

Abstract
Arthrinins E–G (1–3), three new sesquiterpenoids possessing non-isoprenoid botryane skeleton, were isolated from the fermentation of an endophytic fungus named Arthrinium sp. HS66 which colonized in the stems of Isodon xerophilus. Their structures were determined by extensive spectroscopic methods. Furthermore, the structure of 1 was unambiguously confirmed by X-ray diffraction, while those of 2 and 3 were verified through quantum chemical calculation of NMR data and ECD spectra.
Electronic supplementary material
The online version of this article (doi:10.1007/s13659-020-00248-y) contains supplementary material, which is available to authorized users.
Keywords: Isodon xerophilus, Endophytic fungus, Arthrinium, Botryane sesquiterpenoid, Quantum chemical calculation
Introduction
Fungal metabolites have received considerable attention nowadays because their diverse chemical structures and bioactivities greatly facilitate the progress of drug discovery [1]. Botryanes are a class of fungus-derived sesquiterpene metabolites featuring characteristic bicyclic non-isoprenoid system [2–4], and they were found to possess broad spectrum of biological activities, such as cytotoxicity[5, 6], phytotoxicity [7, 8], and antimicrobial property [9, 10]. Furthermore, fascinating by the botryane sesquiterpenoids, researchers have conducted plenty of in-depth investigations about their structure–activity relationships [11], synthesis [12, 13] and biosynthetic pathway [14–16].
Fungal endophytes have become an important source for discovering structurally novel and biologically active secondary metabolites [17]. Over the past several years, our groups have made great efforts to study the secondary metabolites from endophytic fungi inhabiting the Isodon species. As a result, isopenicin A, a potent inhibitor of Wnt signaling [18], as well as several antineoplastic compounds like phomopchalasins A and B [19] have been successfully obtained. In the present research, an endophytic fungus colonizing in the stems of Isodon xerophilus was discovered and identified as Arthrinium sp. HS66. Subsequent large fermentation and chemical investigation on this strain resulted in the isolation of three new botryane sesquiterpenoids named arthrinins E–G (1–3). Notably, compound 2 possesses uncommon 15-nor-botryane skeleton. Herein, details of the isolation, structure elucidation, and cytotoxicity of these compounds were reported (Fig. 1).
Fig. 1.
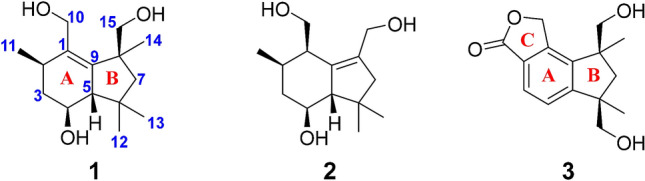
Chemical structures of compounds 1–3
Results and Discussion
Compound 1 was isolated as colorless oil, and it was assigned a molecular formula of C15H26O3 according to its positive HRESIMS ion peak at m/z 277.1775 ([M + Na]+, calcd for 277.1774), which required three degrees of unsaturation. The IR spectrum showed the presence of hydroxyl (3374 cm–1) group. The 1H NMR data (Table 1) exhibited the existence of three singlet methyls (δH 0.97, 1.18 and 1.38), one doublet methyl (δH 1.10, d, J = 6.9 Hz), a pair of nonequivalent oxygenated methylene protons at δH 4.04 (dd, J = 11.9, 2.1 Hz)/4.33 (d, J = 11.9 Hz), one hydroxylated methylene signal at δH 3.30 (overlap), and one hydroxylated methine proton at δH 3.63 (ddd, J = 11.9, 9.6, 3.6 Hz). The analysis of its 13C NMR and DEPT spectra revealed 15 carbon resonances which were assigned as four methyls, four sp3 methylenes, three sp3 methines, two sp3 quaternary carbons and two olefinic carbons (Table 2). These data suggested that compound 1 might be a botryane sesquiterpenoid [20].
Table 1.
