was mediated by both paracrine production from the kidney and endocrine production from the liver, it seems more likely that the vast majority of the effect is from the liver, where transduction and transgene efficiency were markedly higher than in the kidney. Several other studies share the “kidney effector” label. In 2017, Holditch et al. intravenously delivered AAV9 encoding B-type natriuretic peptide (BNP), a guanylyl cyclase A agonist, to polycystic kidney (PCK) rats and showed that this therapy reduced cystic index and glomerular injury, although gene expression came primarily from transduced cells in the heart and secondarily from the liver [156]. More recently, in 2019, Holditch et al. delivered a constitutively hypophosphorylated eukaryotic translation initiation factor 4E-binding protein 1 mutant (4E-BP1F113A) intraperitoneally to a mouse model of autosomal dominant polycystic kidney disease (ADPKD) using AAV9 as a vector, hypothesizing that overexpression of variant would sequester eIF4E and reduce proliferation in cystic epithelial cells. Interestingly, in the four months following AAV9 administration, rather than ameliorate the ADPKD pathophysiology, this experiment resulted in increased total kidney volume, cystic index, number of cysts, size of cysts, and Bcl-2 expression (an anti-apoptotic protein)[157]. While the authors defined 4E-BP1F113A-mediated mitochondrial stress as a mechanism of worsened ADPKD phenotype, it also possible that this resulted from an endocrine effect from the liver and heart, which transduced and showed AAV transgene expression in addition to the kidney. While this study worsened a kidney disease phenotype rather than improve it, it is still more evidence that kidney physiology can be modulated through an endocrine effect of other body tissues. In 2020, Wang et al. documented a distinct endocrine effect by injecting AAV9-miR-29a, an antifibrotic micro RNA, into the tibialias anterior (TA) muscle of UUO mice and observing a decrease in both muscle atrophy and kidney fibrosis markers as well as some prevention of muscle loss and kidney fibrosis[158]. While the AAV injection was made intramuscularly, expression of the GFP reporter gene was found in the injected TA, uninjected TA, left kidney, right kidney, and liver indicating that the vector leaked systemically from the site of injection. Nonetheless, the authors note that miR-29a is present in serum exosomes after UUO injury, and argue that the antifibrotic effect was likely due to exosome-mediated transfer of miR-29a from muscle to kidney. The aforementioned studies show the feasibility of transducing off-target tissues to effect kidney disease in an endocrine fashion.
Cell Therapy
One other notable area of interest for molecular therapy in the kidney is cell therapy, which can be considered as the close cousin of gene therapy. Already mentioned in this review are KYMRIAH® and YESCARTA®, two autologous T cell therapies in which a patient’s own T cells are genetically modified via lentiviral transduction and re-implanted[159]. Similar methodology has been studied in applications that could potentially be therapeutic for kidney disease. In 1994, Kitamura et al. transduced cultured rat mesangial cells in vitro with a retroviral vector and engrafted them back into rat glomeruli through renal artery injection [160]. This method resulted in approximately 60% of glomeruli in the rat kidney to express the beta-galactosidase reporter gene from the modified cells and this expression was sustained for 4 weeks, and 8 weeks when the pre-existing mesangial cells had been depleted beforehand. In 1996, Kitamura et al. accomplished the engraftment of autologous mesangial cells in the glomerulus using stable transfectants rather than retrovirally transduced cells[161]. In 1996, Naito et al. transduced tubule epithelial cells in vitro with a retrovirus expressing colony stimulating factor-1 (CSF-1) and implanted them subcapsularly in a lupus mouse model to induce renal injury [162]. While this procedure was obviously not therapeutic, the authors did observe transgene expression for several months after cell delivery. In a study of more potential therapeutic interest, in 1996, Masanori Kitamura developed a system to stably transfect rat mesangial cells with a reporter gene under control of a tetracycline regulatory system, and that these cells could be engrafted in vivo by left renal artery injection [163]. In the same year, Kitamura and Kawachi developed an in vivo rat mesangial cell cytosensor capable of automatically responding to glomerular inflammation [164]. They achieved this by stably transfecting the mesangial cells with a β-gal expression cassette driven by CArG, a regulatory sequence from alpha-smooth muscle actin promoter, which is active in glomerular mesangial cells during inflammatory states. These studies effectively gave proof of principle that cells therapies could be designed using an on/off switch for expression, triggered by either administration of activating molecules or a response to endogenous conditions [163]. In some cases, cell therapy has been showed to be capable of treating genetic kidney diseases in mice. More recently, in 2009, Syres et al. attenuated disease progression a mouse model of cystinosis (Ctns−/−) by performing bone marrow cell transplants and hematopoietic stem cell transplants from syngeneic cystinosin wild type (Ctns+/+) mice. Although cystinosis is a multisystemic lysosomal storage disorder, the transplanted cells were able to engraft in many tissues in Ctns−/− mice, including the kidney, decrease cystine levels in engrafted tissues, decrease kidney damage by preventing cystine crystal formation, and improve kidney function [165]. In 2013, Harrison et al. expanded upon this work by transducing Ctns−/− mouse hematopoietic stem and progenitor cells with a lentiviral vector expressing human CTNS cDNA then performing autologous transplantation to rescue the mice from cystinosis [166]. Strikingly, this study showed that the functional cystinosin protein product was transferred from the genetically modified hematopoietic cells to Ctns-deficient cells, which was unexpected considering that cystinosin is a lysosomal integral membrane protein. Subsequent work by Naphade et al. in 2015 showed that macrophages derived from the genetically corrected hematopoietic stem cells are capable of transferring cystinosin-bearing lysosomes to Ctns-deficient renal tubule cells, elucidating the mechanism of cellular cross-correction [167]. These studies on cell therapy for cystinosis in mice suggest that other genetic kidney disease may be treatable by cell therapy via a vesicle-mediated mechanism.
Summary
Molecular therapy for kidney disease, i.e. kidney gene therapy, remains a largely untapped field in terms of the science that has thus far been translated to the clinic. However, there is great potential for human genetic kidney disease that still has necessity to be treated. In addition, chronic kidney disease, which is not always genetic in etiology, could also benefit from efficient gene delivery to the kidney.
