

Abstract
The goal of this study was to determine the validity of using N6-etheno-bridged adenine nucleotides to evaluate ecto-nucleotidase activity. We observed that the metabolism of N6-etheno-ATP versus ATP was quantitatively similar when incubated with recombinant CD39, ENTPD2, ENTPD3, or ENPP-1, and the quantitative metabolism of N6-etheno-AMP versus AMP was similar when incubated with recombinant CD73. This suggests that ecto-nucleotidases process N6-etheno-bridged adenine nucleotides similarly to endogenous adenine nucleotides. Four cell types rapidly (t1/2, 0.21 to 0.66 h) metabolized N6-etheno-ATP. Applied N6-etheno-ATP was recovered in the medium as N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and surprisingly N6-etheno-adenine; intracellular N6-etheno compounds were undetectable. This suggests minimal cellular uptake, intracellular metabolism, or deamination of these compounds. N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine had little affinity for recombinant A1, A2A, or A2B receptors, for a subset of P2X receptors (3H-α,β-methylene-ATP binding to rat bladder membranes), or for a subset of P2Y receptors (35S-ATP-αS binding to rat brain membranes), suggesting minimal pharmacological activity. N6-etheno-adenosine was partially converted to N6-etheno-adenine in four different cell types; this was blocked by purine nucleoside phosphorylase (PNPase) inhibition. Intravenous N6-etheno-ATP was quickly metabolized, with N6-etheno-adenine being the main product in naïve rats, but not in rats pretreated with a PNPase inhibitor. PNPase inhibition reduced the urinary excretion of endogenous adenine and attenuated the conversion of exogenous adenosine to adenine in the renal cortex. The N6-etheno-bridge method is a valid technique to assess extracellular metabolism of adenine nucleotides by ecto-nucleotidases. Also, rats express an enzyme with PNPase-like activity that metabolizes N6-etheno-adenosine to N6-etheno-adenine.
Electronic supplementary material
The online version of this article (10.1007/s11302-020-09699-x) contains supplementary material, which is available to authorized users.
Keywords: N6-etheno-ATP, Ecto-nucleotidases, Purine nucleoside phosphorylase, 8-Aminoguanine
Introduction
Most cells release ATP into the extracellular compartment [1–4], and extracellular ATP is converted to ADP, AMP, and adenosine on the surface of cells by various ecto-nucleotidases [5–11]. Since ATP and its metabolites activate cell surface purinergic receptors [12–21], the extracellular metabolism of ATP by ecto-nucleotidases is of considerable interest to researchers who study immunology, cancer biology, inflammation, rheumatoid arthritis, and physiology/pharmacology of the heart, vasculature, kidneys, bladder, gut, peripheral nervous system, central nervous system, and eye (not an exhaustive list) [3, 4, 22–38]. Moreover, because the metabolism of extracellular ATP is a major contributing factor to vascular calcification [39–42], the function of ecto-nucleotidases is an important determinant of atherosclerosis risk.
Evaluating the extracellular metabolism of ATP by ecto-nucleotidases in cells, tissues, and intact organs is, for several reasons, challenging. Two important analytical challenges are assay sensitivity and specificity. For example, accurate measurement of adenine nucleotides and nucleosides using high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection generally requires the injection of greater than 300 nmol/L of these purines to obtain adequate signal-to-noise ratios [43]. Also, biological samples contain numerous other compounds that compromise the specificity of HPLC-UV quantitation of ATP and its metabolites. Although these limitations can be overcome for AMP and adenosine using HPLC coupled to mass spectrometry [33], this method is expensive. Moreover, in our experience, solvent systems that provide sharp HPLC chromatographic peaks for ATP and ADP suppress ionization in the ion source of mass spectrometers (thus reducing sensitivity), and solvent systems that do not attenuate ion production yield poor chromatographic peak morphologies (broad peaks with tailing) for ATP and ADP (thus reducing both sensitivity and specificity). Consequently, optimizing HPLC-mass spectrometry conditions for analysis of ATP, ADP, AMP, and adenosine in a single analytical run is problematic.
Another issue is cellular uptake followed by intracellular metabolism and release. Purine nucleotide and nucleoside transporters may convey extracellular adenine-based compounds to the intracellular compartment, where they can be converted to other compounds that may then exit the cell [44]. Thus, whether metabolism of added ATP and its metabolites is occurring on or within cells cannot be readily established. Yet another confounding factor is that AMP and adenosine can be diverted to other metabolic pathways via deamination by AMP [45] and adenosine [46] deaminases, which complicates interpretation with respect to the activity of ecto-nucleotidases. Finally, since ATP, ADP, AMP, and adenosine can activate purinergic receptors, the application of these compounds to cells and tissues may per se perturb, via signal transduction pathways, the very metabolic pathways under study.
A method that may circumvent the aforementioned technical issues is to employ adenine-based compounds modified with an “etheno bridge” connecting the N6 nitrogen with the nitrogen in the 1 position of the adenine structure (Fig. 1). Creation of the etheno bridge yields adenine nucleotide/nucleoside derivatives that can be monitored by fluorescence detection, thus providing improved sensitivity and selectivity compared with UV detection. Indeed, in 1984, Levitt et al. reported the HPLC-fluorometric method of quantifying adenosine and adenine nucleotides by treating samples with chloroacetaldehyde to form the corresponding etheno-bridged derivatives before HPLC analysis [47], a method that has been refined [48] and used extensively; for examples, see [49–51]. Because the etheno bridge is remote from the ribose, it is possible that the dephosphorylation of etheno-bridged adenine nucleotides by ecto-nucleotidases may proceed with efficiency close to that for native adenine nucleotides. Also, since the etheno bridge connects with the N6 nitrogen of the adenine structure, the etheno bridge likely would block deamination reactions. Moreover, because the purine moiety is critically important for biological activity, it is conceivable that the etheno bridge would prevent interactions of etheno-bridged adenine nucleotides with transporters and receptors. It is not surprising, therefore, that a number of studies used etheno-bridged adenine nucleotides to evaluate the role of ecto-nucleotidases in the metabolism of extracellular adenine nucleotides. For example, this method has been employed to study ecto-nucleotidases in the guinea-pig vas deferens [52], intestinal epithelial cells [53], human microvascular endothelial cells [54], canine mesenteric artery and vein [55], the Langendorff heart preparation [56], the rat vas deferens [57], regulatory T cells [58], melanoma cells [59], macrophages [60], and epicardium-derived cells [61].
Fig. 1.

Chemical structures of N6-etheno derivatives of ATP, ADP, AMP, adenosine (ADO), and adenine (ADE) and the pathways of ATP metabolism by ecto-nucleotidases. Note the etheno bridge (red) connecting the N6 nitrogen with the nitrogen in the 1 position of the adenine structure
Despite the use of etheno-bridged adenine nucleotides for studying ecto-nucleotidases, the method has not been adequately validated. To our knowledge, it remains to be shown that purified (i.e., recombinant) ecto-nucleotidases metabolize etheno-adenine nucleotides versus naturally occurring adenine nucleotides with similar efficiency. Also, whether etheno-adenine nucleotides are mostly restricted to the extracellular compartment, are not subjected to reactions other than ecto-nucleotidase-mediated dephosphorylation, do not interact with purinergic receptors, have minimal biological activity, and can be used to monitor the full pathway of extracellular ATP metabolism remain open questions. Accordingly, here we carefully evaluated the utility of the etheno-bridge method for studying extracellular ATP processing by ecto-nucleotidases and confirmed its validity in this regard.
During the course of this evaluation, we made the unexpected observation that in cultured cells, isolated perfused kidneys, and in vivo, N6-etheno-adenosine was converted to N6-etheno-adenine. Moreover, we observed that although recombinant mammalian purine nucleoside phosphorylase (PNPase) did not metabolize N6-etheno-adenosine or adenosine, the conversion of N6-etheno-adenosine to N6-etheno-adenine could not be attributed to microbiological contamination (a potential source of bacterial PNPase), yet was abrogated by two structurally distinct and potent inhibitors of PNPase. These finding suggest that in intact cells some forms of mammalian PNPase may metabolize extracellular N6-etheno-adenosine to N6-etheno-adenine.
Materials and methods
Animals
Adult (12- to 14-week-old) male normotensive Wistar–Kyoto rats were obtained from Charles River Laboratories (Wilmington, MA). The Institutional Animal Care and Use Committee approved all procedures. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (8th ed., 2011).
Analytical methods
N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine were measured by HPLC analysis using an Agilent (Santa Clara, CA) HPLC system which included a HP Agilent Technologies 1100 series HPLC chromatograph equipped with an Agilent 1260 Infinity fluorescence detector (G1321B). Cell culture, microdialysate, urine samples, and plasma samples were heated to 90 °C for 3 min (to inactivate enzymes), vortexed, and centrifuged at 14,000 rpm for 20 min at 4 °C, and the supernatants were collected. Urine and plasma samples were diluted 1:20 and 1:1, respectively, with buffer A (0.2 M KH2PO4) and all samples were transferred into HPLC vials. Aliquots of samples (plasma, 8 μL; urine, 2 μL; microdialysate and cell culture, 10 μL) were injected onto a C-18 reverse phase column (Agilent Eclipse Plus C18, 5 μm, 4.6 × 250 mm) which was protected by a guard cartridge. N6-etheno-purines were separated by HPLC in gradient mode [buffer A—0.2 M KH2PO4 in water; buffer B—0.2 M KH2PO4 in 15% acetonitrile; linear gradient (% B)—at 0 min 5.0%; from 0 to 5 min, 5.0%; from 5 to 30 min, 100.0%; from 30 to 40 min, 100.0%; from 40 to 41 min, 5.0%; from 41 to 50 min, 5.0%]. The flow rate was 1.0 mL/min. Fluorescence (FL) of adenine nucleotides in the eluate was monitored at an emission of 420 nm, after excitation at 280 nm. Chromatograms were processed and stored in digital form with Agilent OpenLAB CDS software. Standard curves were generated from authentic N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine (BioLog Life Science Institute, Hayward, CA, catalog numbers E 004, E 007, E 005, E 011, and E 012, respectively).
Metabolism of N6-etheno-purines by recombinant ecto-nucleotidases
Recombinant human CD39 (rhCD39), recombinant human CD73 (rhCD73), recombinant human ecto-nucleotide pyrophosphatase/phosphodiesterase family member 1 (rhENPP-1), recombinant human ectonucleoside triphosphate diphosphohydrolase family member 2 (rhENTPD2), and recombinant human ectonucleoside triphosphate diphosphohydrolase family member 3 (rhENTPD3) were obtained from R&D Systems (Minneapolis, MN; catalog numbers 4397-EN-010, 5795-EN-010, 6136-EN-010, 6087-EN-010, and 4400-EN-010, respectively). The ecto-nucleotidases catalyze the following reactions: CD39, ENTPD2, and ENTPD3 metabolize ATP to ADP + orthophosphate (Pi) and ADP to AMP + Pi, with CD39 having a greater catalytic efficiency, compared to ENTPD2 and ENTPD3, for converting ADP to AMP [5]; CD73 metabolizes AMP to adenosine + Pi [5]; and ENPP-1 metabolizes ATP to AMP + pyrophosphate (PPi) [5]. The specific activities (pmol/min/μg) reported by R&D Systems for the recombinant enzymes were as follows: for rhCD39, rhENTPD2, and rhENTPD3, > 5000, > 5000, and > 70,000, respectively, as assessed by measuring Pi production from ATP; for rhENPP-1, > 35,000, as assessed by measuring metabolism of thymidine 5′-monophosphate p-nitrophenyl ester; and for rhCD73, > 15,000, as assessed by measuring Pi production from AMP.
To determine whether these recombinant ecto-nucleotidases can metabolize N6-etheno-purines, we incubated (30 min at 30 °C) N6-etheno-ATP (1 μmol/L) with rhCD39 (20 ng in 50 mmol/L HEPES at pH 7.4 with 5 mmol/L of CaCl2), rhENPP-1 (80 ng in 125 mmol/L NaCl with 25 mmol/L Tris at pH 7.5), rhENTPD2 (40 ng in 50 mmol/L HEPES at pH 7.4 with 5 mmol/L of CaCl2), or rhENTPD3 (11 ng in 50 mmol/L HEPES at pH 7.4 with 5 mmol/L of CaCl2). We also incubated (30 min at 30 °C) N6-etheno-AMP (1 μmol/L) with rhCD73 (40 ng in 25 mmol/L Tris at pH 7.5 with 5 mmol/L of MgCl2). After the incubations, N6-etheno-purines were measured by HPLC-FL. We selected 30 min at 30 °C as the incubation time and temperature because longer times and higher temperatures can lead to enzyme degradation. The amount of each recombinant ecto-nucleotidase was adjusted to completely metabolize the N6-etheno-purines within the 30-min time frame.