1H NMR data (δ in ppm, J in Hz) of compounds 1–3
| No. | 1a | 2a | 3b |
|---|---|---|---|
| δH, mult (J) | δH, mult (J) | δH, mult (J) | |
| 1 | 2.60 (dt, 10.2, 4.4) | ||
| 2 | 2.58 (m) | 1.67 (overlap) | |
| 3 | β 1.97 (ddd, 12.0, 5.8, 3.6) | β 1.31 (m) | 7.72 (d, 7.9) |
| α 1.29 (overlap) | α 1.67 (overlap) | ||
| 4 | 3.63 (ddd, 11.9, 9.6, 3.6) | 3.57 (td, 10.8, 4.5) | 7.43 (d, 7.9) |
| 5 | 2.10 (ddd, 9.6, 3.5, 2.1) | 2.30 (d, 10.8) | |
| 6 | |||
| 7 | β 1.74 (d, 13.0) | β 2.37 (dd, 15.6,2.3) | β 2.36 (d, 13.9) |
| α 1.30 (overlap) | α 2.16 (dd, 15.6, 2.3) | α 1.70 (d, 13.9) | |
| 8 | |||
| 9 | |||
| 10 | 4.33 (d, 11.9) | 3.69 (dd, 10.2, 4.4) | 5.55 (d, 15.7) |
| 4.04 (dd, 11.9, 2.1) | 3.45 (t, 10.2) | 5.46 (d, 15.7) | |
| 11 | 1.10 (d, 6.9) | 1.03 (d, 7.0) | |
| 12 | 0.97 (s) | 1.07 (s) | 3.61 (d, 10.8) |
| 3.58 (d, 10.8) | |||
| 13 | 1.18 (s) | 1.23 (s) | 1.34 (s) |
| 14 | 1.38 (s) | 4.05 (s) | 1.37 (s) |
| 15 | 3.30 (overlap) | 3.64 (s) |
aRecorded at 600 MHz, Recorded in CD3OD
bRecorded at 500 MHz, Recorded in CD3OD
Table 2.
13C NMR data (δ in ppm) of compounds 1–3
| No. | 1a | 2a | 3b |
|---|---|---|---|
| δC, type | δC, type | δC, type | |
| 1 | 136.0, C | 43.5, CH | 145.2, C |
| 2 | 34.4, CH | 33.7, CH | 126.0, C |
| 3 | 44.1, CH2 | 40.8, CH2 | 125.4, CH |
| 4 | 70.0, CH | 71.9, CH | 126.1, CH |
| 5 | 61.4, CH | 60.9, CH | 157.8, C |
| 6 | 40.0, C | 39.7, C | 50.1, C |
| 7 | 56.7, CH2 | 51.7, CH2 | 47.8, CH2 |
| 8 | 47.5, C | 136.9, C | 50.2, C |
| 9 | 145.9, C | 139.8, C | 144.7, C |
| 10 | 58.9, CH2 | 59.9, CH2 | 71.1, CH2 |
| 11 | 20.7, CH3 | 19.4, CH3 | 173.9, C |
| 12 | 23.9, CH3 | 25.6, CH3 | 71.3, CH2 |
| 13 | 30.9, CH3 | 31.6, CH3 | 27.2, CH3 |
| 14 | 26.1, CH3 | 59.2, CH2 | 24.9, CH3 |
| 15 | 72.6, CH2 | 70.8, CH2 |
aRecorded at 150 MHz, Recorded in CD3OD
bRecorded at 125 MHz, Recorded in CD3OD
Specifically, a structural subunit of C-11/C-2/C-3/C-4/C-5 could be established as an isolated spin-system according to the 1H-1H COSY correlations of H3-11/H-2/H2-3/H-4/H-5. Then, the subunit can be assigned to ring A through the HMBC correlations from H-5 to C-9 (δC 145.9), from H2-10 to C-1 (δC 136.0), C-2 (δC 34.4) and C-9, from H3-11 to C-1. Meanwhile, the HMBC correlations from H2-7 to C-13 (δC 30.9) and C-14 (δC 26.1), from H3-12 to C-6 (δC 40.0), from H3-13 to C-5 (δC 61.4) and C-12 (δC 23.9), from H3-14 to C-8 (δC 47.5), from H2-15 to C-9 and C-14, in combination with the remaining one degree of hydrogen deficiency, successfully established the ring B as well as its connection with ring A (Fig. 2). Hence, the planar structure of compound 1 was resolved.
Fig. 2.
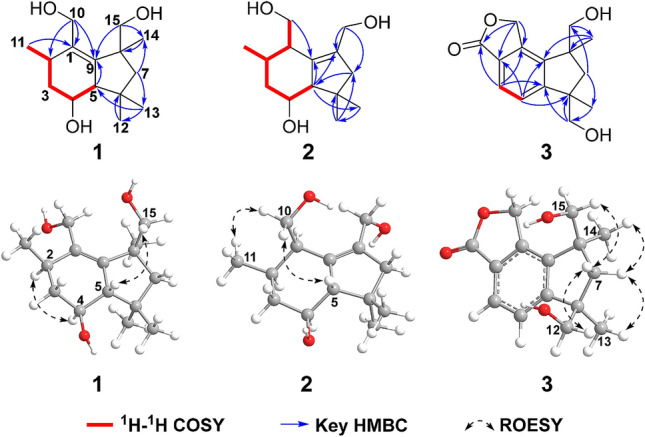
Key 2D NMR correlations of compounds 1–3
The relative configuration of 1 was deduced by analyses of ROESY and 1H NMR data. Randomly assigning H-2 as α-oriented, the cross peaks of H-4/H-2 and H-5/H2-15, together with the coupling constant (J = 9.6 Hz) between H-4 and H-5 indicated that both H-4 and CH3-14 adopted α-orientation, while both H-5 and CH2OH-15 adopted β-orientation (Fig. 2). Fortunately, colorless square crystals of 1 were eventually obtained through slow evaporation of methanol. Then a single-crystal X-ray diffraction experiment with Cu Kα radiation of 1 [Flack parameter = 0.07(3)] unambiguously verified the aforementioned deduction and assigned the absolute configuration of 1 as 2R,4S,5S, and 8S (Fig. 3). Arthrinins A–D from the sponge derived fungus Arthrinium sp. has been reported, so we gave compound 1 the trivial name from arthrinin E [21].