This review not only details past efforts regarding nucleic acid delivery to the kidney, but also defines the current diseases at hand, tools available, and challenges to overcome in the realm of kidney molecular therapy. For example, the genetic carrying capacity of available vectors varies greatly, from the 4.7 kb AAV on the lower end to the 38 kbp HDAd on the upper end (Figure 3). In addition, the diameter and molecular weight of these vectors also vary greatly, could affect the vector’s ability to escape from the tubule lumen to the basolateral side of tubules, or vice versa, after injection (Figures 4 and 5). These aforementioned properties of the chosen vector are of great importance when attempting to treat a particular disease. The COL4A3, COL4A4, and COL4A5 genes, mutations in which cause Alport syndrome, each have cDNA’s greater than 4.7 kb, and thus too large to be packaged in popular AAV vectors (Table 1). The gene therapy strategy is also dependent upon the etiology of the disease. Diseases such as ADPKD, ARPKD, Bartter Syndrome, Von-Hippel Lindau syndrome, Gitelman syndrome, Congenital nephrotic syndrome, Dent’s disease, Papillorenal syndrome, Alport syndrome, and cystinosis are each likely caused by a partial or complete loss of a particular gene, and a gene addition strategy (I.E., delivering wild-type cDNA of the mutant gene) would theoretically be therapeutic for each disease [168–184]. However, other diseases such as Liddle syndrome, ADTKD-MUC1, ADTKD-UMOD, and ADTKD-REN are likely caused by dominant negative mutations and could theoretically be treated by knockdown of the mutant transcript using siRNA, shRNA, or synthetic microRNA (miR), or by knockout of the mutant allele altogether using CRISPR-Cas9 [185–189]. Notably, the aforementioned antisense oligonucleotides are significantly smaller in diameter and molecular weight than viral vectors, and may not be subject to the same size-limiting restriction at the glomerulus. An example of this RGLS4326, a short oligonucleotide inhibitor of miR-17, which preferentially accumulates in the kidneys and ameliorates ADPKD in mice after subcutaneous injection, a type of systemic delivery[190]. Even considering all of these points, one must still consider which cell type within the plethora that exist within the kidney should be genetically corrected to treat a particular disease, and how to target that cell type. Examples such as different serotypes of Ad and AAV, novel capsids, engineered glycoproteins, and tissue-specific promoters are examples of vector targeting discussed in this review.
Efficiency of gene delivery to the kidney is clearly one sticking point currently limiting molecular therapy to this organ. Many of the studies cited in this review demonstrate that their methodology is capable of transducing cells in the kidney in vivo, however, in most cases, it just a small fraction of cells transduced within a large field of view, and quantitation is sparsely discussed. More recent studies that utilize arterial, venous, and ureteral clamps seem to enhance kidney transduction. Future work should include transduction studies that quantify how many cell are expressing reporter genes and which cell types have been transduced so that a given vector type plus a given injection method can yield a concrete, quantitative gene delivery efficiency. One optimistic note is that many of the physical delivery methods detailed in this review already have similar access routes in humans in the clinic. For example, a renal vein injection correlates to renal venography where a catheter is threaded retrograde from the femoral vein[191]. One other interesting point to consider is whether the age of an injected animal modulates level of transduction from a gene therapy vector such as Ad. In 2005, Jerebtsova et al. injected Ad5-β-gal in 14 days old versus adult mice and found that roughly ten to twenty-fold of the Ad stayed in circulation in 14 days old mice versus adult mice up to 90 minutes after injection. In addition, two days post injection, approximately 45% of 14 days old mice glomeruli were positive for β-gal staining while adult glomeruli showed none [192]. Therefore, there is merit in further investigating specifically how the age and developmental phase of a model organism or a potential patient could change the pharmacodynamics of administered viral vectors.
While organs such as the eyes and liver have found efficient routes of vector delivery, the kidney is still being researched as a valuable tissue with a need for consistent and effective modality of delivery. The need for these effective modalities is not purely driven by basic research interest. The Centers for Disease Control and Prevention (CDC) estimates that chronic kidney disease (CKD) affects greater than 30 million people in the US and is the ninth leading cause of death in US. In addition, CKD and end stage renal disease Medicare costs are $114 billion annually, and there are estimated to be over 300 new dialysis patients each day. Society will benefit greatly in terms of health and overall financial healthcare costs by finding robust methods of translational kidney molecular therapy.
Key Points.
Kidney molecular therapy has lagged behind other fields
There are several promising results in the literature using viral and non-viral vectors that should be built upon to improve kidney molecular therapy
There are a plethora of diseases that could potentially be treatable once pre-clinical kidney gene delivery has been improved
Acknowledgements
This work was supported by the Department of Molecular Medicine at Mayo Clinic (J.D.R.), National Institute of Diabetes Digestive and Kidney Diseases Grant Number 1F31DK123858-01 ( J.D.R.), and the Department of Laboratory Medicine and Pathology at Mayo Clinic (M.A.B.).
Footnotes
Author Disclosure Statement
The authors declare they do not have any competing or financial interests.
References
- 1.Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, et al. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PLoS One. 2016;11(7):e0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hildebrandt F Genetic kidney diseases. Lancet. 2010. April 10;375(9722):1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung JC. Inherited renal diseases. Curr Pediatr Rev. 2014;10(2):95–100. [DOI] [PubMed] [Google Scholar]
- 4.Boron WB, Emile. Glomerular Filtration and Renal Blood Flow Medical Physiology. 3rd ed: Elsevier; 2017. p. 739–53.e2. [Google Scholar]
