

SUMMARY
Proteolysis targeting chimeras (PROTACs) is a paradigm shift for small molecule drug discovery. However, limited E3 ubiquitin ligase ligands with cellular activity are available. In-vitro binding assays involve the expression and purification of a large amount of proteins and they often yield ligands that are inactive in cell-based assays due to poor cell permeability, stability, and other reasons. We herein report the development of a practical and efficient cell-based target engagement assay to evaluate the binding affinity of a small library of cereblon ligands to its E3 ligase in cells. Selected cell permeable E3 ligase ligands derived from this assay are then used to construct HDAC6 degraders with cellular protein degradation activity. Because the assay does not involve any genetic engineering, it is relatively easy to transfer from one cell type to a different one.
Keywords: PROTAC, CRBN, E3 ligase, E3 ligand, Targeted protein degradation, Target Engagement
eTOC Blurb
Yang et al developed a cell-based target engagement assay for the identification of cereblon E3 ubiquitin ligase ligands, whose utility was demonstrated for the degradation of HDAC6. This strategy provides a practical and efficient alternative to the biochemical assay to evaluate the ligands of E3 ubiquitin ligases.
INTRODUCTION
Thalidomide and its analogues, such as pomalidomide and lenalidomide belong to the class of immunomodulatory drugs (IMiDs) for the treatment of multiple myeloma(Holstein et al., 2018) (Figure 1A). Thalidomide was once notorious for its teratogenic effect in infants when the drug was prescribed to pregnant women for relieving pregnancy nausea(Matyskiela et al., 2018a). After being withdrawn from the market, thalidomide and its analogues were found to possess multiple other functions such as anti-inflammation, anti-angiogenesis, and immune modulation, including T-cell co-stimulation and NK cell activation(Quach et al., 2010). After years of carefully designed clinical trials, thalidomide and several of its analogues were approved as IMiDs for the treatment of multiple myeloma(Palumbo et al., 2008). However, their molecular target and the mechanism of action of their anticancer effect were uncovered years later.
Figure 1. Experimental Design for Evaluating Binding Affinity of IMiDs.

(A) Structure of IMiDs. (B) Illustration of the principle for cell-based target engagement assay.
Cereblon (CRBN), a component of E3 complex with other proteins such as DDB1, was identified as the cellular target of IMiDs (Ito et al., 2010; Lopez-Girona et al., 2012). Upon binding to CRBN, the IMiDs can induce interactions between the ligand-bound CRBN E3 ligase and some of the physiologically irrelevant protein substrates including IKZF1, IKZF3, SALL4, and GSPT1 (Lu et al., 2014). IMiDs were therefore described as “molecular glues” for this type of mechanism of action and the above protein substrates are often termed as “neo-substrates” of CRBN. Specifically, the degradation of IKZF1/3 will downregulate cMYC, which is an essential transcription factor for the growth of multiple myeloma. The downregulation of IRF4/cMYC was proposed as the mechanism of action for the anti-myeloma activity of IMiDs.(Shaffer et al., 2008; Zhu et al., 2011) The degradation of SALL4 was proposed to be responsible for the teratogenic effect.(Donovan et al., 2018; Matyskiela et al., 2018a)
In addition to being used as “molecular glues”, thalidomide and its derivatives are widely used as the ligand of CRBN E3 ligase for the design of bifunctional proteolysis targeting chimera (PROTAC), which can efficiently recruit CRBN E3 ligase to a protein of interest (POI) and induce the polyubiquitination-followed by degradation of the POI(An et al., 2019; Bondeson et al., 2018; Nabet et al., 2018; Schiedel et al., 2018; Zhou et al., 2018) (Figure 1B). PROTAC is an emerging novel technology for the development of small molecule therapeutics that can induce the degradation of abnormally expressed or mutated disease-associated proteins(Pettersson and Crews, 2019). To expand the toolbox of targeted protein degradation (TPD), it is important to develop novel ligands for well-known and also novel E3 ligases. Due to the ease of synthesis of thalidomide and its derivatives, most reported PROTACs use them as the ligand to recruit CRBN E3 ligase. However, PROTACs based on thalidomide and its derivatives such as pomalidomide and lenalidomide can still degrade some of the neo-substrates, including IKZFs(Matyskiela et al., 2016; Nabet et al., 2018; Nowak et al., 2018; Zorba et al., 2018) or GSPT1(Yang et al., 2019). The off-target effect may lead to downstream effects that are irrelevant to the intended target of PROTACs and complicate many studies.
In-vitro binding assays involve the expression and purification of a large amount of proteins and they often yield ligands that are inactive in cell-based assays due to poor cell permeability, stability, and other reasons. For example, cereblon co-exists with many other protein partners in cells to form the functional E3 ubiquitin ligase complex and the binding mode of small molecule ligands to recombinant cereblon may not be the same as in the protein complex. It is important to ensure the engagement of the native stage of the protein target in the relevant cellular context for the development of chemical probes and therapeutics(Simon et al., 2013). Numerous assays are available for the study of target engagement, such as fluorescence resonance energy transfer (FRET), bioluminescence resonance energy transfer (BRET), affinity-based chemoproteomics, ligand-directed protein labeling, enzyme fragment complementation assay, and cellular thermal shift assays(Schürmann et al., 2016). As for PROTAC, methods to assess the degradation, ubiquitination, and interactions among all participating partners have also been employed to understand the detailed mechanism of action. For example, Promega has applied its NanoLuc technology to study the target engagement for both the targeted protein and the E3 ligase(Riching et al., 2018). However, this method involves transfection, validation, and potential interference with endogenous targets. The transfection requires extensive optimization for each protein target and each cell type. To confirm the binding of a chemical probe (CCT251236) to its putative transcription factor regulator Pirin in living cells, a library of PROTAC small molecules based on this chemical probe was developed to support the intracellular target engagement(Chessum et al., 2018).
We(Yang et al., 2018) recently prepared a small library of PROTACs by tethering a pan histone deacetylase (HDAC) inhibitor as the ligand for all 11 isoforms of HDACs with pomalidomide, a well-known member of the IMiDs as the ligand for CRBN E3 ligase. Then, we discovered that HDAC6 was a viable target for TPD by CRBN E3 ubiquitin ligase. Later, we developed multifunctional HDAC6 selective degraders by tethering Nexturastat A (Next-A), a selective HDAC6 inhibitor, with pomalidomide. (Figure 2A) These PROTACs efficiently depleted HDAC6 and inhibited cell growth of multiple myeloma. A high throughput in-cell ELISA was developed to assess the degradation efficiency of the above HDAC6 degraders. Herein, we describe our efforts on the development of a cellular target engagement assay based on this in-cell ELISA for the evaluation of binding affinity of various analogues of thalidomide including those with partial linkers to the native stage of its CRBN E3 ligase in cells. It has been reported that the type and length of linkers are critical factors for the development of PROTACs(Chan et al., 2017; Cyrus et al., 2011; Smith et al., 2019). When the same pair of the ligand of the POI and the ligand of the E3 ligase was tethered by different linkers, the resulting biological outcomes can be very different. Although the types of linkers can be evaluated by the degradation efficiency of the complete PROTACs in cells, it is difficult to generalize the linker-activity-relationship for the development of PROTACs against different targets. Using the cellular target engagement assay described here, it is possible to systematically evaluate the binding of thalidomide analogues with or without partial linkers to CRBN, which can yield useful information for the design of PROTACs against HDAC6 and possibly many other different targets.
Figure 2. Proof-of-principle Study.
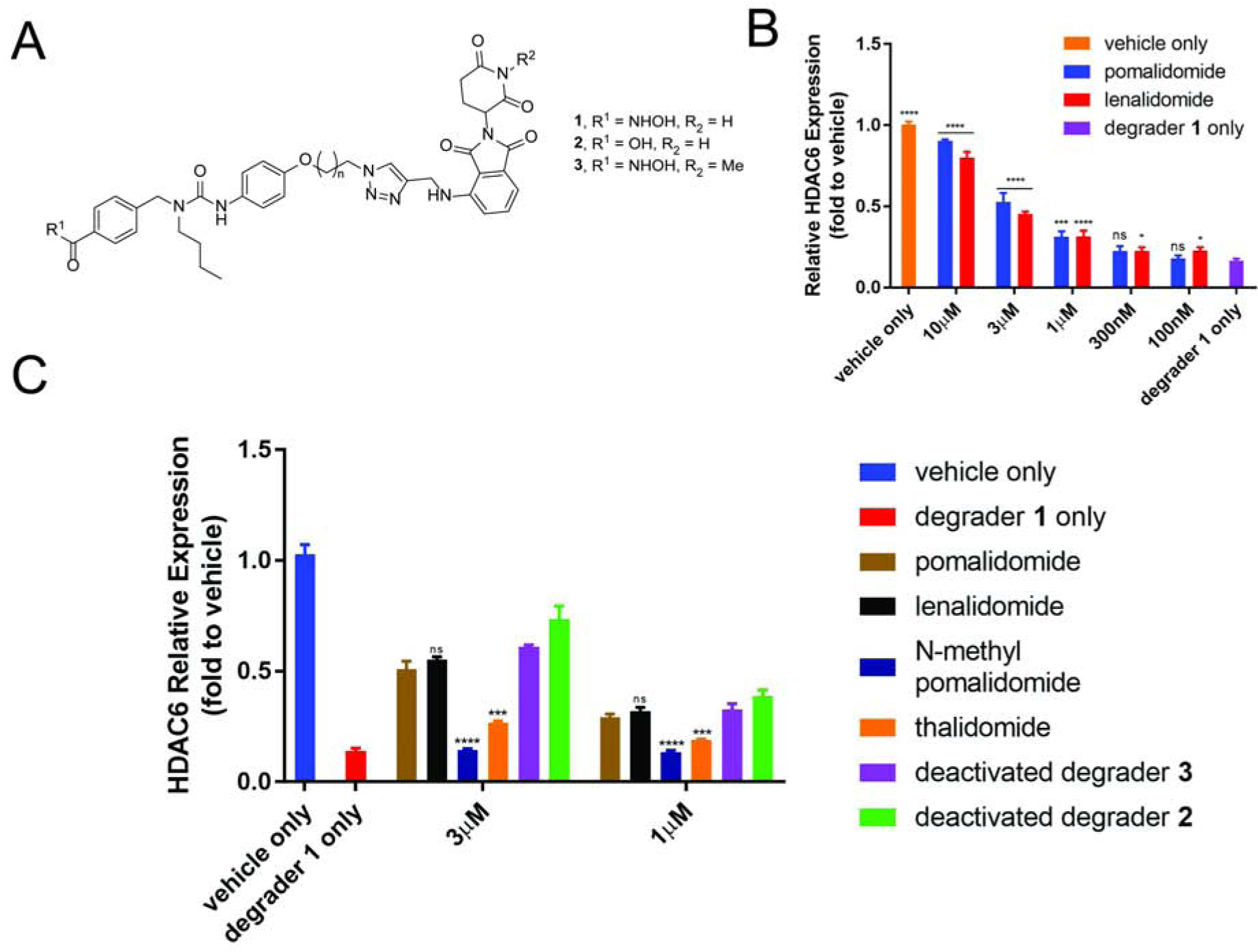
(A) Chemical structures of HDAC6 degrader 1 and deactivated degraders 2 and 3. (B) Assay optimization. (C) Proof-of-concept study. MM1S cells were treated with CRBN ligand or vehicle (1 h), then degrader or vehicle (5 h) and analyzed by in-cell ELISA. See also Figure S1. Bar graphs were generated as means of HDAC6 relative expression (n = 3) with ± SD as the error bar. Variances were analyzed by one-way ANOVA and multiple group comparison with “degrader 1 only” group (B) or unpaired two-tail student t-test in comparison with “pomalidomide” group (C). Not significant (ns) P > 0.05, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001.
We showed previously that pomalidomide or HDAC inhibitor could abolish the degradation of HDAC6 induced by the degraders.(Wu et al., 2019) These ligands could compete with the degraders for the binding sites of either CRBN E3 or HDAC6. This competition led to the formation of fewer ternary complexes when the degraders are introduced to the cellular system. These co-treatment experiments used by us and many others supported the involvement of both POI and E3 ligase in PROTAC-induced degradation. We envisioned that upon further optimization, we might be able to use this type of co-treatment experiments to develop a cell-based assay to systematically evaluate the binding of various potential E3 ligase ligands to the native stage of its cellular target. In theory, ligands with higher affinity to their cellular targets should compete for the binding sites of CRBN more effectively and abolish the effect of PROTAC degrader more efficiently (Figure 1B). Therefore, we hypothesize that we may be able to evaluate the binding affinity of a synthetic library of thalidomide analogues to cellular CRBN E3 ligase using degrader 1 and the in-cell ELISA upon further optimization. For cell-based assays, many other factors such as permeability and stability of the small molecules may also contribute to the observed results, in addition to binding affinity. More active ligands or motifs identified from cell-based assays may be the combination of several factors and should be analyzed carefully in each case.
