

Abstract
Visualizing and manipulating behavior of proteins is crucial to understanding the physiology of the cell. Methods of biorthogonal protein labelling are important tools to attain this goal. In this review, we discuss advances in probe technology specific for self-labelling protein tags, focusing mainly on the application of HaloTag and SNAP-tag systems. We describe the latest developments in small molecule probes that enable fluorogenic (no wash) imaging and super-resolution fluorescence microscopy. In addition, we cover several methodologies that enable the perturbation or manipulation of protein behavior and function towards the control of biological pathways. Thus, the current technical advancement on the HaloTag and SNAP-tag systems are becoming powerful tools to enable the visualization and manipulating of biological processes, providing invaluable scientific insights that are otherwise difficult to obtain using traditional methodologies. As the multiplex self-labelling protein tag systems continue to be developed and expanded, the utility of these protein tags will allow researchers to address previously inaccessible questions at the forefront of biology.
Keywords: HaloTag, SNAP-tag, bio-orthogonal chemistry, molecular probes, chemical biology
Graphical Abstract

We review recent chemical probe development for self-labelling protein tags, focusing on the HaloTag and SNAP-tag systems. Content includes the recently developed small molecule probes for fluorescence microscopy and several methodologies that perturb or manipulate biological processes in live cells. Future development on self-labelling protein tags will allow for enabling methods to address previously inaccessible biological questions.
1. Introduction
Methodologies to probe the behavior and function of biological macromolecules in vivo mandate a certain standard of selectivity to ensure the fidelity of results and minimize off-target background noise. The challenge of rapid, selective, and covalent protein labelling in vivo is exacerbated by the complexity and reactivity of the cellular environment. As such, bioorthogonality has become key to assay design and method development in chemical biology laboratories that aim to visualize and perturb protein-based life processes. While methods in molecular recognition through high affinity binding with antibodies allow for selective enrichment and fluorescence imaging, they fall short in the observation of dynamic systems as these techniques require cell fixation. However, real-time surveillance of protein behavior can be accomplished with the power of genetic fusion. Most notably and wide spread is the utility of fluorescent protein tags, which upon fusion to a protein of interest (POI) can be used to directly report on the expression and localization of a specific protein in vivo. In addition to fluorescent proteins, self-labeling protein tags have become another powerful set of chemical tools that offer flexibility of imaging assays and feasibility of allowing chemical manipulation of POIs.
In this minireview, we focus on the recent developments in methods of visualizing and manipulating biological systems using small molecule probes for self-labelling protein tags. With a focus on HaloTag and SNAP-tag, we describe the fluorescence imaging methods to track protein localization and stability as well as molecular tools and strategies for the manipulation of protein systems. Further we describe how in tandem with these described ligands, HaloTag and SNAP-tag based systems can be used as probes themselves to report on the health of the cellular proteome. Our aim is to provide a comprehensive outlook on the current state of the self-labelling protein-tag toolbox in order to provide a perspective on the future of the field.
2. Self-labelling protein tags
Self-labelling protein tags are small (<40 kDa) suicide proteins designed for covalent conjugation to a small molecule probe functionalized with a biorthogonal linker. In this manner nearly any probe that can be functionalized with appropriate linker can be conjugated to its respective tag and be used to visualize or manipulate the function and dynamics of a fused POI. An ideal self-labelling tag would possess several characteristics; small size to minimize perturbance of the POI, rapid labelling kinetics, low substrate promiscuity, and sufficient thermodynamic stability. Many of such tags have been developed, including HaloTag derived from the haloalkane dehalogenase,[1] SNAP-tag derived from human O6-alkylguanine DNA alkyltransferase,[2] CLIP-tag derived from SNAP-tag,[3] PYP-tag derived from the photoactive yellow protein in purple bacteria,[4] DHFR-tag derived from dihydrofolate reductase,[5] BL-tag derived from β-lactamase,[6] and RA-tag derived from de novo designed retroaldolase.[7] These tags have been introduced and summarized extensively in previous review articles.[8] The scope of this review, however, will focus specifically on the application HaloTag and SNAP-tag provided the versatility and broad commercial availability of probes for these tags.
2.1. HaloTag
Developed by Promega in 2008, HaloTag is a protein tag engineered from a bacterial haloalkane dehalogenase (DhaA, Rhodococcus).[1] The wild-type protein catalyzes the hydrolysis of a chloroalkane substrate via a Glu-His-Asp catalytic triad beginning with SN2 displacement of the substrate halogen by the active-site aspartate residue.[9] The resulting ester intermediate is then hydrolyzed via nucleophilic acyl substitution by a water molecule activated through general-base catalysis via the active-site histidine residue.[9c] Wood and co-workers engineered DhaA to irreversibly capture the covalent ester intermediate by substituting this histidine for a catalytically inactive phenylalanine residue through mutagenesis, yielding HaloTag in the form of the DhaA(H272F) mutant.
With rapid labelling kinetics (2.7×106 M−1s−1) and high stability (ΔGfolding = −5.6 kcal/mol), HaloTag rapidly and reliably conjugates to probes functionalized with a chloroalkane linker (Table 1, Figure 1A).[1,10] Comparatively to other self-labelling protein tags, HaloTagis rather large in size (33 kDa). Fusion of such a large domain may perturb the native function of the protein of interest, but HaloTag’s size and net negative charge at physiological pH (pI ~5.0) allows it to act as a solubility tag in protein purification methods with reduced propensity for aggregation.[11]
Table 1.
Parameters of HaloTag and SNAP-tag.
Figure 1.

Chemical mechanisms of chemical labelling for A) HaloTag and B) SNAP-tag.
2.2. SNAP-tag
Developed by Kai Johnsson’s lab, SNAP-tag is derived from human O6-alkylguanine-DNA alkyltransferase (hAGT).[2,12–13] hAGT is a native DNA repair protein that irreversibly transfers an alkyl group from O6 of an alkylguanine substrate to an active site cysteine residue via an SN2 mechanism (Figure 1B).[14] Johnsson’s efforts utilized directed evolution and random mutagenesis of hAGT to develop SNAP-tag to reduce protein-DNA interactions, improve labelling kinetics and specificity for an O6-benzylguanine substrate, garner resistance to wild-type hAGT inhibitors, and decrease size and remove excess cysteines.[2,12–13] While slower than HaloTag, SNAP-tag rapidly binds to an O6-benzylguanine equipped ligand (k = 2.8×104M−1 s−1) (Table 1).[3] The smaller size of SNAP-tag offers less overall perturbation of the dynamics and function of the POI. Notably, the engineered SNAP-tag is resistant to trypsin digestion upon alkylation and demonstrates superior stability (TM = 67°C) relative to wild-type hAGT (TM = 50°C) (Table 1).
3. Fluorescent probes for cellular imaging
In vivo visualization of protein dynamics is key to discovering the role of proteins in cellular processes. Compared to fixed cell methods of microscopy such as immunofluorescence or standard fluorescent protein tags, self-labelling protein tags offer a means of orthogonal protein labelling in live cells. This probing strategy confers the advantages of small molecule organic fluorophores engineered with favorable photophysical properties for both standard and advanced, super-resolution microscopy[15] while simultaneously expanding experimental methodologies to allow for pulse-chase based assays to observe the fate of a POI following perturbation.[3,16] As such, the development of probes for self-labelling protein tags functionalized for fluorescence microscopy remains an advancing field of research. Provided the extensive progress in fluorescence microscopy over the latest decades, these techniques have been extensively reviewed elsewhere.[17] Instead, here we focus on recent developments in the tools employed within those techniques such as (1) “always on” fluorescent labelling probes, (2) fluorogenic probes for no-wash, live cell imaging, and (3) fluorogenic technologies for the visualization of protein dynamics.
3.1. “Always-on” labelling probes for HaloTag and SNAP-tag
In order to label and track the expression or localization of POI by fluorescence microscopy, an employed label must be constitutively fluorescent, or “always-on”. Fluorescent protein tags, such as GFP, are commonly used for these purposes, but self-labelling tags can accomplish the same goal when combined with an effectively bright fluorescent probe. Furthermore, many of these general labelling probes possess the inherent properties (stochastic blinking or photoswitchability) necessary for the execution of nanometer-resolution microscopic techniques. An array of these types of probes are available commercially for HaloTag(Promega) and SNAP-tag (New England BioLabs), designed for spectral coverage (from ultra-violet to near-infrared excitations), intra- or extracellular labelling, and photophysical properties.
Coumarin based general labelling probes.
Coumarin offers an effective small molecule donor-acceptor type chromophore derived from the naturally occurring umbelliferone. In order to improve stability, the umbelliferone scaffold is methylated at the 4-position. Further, pH sensitivity can be removed by substituting the 7-position hydroxyl for an amino donating group (Figure 2). The resulting, commercially available (Promega) 7-amino-4-methylcoumarin 1 yields a bright constitutively fluorescent dye (quantum yield Φf = 0.75) equipped with a HaloTagligand at the 3-position for conjugation to a fused POI. Coumarin 2 is excited in the UV region (Ex/Em: 362 nm / 450 nm) at a high energy wavelength that yields high background noise due to auto-fluorescence of the sample as well as cellular damage in live-cell imaging experiments.[18] Several strategies to tune the excitation and emission properties of the coumarin scaffold are established, including N-alkylation, introduction of electron withdrawing groups, and extension of π-conjugation, but these strategies often confer a solvatochromic nature to the dye.[19] Likewise, the commercially available coumarin for SNAP-tag, SNAP-Cell 430 (New England BioLabs) 2, utilizes an alkylated 7-amino donor with a SNAP-Tag ligand connected via an electron withdrawing 3-position amide. The resulting bathochromic shift allows for overlap with the common 405 nm laser line for cell imaging (Ex/Em: 430 nm / 490 nm).