To directly compare the catalytic efficiency of metabolism of N6-etheno-ATP with ATP, 1 μmol/L of N6-etheno-ATP and 1 μmol/L ATP were incubated head-to-head in separate tubes for 5 min at 30 °C with either rhCD39, rhENPP-1, rhENTPD2, or rhENTPD3, and the concentrations of N6-etheno-ATP and its downstream metabolites were determined directly (as described above), whereas the concentrations of ATP and its downstream metabolites were determined after conversion to their corresponding etheno-bridge derivatives by incubation (80 °C for 20 min) with 2-chloroacetaldehyde. In these experiments, the amount of recombinant enzyme was adjusted to catalyze only partial metabolism of the substrate. A similar experimental design was used to test the conversion of N6-etheno-AMP versus AMP to adenosine by rhCD73. In additional experiments with CD39 (10 ng) and CD73 (0.25 ng), we employed a range of high concentrations of substrates (25 to 200 μmol/L for 10 min at 30 °C) in order to evaluate initial reaction velocities (i.e., at a constant substrate concentration) versus substrate concentrations.
Culture of rat cardiac fibroblasts
Rat cardiac fibroblasts (CFs) were isolated, cultured, and characterized as described by us [62, 63]. Briefly, left cardiac ventricles were removed and minced, and the minced sections were washed with DMEM and treated with collagenase. After collagenase digestion, the dissociated cells were centrifuged, and the pellet was suspended in complete culture medium (DMEM/F12 supplemented with HEPES 25 mmol/L) containing 20% fetal bovine serum (FBS). Cells were plated in tissue culture flasks (75 cm2) and incubated under standard tissue culture conditions (37 °C, 5% CO2/95% air, and 98% humidity). CFs were then purified by selective plating.
Culture of rat preglomerular vascular smooth muscle cells
Rat preglomerular vascular smooth muscle cells (PGVSMCs) were isolated, cultured, and characterized as recently described by us [64]. Briefly, iron oxide particles (5% suspension in DMEM) were infused into the suprarenal aorta of anesthetized rats. Kidneys were removed and decapsulated, and the cortical tissue was obtained and placed in DMEM containing antibiotics, fungicidals, and HEPES. The cortical tissue was minced and then dispersed using a wire mesh to isolate the microvessel-containing fraction. The microvessel fraction was washed multiple times with ice-cold supplemented DMEM, and a magnet was used to retain the iron-laden vessels after each wash. The microvessel-containing fraction was digested with collagenase and passed through a 20-gauge hypodermic needle to shear off glomeruli. The arteriolar fraction retained after sieving through an 80-μm mesh was suspended in DMEM supplemented with 20% FBS, plated, and incubated at 37 °C in 5% CO2–95% air and 98% humidity. PGVSMCs were repeatedly subjected to selective plating to eliminate residual fibroblast contamination.
Culture of rat glomerular mesangial cells
Rat glomerular mesangial cells (GMCs) were isolated, cultured, and characterized as previously described by us [65]. Briefly, kidneys were obtained from rats and glomeruli were isolated from renal cortical tissue with stainless steel screens of three different pore sizes. The isolated glomeruli were incubated at 37 °C for 20 min in phosphate-buffered saline (PBS) containing 0.5 mg/mL collagenase. After washing several times in PBS, the glomeruli were resuspended in 10 mL RPMI 1640 medium supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), NaHCO3 (2200 mg/L), HEPES (25 mmol/L), and 20% FBS, plated in 75 cm2 tissue culture flasks, and incubated under standard tissue culture conditions (37 °C, 5% CO2/95% air, and 98% humidity). After 2 to 3 days, glomerular epithelial cells started to grow and were numerically dominant for another 5 to 7 days. After this period, the epithelial cells deteriorated for several days and mesangial cells became the prevailing cell type and grew to confluence within 4 to 5 weeks.
Culture of rat renal microvascular endothelial cells
Eight rat kidneys were decapsulated and the cortices were removed, combined, and minced. The mince was passed through a 200-μm mesh screen, and the glomeruli with attached afferent and efferent arterioles were collected from the top of the mesh. This material was placed in a sterile tube containing 1.8 mg/mL of collagenase dissolved in DMEM/F12, and the sample was incubated in a shaking water bath at 37 °C until digestion was complete. Next, the digested sample was filtered through a 70-μm nylon mesh and centrifuged. The pellet was resuspended in DMEM/F12 supplemented with the Lonza Walkersville EGM-2-MV Bulletkit (purchased from ThermoFisher Scientific, Waltham MA), and incubated in a tissue culture plate for 1 h to allow fibroblasts to attach. The medium with suspended cells was collected and placed in another sterile tube. Dynabeads from the Pan Mouse IgG kit (ThermoFisher Scientific; catalog number 11-531-D) coated with mouse anti-rat CD31 (BD Biosciences, San Jose, CA; catalog number 555027) were added to the mixture. The vascular endothelial cells were allowed to attach to the beads, and the beads were then collected with a magnet and nonattached cells were discarded. The beads, now coated with vascular endothelial cells, were then plated in a flask with EGM-2-MV and the cells were allowed to proliferate off the beads.
Testing for Mycoplasma contamination
Cells were tested by PCR followed by agarose gel analysis for bacterial contamination using the Universal Mycoplasma Detection Kit (catalog number 30-1012K; ATCC, Manassas, VA) as directed by the manufacturer. This method detects over 60 species of Mycoplasma, Acholeplasma, Spiroplasma, and Ureaplasma including the eight species most likely to contaminate cell cultures: M. arginini, M. fermentans, M. hominis, M. hyorhinis, M. orale, M. pirum, M. salivarium, and A. laidlawii.
Isolated perfused rat kidney
A Hugo Sachs Elektronik-Harvard Apparatus GmbH (March-Hugstetten, Germany) kidney perfusion system was perfused with 75% ethanol for 1.5 h, followed by perfusion with sterile water for 1.5 h. Then the perfusate was switched to Tyrode’s solution (in mmol/L: NaCl, 137.0; KCl, 2.7; CaCl2, 1.8; MgCl2, 1.1; NaHCO3, 12.0; NaH2PO4, 0.42; and D(+)-glucose, 5.6) that was filtered to 0.22 μm to remove any bacteria. The perfusion rate was 5 mL/min (constant flow) and the Tyrode’s solution was oxygenated (95% O2/5% CO2). Next, rats were anesthetized with thiobutabarbital (100 mg/kg, intraperitoneal injection), and kidneys were isolated under sterile conditions (sterile gloves and instruments) and transferred with minimal interruption of perfusion to the perfusion apparatus. The kidney perfusion system included the following components: Model UP 100 Universal Perfusion System; Model ISM 834 Channel Reglo Digital Roller Pump; a glass double-walled perfusate reservoir maintained at 37 °C and oxygenated; a R 120144 glass-oxygenator maintained at 37 °C; mechanical integration of the oxygenator with the Universal Perfusion System UP 100; a Windkessel for absorption of pulsations; an inline holder for disc particle filters (80 μm); a temperature-controlled plexiglass kidney chamber integrated with the UP 100; and a thermostatic circulator. The plexiglass chamber contained a heat exchanger to maintain the temperature of the perfusate at 37 °C at the point of entry into the tissue, and also contained a device to extract bubbles from the perfusate just before the perfusate entered the kidney. Perfusion pressure was monitored using a PM-4 Perfusion Pressure Monitor (Living Systems Instrumentation, St. Albans, VT) and recorded using LabChart software (ADInstruments, Colorado Springs, CO).
In vivo experiments
After inducing anesthetized with thiobutabarbital (90 mg/kg, i.p.), each rat was positioned on an isothermal platform, and a thermometer probe was inserted into the rectum, and the rat’s body temperature was continuously monitored and maintained at 37 °C by adjusting the distance of a heat lamp from the animal’s body. Using a polyethylene (PE)-240 tubing, the trachea was cannulated to aid respiration. Next, a PE-50 cannula was inserted into the carotid artery and connected to a digital blood pressure analyzer (Micro-Med, Inc., Louisville, KY) for continuous measurement of mean arterial blood pressure (MABP) and heart rate (HR). A PE-50 cannula was placed in the jugular vein, and an infusion of 0.9% saline (100 μL/min) was begun, and a 1-mm transit-time flow probe was positioned on the left renal artery and connected to a flowmeter (model T402, Transonic Systems, Inc., Ithaca, NY) for continuous measurement of renal blood flow (RBF). A microdialysis probe was inserted into the renal cortex of the right kidney and was perfused at 2 μL/min with 0.9% saline. The microdialysis probes were from Bioanalytical Systems (West Lafayette, IN; CMA/20 microdialysis probe 4 mm; outer diameter of 0.5 mm; 20,000 Da membrane cut-off). Finally, a PE-10 cannula was inserted into the left ureter for timed collections of urine. After a 30-min stabilization period, the experimental protocol was begun. Urine and microdialysate were collected for 10 min, and a 0.5-mL blood sample (in heparin) was taken and immediately centrifuged at 4 °C to obtain plasma. Next, N6-etheno-ATP was infused intravenously at 0.1 μmol/kg/min for 30 min. Urine and microdialysate were collected over each 10-min interval, and plasma samples were obtained just before and 30 min after stopping the infusion of N6-etheno-ATP. Urine, microdialysate, and plasma samples were stored at − 80 °C until analyzed for N6-etheno-purines by HPLC-FL.
In another set of in vivo experiments, rats were prepared similarly to those described above except that a flow probe was not placed on the renal artery and a microdialysis probe was inserted into the renal cortex of both the right and left kidneys. After a 1-h stabilization period, microdialysate was collected for 45 min, and then the following drugs were added to the microdialysis perfusate of both kidneys: 10 μmol/L of erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA; to block adenosine deaminase), 0.1 μmol/L of 5-iodotubericidin (IDO; to block adenosine kinase), and 10 μmol/L of S-(4-nitrobenzyl)-6-thioinosine (NBTI; to block adenosine uptake). After 15 min for the drugs to reach the microdialysate probe tip, microdialysate was again collected for another 45 min. Next, in addition to the drugs listed above, adenosine (30 μmol/L) was added to the left kidney perfusate and adenosine (30 μmol/L) plus forodesine (5 μmol/L) was added to the right kidney perfusate. After 15 min, another 45-min collection of microdialysate was obtained. The microdialysate samples were assayed for adenine using ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) as previously described by us [66].
Purinergic receptor binding studies
The binding of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine to a panel of purinergic receptors was tested by Eurofins Panlabs Discovery Services (Taipei, Taiwan, R.O.C.) using classical competitive receptor binding experiments as described below. Each N6-etheno-purine was tested in triplicate at 10 μmol/L to determine the degree to which the N6-etheno-purine displaced the radiolabeled ligand from its cognate receptor.
A1 receptors
Source, human recombinant CHO-K1 cells; ligand, 1.0 nmol/L of 3H-DPCPX (Kd of 1.40 nmol/L); incubation time and temperature, 90 min at 25 °C; incubation buffer, HEPES (20 mmol/L, pH 7.4) + MgCl2 (10 mmol/L) + NaCl (100 mmol/L); assessment of nonspecific binding, 100 μmol/L of R(−)-PIA; specific binding, 85%; Bmax, 2.70 pmol/mg protein.
A2A receptors
Source, human recombinant HEK-293 cells; ligand, 50 nmol/L of 3H-CGS-21680 (Kd of 64 nmol/L); incubation time and temperature, 90 min at 25 °C; incubation buffer, Tris-HCl (50 mmol/L, pH 7.4) + MgCl2 (10 mmol/L) + EDTA (1 mmol/L) + adenosine deaminase (2 units); assessment of nonspecific binding, 50 μmol/L of NECA; specific binding, 85%; Bmax, 7.0 pmol/mg protein.
A2B receptors
Source, human recombinant HEK-293 cells; ligand, 1.60 nmol/L of 3H-MRS1754 (Kd of 0.77 nmol/L); incubation time and temperature, 90 min at 25 °C; incubation buffer, Tris-HCl (50 mmol/L, pH 6.5) + MgCl2 (5 mmol/L) + EDTA (1 mmol/L) + bacitracin (0.01%); assessment of nonspecific binding, 100 μmol/L of NECA; specific binding, 77%; Bmax, 6.20 pmol/mg protein.
Purinergic P2X receptors
Source, Wistar rat urinary bladder membranes; ligand, 3.0 nmol/L of 3H-α,β-methylene-ATP (Kd of 2.60 nmol/L); incubation time and temperature, 120 min at 4 °C; incubation buffer, Tris-HCl (50 mmol/L, pH 7.4) + leupeptin (1 μg/mL) + pepstatin A (1 μmol/L) + trypsin inhibitor (10 μg/mL) + bacitracin (0.1 mg/mL) + aprotinin (0.02 mg/mL); assessment of nonspecific binding, 10 μmol/L of α,β-methylene-ATP; specific binding, 90%; Bmax, 3.65 pmol/mg protein.
Purinergic P2Y receptors
Source, Wistar rat brain membranes; ligand, 0.10 nmol/L of 35S-ATP-αS (Kd of 2.60 nmol/L); incubation time and temperature, 60 min at 4 °C; incubation buffer, Tris-HCl (50 mmol/L, pH 7.4); assessment of nonspecific binding, 10 μmol/L of ADP-βS; specific binding, 90%; Bmax, 16 pmol/mg protein.