RESULTS
Relevant Binding Affinity of IMiDs in Cellular Target Engagement Assay
To optimize the assay condition, we pre-treated MM1S cells with different concentrations of pomalidomide, lenalidomide, and Next-A (HDAC6 inhibitor) for 1 h and then added 100 nM degrader 1 for 5 h of treatment. The resulting cells were analyzed by in-cell ELISA. (Figure 2B) We observed the expected dose responses for all three compounds. Compare to the HDAC6 inhibitor, pomalidomide and lenalidomide are less effective to abolish the effect of degrader-induced degradation of HDAC6. With pre-treatment of 10 μM of the CRBN ligand (100 times of the degrader concentration), there was still 10–20% of less HDAC6 than vehicle control. At 3 μM of pomalidomide or lenalidomide, about 50% of HDAC6 remained compared to the vehicle control. We decided to use this condition for the screening of analogues of thalidomide because this concentration provides an ideal window to compare the analogues with the parent compounds. At this concentration, an analogue that is more potent than pomalidomide/lenalidomide for binding to cellular CRBN will abolish more HDAC6 degradation and have over 50% of HDAC6 level. A weaker ligand would have lower than 50% of the HDAC6 level, assuming other properties of the analogues are similar to pomalidomide/lenalidomide.
The proof-of-concept study for cellular target engagement assay is shown in Figure 2C. Ligands that can bind to either CRBN (e.g. deactivated degrader 2, thalidomide, lenalidomide, and pomalidomide) or HDAC6 (e.g. deactivated degrader 3) can abolish the degradation effect of HDAC6 induced by degrader 1. The HDAC6 level for cells co-treated with any one of these ligands and degrader 1 was, therefore, higher than the HDAC6 level in cells that were treated only with degrader 1. The highest level of HDAC6 was observed for compound 2, while the lowest level of HDAC6 was observed for N-Methyl pomalidomide.
The binding affinities of thalidomide, pomalidomide, and lenalidomide to CRBN have been reported in literatures(Chamberlain et al., 2014; Fischer et al., 2014; Lopez-Girona et al., 2012; Sievers et al., 2018). Pomalidomide is slightly more potent than lenalidomide, while thalidomide is the weakest binder among the three. This potency order was confirmed by our cellular assay in Figure 2C. Thalidomide had the least HDAC6 recovery compared to the other two at both 3 μM and 1 μM concentrations. Our results also indicated that pomalidomide and lenalidomide were almost equally potent in cell-based assays. Deactivated degrader 2 can still bind to CRBN but not to HDAC6. The strongest effect for abolishing HDAC6 degradation by 2 suggests that the linker part may contribute to the binding of 2 to CRBN. The known negative control, N-methylated pomalidomide, could not bind to CRBN E3(Lu et al., 2015). As expected, this compound has almost no effect on the degrader 1 induced degradation of HDAC6. Deactivated degrader 3 cannot bind to CRBN, but it can still bind to HDAC6 and abolish part of degrader 1 induced HDAC6 degradation.
Then, we evaluated several other known thalidomide analogues including CC-122 (Avadomide)(Hagner et al., 2015), CC-220 (Iberdomide)(Matyskiela et al., 2018b) and CC-885(Matyskiela et al., 2016) (Figure 3A and 3B). An IC50 of 60 nM was reported for CC-220 in the TR-FRET binding assay to CRBN, while the IC50s for pomalidomide and lenalidomide were 1.2 μM and 1.5 μM, respectively, from the same assay(Matyskiela et al., 2018b). We evaluated these three compounds in our cell-based assay. CC-122 was found to be less potent than pomalidomide. Both CC-220 and CC-885 remarkably reduced HDAC6 degradation, which is consistent with their high affinity to CRBN, as reported in the literature. However, CC-885 can recruit both IKZFs and GSPT1 as neo-substrates to CRBN and promote their degradation(Ishoey et al., 2018; Matyskiela et al., 2016), suggesting that using CC-885 as the ligand for PROTACs will have off-target effects.
Figure 3. Validation of Cellular Target Engagement Assay and Preliminary Screening.
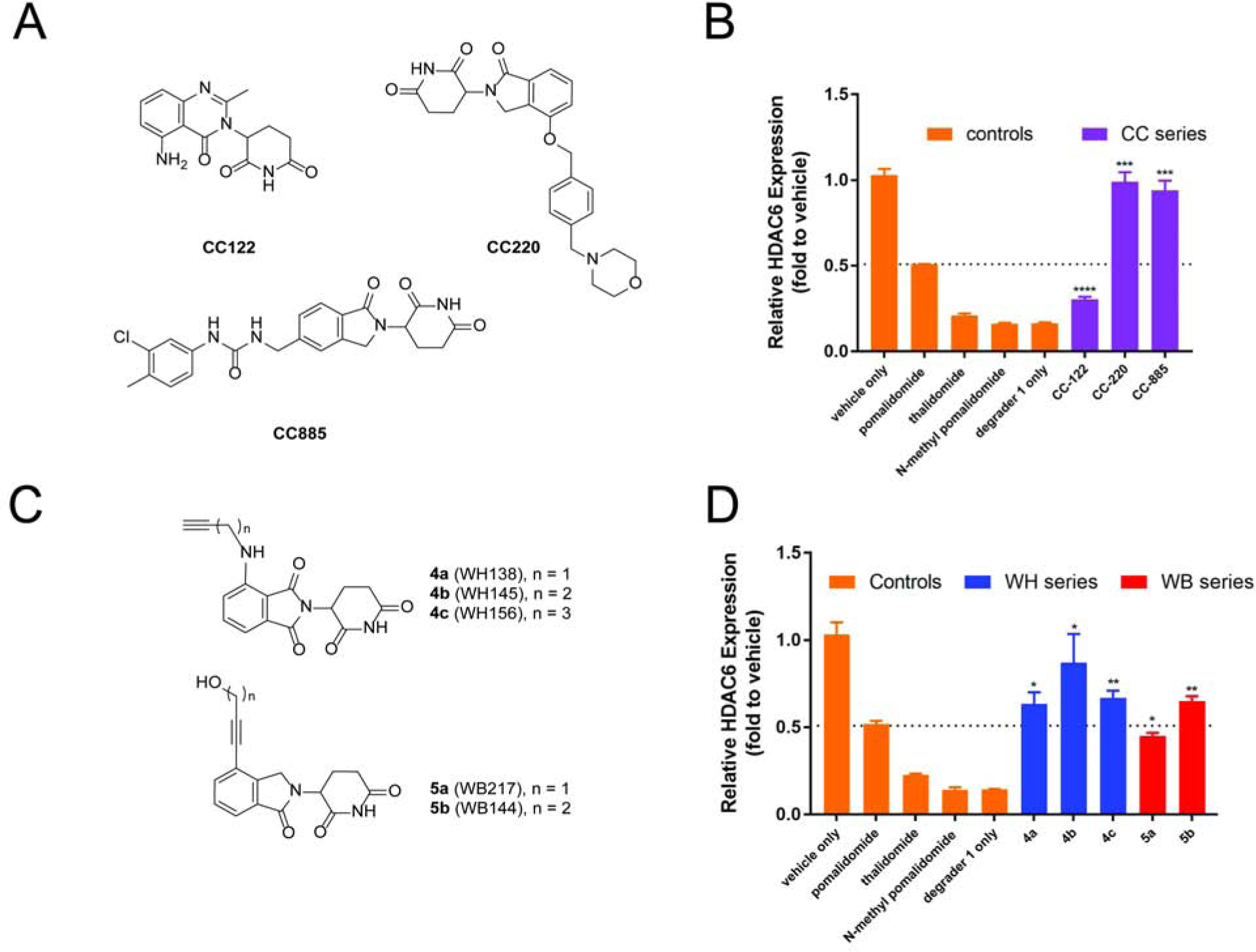
(A) and (B) Chemical structures and bioactivity test of known IMiDs through cellular target engagement. (C) and (D) Chemical structures and bioactivity test of selected thalidomide derivatives with partial linkers through cellular target engagement. See also Figure S2 MM1S cells were treated with CRBN ligand or vehicle (1 h), then degrader or vehicle (5 h) and analyzed by in-cell ELISA. Bar graphs were generated as means of HDAC6 relative expression (n = 3) with ± SD as the error bar. Variances were analyzed by unpaired two-tail student t-test in comparison with “pomalidomide” group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Because the above cell-based assay does not involve genetic manipulations, it should be relatively easy to transform from one cell to another one. To further demonstrate the utility and generality of this assay, we applied it to a commonly used T-cell model, Jurkat cell line, which is difficult to undergo efficient transfection due to low content of proteoglycans(Riedl et al., 2018). We treated the Jurkat cells with different concentrations of reported IMiDs and then 100 nM of degrader 1 (Figure S1). As expected, we observed a similar trend of potency for these compounds as in MM1S cells. One exception was that the CC885 recovered less HDAC6 degradation than what it did in MM1S. Considering CC220 and CC885 were reported with similar in-vitro binding affinity to CRBN and their similar activities in MM1S cells (Figure 3B), the discrepancy may be due to the compound’s other properties, such as cell permeability, metabolic stability or in-cell retention time, in this particular cell line. This observation further highlights the utility of the cell-bases target engagement assay for the development of E3 ligases ligands in its relevant cellular context.
Overall, the above results indicated that the optimized in-cell ELISA was able to differentiate the binding affinity of thalidomide derivatives through cellular target engagement, assuming other properties of these thalidomide derivatives are close to the parent pomalidomide/lenalidomide in a given cellular context. The higher potency of any analogue than its parent compounds is the reflection of its overall properties including the affinity to its cellular target at the native conformation, permeability, stability, and other factors.
CRBN E3 Ligands Are Identified and Characterized
With a validated cell-based assay in hand, we then screened a library of synthetic thalidomide analogues including those with partial linkers. (Figure 3C, 3D, Figure S2) Among them, we discovered several classes of promising ligands for CRBN E3 ligase. Compounds 4a-c were pomalidomide derivatives linked with a terminal alkyne via methylene linkers of different lengths (Figure 3C). All of them showed more HDAC6 recovery than pomalidomide. Compound 4b with a linker of two methylene units had the highest potency. These results indicate that the linker part of the ligand may contribute to its binding to the CRBN E3 ligase (Figure 3D), assuming their other properties are similar. Compounds 5a and 5b are lenalidomide derivatives with an alkyne motif directly attached to the benzene ring and an alcohol at the end of the partial linker. Both of them showed promising activity. Interestingly, compound 5b, which is only one methylene different from 5a, showed much higher activity than the parent thalidomide. Compound 5b, which lacks the aniline nitrogen compared to pomalidomide, is even more active than pomalidomide. These results suggest that the distance between the terminal hydroxyl group and the phthalimidine ring is critical for their cellular activity, which is likely due to different binding affinity to CRBN, considering their overall structural similarity to pomalidomide and lenalidomide. Overall, the in-cell ELISA allowed us to systematically evaluate the effect of modifications on thalidomide including the linker part for the degradation of HDAC6 in relevant cellular context.
Based on our preliminary results with 4a-c and 5a-b, we synthesized more lenalidomide derivatives with an alkyne or a phenyl group directly attached to the benzene ring of lenalidomide (compound 6a-i, 7a-b, 8, 9a-f and 10) and other types of analogues as shown in Figure 4A. Compared to pomalidomide, several of them have a higher activity (Figure 4B). Compounds 6a-i all have an alkyne attachment and they differ in the linker length and the bridging atom (nitrogen or oxygen) between the alkyne and the terminal phenyl ring. Compound 6d-f (X = O) appear to have decreased activity as the linker lengths increase. Aldehyde-substitution enhanced the activity as suggested by the higher activity of 6g and 6i over 6f and 6h against HDAC6, respectively. Compounds 6g and 6i showed the highest recovery of HDAC6 protein level and both of them are more potent than pomalidomide. The HDAC6 recovery percentage in response to various concentrations of compounds 6g and 6i was also examined (Figure 4C). The EC50s (concentration at which half of degraded HDAC6 was recovered) were 6.4, 1.3 and 1.5 μM for pomalidomide, 6g and 6i, respectively. It confirmed the improved activities of these ligands to CRBN in cells. Compounds 7a-b have an additional hydroxyl group between the alkyne and the terminal phenyl group. Both of them are comparable to 6i for the recovery of the HDAC6 protein level in the competition experiment. Surprisingly, compound 8 failed to recover the HDAC6 protein level induced by degrader 1, suggesting that the terminal phenyl ring in 7a-b played an important role in enhancing their cellular activity. In addition, a series of glutarimide compounds, such as R2, R5, and R7, was reported as ligands of CRBN E3 ligase in the in-vitro binding assay.(Goergler, Annick; Norcross, Roger; Schmid, Philipp; Dey, Fabian; Kusznir, 2019) Kds of these compounds were obtained and they ranged from less than 1 nM to 19 nM in a fluorescence direct binding assay. We synthesized these compounds and some related analogues (R1-13) to explore the activity of these compounds in our cell-based assay (Figure 4A). However, to our surprise, none of those compounds showed improved binding affinity than pomalidomide (Figure 4B). Our results suggest that the complete structure of phthalimidine is important to maintain the cellular activity of lenalidomide.