Fig. 3.
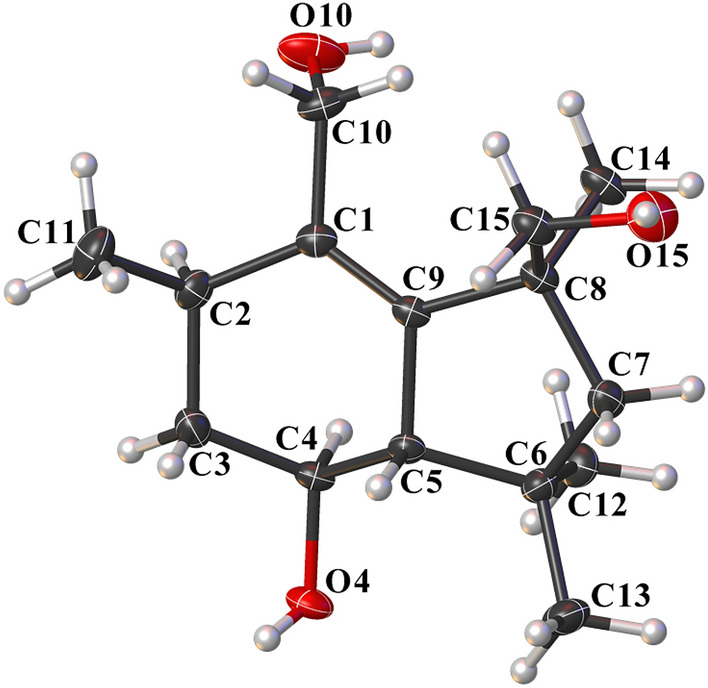
X-ray crystallographic structure of 1
Compound 2 was obtained as colorless oil. Its molecular formula C14H24O3 was ascertained by the positive HRESIMS ion peak (m/z 263.1618, [M + Na]+, calcd for 263.1618), indicating three degrees of unsaturation. Comparison of the 1H NMR spectrum of 2 with that of 1 disclosed that 2 was structurally analogous to compound 1 (Table 1). Furthermore, the 13C NMR and DEPT spectra, which demonstrated 14 carbons resonances involving three methyls, four sp3 methylenes, four sp3 methines, one quaternary carbon and two olefinic carbons (Table 2), suggested that the structure of 2 was highly similar to that of boledulin C [22], a 15-nor-botryane sesquiterpenoid.
More credible evidence was obtained from 2D NMR spectra (Figs. S20-23) of 2. On one hand, the structure of ring A was established by the 1H-1H COSY correlations of H2-10/H-1/H-2/H2-3/H-4/H-5 and H-2/H3-11 and HMBC correlations from H-5 to C-9 (δC 139.8), from H2-10 to C-9. On the other hand, the HMBC correlations from H-5 to C-8 (δC 136.9) and C-13 (δC 31.6), from H2-7 to C-5 (δC 60.9), from H3-12 to C-6 (δC 39.7) and C-7 (δC 51.7), from H3-13 to C-12 (δC 25.6), from H2-14 to C-7 and C-8 defined the ring B fragment and planar structure of 2 ultimately (Fig. 2).
As for the configuration of 2, randomly assigning H-5 as β-oriented, the observed correlations of H2-10/H3-11/H-5 in the ROESY spectrum, together with the coupling constant (J = 4.4 Hz) between H-1 and H-2 implied that both CH2OH-10 and CH3-11 adopted β-orientation, while the analysis of coupling constant (J = 10.8 Hz) between H-4 and H-5 suggested that H-4 was α-oriented (Fig. 2). Then, (1R*,2R*,4S*,5S*)-2 was subjected to quantum chemical calculation of NMR chemical shifts and spin–spin coupling constants at mPW1PW91-SCRF/6–31 + G(d,p)//B3LYP-D3BJ-SCRF/6-31G(d) and B972-SCRF/pcJ-1//B3LYP-D3BJ-SCRF/6-31G(d) level of theory (both with methanol as solvent and SMD solvent model), respectively, and the calculated results matched their experimental counterparts very well (Tables 3 and S3), which verified the established planar structure and relative configuration of 2. Moreover, TDDFT ECD calculation of (1R,2R,4S,5S)-2 was run at CAM-B3LYP-SCRF/def2-SVP//B3LYP-D3BJ-SCRF/6-31G(d) level of theory in MeOH with SMD solvent model, and the obtained curve, which is entirely consistent with its experimental counterpart, supported the absolute configuration of 2 to be 1R,2R,4S, and 5S (Fig. 4). Therefore, compound 2 was confirmed to possess 15-nor-botryane skeleton and named as arthrinin F.
