

Summary
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spillover infection in December 2019 has caused an unprecedented pandemic. SARS-CoV-2, as other coronaviruses, binds its target cells through the angiotensin-converting enzyme 2 (ACE2) receptor. Accordingly, this makes ACE2 research essential for understanding the zoonotic nature of coronaviruses and identifying novel drugs. Here we present a systematic analysis of the ACE2 conservation and co-evolution protein network across 1,671 eukaryotes, revealing an unexpected conservation pattern in specific metazoans, plants, fungi, and protists. We identified the co-evolved protein network and pinpointed a list of drugs that target this network by using data integration from different sources. Our computational analysis found widely used drugs such as nonsteroidal anti-inflammatory drugs and vasodilators. These drugs are expected to perturb the ACE2 network affecting infectivity as well as the pathophysiology of the disease.
Subject Areas: Classification of Proteins, Evolutionary Mechanisms, Virology
Graphical Abstract
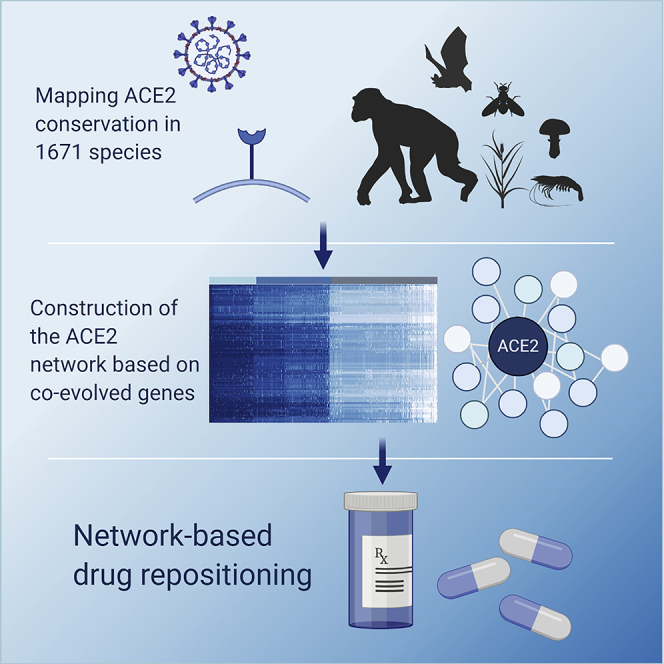
Highlights
-
•
Mapping the ACE2 conservation pattern in 146 mammal reservoirs and 1,671 eukaryotes
-
•
Identification of genes that show a similar evolutionary pattern as ACE2
-
•
Generation of an ACE2 protein network using co-evolution and data integration
-
•
Drugs-to-network analyses mapped 145 drugs that perturbed the ACE2 network
Classification of Proteins; Evolutionary Mechanisms; Virology
Introduction
Coronaviruses are a family of enveloped positive-stranded RNA viruses that have affected humans and many other mammals for over the last half century. To date, there are seven coronaviruses known to infect humans, four of which are circulating strains that are responsible for approximately 10%–30% of the cases of common cold (Paules et al., 2020). The other three strains can cause a potentially fatal disease in humans (severe acute respiratory syndrome coronavirus [SARS-CoV], Middle East respiratory syndrome coronavirus [MERS-CoV], and SARS-CoV-2). These seven strains originate from a common ancestor in bat viruses and are transferred to humans through intermediate animal hosts. SARS-CoV, the cause of a serious respiratory disease outbreak in 2002–2003, was transmitted through civet cats, whereas MERS-CoV, the cause of the 2012 outbreak, was transmitted through dromedary camels (Cui et al., 2019). Overall, more than 500 coronaviruses have been identified in bats in China, with estimates of more than 3,000 unknown bat coronavirus strains (Anthony et al., 2017).