Figure 2.

Structures of commonly employed A) coumarin, B) triarylmethane, and C) cyanine based fluorophores for general labelling of HaloTag and SNAP-tag fused proteins in fluorescence-based microscopy. R1 indicates common positions for coupling chloroalkane and O6-benzyl guanine ligands for HaloTag and SNAP-tag ligands. D) Spectral coverage of commonly employed fluorescent probes. Excitation (top value) and emission (bottom value) maxima for the included fluorophores.
Fluorescein based general labelling probes
Emitting in the green range of the visible spectrum is the commonly used fluorophore, fluorescein 3 (Ex/Em: 492nm / 521nm, Φf 0.95) (Figure 2). Fluorescein is based on an oxidized triarylmethane scaffold, with oxygen donors located symmetrically at the 3’ and 6’-positions. With a pKa of 6.4, fluorescein remains deprotonated at physiological pH, with equilibrium favouring the open, fluorescent quinone form (Scheme 1). However, this charged conformation is impermeant to the cell membrane. Permeability can be recovered through acylation of the hydroxyl substituents (compound 4), which shifts equilibrium toward favoring the closed, non-fluorescent lactone form due to poor electron donation into the xanthene core. The acyl groups are hydrolyzed in the cellular environment by endogenous esterases, restoring fluorescence. Alternatively, to improve permeability Nagano and co-workers removed the carboxylate moiety of fluorescein, resulting in the more hydrophobic derivative, Tokyo Green 6.[20] Fluorination of fluorescein at the 2’ and 7’ (compound 5, Oregon Green) positions reduces the pKa to 4.7, ensuring near quantitative deprotonation under physiological conditions and removing pH sensitivity of the fluorophore.[21] Combining the two strategies, Peterson and co-workers developed a more cell permeable, pH insensitive fluorescein derivative known as Pennsylvania Green 7.[22]
Scheme 1.

Equilibrium of 2-carboxyphenyl xanthene-based chromophores between open quinoid and closed lactone forms.
Rhodamine based general labelling probes
Substitution of the fluorescein 3’ and 6’ oxygens for amine based donating groups yields another class of fluorophore, rhodamine. Similarly, to fluorescein, rhodamine exists in equilibrium between a fluorescent, zwitterionic quinoid form and a dark, neutral lactone form (Scheme 1). This equilibrium is polarity sensitive, with the closed, lactone form favored in hydrophobic environments and the open, zwitterionic form in hydrophilic ones. Notably, high intensity excitation of rhodamine derivatives allows for intersystem crossing to achieve the triplet state that is susceptible to reduction by thiols and thiolates in solution (Scheme 2).[23] The resulting radical anion species is non-fluorescent with absorption in the UV–blue range. In the absence of oxygen, the rhodamine radical anion is long lived (lifetime ~7 hours). This reduced rhodamine derivative can be converted back to the fluorescent state upon irradiation near its absorbance maxima with minimal photobleaching over several cycles. The photoswitchable properties of rhodamine fluorophores enable their application in single molecule localization based imaging techniques.[24] Given this application of rhodamine for tracking the localization and expression of proteins in super-resolution microscopy, much effort has been made to control the photophysical properties of rhodamine based fluorophores.
Scheme 2.

Photoredox mechanism of rhodamine photoswitching. (T1 = triplet state, β-ME = β-mercaptoethanol)
The most basic rhodamine, Rhodamine 110 8, exhibits high fluorescence quantum yield, but emits a green fluorescence (Ex/Em: 498nm / 528nm, Φf: 0.88). Alkylation of the amino donating groups, such as that of tetramethylrhodamine (TMR) 9 results in a significant red shift of the fluorophore but reduces the overall brightness of the dye (Ex/Em: 552nm / 578nm, Φf: 0.41). Alkylation creates allylic strain, forcing the donor to adopt a more twisted conformation in the ground state. In the excited state, this steric clash exacerbates a mechanism of internal energy conversion in which the donating group rotates 90° out of the plane of xanthene core π system. This process, known as twisted intramolecular charge transfer (TICT), leads to rapid non-radiative decay and photooxidation of the fluorophore.[25] Lavis and co-workers developed a strategy to overcome this steric interaction and alleviate allylic strain of alkylated donors by increasing the C–N–C bond angle of the donor in the form of azetidine.[26] In doing so, the donor adopts a more co-planar orientation with respect to the xanthene core. Named Janelia Fluor™ 549 10, the azetidino functionalized rhodamine dye displays enhanced brightness and photostability (Ex/Em: 550nm / 570nm, Φf: 0.88).
Furthermore, the spectral properties of rhodamine dyes can be extended to the far red region by decreasing the HOMO-LUMO energy gap with installation of a silicon heteroatom to the xanthene core, as in that of Janelia Fluor™ 646 11.[26–27] Notably, Si-rhodamines favour the closed, lactone form in the ground state, conferring an inherent blinking property to the dye that allows for single molecule localization imaging without a need for reducing agent spiked buffers and anoxic conditions.[28] Further efforts showed that finetuning of these spectral properties across both oxygen and silicon based rhodamines can be achieved with addition of various electron withdrawing substituents on the azetidino donor.[29] In addition to color and brightness, other efforts to control the photophysical properties of rhodamines have aimed to improve photostability as Hell and co-workers have developed a novel method to reduce photobleaching with t-butyl amino donors.[30]
Cyanine based general labelling probes
Cyanine dyes are based on a cationic polymethine structure, wherein the conjugated π system allows for the absorption of visible light. The absorption and emission wavelengths can be facilely controlled through the length of the π system, as increasing π conjugation lowers the HOMO-LUMO energy gap. The shortest cyanine derivative, Cy3 12, is excited with green light (Ex/Em: 550nm / 570nm) and the extended derivatives, Cy5 13 and Cy7 14, absorb in the red and near-IR regions (Ex/Em: 646nm / 662nm and 750nm / 773nm, respectively).[31]
These photophysical properties of cyanine dyes can be tuned through chemical modification of the terminal heterocycles of the dye. Larger benzene fusions further reduce the excitation band gap, yielding red-shifted cyanines 15–17.[32] In order to prevent aggregation and improve aqueous solubility of cyanine dyes, a common strategy is to append solubilizing sulfonate moieties.[33] Furthermore, fluorination of the cyanine derivatives can be used to reduce reactivity with oxygen, improving photostability.[34]
Similar to rhodamines, cyanine dyes possess an inherent photoswitchability making the dye broadly employed in single molecule localization microscopy.[24,35] Excitation of Cy5 13 with red light in the presence of a thiolate nucleophile leads to attack at the γ-carbon of the polymethine conjugate.[35a] The resulting product of this excited state reaction is left in a non-fluorescent dark state, but excitation with high energy, UV light can recover the bright state of the dye. This photoswitching mechanism is applicable for extended cyanine derivates (13–14 and 16–17) but not for the shorter Cy3 12 and Cy3.5 15 dyes.
3.2. Fluorogenic, “no-wash” labelling probes for HaloTag and SNAP-tag
A major disadvantage of “always-on” probes is the need for removal of unbound fluorophores through washing. By adding several minutes to the imaging procedure, washing can impede time-dependent assays. Further, inefficient washing technique can result in high background fluorescence or damage to the sample prior to analysis. As such, no wash methods for cellular labelling are highly desired. The application of fluorogen activating proteins in which fluorescence is activated upon reversible, non-covalent binding to a small molecule fluorogen represents one such method. The utility of FAPs is well reviewed elsewhere.[36] Alternatively, several groups have devised unique strategies for labelling of HaloTagand SNAP-tag using fluorogenic probes to remove the requisite washing step. Typical fluorogenic probes are designed for fluorescent response to small molecule species through chemical reaction to activate fluorescence.[37] Similarly, several unique strategies have been employed to develop probes that activate fluorescence upon covalent conjugation to HaloTag and SNAP-tag specifically. In this section we review three of these fluorogenic strategies for “no-wash” labelling of HaloTag and SNAP-tag: 1) quencher cleavage and release 2) lactone ring opening of rhodamine-based fluorophores or 3) environmental sensitive fluorophores (Figure 3). The structures of many of these established fluorogenic probes for Halo- and SNAP-tag are presented in Figure 4.
Figure 3.