- 5.Murphy JJ, Myint MK, Rattner WH, Klaus R, Shallow J. The lymphatic system of the kidney. J Urol. 1958. July;80(1):1–6. [DOI] [PubMed] [Google Scholar]
- 6.McIntosh GH, Morris B. The lymphatics of the kidney and the formation of renal lymph. J Physiol. 1971. May;214(3):365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003. Sep-Oct;80(1–2):148–58. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Ertl HC, Wilson JM. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity. 1994. August;1(5):433–42. [DOI] [PubMed] [Google Scholar]
- 9.Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. 2009. September;20(9):975–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry MA, Weaver EA, Chen CY. Mining the adenovirus “virome” for systemic oncolytics. Curr Pharm Biotechnol. 2012. July;13(9):1804–8. [DOI] [PubMed] [Google Scholar]
- 11.Lieber A, He CY, Meuse L, Schowalter D, Kirillova I, Winther B, et al. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J Virol. 1997. November;71(11):8798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Worgall S, Wolff G, Falck-Pedersen E, Crystal RG. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum Gene Ther. 1997. January 1;8(1):37–44. [DOI] [PubMed] [Google Scholar]
- 13.Zhu G, Nicolson AG, Zheng XX, Strom TB, Sukhatme VP. Adenovirus-mediated beta-galactosidase gene delivery to the liver leads to protein deposition in kidney glomeruli. Kidney Int. 1997. October;52(4):992–9. [DOI] [PubMed] [Google Scholar]
- 14.Ye X, Jerebtsova M, Ray PE. Liver bypass significantly increases the transduction efficiency of recombinant adenoviral vectors in the lung, intestine, and kidney. Hum Gene Ther. 2000. March 1;11(4):621–7. [DOI] [PubMed] [Google Scholar]
- 15.Moullier P, Friedlander G, Calise D, Ronco P, Perricaudet M, Ferry N. Adenoviral-mediated gene transfer to renal tubular cells in vivo. Kidney Int. 1994. April;45(4):1220–5. [DOI] [PubMed] [Google Scholar]
- 16.Heikkila P, Parpala T, Lukkarinen O, Weber M, Tryggvason K. Adenovirus-mediated gene transfer into kidney glomeruli using an ex vivo and in vivo kidney perfusion system - first steps towards gene therapy of Alport syndrome. Gene Ther. 1996. January;3(1):21–7. [PubMed] [Google Scholar]
- 17.McDonald GA, Zhu G, Li Y, Kovesdi I, Wickham TJ, Sukhatme VP. Efficient adenoviral gene transfer to kidney cortical vasculature utilizing a fiber modified vector. J Gene Med. 1999. Mar-Apr;1(2):103–10. [DOI] [PubMed] [Google Scholar]
- 18.Ye X, Liu X, Li Z, Ray PE. Efficient gene transfer to rat renal glomeruli with recombinant adenoviral vectors. Hum Gene Ther. 2001. January 20;12(2):141–8. [DOI] [PubMed] [Google Scholar]
- 19.Chetboul V, Klonjkowski B, Lefebvre HP, Desvaux D, Laroute V, Rosenberg D, et al. Short-term efficiency and safety of gene delivery into canine kidneys. Nephrol Dial Transplant. 2001. March;16(3):608–14. [DOI] [PubMed] [Google Scholar]
- 20.Choi YK, Kim YJ, Park HS, Choi K, Paik SG, Lee YI, et al. Suppression of glomerulosclerosis by adenovirus-mediated IL-10 expression in the kidney. Gene Ther. 2003. April;10(7):559–68. [DOI] [PubMed] [Google Scholar]
- 21.Fujishiro J, Takeda S, Takeno Y, Takeuchi K, Ogata Y, Takahashi M, et al. Gene transfer to the rat kidney in vivo and ex vivo using an adenovirus vector: factors influencing transgene expression. Nephrol Dial Transplant. 2005. July;20(7):1385–91. [DOI] [PubMed] [Google Scholar]
- 22.Heikkila P, Tibell A, Morita T, Chen Y, Wu G, Sado Y, et al. Adenovirus-mediated transfer of type IV collagen alpha5 chain cDNA into swine kidney in vivo: deposition of the protein into the glomerular basement membrane. Gene Ther. 2001. June;8(11):882–90. [DOI] [PubMed] [Google Scholar]
- 23.Rubin J, Nguyen TV, Allen K, Ayasoufi K, Barry MA. Comparison of Gene Delivery to the Kidney by Adenovirus, Adeno-associated Virus, and Lentiviral Vectors after Intravenous and Direct Kidney Injections. Hum Gene Ther. 2019. October 22. [DOI] [PMC free article] [PubMed]
- 24.Watanabe S, Ogasawara T, Tamura Y, Saito T, Ikeda T, Suzuki N, et al. Targeting gene expression to specific cells of kidney tubules in vivo, using adenoviral promoter fragments. PLoS One. 2017;12(3):e0168638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rux JJ, Burnett RM. Adenovirus structure. Hum Gene Ther. 2004. December;15(12):1167–76. [DOI] [PubMed] [Google Scholar]
- 26.Shayakhmetov DM, Gaggar A, Ni S, Li ZY, Lieber A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol. 2005. June;79(12):7478–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parker AL, Waddington SN, Nicol CG, Shayakhmetov DM, Buckley SM, Denby L, et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood. 2006. October 15;108(8):2554–61. [DOI] [PubMed] [Google Scholar]
- 28.Xu Z, Tian J, Smith JS, Byrnes AP. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol. 2008. December;82(23):11705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krasnykh V, Dmitriev I, Mikheeva G, Miller CR, Belousova N, Curiel DT. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J Virol. 1998. March;72(3):1844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G, et al. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol. 1998. December;72(12):9706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koizumi N, Mizuguchi H, Hosono T, Ishii-Watabe A, Uchida E, Utoguchi N, et al. Efficient gene transfer by fiber-mutant adenoviral vectors containing RGD peptide. Biochim Biophys Acta. 2001. November 7;1568(1):13–20. [DOI] [PubMed] [Google Scholar]
- 32.Mizuguchi H, Koizumi N, Hosono T, Utoguchi N, Watanabe Y, Kay MA, et al. A simplified system for constructing recombinant adenoviral vectors containing heterologous peptides in the HI loop of their fiber knob. Gene Ther. 2001. May;8(9):730–5. [DOI] [PubMed] [Google Scholar]
- 33.Wickham TJ, Tzeng E, Shears LL 2nd, Roelvink PW, Li Y, Lee GM, et al. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J Virol. 1997. November;71(11):8221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michael SI, Hong JS, Curiel DT, Engler JA. Addition of a short peptide ligand to the adenovirus fiber protein. Gene Ther. 1995. November;2(9):660–8. [PubMed] [Google Scholar]