In December 2019, a novel coronavirus was identified as the pathogen responsible for an outbreak of a severe infectious respiratory disease in Wuhan, China (Zhu et al., 2020). The virus was named SARS-CoV-2, causing the coronavirus disease COVID-19. In March 2020, the World Health Organization declared the COVID-19 outbreak as a pandemic, and immense efforts around the world are in progress to develop therapeutics. Due to the association of the earliest COVID-19 cases with the Huanan market in Wuhan, it is most plausible that the SARS-CoV-2 also emerged from an animal source. The intermediate hosts and the transmission chain in the SARS-CoV-2 spillover are yet to be elucidated. The closest SARS-like coronavirus found was sampled from the intermediate horseshoe bat (Rhinolophus affinis), with an ∼97% identity to SARS-CoV-2. However, it displayed lesser affinity for the angiotensin-converting enzyme 2 (ACE2) receptor, identified as the key player in the SARS-CoV-2 cell entry mechanism (Hoffmann et al., 2020; Zhou et al., 2020), when compared with the coronavirus isolated from the Malayan pangolin (Manis Javanica) found in the Guangdong province, which also displays a high homology to the human virus (Andersen et al., 2020; Zhang et al., 2020).
To date, there is no approved treatment or vaccine against SARS-CoV-2. In addition, the variability in disease severity, coupled with the fatality rates among people and in different human populations, raises the possibility that there are genetic, environmental, nutritional, or medication usage differences (in addition to age at infection) that affect the pathophysiology of the infection. Although the complex mechanism of the disease remains an important unanswered question, substantial evidence identifies pathways of inflammation and circulation as the main drivers of morbidity (Marini and Gattinoni, 2020; Varga et al., 2020). There is an urgent need to identify other risk factors, including chronic use of certain medications, in addition to development of novel drugs. The development of new drugs is a slow, costly process, and antiviral medications are especially challenging due to the rapid evolution of virus genomes. One effective strategy is drug repurposing, which involves finding new uses for existing Food and Drug Administration (FDA)-approved drugs (Strittmatter, 2014; Pushpakom et al., 2018; Pizzorno et al., 2019). This strategy can reduce both the cost and time it would take to develop and acquire approval for COVID-19 treatments when compared with de novo drug discovery. Equally important, the study of known drugs might reveal those that can increase the risk for infection and worsen the outcome (Berghauser Pont et al., 2015; Thackray et al., 2018). This has extraordinary significance for drugs that are commonly used by millions of people in the world during this pandemic.
Viruses depend on their hosts to reproduce and can only survive by hijacking and rewiring the human cells' protein and gene networks. Specifically, SARS-CoV-2 binds to the cell membrane ACE2 receptor to enter human cells (Hoffmann et al., 2020; Zhou et al., 2020) and subsequently replicates inside, thus marking ACE2 as a potential target for therapeutic intervention. It may also be logically assumed that cellular pathways that were conserved during the evolution in association with the ACE2 receptor might have a role in the pathogenesis of the disease. Drugs that perturb the ACE2 network might affect the coronavirus infectivity or the severity of the diseases.
Recent work analyzed the ACE2 network using different experimental methodologies and focused on establishing the protein network (Gordon et al., 2020; Sinha et al., 2020). Although integration of this information is very beneficial, a comparative genomics approach can identify functional associations that occur across the tree of life and are not only limited to humans. Our approach to map the ACE2 network and evolution using phylogenetic profiling (PP) is independent and complementary to protein-protein interactions (PPI) (Gordon et al., 2020), or expression-based approaches (Sinha et al., 2020).
PP is an unbiased method for identifying genetic interactions through comparative genomics of thousands of organisms (Pellegrini et al., 1999). The PP of a gene describes its evolutionary conservation (in terms of sequence similarity) throughout the tree of life. If a set of genes has similar PPs, meaning their loss (sequence divergence) and retention patterns are co-dependent, they are likely to interact (Tabach et al., 2013a, 2013b; Sadreyev et al., 2015; Sherill-Rofe et al., 2019; Bloch et al., 2020).
Here we offer a novel approach for drug selection and contraindications for COVID-19. Co-evolution between proteins suggests that they are functionally coupled (Pellegrini et al., 1999). Complementary to PPI maps, perturbation of genes co-evolved with ACE2 might influence the expression, activity, or downstream target of ACE2. Consequently, viral infectivity and disease progression may be affected. Our analysis covers thousands of FDA-approved drugs to optimize the list of drugs ready for validation that can rapidly be deployed to treat the COVID-19 epidemic.