Statistics
Statistical analysis was performed using unpaired Student’s t test (equal or unequal variances as appropriate) or 2-factor repeated measures analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) test if the effects in the ANOVA were significant. The criterion of significance was P < 0.05. For values less than the assay detection limit, statistics were performed with such values set either to zero or the assay detection limit; however, the statistical significance was the same either way. For some graphs, data are presented as scatter plots in which every data point is represented as well as the means ± standard deviations. In some graphs, however, the data points are so similar that the symbols from individual points would be superimposed. In this case, only the means ± standard deviations are presented; however, in some cases, the standard deviation lies within the symbol. The results for in vivo studies are presented as means ± SEM.
Results
Figure 2 shows typical standard curves for all five N6-etheno-purines, as well as a typical HPLC-FL chromatogram obtained by injecting a solution containing N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine. Note that for all of the N6-etheno-purines, standard curves were highly linear with R2 values of 0.999 and detection limits less than 1 pmol. As illustrated, this HPLC configuration provided baseline separation of all five N6-etheno-purines. Also, injections of 2, 4, and 8 pmol of all five compounds (n = 6 for each compound at each amount) gave average assay results close to the nominal amounts with coefficients of variation of less than 2% (Table 1).
Fig. 2.

Line graphs show typical standard curves for a N6-etheno-ATP, b N6-etheno-ADP, c N6-etheno-AMP, d N6-etheno-adenosine (ADO), and e N6-etheno-adenine (ADE) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. Chromatogram (f) shows the baseline separation of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenine, and N6-etheno-adenosine using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section
Table 1.
Summary of accuracy
| N6-etheno-purine | Amount detected by assay (mean of n = 6) | Standard deviation | Coefficient of variation (%) |
|---|---|---|---|
| Amount injected on column = 2 pmol | |||
| N6-etheno-ATP | 2.00 | 0.02 | 0.95 |
| N6-etheno-ADP | 2.00 | 0.02 | 0.79 |
| N6-etheno-AMP | 1.99 | 0.02 | 1.01 |
| N6-etheno-ADO | 2.04 | 0.04 | 1.97 |
| N6-etheno-ADE | 2.01 | 0.02 | 1.02 |
| Amount injected on column = 4 pmol | |||
| N6-etheno-ATP | 4.00 | 0.03 | 0.75 |
| N6-etheno-ADP | 4.00 | 0.03 | 0.71 |
| N6-etheno-AMP | 4.00 | 0.03 | 0.66 |
| N6-etheno-ADO | 3.97 | 0.02 | 0.55 |
| N6-etheno-ADE | 4.01 | 0.01 | 0.66 |
| Amount injected on column = 8 pmol | |||
| N6-etheno-ATP | 8.10 | 0.06 | 0.08 |
| N6-etheno-ADP | 8.10 | 0.07 | 0.08 |
| N6-etheno-AMP | 8.10 | 0.06 | 0.08 |
| N6-etheno-ADO | 7.99 | 0.08 | 0.10 |
| N6-etheno-ADE | 8.05 | 0.05 | 0.07 |
To be a useful assay system to track the metabolism of extracellular ATP by ecto-nucleotidases, N6-etheno-purines should be substrates for well-defined ecto-nucleotidases of interest. To test this, we obtained recombinant forms of five representative and widely expressed ecto-nucleotidases known to be involved in the extracellular metabolism of adenine nucleotides [5]. As illustrated in Fig. 3a when N6-etheno-ATP was incubated for 30 min at 30 °C in the absence of ecto-nucleotidases, the only chromatographic peak observed was intact N6-etheno-ATP. Similarly, when N6-etheno-AMP was incubated for 30 min at 30 °C in the absence of ecto-nucleotidases, the only chromatographic peak observed was intact N6-etheno-AMP (Fig. 3b). These findings indicated that N6-etheno-ATP and N6-etheno-AMP were chemically stable under these test conditions; therefore, any changes in the presence of an ecto-nucleotidase could be attributed to the enzymatic action of the added ecto-nucleotidase. In this regard, when N6-etheno-ATP was incubated for 30 min at 30 °C in the presence of either 20 ng of rhCD39 (Fig. 3c), 80 ng of rhENPP-1 (Fig. 3d), 40 ng of rhENTPD2 (Fig. 3e), or 11 ng of rhENTPD3 (Fig. 3f), N6-etheno-ATP was converted to N6-etheno-AMP. This is consistent with the known facts that CD39, ENTPD2, and ENTPD3 convert ATP to ADP and then ADP to AMP and that ENPP-1 metabolizes ATP directly to AMP [5]. Although it is known that CD39, compared with ENTPD2 and ENTPD3, more efficiently converts ADP to AMP, under the assay conditions here all three ecto-nucleotidases converted, essentially quantitatively, N6-etheno-ATP to N6-etheno-AMP. CD73 is an ecto-5′-nucleotidase that metabolizes AMP to adenosine [5]. As shown in Fig. 3g , when incubated (30 min at 30 °C) in the presence of rhCD73 (40 ng), all of the N6-etheno-AMP was recovered as N6-etheno-adenosine. In these experiments, we selected 30 min at 30 °C as the incubation time and temperature because longer times and higher temperatures can lead to enzyme degradation. The amount of each recombinant ecto-nucleotidase used in these experiments was adjusted to completely metabolize the N6-etheno-purines within the 30-min time frame.
Fig. 3.

When N6-etheno-ATP (a) and N6-etheno-AMP (b) were incubated for 30 min at 30 °C in the absence of ecto-nucleotidases, the only chromatographic peaks observed were intact N6-etheno-ATP and intact N6-etheno-AMP, respectively, thus indicating that N6-etheno-ATP and N6-etheno-AMP were chemically stable under these test conditions. When N6-etheno-ATP was incubated for 30 min at 30 °C in the presence of either 20 ng of rhCD39 (c), 80 ng of rhENPP-1 (d), 40 ng of rhENTPD2 (e), or 11 ng of rhENTPD3 (f), N6-etheno-ATP was essentially quantitatively converted to N6-etheno-AMP. When N6-etheno-AMP (g) was incubated for 30 min at 30 °C in the presence of rhCD73 (40 ng), all of the N6-etheno-AMP was recovered as N6-etheno-adenosine (ADO)
The aforementioned experiments demonstrate that well-defined ecto-nucleotidases that process extracellular adenine nucleotides can efficiently metabolize N6-etheno-adenine nucleotides. To investigate whether the efficiencies of these ecto-nucleotidases for processing very low concentrations of N6-etheno-adenine nucleotides are similar to the efficiencies of these enzymes for metabolizing very low (physiological) levels of naturally occurring adenine nucleotides, we incubated the five recombinant ecto-nucleotidases for 5 min at 30 °C with either their natural substrates (1 μmol/L) or the corresponding etheno-bridged substrates (1 μmol/L). Since in these experiments we used very low levels of substrates, the amount of ecto-nucleotidases employed was adjusted such that the metabolism of the natural substrates was only partial so that the enzymatic efficiencies of the ecto-nucleotides for natural substrates versus etheno-bridged substrates could be directly compared. After 5 min, the percent of substrate remaining and the percent conversion to products were determined by HPLC-FL. In this regard, the levels of N6-etheno-adenine nucleotides/nucleosides were assessed directly by HPLC-FL, whereas the concentrations of ATP and its downstream metabolites were determined after conversion to their corresponding etheno-bridged derivatives by incubation (80 °C for 20 min) with 2-chloroacetaldehyde. rhENPP-1 converted ATP to AMP and N6-etheno-ATP to N6-etheno-AMP with similar efficiency (Fig. 4a); rhENTPD2 metabolized ATP to ADP and N6-etheno-ATP to N6-etheno-ADP with similar efficiency (Fig. 4b); rhENTPD3 metabolized ATP to ADP plus AMP and N6-etheno-ATP to N6-etheno-ADP plus N6-etheno-AMP with similar efficiency (Fig. 4c); and rhCD73 converted AMP to adenosine and N6-etheno-AMP to N6-etheno-adenosine with similar efficiency (Fig. 4d). As shown in Fig. 4e , rhCD39 also converted ATP to ADP and N6-etheno-ATP to N6-etheno-ADP with similar efficiency. Overall, these data show that rhENPP-1, rhENTPD2, rhENTPD3, rhCD73, and rhCD39 process natural adenine nucleotides versus N6-etheno-adenine nucleotides with similar efficiency when substrate concentrations are low.
Fig. 4.

Scatter plots show the percentage (%) of applied substrate (either the natural adenine nucleotide substrate or the corresponding etheno-bridged adenine nucleotide substrate, both at 1 μmol/L) that remained or was recovered as product (either the natural product or corresponding etheno-bridged product) after incubation (5 min at 30 °C) with recombinant human (rh) ecto-nucleotidases a rhENPP-1, b rhENTPD2, c rhENTPD3, d rhCD73, or e rhCD39. For each ecto-nucleotidase, the amount of enzyme incubated with substrate was selected to only partially metabolize the natural adenine nucleotide substrate. eATP = N6-etheno-ATP; eADP = N6-etheno-ADP; eAMP = N6-etheno-AMP; eADO = N6-etheno-adenosine (eADO). *P < 0.05 versus corresponding natural substrate. All individual data points are provided along with the means and SDs
At low levels of substrates, initial reaction velocities are difficult to assess accurately because even with short incubation times the levels of substrates change during the reaction time. Therefore, to examine initial reaction rates, we incubated CD39 and CD73 with high concentrations of substrates (25 to 200 μmol/L). CD39 and CD73 were chosen for these experiments because they are considered the major enzymes involved in processing endogenous ATP to adenosine in vivo. As shown in Fig. 5a , CD39 metabolized ATP and N6-etheno-ATP with similar initial reaction velocities for concentration ranges up to 50 μmol/L. At higher concentrations of substrates, CD39 tended to metabolize N6-etheno-ATP faster than ATP; however, these differences were variable and quantitatively small. Similar initial reaction velocities were observed when CD39 was incubated with ADP versus N6-etheno-ADP (Fig. 5b). Likewise, CD73 metabolized high concentrations of N6-etheno-AMP at initial reaction rates similar to those for AMP (Fig. 5c). Together, these data support the conclusion that for all intents and purposes N6-etheno-adenine nucleotides are metabolized by ecto-nucleotidases with efficiencies similar to natural substrates, a conclusion consistent with the fact that ecto-nucleotidases act on phosphate groups remote from the etheno bridge.
Fig. 5.