Table 3.
Analyses of the NMR computation results of (1R*,2R*,4S*,5S*)-2 and (6R*,8S*)-3
| Parameters | R2 | MAE (ppm) | CMAE (ppm) |
|---|---|---|---|
| (1R*,2R*,4S*,5S*)-2 | |||
| 13C | 0.9992 | 1.3 | 0.8 |
| 1H | 0.9938 | 0.1 | 0.06 |
| (6R*,8S*)-3 | |||
| 13C | 0.9989 | 1.5 | 1.4 |
| 1H | 0.9948 | 0.21 | 0.13 |
Fig. 4.
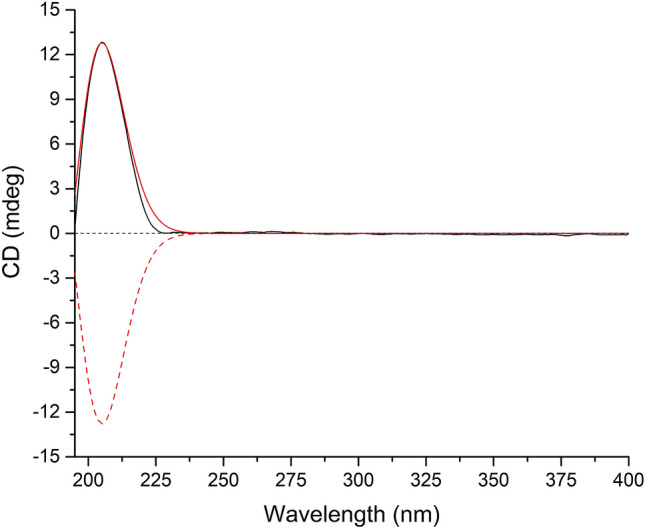
Experimental ECD spectrum of 2 (black); Calculated ECD spectra of (1R,2R,4S,5S)-2 (shift = 12.5 nm, red) and (1S,2S,4R,5R)-2 (shift = 12.5 nm, red dash)
Compound 3 was isolated as a colorless oil. The [M+Na]+ peak at m/z 285.1100 (calcd for 285.1097) existing in the positive HRESIMS spectrum revealed the molecular formula of 3 to be C15H18O4, which implied seven indices of hydrogen deficiency. Its IR spectrum indicated the presence of hydroxyl (3419 cm–1), methyl (2932 and 2869 cm–1), phenyl (1607 and 1467 cm–1) and ester (1743 and 1040 cm–1) groups. The 1H NMR spectrum gave two characteristic aromatic protons at δH 7.43 (d, J = 7.9 Hz), 7.72 (d, J = 7.9 Hz) and two singlet methyl groups (δH 1.37, 1.34) (Table 1). The 13C NMR and DEPT spectra displayed 15 carbon signals, including one ester carbonyl carbon, six aromatic carbons, two sp3 quaternary carbons, four sp3 methylenes and two methyls (Table 2). The above data suggested that the structure of 3 resembled to that of dehydrobotrylactone with a botryane scaffold [23].
The 1H-1H COSY correlation between H-3 and H-4, along with the HMBC correlations from H-3 to C-1 (δC 145.2), C-5 (δC 157.8) and C-11 (δC 173.9), from H-4 to C-2 (δC 126.0) and C-9 (δC 144.7), from H2-10 to C-1, C-2 and C-11 established the structure of the butyrolactone motif (ring C), and its connection to a tetra-substituted benzene ring (ring A). Moreover, the HMBC cross peaks from H-4 to C-6 (δC 50.1), from H2-7 to C-13 (δC 27.2) and C-15 (δC 70.8), from H2-12 to C-5 and C-6, from H3-13 to C-12 (δC 71.3), from H3-14 to C-8 (δC 50.2) and C-9, from H2-15 to C-14 (δC 24.9) determined the existence of a cyclopentane fragment (ring B) and its linkage with ring A. As a result, the planar structure of compound 3 was elucidated as shown in Fig. 2.
As for the stereochemistry of 3, randomly assigning H-7a as α-oriented, according to the ROESY correlations observed from H-7α to both H3-13 and H3-14, as well as from H-7β to both H2-12 and H2-15, it can be concluded that both CH3-13 and CH3-14 adopted α-orientation. Then, the NMR chemical shifts of (6R*,8S*)-3 were calculated at mPW1PW91-SCRF/6–31 + G(d,p)//B3LYP-D3BJ-SCRF/6-31G(d) level of theory with methanol as solvent and SMD solvent model, the predicted chemical shifts matched their experimental counterparts very well (Table 3), which supported the above deduction concerning the relative configuration of 3. Furthermore, the theoretical ECD spectrum of (6R,8S)-3 was obtained at CAM-B3LYP-SCRF/def2-SVP//B3LYP-D3BJ-SCRF/6-31G(d) level of theory in MeOH with SMD solvent model, and the calculated curve matched the experimental one very well and thus supported the absolute configuration of 3 to be 6R,8S. (Fig. 5). Compound 3 was given the trivial name arthrinin G.