Results
Characterization of ACE2 Phylogenetic Profiling and Conservation Pattern along 1,671 Eukaryotes
We mapped the ACE2 best BLASTP bit-score homologs across 1,671 eukaryotes, calculated their sequence conservation, and analyzed their protein domains compared with the human ACE2 (Figure 1) (Altschul et al., 1990). The domain analysis was conducted using the NCBI Batch Conserved Domains-Search Tool (Figures S1 and S2 and Table S1) to further assess the changes in domain structure or conservation among the species and achieve a more precise evolutionary analysis (Marchler-Bauer and Bryant, 2004).
Figure 1.

Evolutionary Pattern of ACE2 Proteins across 1,671 Species
The percentage of sequence similarity (y axis) to the human ACE2 protein when compared with the most similar orthologs across all eukaryotes (x axis) is presented. The Conservation score is defined as Pab/Paa, where Pab is the best BLASTP bit-score between a Homo sapiens protein “a” and all open reading frames of a eukaryote genome “b.” Paa is marked as the self-similarity score of the H. sapiens protein “a” when blasted against itself. Organisms with scores that deviated from their clade-average conservation rate are highlighted with the corresponding name. See also Figures S1 and S2, and Table S1.
Overall, ACE2 is conserved across the majority of the animal kingdom, with a relatively high conservation percent within vertebrates, especially mammals. As expected, the conservation rate of ACE2 decreases as the evolutionary distance increases in relation to Homo sapiens. This suggests the majority of the animal kingdom as potential intermediate hosts for SARS-CoV-2. Interestingly, more than 10 mammals show high divergence when compared with their clade, implying a possible protective effect from SARS-CoV-2 that interacts with ACE2. These species include Odobenus rosmarus, Castor canadensis, Odocoileus virginianus, Bison bison, Galeopterus variegatus, Meriones unguiculatus, Leptonychotes weddellii, Ornithorhynchus anatinus, Procavia capensis, Cavia aperea, Tupaia belangeri, Notamacropus eugenii, and Choloepus hoffmanni. Importantly, as far as we are aware, none of these species are known to be infected by SARS-CoV-2 through ACE2. An additional interesting observation is the lower conservation of Ciona savignyi within the chordates' clade, which may also suggest divergence from the known human ACE2 function in this species. When exploring eukaryotes that further diverge from human-like fungi, plants, and different protists, there are no orthologs for ACE2. The exceptions are one green algae (Ostreococcus tauri), five fungi (Spizellomyces punctatus, Gonapodya prolifera, Allomyces macrogynus, Catenaria anguillulae, Puccinia striiformis), and two protists (Capsaspora owczarzaki and Thecamonas trahens). This observation was unexpected as ACE2 generally exists almost only in metazoans and may imply a horizontal gene transfer along the evolution. Taken together, these results point to some species for further analysis as suspected SARS-CoV-2 intermediate hosts.
Mapping Proteins that Show Similar Phylogenetic Profiling with ACE2 and Generating the ACE2 Co-evolved Network
Co-evolution across the tree of life can predict proteins that are associated with the same function, share common pathways, and contribute to corresponding diseases (Tabach et al., 2013a, 2013b; Omar et al., 2018; Arkadir et al., 2019; Bauer et al., 2019; Sherill-Rofe et al., 2019). To map the proteins that co-evolved with ACE2, we generated an extensive database of 1,671 eukaryotic proteomes and identified the set of genes that showed similar phylogenetic profiles to ACE2 (Figures 2A and S3). For each human protein, we identified the genes with the best BLASTP bit-score in each of the organisms in the database. Then we normalized the data to account for protein length and phylogenetic distance from humans (see Experimental Procedures) to generate the normalized phylogenetic profiling (NPP). We ranked all genes according to their similarity to the ACE2 profile as measured by Pearson correlation after normalization. As ACE2 is mainly conserved in vertebrates we used a clade-specific approach, which showed a higher sensitivity in detecting gene function when compared with global PP approaches (Sherill-Rofe et al., 2019). We identified ACE2 co-evolved genes in eukaryotes, metazoans, chordates, and mammals clades that contain 1,671, 688, 369, and 146 organisms, respectively (Tables S2 and S3). Finally, the top genes were further analyzed for functional enrichment.