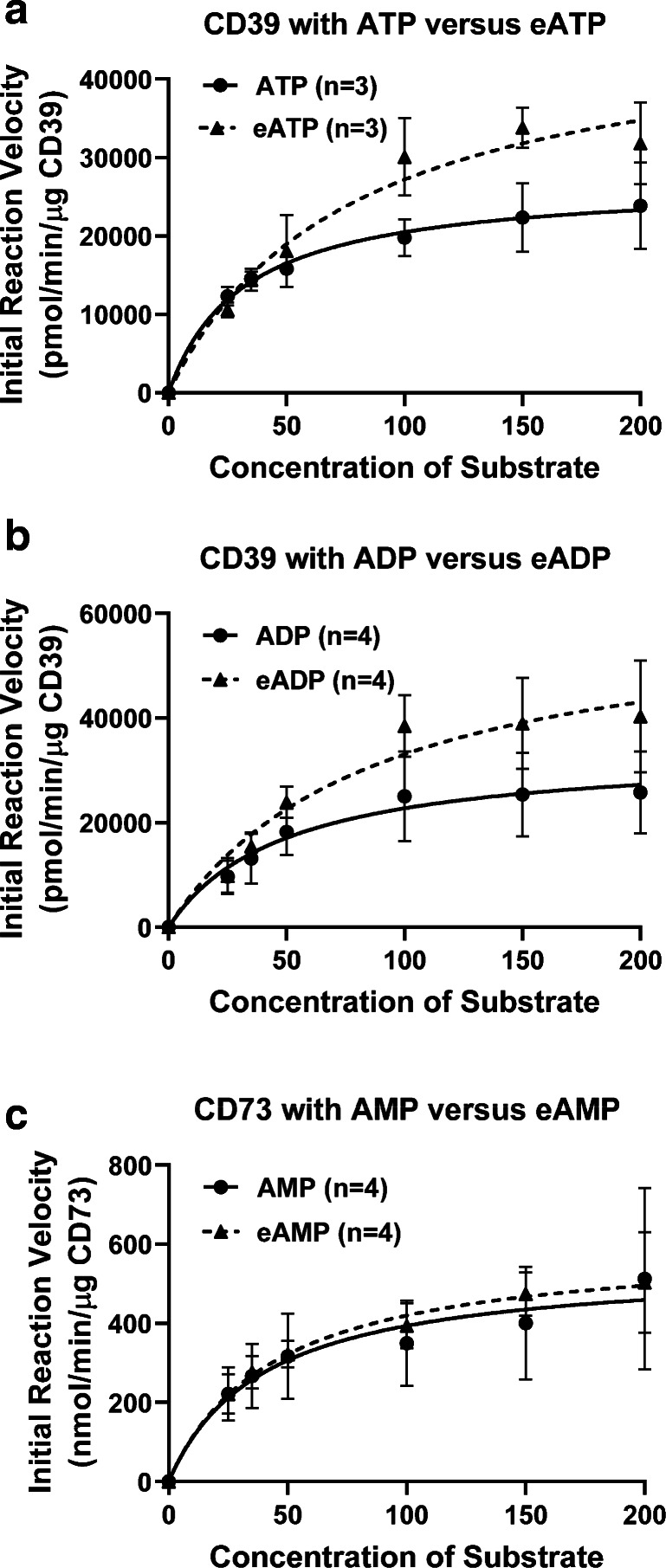
To determine initial reaction velocities, CD39 (10 ng) was incubated with high concentrations of substrates (25 to 200 μmol/L) for 10 min at 30 °C. In panel a, substrates were either ATP or N6-etheno-ATP and the downstream products (ADP + AMP or N6-etheno-ADP + N6-etheno-AMP) were measured. In panel b, substrates were either ADP or N6-etheno-ADP and the downstream products (AMP or N6-etheno-AMP) were measured. The experiment in panel c was similar to that described for panel b with the exception that the substrates were AMP or N6-etheno-AMP, the enzyme was CD73 (0.25 ng), and the measured products were adenosine and N6-etheno-ADO. eATP = N6-etheno-ATP; eADP = N6-etheno-ADP; eAMP = N6-etheno-AMP. Values represent means ± SDs
To further test the utility of the etheno-bridge method to track the extracellular metabolism of ATP, we applied 10 μmol/L of N6-etheno-ATP in PBS to monolayers of CFs, GMCs, renal microvascular endothelial cells (RMVECs), and PGVSMCs in cell culture. We also applied N6-etheno-ATP to empty culture wells to test for stability of the N6-etheno-ATP over the observation period. In some culture wells, the N6-etheno-ATP-containing PBS was applied, and then the medium was immediately removed (time 0 point). In other culture wells, the medium was collected at 0.5, 1, 4, 6, or 8 h after the N6-etheno-ATP was applied. All culture wells were maintained in an incubator under standard cell culture conditions (5% CO2; 37 °C). Samples were then analyzed for concentrations of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine by HPLC-FL. In culture wells (n = 6) devoid of cells, 98% of the added N6-etheno-ATP was recovered as N6-etheno-ATP. This indicated that N6-etheno-ATP was stable under the conditions of these experiments. As shown in Figs. 6, 7, 8, and 9, in all four cell types, N6-etheno-ATP was very rapidly metabolized and disappeared with a half-life of 0.25, 0.66, 0.21, and 0.38 h in CFs, GMCs, RMVECs, and PGVSMCs, respectively. In CFs, the processing of the intermediate metabolites, N6-etheno-ADP and N6-etheno-AMP, was so rapid that only a small concentration (less than 1 μmol/L) of either metabolite could be detected and then only during the early (0.5 and 1 h) time points (Fig. 6b and c). In contrast to the time profiles for N6-etheno-ADP and N6-etheno-AMP, N6-etheno-adenosine rapidly accumulated to nearly 8 μmol/L (Fig. 6d), then slowly declined as the N6-etheno-adenosine was converted to the final product N6-etheno-adenine (Fig. 6e). Figure 6f shows the summation of all five N6-etheno-purines (N6-etheno-Total) at all time points. Notably, the concentration of N6-etheno-Total was close to the concentration of added N6-etheno-ATP throughout the 8-h period, suggesting that N6-etheno-ATP and its metabolites remained in the medium and were not taken up by cells nor shunted to alternative metabolic pathways such as deamination.
Fig. 6.

Cardiac fibroblasts were treated with N6-etheno-ATP (10 μmol/L) for up to 8 h, and at the indicated time points, the medium was collected and analyzed for a N6-etheno-ATP, b N6-etheno-ADP, c N6-etheno-AMP, d N6-etheno-adenosine (ADO), and e N6-etheno-adenine (ADE) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. The sum (N6-etheno-Total) of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine is shown in f. In a, concentrations of N6-etheno-ATP were fitted to a mono-exponential decay function and the half-life (t1/2) was calculated. Values represent means and SDs. For some points, SD bars reside within the symbol. Individual data points are not shown because the points overlap
Fig. 7.

Glomerular mesangial cells were treated with N6-etheno-ATP (10 μmol/L) for up to 8 h, and at the indicated time points, the medium was collected and analyzed for a N6-etheno-ATP, b N6-etheno-ADP, c N6-etheno-AMP, d N6-etheno-adenosine (ADO), and e N6-etheno-adenine (ADE) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. The sum (N6-etheno-Total) of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine is shown in f. In a, concentrations of N6-etheno-ATP were fitted to a mono-exponential decay function and the half-life (t1/2) was calculated. Values represent means and SDs. For some points, SD bars reside within the symbol. Individual data points are not shown because the symbols would overlap
Fig. 8.

Renal microvascular endothelial cells were treated with N6-etheno-ATP (10 μmol/L) for up to 8 h, and at the indicated time points, the medium was collected and analyzed for a N6-etheno-ATP, b N6-etheno-ADP, c N6-etheno-AMP, d N6-etheno-adenosine (ADO), and e N6-etheno-adenine (ADE) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. The sum (N6-etheno-Total) of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine is shown in f. In a, concentrations of N6-etheno-ATP were fitted to a mono-exponential decay function and the half-life (t1/2) was calculated. Values represent means and SDs. For some points, SD bars reside within the symbol. Individual data points are not shown because the symbols overlap
Fig. 9.

Preglomerular vascular smooth muscle cells were treated with N6-etheno-ATP (10 μmol/L) for up to 8 h, and at the indicated time points, the medium was collected and analyzed for a N6-etheno-ATP, b N6-etheno-ADP, c N6-etheno-AMP, d N6-etheno-adenosine (ADO), and e N6-etheno-adenine (ADE) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. The sum (N6-etheno-Total) of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine is shown in f. In a, concentrations of N6-etheno-ATP were fitted to a mono-exponential decay function and the half-life (t1/2) was calculated. Values represent means and SDs. For some points, SD bars reside within the symbol. Individual data points are not shown because the symbols overlap
The time–concentration profiles for the five N6-etheno-purines in GMCs (Fig. 7), RMVECs (Fig. 8), and PGVSMCs (Fig. 9) differed from those of CFs (Fig. 6). In this regard, the peak accumulations of N6-etheno-ADP and N6-etheno-AMP were higher in GMCs, RMVECs, and PGVSMCs compared with CFs. RMVECs, in particular, had a sluggish metabolism of N6-etheno-ADP and N6-etheno-AMP such that N6-etheno-adenosine barely accumulated because it was converted to N6-etheno-adenine at a rate approximately equal to its formation (Fig. 8). As was the case with CFs, in PGVSMCs, GMCs, and RMVECs, N6-etheno-Total was close to the concentration of added N6-etheno-ATP throughout the 8-h period, suggesting that N6-etheno-ATP and its metabolites remained in the medium and were not taken up by cells or shunted to alternative metabolic pathways.
To further test the conclusion that N6-etheno-purines remained in the extracellular compartment, we treated CFs with 10 μmol/L of N6-etheno-ATP for 8 h, washed the cells with PBS, extracted intracellular purines with acetonitrile and methanol, and assayed for intracellular levels of N6-etheno-purines. Intracellular levels of N6-etheno-purines were below detection limit of our assay.
An unanticipated finding of the cell culture experiments was the accumulation of N6-etheno-adenine. The conventional view is that adenosine is not metabolized by mammalian PNPase to adenine, but rather is metabolized by adenosine deaminase (ADA) to inosine [67]. Therefore, we conjectured that the most likely explanation for N6-etheno-adenine formation in our cell culture models was that our cultured cells were infected with Mycoplasma (an intracellular microorganism that often infects cell culture labs and often goes unnoticed). Although mammalian PNPase is thought not to metabolize adenosine to adenine, bacterial PNPase is well known to convert adenosine to adenine [67]. To test whether our cells were infected with Mycoplasma, we applied the most sensitive and broad spectrum Mycoplasma detection method (PCR-based method) available to all four cell types used in the present study. We included quality control samples (positive and negative controls) for all batches of cells tested. There was no evidence whatsoever for Mycoplasma contamination (see Figure in the Online supplement).
Next, we tested whether recombinant mammalian (human in this case) PNPase (rhPNPase; R&D Systems, catalog number 6486-NP-010; specific activity, > 35,000 pmol/min/μg, as measured by phosphorolysis of 7-methyl-6-thioguanosine) metabolizes adenosine to adenine or N6-etheno-adenosine to N6-etheno-adenine. Figure 10a shows a chromatogram obtained by HPLC-UV of a sample containing both 50 μmol/L of inosine and hypoxanthine incubated (30 min at 30 °C) in the absence of rhPNPase. Figure 10b shows a HPLC-UV chromatogram of a sample containing 50 μmol/L of inosine incubated (30 min at 30 °C) with rhPNPase (640 ng). As demonstrated, when inosine was incubated with rhPNPase, only the hypoxanthine peak was observed, indicating complete conversion of inosine to hypoxanthine and thus confirming the activity of the rhPNPase. Figure 10c and d show HPLC-UV chromatograms from samples containing 50 μmol/L of adenosine incubated (30 min at 30 °C) without and with, respectively, rhPNPase. As shown in Fig. 10d, all of the added adenosine was recovered as adenosine. Figure 10e and f show the results of the corresponding experiment with N6-etheno-adenosine incubated (30 min at 30 °C) without and with, respectively, rhPNPase. These HPLC-FL chromatograms show that none of the N6-etheno-adenosine was metabolized to N6-etheno-adenine. Thus, recombinant PNPase, which is likely in the monomeric form, does not metabolize either adenosine or N6-etheno-adenosine, a finding consistent with a recent report by Stachelska-Wierzchowska and coworkers [68].
Fig. 10.

a A chromatogram obtained by HPLC-UV of a sample containing both 50 μmol/L of inosine and hypoxanthine incubated (30 min at 30 °C) in the absence of rhPNPase. b A HPLC-UV chromatogram of a sample containing 50 μmol/L of inosine incubated (30 min at 30 °C) with rhPNPase (640 ng). When inosine was incubated with rhPNPase, only a hypoxanthine peak was observed, indicating complete conversion of inosine to hypoxanthine and thus confirming the activity of the rhPNPase. c, d HPLC-UV chromatograms from samples containing 50 μmol/L of adenosine incubated (30 min at 30 °C) without and with, respectively, rhPNPase. As shown, in the presence of rhPNPase, all of the added adenosine was recovered as adenosine. e, f The results of the corresponding experiment with N6-etheno-adenosine (ADO) incubated (30 min at 30 °C) without and with, respectively, rhPNPase. These HPLC-FL chromatograms show that none of the N6-etheno-adenosine was metabolized to N6-etheno-adenine
Since rhPNPase did not metabolize N6-etheno-adenosine, we considered that the formation of N6-etheno-adenine by cultured cells was due to an enzyme other than PNPase. To test this, we incubated CFs, PGVSMCs, GMCs, and RMVECs for 4 h with N6-etheno-adenosine and with and without 8-aminoguanine (potent and selective inhibitor of PNPase [69]) and measured both N6-etheno-adenosine and N6-etheno-adenine. N6-etheno-adenosine was partially converted to N6-etheno-adenine by CFs (Fig. 11a, b, and c), GMCs (Fig. 12a, b, and c), PGVSMCs (Fig. 12d, e, and f), and RMVECs (Fig. 12g, h, and i), and in all four cell types, this reaction was abolished by 8-aminoguanine (100 μmol/L). To rule out an off-target action of 8-aminoguanine, we also examined the effects of forodesine (5 μmol/L), an alternative and highly selective and potent inhibitor of PNPase [70] approved for treatment of T cell lymphoma in Japan, on metabolism of N6-etheno-adenosine in CFs (Fig. 11d, e, and f). As with 8-aminoguanine, forodesine also abolished the metabolism of N6-etheno-adenosine to N6-etheno-adenine in CFs.
Fig. 11.

Cardiac fibroblasts (CFs) were thoroughly washed with phosphate-buffered saline and incubated with N6-etheno-adenosine (10 μmol/L) in the absence or presence of the purine nucleoside phosphorylase inhibitor 8-aminoguanine (100 μmol/L) or forodesine (5 μmol/L). After 4 h, the medium was collected and assayed for N6-etheno-adenine (a, d, g) and N6-etheno-adenosine (b, e, h) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. The sum (N6-etheno-Total) of N6-etheno-adenine and N6-etheno-adenosine is shown in c, f, and i. In a, b, c, d, e, and f, cells were expanded using bovine calf serum. In g, h, and i, cells were expanded using platelet-derived growth factor rather than serum to eliminate any possibility of contaminating the cells with purine nucleoside phosphorylase from serum. Both 8-aminoguanine (a, b, c, g, h, i) and forodesine (d, e, f) abolished the formation of N6-etheno-adenine. Results were the same in cells expanded with versus without bovine calf serum. All individual data points are provided along with the means and SDs. *P < 0.05 versus control. ADE, adenine; ADO, adenosine; DL, detection limit
Fig. 12.