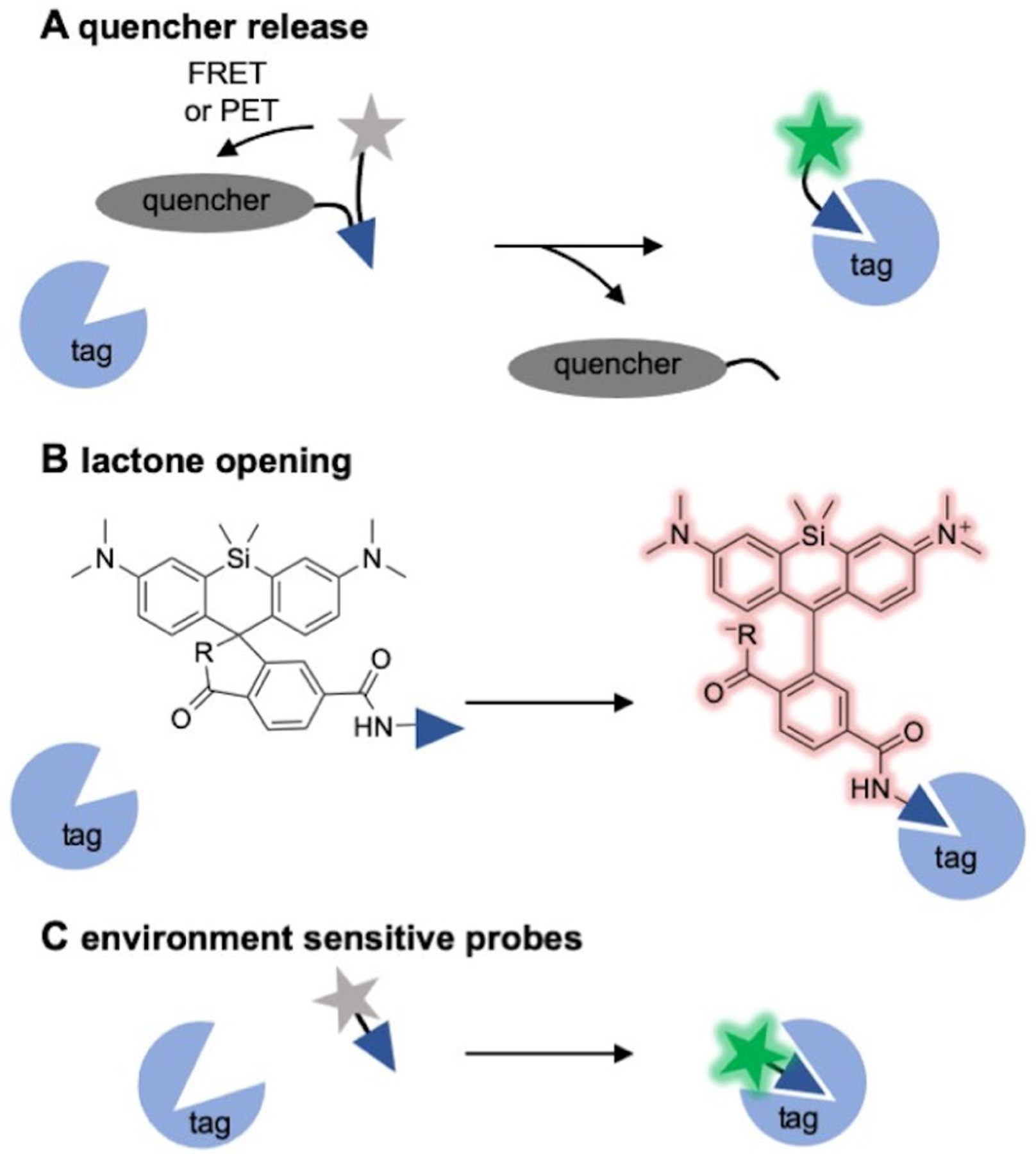
No-wash labelling strategies for HaloTag and SNAP-tag. A) Release of a PET or FRET based quencher upon conjugation. B) Fluorogenic rhodamines, lactone ring opening upon conjugation. C) Embedding of environmentally-sensitive fluorophores into the substrate binding tunnel.
Figure 4.
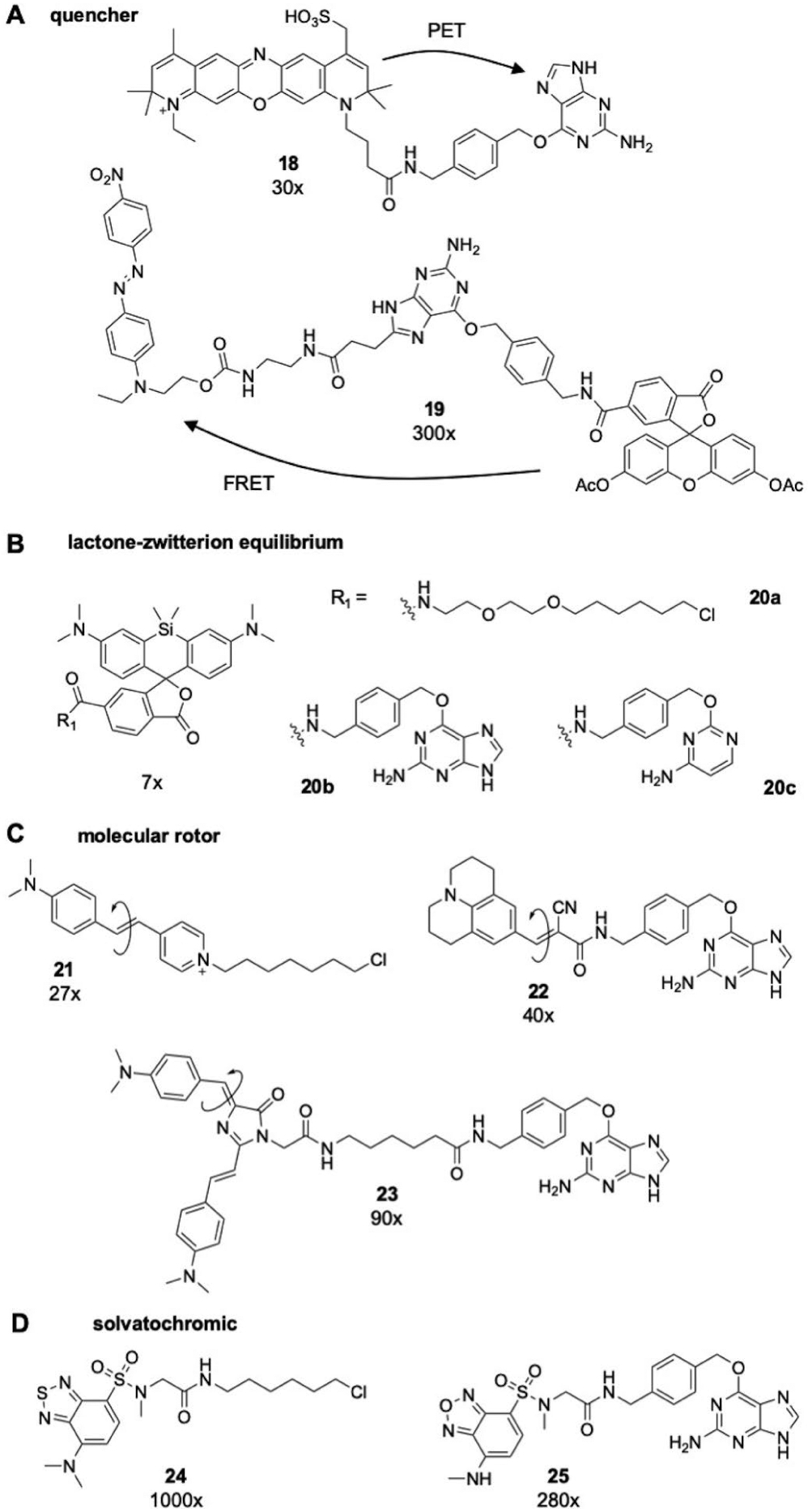
Structures of A) PET and FRET, B) Si-rhodamine, C) molecular rotor, or D) solvatochromic based fluorogenic, wash-free labelling probes for self-labelling protein tags and their corresponding fold-change upon binding to SNAP-tag or HaloTag in vitro.
Quencher release strategies for no-wash labelling
Fluorescence quenching of dyes can be accomplished through Förster resonance energy transfer (FRET) or photoinduced electron transfer (PET) to an adjacent acceptor or quencher.[38] This principle enables one strategy for fluorogenicity with self-labelling protein tags through cleavage and release of the quencher upon conjugation (Figure 3A). For SNAP-tag ligands, O6-benzylguanine itself can serve as an electron acceptor, acting to quench fluorescence of labelling dyes prior to conjugation to SNAP-tagby PET. Herten and co-workers observed greater than 30-fold fluorescence enhancement upon conjugation of O6-benzylguanine functionalized ATTO 700 (compound 18) to SNAP-tag.[39] Other groups have designed fluorogenic probes based on FRET quenching strategies. Urano and coworkers engineered one such probe with a fluorescein based dye coupled to an O6-benzylguanine ligand functionalized at the C-8 position with a Disperse Red-1 FRET acceptor to quench fluorescence (compound 19).[40] Upon conjugation to SNAP-tag the guanine—Disperse Red-1 motif is released, yielding a 300-fold increase in emission intensity of the fluorescein dye. Concomitantly, Yao and co-workers published a similar strategy with the quencher attached at the N-9 position.[41] Correa furthered this work by expanding the palette of fluorogenic dyes, using an O6-benzylguanine functionalized with cyanine derivatives as FRET quenchers.[42]
Notably, no-wash labelling strategies that employ FRET or PET quenching mechanisms are limited to small tags with active sites permissible to large probes such as SNAP-, CLIP-, BL- and TMP-tags.[40–43] To date, this strategy has not been applied to HaloTag. The long, narrow binding tunnel prohibits large quencher substrates from entering to be cleaved by nucleophilic substitution. Xiao’s work with thiocarbonyl based PET quenching offers one means of reducing the size of the quencher for this purpose,[44] but still, FRET and PET methods for fluorogenic labelling of HaloTagremain a niche to be filled.