Fig. 5.
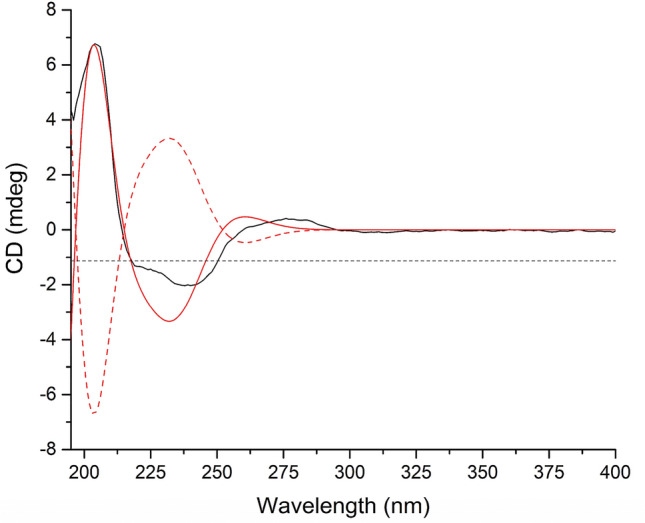
Experimental ECD spectrum of 3 (black); Calculated ECD spectra of (6R,8S)-3 (shift = 12 nm, red) and (6S,8R)-3 (shift = 12 nm, red dash)
Additionally, compounds 1–3 were evaluated for their cytotoxicity against five human cancer cell lines (HL-60, A549, SMMC-7721, MCF-7, SW480), with cis-platin and paclitaxel as positive controls, however, no compounds showed activity against the tested cell lines (Fig. S2).
Experimental
General Experimental Procedures
Optical rotations were measured with a JASCO P-1020 polarimeter. UV spectra were obtained using a Shimadzu UV-2401 PC spectrophotometer. A Tensor 27 spectrophotometer was used for scanning IR spectroscopy with KBr pellets. 1D and 2D NMR spectra were recorded on Bruker DRX-600 and 500 spectrometers with TMS as internal standard. Chemical shifts (δ) are expressed in parts per million (ppm) with reference to the solvent signals. HRESIMS was performed on an API QSTAR spectrometer. Semipreparative HPLC was performed on an Agilent 1200 liquid chromatograph with a Zorbax SB-C18 (9.4 mm × 250 mm) column. LC–MS/MS was performed on Agilent 6530 Accurate-Mass Q-TOF spectrometer coupled to an Agilent 1290 LC system with Zorbax SB-C18 (9.4 mm × 250 mm) column. Column chromatography was performed with silica gel (100–200 mesh, Qingdao Marine Chemical, Inc., Qingdao, People’s Republic of China). Fractions were monitored by TLC, and spots were visualized by heating silica gel plates sprayed with 10% H2SO4 in EtOH.
Fungal Material, Identification and Fermentation
The fungal strain of Arthrinium sp. HS66 was isolated from the fresh stems of Isodon xerophilus that was collected from Kunming Botanical Garden, Kunming City, Yunnan Province, People’s Republic of China, in August 2018. The isolate was identified based on sequence (GenBank Accession No. MT355097) analysis of the ITS region of the rDNA. The fungal strain was cultured on slants of potato dextrose agar at 28 °C for 7 days. Agar plugs were cut into small pieces (about 0.5 × 0.5 × 0.5 cm3) under aseptic conditions, and 15 pieces were used to inoculate three Erlenmeyer flasks (500 mL), each containing 200 mL of media (0.4% glucose, 1% malt extract, and 0.4% yeast extract); the final pH of the media was adjusted to 7.0, and the flasks were sterilized by autoclave. Three flasks of the inoculated media were incubated at 28 °C on a rotary shaker at 170 rpm for 7 days to prepare the seed culture. The detailed lager fermentation procedure was as following: Fermentation was carried out on solid rice medium in 125 Fernbach flasks (500 mL, 90 mL distilled water was added to 80 g rice and kept overnight before autoclaving). Each flask was inoculated with 5.0 mL of the spore inoculum and incubated for 30 days at 28 °C in a static incubator.