Glomerular mesangial cells (GMCs; a, b, c), preglomerular vascular smooth muscle cells (PGVSMCs; d, e, f), and renal microvascular endothelial cells (RMVECs; g, h, i) were thoroughly washed with phosphate-buffered saline and incubated with a N6-etheno-adenosine (10 μmol/L) in the absence or presence of the purine nucleoside phosphorylase inhibitor 8-aminoguanine (100 μmol/L). After 4 h, the medium was collected and assayed for N6-etheno-adenine (a, d, g) and N6-etheno-adenosine (b, e, h) using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. The sum (N6-etheno-Total) of N6-etheno-adenine and N6-etheno-adenosine is shown in c, f, and i. In all three cell types, 8-aminoguanine abolished the formation of N6-etheno-adenine. All individual data points are provided along with the means and SDs. *P < 0.05 versus control. ADE, adenine; ADO, adenosine; DL, detection limit
We also considered the possibility that since our cells were expanded in medium containing fetal bovine serum, PNPase in the serum may have been delivered to the cells. To test this, we maintained CFs in serum-free medium for 4 days, washed them thoroughly with PBS, and repeated the experiment. As shown in Fig. 11g, h, and i , the metabolism of N6-etheno-adenosine to N6-etheno-adenine was similar in CFs expanded in the presence or absence of fetal bovine serum. Again, the conversion of N6-etheno-adenosine to N6-etheno-adenine was abrogated by 8-aminoguanine (Fig. 11g, h, and i).
To determine whether N6-etheno-ADO is converted to N6-etheno-ADE in an intact mammalian organ system, we isolated and perfused rat kidneys while taking care to avoid bacterial contamination of the perfusion system, the perfusate, or kidneys. In this regard, the kidney perfusion system was perfused with 75% ethanol for 1.5 h followed by perfusion with sterile water for 1.5 h, the perfusate was filtered to 0.22 μm, and the kidney was isolated under sterile conditions. Further, we immediately started the experiment after initiating kidney perfusion in order to reduce any chance of bacteria proliferating within the system over time. In these experiments, we infused 3 μmol/L of either N6-etheno-ATP or N6-etheno-adenosine (final concentration in perfusate) for 5 min and collected the venous outflow from 4 to 5 min into the infusion. We also did a series of sham perfusions in which the kidney was not placed in the perfusion system. The collected renal venous samples were analyzed for N6-etheno-purines by HPLC-FL. As shown in Fig. 13a , when kidneys were not inserted into the perfusion system, N6-etheno-ATP was not metabolized. In contrast, when kidneys were inserted into the perfusion system, N6-etheno-ATP was completely metabolized during a single pass through the kidney, with 13% of the N6-etheno-Total recovered as N6-etheno-adenine (Fig. 13b). Also, 16% of infused N6-etheno-adenosine was converted to N6-etheno-adenine during a single pass through the kidney (Fig. 13c).
Fig. 13.
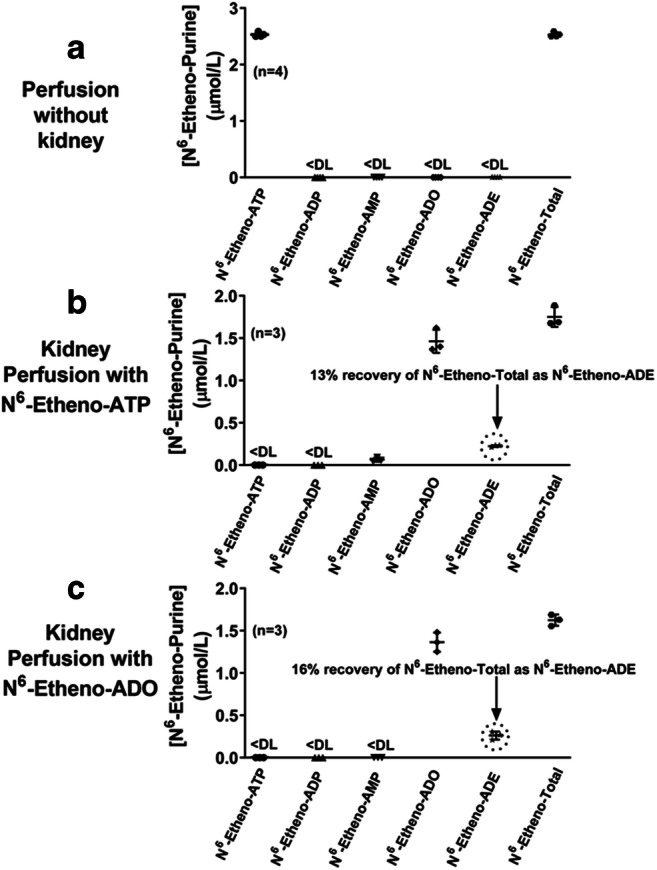
The metabolism of N6-etheno-ATP (3 μmol/L) and N6-etheno-adenosine (ADO) (3 μmol/L) was examined in the isolated, perfused rat kidney. The perfusion system was sterilized in all experiments to prevent bacterial contamination. In a, the perfusion system without the kidney in place received N6-etheno-ATP. In b, N6-etheno-ATP was infused into the renal artery, and in c, N6-etheno-adenosine was infused into the renal artery. In all experiments, perfusate from the venous outflow was collected and analyzed for N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. Also, the sum (N6-etheno-Total) of N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine was determined. All individual data points are provided along with the means and SDs. ADE, adenine; ADO, adenosine; DL, detection limit
To test whether N6-etheno-ADO is converted to N6-etheno-ADE in vivo, we infused into anesthetized rats 0.1 μmol/kg/min of N6-etheno-ATP and measured in renal microdialysate, urine, and plasma all five N6-etheno-purines by HPLC-FL. In these experiments, N6-etheno-ATP, N6-etheno-ADP, and N6-etheno-AMP were below the detection limit of our assay system. However, both N6-etheno-adenosine and N6-etheno-adenine were readily detected in all samples. Surprisingly, in microdialysate (Fig. 14a), urine (Fig. 14b), and plasma (Fig. 14c), in most samples, the levels of N6-etheno-adenine exceeded the levels of N6-etheno-adenosine. To confirm this finding and to determine the role of PNPase in producing N6-etheno-adenine in vivo, we repeated the in vivo experiment in two additional groups of rats, one pretreated with 8-aminoguanine (33.5 μmol/kg, intravenous bolus) and the other pretreated with the vehicle for 8-aminoguanine (vehicle-control group). As before, in the control group, 0.1 μmol/kg/min of N6-etheno-ATP increased N6-etheno-adenine more than N6-etheno-adenosine in most samples of microdialysate (Fig. 15a), urine (Fig. 15d), and plasma (Fig. 15g). In contrast, in the group treated with 8-aminoguanine, 0.1 μmol/kg/min of N6-etheno-ATP increased N6-etheno-adenosine more than N6-etheno-adenine in most samples of microdialysate (Fig. 15b), urine (Fig. 15e), and plasma (Fig. 15h). Also, the ratio of N6-etheno-adenine to N6-etheno-adenosine in microdialysate (Fig. 15c), urine (Fig. 15f), and plasma (Fig. 15i) was manyfold higher in the control group versus the 8-aminoguanine group. To confirm that the dose of 8-aminoguanine blocked PNPase, we also measured the urinary excretion rate of endogenous inosine and adenine using UPLC-tandem mass spectrometry as previously described by us [66]. The urinary excretion rate of inosine in the 8-aminoguanine-treated group was 8-fold higher (P = 0.006) compared with the control group (287 ± 71 versus 35 ± 6 ng/min, respectively; mean ± SEM), indicating blockade of PNPase. Correspondingly, the urinary excretion of endogenous adenine was reduced in the 8-aminoguanine-treated group by 73% (P = 0.007) compared with the control group (4.8 ± 1.2 versus 17.7 ± 4.7 ng/min, respectively; mean ± SEM).
Fig. 14.

N6-etheno-ATP (0.1 μmol/kg/min for 30 min) was infused intravenously into anesthetized rats, and renal microdialysate (a), urine (b), and plasma (c) were analyzed at different time points for N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. Only N6-etheno-adenosine and N6-etheno-adenine were detected in the samples. Other panels show mean arterial blood pressure (d), heart rate (e), and renal blood flow (f) during the experiment. *P < 0.05, N6-etheno-adenosine versus N6-etheno-adenine at the same time point. Values are means ± SEM. ADE, adenine; ADO, adenosine
Fig. 15.

N6-etheno-ATP (0.1 μmol/kg/min for 30 min) was infused intravenously into anesthetized rats, and renal microdialysate (a, b), urine (d, e), and plasma (g, h) were analyzed at different time points for N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine using reverse phase high-performance liquid chromatography as described in detail in the “Analytical methods” section. Some rats (b, e, h) were pretreated with an intravenous bolus of the purine nucleoside phosphorylase inhibitor 8-aminoguanine (33.5 μmol/kg), while others received only the vehicle for 8-aminoguanine (a, b, g). Only N6-etheno-adenosine and N6-etheno-adenine were detected in the samples. In the vehicle-control rats, in most samples, N6-etheno-adenine exceeded N6-etheno-adenosine (a, d, g). In contrast, in the 8-aminoguanine-treated rats, in most samples, N6-etheno-adenosine exceeded N6-etheno-adenine (b, e, h). Also shown is the ratio of N6-etheno-adenine to N6-etheno-adenosine in microdialysate (c), urine (f), and plasma (i) of vehicle-treated versus 8-aminoguanine-treated rats. This ratio was higher in the vehicle-treated group versus the 8-aminoguanine-treated group. *P < 0.05, N6-etheno-adenosine versus N6-etheno-adenine or vehicle group versus 8-aminoguanine group at the same time point. Values are means ± SEM. ADE, adenine; ADO, adenosine
To further explore the concept that endogenous PNPase can metabolize authentic adenosine to adenine under certain conditions (for example, when metabolism and uptake of adenosine is impaired, as it would be for N6-etheno-adenosine), we examined the effects of local delivery of adenosine into the renal cortical interstitium using “reverse microdialysis” on renal cortical interstitial levels of adenine. These experiments were conducted in the presence of inhibitors of adenosine deaminase, adenosine kinase, and adenosine uptake to block alternative pathways of adenosine disposition. As shown in Fig. 16, under these experimental conditions, renal interstitial delivery of adenosine increased renal interstitial levels of adenine, a response that was blocked by the PNPase inhibitor forodesine.
Fig. 16.
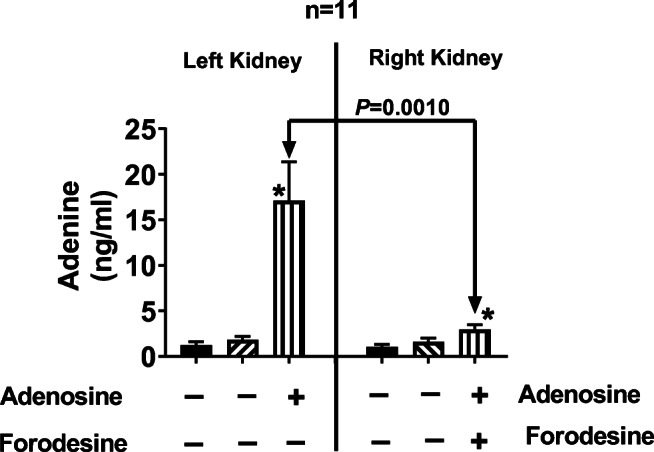
Bar graphs show cortical microdialysate levels of adenine in the left and right kidneys during three experimental periods. After a 1-h stabilization period, microdialysate was collected for 45 min (period 1), and then the following drugs were added to the microdialysis perfusate of both kidneys: 10 μmol/L of erythro-9-(2-hydroxy-3-nonyl) adenine (blocks adenosine deaminase), 0.1 μmol/L of 5-iodotubericidin (blocks adenosine kinase), and 10 μmol/L of S-(4-nitrobenzyl)-6-thioinosine (blocks adenosine uptake). After 15 min for the drugs to reach the microdialysate probe tip, microdialysate was again collected for another 45 min (period 2). Next, in addition to the drugs listed above, adenosine (30 μmol/L) was added to the left kidney perfusate (period 3) and adenosine (30 μmol/L) plus forodesine (5 μmol/L; blocks purine nucleoside phosphorylase) was added to the right kidney perfusate (period 3). *P < 0.05, versus periods 1 and 2. Values are means ± SEM
Importantly, intravenous infusions of N6-etheno-ATP had no discernable effects on mean arterial blood pressure (Fig. 14d), heart rate (Fig. 14e), renal blood flow (Fig. 14f), or urine excretion rate (data not shown) suggesting that N6-etheno-ATP and its downstream metabolites were devoid of pharmacological activity. To further test this conclusion, we examined using competitive receptor binding experiments (in triplicate) whether N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, or N6-etheno-adenine interacted with the orthosteric binding site on recombinant A1, A2A, and A2B receptors, on P2X receptors in membranes from rat urinary bladder, and on P2Y receptors in membranes from rat brain (see “Analytical methods” section for details). In these competitive receptor binding experiments, at 10 μmol/L, neither N6-etheno-ATP, N6-etheno-ADP, N6-etheno-AMP, N6-etheno-adenosine, nor N6-etheno-adenine inhibited radioactive-ligand binding to recombinant A1, A2A, or A2B receptors or to bladder membrane P2X receptors. Also, at 10 μmol/L, N6-etheno-AMP, N6-etheno-adenosine, and N6-etheno-adenine did not affect radioactive-ligand binding to brain P2Y receptors. N6-etheno-ATP and N6-etheno-ADP interacted with brain P2Y receptors but with weak affinities. The calculated Kds of N6-etheno-ATP for brain P2Y receptors in three separate experiments were 5.9, 4.7, and 4.5 μmol/L (mean ± SEM, 5.1 ± 0.4 μmol/L). The calculated Kds of N6-etheno-ADP for brain P2Y receptors in three separate experiments were 5.8, 2.6, and 3.6 μmol/L (mean ± SEM, 4.0 ± 0.9 μmol/L).