Lactone opening for rhodamine-based fluorophores
As described in Section 3.1, rhodamine based dyes exist in equilibrium between an open, zwitterionic and closed lactone form. Johnsson and co-workers first exploited the polarity sensitivity of this equilibrium to develop a fluorogenic labelling method for super-resolution microscopy with Si-rhodamines, such as compounds 11 and 20a–c.[28b] Si-rhodamines exist predominately in the closed, non-fluorescent lactone form in aqueous solutions, likely due to a reversible aggregation of the fluorophore. Conjugation to a self-labelling protein tag rescues fluorescence in aqueous solutions, as demonstrated for Halo-, CLIP-, and SNAP-Tag with approximately 7-fold change in fluorescence intensity, a behaviour that can also be applied to carbon-rhodamine derivatives.[45] Furthermore, background noise with Si-rhodamine probes is minimized as the equilibrium further shifts to closed structure with non-specific binding of the dye to hydrophobic surfaces in the cell. Lavis and co-workers recently demonstrated that the equilibrium constant of the lactone-zwitterion process can be used to predict fluorogenicity of rhodamines, allowing for rational design of probes for no-wash labelling in vivo.[28a] Johnsson and co-workers furthered their initial work by demonstrating control over the lactone-equilibrium by substitution of the carboxyl moiety for electron deficient amides.[46]
Environment sensitive probes
In contrast to the “always-on” labelling probes described in Section 3.1, many donor-acceptor type fluorophores possess an inherent sensitivity to the polarity and rigidity of their local environment. In aqueous environments these environmentally sensitive dyes are largely non-fluorescent due to both inter- and intramolecular non-radiative decay mechanisms, such as external conversion and TICT (Figure 5).[47] However, if fixed in non-polar or rigid environments, fluorescence intensity is recovered. This environmental sensitivity enables a strategy for no-wash labelling of tagged proteins in vivo by embedding the fluorophore into the rigid, non-polar binding pocket or tunnel of SNAP-tag or HaloTag, typically accomplished through truncation of the linker moiety (Figure 3C).
Figure 5.

Mechanisms of viscosity and polarity dependent excited state non-radiative decay in donor-acceptor type chromophores. A) Twisted intramolecular charge transfer. B) Solvent-mediated external conversion.
Kool and co-workers pioneered the application of environmentally sensitive dyes for wash-free labelling of HaloTag with a molecular rotor type probe.[47a,48] A dimethylaminostilbazolium chromophore (compound 21) was able to be fixed in the narrow binding tunnel of HaloTag upon conjugation, yielding greater than 25-fold change in fluorescence intensity. A similar approach was utilized by our group as well as Tan and co-workers whereby coupling of a CCVJ (compound 22) or Kaede mimetic (compound 23) derived fluorophore to SNAP- tag led to a 40-fold and 90-fold increases in emission respectively.[49] Again it was found that fluorogenicity was dependent on the length of the linker employed.
In addition to molecular rotor type fluorophores, a breadth of polarity sensitive probes has also been employed in environmental sensitive strategies for wash-free labelling. Fluorogenicity upon conjugation to SNAP-tag using a polarity-sensitive sulfonylbenzoxadiazole (SBD) fluorophore (compound 24) that remains quenched in aqueous solutions yielded a 280-fold increase in fluorescence intensity.[50] Our group has also utilized SBD based fluorogenic probes for no-wash imaging of HaloTag (compound 25), achieving nearly 1000-fold fluorescence turn-on upon conjugation.[51] Interestingly, this drastic fold change was attributed to a novel excited state cation-π interaction as the electron donating amine of the SBD chromophore was found to align with the aromaticity of a binding-tunnel tryptophan residue. Extrapolation of this interaction from the protein conjugate system into small molecule scaffold remains to be achieved; but promises to yield a better understanding of the influence that electrostatic interactions have on excited state behaviour. Other groups have employed an array of solvatochromic dyes for no-wash labelling of SNAP-tag, including napthalimide, Nile-red, and merocyanine.[52]
3.3. Environmental sensitive labelling probes to visualize protein structural changes.
While the previously described ‘always-on’ and fluorogenic probes offer several means to track a HaloTag or SNAP-tag fused POI, the advantage of these approaches over conventional fluorescent protein tagging is minimal. However, the versatility of self-labelling protein tags allows for the incorporation of functional probes to report on more than just in vivo localization and expression, but rather can allow for visualization of protein dynamics and conformation. The inherent polarity and viscosity sensitivity of many fluorophores allows for visualization of changes in the physicochemical properties of the local microenvironment that arise with transition between protein folded states.[47] As such, many probes have been developed for the in situ recognition of misfolded and aggregate protein species without a need for direct conjugation. However, with the application of a self-labelling protein tag, greater specificity to track the dynamics of single POI with a membrane permeable, environmental sensitive fluorophore in live cells can be accomplished. In contrast to the application of environmental sensitive probes for no-wash labelling, this strategy mandates that the employed fluorophore remain non-fluorescent upon conjugation to the labelling tag. This is accomplished often by incorporating spacer moieties on the linker between the ligand and the chromophore, extending it out of the binding tunnel or pocket, and away from the protein surface.
Our group has initiated the application of environmental sensitive probes to monitor protein folded states. We developed a thermally destabilized mutant (K73T) of HaloTag, termed “AgHalo”, and resolved the conformational species from folded monomers to soluble oligomers and insoluble aggregates as a function of temperature (Figure 6A).[10,53] Conjugation of AgHalo to a polarity sensitive SBD based probe 26 led to a non-fluorescent conjugate with the folded protein. Heating the conjugate to 59°C yielded a 10-fold change in fluorescence intensity as protein formed insoluble aggregates. The destabilization of AgHalo (ΔGfolding = −2.0 kcal/mol) yielded a protein representative of the mammalian cell proteome. In conjugation with the SBD probe, the onset of chemical and thermally induced proteome stress was resolved in live cells with spatiotemporal resolution. Furthering this work we additionally developed a molecular rotor based probe for AgHalo based on a 9-(2-carboxy-2-cyanovinyl)julolidine derivative 27.[54] With this tool, soluble misfolded conformers could be distinguished from insoluble aggregates as a 12-fold change in emission intensity was observed at 45.6°C where soluble oligomeric species formed.
Figure 6.

The A) AgHalo and B) AggTag methods to monitor protein conformational dynamics and proteome stress onset.
With the AgHalo method established, our group aimed to observe the misfolding and aggregation of selected POIs through the use of an environmental sensitive fluorophore conjugated to a fused self-labelling tag, a method we termed “AggTag” (Figure 6B).[55] This method features environmental sensitive HaloTag or SNAP-tag probes that turn on their fluorescence only upon interaction with misfolded oligomers and insoluble aggregates, due to the increased surface hydrophobicity and/or elevated intermolecular rigidity of these species compared to folded proteins. When these probes are covalently conjugated to a protein tag that is genetically fused to a folded POI, fluorescence is quenched. Formation of misfolded oligomers and/or insoluble aggregates of the POI, however, inhibits quenching and yields turn-on fluorescence via intra-molecular interactions with probes. Unlike conventional FP-fusion methods that track the location of late-stage aggregates via appearance of fluorescent puncta, these probes specifically fluoresce in response to changes in protein conformation and thus report on early events during protein aggregation. We utilized a derivative of the red fluorescent protein chromophore, Kaede, functionalized with a rigid, extended chloroalkane ligand for HaloTag (compound 28).The Kaede mimetic probe was capable of detecting the misfolded soluble oligomeric conformers of the Parkinson’s and amyotrophic lateral sclerosis associated proteins, α-synuclein and mutant superoxide dismutase-1 (V31A) as fluorescence intensity increases were observed prior to the formation of fibrils and aggregates as monitored by Thioflavin-T and turbidity. We further expanded the AggTag method with the development of an orthogonal probe for SNAP-tag utilizing an SBD based probe with an extended O6-benzylguanine ligand (compound 29).[56] As such the AggTag method can be used to monitor the misfolding and aggregation of two independent proteins via multicolour imaging.
4. HaloTag and SNAP-tag probes for manipulation of protein systems
While visualizing protein localization, expression, and dynamics in vivo is crucial to understanding their behavior and function, only very rarely does a protein function independently within a cell. Instead, cellular machinery operates via a network of integrated and interconnected systems of interactions between small molecules, nucleic acids, carbohydrates, lipids, and proteins. Thus, in order to understand the physiological role of a POI in the cell, it is necessary to understand how it intercalates into this network and how perturbations to its behavior effect cellular function. Several technologies have been developed utilizing the bioorthogonality and versatility of self-labelling protein tags to allow for the manipulation of proteins in vivo via immobilization, expression-level control, or photoactivation.[57]
4.1. Immobilization of Halo- and SNAP-Tagged POIs.
Immobilization of proteins and protein-based complexes is in an important procedure in many analytical and preparatory biochemical techniques. Isolated proteins can be characterized in a controlled system in vitro to resolve their biochemical and biophysical properties. To this end, protein affinity tags have been widely developed and are employed near universally in standard biochemical techniques. Protein self-labelling tags offer one major advantage over typical affinity tags in that they don’t rely on a favourable dissociation constant to tightly bind the tagged POI, but rather they form irreversible, covalent linkages that enable more rigorous washing to remove unwanted contaminants.