Figure 4. Screening of Lenalidomide Analogues with An Attachment of An Alkyne- or A Phenyl Group.

(A) Chemical structures of lenalidomide derivatives. (B) Target engagement and in-cell ELISA results. (C) Comparison of HDAC6 recovery under the treatment of 6g, 6i or pomalidomide at different concentrations. MM1S cells were treated with CRBN ligand or vehicle (1 h), then degrader or vehicle (5 h) and analyzed by in-cell ELISA. Bar graphs were generated as means of HDAC6 relative expression (n = 3) with ± SD as the error bar. Variances were analyzed by unpaired two-tail student t-test in comparison with “pomalidomide” group. Not significant (ns) P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Encouraged by the improved activity for the thalidomide analogues with an alkyne attachment, we then prepared compounds 9a-f and 10 by directly introducing a substituted phenyl ring to thalidomide. We were pleased to find that most of them have either comparable or better activity than pomalidomide. Although compound 9a with an unsubstituted phenyl group has the highest activity, the introduction of substituents to the ortho, meta, or para positions did not decrease the activity much, suggesting that it is possible to place a linker at any of these positions for the development of PROTACs against HDAC6 and possibly other protein targets.
We next examined the degradation of activity of selected CRBN ligands for several well-known neo-substrates of IMiDs, such as IKZFs and GSPT1(Nabet et al., 2018; Yang et al., 2019). We tested these compounds in MM1S cell lines by both in-cell ELISA and Western blot (Figure 5). No significant degradation of IKZFs was observed at 100 nM concentration of most ligands in the in-cell ELISA. (Figures 5A and 5B). Selected ligands were also examined with higher concentrations (300 nM and 1 μM) and showed no obvious effects (Figure 5C). Western blot analysis of these ligands confirmed no degradation of IKZFs occurred at 1 μM concentration. Since the degradation of IKZFs by IMiDs was responsible for the anti-proliferative effects in multiple myeloma,(Lu et al., 2014) we then treated MM1S cells with the CRBN ligands developed here for 72 h and analyzed the cell viability (Figure S3). The proliferation of myeloma cells was not affected by the CRBN ligands developed here, while pomalidomide and lenalidomide have a significant anti-proliferation effect. Compounds 6g and 9d were examined at a wide range of concentrations (30 nM to 3 μM) by Western blot (Figure S4). No related HDACs or well- known neo-substrates of CRBN was affected by these two CRBN ligands. All of our results suggest that the CRBN ligands developed here, such as 6d/g, 6h/i and 9a/d, can be used for the development of more selective PROTACs. However, the neo-substrate is often dependent on the entire structure of the PROTAC. Additional studies will be necessary in order to define the selectivity of any given PROTAC.
Figure 5. Lenalidomide Analogues Did Not Induce Degradation of Selected CRBN Neo-substrates.
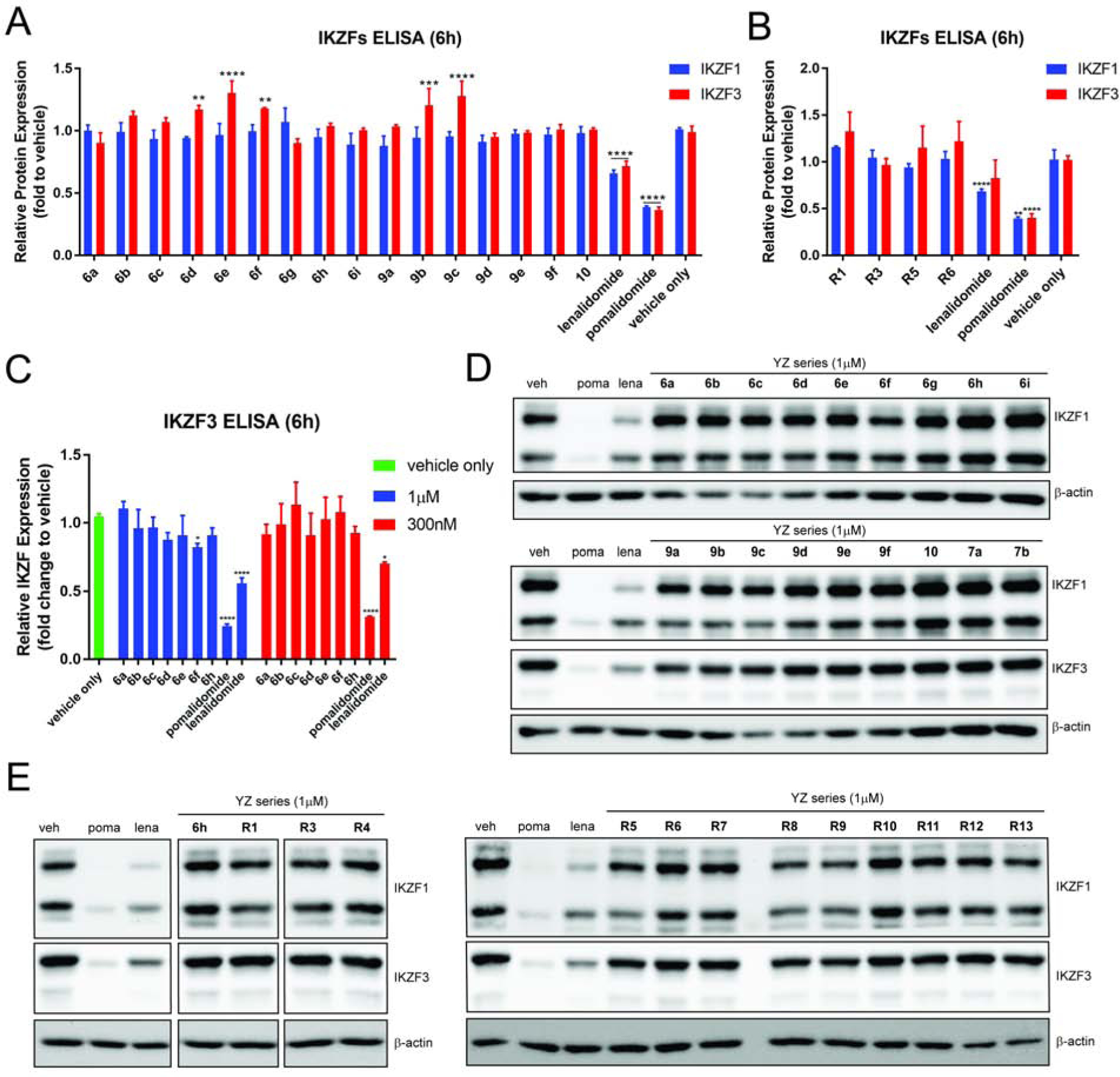
(A)-(E) MM1S cells were treated with CRBN ligands or vehicle (6 h) and analyzed by in-cell ELISA or Western blot. For anti-proliferation results and additional blots, see Figure S3 and S4. Bar graphs were generated as means of protein (IKZF1 or IKZF3) relative expression (n = 3) with ± SD as the error bar. Variances were analyzed by one-way ANOVA and multiple group comparison with corresponding “vehicle only” group were followed Dunnett correction. Not significant (ns) P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. “ns” was obtained for any bar without asterisk rating.
HDAC6 PROTAC Degraders Are Developed Based on E3 Ligands Developed Here
To investigate the feasibility of the CRBN ligands discovered here for the development of HDAC6 degraders and possibly PROTACs against other targets, we followed our previously developed protocol(Tang et al., 2011; Yang et al., 2018) to conjugate a HDAC biasing reagent to 6g, 6i and 9d and generated bifunctional molecules 11a-b and 12 to target the HDAC family (Figure 6A). After profiling the expression of representative HDACs, we found that only HDAC6 was degraded and other HDACs were left intact when the MM1S cells were treated 11a and 11b (Figure 6B). No obvious effect was observed for 12. Degrader 11a also showed a narrower working window (concentration range) than 11b. By examining the dose response of 11b, we observed significant degradation of HDAC6 at a concentration as low as 3 nM. The “hook effect” was observed at around 300 nM (Figure 6C). Neither HDAC4, another member of class II HDACs, nor IKZFs were targeted by 11b for degradation. DC50 of 1 and 11b for the degradation of HDAC6 in the in-cell ELISA are 1.5 nM and 1.9 nM, respectively (Figure 6D). Comparing with the previous HDAC6 degraders with multiple ethylene glycol units(Yang et al., 2018) or HDAC6 degrader 1 with a triazole ring and more than six methylene units(Wu et al., 2019), HDAC6 degrader 11b has a much shorter linker (an alkyne and three methylene units), and a potency that is similar to 1. It is likely that 11b and 1 may form distinct ternary complexes. It is generally more desirable to have a shorter linker with less flexibility and lower molecular weight for the development of any therapeutics. The discovery of HDAC6 degrader 11b based on CRBN ligand 6i highlights the potential utility of our cell-based target engagement assay.
Figure 6. Design and Development of HDAC6 Degraders Based on Pan HDAC Inhibitor and CRBN Ligands.
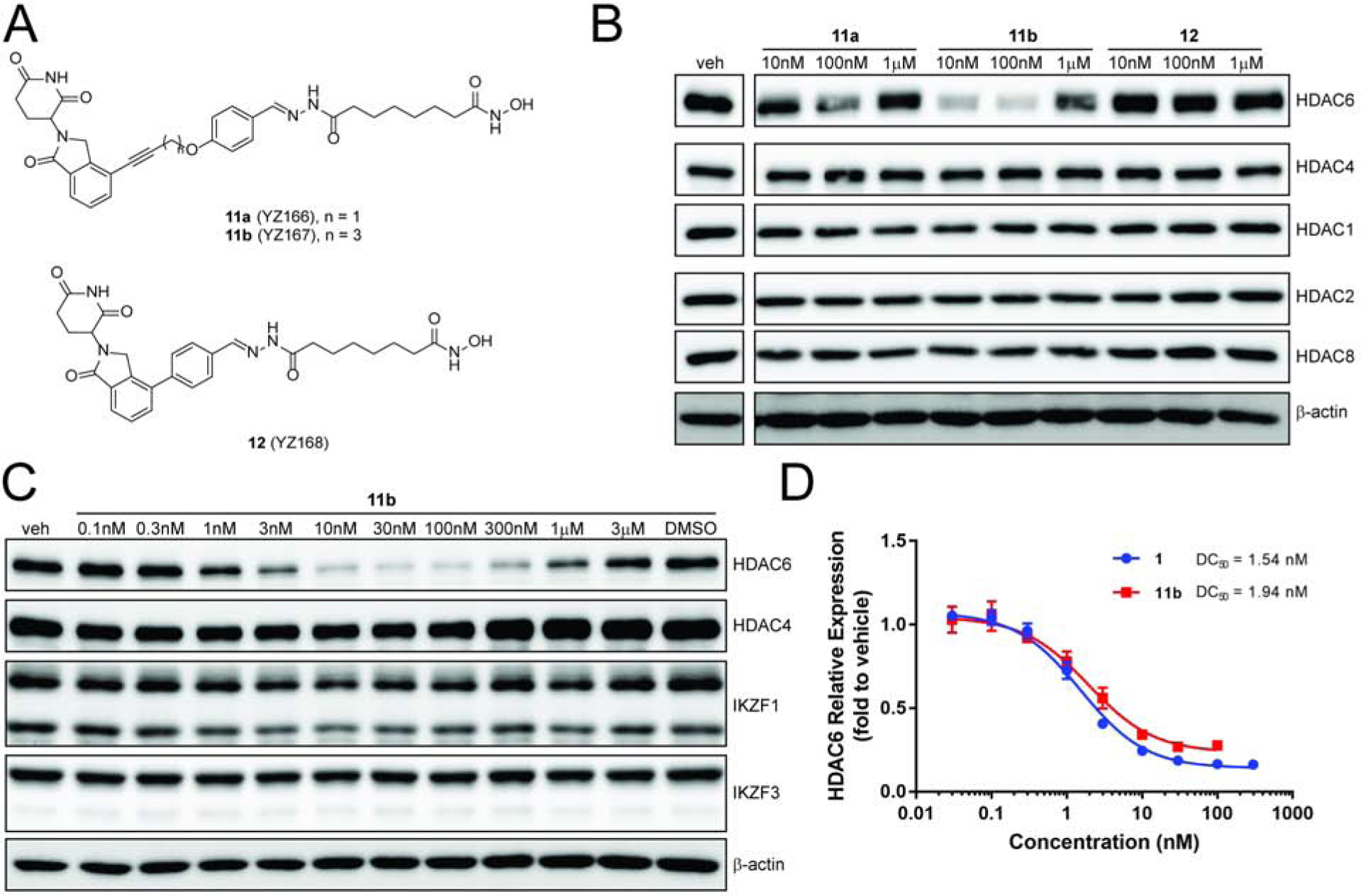
(A) Chemical structures of HDAC degraders 6g, 6i and 9d. (B) and (C) Western blot analysis of proteins expression of MM1S cells treated with degraders or vehicle for 6 h. (D) In-cell ELISA analysis of HDAC6 expression undertreatment of degraders. MM1S cells were treated with degraders or vehicle (6 h) and then analyzed by Western blot or in-cell ELISA. Bar graphs were generated as means of protein (IKZF1 or IKZF3) relative expression (n = 3) with ± SD as the error bar. Dose response were nonlinear fitted by [Inhibitor] vs. response (three parameters) and fitted curve was generated in GraphPad Prism.