Cytotoxicity Assay
Five human cancer cell lines, human myeloid leukemia HL-60, lung cancer A-549 cells, hepatocellular carcinoma SMMC-7721, breast cancer MCF-7, and colon cancer SW480, were purchased from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Cells were cultured according to the manufacturer’ recommendations. All mediums were supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin G sodium and 100 μg/ml streptomycin (HyClone). All the cells were incubated at 37 ℃, 5% CO2 in a humidified atmosphere. Cytotoxicity of compounds was determined by MTS method. Briefly, 5 × 103 cells were plated in 96-well plates 12 h before treatment and continuously exposed to test compounds for 48 h. Then MTS (Promega) was added to each well. The samples were incubated at 37 ℃ for 1–4 h and the optical density (OD) was measured at 490 nm using a microplate reader (Bio-Rad Laboratories). The IC50 values were calculated by Reed and Muench’s method [24].
Extraction and Isolation of Compounds 1–3
The culture medium was overlaid and extracted with MeOH by maceration. With filtration and concentration, the resultant extract was partitioned with EtOAc. Then the solvent was evaporated in vacuo to afford a crude extract (130 g). The extraction was subjected to column chromatography on silica gel with a CHCl3/Me2CO gradient system (1:0, 9:1, 8:2, 7:3, 6:4, 1;1, 0:1) to yield seven fractions, A-G. Fraction C (CHCl3/Me2CO 8:2, 9 g) was chromatographed on a RP-18 column with a methanol/ H2O gradient system (from 30:70 to 100:0) to afford fractions C1-C8. Fraction C2 (methanol/ H2O, 40:60, 1.4 g) was subjected to chromatography over silica gel (chloroform/ Me2CO, from 80:1 to 0:1) to yield subfractions C2/1–11, subfraction C2-9 was purified by semipreparative HPLC (3 ml/min, detector UV λmax = 195 nm, MeCN/H2O 22.5:77.5) to yield 1 (3.1 mg, tR = 18.7 min) and 2 (1.8 mg, tR = 20.2 min), subfraction C2-10 was purified by semipreparative HPLC (3 ml/min, detector UV λmax = 195 nm, MeCN/H2O 40:60) to yield 3 (3.6 mg, tR = 25.8 min).
Physical Constants and Spectroscopic Data of Compounds 1–3
Arthrinin E (1): initially obtained as a colorless oil, by using slow evaporation of methanol in a closed tube, the colorless square crystals were obtained eventually; mp: 135–140 °C; [α]22.2D: + 66.6 (MeOH, c 0.100); ECD (MeOH) λmax (Δε): 220 (0.02) nm; UV (MeOH) λmax (log ε): 204 (3.03) nm; IR (νmax): 3374, 2927, 2869, 1630, 1607, 1460, 1384, 1366, 1093, 1057, 1026, 1010, 986, 971 cm–1. HRESIMS at m/z 277.1775 ([M+Na]+, calcd for 277.1774). 1H and 13C NMR data, see Tables 1 and 3.
Arthrinin F (2): obtained as a colorless oil; [α]22.4D: + 107.00 (MeOH, c 0.060); ECD (MeOH) λmax (Δε): 205 (0.68) nm; UV (MeOH) λmax (log ε): 201 (3.14) nm; IR (νmax): 3383, 2955, 2927, 2890, 2871, 1631, 1609, 1465, 1454, 1384, 1361, 1082, 1026, 994, 978 cm–1. HRESIMS at m/z 263.1618 ([M + Na]+, calcd for 263.1618). 1H and 13C NMR data, see Tables 1 and 3.
Arthrinin G (3): isolated as a colorless oil; [α]22.8D: + 12.55 (MeOH, c 0.110); ECD (MeOH) λmax (Δε): 204 (0.21), 240 (– 0.06), 276 (0.01) nm; UV (MeOH) λmax (log ε): 208 (3.22), 241 (2.74), 283 (1.98) nm; IR (νmax): 3419, 2957, 2932, 2869, 1743, 1607, 1467, 1384, 1367, 1040, 1017, 744 cm–1. HRESIMS at m/z 285.1100 ([M+Na]+, calcd for 285.1097). 1H and 13C NMR data, see Tables 1 and 3.
Crystallographic data for the structures of arthrinin E (1, deposition number CCDC 1997674) has been deposited in the Cambridge Crystallographic Data Centre database. Copies of the data can be obtained free of charge from the CCDC at www.ccdc.cam.ac.uk.
Crystal data for1: C15H26O3, M = 254.36, a = 8.8861(2) Å, b = 8.8861(2) Å, c = 31.7909(6) Å, α = 90°, β = 90°, γ = 120°, V = 2173.98(11) Å3, T = 100.(2) K, space group P3221, Z = 6, μ(Cu Kα) = 0.629 mm–1, 29,180 reflections measured, 2838 independent reflections (Rint = 0.0291). The final R1 values were 0.0351 (I > 2σ(I)). The final wR(F2) values were 0.0969 (I > 2σ(I)). The final R1 values were 0.0353 (all data). The final wR(F2) values were 0.0970 (all data). The goodness of fit on F2 was 1.142. Flack parameter = 0.07(3).