- 35.Nicklin SA, Wu E, Nemerow GR, Baker AH. The influence of adenovirus fiber structure and function on vector development for gene therapy. Mol Ther. 2005. September;12(3):384–93. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh D, Barry MA. Selection of muscle-binding peptides from context-specific peptide-presenting phage libraries for adenoviral vector targeting. J Virol. 2005. November;79(21):13667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barry MA, Dower WJ, Johnston SA. Toward cell-targeting gene therapy vectors: selection of cell-binding peptides from random peptide-presenting phage libraries. Nat Med. 1996;2(3):299–305. [DOI] [PubMed] [Google Scholar]
- 38.Wu CH, Liu IJ, Lu RM, Wu HC. Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci. 2016. January 19;23:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barry MA, Takahashi S, Parrott MB. Selection of peptides on phage In: Douglas JaC DT, editor. Vector Targeting Strategies for Gene Therapy. Hoboken, NJ: Wiley-Liss, Inc.; 2002. p. 549–79. [Google Scholar]
- 40.Denby L, Work LM, Seggern DJ, Wu E, McVey JH, Nicklin SA, et al. Development of renal-targeted vectors through combined in vivo phage display and capsid engineering of adenoviral fibers from serotype 19p. Mol Ther. 2007. September;15(9):1647–54. [DOI] [PubMed] [Google Scholar]
- 41.Diaconu I, Denby L, Pesonen S, Cerullo V, Bauerschmitz GJ, Guse K, et al. Serotype chimeric and fiber-mutated adenovirus Ad5/19p-HIT for targeting renal cancer and untargeting the liver. Hum Gene Ther. 2009. June;20(6):611–20. [DOI] [PubMed] [Google Scholar]
- 42.Campos SK, Barry MA. Current advances and future challenges in Adenoviral vector biology and targeting. Curr Gene Ther. 2007. June;7(3):189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parrott MB, Adams KE, Mercier GT, Mok H, Campos SK, Barry MA. Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol Ther. 2003. October;8(4):688–700. [DOI] [PubMed] [Google Scholar]
- 44.Campos SK, Parrott MB, Barry MA. Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol Ther. 2004. June;9(6):942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campos SK, Barry MA. Comparison of adenovirus fiber, protein IX, and hexon capsomeres as scaffolds for vector purification and cell targeting. Virology. 2006. June 5;349(2):453–62. [DOI] [PubMed] [Google Scholar]
- 46.Lanciotti J, Song A, Doukas J, Sosnowski B, Pierce G, Gregory R, et al. Targeting adenoviral vectors using heterofunctional polyethylene glycol FGF2 conjugates. Mol Ther. 2003. July;8(1):99–107. [DOI] [PubMed] [Google Scholar]
- 47.Romanczuk H, Galer CE, Zabner J, Barsomian G, Wadsworth SC, O’Riordan CR. Modification of an adenoviral vector with biologically selected peptides: a novel strategy for gene delivery to cells of choice. Hum Gene Ther. 1999. November 1;10(16):2615–26. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi S, Mok H, Parrott MB, Marini FC 3rd, Andreeff M, Brenner MK, et al. Selection of chronic lymphocytic leukemia binding peptides. Cancer Res. 2003. September 1;63(17):5213–7. [PubMed] [Google Scholar]
- 49.Menezes KM, Mok HS, Barry MA. Increased transduction of skeletal muscle cells by fibroblast growth factor-modified adenoviral vectors. Hum Gene Ther. 2006. March;17(3):314–20. [DOI] [PubMed] [Google Scholar]
- 50.O’Riordan CR, Lachapelle A, Delgado C, Parkes V, Wadsworth SC, Smith AE, et al. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum Gene Ther. 1999. May 20;10(8):1349–58. [DOI] [PubMed] [Google Scholar]
- 51.Mok H, Palmer DJ, Ng P, Barry MA. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol Ther. 2005. January;11(1):66–79. [DOI] [PubMed] [Google Scholar]
- 52.Hofherr SE, Shashkova EV, Weaver EA, Khare R, Barry MA. Modification of adenoviral vectors with polyethylene glycol modulates in vivo tissue tropism and gene expression. Mol Ther. 2008. July;16(7):1276–82. [DOI] [PubMed] [Google Scholar]
- 53.Atchison RW, Casto BC, Hammon WM. Adenovirus-Associated Defective Virus Particles. Science. 1965. August 13;149(3685):754–6. [DOI] [PubMed] [Google Scholar]
- 54.Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008. October;21(4):583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008. June;16(6):1073–80. [DOI] [PubMed] [Google Scholar]
- 56.Hillestad ML, Guenzel AJ, Nath KA, Barry MA. A vector-host system to fingerprint virus tropism. Hum Gene Ther. 2012. October;23(10):1116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lang JF, Toulmin SA, Brida KL, Eisenlohr LC, Davidson BL. Standard screening methods underreport AAV-mediated transduction and gene editing. Nat Commun. 2019. July 30;10(1):3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lipkowitz MS, Hanss B, Tulchin N, Wilson PD, Langer JC, Ross MD, et al. Transduction of renal cells in vitro and in vivo by adeno-associated virus gene therapy vectors. J Am Soc Nephrol. 1999. September;10(9):1908–15. [DOI] [PubMed] [Google Scholar]
- 59.Chen S, Agarwal A, Glushakova OY, Jorgensen MS, Salgar SK, Poirier A, et al. Gene delivery in renal tubular epithelial cells using recombinant adeno-associated viral vectors. J Am Soc Nephrol. 2003. April;14(4):947–58. [DOI] [PubMed] [Google Scholar]
- 60.Takeda S, Takahashi M, Mizukami H, Kobayashi E, Takeuchi K, Hakamata Y, et al. Successful gene transfer using adeno-associated virus vectors into the kidney: comparison among adeno-associated virus serotype 1–5 vectors in vitro and in vivo. Nephron Exp Nephrol. 2004;96(4):e119–26. [DOI] [PubMed] [Google Scholar]
- 61.Ito K, Chen J, Khodadadian JJ, Vaughan ED Jr., Lipkowitz M, Poppas DP, et al. Adeno-associated viral vector transduction of green fluorescent protein in kidney: effect of unilateral ureteric obstruction. BJU Int. 2008. February;101(3):376–81. [DOI] [PubMed] [Google Scholar]
- 62.Qi YF, Li QH, Shenoy V, Zingler M, Jun JY, Verma A, et al. Comparison of the transduction efficiency of tyrosine-mutant adeno-associated virus serotype vectors in kidney. Clin Exp Pharmacol Physiol. 2013. January;40(1):53–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung DC, Fogelgren B, Park KM, Heidenberg J, Zuo X, Huang L, et al. Adeno-Associated Virus-Mediated Gene Transfer to Renal Tubule Cells via a Retrograde Ureteral Approach. Nephron Extra. 2011. January;1(1):217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen X, Xu Y, Bai Z, Ma D, Niu Q, Meng J, et al. Transparenchymal Renal Pelvis Injection of Recombinant Adeno-Associated Virus Serotype 9 Vectors Is a Practical Approach for Gene Delivery in the Kidney. Hum Gene Ther Methods. 2018. December;29(6):251–8. [DOI] [PubMed] [Google Scholar]