In addition to HDAC degraders derived from pan HDAC inhibitors, we also prepared a series of HDAC degraders based on HDAC6-selective inhibitor Next-A. Degraders 13a-c were synthesized by tethering CRBN ligand 9d (or 9a) and Next-A with various lengths of linkers (Figure 7A). Initial Western blot analysis showed that 13b and 13c at a concentration of 1 μM decreased HDAC6 protein level, while 13a with shortest linker was almost inactive (Figure S5). We then elongated the linker and used in-cell ELISA to analyze cells treated by 13a-f. We found that the potency of these degraders increased in a linker-length-dependent manner (Figure 7B). Degrader 13f with the longest linker was the most potent one and its activity is close to 1, the most potent degrader we previously discovered after extensive optimization of the lengths and types of the linker.(Wu et al., 2019) Our data indicate that compounds 9a or 9d are indeed viable CRBN ligands for the development of PROTACs.
Figure 7. Design and Development of HDAC6 Degrader Based on CRBN Ligand and HDAC6-selective Inhibitor.
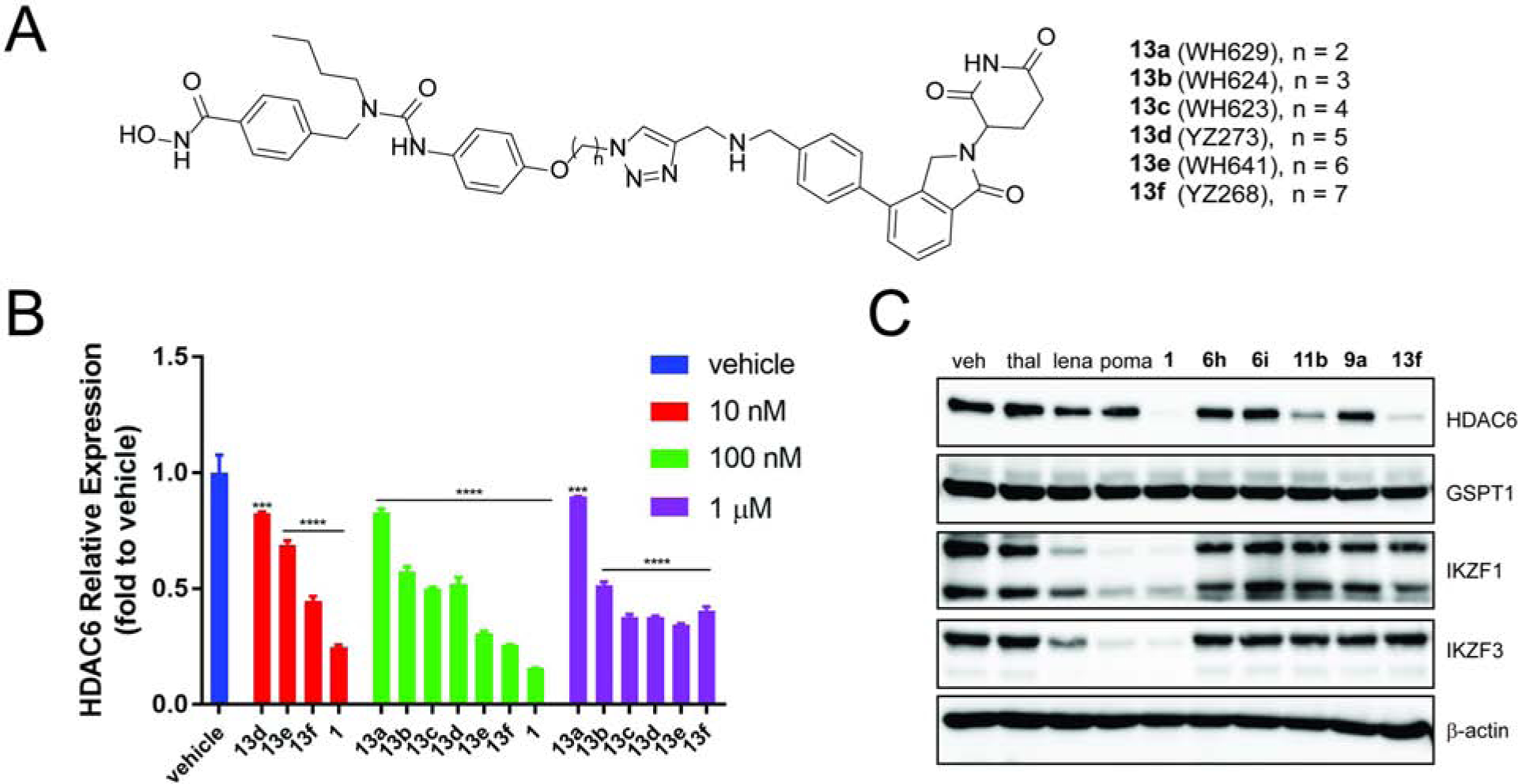
(A) Chemical structures of Next-A based degraders. (B) In-cell ELISA analysis of HDAC6 level under treatment of degraders. For western blot analysis, see Figure S5. (C) Western blot analysis of proteins level under treatment of CRBN ligands and degraders. MM1S cells were treated with ligands or degraders or vehicle (6 h) and then analyzed by Western blot or in-cell ELISA. Bar graphs were generated as means of HDAC6 relative expression (n = 3) with ± SD as the error bar. Variances were analyzed by one-way ANOVA and multiple group comparison with corresponding “vehicle” group were followed Dunnett correction. ***P ≤ 0.001, ****P ≤ 0.0001.
We then further compared the activity of CRBN ligands 6h, 6i, and 9a with HDAC6 degraders derived from them, 11b and 13f, together with other known IMiDs and previously developed HDAC6 degrader 1. We treated MM1S cells with these compounds at 500 nM concentration (Figure 7C). The Western blot results showed that while degraders 1, 11b and 13f all induced the degradation of HDAC6, the latter two based on CRBN ligands 6h/i and 9a/9d had no effect on neo-substrates IKZFs. No IMiDs or lenalidomide analogues affected HDAC6. Lenalidomide, pomalidomide, and degrader 1 induced obvious degradation of IKZFs. This result confirmed that IKZFs were not degraded when the CRBN ligands developed here were incorporated into the full PROTACs. In addition, all HDAC6 degraders did not induce the degradation of GSPT1. Comparing to compound 1, which degrades HDAC6, IKZF1, and IKZF3, degrader 13f can be used as a more specific chemical probe to knock-down HDAC6. The potency of compound 1 was extensively optimized by systematically varying the lengths and types of the linker. Further optimization will be necessary to improve the potency of degraders derived from the CRBN ligands developed here.
Discussion
Thalidomide and its analogues have been widely used for the development of PROTACs for TPD. The E3 ligand moiety is crucial in successful and efficient recruitment of E3 ligase to the targeted protein. However, the activity and selectivity of the E3 ligase ligand without and with partial linkers have not been investigated systematically in the relevant cellular context. In addition to the ligand of POI and the full linker, the structure of E3 ligase ligand and the partial linker attached to the ligand may have a significant impact on the activity and selectivity of PROTACs. Several cell-based methods have been developed to assess the binding and permeability of PROTACs, such NanoLuc system(Riching et al., 2018), ligand-displacement AlphaScreen assay(Nabet et al., 2018), cellular thermal shift assay (CETSA)(Winter et al., 2017) and chloroalkane cell penetration assay(Foley et al., 2020). However, most of them involve genetic manipulation of the protein target and it is not straightforward to transfer the assay from one cellular system to another one. Although the chloroalkane penetration assay represents a significant advancement over in-vitro permeability assays, it requires the attachment of a chloroalkane motif to PROTACs, which is not trivial and may change the property of the PROTACs. Our cellular target engagement assay reported here can be transformed from one cell to another one relatively easily. One can use existing PROTACs for the optimization of the E3 ligase ligands with relatively high throughputs. Our data clearly indicated that the structure of E3 ligase ligand and the partial linker attached to it can both impact the cellular activities against HDAC6, which are most likely due to their binding affinity to the E3 ligase in its native conformation and also the structure of the resulting ligand-E3 complexes, which determine the selectivity for the degradation of potential neo-substrates.
In the present study, HDAC6 degradation was used as the readout to evaluate the potency of ligands to competitively occupy the binding pocket of an E3 ligase in the relevant cellular context. We first validated the assay using thalidomide analogues with known in-vitro binding data and cellular activity. The results from our cell-based assay are consistent with the binding affinity of these known thalidomide analogues in most cases. Given the overall structural similarity between the thalidomide analogues described here and the parent pomalidomide/lenalidomide, the cellular activity difference is most likely due to their binding affinity to CRBN E3 ligase. However, without additional extensive studies, we cannot rule out other possibilities. For example, the increased HDAC6 recovery by the CRBN ligands developed here can be caused by the increased permeability, stability, in-cell retention time, or other mechanisms such as allosteric inhibition.
After profiling several diverse sets of thalidomide analogues, we identified candidates with promising cellular activity. Ligands 6d/g, 6h/I and 9a/d could efficiently compete off the full PROTAC and rescue the HDAC6 from degradation. These ligands could be used for the development of HDAC degraders. We also found that these ligands might have a different mode of actions compared to thalidomide. For example, 11b with a very short linker resulted in similar potency as degrader 1. Meanwhile, the series of PROTACs based on ligand 13 bearing the phenyl substituent on the phthalimidine require longer linker lengths compared to degrader 1.
Gratifyingly, the ligands we identified from the cell-based assay did not promote the degradation of several well-known neo-substrates of CRBN. Our results indicated that the alkynyl or the phenyl group could impact both the potency and selectivity. It has been reported that the amino group of pomalidomide or lenalidomide can facilitate the interactions between CRBN and IKZFs(Sievers et al., 2018). Replacing the amino group by an alkyne or phenyl group may significantly decrease the interaction between CRBN and IKZFs, which may explain the lack of reactivity for the degradation of several known neo-substrates. However, comprehensive proteomic studies will be necessary for each PROTAC to gain better understanding of the selectivity, which depends on the entire structure of the PROTAC.
It is known that the linker moiety of PROTACs can promote the interactions between the E3 ligase and the targeted protein, which will increase the cooperativity(Gadd et al., 2017). Both alkynyl and phenyl substituent on the phthalimidine ring are relatively rigid. They can restrict the orientation of the CRBN E3 ligase towards either the more or less cooperative conformations compared to the more flexible functional groups. PROTACs 11b and 13f may represent examples for each of these two scenarios. The former may result in a more cooperative conformation at least for HDAC6 and therefore it requires a shorter linker than 1, while the latter may result in a less cooperative conformation for HDAC6 and therefore it requires longer linker than 1. In addition to the identification of cell permeable CRBN ligands for the development of PROTACs against HDAC6 and possibly other targets, we also found that previously reported potent ligands for CRBN in the biochemical assay could not compete off the PROTACs effectively in our cell-based assay. The lack of activity of these ligands is likely due to low binding affinity to CRBN in its native stage, though we cannot rule out the possibility of other factors, such as poor cell permeability. This further highlights the advantage of the cell-based target engagement assay, which allows the optimization of the combined property of the E3 ligase ligands for the development of PROTACs against HDAC6 and possibly other protein targets.