As such, the commercially available HaloLink™ (Promega) has been engineered with the chloroalkane ligand for HaloTag surface-bound to a Sepharose resin (Figure 7A).[58] HaloLink™ allows for capture of a HaloTag fused POI expressed in vivo following lysis. The resulting resin bound protein can be eluted by treatment with a sequence-specific protease targeting the linker between the POI and HaloTag.[11,59] Alternatively the POI can be left bound to the resin, allowing for surface-based biochemical methods including protein-protein interaction identifications or enzyme activity analysis (Figure 7B).[58,60] A similar tool is commercially available for SNAP-tag fused proteins, named SNAP-Capture™ (New England BioLabs; Figure 7A), which can applied to any of the mentioned techniques and analyses for HaloLink™. Additionally, the biorthogonality of SNAP-tag and HaloTag offers the ability to covalently attach an affinity label, such as a biotinylated probe, to a POI to allow for standard pull-down assays.
Figure 7.

A) Immobilization strategy for Halo- and SNAP-Tag fused proteins of interest and B) downstream applications following immobilization.
Immobilization of self-labelling tag fused proteins can also allow for high-throughput analysis of protein-protein and protein-DNA interactions using fluorescence-based microarray methodologies. The throughput of standard protein microarrays is hindered by a need for large scale manual purification of thousands of proteins to be fixed to the array, but more recent developments in self-assembly of protein microarrays using in situ translation of pre-assembled DNA microarrays and binding of the nascent bait protein allows for proteome-wide screens against any recombinant query protein. So called nucleic acid programmable protein arrays (NAPPAs) typically utilize antibody based methods to capture and adhere the nascently translated bait protein the surface.[61] However, the covalent nature of a self-labelling protein tag allows for greater density of surface bound proteins, yielding greater signal-to-noise ratios for fluorescence based readout methods. Developed by Braun, Ecker, and co-workers, the HaloTag-NAPPA was used to identify the protein interacting partners of 38 Arabidopsis transcription factors in a screen against 12,000 unique open-reading frames.[62]
4.2. Proteasomal Targeting Chimeras (PROTACs).
Most small molecule drug therapies target proteins to downregulate activity, relying on a favourable dissociation constant to tightly bind to a protein of interest and inhibit function. Alternatively, Crews and co-workers have introduced a novel strategy of targeting overexpressed, hyperactive, or aberrantly behaving protein targets for proteasomal degradation.[63] This strategy uses small molecule recognition motifs to recruit an E3 ligase for ubiquitination of the target and subsequent proteasomal degradation. In doing so, the knock-down of a particular POI can be accomplished without need for excessive dosages. With the major challenge of targeting a POI with these PROteasomal TArgeting Chimeras (PROTACs) being the reliance on a particular binding motif, Crews and co-workers expanded the application of PROTACs to knockdown stably expressed HaloTag fused proteins in vivo (Figure 8). So called HaloPROTACs utilizes a recognition motif for the von Hippel-Lindau (VHL) E3 ligase functionalized with a chloroalkane linker (compound 30).[64] The efficiency of HaloPROTACs induced protein degradation was shown to be dependent on the affinity of the recognition motif for VHL. Compound 30 was shown to be the most effective, achieving approximately 10-fold reduction of GFP fused HaloTag in expression HEK293 cells. Replacing the terminal 1-isoindolinone motif of 30 with an isoxazole moiety yielded only a 2-fold reduction in expression level.[65] Furthermore, degradation efficiency is concentration dependent, with optimal degradation achieved in high nano- to low micromolar range. In the application of HaloPROTACs, thus, it is important to consider the structure of the probe and the employed concentration so as to desirably tune the extent of protein knockdown. It has also been demonstrated that degradation of HaloTagfusion proteins can be induced upon labelling with hydrophobic adamantyl groups (compound 31).[66] HaloPROTACs offers a novel strategy to observe the effects of pharmacological manipulation of protein expression levels in vivo, allowing for the identification of protein targets for PROTACs based therapies.
Figure 8.

HaloPROTACs strategy for induced proteasomal degradation of HaloTag fused POIs.
4.3. SNAP-tag and HaloTag strategies for optochemical control of protein systems
Light-induced activation of small molecule probes offers a unique level of spatiotemporal control over biological systems. The application of photolabile protecting groups enable this control, allowing researchers to deliver small molecule perturbations or activate probes on-demand.[67] Several UV-light activated photocaging systems can be used to supress the function of biological probes including the trimethyl-lock,[68] coumarinyl methyl,[69] o-nitrobenzyl,[70] BODIPY,[71] and anthraquinone systems.[72] Here we review how these photolabile protecting groups can be used in tandem with SNAP- or HaloTag to manipulate a protein system.
Targeted reactive electrophiles and oxidants (T-REX).
Protein behaviour and function can be non-pathogenically modulated through reaction with electrophilic and oxidative species operating as second messengers. In order to understand how these signals are translated through the cell, Aye and co-workers have developed a means of selective delivery of small molecule reactive electrophiles and oxidants to modify a specific POI in vivo.[73] In this strategy a POI is fused to HaloTag to allow for biorthogonal labelling with chloroalkane ligand attached to a photocaged 4-hydroxynonenal probe (compound 32). Upon labelling of HaloTag, irradiation of the anthraquinone photolabile protecting group can be used to eliminate the α,β-unsaturated aldehydes for labelling of the POI (Figure 9A). Experimental results demonstrated that this labelling strategy is selective for the fused POI in vivo, and further that by incorporating an alkyne motif on the labelling electrophile, that nearly any functionality can be attached via a downstream click reaction. Aye’s lab was able to show that the T-REX platform is able to accommodate α,β-unsaturated aldehydes of variable structure and sterics.[74] Through this they were elucidate the promiscuity of the signal transductor factor, Keap1, as a trigger of the antioxidant response element.
Figure 9.
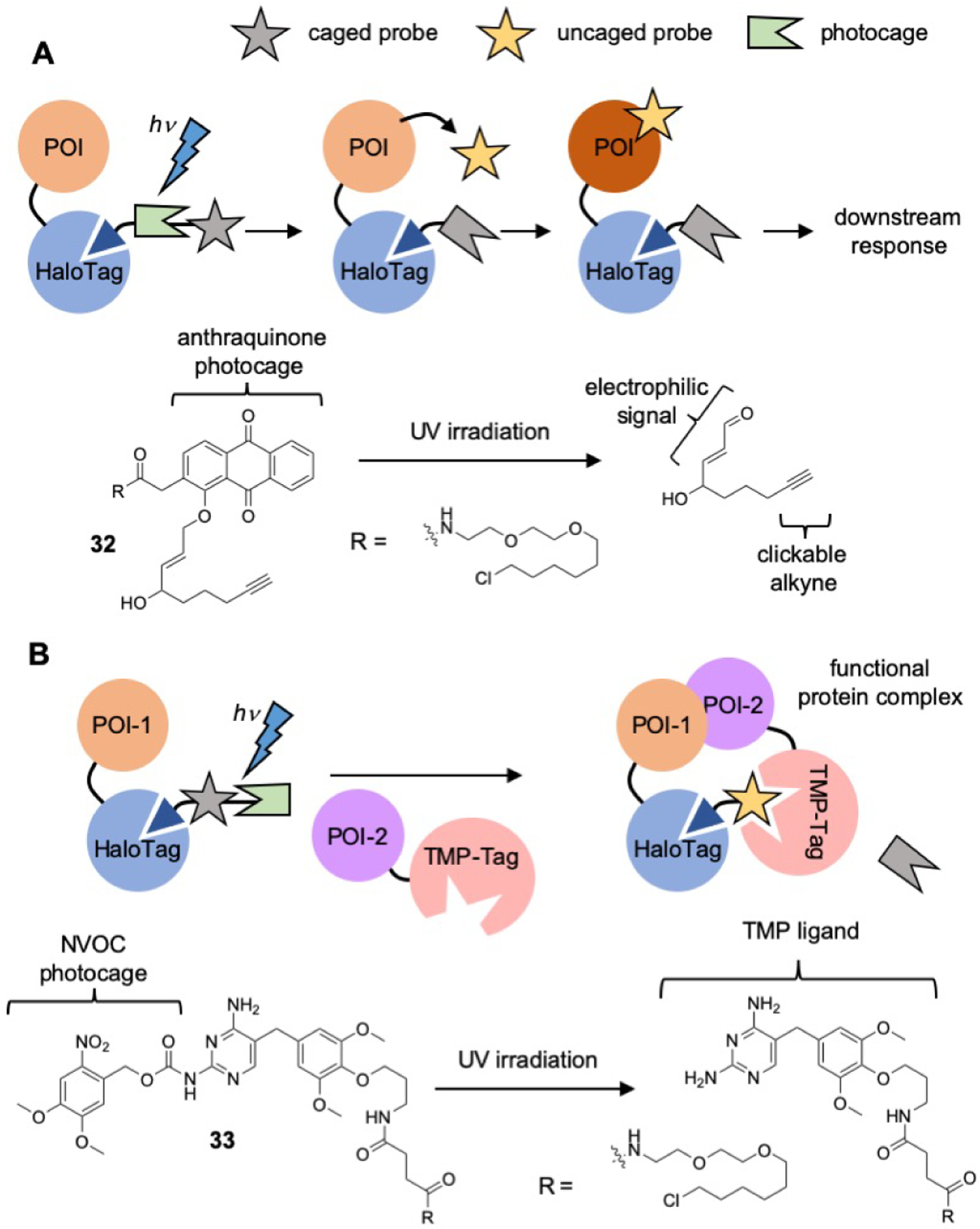
A) T-REX and B) CID strategies of optochemical control and perturbation of protein systems.