- 65.Rocca CJ, Ur SN, Harrison F, Cherqui S. rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: conclusion of a comparative study. Gene Ther. 2014. June;21(6):618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corridon PR, Rhodes GJ, Leonard EC, Basile DP, Gattone VH 2nd, Bacallao RL, et al. A method to facilitate and monitor expression of exogenous genes in the rat kidney using plasmid and viral vectors. Am J Physiol Renal Physiol. 2013. May 1;304(9):F1217–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saito S, Ohno SI, Harada Y, Oikawa K, Fujita K, Mineo S, et al. rAAV6-mediated miR-29b delivery suppresses renal fibrosis. Clin Exp Nephrol. 2019. December;23(12):1345–56. [DOI] [PubMed] [Google Scholar]
- 68.Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015. February;125(2):870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nault JC, Datta S, Imbeaud S, Franconi A, Mallet M, Couchy G, et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. 2015. October;47(10):1187–93. [DOI] [PubMed] [Google Scholar]
- 70.Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther. 2018. March;29(3):285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perabo L, Endell J, King S, Lux K, Goldnau D, Hallek M, et al. Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J Gene Med. 2006. February;8(2):155–62. [DOI] [PubMed] [Google Scholar]
- 72.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and Selection of Shuffled AAV Genomes: A New Strategy for Producing Targeted Biological Nanoparticles. Mol Ther. 2008. July;16(7):1252–60. [DOI] [PubMed] [Google Scholar]
- 73.Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, Andres-Mateos E, et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015. August 11;12(6):1056–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ikeda Y, Sun Z, Ru X, Vandenberghe LH, Humphreys BD. Efficient Gene Transfer to Kidney Mesenchymal Cells Using a Synthetic Adeno-Associated Viral Vector. J Am Soc Nephrol. 2018. September;29(9):2287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buchholz CJ, Friedel T, Buning H. Surface-Engineered Viral Vectors for Selective and Cell Type-Specific Gene Delivery. Trends Biotechnol. 2015. December;33(12):777–90. [DOI] [PubMed] [Google Scholar]
- 76.Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007. July 27;317(5837):477. [DOI] [PubMed] [Google Scholar]
- 77.Russell DW, Grompe M. Adeno-associated virus finds its disease. Nat Genet. 2015. October;47(10):1104–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berns KI, Byrne BJ, Flotte TR, Gao G, Hauswirth WW, Herzog RW, et al. Adeno-Associated Virus Type 2 and Hepatocellular Carcinoma? Hum Gene Ther. 2015. December;26(12):779–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol Ther. 2018. March 7;26(3):664–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J. 2012. May 1;443(3):603–18. [DOI] [PubMed] [Google Scholar]
- 81.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013. April 30;110(18):7306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A. 1993. September 1;90(17):8033–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B, et al. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat Med. 2005. March;11(3):346–52. [DOI] [PubMed] [Google Scholar]
- 84.Gennari F, Lopes L, Verhoeyen E, Marasco W, Collins MK. Single-chain antibodies that target lentiviral vectors to MHC class II on antigen-presenting cells. Hum Gene Ther. 2009. June;20(6):554–62. [DOI] [PubMed] [Google Scholar]
- 85.Hashiguchi T, Maenaka K, Yanagi Y. Measles virus hemagglutinin: structural insights into cell entry and measles vaccine. Front Microbiol. 2011;2:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kneissl S, Abel T, Rasbach A, Brynza J, Schneider-Schaulies J, Buchholz CJ. Measles virus glycoprotein-based lentiviral targeting vectors that avoid neutralizing antibodies. PLoS One. 2012;7(10):e46667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Munch RC, Muhlebach MD, Schaser T, Kneissl S, Jost C, Pluckthun A, et al. DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther. 2011. April;19(4):686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou Q, Uhlig KM, Muth A, Kimpel J, Levy C, Munch RC, et al. Exclusive Transduction of Human CD4+ T Cells upon Systemic Delivery of CD4-Targeted Lentiviral Vectors. J Immunol. 2015. September 1;195(5):2493–501. [DOI] [PubMed] [Google Scholar]
- 89.Bosch RJ, Woolf AS, Fine LG. Gene transfer into the mammalian kidney: direct retrovirus-transduction of regenerating tubular epithelial cells. Exp Nephrol. 1993. Jan-Feb;1(1):49–54. [PubMed] [Google Scholar]
- 90.Gusella GL, Fedorova E, Hanss B, Marras D, Klotman ME, Klotman PE. Lentiviral gene transduction of kidney. Hum Gene Ther. 2002. February 10;13(3):407–14. [DOI] [PubMed] [Google Scholar]
- 91.Asico LD, Cuevas S, Ma X, Jose PA, Armando I, Konkalmatt PR. Nephron segment-specific gene expression using AAV vectors. Biochem Biophys Res Commun. 2018. February 26;497(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu X, Tan X, Tampe B, Wilhelmi T, Hulshoff MS, Saito S, et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat Commun. 2018. August 29;9(1):3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W, et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016;2:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pulecio J, Verma N, Mejia-Ramirez E, Huangfu D, Raya A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell. 2017. October 5;21(4):431–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lavender P, Kelly A, Hendy E, McErlean P. CRISPR-based reagents to study the influence of the epigenome on gene expression. Clin Exp Immunol. 2018. October;194(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim M, Chen SW, Park SW, Kim M, D’Agati VD, Yang J, et al. Kidney-specific reconstitution of the A1 adenosine receptor in A1 adenosine receptor knockout mice reduces renal ischemia-reperfusion injury. Kidney Int. 2009. April;75(8):809–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Selective intrarenal human A1 adenosine receptor overexpression reduces acute liver and kidney injury after hepatic ischemia reperfusion in mice. Lab Invest. 2010. March;90(3):476–95. [DOI] [PubMed] [Google Scholar]