Figure 2.

Evolution of the ACE2 Protein
(A) The top 100 proteins that co-evolved with ACE2, and its phylogenetic profiles across 1,671 eukaryotes. The heatmap represents the top 100 genes most correlated with ACE2, ranked by Pearson correlation of the normalized profile (log2 transformation and Z scoring). The rows are ordered based on hierarchical clustering (by the UPGMA method). Each row represents a single gene across 1,671 eukaryotes ordered by their phylogenetic distance from Homo sapiens. Each column represents a species. The colors indicate the relative degree of conservation from high similarity (dark blue) to not conserved (white).
(B) Enrichment analysis of the top 100 genes most correlated with ACE2 in eukaryotes, metazoans, chordates, and mammals. The top gene ontology terms and biological processes correlated in at least two clades were chosen for visualization. The color scale shows the p value cutoff levels for each biological process or disease. Deeper colors represent higher significance.
Analysis of the top 100 most correlated genes with ACE2 in each clade revealed enrichment of common biological pathways, gene ontology (GO) biological processes, and diseases (Figure 2B) (Ben-Ari Fuchs et al., 2016). As expected, the results substantially overlapped between the four groups, stressing the similarity between them. Among the biological annotations were metabolism, membranes, endothelial barrier, ion transport, and mTOR signaling pathway. Genetic associations with the following conditions were identified as: hypertension, diabetes mellitus, myocardial infarction, and lipid metabolism disorder. All the aforementioned conditions are suspected risk factors for the severe manifestation of COVID-19 (Chen et al., 2020). These results demonstrate our ability to recapitulate the function of ACE2 and risk factors for COVID-19 infection disease using an unbiased phylogenetic profile analysis.
Identifying Drugs that Can Target the ACE2 Co-evolved Network
After establishing our ability to map disease risk factors in relation to COVID-19 pandemic, we then identified drugs that could affect the ACE2 network. For this purpose, we cross-referenced the 400 highest scoring genes (top 100 for each clade) with databases of drug-gene interactions (DGIdb, Cotto et al., 2018; DrugBank, Wishart et al., 2018) to identify druggable genes. We obtained 729 optional drugs already approved by the FDA, and we further prioritized drugs according to clinical consideration (see Experimental Procedures), which resulted in 145 molecular entities (Table S4). Representative connections of the network clade-gene-drug condition are depicted in Figure 3A. Analysis of the ACE2 network using STRING (Szklarczyk et al., 2019) shows that a large number of the co-evolved interactions also have supporting data for expression and proteomics in the literature (Figure 3B).
Figure 3.

Representative Connections between Clade-Gene-Drug-Condition
(A) Sankey diagram of the genes that are co-evolved with ACE2 in four clades and the drugs targeting them.
(B) A subnetwork of ACE2 highlighting the genes with drug repositioning options. Node fill colors indicate subgroup affiliations inside the network: genes related to inflammation, drug metabolism, glucose metabolism, and vasodilators. Edge stroke colors represent six types of experimental evidence of the protein-protein interactions.
See also Table S4.
The identified 145 drugs were divided into four main categories: anti-inflammatory drugs, vasodilators, diabetes medications, and drugs targeting drug metabolism pathways, according to their clinical indication. The first group was identified with various genes playing a major role in pathways of immune response and inflammation including COX-2 (PTGS2), SLC5A8, PIK3R3, ADAMTS1, and ABCG2, which tightly co-evolved in eukaryotes and metazoans with ACE2 (Figure 3). Among the drugs targeting the aforementioned genes are the commonly used nonsteroidal anti-inflammatory drugs (NSAIDs) and inhibitors of COX-2, such as ibuprofen, aspirin, and etoricoxib. Interestingly, ADAMTS1 is targeted by statins, which are widely prescribed medications also known to have an immunomodulatory effect (Vaughan et al., 1996). In addition, ABCG2 is targeted by telaprevir and cyclosporine. The former is a direct-acting antiviral drug classified as a protease inhibitor used to treat hepatitis C (Zeuzem et al., 2011), whereas the latter inhibits viral replication of the human immunodeficiency virus and hepatitis C virus (Xue et al., 2018).