Chemically induced dimerization (CID)
Chemical control over the assembly of protein complexes is a powerful tool to directly observe the mechanisms and effects of protein-protein interactions in cells. Chenoweth and co-workers developed a strategy for the optochemical control of protein assembly using a dual ligand strategy with a HaloTagchloroalkane functionalized with photocage protect a TMP substrate (compound 33, Figure 9B).[75] The photocage, 6-nitroveratryl carbamate (NVOC), was used to protect the pyrimidine ring of TMP, preventing substrate binding to DHFR. Upon irradiation, the photocage is removed and the DHFR substrate is able to bind to the TMP probe. This binding event results in the rapid dimerization of two POIs, one fused to TMP-Tag and the other to HaloTag. An additional value of this strategy is the amenability of the probe employed, allowing for use of nearly any photocage or ligand, connected in any order that is chemically feasible. Wymann and co-workers took advantage of this malleability and developed a CID probe that could be readily cleaved with UV irradiation by placing the photo-uncaging moiety between the two ligands.[76]
5. Summary and Outlook
Self-labelling protein tags have enabled chemical biology studies in a wide range of biological pathways and cellular models. The versatility of these systems for protein labelling allows chemical biologists to incorporate nearly any functional probe into a fused POI. This unique capability has made HaloTag and SNAP-tag technologies into critical components of the chemical biology toolbox, allowing researchers to label a POI with small molecule probes to observe and manipulate protein behaviour. To date the field is well populated with imaging methods to visualize protein expression, localization, and dynamics. The use of self-labelling protein tags allows for incorporation of more diverse fluorophores with compatibility for super-resolution techniques or sensitization to protein conformation. Other probes are used to manipulate protein behaviour, through binding and immobilization, expression level control, complex formation, or small molecule signalling. We envision that the future of the field is defined by the power of self-labelling tags to enable the understanding and regulation of biological processes. Thus, we will provide an outlook with a focus on two aspects: the further development of multiplex tagging systems for targeting multiple proteins simultaneously and the extended chemical space of labelling probes to target more biological processes.
The existing multiplex of tagging systems has enabled a wide range of applications, featured by multiple-color imaging of proteins that are individually fused to HaloTag or SNAP-tag in the same cells. We envision that future efforts of expansion can be focused on developing protein tags with the following aspects; smaller molecular weight, faster labelling kinetics, and more bio-orthogonal chemistry for labelling. Smaller molecular weight is highly desired for the purpose with minimal perturbation of POI’s folding, conformational changes, interactome and function. Although protein-based tags are self-sufficient for covalent labelling without the need of an external co-factor, their sizes are as small as 14 kDa for the PYP tag. Faster labelling kinetics is beneficial to enable time-sensitive applications. So far, HaloTag serves as one of the most rapid self-labelling proteins tags with a labelling rate constant of 2.7•106 M−1•s−1 and expanding this property to other self-labelling protein tags remains a persistent task. Further, labelling in vivo depends on factors beyond the second order rate constant of the protein tag itself. Thus, to engineer more desired protein tags factors such as membrane permeability, solubility, toxicity, and compartmental localization should be considered in the design of probes and ligands for labelling.
Multiplex tagging system relies on the inherent orthogonality of probes’ labelling chemistry. Expanding this chemical space and harbouring more types of bio-orthogonal reaction could increase the capacity of this toolbox. In addition to protein tags, peptide-based tags have been developed based on sequence-specific chemical reactions, including tetracysteine tags (represented by the CCPGCC motif), sortase tag (represented by the commonly used C-terminal LPXTG sequence), biotin-tag (15 a.a. sequence, also named as AviTag) and others. These tags can also be used in tandem with self-labelling proteins for multiplex labelling systems.
With self-labelling protein tag technology, we envision that researchers can harness this powerful toolbox to address questions in a wider range of biological processes. At present, we have reviewed the ongoing efforts to understand protein aggregation (AgHalo and AggTag), redox signalling (T-REX), targeted protein degradation (PROTAC), and protein complex formation (CID). The palette of biological systems that have been investigated using self-labelling protein tag technology is far too expansive to be covered under the aim of this review. However, the utility of the HaloTag and SNAP-tag systems is nearly ubiquitous in the address of existing biological questions. As such we anticipate that researchers will continue to develop unique chemical strategies to probe protein-based processes at the subcellular level (i.e. membranes, organelles, and membraneless organelles) and in fields not discussed here (i.e. immunology, oncology, genetics).
Scheme 3.

Nucleophilic mechanism of cyanine photo-switching.
Acknowledgements
This work was supported by the Burroughs Wellcome Fund Career Award at the Scientific Interface (X.Z.), Paul Berg Early Career Professorship (X.Z.), Lloyd and Dottie Huck Early Career Award (X.Z.), Sloan Research Fellowship (X.Z.), Pew Biomedical Scholars Program (X.Z.) and National Institute of Health R35 GM133484 (X.Z.).
Biographies

Prof. Xin Zhang is the holder of the Paul Berg Early Career Professorship and an Assistant Professor of Chemistry and of Biochemistry and Molecular Biology at Penn State. Zhang’s independent work at Penn State has received multiple awards, including NSF CAREER Award, NIGMS MIRA, Pew Scholar in the Biomedical Sciences, Sloan Research Fellowship in Chemistry, the Lloyd and Dottie Huck Early Career Award, the Burroughs Wellcome Fund Career Award at the Scientific Interface.

Conner Hoelzel is a doctoral candidate in the Department of Chemistry at Penn State University. His work in Prof. Zhang’s lab aims to develop novel fluorescent probes for visualization of protein based life processes. Hoelzel completed his Bachelor’s at the College of Wooster (Wooster, OH) in 2016. His undergraduate research was focused on the development of new tools for auxiliary mediated peptide ligation.
References
- [1].Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV, ACS Chem. Biol 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- [2] a).Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K, Nat. Biotechnol 2003, 21, 86–89; [DOI] [PubMed] [Google Scholar]; b) Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, Johnsson K, Chem. Biol 2003, 10, 313–317. [DOI] [PubMed] [Google Scholar]
- [3].Gautier A, Juillerat A, Heinis C, Correa IR, Kindermann M, Beaufils F, Johnsson K, Chem. Biol 2008, 15, 128–136. [DOI] [PubMed] [Google Scholar]
- [4].Hori Y, Ueno H, Mizukami S, Kikuchi K, J. Am. Chem. Soc 2009, 131, 16610–16611. [DOI] [PubMed] [Google Scholar]
- [5] a).Miller LW, Sable J, Goelet P, Sheetz MR, Cornish VW, Angew. Chem., Int. Ed 2004, 43, 1672–1675; [DOI] [PubMed] [Google Scholar]; b) Miller LW, Cai YF, Sheetz MP, Cornish VW, Nat. Methods 2005, 2, 255–257. [DOI] [PubMed] [Google Scholar]
- [6] a).Watanabe S, Mizukami S, Hori Y, Kikuchi K, Bioconjugate Chem. 2010, 21, 2320–2326; [DOI] [PubMed] [Google Scholar]; b) Mizukami S, Watanabe S, Hori Y, Kikuchi K, J. Am. Chem. Soc 2009, 131, 5016–5017. [DOI] [PubMed] [Google Scholar]