- 98.Park SW, Chen SW, Kim M, Brown KM, D’Agati VD, Lee HT. Protection against acute kidney injury via A(1) adenosine receptor-mediated Akt activation reduces liver injury after liver ischemia and reperfusion in mice. J Pharmacol Exp Ther. 2010. June;333(3):736–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D’Agati VD, et al. Selective renal overexpression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol. 2010. August;299(2):F347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Espana-Agusti J, Tuveson DA, Adams DJ, Matakidou A. A minimally invasive, lentiviral based method for the rapid and sustained genetic manipulation of renal tubules. Sci Rep. 2015. June 5;5:11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yuzefovych Y, Valdivia E, Rong S, Hack F, Rother T, Schmitz J, et al. Genetic Engineering of the Kidney to Permanently Silence MHC Transcripts During ex vivo Organ Perfusion. Front Immunol. 2020;11:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharma A, Li X, Bangari DS, Mittal SK. Adenovirus receptors and their implications in gene delivery. Virus Res. 2009. August;143(2):184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Molinier-Frenkel V, Gahery-Segard H, Mehtali M, Le Boulaire C, Ribault S, Boulanger P, et al. Immune response to recombinant adenovirus in humans: capsid components from viral input are targets for vector-specific cytotoxic T lymphocytes. J Virol. 2000. August;74(16):7678–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suzuki M, Cela R, Bertin TK, Sule G, Cerullo V, Rodgers JR, et al. NOD2 signaling contributes to the innate immune response against helper-dependent adenovirus vectors independently of MyD88 in vivo. Hum Gene Ther. 2011. September;22(9):1071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006. March;17(3):253–63. [DOI] [PubMed] [Google Scholar]
- 106.Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004. December;15(12):1157–66. [DOI] [PubMed] [Google Scholar]
- 107.Ertl HCJ, High KA. Impact of AAV Capsid-Specific T-Cell Responses on Design and Outcome of Clinical Gene Transfer Trials with Recombinant Adeno-Associated Viral Vectors: An Evolving Controversy. Hum Gene Ther. 2017. April;28(4):328–37. [DOI] [PubMed] [Google Scholar]
- 108.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997. March;71(3):1842–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999. July;6(7):1258–66. [DOI] [PubMed] [Google Scholar]
- 110.Liu F, Huang L. Development of non-viral vectors for systemic gene delivery. J Control Release. 2002. January 17;78(1–3):259–66. [DOI] [PubMed] [Google Scholar]
- 111.Zhdanov RI, Podobed OV, Vlassov VV. Cationic lipid-DNA complexes-lipoplexes-for gene transfer and therapy. Bioelectrochemistry. 2002. November;58(1):53–64. [DOI] [PubMed] [Google Scholar]
- 112.Zhang S, Xu Y, Wang B, Qiao W, Liu D, Li Z. Cationic compounds used in lipoplexes and polyplexes for gene delivery. J Control Release. 2004. November 24;100(2):165–80. [DOI] [PubMed] [Google Scholar]
- 113.Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006. November 28;116(2):255–64. [DOI] [PubMed] [Google Scholar]
- 114.Tros de Ilarduya C, Sun Y, Duzgunes N. Gene delivery by lipoplexes and polyplexes. Eur J Pharm Sci. 2010. June 14;40(3):159–70. [DOI] [PubMed] [Google Scholar]
- 115.Zhang XX, McIntosh TJ, Grinstaff MW. Functional lipids and lipoplexes for improved gene delivery. Biochimie. 2012. January;94(1):42–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scholz C, Wagner E. Therapeutic plasmid DNA versus siRNA delivery: common and different tasks for synthetic carriers. J Control Release. 2012. July 20;161(2):554–65. [DOI] [PubMed] [Google Scholar]
- 117.Tomita N, Higaki J, Morishita R, Kato K, Mikami H, Kaneda Y, et al. Direct in vivo gene introduction into rat kidney. Biochem Biophys Res Commun. 1992. July 15;186(1):129–34. [DOI] [PubMed] [Google Scholar]
- 118.Imai E, Isaka Y, Akagi Y, Kaneda Y. Gene transfer into the glomerulus by the hemagglutinating virus of Japan-liposome method. Exp Nephrol. 1997. Mar-Apr;5(2):112–7. [PubMed] [Google Scholar]
- 119.Isaka Y, Fujiwara Y, Ueda N, Kaneda Y, Kamada T, Imai E. Glomerulosclerosis induced by in vivo transfection of transforming growth factor-beta or platelet-derived growth factor gene into the rat kidney. J Clin Invest. 1993. December;92(6):2597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Isaka Y, Akagi Y, Ando Y, Tsujie M, Sudo T, Ohno N, et al. Gene therapy by transforming growth factor-beta receptor-IgG Fc chimera suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1999. February;55(2):465–75. [DOI] [PubMed] [Google Scholar]
- 121.Koike H, Tomita N, Azuma H, Taniyama Y, Yamasaki K, Kunugiza Y, et al. An efficient gene transfer method mediated by ultrasound and microbubbles into the kidney. J Gene Med. 2005. January;7(1):108–16. [DOI] [PubMed] [Google Scholar]
- 122.Kim HJ, Greenleaf JF, Kinnick RR, Bronk JT, Bolander ME. Ultrasound-mediated transfection of mammalian cells. Hum Gene Ther. 1996. July 10;7(11):1339–46. [DOI] [PubMed] [Google Scholar]
- 123.Bao S, Thrall BD, Miller DL. Transfection of a reporter plasmid into cultured cells by sonoporation in vitro. Ultrasound Med Biol. 1997;23(6):953–9. [DOI] [PubMed] [Google Scholar]
- 124.Lauer U, Burgelt E, Squire Z, Messmer K, Hofschneider PH, Gregor M, et al. Shock wave permeabilization as a new gene transfer method. Gene Ther. 1997. July;4(7):710–5. [DOI] [PubMed] [Google Scholar]
- 125.Wyber JA, Andrews J, D’Emanuele A. The use of sonication for the efficient delivery of plasmid DNA into cells. Pharm Res. 1997. June;14(6):750–6. [DOI] [PubMed] [Google Scholar]
- 126.Tachibana K, Uchida T, Ogawa K, Yamashita N, Tamura K. Induction of cell-membrane porosity by ultrasound. Lancet. 1999. April 24;353(9162):1409. [DOI] [PubMed] [Google Scholar]
- 127.Sorace AG, Warram JM, Mahoney M, Zinn KR, Hoyt K. Enhancement of adenovirus delivery after ultrasound-stimulated therapy in a cancer model. Ultrasound Med Biol. 2013. December;39(12):2374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xing Y, Pua EC, Lu X, Zhong P. Low-amplitude ultrasound enhances hydrodynamic-based gene delivery to rat kidney. Biochem Biophys Res Commun. 2009. August 14;386(1):217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kurosaki T, Kawakami S, Higuchi Y, Suzuki R, Maruyama K, Sasaki H, et al. Kidney-selective gene transfection using anionic bubble lipopolyplexes with renal ultrasound irradiation in mice. Nanomedicine. 2014. November;10(8):1829–38. [DOI] [PubMed] [Google Scholar]
- 130.Tsujie M, Isaka Y, Nakamura H, Imai E, Hori M. Electroporation-mediated gene transfer that targets glomeruli. J Am Soc Nephrol. 2001. May;12(5):949–54. [DOI] [PubMed] [Google Scholar]