The second appealing group of drugs in our analysis was the vasoactive drugs. Notably, a group of genes closely correlated with ACE2 in our analysis were PDE6, KCNA10, SLC15A2, and ROCK2 (Figure 3). These genes are targeted by a group of drugs with vasodilatory properties and are in widespread clinical use for hypertension, the calcium channel blockers, such as nisoldipine, verapamil, and fasudil. An additional class of drugs targeting the aforementioned genes is commonly used to treat pulmonary arterial hypertension, the phosphodiesterase (PDE) inhibitors, such as sildenafil, dypridamole, and pentoxifyilline. Notably, nitric oxide (NO), a potent vasodilator, plays an important role in regulating airway function and in treating inflammatory airway diseases (Barnes, 1995) and is utilized in the treatment of COVID-19 infections.
Overall, a systematic analysis of the top 100 highest scoring genes in each clade with databases of drug-gene interactions identified 145 known drug targets of FDA-approved drugs (Table S4). The connectivity of gene-drug interactions enabled the generation of a list of drugs to target the ACE2 network with potential impacts on medical care in fighting the current evolving COVID-19 epidemic.
Discussion
A major problem with coronaviruses is the repeated spillovers from the extensive animal reservoir of the virus. The conservation of ACE2 across animals probably allows high cross-species infection, which can lead to a variety of different habitats for the virus to evolve and interact with the human population. Here we present a comprehensive evolutionary analysis of ACE2. Among 1,671 organisms, we identified those that have ACE2 and the ones that are more conserved and are more likely to serve as a potential reservoir for the virus. In addition, using NPP of hundreds of species, we identified for the first time the proteins that co-evolved with ACE2. These proteins that coordinately evolve with ACE2 fit our understanding of the mechanism of action and the chronic diseases increasing the risk of COVID-19. These genes may serve as a starting point for future research to determine the modulation of ACE2 expression and function, and eventually ways to intervene pharmacologically.
PP is a robust method that identifies strong associations between genes based on their co-evolutionary course. Although the co-evolutionary signal is highly potent, PP cannot provide information about the context, type of interaction, or the directionality of the association between the genes. Future work integrating data from additional sources is needed, such as PPIs and gene expression analysis in SARS-CoV-2-infected cells with extensive validation. Nevertheless, considering the urgency to better understand the COVID-19 pandemic, and the fact that benchwork with SARS-CoV-2 is very limited, different computational analysis are a necessity.
Using an unbiased method to investigate our comparative genomics data, we identified an association between ACE2 and various cellular networks. Within these networks we found druggable targets that may influence susceptibility to poor outcomes of viral infection. Among the drugs are COX inhibitors and vasodilators, medications that might be particularly relevant as the severe course of COVID-19-related disease is characterized by uncontrolled inflammation and circulatory collapse. Indeed, with almost 10 million infected patients in the world, and extensive descriptions of the clinical course and the pathologic changes in the disease, it became more evident that endothelial damage and thromboembolic complications are involved in the severe course of the disease. The vasomotor and thromboembolic pathogenesis of severe COVID-19 probably accounts for the prevalent risk factors of obesity, diabetes, and hypertension, all of them being associated with endothelial dysfunction. These pathways were correctly identified by our analysis before the above data became available. These pathways are targeted by aspirin, statins, and vasodilators. The aforementioned classes of drugs are in extensive clinical use to treat cardiovascular and inflammatory conditions (e.g., over 30 billion doses of NSAIDs are consumed annually in the United States alone).
Awareness of the possible association between these medications and the pathways involved in COVID-19 infection might lead to significant clinical impacts to maximize their positive effect. In addition, an antiviral medication was found to be linked to the network and might be applied to the treatment of COVID-19, subject to appropriate clinical testing.