- [7].Liu Y, Zhang X, Tan YL, Bhabha G, Ekiert DC, Kipnis Y, Bjelic S, Baker D, Kelly JW, J. Am. Chem. Soc 2014, 136, 13102–13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8] a).Jing C, Cornish VW, Acc. Chem. Res 2011, 44, 784–792; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gronemeyer T, Godin G, Johnsson K, Curr. Opin. Biotechnol 2005, 16, 453–458; [DOI] [PubMed] [Google Scholar]; c) Chen I, Ting AY, Curr. Opin. Biotechnol 2005, 16, 35–40; [DOI] [PubMed] [Google Scholar]; d) Marks KM, Nolan GP, Nat. Methods 2006, 3, 591–596; [DOI] [PubMed] [Google Scholar]; e) Zhang J, Campbell RE, Ting AY, Tsien RY, Nat. Rev. Mol. Cell. Biol 2002, 3, 906–918; [DOI] [PubMed] [Google Scholar]; f) Miller LW, Cornish VW, Curr. Opin. Chem. Biol 2005, 9, 56–61. [DOI] [PubMed] [Google Scholar]
- [9] a).Janssen DB, Curr. Opin. Chem. Biol 2004, 8, 150–159; [DOI] [PubMed] [Google Scholar]; b) Krooshof GH, Ridder IS, Tepper AWJW, Vos GJ, Rozeboom HJ, Kalk KH, Dijkstra BW, Janssen DB, Biochemistry 1998, 37, 15013–15023; [DOI] [PubMed] [Google Scholar]; c) Pries F, Kingma J, Krooshof GH, Jeronimusstratingh CM, Bruins AP, Janssen DB, J. Biol. Chem 1995, 270, 10405–10411. [DOI] [PubMed] [Google Scholar]
- [10].Liu Y, Fares M, Dunham NP, Gao Z, Miao K, Jiang XY, Bollinger SS, Boal AK, Zhang X, Angew. Chem., Int. Ed 2017, 56, 8672–8676. [DOI] [PubMed] [Google Scholar]
- [11] a).Ohana RF, Encell LP, Zhao K, Simpson D, Slater MR, Urh M, o KV, Protein Expression Purif. 2009, 68, 110–120; [DOI] [PubMed] [Google Scholar]; b) Peterson SN, Kwon K, Curr. Chem. Genomics 2012, 6, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mollwitz B, Brunk E, Schmitt S, Pojer F, Bannwarth M, Schiltz M, Rothlisberger U, Johnsson K, Biochemistry 2012, 51, 986–994. [DOI] [PubMed] [Google Scholar]
- [13] a).Gronemeyer T, Chidley C, Juillerat A, Heinis C, Johnsson K, Protein Eng., Des. Sel 2006, 19, 309–316; [DOI] [PubMed] [Google Scholar]; b) Juillerat A, Heinis C, Sielaff I, Barnikow J, Jaccard H, Kunz B, Terskikh A, Johnsson K, Chembiochem 2005, 6, 1263–1269; [DOI] [PubMed] [Google Scholar]; c) Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K, Proc. Natl. Acad. Sci. U. S. A 2004, 101, 9955–9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14] a).Duguid EM, Rice PA, He C, J. Mol. Biol 2005, 350, 657–666; [DOI] [PubMed] [Google Scholar]; b) Meyer AS, McCain MD, Fang Q, Pegg AE, Spratt TE, Chem. Res. Toxicol 2003, 16, 1405–1409; [DOI] [PubMed] [Google Scholar]; c) Pegg AE, Mutation Res. 2000, 462, 83–100; [DOI] [PubMed] [Google Scholar]; d) Pegg AE, Dolan ME, Scicchitano D, Morimoto K, Environ. Health Perspect 1985, 62, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Erdmann RS, Baguley SW, Richens JH, Wissner RF, Xi ZQ, Allgeyer ES, Zhong S, Thompson AD, Lowe N, Butler R, Bewersdorf J, Rothman JE, St Johnston D, Schepartz A, Toomre D, Cell Chem. Biol 2019, 26, 584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu Y, Miao K, Li YH, Fares M, Chen SY, Zhang X, Biochemistry 2018, 57, 4663–4674. [DOI] [PubMed] [Google Scholar]
- [17] a).Shashkova S, Leake MC, Biosci. Rep 2017, 37; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) von Diezmann A, Shechtman Y, Moerner WE, Chem. Rev 2017, 117, 7244–7275; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sahl SJ, Hell SW, Jakobs S, Nat. Rev. Mol. Cell. Biol 2017, 18, 685–701; [DOI] [PubMed] [Google Scholar]; d) Lavis LD, Raines RT, ACS Chem. Biol 2014, 9, 855–866; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Leung BO, Chou KC, Appl. Spectrosc 2011, 65, 967–980; [DOI] [PubMed] [Google Scholar]; f) Huang B, Bates M, Zhuang X, Annu. Rev. Biochem 2009, 78, 993–1016; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lavis LD, Raines RT, ACS Chem. Biol 2008, 3, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18] a).Waldchen S, Lehmann J, Klein T, van de Linde S, Sauer M, Sci. Rep 2015, 5, 15348; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Clement M, Daniel G, Trelles M, J. Cosmet. Laser Ther 2005, 7, 177–189. [DOI] [PubMed] [Google Scholar]
- [19] a).Gandioso A, Bresoli-Obach R, Nin-Hill A, Bosch M, Palau M, Galindo A, Contreras S, Rovira A, Rovira C, Nonell S, Marchan V, J. Org. Chem 2018, 83, 1185–1195; [DOI] [PubMed] [Google Scholar]; b) Nizamov S, Sednev MV, Bossi ML, Hebisch E, Frauendorf H, Lehnart SE, Belov VN, Hell SW, Chem. - Eur. J 2016, 22, 11631–11642; [DOI] [PubMed] [Google Scholar]; c) Schill H, Nizamov S, Bottanelli F, Bierwagen J, Belov VN, Hell SW, Chem. - Eur. J 2013, 19, 16556–16565; [DOI] [PubMed] [Google Scholar]; d) Nizamov S, Willig KI, Sednev MV, Belov VN, Hell SW, Chem. - Eur. J 2012, 18, 16339–16348; [DOI] [PubMed] [Google Scholar]; e) Jin X, Uttamapinant C, Ting AY, Chembiochem 2010, 12, 65–70; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Hoelzel CA, Hu H, Wolstenholme CH, Karim BA, Munson KT, Jung K, Liu Y, Yennawar HP, Asbury JB, Li X, Zhang X, Angew. Chem., Int. Ed 2020, 10.1002/anie.201915744. [DOI] [PubMed] [Google Scholar]
- [20].Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T, J. Am. Chem. Soc 2005, 127, 4888–4894. [DOI] [PubMed] [Google Scholar]
- [21].Sun WC, Gee KR, Klaubert DH, Haugland RP, J. Org. Chem 1997, 62, 6469–6475. [Google Scholar]
- [22].Mottram LF, Boonyarattanakalin S, Kovel RE, Peterson BR, Org. Lett 2006, 8, 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].van de Linde S, Krstic I, Prisner T, Doose S, Heilemann M, Sauer M, Photochem. Photobiol. Sci 2011, 10, 499–506. [DOI] [PubMed] [Google Scholar]
- [24].Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang XW, Nat. Methods 2011, 8, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Grabowski ZR, Dobkowski J, Pure Appl. Chem 1983, 55, 245–252. [Google Scholar]
- [26].Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, Lionnet T, Lavis LD, Nat. Methods 2015, 12, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T, ACS Chem. Biol 2011, 6, 600–608. [DOI] [PubMed] [Google Scholar]
- [28] a).Zheng QS, Ayala AX, Chung I, Weigel AV, Ranjan A, Falco N, Grimm JB, Tkachuk AN, Wu C, Lippincott-Schwartz J, Singer RH, Lavis LD, Acs Cent. Sci 2019, 5, 1602–1613; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lukinavicius G, Umezawa K, Olivier N, Honigmann A, Yang GY, Plass T, Mueller V, Reymond L, Correa IR, Luo ZG, Schultz C, Lemke EA, Heppenstall P, Eggeling C, Manley S, Johnsson K, Nat. Chem 2013, 5, 132–139. [DOI] [PubMed] [Google Scholar]
- [29].Grimm JB, Muthusamy AK, Liang YJ, Brown TA, Lemon WC, Patel R, Lu RW, Macklin JJ, Keller PJ, Ji N, Lavis LD, Nat. Methods 2017, 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Butkevich AN, Bossi ML, Lukinavičius G, Hell SW, J. Am. Chem. Soc 2019, 141, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS, Bioconjugate Chem. 1993, 4, 105–111. [DOI] [PubMed] [Google Scholar]
- [32].Mujumdar SR, Mujumdar RB, Grant CM, Waggoner AS, Bioconjugate Chem. 1996, 7, 356–362. [DOI] [PubMed] [Google Scholar]
- [33].van der Wal S, Kuil J, Valentijn ARPM, van Leeuwen FWB, Dyes Pigments 2016, 132, 7–19. [Google Scholar]
- [34].Renikuntla BR, Rose HC, Eldo J, Waggoner AS, Armitage BA, Org. Lett 2004, 6, 909–912. [DOI] [PubMed] [Google Scholar]
- [35] a).Dempsey GT, Bates M, Kowtoniuk WE, Liu DR, Tsien RY, Zhuang X, J. Am. Chem. Soc 2009, 131, 18192–18193; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rust MJ, Bates M, Zhuang XW, Nat. Methods 2006, 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gallo E, Bioconjugate Chem. 2020, 31, 16–27. [DOI] [PubMed] [Google Scholar]
- [37] a).Knox HJ, Chan J, Acc. Chem. Res 2018, 51, 2897–2905; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pino NW, Davis J, Yu ZX, Chan J, J. Am. Chem. Soc 2017, 139, 18476–18479; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Roth A, Li H, Anorma C, Chan J, J. Am. Chem. Soc 2015, 137, 10890–10893; [DOI] [PubMed] [Google Scholar]; d) Chan J, Dodani SC, Chang CJ, Nat. Chem 2012, 4, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen YC, Tsao K, Keillor JW, Can. J. Chem 2015, 93, 389–398. [Google Scholar]