STAR METHOD
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Weiping Tang (weiping.tang@wisc.edu).
Materials Availability
All unique/stable reagents generated in this study will be provided without restriction as long as stocks remain available and reasonable compensation is provided by requestor to cover processing and shipment.
Data and Code Availability
The published article includes all biological and chemical data (1H NMR, 13CNMR and LC-MS of all compounds) generated during in this study. No unique code was generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line and Cell Culture
Human cell lines MM1S (CRL-2974, derived from female) and Jurkar (Clone E6–1, TIB-152, derived from male) was obtained from the American Type Culture Collection (ATCC). Bothe cell lines were cultured in RPMI-1640 medium (Corning) supplemented with 10% FBS, 1% Sodium Pyruvate, and 1% Penicillin/Streptomycin, 10mM HEPES. Cells were grown at 37°C in a humidified 5% CO2 atmosphere.
METHDO DETAILS
Cell Seeding and Treatment Method for In-cell ELISA
MM1S cells were harvested and plated with 5×105 cells in 100μL media per well 96-wells plate. After overnight seeding, 25 μL media containing 5X dosing concentration of the compounds or vehicle were added to each well. For target engagement experiment, 12.5 μL media containing 10X dosing concentration of the thalidomide derivative or vehicle were added to each well, followed by 1-hour incubation and then addition of 12.5 μL media containing 10X dosing concentration of WH181 (1) or vehicle.
In-cell ELISA
After 5-hour treatment at 37°C in a humidified 5% CO2 atmosphere, cells were fixed by adding 125 μL 8% formaldehyde in TBS buffer (137 mM NaCl, 25 mM Tris, 2.7 mM potassium chloride, pH 7.6). and incubated at room temperature (RT) for 15 minutes. Removal of fixing solution was followed by once rinse and twice washes with TBS-T washing buffer (137 mM NaCl, 20 mM Tris, 0.1% Tween, pH 7.6). Cells were then permeabilized by adding 100 μL 0.1% Triton-X in TBS and incubated at RT for 15 minutes. Removal of permeabilizing solution was followed by once rinse and once wash with TBS-T. Cellular endogenous peroxidases were quenched by adding 100 μL 1% H2O2 in TBS and incubation at RT for 20 minutes. Removal of quenching solution was followed by once rinse and once wash with TBS-T. Non-specific binding sites were blocked by adding 200 μL 5% BSA in TBS-T (with 0.02% NaN3) and incubation at 4 °C overnight. Removal of blocking solution was followed by adding 50 μL primary antibody solution (HDAC6 Rabbit mAb, CST #7558, 1:1000 dilution in 5% BSA in TBS-T with 0.02% NaN3) and incubation at RT for 2 hours. In-cell ELISA was also applied to other proteins by using antibodies against IKZF1 (#14859s, 1:1000 dilution) and IKZF3 (#15103s, 1:2000 dilution) from CST.
Two or more wells treated with DMSO or untreated were added blocking solution without antibody as background control. Removal of primary antibody solution was followed by once rinse and three times washes with TBS-T. Secondary antibody solution (Anti-rabbit IgG, HRP-linked Antibody, CST #7074, 1:2000 in 1% BSA in TBS-T) was added into cells and incubated at RT for 1 hour. Removal of secondary antibody solution was followed by once rinse and four times washes with TBS-T. TMB substrates were premixed, added into cells and incubated in dark at RT for 20 minutes. Stop solution (2N H2SO4 in ddH2O) was added into reaction mixture and incubated at RT for 5 minutes with gentle shaking. The optical density (OD) of each well was read at 450 nm and 570 nm by FLUOstar Omega microplate reader (BMG LABTECH). ELISA OD was obtained by subtracting the OD570 from OD450.
Janus Green Stain
Normalization of ELISA OD to cell number was processed by Janus Green Stain(Andersen and Vermette, 2016). Removal of stop solution from last step was followed by once rinse and three times washes with TBS-T. 50 μL of 0.3% Janus Green B in TBS was added to cells and incubated for 5 minutes. Removal of stain solution was followed by twice rinses and three times washes with TBS-T. Cells were quickly rinsed with 100 μL 140 mM NaCl solution and then exposed to 0.5 M HCl in 100 μL 140 mM NaCl. Cells were incubated at RT for 5 minutes with medium shaking. The Janus Green OD of each well was read at 595 nm.
Immunoblot
When the cells reached 90% confluence, they were harvested and plated 4×105 cells per well in 12-well plate. After overnight seeding, the cells were treated with a solution of compounds or vehicle in culture medium. The culture medium was removed after treatment and then washed twice with cold PBS. To obtain whole cell lysate, all cells were treated with RIPA lysis buffer (25mM Tris, pH 7–8, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, protease inhibitor cocktail (Roche, 1 tablet per 10 mL) and 1mM PMSF) on ice for 10 minutes. Supernatant was collected after spinning down at 16,000g at 4 °C for 15 minutes. Protein concentration was measured by using the Pierce BCA protein assay (Thermo Fisher Scientific). About 10–40 μg of total protein was mixed with 4X Laemmli Loading Dye (250 mM Tris, pH 6.8, 40% glycerol, 5% SDS, 0.005% bromophenol blue, 4% BME) and heated at 95–100°C for 5 minutes. The heated sample was then subjected to 7.5–12% SDS-PAGE and transferred to PVDF membrane (Bio-Rad). The membrane was blocked in 5% non-fat milk (Bio-Rad) in TBS-T washing buffer (137 mM NaCl, 20 mM Tris, 0.1% Tween) and then incubated with primary antibodies at 4 °C overnight. The membrane was washed 3 times with TBS-T, incubated with secondary horseradish peroxidase (HRP) linked antibodies for 1 hour, then washed 3 more times with TBS-T. Clarity ECL substrate (Bio-Rad) was incubated with membrane for 5 minutes. The Immunoblot was generated by ChemiDoc MP Imaging Systems (Bio-Rad) and analyzed by Image J software. A band intensity bar graph was generated, and the curve was fitted using “log(inhibitor) vs. response (three parameters)” by GraphPad Prism. Primary antibody against HDAC1, HDAC2, HDAC4, HDAC6, IKZF1, IKZF3 and GSPT1 were purchased from Cell Signaling Technology. Primary antibody against β-actin were purchased from R&D system. Primary antibody against HDAC8 were purchased from Santa Cruz Biotechnology. Figure 5E and 6B were prepared by deletion of lanes of blank samples or samples treated with compounds irrelevant with this work.
Cell Viability Assays
MM1S cells were harvested and plated with 1×104 cells in 100μL media per well in 96-well plate. After overnight seeding, 25 μL media containing 5X dosing concentration of the compounds or vehicle was added to each well. After 72-hour treatment at 37°C in a humidified 5% CO2 atmosphere, 12.5 μL 10X resazurin solution (0.5 mg/mL) was added to each well. Then cells were incubated at 37°C overnight. The optical density was read at 570 nm and 600 nm by platereader.
Chemical Synthesis and Compound Characterization
All reactions were conducted under a positive pressure of dry argon in a glassware that had been oven-dried prior to use. Anhydrous solutions of reaction mixtures were transferred via an oven-dried syringe or cannula. All solvents were dried prior to use unless noted otherwise. Thin-layer chromatography (TLC) was performed using precoated silica gel plates. Flash column chromatography was performed with the silica gel. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker 400 spectrometers. 1H NMR spectra were reported in parts per million (ppm) referenced to 7.26 ppm of CDCl3 or referenced to the center line of a septet at 2.50 ppm of DMSO-d6. Signal splitting patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), or multiplet (m), with coupling constants (J) in hertz. The liquid chromatography–mass spectrometry (LC–MS) analysis of final products was processed on an Agilent 1290 Infinity II LC system using a Poroshell 120 EC-C18 column (5 cm × 2.1 mm, 1.9 μm) for chromatographic separation. Agilent 6120 Quadrupole LC/MS with multimode electrospray ionization plus atmospheric pressure chemical ionization was used for detection. The mobile phases were 5.0% methanol and 0.1% formic acid in purified water (A) and 0.1% formic acid in methanol (B). The gradient was held at 5% (0–0.2 min), increased to 100% in 2.5 min, then held at isocratic 100% B for 0.4 min, and then immediately stepped back down to 5% for 0.1 min re-equilibration. The flow rate was set at 0.8 mL/min. The column temperature was set at 40 °C. All commercial and synthesized compounds were checked by HPLC for over 95% purity and then prepared stock solution in DMSO. Synthesis route were provided in Supplemental Information Scheme S1 and S2. Synthetic intermediate showed in Figure S2B was all reported previously.(Wang et al., 2019; Wu et al., 2019; Yang et al., 2018) Intermediate S1 and S2 were synthesized according to the literature reported method. (Li et al., 2019)
General procedure for the synthesis of 6, 7 and 8:
To a mixture of halogenated-lenalidomide S1 or S2 (1.0 equiv.) and alkyne (4.0 equiv.) in dry DMF was added copper iodide (0.2 equiv.) and Pd(PPh3)Cl2 (0.1 equiv.). The suspension was degassed and refilled with Argon three times. Triethylamine (same volume of DMF) was then added and the solution was purged again with Argon. The reaction mixture was stirred at 70°C overnight. After being cooled down to room temperature, the solvent was removed under reduced pressure and the residue was submitted to the flash column chromatography (eluted with 5–10% methanol in DCM), giving the corresponding desired compound as white or light yellow solid.
3-(1-oxo-4-(3-(phenylamino)prop-1-yn-1-yl)isoindolin-2-yl)piperidine-2,6-dione 6a:
Compound 6a was prepared using S1 as the starting material. White solid (39 mg, 53% yield). 1H NMR (400 MHz, CDCl3): δ 8.00 (s, 1H), 7.80 (dd, J = 7.6, 1.1 Hz, 1H), 7.54 (dd, J = 7.6, 1.1 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.25–7.13 (m, 2H), 6.87–6.67 (m, 3H), 5.18 (dd, J = 13.2, 5.4 Hz, 1H), 4.21 (s, 1H), 4.18 (d, J = 17.0 Hz, 2H), 3.98 (d, J = 17.0 Hz, 2H), 2.95–2.89 (m, 1H), 2.86–2.77 (m, 1H), 2.21 (qd, J = 13.0, 5.1 Hz, 1H), 2.16–2.09 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 170.88, 169.19, 168.81, 146.82, 144.18, 134.07, 131.57, 129.22, 128.41, 123.88, 118.69, 118.44, 114.17, 92.39, 78.99, 51.63, 46.58, 34.42, 31.48, 23.26. LC-MS (M+H: 374.4): 98% purity.
3-(4-(3-(benzylamino)prop-1-yn-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 6b:
Compound 6b was prepared using S1 as the starting material. White solid (63 mg, 51% yield). 1H NMR (400 MHz, CDCl3): δ 8.33 (brs, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.38–7.31 (m, 4H), 7.30–7.28 (m, 1H), 5.24 (dd, J = 13.3, 5.1 Hz, 1H), 4.43 (dd, J = 62.3, 16.7 Hz, 2H), 3.94 (s, 2H), 3.70 (s, 2H), 2.96–2.75 (m, 2H), 2.41 (qd, J = 13.0, 5.1 Hz, 1H), 2.20–2.10 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 171.08, 169.49, 168.93, 143.59, 139.22, 134.83, 131.64, 128.55, 128.49, 128.37, 127.34, 123.81, 118.78, 93.15, 79.36, 52.61, 51.82, 46.95, 38.24, 31.55, 23.43. LC-MS (M+H: 388.5): 97% purity.