Discussion
Extracellular adenine nucleotides and adenosine initiate purinergic signaling, and interest in purinergic signaling is increasing exponentially [8, 71–84]. This escalation of interest in purinergic signaling is the result of discoveries of numerous subtypes of purinergic receptors for extracellular nucleotides and adenosine. In this regard, currently, there are seven known P2X receptor subunits, which form homo-oligomers or hetero-oligomers [85], eight known P2Y receptors [84], and four known P1 receptors [86]. Because P1- and P2-receptor signaling occurs in virtually every organ system and plays a role in many diseases, attention to the extracellular metabolism of ATP is imperative.
In parallel with the explosion of knowledge in purinergic signaling, there is an increasing appreciation of the complexity of extracellular ATP metabolism. Indeed, there is a large array of known ecto-nucleotidases that can participate in the metabolism of ATP to its downstream purinergic metabolites (ATP ➔ ADP ➔ AMP ➔ adenosine) [5]. Unraveling the complexity of extracellular ATP metabolism would be facilitated by a sensitive, specific, inexpensive, and rapid method of measuring extracellular adenine nucleotides and adenosine without confounding by cellular uptake, pharmacologically induced changes in ecto-enzyme amount or activity, or deamination, which would direct adenine-based compounds to side reactions.
Although we cannot conclude that the metabolism of etheno-bridged adenine nucleotides/nucleosides is identical to that of the corresponding natural compounds, we can conclude that the metabolism is sufficiently similar such that etheno-bridged compounds can accurately assess the extracellular metabolism of adenine nucleotides/nucleosides. This conclusion is based on several considerations. First, our preliminary experiments demonstrated that N6-etheno-ATP was processed rapidly by human recombinant CD39, ENTPD2, ENTPD3, and ENPP-1 to the expected final downstream product (N6-etheno-AMP). Also, N6-etheno-AMP was readily converted to etheno-ADO by human recombinant CD73. These findings provide solid support for the conclusion that the etheno bridge, which is located remotely from the phosphate groups in ATP, ADP, and AMP, does not impede the enzymatic processing of etheno-bridged adenine nucleotides. Second, and in confirmation of the first point, human recombinant CD39, ENTPD2, ENTPD3, and ENPP-1 metabolized very low concentrations (1 μmol/L) of N6-etheno-ATP and ATP with similar efficiencies, and human recombinant CD73 metabolized low concentrations (1 μmol/L) of N6-etheno-AMP and AMP with similar efficiencies. These results suggest that at physiological concentrations of naturally occurring adenine nucleotides, CD39 and CD73 metabolize ATP and AMP, respectively, at efficiencies similar to those for correspondingly low concentrations of N6-etheno-ATP and N6-etheno-AMP. Third, initial reaction velocities for CD39 were similar for N6-etheno-ATP versus ATP and for N6-etheno-ADP versus ADP, and initial reaction velocities for CD73 were similar for N6-etheno-AMP versus AMP. For pragmatic reasons, these initial reaction velocities had to be assessed at high substrate concentrations (25 to 200 μmol/L); nonetheless, the results are consistent with the concept that ecto-nucleotidases do not meaningfully discriminate between naturally occurring adenine nucleotides versus etheno-bridged adenine nucleotides. Fourth, in four different primary cell types, N6-etheno-ATP was extremely rapidly metabolized to N6-etheno-ADP, N6-etheno-AMP, and N6-etheno-ADO. Also, the differences among cell types with regard to the accumulation of N6-etheno-ADP, N6-etheno-AMP, and N6-etheno-ADO following administration of N6-etheno-ATP showed that this method detected differences in the relative activities of ecto-nucleotidases present in these cells. Fifth, in intact, isolated, perfused rat kidneys, N6-etheno-ATP was completely metabolized to N6-etheno-ADO plus N6-etheno-ADE in a single pass across the rat renal vasculature. With a kidney perfusion rate of 5 mL/min across a kidney with a total volume of approximately 2 mL, the residence time of the applied N6-etheno-ATP in the renal vasculature would be seconds. Yet, practically all of the N6-etheno-ATP was metabolized during this brief exposure to the renal microcirculation. Sixth, following intravenous infusion of N6-etheno-ATP, the metabolism of N6-etheno-ATP was so rapid that only N6-etheno-ADO plus N6-etheno-ADE was detected in plasma, urine, and renal microdialyate. Together, these data support the conclusion that for all intents and purposes, N6-etheno-adenine nucleotides are metabolized by ecto-nucleotidases with efficiencies similar to natural substrates, a conclusion consistent with the fact that ecto-nucleotidases act on phosphate groups remote from the etheno bridge.
It is worth noting that N6-etheno-ATP did not invoke any observable changes in mean arterial blood pressure, heart rate, renal blood flow, or urine volume, suggesting that N6-etheno-ATP and its downstream metabolites interact poorly with purinergic receptors. This conclusion is further supported by our receptor binding studies that showed undetectable or very low affinities of N6-etheno-ATP and its downstream metabolites to an array of purinergic receptors. A limitation here is that binding assays using rat urinary bladder and brain membranes sampled only a subset of P2X and P2Y receptors. Thus, it remains possible that these compounds interact with some P2X or P2Y receptors. In this regard, although Durnin and colleagues noted that N6-etheno-ATP relaxed the murine colon, these responses were achieved with very high (30 μmol/L) concentrations of N6-etheno-ATP [87]. Taken together, the evidence supports the conclusion that N6-etheno-ATP and its downstream metabolites are processed by ecto-nucleotidases as readily as unmodified ATP and its downstream metabolites, yet the etheno bridge-modified compounds are for all intents and purposes pharmacologically inert at concentrations required to assess extracellular metabolism of adenine nucleotides by ecto-nucleotidases.
Another important conclusion supported by the current study is that most of the applied N6-etheno-ATP and its downstream metabolites remain in the extracellular compartment. The evidence for this conclusion is that the concentration of N6-etheno-Total in the culture medium is approximately the same as the applied concentration of N6-etheno-ATP. Although the match is not perfect, the slight discrepancy is likely due to the fact that not all of the medium can be removed from culture wells before replacing it with medium containing N6-etheno-ATP, and therefore, any remaining medium that adheres to the culture well or cells would dilute the added N6-etheno-ATP, thus giving rise to a slightly lower concentration of N6-etheno-Total compared with applied N6-etheno-ATP. This conclusion is further confirmed by the lack of N6-etheno-ATP or its metabolites in cells exposed to N6-etheno-ATP for 8 h. Although it is possible that trace amounts of N6-etheno-ATP and its metabolites entered cells, it is unlikely that intracellular metabolism makes a significant contribution to the cellular metabolism of applied N6-etheno-ATP.
Yet another important finding of the present study is that N6-etheno-ATP and its downstream metabolites are not diverted from the main ecto-nucleotidase pathway: ATP ➔ ADP ➔ AMP ➔ adenosine. This conclusion is also based on the observation that nearly all of the applied moles of N6-etheno-ATP are accounted for in the detected moles of N6-etheno-Total. Given the fact that the etheno bridge blocks the N6 nitrogen, it is to be expected that deaminases would not process ATP, ADP, AMP, or adenosine to their corresponding inosine metabolites. Thus, the etheno-bridge method eliminates not only confounding due to cellular uptake, but also confounding due to deamination.
Mammalian PNPase is an enzyme that converts inosine to hypoxanthine and guanosine to guanine. The conventional view is that adenosine is not a substrate for mammalian PNPase and is therefore deaminated first by adenosine deaminase to inosine, which is subsequently metabolized to hypoxanthine by PNPase. Therefore, we were surprised that in all four cell types investigated here, the terminal metabolite of N6-etheno-ATP was not N6-etheno-ADO, but rather N6-etheno-ADE. Our initial reaction to this finding was to suspect that our cells were contaminated with the intracellular bacterium, Mycoplasma. Mycoplasma is the bane of cell culturists because it commonly infects cell culture laboratories, hides within cells, is difficult to detect, and modifies the biochemistry of cultured cells. Indeed, there are estimates that 70% of cell cultures are contaminated with Mycoplasma [88]. Because bacterial PNPase, unlike mammalian PNPase, can metabolize adenosine to adenine [67], contamination with Mycoplasma provides a simple explanation for why the end product of N6-etheno-ATP metabolism was N6-etheno-ADE, rather than N6-etheno-ADO.
To test the possibility that our cell cultures were contaminated by Mycoplasma or other intracellular bacteria, we employed the Universal Mycoplasma Detection Kit, a method that uses 32 rounds of PCR to detect femtogram levels of bacterial DNA and covers over 60 species of Mycoplasma, Acholeplasma, Spiroplasma, and Ureaplasma including the eight species most likely to contaminate cell cultures: M. arginini, M. fermentans, M. hominis, M. hyorhinis, M. orale, M. pirum, M. salivarium, and A. laidlawii. The results were negative. Thus, we could not attribute the accumulation of N6-etheno-ADE to bacterial contamination.
Still concerned that production of N6-etheno-ADE was an artifact, we considered the possibility that the bovine fetal serum used in expanding our cells may have contained PNPase (either mammalian or bacterial) and that this was the source of PNPase that metabolized N6-etheno-ADO to N6-etheno-ADE. To test this, we expanded CFs for 4 days in the absence of bovine serum, washed the cells thoroughly, and examined the metabolism of N6-etheno-ADO to N6-etheno-ADE. Importantly, the metabolism of N6-etheno-ADO to N6-etheno-ADE was essentially identical in cells expanded in the absence or presence of bovine serum.
We also considered the hypothesis that N6-etheno-ADO was converted to N6-etheno-ADE by an unknown enzyme that is not PNPase, yet has “PNPase-like” activity. To test this, we examined in all four cell types the ability of 8-aminoguanine, a well-known inhibitor of PNPase, to block the conversion of N6-etheno-ADO to N6-etheno-ADE. Indeed, in all four cell types, 8-aminoguanine abolished the metabolism of N6-etheno-ADO to N6-etheno-ADE. It is conceivable that 8-aminoguanine blocks, in addition to PNPase, other enzymes with “PNPase-like” activity. To test this, we also examined the effects of forodesine, a structurally distinct and highly potent and selective PNPase inhibitor, on the metabolism of N6-etheno-ADO to N6-etheno-ADE in CFs. Consistent with the results with 8-aminoguanine, forodesine too abolished the formation of N6-etheno-ADE in CFs.
Still skeptical, we considered that somehow the metabolism of N6-etheno-ADO to N6-etheno-ADE was an artifact of our cell culture models. Therefore, next we investigated whether N6-etheno-ATP or N6-etheno-ADO is metabolized to N6-etheno-ADE in an intact organ, in this case the isolated, perfused kidney. As with our cell culture models, we were concerned that bacterial contamination of the perfusion system or the perfused kidneys could confound interpretation. To make sure the perfusion system per se was not contaminated, we determined whether N6-etheno-ATP was metabolized across the perfusion system in the absence of an intervening kidney. Essentially, no N6-etheno-ATP was metabolized in the absence of an intervening kidney. However, in the presence of an intervening kidney, 13% of applied N6-etheno-ATP and 16% of applied N6-etheno-ADO were converted to N6-etheno-ADE in a single pass (taking only seconds) across the kidney. In these experiments, the perfusion system was thoroughly flushed with ethanol and sterile water, the perfusate was filtered to 0.22 μm, and the kidney was isolated under sterile conditions.
Next, we investigated whether N6-etheno-ATP could be converted to N6-etheno-ADE in vivo. In these experiments, renal microdialysate, urine, and plasma were collected before, during, and after an intravenous infusion of N6-etheno-ATP. Again, to our surprise, the major observable product of N6-etheno-ATP metabolism in vivo was not N6-etheno-ADP, N6-etheno-AMP, or N6-etheno-ADO, but rather N6-etheno-ADE. To confirm and extend these in vivo observations, we repeated the in vivo study in two groups, one treated with 8-aminoguanine and one treated with the vehicle for 8-aminoguanine. In the control group, once again we observed that the major observable metabolite was N6-etheno-ADE with lesser amounts of N6-etheno-ADO; yet in the 8-aminoguanine-treated group, the reverse was true (N6-etheno-ADO exceeded N6-etheno-ADE). This study confirms that in vivo an enzyme with PNPase-like activity that is sensitive to inhibition by 8-aminoguanine metabolizes N6-etheno-ADO to N6-etheno-ADE. We also measured by UPLC-MS/MS urine levels of endogenous inosine and observed that 8-aminoguanine increased urinary inosine excretion by 8-fold, suggesting the dose of 8-aminoguanine used did indeed inhibit PNPase.