- [39].Stohr K, Siegberg D, Ehrhard T, Lymperopoulos K, Oz S, Schulmeister S, Pfeifer AC, Bachmann J, Klingmuller U, Sourjik V, Herten DP, Anal. Chem 2010, 82, 8186–8193. [DOI] [PubMed] [Google Scholar]
- [40].Komatsu T, Johnsson K, Okuno H, Bito H, Inoue T, Nagano T, Urano Y, J. Am. Chem. Soc 2011, 133, 6745–6751. [DOI] [PubMed] [Google Scholar]
- [41].Zhang CJ, Li L, Chen GYJ, Xu QH, Yao SQ, Org. Lett 2011, 13, 4160–4163. [DOI] [PubMed] [Google Scholar]
- [42].Sun XL, Zhang AH, Baker B, Sun L, Howard A, Buswell J, Maurel D, Masharina A, Johnsson K, Noren CJ, Xu MQ, Correa IR, Chembiochem 2011, 12, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43] a).Jing C, Cornish VW, ACS Chem. Biol 2013, 8, 1704–1712; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mizukami S, Watanabe S, Akimoto Y, Kikuchi K, J. Am. Chem. Soc 2012, 134, 1623–1629. [DOI] [PubMed] [Google Scholar]
- [44].Tang J, Robichaux MA, Wu KL, Pei JQ, Nguyen NT, Zhou YB, Wensel TG, Xiao H, J. Am. Chem. Soc 2019, 141, 14699–14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45] a).Butkevich AN, Mitronova GY, Sidenstein SC, Klocke JL, Kamin D, Meineke DNH, D’Este E, Kraemer PT, Danzl JG, Belov VN, Hell SW, Angew. Chem., Int. Ed 2016, 55, 3290–3294; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Grimm JB, Sung AJ, Legant WR, Hulamm P, Matlosz SM, Betzig E, Lavis LD, ACS Chem. Biol 2013, 8, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang L, Tran M, D’Este E, Roberti J, Koch B, Xue L, Johnsson K, Nat. Chem 2019. [DOI] [PubMed] [Google Scholar]
- [47] a).Haidekker MA, Theodorakis EA, J. Biol. Eng 2010, 4, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reichardt C, Chem. Rev 1994, 94, 2319–2358. [Google Scholar]
- [48].Clark SA, Singh V, Vega Mendoza D, Margolin W, Kool ET, Bioconjugate Chem. 2016, 27, 2839–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49] a).Jung KH, Fares M, Grainger LS, Wolstenholme CH, Hou A, Liu Y, Zhang X, Org. Biomol. Chem 2019, 17, 1906–1915; [DOI] [PubMed] [Google Scholar]; b) Yu WT, Wu TW, Huang CL, Chen IC, Tan KT, Chem. Sci 2016, 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu TK, Hsieh PY, Zhuang YD, Hsia CY, Huang CL, Lai HP, Lin HS, Chen IC, Hsu HY, Tan KT, ACS Chem. Biol 2014, 9, 2359–2365. [DOI] [PubMed] [Google Scholar]
- [51].Liu Y, Miao K, Dunham NP, Liu HB, Fares M, Boal AK, Li XS, Zhang X, Biochemistry 2017, 56, 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52] a).Leng S, Qiao Q, Miao L, Deng W, Cui J, Xu Z, Chem. Commun. (Cambridge, U. K.) 2017, 53, 6448–6451; [DOI] [PubMed] [Google Scholar]; b) Chen HJ, Chew CY, Chang EH, Tu YW, Wei LY, Wu BH, Chen CH, Yang YT, Huang SC, Chen JK, Chen IC, Tan KT, J. Am. Chem. Soc 2018, 140, 5224–5234; [DOI] [PubMed] [Google Scholar]; c) Qiao QL, Liu WJ, Chen J, Zhou W, Yin WT, Miao L, Cui JN, Xu ZC, Dyes Pigments 2017, 147, 327–333; [Google Scholar]; d) Hong YR, Lam CH, Tan KT, Bioconjugate Chem. 2017, 28, 2895–2902. [DOI] [PubMed] [Google Scholar]
- [53].Fares M, Zhang X, Curr. Protoc. Chem. Biol 2019, 11, e58. [DOI] [PubMed] [Google Scholar]
- [54].Fares M, Li Y, Liu Y, Miao K, Gao Z, Zhai Y, Zhang X, Bioconjugate Chem. 2018, 29, 215–224. [DOI] [PubMed] [Google Scholar]
- [55].Liu Y, Wolstenholme CH, Carter GC, Liu HB, Hu H, Grainger LS, Miao K, Fares M, Hoelzel CA, Yennawar HP, Ning G, Du MY, Bai L, Li XS, Zhang X, J. Am. Chem. Soc 2018, 140, 7381–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jung KH, Kim SF, Liu Y, Zhang X, Chembiochem 2019, 20, 1078–1087. [DOI] [PubMed] [Google Scholar]
- [57].England CG, Luo HM, Cai WB, Bioconjugate Chem. 2015, 26, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Urh M, Simpson D, Sankbeil J, Hartzell D, Karassina N, Nassif N, English J, Zhu J, Meisenheimer P, Klaubert D, Bulleit B, Wood KV, Promega cell notes 2006, 14, 15–19. [Google Scholar]
- [59] a).Ohana RF, Hurst R, Curr. Protoc. Mol. Biol 2015, 110, 10 31 11–10 31 15; [DOI] [PubMed] [Google Scholar]; b) Kimple ME, Brill AL, Pasker RL, Curr. Protoc. Protein. Sci 2013, 73, 9 9 1–9 9 23; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Locatelli-Hoops S, Sheen FC, Zoubak L, Gawrisch K, Yeliseev AA, Protein Expression Purif. 2013, 89, 62–72; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chumanov RS, Kuhn PA, Xu W, Burgess RR, Protein Expression Purif. 2011, 76, 145–153; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ohana RF, Hurst R, Vidugiriene J, Slater MR, Wood KV, Urh M, Protein Expression Purif. 2011, 76, 154–164. [DOI] [PubMed] [Google Scholar]
- [60].Urh M, Hartzell D, Mendez J, Klaubert DH, Wood K, in Affinity Chromatography: Methods and Protocols (Ed.: Zachariou M), Humana Press, Totowa, NJ, 2008, pp. 191–210. [Google Scholar]
- [61] a).Ramachandran N, Hainsworth E, Bhullar B, Eisenstein S, Rosen B, Lau AY, Walter JC, LaBaer J, Science 2004, 305, 86–90; [DOI] [PubMed] [Google Scholar]; b) Ramachandran N, Raphael JV, HainsworthV E, Demirkan G, Fuentes MG, Rolfs A, HuV Y, LaBaerV J, Nat. Methods 2008, 5, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yazaki J, Galli M, Kim AY, Nito K, Aleman F, Chang KN, Carvunis AR, Quan R, Nguyen H, Song L, Alvarez JM, Huang SSC, Chen HM, Ramachandran N, Altmann S, Gutierrez RA, Hill DE, Schroeder JI, Chory J, LaBaer J, Vidal M, Braun P, Ecker JR, P. Natl. Acad. Sci. U. S. A 2016, 113, E4238–E4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Buckley DL, Crews CM, Angew. Chem., Int. Ed. Engl 2014, 53, 2312–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, Crews CM, ACS Chem. Biol 2015, 10, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM, J. Am. Chem. Soc 2012, 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM, Nat Chem Biol 2011, 7, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67] a).Bardhan A, Deiters A, Curr. Opin. Struct. Biol 2019, 57, 164–175; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ankenbruck N, Courtney T, Naro Y, Deiters A, Angew. Chem., Int. Ed 2018, 57, 2768–2798; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Klán P, Šolomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J, Chem. Rev 2013, 113, 119–191; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Paola Pelliccioli A, Wirz J, Photochem. Photobiol. Sci 2002, 1, 441–458. [DOI] [PubMed] [Google Scholar]
- [68] a).Walton DP, Dougherty DA, J. Am. Chem. Soc 2017, 139, 4655–4658; [DOI] [PubMed] [Google Scholar]; b) Yatzeck MM, Lavis LD, Chao TY, Chandran SS, Raines RT, Bioorg. Med. Chem. Lett 2008, 18, 5864–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bassolino G, Nançoz C, Thiel Z, Bois E, Vauthey E, Rivera-Fuentes P, Chem. Sci 2018, 9, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70] a).Amit B, Patchornik A, Tetrahedron Lett. 1973, 2205–2208; [Google Scholar]; b) Patchornik A, Amit B, Woodward RB, J. Am. Chem. Soc 1970, 92, 6333–6335. [Google Scholar]
- [71].Goswami PP, Syed A, Beck CL, Albright TR, Mahoney KM, Unash R, Smith EA, Winter AH, J. Am. Chem. Soc 2015, 137, 3783–3786. [DOI] [PubMed] [Google Scholar]
- [72].Furuta T, Torigai H, Sugimoto M, Iwamura M, J. Org. Chem 1995, 60, 3953–3956. [Google Scholar]
- [73] a).Parvez S, Long MJ, Lin HY, Zhao Y, Haegele JA, Pham VN, Lee DK, Aye Y, Nat Protoc 2016, 11, 2328–2356; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Long MJ, Poganik JR, Aye Y, J. Am. Chem. Soc 2016, 138, 3610–3622; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Parvez S, Fu Y, Li J, Long MJ, Lin HY, Lee DK, Hu GS, Ay Y, J. Am. Chem. Soc 2015, 137, 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lin HY, Haegele JA, Disare MT, Lin Q, Aye Y, J. Am. Chem. Soc 2015, 137, 6232–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75] a).Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, Chenoweth DM, Nat. Commun 2014, 5; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H, Aonbangkhen C, Tarasovetc EV, Ballister ER, Chenoweth DM, Lampson MA, Nat. Chem. Biol 2017, 13, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zimmermann M, Cal R, Janett E, Hoffmann V, Bochet CG, Constable E, Beaufils F, Wymann MP, Angew. Chem., Int. Ed. Engl 2014, 53, 4717–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]