- 131.Lai LW, Chan DM, Erickson RP, Hsu SJ, Lien YH. Correction of renal tubular acidosis in carbonic anhydrase II-deficient mice with gene therapy. J Clin Invest. 1998. April 1;101(7):1320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Maruyama H, Higuchi N, Nishikawa Y, Hirahara H, Iino N, Kameda S, et al. Kidney-targeted naked DNA transfer by retrograde renal vein injection in rats. Hum Gene Ther. 2002. February 10;13(3):455–68. [DOI] [PubMed] [Google Scholar]
- 133.Maruyama H, Higuchi N, Kameda S, Nakamura G, Iguchi S, Miyazaki J, et al. Rat kidney-targeted naked plasmid DNA transfer by retrograde injection into the renal vein. Mol Biotechnol. 2004. May;27(1):23–31. [DOI] [PubMed] [Google Scholar]
- 134.Kameda S, Maruyama H, Higuchi N, Iino N, Nakamura G, Miyazaki J, et al. Kidney-targeted naked DNA transfer by retrograde injection into the renal vein in mice. Biochem Biophys Res Commun. 2004. February 6;314(2):390–5. [DOI] [PubMed] [Google Scholar]
- 135.Woodard LE, Cheng J, Welch RC, Williams FM, Luo W, Gewin LS, et al. Kidney-specific transposon-mediated gene transfer in vivo. Sci Rep. 2017. March 20;7:44904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tomita N, Morishita R, Yamamoto K, Higaki J, Dzau VJ, Ogihara T, et al. Targeted gene therapy for rat glomerulonephritis using HVJ-immunoliposomes. J Gene Med. 2002. Sep-Oct;4(5):527–35. [DOI] [PubMed] [Google Scholar]
- 137.Uchida M, Maier B, Waghwani HK, Selivanovitch E, Pay SL, Avera J, et al. The archaeal Dps nanocage targets kidney proximal tubules via glomerular filtration. J Clin Invest. 2019. September 3;129(9):3941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kulkarni JA, Cullis PR, van der Meel R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018. June;28(3):146–57. [DOI] [PubMed] [Google Scholar]
- 139.Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018. July 5;379(1):11–21. [DOI] [PubMed] [Google Scholar]
- 140.Suga H, Nagasaki H, Kondo TA, Okajima Y, Suzuki C, Ozaki N, et al. Novel treatment for lithium-induced nephrogenic diabetes insipidus rat model using the Sendai-virus vector carrying aquaporin 2 gene. Endocrinology. 2008. November;149(11):5803–10. [DOI] [PubMed] [Google Scholar]
- 141.Bajimaya S, Hayashi T, Takimoto T. Rescue of Sendai Virus from Cloned cDNA. Methods Mol Biol. 2017;1602:103–10. [DOI] [PubMed] [Google Scholar]
- 142.Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J Pathol. 2012. January;226(2):365–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Agrawal S, Temsamani J, Tang JY. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc Natl Acad Sci U S A. 1991. September 1;88(17):7595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Goodarzi G, Watabe M, Watabe K. Organ distribution and stability of phosphorothioated oligodeoxyribonucleotides in mice. Biopharm Drug Dispos. 1992. April;13(3):221–7. [DOI] [PubMed] [Google Scholar]
- 145.Cossum PA, Truong L, Owens SR, Markham PM, Shea JP, Crooke ST. Pharmacokinetics of a 14C-labeled phosphorothioate oligonucleotide, ISIS 2105, after intradermal administration to rats. J Pharmacol Exp Ther. 1994. April;269(1):89–94. [PubMed] [Google Scholar]
- 146.Oberbauer R, Schreiner GF, Meyer TW. Renal uptake of an 18-mer phosphorothioate oligonucleotide. Kidney Int. 1995. October;48(4):1226–32. [DOI] [PubMed] [Google Scholar]
- 147.Rappaport J, Hanss B, Kopp JB, Copeland TD, Bruggeman LA, Coffman TM, et al. Transport of phosphorothioate oligonucleotides in kidney: implications for molecular therapy. Kidney Int. 1995. May;47(5):1462–9. [DOI] [PubMed] [Google Scholar]
- 148.Noiri E, Peresleni T, Miller F, Goligorsky MS. In vivo targeting of inducible NO synthase with oligodeoxynucleotides protects rat kidney against ischemia. J Clin Invest. 1996. May 15;97(10):2377–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Akagi Y, Isaka Y, Arai M, Kaneko T, Takenaka M, Moriyama T, et al. Inhibition of TGF-beta 1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1996. July;50(1):148–55. [DOI] [PubMed] [Google Scholar]
- 150.Daniel C, Takabatake Y, Mizui M, Isaka Y, Kawashi H, Rupprecht H, et al. Antisense oligonucleotides against thrombospondin-1 inhibit activation of tgf-beta in fibrotic renal disease in the rat in vivo. Am J Pathol. 2003. September;163(3):1185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tomita N, Kashihara N, Morishita R. Transcription factor decoy oligonucleotide-based therapeutic strategy for renal disease. Clin Exp Nephrol. 2007. March;11(1):7–17. [DOI] [PubMed] [Google Scholar]
- 152.Molitoris BA, Dagher PC, Sandoval RM, Campos SB, Ashush H, Fridman E, et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol. 2009. August;20(8):1754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Morishita Y, Yoshizawa H, Watanabe M, Ishibashi K, Muto S, Kusano E, et al. siRNAs targeted to Smad4 prevent renal fibrosis in vivo. Sci Rep. 2014. September 19;4:6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alidori S, Akhavein N, Thorek DL, Behling K, Romin Y, Queen D, et al. Targeted fibrillar nanocarbon RNAi treatment of acute kidney injury. Sci Transl Med. 2016. March 23;8(331):331ra39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schievenbusch S, Strack I, Scheffler M, Nischt R, Coutelle O, Hosel M, et al. Combined paracrine and endocrine AAV9 mediated expression of hepatocyte growth factor for the treatment of renal fibrosis. Mol Ther. 2010. July;18(7):1302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Holditch SJ, Schreiber CA, Harris PC, LaRusso NF, Ramirez-Alvarado M, Cataliotti A, et al. B-type natriuretic peptide overexpression ameliorates hepatorenal fibrocystic disease in a rat model of polycystic kidney disease. Kidney Int. 2017. September;92(3):657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Holditch SJ, Brown CN, Atwood DJ, Pokhrel D, Brown SE, Lombardi AM, et al. The consequences of increased 4E-BP1 in polycystic kidney disease. Hum Mol Genet. 2019. December 15;28(24):4132–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang B, Wang J, He W, Zhao Y, Zhang A, Liu Y, et al. Exogenous miR-29a Attenuates Muscle Atrophy and Kidney Fibrosis in Unilateral Ureteral Obstruction Mice. Hum Gene Ther. 2020. February 21. [DOI] [PMC free article] [PubMed]
- 159.Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. 2018. October;53:164–81. [DOI] [PubMed] [Google Scholar]
- 160.Kitamura M, Taylor S, Unwin R, Burton S, Shimizu F, Fine LG. Gene transfer into the rat renal glomerulus via a mesangial cell vector: site-specific delivery, in situ amplification, and sustained expression of an exogenous gene in vivo. J Clin Invest. 1994. August;94(2):497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kitamura M, Burton S, Yokoo T, Fine LG. Gene delivery into the renal glomerulus by transfer of genetically engineered, autologous mesangial cells. Exp Nephrol. 1996. Jan-Feb;4(1):56–9. [PubMed] [Google Scholar]