A limitation of our in vivo experiments is that the metabolism of N6-etheno-ADO to N6-etheno-ADE could have been mediated by the gut microbiota. However, in view of the rapid (first 10 min) detection of N6-etheno-ADE in the kidney microdialysate during intravenous infusions of N6-etheno-ATP, it would seem that at least a portion of the metabolism to N6-etheno-ADO to N6-etheno-ADE occurred outside the gut. It is also conceivably that in vivo the enzyme that metabolizes N6-etheno-ADO to N6-etheno-ADE does not metabolize authentic adenosine to adenine, although this too seems unlikely. To test these conjectures, we delivered authentic adenosine directly into the renal cortex by reverse microdialysis and assessed renal cortical levels of adenine by measuring levels of adenine in the perfusate exiting the microdialysis probe. Importantly, direct delivery of adenosine to the renal cortex resulted in detectable increases in renal levels of adenine, a response that was blocked by the PNPase inhibitor, forodesine.
The molecular form of the enzyme that conferred PNPase activity in our mammalian experimental paradigms is unknown and deserves additional investigation. In this regard, we also examined the effects of authentic human recombinant PNPase on the metabolism of both adenosine and N6-etheno-ADO. This recombinant PNPase did not metabolize either of these substrates. Importantly, PNPase (both mammalian and bacterial) can exist in multiple forms (monomers, dimers, trimers, tetramers, pentamers, and hexamers) that have different enzymatic properties and preferred substrates [67]. The molecular form of human recombinant PNPase in solution is unknown (although likely a monomer) and may not have the same enzymatic properties as various multimers of PNPase in vivo. Moreover, it is conceivable that the enzymatic properties of PNPase in vivo may be modified further by interactions with other protein-binding partners. Thus, one explanation for the present results is that in vivo some forms of mammalian PNPase do accept N6-etheno-ADO and adenosine as substrates. An alternative explanation is that there exists an enzyme other than PNPase that has “PNPase-like” activity and is inhibited both by 8-aminoguanine and forodesine. Either way, our results show that N6-etheno-ADO can be used to interrogate the presence and activity levels of this form of PNPase or “PNPase-like” enzyme and could provide important insights into the role of PNPase or a “PNPase-like” enzyme in health and disease. However, whether this enzyme activity contributes to regulating in vivo the levels of adenosine is unknown and merits additional investigation.
In conclusion, this experimental series establishes the validity of using N6-etheno-ATP to conveniently assess the processing of extracellular ATP using a sensitive, specific, and precise methodology that is not confounded by uptake of ATP or its metabolites, is not confounded by deamination, and is likely not confounded by pharmacological activation of purinergic receptors. This technique could prove quite useful not only in in vitro studies, but in in vivo studies as well. For example, in cancer biology, this method could be applied to identify which ecto-nucleotidases are involved in adenosine production within a tumor and therefore could direct personal medicine targeted at the most important ecto-nucleotidase to prevent the production of immunosuppressive and tumor-promoting adenosine. Finally, the current findings suggest that some form of PNPase or “PNPase-like” enzyme is involved in the in vivo metabolism of adenosine to adenine. The metabolism of N6-etheno-ADO to N6-etheno-ADE may prove useful as a method to investigate the activity and importance of this enzyme.
Electronic supplementary material
(DOCX 374 kb)
Funding information
This study was supported by grants from the NIH (NS087978, DK091190, HL109002, HL069846, DK068575, and DK079307).
Compliance with ethical standards
Conflicts of interest
Edwin K. Jackson declares that he has no conflict of interest.
Delbert G. Gillespie declares that he has no conflict of interest.
Dongmei Cheng declares that he has no conflict of interest.
Zaichuan Mi declares that he has no conflict of interest.
Elizabeth V. Menshikova declares that she has no conflict of interest.
Ethical approval
The Institutional Animal Care and Use Committee approved all procedures.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Edwin K. Jackson, Email: edj@pitt.edu
Delbert G. Gillespie, Email: dgg3@pitt.edu
Dongmei Cheng, Email: doc14@pitt.edu.
Zaichuan Mi, Email: zmi8@pitt.edu.
Elizabeth V. Menshikova, Email: evm3@pitt.edu
References
- 1.Li A, Banerjee J, Leung CT, Peterson-Yantorno K, Stamer WD, Civan MM (2011) Mechanisms of ATP release, the enabling step in purinergic dynamics, Cellular Physiology & Biochemistry. 28(6):1135–1144. 10.1159/000335865 [DOI] [PMC free article] [PubMed]
- 2.Fields RD. Nonsynaptic and nonvesicular ATP release from neurons and relevance to neuron-glia signaling. Semin Cell Dev Biol. 2011;22(2):214–219. doi: 10.1016/j.semcdb.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cisneros-Mejorado A, Perez-Samartin A, Gottlieb M, Matute C. ATP signaling in brain: release, excitotoxicity and potential therapeutic targets. Cell Mol Neurobiol. 2015;35(1):1–6. doi: 10.1007/s10571-014-0092-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohman AW, Billaud M, Isakson BE. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc Res. 2012;95(3):269–280. doi: 10.1093/cvr/cvs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmermann H, Zebisch M, Strater N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012;8(3):437–502. doi: 10.1007/s11302-012-9309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westfall DP, Todorov LD, Mihaylova-Todorova ST. ATP as a cotransmitter in sympathetic nerves and its inactivation by releasable enzymes. J Pharmacol Exp Ther. 2002;303(2):439–444. doi: 10.1124/jpet.102.035113. [DOI] [PubMed] [Google Scholar]
- 7.Matsuoka I, Ohkubo S. ATP- and adenosine-mediated signaling in the central nervous system: adenosine receptor activation by ATP through rapid and localized generation of adenosine by ecto-nucleotidases. J Pharmacol Sci. 2004;94(2):95–99. doi: 10.1254/jphs.94.95. [DOI] [PubMed] [Google Scholar]
- 8.Gendron FP, Benrezzak O, Krugh BW, Kong Q, Weisman GA, Beaudoin AR. Purine signaling and potential new therapeutic approach: possible outcomes of NTPDase inhibition. Curr Drug Targets. 2002;3(3):229–245. doi: 10.2174/1389450023347713. [DOI] [PubMed] [Google Scholar]
- 9.Cunha RA, Sebastiao AM, Ribeiro JA (1998) Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J Neurosci 18(6):1987–1995 [DOI] [PMC free article] [PubMed]
- 10.Baqi Y. Ecto-nucleotidase inhibitors: recent developments in drug discovery. Mini-Rev Med Chem. 2015;15(1):21–33. doi: 10.2174/1389557515666150219115141. [DOI] [PubMed] [Google Scholar]
- 11.al-Rashida M, Iqbal J. Therapeutic potentials of ecto-nucleoside triphosphate diphosphohydrolase, ecto-nucleotide pyrophosphatase/phosphodiesterase, ecto-5′-nucleotidase, and alkaline phosphatase inhibitors. Med Res Rev. 2014;34(4):703–743. doi: 10.1002/med.21302. [DOI] [PubMed] [Google Scholar]
- 12.Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci. 2015;191:117–123. doi: 10.1016/j.autneu.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Burnstock G, Vaughn B, Robson SC. Purinergic signalling in the liver in health and disease. Purinergic Signal. 2014;10(1):51–70. doi: 10.1007/s11302-013-9398-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burnstock G, Ralevic V. Purinergic signaling and blood vessels in health and disease. Pharmacol Rev. 2014;66(1):102–192. doi: 10.1124/pr.113.008029. [DOI] [PubMed] [Google Scholar]
- 15.Burnstock G, Pelleg A. Cardiac purinergic signalling in health and disease. Purinergic Signal. 2015;11(1):1–46. doi: 10.1007/s11302-014-9436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burnstock G, Evans LC, Bailey MA. Purinergic signalling in the kidney in health and disease. Purinergic Signal. 2014;10(1):71–101. doi: 10.1007/s11302-013-9400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal. 2014;10(4):529–564. doi: 10.1007/s11302-014-9427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnstock G. Purinergic signaling in the cardiovascular system. Circ Res. 2017;120(1):207–228. doi: 10.1161/CIRCRESAHA.116.309726. [DOI] [PubMed] [Google Scholar]
- 19.Burnstock G. Blood cells: an historical account of the roles of purinergic signalling. Purinergic Signal. 2015;11(4):411–434. doi: 10.1007/s11302-015-9462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burnstock G. Purinergic signalling in the urinary tract in health and disease. Purinergic Signal. 2014;10(1):103–155. doi: 10.1007/s11302-013-9395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burnstock G. Purinergic signalling in endocrine organs. Purinergic Signal. 2014;10(1):189–231. doi: 10.1007/s11302-013-9396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson KE. Purinergic signalling in the urinary bladder. Auton Neurosci. 2015;191:78–81. doi: 10.1016/j.autneu.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Burnstock G. Purinergic signalling in the gut. Adv Exp Med Biol. 2016;891:91–112. doi: 10.1007/978-3-319-27592-5_10. [DOI] [PubMed] [Google Scholar]
- 24.Sanderson J, Dartt DA, Trinkaus-Randall V, Pintor J, Civan MM, Delamere NA, Fletcher EL, Salt TE, Grosche A, Mitchell CH. Purines in the eye: recent evidence for the physiological and pathological role of purines in the RPE, retinal neurons, astrocytes, Muller cells, lens, trabecular meshwork, cornea and lacrimal gland. Exp Eye Res. 2014;127:270–279. doi: 10.1016/j.exer.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwiebert EM, Kishore BK. Extracellular nucleotide signaling along the renal epithelium. Am J Physiol Ren Physiol. 2001;280(6):F945–F963. doi: 10.1152/ajprenal.2001.280.6.F945. [DOI] [PubMed] [Google Scholar]
- 26.Vallon V, Rieg T (2011) Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Ren Physiol 301(3):F463–F475. 10.1152/ajprenal.00236.2011 [DOI] [PMC free article] [PubMed]
- 27.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367(24):2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sitkovsky M, Ohta A. Targeting the hypoxia-adenosinergic signaling pathway to improve the adoptive immunotherapy of cancer. J Mol Med. 2013;91(2):147–155. doi: 10.1007/s00109-013-1001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cronstein BN, Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol. 2017;13(1):41–51. doi: 10.1038/nrrheum.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasko G, Sitkovsky MV, Szabo C. Immunomodulatory and neuroprotective effects of inosine. Trends Pharmacol Sci. 2004;25(3):152–157. doi: 10.1016/j.tips.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Ohta A, Sitkovsky M (2011, 200) Methylxanthines, inflammation, and cancer: fundamental mechanisms. Handb Exp Pharmacol:469–481. 10.1007/978-3-642-13443-2_19 [DOI] [PubMed]
- 32.Schuler PJ, Westerkamp AM, Kansy BA, Bruderek K, Dissmann PA, Dumitru CA, Lang S, Jackson EK, Brandau S. Adenosine metabolism of human mesenchymal stromal cells isolated from patients with head and neck squamous cell carcinoma. Immunobiology. 2017;222(1):66–74. doi: 10.1016/j.imbio.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 33.Jackson EK, Kotermanski SE, Menshikova EV, Dubey RK, Jackson TC, Kochanek PM. Adenosine production by brain cells. J Neurochem. 2017;141(5):676–693. doi: 10.1111/jnc.14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norton ED, Jackson EK, Virmani R, Forman MB. Effect of intravenous adenosine on myocardial reperfusion injury in a model with low myocardial collateral blood flow. Am Heart J. 1991;122(5):1283–1291. doi: 10.1016/0002-8703(91)90567-2. [DOI] [PubMed] [Google Scholar]
- 35.Jackson EK, Dubey RK. Role of the extracellular cAMP-adenosine pathway in renal physiology. Am J Physiol Ren Physiol. 2001;281(4):F597–F612. doi: 10.1152/ajprenal.2001.281.4.F597. [DOI] [PubMed] [Google Scholar]
- 36.Dubey RK, Fingerle J, Gillespie DG, Mi Z, Rosselli M, Imthurn B, Jackson EK. Adenosine attenuates human coronary artery smooth muscle cell proliferation by inhibiting multiple signaling pathways that converge on cyclin D. Hypertension. 2015;66(6):1207–1219. doi: 10.1161/hypertensionaha.115.05912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson EK, Cheng D, Tofovic SP, Mi Z. Endogenous adenosine contributes to renal sympathetic neurotransmission via postjunctional A1 receptor-mediated coincident signaling. Am J Physiol Ren Physiol. 2012;302(4):F466–F476. doi: 10.1152/ajprenal.00495.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson EK, Cheng D, Mi Z, Verrier JD, Janesko-Feldman K, Kochanek PM. Role of A1 receptors in renal sympathetic neurotransmission in the mouse kidney. Am J Physiol Ren Physiol. 2012;303(7):F1000–F1005. doi: 10.1152/ajprenal.00363.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villa-Bellosta R, Hamczyk MR, Andres V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am J Phys Cell Phys. 2016;310(10):C788–C799. doi: 10.1152/ajpcell.00370.2015. [DOI] [PubMed] [Google Scholar]