3-(1-oxo-4-(3-(phenethylamino)prop-1-yn-1-yl)isoindolin-2-yl)piperidine-2,6-dione 6c:
Compound 6c was prepared using S1 as the starting material. White solid (57 mg, 88% yield). 1H NMR (400 MHz, CDCl3): δ 8.59 (brs, 1H), 7.82 (d, J = 7.5Hz, 1H), 7.58 (d, J = 7.5 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.33–7.27 (m, 2H), 7.24–7.19 (m, 3H), 5.21 (dd, J = 13.3, 5.2 Hz, 1H), 4.38 (dd, J = 62.3, 16.7 Hz, 2H), 3.70 (s, 2H), 3.11–2.99 (m, 2H), 2.90–2.76 (m, 4H), 2.29 (qd, J = 12.9, 5.3 Hz, 1H), 2.17–1.93 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 171.27, 169.60, 168.93, 143.60, 139.63, 134.78, 131.64, 128.78, 128.71, 128.62, 128.56, 128.51, 128.49, 128.45, 126.33, 126.27, 123.79, 118.74, 51.79, 46.93, 31.58, 23.38. LC-MS (M+H: 402.5): 97% purity.
3-(1-oxo-4-(3-phenoxyprop-1-yn-1-yl)isoindolin-2-yl)piperidine-2,6-dione 6d:
Compound 6d was prepared using S2 as the starting material. White solid (38 mg, 76% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.05 (s, 1H), 7.77 (d, J = 7.5 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.34 (t, J = 7.9 Hz, 2H), 7.09 (d, J = 8.1 Hz, 2H), 6.98 (t, J = 7.3 Hz, 1H), 5.16 (d, J = 5.1 Hz, 1H), 5.12 (s, 2H), 4.22 (dd, J = 64.3, 17.8 Hz, 2H), 2.93 (ddd, J = 18.3, 13.6, 5.2 Hz, 1H), 2.68–2.58 (m, 1H), 2.32 (qd, J = 13.2, 4.4 Hz, 1H), 2.04–1.92 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 178.11, 176.11, 172.62, 162.25, 149.51, 139.23, 137.27, 134.71, 134.01, 128.94, 126.59, 122.35, 120.47, 95.64, 87.51, 61.04, 56.69, 51.81, 36.35, 27.67. LC-MS (M+H: 375.4): 98% purity.
3-(4-(3-(benzyloxy)prop-1-yn-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 6e:
Compound 6e was prepared using S2 as the starting material. White solid (52 mg, 85% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.02 (s, 1H), 7.78 (dd, J = 7.6, 1.0 Hz, 1H), 7.74 (dd, J = 7.6, 1.1 Hz, 1H), 7.58 (t, = 7.6 Hz, 1H), 7.41–7.33 (m, 4H), 7.35–7.28 (m, 1H), 5.16 (dd, J = 13.3, 5.1 Hz, 1H), 4.64 (s, 2H), 4.56–4.18 (m, 4H), 2.92 (ddd, J = 17.2, 13.6, 5.4 Hz, 1H), 2.66–2.55 (m, 1H), 2.42 (qd, J = 13.3, 4.5 Hz, 1H), 2.05–1.99 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.33, 171.42, 167.96, 144.48, 138.11, 134.85, 132.57, 129.21, 128.81, 128.38, 128.19, 123.99, 117.97, 91.73, 82.07, 71.54, 57.96, 52.11, 47.38, 31.66, 22.84. LC-MS (M+H: 389.4): 95% purity.
3-(1-oxo-4-(3-phenethoxyprop-1-yn-1-yl)isoindolin-2-yl)piperidine-2,6-dione 6f:
Compound 6f was prepared using S2 as the starting material. White solid (47 mg, 87% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.02 (s, 1H), 7.77 (dd, J = 7.6, 1.1 Hz, 1H), 7.71 (dd, J = 7.7, 1.1 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.33–7.24 (m, 3H), 7.21–7.17 (m, 1H), 5.15 (dd, J = 13.3, 5.1 Hz, 1H), 4.54–4.15 (m, 4H), 3.77 (t, J = 6.8 Hz, 2H), 2.98–2.91 (m, 1H), 2.88 (t, J = 6.8 Hz, 2H), 2.60 (dd, J = 17.3, 3.8 Hz, 1H), 2.39 (qd, J = 13.3, 4.5 Hz, 1H), 2.05–1.99 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.32, 171.41, 167.97, 144.41, 139.34, 134.81, 132.55, 129.26, 129.21, 128.70, 126.55, 123.95, 117.99, 91.88, 81.80, 70.66, 58.35, 52.08, 47.32, 35.72, 31.67, 22.84. LC-MS (M+H: 403.5): 98% purity.
4-((3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)prop-2-yn-1-yl)oxy)benzaldehyde 6g:
Compound 6g was prepared using S2 as the starting material. White solid (49 mg, 90% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.02 (s, 1H), 9.89 (s, 1H), 7.99–7.85 (m, 2H), 7.78 (dd, J = 7.6, 1.1 Hz, 1H), 7.72 (dd, J = 7.7, 1.0 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.35–7.22 (m, 2H), 5.27 (s, 2H), 5.14 (dd, J = 13.3, 5.1 Hz, 1H), 4.29 (dd, J = 64.3, 17.8 Hz, 2H), 2.97–2.88 (m, 1H), 2.68–2.57 (m, 1H), 2.33 (qd, J = 13.2, 4.4 Hz, 1H), 2.05–1.89 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 191.85, 173.26, 171.36, 167.85, 162.41, 144.79, 134.73, 132.57, 132.17, 130.73, 129.29, 124.37, 117.37, 116.08, 89.92, 83.30, 56.85, 52.02, 47.20, 31.65, 22.90. LC-MS (M+H: 403.4): 94% purity.
3-(1-oxo-4-(5-phenoxypent-1-yn-1-yl)isoindolin-2-yl)piperidine-2,6-dione 6h:
Compound 6h was prepared using S2 as the starting material. White solid (50 mg, 84% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.01 (s, 1H), 7.71 (dd, J = 7.6, 1.1 Hz, 1H), 7.65 (dd, J = 7.6, 1.1 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.36–7.20 (m, 2H), 7.05–6.96 (m, 2H), 6.95–6.89 (m, 1H), 5.12 (dd, J = 13.4, 5.1 Hz, 1H), 4.33 (dd, J = 64.3, 17.8 Hz, 2H), 4.12 (t, J = 6.1 Hz, 2H), 2.92 (ddd, J = 17.3, 13.6, 5.4 Hz, 1H), 2.68 (t, J = 7.0 Hz, 2H), 2.58 (ddd, J = 17.2, 4.6, 2.2 Hz, 1H), 2.31 (qd, J = 13.2, 4.5 Hz, 1H), 2.08–1.87 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ 173.32, 171.38, 168.11, 158.99, 144.28, 134.53, 132.43, 129.99, 129.08, 123.18, 121.03, 119.13, 114.87, 95.80, 77.24, 66.31, 52.01, 47.30, 31.68, 28.17, 22.79, 16.09. LC-MS (M+H: 403.5): 96% purity.
4-((5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pent-4-yn-1-yl)oxy)benzaldehyde 6i:
Compound 6i was prepared using S2 as the starting material. White solid (50 mg, 78% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.00 (s, 1H), 9.87 (s, 1H), 7.95–7.83 (m, 2H), 7.72 (dd, J = 7.6, 1.1 Hz, 1H), 7.65 (dd, J = 7.7, 1.1 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.37 (dd, J = 64.3, 17.8 Hz, 2H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.47–4.27 (m, 2H), 4.25 (t, J = 6.2 Hz, 2H), 2.92 (ddd, J = 17.2, 13.6, 5.4 Hz, 1H), 2.69 (t, J = 7.0 Hz, 2H), 2.60 (dd, J = 4.6, 2.3 Hz, 1H), 2.55 (dd, J = 4.5, 2.2 Hz, 1H), 2.37 (qd, J = 13.2, 4.5 Hz, 1H), 2.08 (p, J = 6.6 Hz, 2H), 2.02–1.96 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 191.76, 173.30, 171.40, 168.11, 163.97, 144.30, 134.55, 132.44, 132.31, 130.15, 129.08, 123.21, 119.08, 115.40, 95.64, 77.31, 67.16, 52.05, 47.36, 31.66, 28.04, 22.83, 16.04. LC-MS (M+H: 431.5): 95% purity.
3-(4-(3-hydroxy-3-phenylprop-1-yn-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 7a:
Compound 7a was prepared using S2 as the starting material. White solid (45 mg, 90% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.02 (s, 1H), 7.74 (ddd, J = 23.3, 7.6, 1.1 Hz, 2H), 7.63–7.50 (m, 3H), 7.44–7.36 (m, 2H), 7.36–7.23 (m, 1H), 6.25 (dd, J = 6.0, 1.1 Hz, 1H), 5.67 (d, J = 5.9 Hz, 1H), 5.16 (dd, J = 13.3, 5.1 Hz, 1H), 4.28 (dd, J = 64.3, 17.8 Hz, 2H), 3.00–2.82 (m, 1H), 2.61 (ddd, J = 17.2, 4.5, 2.2 Hz, 1H), 2.49–2.36 (m, 1H), 2.07–2.01 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.47, 167.99, 144.43, 142.23, 134.58, 132.55, 129.21, 128.84, 128.23, 127.02, 126.99, 123.80, 118.26, 97.17, 80.80, 63.47, 63.45, 52.13, 47.42, 31.66, 22.87. LC-MS (M+H: 375.4): 97% purity.
3-(4-(3-hydroxy-3-phenylbut-1-yn-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 7b:
Compound 7b was prepared using S2 as the starting material. White solid (51 mg, 82% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.02 (s, 1H), 7.77 (d, J = 7.5 Hz, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.69–7.64 (m, 2H), 7.57 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 7.5 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 6.30 (d, J = 1.8 Hz, 1H), 5.16 (dd, J = 13.3, 5.1 Hz, 1H), 4.42 (dd, J = 64.3, 17.8 Hz, 2H), 2.93 (ddd, J = 18.2, 13.5, 5.3 Hz, 1H), 2.62 (d, J = 17.6 Hz, 1H), 2.45 (d, J = 13.3 Hz, 1H), 2.22–1.94 (m, 1H), 1.76 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 173.35, 171.48, 168.04, 168.03, 146.81, 144.49, 134.42, 132.56, 129.21, 128.56, 127.68, 125.38, 123.70, 118.38, 100.55, 79.22, 69.00, 52.15, 47.44, 34.17, 34.14, 31.66, 22.88. LC-MS (M+H: 389.2): 95% purity.
3-(4-((4-hydroxypiperidin-4-yl)ethynyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione hydrochloride 8:
The precursor of Compound 8 was prepared using S2 as the starting material. The intermediate was suspended in 4M hydrochloric acid in dioxane and stirred for 2 h at room temperature. Remove the solvent using rotary evaporators to afford the target product. White solid (15 mg, 86% yield over two steps). 1H NMR (400 MHz, DMSO-d6): δ 11.02 (s, 1H), 9.12 (s, 1H), 9.00 (s, 1H), 7.78 (dd, J = 7.6, 1.1 Hz, 1H), 7.71 (dd, J = 7.6, 1.1 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 5.97 (s, 1H), 5.17 (dd, J = 13.3, 5.1 Hz, 1H), 4.43 (dd, J = 64.3, 17.8 Hz, 2H) 3.20–3.17 (m, 2H), 3.11–3.07 (m, 2H), 2.94 (ddd, J = 17.1, 13.4, 5.3 Hz, 1H), 2.66–2.58 (m, 1H), 2.54–2.43 (m, 2H), 2.14–2.09 (m, 2H), 2.07–1.97 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.44, 167.98, 144.58, 134.56, 132.57, 129.22, 123.95, 117.93, 98.18, 79.75, 72.64, 70.99, 63.71, 60.65, 52.10, 47.40, 35.72, 35.70, 31.66, 22.88. LC-MS (M+H: 368.2): 98% purity.
General procedure for the synthesis of 9:
To a mixture of 3-(4-iodo-1-oxoisoindolin-2-yl)-1-methyl piperidine-2,6-dione (S2, 0.2 mmol, 1.0 equiv.) and arylboronic acid (0.3 mmol, 1.5 equiv.) in dry DMF was added sodium hydroxide (8 mg, 0.2 mmol, 1.0 equiv.), N,N-Dicyclohexylmethylamine (0.2 mmol, 1.0 equiv.) and Pd(PPh3)Cl2 (14 mg, 0.02 mmol, 0.1 equiv.). The resulting solution was degassed and refilled with Argon three times. The reaction mixture was stirred at 80°C overnight. After being cooled down to room temperature, the solvent was removed under reduced pressure and the residue was submitted to the flash column chromatography (eluted with 5–10% methanol in DCM), giving the corresponding desired compound as white or light yellow solid, which was further purified by recrystallization in acetone.