- 162.Naito T, Yokoyama H, Moore KJ, Dranoff G, Mulligan RC, Kelley VR. Macrophage growth factors introduced into the kidney initiate renal injury. Mol Med. 1996. May;2(3):297–312. [PMC free article] [PubMed] [Google Scholar]
- 163.Kitamura M Creation of a reversible on/off system for site-specific in vivo control of exogenous gene activity in the renal glomerulus. Proc Natl Acad Sci U S A. 1996. July 9;93(14):7387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kitamura M, Kawachi H. Creation of an In vivo cytosensor using engineered mesangial cells. Automatic sensing of glomerular inflammation controls transgene activity. J Clin Invest. 1997. September 15;100(6):1394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Syres K, Harrison F, Tadlock M, Jester JV, Simpson J, Roy S, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009. September 17;114(12):2542–52. [DOI] [PubMed] [Google Scholar]
- 166.Harrison F, Yeagy BA, Rocca CJ, Kohn DB, Salomon DR, Cherqui S. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol Ther. 2013. February;21(2):433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Naphade S, Sharma J, Gaide Chevronnay HP, Shook MA, Yeagy BA, Rocca CJ, et al. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells. 2015. January;33(1):301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018. December 6;4(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997. October;17(2):171–8. [DOI] [PubMed] [Google Scholar]
- 170.Konrad M, Vollmer M, Lemmink HH, van den Heuvel LP, Jeck N, Vargas-Poussou R, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000. August;11(8):1449–59. [DOI] [PubMed] [Google Scholar]
- 171.Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001. November;29(3):310–4. [DOI] [PubMed] [Google Scholar]
- 172.Nozu K, Fu XJ, Kaito H, Kanda K, Yokoyama N, Przybyslaw Krol R, et al. A novel mutation in KCNJ1 in a Bartter syndrome case diagnosed as pseudohypoaldosteronism. Pediatr Nephrol. 2007. August;22(8):1219–23. [DOI] [PubMed] [Google Scholar]
- 173.Sun M, Ning J, Xu W, Zhang H, Zhao K, Li W, et al. Genetic heterogeneity in patients with Bartter syndrome type 1. Mol Med Rep. 2017. February;15(2):581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Seys E, Andrini O, Keck M, Mansour-Hendili L, Courand PY, Simian C, et al. Clinical and Genetic Spectrum of Bartter Syndrome Type 3. J Am Soc Nephrol. 2017. August;28(8):2540–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Findeis-Hosey JJ, McMahon KQ, Findeis SK. Von Hippel-Lindau Disease. J Pediatr Genet. 2016. June;5(2):116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Enriquez R, Adam V, Sirvent AE, Garcia-Garcia AB, Millan I, Amoros F. Gitelman syndrome due to p.A204T mutation in CLCNKB gene. Int Urol Nephrol. 2010. December;42(4):1099–102. [DOI] [PubMed] [Google Scholar]
- 177.Lee JW, Lee J, Heo NJ, Cheong HI, Han JS. Mutations in SLC12A3 and CLCNKB and Their Correlation with Clinical Phenotype in Patients with Gitelman and Gitelman-like Syndrome. J Korean Med Sci. 2016. January;31(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Blanchard A, Bockenhauer D, Bolignano D, Calo LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017. January;91(1):24–33. [DOI] [PubMed] [Google Scholar]
- 179.Kong Y, Xu K, Yuan K, Zhu J, Gu W, Liang L, et al. Digenetic inheritance of SLC12A3 and CLCNKB genes in a Chinese girl with Gitelman syndrome. BMC Pediatr. 2019. April 18;19(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Patrakka J, Kestila M, Wartiovaara J, Ruotsalainen V, Tissari P, Lenkkeri U, et al. Congenital nephrotic syndrome (NPHS1): features resulting from different mutations in Finnish patients. Kidney Int. 2000. September;58(3):972–80. [DOI] [PubMed] [Google Scholar]
- 181.Cox JP, Yamamoto K, Christie PT, Wooding C, Feest T, Flinter FA, et al. Renal chloride channel, CLCN5, mutations in Dent’s disease. J Bone Miner Res. 1999. September;14(9):1536–42. [DOI] [PubMed] [Google Scholar]
- 182.Alur RP, Vijayasarathy C, Brown JD, Mehtani M, Onojafe IF, Sergeev YV, et al. Papillorenal syndrome-causing missense mutations in PAX2/Pax2 result in hypomorphic alleles in mouse and human. PLoS Genet. 2010. March 5;6(3):e1000870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kruegel J, Rubel D, Gross O. Alport syndrome--insights from basic and clinical research. Nat Rev Nephrol. 2013. March;9(3):170–8. [DOI] [PubMed] [Google Scholar]
- 184.Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Cystinosis: a review. Orphanet J Rare Dis. 2016. April 22;11:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tetti M, Monticone S, Burrello J, Matarazzo P, Veglio F, Pasini B, et al. Liddle Syndrome: Review of the Literature and Description of a New Case. Int J Mol Sci. 2018. March 11;19(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yu SM, Bleyer AJ, Anis K, Herlitz L, Zivna M, Hulkova H, et al. Autosomal Dominant Tubulointerstitial Kidney Disease Due to MUC1 Mutation. Am J Kidney Dis. 2018. April;71(4):495–500. [DOI] [PubMed] [Google Scholar]
- 187.Gast C, Marinaki A, Arenas-Hernandez M, Campbell S, Seaby EG, Pengelly RJ, et al. Autosomal dominant tubulointerstitial kidney disease-UMOD is the most frequent non polycystic genetic kidney disease. BMC Nephrol. 2018. October 30;19(1):301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Clissold RL, Clarke HC, Spasic-Boskovic O, Brugger K, Abbs S, Bingham C, et al. Discovery of a novel dominant mutation in the REN gene after forty years of renal disease: a case report. BMC Nephrol. 2017. July 12;18(1):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, et al. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019. September 5;5(1):60. [DOI] [PubMed] [Google Scholar]
- 190.Lee EC, Valencia T, Allerson C, Schairer A, Flaten A, Yheskel M, et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat Commun. 2019. September 12;10(1):4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rocca CJ, Cherqui S. Gene Transfer to Mouse Kidney In Vivo. Methods Mol Biol. 2019;1937:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jerebtsova M, Liu XH, Ye X, Ray PE. Adenovirus-mediated gene transfer to glomerular cells in newborn mice. Pediatr Nephrol. 2005. October;20(10):1395–400. [DOI] [PubMed] [Google Scholar]