- 40.Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acin-Perez R, Enriquez JA, Lopez-Otin C, Andres V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127(24):2442–2451. doi: 10.1161/CIRCULATIONAHA.112.000571. [DOI] [PubMed] [Google Scholar]
- 41.Villa-Bellosta R, Sorribas V. Prevention of vascular calcification by polyphosphates and nucleotides—role of ATP. Circ J. 2013;77(8):2145–2151. doi: 10.1253/circj.CJ-13-0016. [DOI] [PubMed] [Google Scholar]
- 42.Fish RS, Klootwijk E, Tam FW, Kleta R, Wheeler DC, Unwin RJ, Norman J. ATP and arterial calcification. Eur J Clin Investig. 2013;43(4):405–412. doi: 10.1111/eci.12055. [DOI] [PubMed] [Google Scholar]
- 43.Coolen EJ, Arts IC, Swennen EL, Bast A, Stuart MA, Dagnelie PC. Simultaneous determination of adenosine triphosphate and its metabolites in human whole blood by RP-HPLC and UV-detection. J Chromatography B: Analytical Technol Biomed Life Sci. 2008;864(1-2):43–51. doi: 10.1016/j.jchromb.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 44.Ipata PL, Camici M, Micheli V, Tozz MG. Metabolic network of nucleosides in the brain. Curr Top Med Chem. 2011;11(8):909–922. doi: 10.2174/156802611795347555. [DOI] [PubMed] [Google Scholar]
- 45.Zabielska MA, Borkowski T, Slominska EM, Smolenski RT. Inhibition of AMP deaminase as therapeutic target in cardiovascular pathology. Pharmacol Rep. 2015;67(4):682–688. doi: 10.1016/j.pharep.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Moriwaki Y, Yamamoto T, Higashino K. Enzymes involved in purine metabolism—a review of histochemical localization and functional implications. Histol Histopathol. 1999;14(4):1321–1340. doi: 10.14670/HH-14.1321. [DOI] [PubMed] [Google Scholar]
- 47.Levitt B, Head RJ, Westfall DP. High-pressure liquid chromatographic-fluorometric detection of adenosine and adenine nucleotides: application to endogenous content and electrically induced release of adenyl purines in guinea pig vas deferens. Anal Biochem. 1984;137(1):93–100. doi: 10.1016/0003-2697(84)90352-X. [DOI] [PubMed] [Google Scholar]
- 48.Haink G, Deussen A. Liquid chromatography method for the analysis of adenosine compounds. Journal of Chromatography B: Analytical Technologies in the Biomedical & Life Sciences. 2003;784(1):189–193. doi: 10.1016/S1570-0232(02)00752-3. [DOI] [PubMed] [Google Scholar]
- 49.Tatur S, Kreda S, Lazarowski E, Grygorczyk R. Calcium-dependent release of adenosine and uridine nucleotides from A549 cells. Purinergic Signal. 2008;4(2):139–146. doi: 10.1007/s11302-007-9059-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackson EK, Mi Z, Koehler MT, Carcillo J, Herzer WA. Injured erythrocytes release adenosine deaminase into the circulation. J Pharmacol Exp Ther. 1996;279(3):1250–1260. [PubMed] [Google Scholar]
- 51.Clark RS, Carcillo JA, Kochanek PM, Obrist WD, Jackson EK, Mi Z, Wisneiwski SR, Bell MJ, Marion DW. Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans. Neurosurgery. 1997;41(6):1284–1293. doi: 10.1097/00006123-199712000-00010. [DOI] [PubMed] [Google Scholar]
- 52.Todorov LD, Mihaylova-Todorova S, Westfall TD, Sneddon P, Kennedy C, Bjur RA, Westfall DP. Neuronal release of soluble nucleotidases and their role in neurotransmitter inactivation. Nature. 1997;387(6628):76–79. doi: 10.1038/387076a0. [DOI] [PubMed] [Google Scholar]
- 53.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110(7):993–1002. doi: 10.1172/jci15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP (2003) Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med 198(5):783–796. 10.1084/jem.20030891 [DOI] [PMC free article] [PubMed]
- 55.Bobalova J, Mutafova-Yambolieva VN. Membrane-bound and releasable nucleotidase activities: differences in canine mesenteric artery and vein. Clin Exp Pharmacol Physiol. 2003;30(3):194–202. doi: 10.1046/j.1440-1681.2003.03808.x. [DOI] [PubMed] [Google Scholar]
- 56.Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sevigny J, Gear A, Weber AA, Molojavyi A, Ding Z, Weber C, Ley K, Zimmermann H, Godecke A, Schrader J. Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95(8):814–821. doi: 10.1161/01.RES.0000144796.82787.6f. [DOI] [PubMed] [Google Scholar]
- 57.Diniz C, Fresco P, Goncalves J. Regional differences in extracellular purine degradation in the prostatic and epididymal portions of the rat vas deferens. Clin Exp Pharmacol Physiol. 2005;32(9):721–727. doi: 10.1111/j.1440-1681.2005.04252.x. [DOI] [PubMed] [Google Scholar]
- 58.Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am J Phys Cell Phys. 2011;301(2):C530–C539. doi: 10.1152/ajpcell.00385.2010. [DOI] [PubMed] [Google Scholar]
- 59.Burghoff S, Gong X, Viethen C, Jacoby C, Flogel U, Bongardt S, Schorr A, Hippe A, Homey B, Schrader J. Growth and metastasis of B16-F10 melanoma cells is not critically dependent on host CD73 expression in mice. BMC Cancer. 2014;14:898. doi: 10.1186/1471-2407-14-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caiazzo E, Maione F, Morello S, Lapucci A, Paccosi S, Steckel B, Lavecchia A, Parenti A, Iuvone T, Schrader J, Ialenti A, Cicala C. Adenosine signalling mediates the anti-inflammatory effects of the COX-2 inhibitor nimesulide. Biochem Pharmacol. 2016;112:72–81. doi: 10.1016/j.bcp.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 61.Hesse J, Leberling S, Boden E, Friebe D, Schmidt T, Ding Z, Dieterich P, Deussen A, Roderigo C, Rose CR, Floss DM, Scheller J, Schrader J. CD73-derived adenosine and tenascin-C control cytokine production by epicardium-derived cells formed after myocardial infarction. FASEB J. 2017;31(7):3040–3053. doi: 10.1096/fj.201601307R. [DOI] [PubMed] [Google Scholar]
- 62.Dubey RK, Gillespie DG, Mi Z, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation. 1997;96:2656–2666. doi: 10.1161/01.CIR.96.8.2656. [DOI] [PubMed] [Google Scholar]
- 63.Zhu X, Gillespie DG, Jackson EK. NPY1-36 and PYY1-36 activate cardiac fibroblasts: an effect enhanced by genetic hypertension and inhibition of dipeptidyl peptidase 4. Am J Physiol Heart Circ Physiol. 2015;309(9):H1528–H1542. doi: 10.1152/ajpheart.00070.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu X, Jackson EK. RACK1 regulates angiotensin II-induced contractions of SHR preglomerular vascular smooth muscle cells. Am J Physiol Ren Physiol. 2017;312(4):F565–f576. doi: 10.1152/ajprenal.00547.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Inoue T, Mi Z, Gillespie DG, Jackson EK. Cyclooxygenase inhibition reveals synergistic action of vasoconstrictors on mesangial cell growth. Eur J Pharmacol. 1998;361(2–3):285–291. doi: 10.1016/S0014-2999(98)00720-1. [DOI] [PubMed] [Google Scholar]
- 66.Jackson EK, Mi Z, Kleyman TR, Cheng D. 8-Aminoguanine induces diuresis, natriuresis, and glucosuria by inhibiting purine nucleoside phosphorylase and reduces potassium excretion by inhibiting Rac1. J Am Heart Assoc. 2018;7(21):e010085. doi: 10.1161/jaha.118.010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Ther. 2000;88(3):349–425. doi: 10.1016/S0163-7258(00)00097-8. [DOI] [PubMed] [Google Scholar]
- 68.Stachelska-Wierzchowska A, Wierzchowski J, Bzowska A, Wielgus-Kutrowska B. Tricyclic nitrogen base 1,N6-ethenoadenine and its ribosides as substrates for purine-nucleoside phosphorylases: spectroscopic and kinetic studies. Nucleosides Nucleotides Nucleic Acids. 2018;37(2):89–101. doi: 10.1080/15257770.2017.1419255. [DOI] [PubMed] [Google Scholar]
- 69.Chern JW, Lee HY, Chen CS, Shewach DS, Daddona PE, Townsend LB. Nucleosides. 5. Synthesis of guanine and formycin B derivatives as potential inhibitors of purine nucleoside phosphorylase. J Med Chem. 1993;36(8):1024–1031. doi: 10.1021/jm00060a010. [DOI] [PubMed] [Google Scholar]
- 70.Gandhi V, Kilpatrick JM, Plunkett W, Ayres M, Harman L, Du M, Bantia S, Davisson J, Wierda WG, Faderl S, Kantarjian H, Thomas D. A proof-of-principle pharmacokinetic, pharmacodynamic, and clinical study with purine nucleoside phosphorylase inhibitor immucillin-H (BCX-1777, forodesine) Blood. 2005;106(13):4253–4260. doi: 10.1182/blood-2005-03-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bjelobaba I, Janjic MM, Stojilkovic SS. Purinergic signaling pathways in endocrine system. Auton Neurosci. 2015;191:102–116. doi: 10.1016/j.autneu.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36(3):293–303. doi: 10.1038/onc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gohar EY, Kasztan M, Pollock DM. Interplay between renal endothelin and purinergic signaling systems. Am J Physiol Ren Physiol. 2017;313(3):F666–F668. doi: 10.1152/ajprenal.00639.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kling L, Kramer BK, Yard BA, Kalsch AI. The adenosinergic system: a potential player in the pathogenesis of ANCA-associated vasculitis? Clin Exp Rheumatol. 2018;36 Suppl 111(2):143–151. [PubMed] [Google Scholar]
- 75.Koles L, Furst S, Illes P. Purine ionotropic (P2X) receptors. Curr Pharm Des. 2007;13(23):2368–2384. doi: 10.2174/138161207781368747. [DOI] [PubMed] [Google Scholar]
- 76.Longhi MS, Moss A, Jiang ZG, Robson SC. Purinergic signaling during intestinal inflammation. J Mol Med. 2017;95(9):915–925. doi: 10.1007/s00109-017-1545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinez-Ramirez AS, Diaz-Munoz M, Butanda-Ochoa A, Vazquez-Cuevas FG. Nucleotides and nucleoside signaling in the regulation of the epithelium to mesenchymal transition (EMT) Purinergic Signal. 2017;13(1):1–12. doi: 10.1007/s11302-016-9550-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ochoa-Cortes F, Linan-Rico A, Jacobson KA, Christofi FL. Potential for developing purinergic drugs for gastrointestinal diseases. Inflamm Bowel Dis. 2014;20(7):1259–1287. doi: 10.1097/MIB.0000000000000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Strazzulla LC, Cronstein BN. Regulation of bone and cartilage by adenosine signaling. Purinergic Signal. 2016;12(4):583–593. doi: 10.1007/s11302-016-9527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun K, Liu H, Song A, Manalo JM, D'Alessandro A, Hansen KC, Kellems RE, Eltzschig HK, Blackburn MR, Roach RC, Xia Y. Erythrocyte purinergic signaling components underlie hypoxia adaptation. J Appl Physiol. 2017;123(4):951–956. doi: 10.1152/japplphysiol.00155.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeiser R, Robson SC, Vaikunthanathan T, Dworak M, Burnstock G. Unlocking the potential of Purinergic signaling in transplantation. Am J Transplant. 2016;16(10):2781–2794. doi: 10.1111/ajt.13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Menzies RI, Tam FW, Unwin RJ, Bailey MA. Purinergic signaling in kidney disease. Kidney Int. 2017;91(2):315–323. doi: 10.1016/j.kint.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 83.Lee JS, Yilmaz O (2018) Unfolding role of a danger molecule adenosine signaling in modulation of microbial infection and host cell response. Int J Mol Sci 19(1). 10.3390/ijms19010199 [DOI] [PMC free article] [PubMed]
- 84.Nishimura A, Sunggip C, Oda S, Numaga-Tomita T, Tsuda M, Nishida M. Purinergic P2Y receptors: molecular diversity and implications for treatment of cardiovascular diseases. Pharmacol Ther. 2017;180:113–128. doi: 10.1016/j.pharmthera.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 85.Hausmann R, Kless A, Schmalzing G. Key sites for P2X receptor function and multimerization: overview of mutagenesis studies on a structural basis. Curr Med Chem. 2015;22(7):799–818. doi: 10.2174/0929867322666141128163215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trincavelli ML, Daniele S, Martini C. Adenosine receptors: what we know and what we are learning. Curr Top Med Chem. 2010;10(9):860–877. doi: 10.2174/156802610791268756. [DOI] [PubMed] [Google Scholar]
- 87.Durnin L, Moreland N, Lees A, Mutafova-Yambolieva VN. A commonly used ecto-ATPase inhibitor, ARL-67156, blocks degradation of ADP more than the degradation of ATP in murine colon. Neurogastroenterology and Motility : the official journal of the European Gastrointestinal Motility Society. 2016;28(9):1370–1381. doi: 10.1111/nmo.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wehbe K, Vezzalini M, Cinque G. Detection of mycoplasma in contaminated mammalian cell culture using FTIR microspectroscopy. Anal Bioanal Chem. 2018;410(12):3003–3016. doi: 10.1007/s00216-018-0987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 374 kb)