In conclusion, this is the first study using PP to identify ACE2 conservation and its co-evolved proteins across millions of years of evolution. Many of the drugs identified show a clear association with the disease, and their effect on patients should be examined. Overall, due to the urgent need for a comprehensive approach to treat COVID-19 during the current epidemic, we believe that our work provides a list of existing drugs to be evaluated. Nevertheless, additional data are needed to prove these observations.
Limitations of the Study
Computational and high-throughput approaches including PP analysis can provide a hint and suggest a mechanism. However, these observations, even when biologically sound, still may not be clinically relevant, and further extensive work is required.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Yuval Tabach (yuvaltab@ekmd.huji.ac.il).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Original data have been deposited to Mendeley Data: http://doi.org/10.17632/zrzh6x74th.4.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Funding was received from the Israel Science Foundation (grant agreement 1591/19), and the Israel Innovation Authority under the R&D Plans of Industrial Products for the Prevention and Treatment of the COVID-19 (grant agreement 70273). The graphical abstract was created by BioRender.com.
Author Contributions
Conceptualization, M.B. and Y.T.; Methodology, M.B., E.S., I.U., and Y.T.; Software, E.S. and I.U.; Formal Analysis, M.B.; Investigation, M.B., M.M., E.S., and I.U.; Resources, A.M.S. and A.V. Writing – Original Draft, M.B., E.S., and M.M.; Writing – Review & Editing, M.B., M.M., S.B., and Y.T.; Visualization, I.U., E.S., M.B., and M.M.; Supervision, Y.T.
Declaration of Interests
The authors declare no competing interests.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101384.
Supplemental Information
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990 doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andersen K.G., Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;89:44–48. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony S.J., Johnson C.K., Greig D.J., Kramer S., Che X., Wells H., Hicks A.L., Joly D.O., Wolfe N.D., Daszak P. Global patterns in coronavirus diversity. Virus Evol. 2017;3:1–15. doi: 10.1093/ve/vex012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkadir D., Lossos A., Rahat D., Abu Snineh M., Schueler-Furman O., Nitschke S., Minassian B.A., Sadaka Y., Lerer I., Tabach Y., Meiner V. MYORG is associated with recessive primary familial brain calcification. Ann. Clin. Transl. Neurol. 2019 doi: 10.1002/acn3.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P.J. Nitric oxide and airway disease. Ann. Med. 1995 doi: 10.3109/07853899509002592. [DOI] [PubMed] [Google Scholar]
- Bauer M., Rahat D., Zisman E., Tabach Y., Lossos A., Meiner V., Arkadir D. MYORG mutations: a major cause of recessive primary familial brain calcification. Curr. Neurol. Neurosci. Rep. 2019 doi: 10.1007/s11910-019-0986-z. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Fuchs S., Lieder I., Stelzer G., Mazor Y., Buzhor E., Kaplan S., Bogoch Y., Plaschkes I., Shitrit A., Rappaport N. GeneAnalytics: an integrative gene set analysis tool for next generation sequencing, RNAseq and microarray data. OMICS. 2016 doi: 10.1089/omi.2015.0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghauser Pont L.M., Balvers R.K., Kloezeman J.J., Nowicki M.O., van den Bossche W., Kremer A., Wakimoto H., van den Hoogen B.G., Leenstra S., Dirven C.M. In vitro screening of clinical drugs identifies sensitizers of oncolytic viral therapy in glioblastoma stem-like cells. Gene Ther. 2015 doi: 10.1038/gt.2015.72. [DOI] [PubMed] [Google Scholar]
- Bloch I., Sherill-Rofe D., Stupp D., Unterman I., Beer H., Sharon E., Tabach Y. Optimization of Co-evolution analysis through phylogenetic profiling reveals pathway-specific signals. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa281. [DOI] [PubMed] [Google Scholar]
- Chen T., Wu D., Chen H., Yan W., Yang D., Chen G., Ma K., Xu D., Yu H., Wang H. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020 doi: 10.1136/bmj.m1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotto K.C., Wagner A.H., Feng Y.Y., Kiwala S., Coffman A.C., Spies G., Wollam A., Spies N.C., Griffith O.L., Griffith M. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018 doi: 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019 doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020 doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically-proven protease inhibitor. Cell. 2020:1–10. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A., Bryant S.H. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004 doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini J.J., Gattinoni L. Management of COVID-19 respiratory distress. JAMA. 2020 doi: 10.1001/jama.2020.6825. [DOI] [PubMed] [Google Scholar]