Computational Method
Conformational searching of (1R*,2R*,4S*,5S*)-2 and (6R*,8S*)-3 were undertaken with the CREST code (version 2.8) using the default iMTD-GC procedure [25]. The first 20 conformers of (1R*,2R*,4S*,5S*)-2 and (6R*,8S*)-3 were subjected to DFT geometry optimization at B3LYP-D3BJ-SCRF/6-31G(d) level of theory (with MeOH as solvent and SMD solvent model). Frequency analyses of all optimized conformers were undertaken at the same level of theory to ensure that no imaginary frequency exists. Then, thermal correction to Gibbs free energies obtained by frequency analyses were added to the electronic energies obtained at B3LYP-D3BJ-SCRF/6–311 + G(d,p) level of theory (with MeOH as solvent and SMD solvent model) to get the Gibbs free energies of each conformer. Subsequently, Room-temperature (298.15 K) equilibrium populations were calculated according to Boltzmann distribution law:
where Pi is the population of the ith conformer; ni the number of molecules in ith conformer; ΔG is the relative Gibbs free energy (kcal/mol); T is room temperature (298.15 K) here; R is the ideal gas constant (0.0019858995). Those conformers with a population of over 2% were subjected to subsequent NMR and ECD calculations.
NMR shielding constants were calculated with the GIAO method at mPW1PW91-SCRF/6–31 + G(d,p) level (with MeOH and SMD solvent model). The obtained shielding constants were converted into chemical shifts by referencing to TMS at 0 ppm (δcal = σTMS – σcal), where the σTMS was the shielding constant of TMS calculated at the same level of theory. The parameters a and b of the linear regression δcal = aδexp + b; the correlation coefficient, R2; the mean absolute error (MAE) defined as Σn|δcal – δexp|/n; the corrected mean absolute error (CMAE) defined as Σn|δcorr – δexp|/n, where δcorr = (δcal – b)/a were calculated [26, 27]. Calculation of coupling constants were run at B972/pcJ-1 level of theory (with MeOH as solvent and SMD solvent model) [28].
TDDFT ECD calculations were run at CAM-B3LYP/def2-SVP level of theory (with MeOH as solvent and SMD solvent model) [29]. For each conformer, 30 excited states were calculated. The calculated ECD curves were generated using the Multiwfn software (version 3.7) [30].
The geometry optimization, single-point energy calculation, NMR shielding constant calculation, coupling constant calculation, and TDDFT ECD calculation were all completed in Gaussian 09 program [31].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material 1 (PDF 3960 kb)
Acknowledgements
This project was supported financially by the National Natural Science Foundation of China (No. 81874298), the CAS “Light of West China” program (Pema-Tenzin Puno), and the Yunnan Science Fund for Distinguished Young Scholars (2019FJ002).
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Barbero M, Artuso E, Prandi C. Curr. Med. Chem. 2018;25:141–185. doi: 10.2174/0929867324666170511112815. [DOI] [PubMed] [Google Scholar]
- 2.Fehlhaber HW, Geipel R, Mercker HJ, Tschesche R, Welmar K, Schoenbeck F. Chem. Ber. 1974;107:1720–1730. doi: 10.1002/cber.19741070530. [DOI] [Google Scholar]
- 3.Kimata T, Natsume M, Marumo S. Tetrahedron Lett. 1985;26:2097–2100. doi: 10.1016/S0040-4039(00)94788-9. [DOI] [Google Scholar]
- 4.Qin XD, Shao HJ, Dong ZJ, Liu JK. J. Antibiot. 2008;61:556–562. doi: 10.1038/ja.2008.74. [DOI] [PubMed] [Google Scholar]
- 5.Medina RP, Araujo AR, Batista JM, Cardoso CL, Seidl C, Vilela AFL, Domingos HV, Costa-Lotufo LV, Andersen RJ, Silva DHS. Sci. Rep. 2019;9:12318. doi: 10.1038/s41598-019-48655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan YF, Feng Y, Ren FX, Niu SB, Liu XZ, Che YS. Org. Lett. 2013;15:6050–6053. doi: 10.1021/ol402953k. [DOI] [PubMed] [Google Scholar]