3-(1-oxo-4-phenylisoindolin-2-yl)piperidine-2,6-dione 9a:
White solid (15 mg, 34% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 1H), 7.77 (dd, J = 7.3, 1.2 Hz, 1H), 7.72 (dd, J = 7.7, 1.2 Hz, 1H), 7.66 (d, J = 7.5 Hz, 1H), 7.64–7.58 (m, 2H), 7.52 (dd, J = 8.4, 6.6 Hz, 1H), 7.48–7.39 (m, 1H), 5.15 (dd, J = 13.3, 5.2 Hz, 1H), 4.45 (dd, J = 64.3, 17.8 Hz, 2H), 2.98–2.84 (m, 1H), 2.58 (ddd, J = 17.1, 4.5, 2.1 Hz, 1H), 2.44 (td, J = 13.2, 4.4 Hz, 1H), 2.03–1.97 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.43, 168.37, 139.88, 138.48, 137.24, 132.91, 132.14, 129.42, 128.54, 128.49, 122.68, 52.08, 47.71, 31.65, 22.85. LC-MS (M+H: 321.1): >99% purity.
3-(4-(4-fluorophenyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 9b:
White solid (18 mg, 25% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 1H), 7.77 (dd, J = 7.3, 1.3 Hz, 1H), 7.71 (dd, J = 7.7, 1.3 Hz, 1H), 7.67 (dd, J = 6.1, 2.6 Hz, 2H), 7.65–7.61 (m, 1H), 7.40–7.25 (m, 2H), 5.15 (dd, J = 13.3, 5.1 Hz, 1H), 4.48 (dd, J = 64.3, 17.8 Hz, 2H), 2.91 (ddd, J = 17.3, 13.6, 5.4 Hz, 1H), 2.59 (ddd, J = 17.2, 4.6, 2.3 Hz, 1H), 2.42 (td, J = 13.2, 4.5 Hz, 1H), 2.03–1.96 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.41, 168.32, 163.67, 161.23, 139.91, 136.25, 134.89, 134.86, 132.92, 132.17, 130.75, 130.67, 129.42, 122.75, 116.37, 116.16, 52.07, 47.63, 31.64, 22.87. LC-MS (M+H: 339.1): 99% purity.
3-(4-(4-acetylphenyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 9c:
White solid (10 mg, 14% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.98 (s, 1H), 8.11–7.99 (m, 2H), 7.81 (td, J = 7.1, 1.2 Hz, 2H), 7.79–7.75 (m, 2H), 7.69 (t, J = 7.5 Hz, 1H), 5.16 (dd, J = 13.3, 5.1 Hz, 1H), 4.49 (J = 64.3, 17.8 Hz, 2H), 2.91 (ddd, J = 17.2, 13.6, 5.4 Hz, 1H), 2.64 (s, 3H), 2.59 (d, J = 17.8 Hz, 1H), 2.42 (td, J = 13.2, 4.5 Hz, 1H), 2.05–1.94 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 198.02, 173.34, 171.39, 168.21, 142.99, 140.08, 136.57, 136.18, 133.05, 132.23, 129.55, 129.25, 128.92, 123.42, 52.09, 47.67, 31.63, 27.30, 22.86. LC-MS (M+H: 363.1): 98% purity.
4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzaldehyde 9d:
White solid (21 mg, 30% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.98 (s, 1H), 10.10 (s, 1H), 8.21–7.97 (m, 2H), 7.91–7.83 (m, 2H), 7.83–7.78 (m, 2H), 7.70 (t, J = 7.6 Hz, 1H), 5.16 (dd, J = 13.3, 5.1 Hz, 1H), 4.57 (dd, J = 64.3, 17.8 Hz, 2H), 3.01–2.80 (m, 1H), 2.59 (dt, J = 17.2, 2.9 Hz, 1H), 2.43 (td, J = 13.3, 4.5 Hz, 1H), 2.03–1.97 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 193.32, 173.34, 171.37, 168.17, 144.31, 140.14, 136.06, 135.94, 133.10, 132.33, 130.48, 129.58, 129.41, 123.59, 52.10, 47.68, 31.63, 22.85. LC-MS (M+H: 349.1): 91% purity.
3-(4-(3-hydroxyphenyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 9e:
White solid (19 mg, 28% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.98 (s, 1H), 9.63 (d, J = 1.3 Hz, 1H), 7.75 (dt, J = 7.1, 1.5 Hz, 1H), 7.69–7.57 (m, 2H), 7.30 (td, J = 7.9, 1.2 Hz, 1H), 7.03–6.98 (m, 1H), 6.94 (q, J = 1.8 Hz, 1H), 6.89–6.81 (m, 1H), 5.15 (dd, J = 13.2, 5.0 Hz, 1H), 4.49 (dd, J = 64.3, 17.8 Hz, 2H), 2.91 (ddd, J = 17.9, 13.4, 5.3 Hz, 1H), 2.58 (d, J = 18.8 Hz, 1H), 2.43 (td, J = 13.3, 4.6 Hz, 1H), 2.06–1.92 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.48, 168.37, 158.21, 139.78, 139.73, 137.39, 132.85, 131.95, 130.49, 129.36, 122.60, 119.17, 115.46, 115.26, 52.05, 47.69, 31.67, 22.85. LC-MS (M+H: 337.1): 97% purity.
3-(4-(2-aminophenyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 9f:
White solid (11 mg, 17% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.98 (s, 1H), 7.73 (dd, J = 6.3, 2.4 Hz, 1H), 7.66–7.44 (m, 2H), 7.13 (t, J = 7.8 Hz, 1H), 6.75 (t, J = 2.0 Hz, 1H), 6.72–6.66 (m, 1H), 6.65–6.55 (m, 1H), 5.22 (s, 2H), 5.15 (dd, J = 13.3, 5.1 Hz, 1H), 4.48 (dd, J = 64.3, 17.8 Hz, 2H), 2.91 (ddd, J = 17.3, 13.6, 5.4 Hz, 1H), 2.63–2.55 (m, 1H), 2.49–2.36 (m, 1H), 2.05–1.88 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.48, 168.45, 149.64, 139.61, 139.11, 138.15, 132.77, 131.78, 129.91, 129.25, 122.31, 115.84, 114.01, 113.70, 52.06, 47.78, 31.67, 22.88. LC-MS (M+H: 336.1): 95% purity.
3-(4-(1H-indazol-5-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 10:
White solid (27 mg, 37% yield). 1H NMR (400 MHz, DMSO-d6): δ 13.20 (s, 1H), 10.97 (s, 1H), 8.15–8.08 (m, 1H), 7.99–7.96 (m, 1H), 7.81–7.71 (m, 2H), 7.67 (d, J = 7.9 Hz, 2H), 7.58 (d, J = 8.7 Hz, 1H), 5.16 (dd, J = 13.9, 5.1 Hz, 1H), 4.78–4.37 (m, 2H), 2.99–2.73 (m, 1H), 2.68–2.55 (m, 1H), 2.47–2.33 (m, 1H), 2.03–1.89 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 174.11, 172.22, 169.25, 140.78, 140.61, 138.65, 135.28, 133.63, 133.17, 131.45, 130.11, 127.80, 124.49, 122.92, 121.25, 111.85, 52.83, 48.56, 32.41, 23.63. LC-MS (M+H: 361.1): 97% purity.
General procedure for the synthesis of R1–13:
To a mixture of aryl carboxylic acid (0.2 mmol, 1.0 equiv.) and HATU (114 mg, 0.3 mmol, 1.5 equiv.) in dry DMF (5 mL) was added N,N-diisopropylethylamine (0.1 mL, 0.6 mmol, 3 equiv.). The resulting solution was stirred for 1 h at room temperature. Then 2,6-Dioxopiperidine-3-ammonium chloride (40 mg, 0.24 mmol, 1.2 equiv.) was added to the solution. The mixture was stirred overnight. After the aryl carboxylic acid was consumed, the solvent was removed under reduced pressure and the residue was submitted to the flash column chromatography (eluted with 5–10% methanol in DCM), giving the corresponding desired compound as white solid.
(4-((2,6-dioxopiperidin-3-yl)carbamoyl)phenyl)methanaminium chloride R1:
The synthetic intermediate was prepared follow the general procedure, then treat the precursor with 4 M HCl solution in 1,4-dioxane to get the target product. White solid (39 mg, 53% yield over two steps). 1H NMR (400 MHz, DMSO-d6): δ 10.88 (s, 1H), 8.86 (d, J = 8.3 Hz, 1H), 8.49 (s, 2H), 7.92 (d, J = 8.3 Hz, 2H), 7.60 (d, J = 8.2 Hz, 2H), 4.79 (ddd, J = 12.4, 8.3, 5.3 Hz, 1H), 4.10 (s, 1H), 2.81 (ddd, J = 17.2, 13.3, 5.5 Hz, 1H), 2.61–2.52 (m, 1H), 2.15 (qd, J = 12.9, 4.5 Hz, 1H), 2.02–1.95 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.51, 172.64, 166.09, 137.88, 134.28, 129.23, 128.00, 50.02, 42.26, 31.47, 24.65. LC-MS (M+H: 262.1): 87% purity.
4-bromo-N-(2,6-dioxopiperidin-3-yl)benzamide R2:
White solid (53 mg, 85% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.88 (s, 1H), 8.87 (d, J = 8.3 Hz, 1H), 7.91–7.76 (m, 2H), 7.75–7.65 (m, 2H), 4.79 (ddd, J = 13.1, 8.3, 5.4 Hz, 1H), 2.81 (ddd, J = 18.1, 13.2, 5.5 Hz, 1H), 2.57 (d, J = 3.9 Hz, 1H), 2.12 (qd, J = 12.9, 4.4 Hz, 1H), 2.04–1.88 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.49, 172.57, 165.68, 133.47, 131.90, 129.91, 125.73, 50.04, 31.44, 24.59. LC-MS (M+H: 311.0): >99% purity.
4-amino-N-(2,6-dioxopiperidin-3-yl)benzamide R3:
White solid (43 mg, 87% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.80 (s, 1H), 8.27 (d, J = 8.3 Hz, 1H), 7.59 (d, J = 8.3 Hz, 2H), 6.56 (d, J = 8.2 Hz, 2H), 5.66 (s, 2H), 4.72 (ddd, J = 12.8, 8.1, 5.5 Hz, 1H), 2.78 (ddd, J = 17.2, 12.7, 5.1 Hz, 1H), 2.54 (d, J = 3.7 Hz, 1H), 2.24–2.02 (m, 1H), 1.95–1.91 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.59, 173.14, 166.50, 152.34, 129.37, 120.95, 112.98, 49.74, 31.52, 24.92. LC-MS (M+H: 248.1): 98% purity.
N-(2,6-dioxopiperidin-3-yl)-2-iodobenzamide R4:
White solid (52 mg, 73% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.86 (s, 1H), 8.72 (d, J = 8.4 Hz, 1H), 7.90 (dd, J = 7.9, 1.1 Hz, 1H), 7.48 (td, J = 7.5, 1.1 Hz, 1H), 7.38 (dd, J = 7.6, 1.7 Hz, 1H), 4.74 (dt, J = 10.6, 8.4 Hz, 1H), 2.93– 2.70 (m, 1H), 2.56 (dt, J = 17.7, 4.1 Hz, 1H), 2.24–1.77 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.45, 172.18, 169.19, 142.68, 139.71, 131.48, 128.59, 128.50, 93.90, 49.76, 31.23, 24.54. LC-MS (M+H: 359.0): 95% purity.
N-(2,6-dioxopiperidin-3-yl)-1,2,3,4-tetrahydroquinoline-4-carboxamide R5:
White solid (46 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.25 (t, J = 8.6 Hz, 1H), 7.11–6.75 (m, 2H), 6.46 (dd, J = 8.2, 1.2 Hz, 1H), 6.42 (tdd, J = 7.4, 4.1, 1.2 Hz, 1H), 5.73 (d, J = 3.1 Hz, 1H), 4.67–4.47 (m, 1H), 3.60 (t, J = 6.1 Hz, 1H), 3.15–3.08 (m, 1H), 2.80–2.71 (m, 1H), 2.04–1.94 (m, 3H), 1.93–1.83 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 174.44, 174.35, 173.48, 173.45, 172.70, 145.91, 145.88, 129.40, 129.27, 127.62, 118.46, 118.35, 115.50, 115.45, 114.25, 114.21, 60.23, 49.62, 49.58, 43.28, 43.05, 31.40, 25.73, 24.80, 24.74, 21.24, 14.56. LC-MS (M+H: 288.1): 99% purity.