- Omar I., Guterman-Ram G., Rahat D., Tabach Y., Berger M., Levaot N. Schlafen2 mutation in mice causes an osteopetrotic phenotype due to a decrease in the number of osteoclast progenitors. Sci. Rep. 2018 doi: 10.1038/s41598-018-31428-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules C.I., Marston H.D., Fauci A.S. Coronavirus infections-more than just the common cold. JAMA. 2020 doi: 10.1001/jama.2020.0757. [DOI] [PubMed] [Google Scholar]
- Pellegrini M., Marcotte E.M., Thompson M.J., Eisenberg D., Yeates T.O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. U S A. 1999 doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorno A., Padey B., Terrier O., Rosa-Calatrava M. Drug repurposing approaches for the treatment of influenza viral infection: reviving old drugs to fight against a long-lived enemy. Front. Immunol. 2019 doi: 10.3389/fimmu.2019.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018 doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- Sadreyev I.R., Ji F., Cohen E., Ruvkun G., Tabach Y. PhyloGene server for identification and visualization of co-evolving proteins using normalized phylogenetic profiles. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherill-Rofe D., Sherill-Rofe D., Rahat D., Findlay S., Mellul A., Guberman I., Braun M., Bloch I., Lalezari A., Samiei A. Mapping global and local coevolution across 600 species to identify novel homologous recombination repair genes. Genome Res. 2019 doi: 10.1101/gr.241414.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S., Cheng K., Aldape K., Schiff E., Ruppin E. Systematic cell line-based identification of drugs modifying ACE2 expression. Preprints. 2020 doi: 10.20944/PREPRINTS202003.0446.V1. [DOI] [Google Scholar]
- Strittmatter S.M. Overcoming drug development bottlenecks with repurposing: old drugs learn new tricks. Nat. Med. 2014 doi: 10.1038/nm.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019 doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y., Golan T., Hernández-Hernández A., Messer A.R., Fukuda T., Kouznetsova A., Liu J.G., Lilienthal I., Levy C., Ruvkun G. Human disease locus discovery and mapping to molecular pathways through phylogenetic profiling. Mol. Syst. Biol. 2013;9:1–17. doi: 10.1038/msb.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y., Billi A.C., Hayes G.D., Newman M.A., Zuk O., Gabel H., Kamath R., Yacoby K., Chapman B., Garcia S.M. Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature. 2013 doi: 10.1038/nature11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray L.B., Handley S.A., Gorman M.J., Poddar S., Bagadia P., Briseño C.G., Theisen D.J., Tan Q., Hykes B.L., Jr., Lin H. Oral antibiotic treatment of mice exacerbates the disease severity of multiple flavivirus infections. Cell Rep. 2018 doi: 10.1016/j.celrep.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., Mehra M.R., Schuepbach R.A., Ruschitzka F., Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020 doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan C.J., Murphy M.B., Buckley B.M. Statins do more than just lower cholesterol. Lancet. 1996 doi: 10.1016/S0140-6736(96)05190-2. [DOI] [PubMed] [Google Scholar]
- Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018 doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C., Sowden M.P., Berk B.C. Extracellular and intracellular cyclophilin A, native and post-translationally modified, show diverse and specific pathological roles in diseases. Arterioscler. Thromb. Vasc. Biol. 2018 doi: 10.1161/ATVBAHA.117.310661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuzem S., Andreone P., Pol S., Lawitz E., Diago M., Roberts S., Focaccia R., Younossi Z., Foster G.R., Horban A. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 2011 doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- Zhang T., Wu Q., Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020:1–6. doi: 10.1016/j.cub.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020 doi: 10.1056/nejmoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to Mendeley Data: http://doi.org/10.17632/zrzh6x74th.4.