- 7.Rossi FR, Garriz A, Marina M, Romero FM, Gonzalez ME, Collado IG, Pieckenstain FL. Mol. Plant Microbe Interact. 2011;24:888–896. doi: 10.1094/MPMI-10-10-0248. [DOI] [PubMed] [Google Scholar]
- 8.Temme N, Oeser B, Massaroli M, Heller J, Simon A, Collado IG, Viaud M, Tudzynski P. Mol. Plant Pathol. 2012;13:704–718. doi: 10.1111/j.1364-3703.2011.00778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krohn K, Dai JQ, Florke U, Aust HJ, Drager S, Schulz B. J. Nat. Prod. 2005;68:400–405. doi: 10.1021/np0498206. [DOI] [PubMed] [Google Scholar]
- 10.Liang CQ, Shi YM, Li XY, Luo RH, Kuhnert YE, Surup F, Wiebach V, Bernecker S, Stadler M. Phytochemistry. 2015;117:116–122. doi: 10.1016/j.phytol.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Duran-Patron R, Hernandez-Galin R, Rebordinos LG, Cantoral JM, Collado IG. Tetrahedron. 1999;55:2389–2400. doi: 10.1016/S0040-4020(99)00032-0. [DOI] [Google Scholar]
- 12.Qiao C, Zhang W, Han JC, Li CC. Org. Lett. 2016;18:4932–4935. doi: 10.1021/acs.orglett.6b02414. [DOI] [PubMed] [Google Scholar]
- 13.Qiao C, Zhang W, Han JC, Dai WM, Li CC. Tetrahedron. 2019;75:1739–1745. doi: 10.1016/j.tet.2018.11.019. [DOI] [Google Scholar]
- 14.J.R. Hanson, R. Nyfeler, J. Chem. Soc., Chem. Commun. 72–73 (1976)
- 15.Collado IG, Sanchez AJM, Hanson JR. Nat. Prod. Rep. 2007;24:674–686. doi: 10.1039/b603085h. [DOI] [PubMed] [Google Scholar]
- 16.Moraga J, Dalmais B, Izquierdo-Bueno I, Aleu J, Hanson JR, Hernandez-Galan R, Viaud M, Collado IG. ACS Chem Biol. 2016;11:2838–2846. doi: 10.1021/acschembio.6b00581. [DOI] [PubMed] [Google Scholar]
- 17.Toghueo RMK. Nat. Prod. Bioprospect. 2019;9:311–328. doi: 10.1007/s13659-019-00220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang JW, Kong LM, Zu WY, Hu K, Li XN, Yan BC, Wang WG, Sun HD, Li Y, Puno PT. Org. Lett. 2019;21:771–775. doi: 10.1021/acs.orglett.8b04020. [DOI] [PubMed] [Google Scholar]
- 19.Yan BC, Wang WG, Hu DB, Sun X, Kong LM, Li XN, Du X, Luo SH, Liu Y, Li Y, Sun HD, Pu JX. Org. Lett. 2016;18:1108–1111. doi: 10.1021/acs.orglett.6b00214. [DOI] [PubMed] [Google Scholar]
- 20.Chen BS, Li EW, Liu L, Liao MF, Zhu ZX, Zhuang WY, Bao L, Liu HW. Planta Med. 2018;84:1055–1063. doi: 10.1055/a-0593-6030. [DOI] [PubMed] [Google Scholar]
- 21.Ebada SS, Schulz B, Wray V, Totzke F, Kubbutat MHG, Mueller WEG, Hamacher A, Kassack MU, Lin W, Proksch P. Bioorg. Med. Chem. 2011;19:4644–4651. doi: 10.1016/j.bmc.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Feng T, Li ZH, Dong ZJ, Su J, Li Y, Liu JK. Nat. Prod. Bioprospect. 2011;1:29–32. doi: 10.1007/s13659-011-0005-9. [DOI] [Google Scholar]
- 23.Bao YR, Chen GD, Gao H, He RR, Wu YH, Li XX, Hu D, Wang CX, Liu XZ, Li Y, Yao XS. RSC Adv. 2015;5:46252–46259. doi: 10.1039/C5RA07122D. [DOI] [Google Scholar]
- 24.Gao ZH, Kong LM, Zou XS, Shi YM, Shang SZ, Luo HR, Liang CQ, Li XN, Li Y, Du X, Xiao WL, Sun HD. Nat. Prod. Bioprospect. 2013;2:249–254. doi: 10.1007/s13659-012-0082-4. [DOI] [Google Scholar]
- 25.Pracht P, Bohle F, Grimme S. Phys. Chem. Chem. Phys. 2020;22:7169–7192. doi: 10.1039/C9CP06869D. [DOI] [PubMed] [Google Scholar]
- 26.Lodewyk MW, Siebert MR, Tantillo DJ. Chem. Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 27.Willoughby PH, Jansma MJ, Hoye TR. Nat. Protoc. 2014;9:643–660. doi: 10.1038/nprot.2014.042. [DOI] [PubMed] [Google Scholar]
- 28.Jensen F. Theor. Chem. Acc. 2010;126:371–382. doi: 10.1007/s00214-009-0699-5. [DOI] [Google Scholar]
- 29.Pescitelli G, Bruhn T. Chirality. 2016;28:466–474. doi: 10.1002/chir.22600. [DOI] [PubMed] [Google Scholar]
- 30.Lu T, Chen F. J. Comput. Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision E.01. Wallingford, CT: Gaussian Inc; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electronic supplementary material 1 (PDF 3960 kb)