N-(2,6-dioxopiperidin-3-yl)-1H-indole-2-carboxamide R6:
White solid (49 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.64 (d, J = 2.2 Hz, 1H), 10.90 (s, 1H), 8.77 (d, J = 8.5 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.45 (dd, J = 8.4, 1.1 Hz, 1H), 7.20 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.16 (d, J = 2.1 Hz, 1H), 7.05 (ddd, J = 8.1, 6.9, 1.0 Hz, 1H), 4.83 (ddd, J = 12.5, 8.5, 5.3 Hz, 1H), 2.84 (ddd, J = 17.3, 13.2, 5.5 Hz, 1H), 2.57 (dt, J = 17.3, 3.8 Hz, 1H), 2.15 (qd, J = 12.9, 4.5 Hz, 1H), 2.06–1.99 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.52, 172.75, 161.55, 137.00, 131.66, 127.48, 123.94, 122.07, 120.25, 112.81, 103.42, 49.64, 31.49, 24.79. LC-MS (M+H: 272.2): 99% purity.
N-(2,6-dioxopiperidin-3-yl)-1H-indole-3-carboxamide R7:
White solid (46 mg, 83% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.60 (d, J = 2.8 Hz, 1H), 10.84 (s, 1H), 8.22–8.11 (m, 2H), 8.05 (d, J = 2.9 Hz, 1H), 7.45 (dt, J = 8.0, 1.1 Hz, 1H), 7.19–7.10 (m, 2H), 4.79 (ddd, J = 12.2, 8.3, 5.4 Hz, 1H), 2.81 (ddd, J = 17.3, 13.1, 5.6 Hz, 1H), 2.56 (dt, J = 17.4, 3.8 Hz, 1H), 2.13 (qd, J = 12.8, 4.4 Hz, 1H), 2.06–1.90 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.62, 173.32, 164.84, 136.59, 128.64, 126.51, 122.41, 121.40, 120.91, 112.35, 110.58, 49.33, 31.59, 25.16. LC-MS (M+H: 272.1): 97% purity.
N-(2,6-dioxopiperidin-3-yl)-1H-indole-6-carboxamide R8:
White solid (36 mg, 67% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.41 (d, J = 2.8 Hz, 1H), 10.85 (s, 1H), 8.66 (d, J = 8.4 Hz, 1H), 7.99 (s, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.55 (dd, J = 8.3, 1.5 Hz, 1H), 7.53 (t, J = 2.8 Hz, 1H), 6.50 (ddd, J = 3.0, 1.9, 0.9 Hz, 1H), 4.81 (ddd, J = 12.4, 8.4, 5.3 Hz, 1H), 2.82 (ddd, J = 17.3, 13.3, 5.5 Hz, 1H), 2.56 (ddd, J = 17.2, 4.3, 2.9 Hz, 1H), 2.16 (qd, J = 12.9, 4.5 Hz, 1H), 2.03–1.97 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.59, 173.02, 167.53, 135.68, 130.46, 128.62, 127.19, 119.92, 118.52, 111.82, 101.74, 49.99, 31.53, 24.84. LC-MS (M+H: 272.1): >99% purity.
N-(2,6-dioxopiperidin-3-yl)-1H-indole-7-carboxamide R9:
White solid (39 mg, 72% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.13 (s, 1H), 10.89 (s, 1H), 8.85 (d, J = 8.3 Hz, 1H), 7.91–7.74 (m, 1H), 7.70 (dd, J = 7.6, 1.0 Hz, 1H), 7.36 (t, J = 2.8 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 6.51 (dd, J = 3.1, 2.0 Hz, 1H), 4.82 (ddd, J = 12.4, 8.3, 5.3 Hz, 1H), 2.84 (ddd, J = 17.3, 13.3, 5.4 Hz, 1H), 2.59 (dt, J = 17.2, 3.8 Hz, 1H), 2.23 (qd, J = 12.9, 4.5 Hz, 1H), 2.05–1.97 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.58, 172.83, 167.35, 134.70, 129.71, 127.21, 124.59, 120.46, 118.53, 116.76, 101.51, 49.79, 31.52, 24.67. LC-MS (M+H: 272.1): 98% purity.
N-(2,6-dioxopiperidin-3-yl)-7-fluoro-1H-indazole-3-carboxamide R10:
White solid (31 mg, 54% yield). 1H NMR (400 MHz, DMSO-d6): δ 14.24 (s, 1H), 10.86 (s, 1H), 8.77 (d, J = 8.5 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.63–6.63 (m, 2H), 4.83 (ddd, J = 12.5, 8.5, 5.3 Hz, 1H), 2.82 (ddd, J = 17.2, 13.6, 5.5 Hz, 1H), 2.57 (t, J = 3.1 Hz, 1H), 2.25 (qd, J = 13.0, 4.4 Hz, 1H), 2.05–1.99 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.53, 172.70, 162.22, 154.78, 131.14, 125.81, 123.41, 118.19, 118.14, 111.30, 49.41, 31.50, 24.57. LC-MS (M+H: 291.1): 94% purity.
4-chloro-N-(2,6-dioxopiperidin-3-yl)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide R11:
White solid (38 mg, 61% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.26 (s, 1H), 8.64 (s, 2H), 4.21 (dd, J = 13.1, 5.2 Hz, 1H), 2.72 (ddd, J = 18.4, 13.4, 5.5 Hz, 1H), 2.64–2.55 (m, 1H), 2.22 (ddd, J = 10.6, 6.6, 4.3 Hz, 1H), 2.03 (qd, J = 13.1, 4.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ 172.78, 170.80, 165.18, 151.60, 124.85, 112.39, 104.16, 90.82, 70.18, 49.50, 30.68, 22.60. LC-MS (M+H: 291.1): 94% purity. LC-MS (M+H: 308.6): 98% purity.
N-(2,6-dioxopiperidin-3-yl)-5-fluoro-1H-indole-2-carboxamide R12:
White solid (47 mg, 82% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.76 (d, J = 2.2 Hz, 1H), 10.90 (s, 1H), 8.82 (d, J = 8.5 Hz, 1H), 7.51–7.35 (m, 2H), 7.15 (dd, J = 2.2, 0.9 Hz, 1H), 7.06 (td, J = 9.3, 2.6 Hz, 1H), 4.83 (ddd, J = 12.4, 8.5, 5.3 Hz, 1H), 2.83 (ddd, J = 17.2, 13.2, 5.5 Hz, 1H), 2.57 (dt, J = 17.4, 3.8 Hz, 1H), 2.15 (qd, J = 12.9, 4.5 Hz, 1H), 2.05–1.97 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.51, 172.68, 161.25, 158.79, 156.48, 127.61, 127.51, 114.05, 113.95, 112.81, 112.54, 106.36, 106.13, 103.47, 103.41, 49.66, 31.48, 24.74. LC-MS (M+H: 290.1): 98% purity.
N-(2,6-dioxopiperidin-3-yl)-1H-indole-4-carboxamide R13:
White solid (46 mg, 82% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.32 (s, 1H), 10.86 (s, 1H), 8.46 (d, J = 8.4 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.47 (dd, J = 5.3, 2.5 Hz, 2H), 7.16 (t, J = 7.7 Hz, 1H), 6.93 (d, J = 2.7 Hz, 1H), 4.85 (ddd, J = 12.2, 8.4, 5.3 Hz, 1H), 2.82 (ddd, J = 17.2, 13.3, 5.5 Hz, 1H), 2.56 (dt, J = 19.3, 4.6 Hz, 1H), 2.16 (qd, J = 12.9, 4.4 Hz, 1H), 2.06–2.00 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.58, 173.02, 168.06, 137.05, 127.06, 126.43, 126.36, 120.59, 119.19, 114.89, 102.30, 49.88, 31.52, 24.84. LC-MS (M+H: 272.1): 99% purity.
General procedure for the synthesis of HDAC6 degraders 11a, 11b and 12:
To a solution of aldehyde (6g, 6i or 9d, 0.05 mmol) and 8-hydrazineyl-N-hydroxy-8-oxooctanamide (10mg, 0.05 mmol) in 2 mL DMF was added a drop of Acetic acid. The resulting solution was stirred at 80°C for 3 h. After being cooled down to room temperature, the solvent was removed under reduced pressure and the residue was submitted to the flash column chromatography (eluted with 10–20% methanol in DCM), giving the corresponding desired compound as white solid.
8-(2-(4-((3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)prop-2-yn-1-yl)oxy)benzylidene) hydrazineyl)-N-hydroxy-8-oxooctanamide 11a (99% yield).
1H NMR (400 MHz, DMSO-d6): δ 11.13 (s, 1H), 10.96 (s, 1H), 10.33 (s, 1H), 8.68 (s, 1H), 8.09 (s, 1H), 7.91 (s, 1H), 7.74 (dd, J = 13.8, 7.7 Hz, 1H), 7.70 (dd, J = 13.8, 7.7 Hz, 1H), 7.63 (dd, J = 11.9, 8.5 Hz, 2H, 7.55 (d, J = 7.5 Hz, 1H), 7.14 (dd, J = 8.5, 5.3 Hz, 2H), 5.33–4.98 (m, 3H), 4.35 (d, J = 17.8 Hz, 1H), 4.20 (dd, J = 17.8, 10.6 Hz, 1H), 2.91 (ddt, J = 18.6, 12.3, 5.4 Hz, 1H), 2.60 (q, J = 10.1, 7.4 Hz, 2H), 2.32 (dtt, J = 22.3, 11.0, 5.9 Hz, 1H), 2.18 (d, J = 7.3 Hz, 1H), 2.07–1.84 (m, 3H), 1.55 (q, J = 7.4 Hz, 2H), 1.48 (q, J = 7.2 Hz, 2H), 1.35–1.18 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 174.62, 173.24, 171.35, 169.57, 168.95, 167.87, 158.85, 158.70, 145.83, 144.79, 144.73, 142.47, 134.67, 132.57, 129.27, 128.89, 128.54, 128.27, 124.27, 117.51, 115.97, 90.45, 82.98, 56.55, 52.04, 47.22, 34.64, 32.72, 32.32, 31.63, 29.01, 28.89, 25.50, 25.41, 24.60, 22.89. LC-MS (M+H: 588.3): 96% purity.
8-(2-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pent-4-yn-1-yl)oxy)benzylidene) hydrazineyl)-N-hydroxy-8-oxooctanamide 11b (99% yield).
1H NMR (400 MHz, DMSO-d6): δ 11.12 (s, 1H), 10.99 (s, 1H), 10.33 (s, 1H), 8.65 (s, 1H), 8.00 (d, J = 71.1 Hz, 1H), 7.72 (d, J = 7.5 Hz, 1H), 7.66–7.51 (m, 3H), 7.03 (d, J = 8.3 Hz, 1H), 5.12 (dd, J = 13.3, 5.1 Hz, 1H), 4.48–4.26 (m, 2H), 4.17 (q, J = 5.5 Hz, 2H), 3.00–2.84 (m, 1H), 2.68 (t, J = 7.0 Hz, 2H), 2.64–2.54 (m, 3H), 2.44–2.31 (m, 1H), 2.17 (t, J = 7.4 Hz, 1H), 2.05 (t, J = 6.5 Hz, 2H), 1.94 (t, J = 7.4 Hz, 2H), 1.58–1.53 (m, 2H), 1.49 (t, J = 6.89Hz, 2H), 1.30–1.24 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 174.58, 173.28, 171.40, 168.10, 146.00, 144.30, 132.45, 129.08, 128.62, 127.51, 123.20, 119.11, 115.22, 95.77, 77.25, 66.72, 52.06, 47.39, 32.72, 32.34, 31.66, 28.90, 28.85, 28.18, 25.50, 25.50, 25.44, 24.62, 24.59, 22.82, 22.81, 20.01, 16.09, 14.35. LC-MS (M+H: 616.3): 99% purity.
8-(2-(4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzylidene)hydrazineyl)-N-hydroxy-8-oxooctanamide 12 (99% yield).
1H NMR (400 MHz, DMSO-d6): δ 11.33 (s, 1H), 10.98 (s, 1H), 10.33 (s, 1H), 8.65 (s, 1H), 8.14 (d, J = 80.1 Hz, 1H), 7.80–7.74 (m, 4H), 7.70–7.58 (m, 3H), 5.15 (ddd, J = 13.3, 5.1, 2.5 Hz, 1H), 4.65 (dd, J = 17.4, 2.3 Hz, 1H), 4.44 (dd, J = 17.4, 4.9 Hz, 1H), 2.91 (ddd, J = 17.9, 13.5, 5.4 Hz, 1H), 2.66–2.53 (m, 2H), 2.49–2.37 (m, 2H), 2.19 (dt, J = 14.9, 7.5 Hz, 1H), 2.04–1.97 (m, 1H), 1.94 (t, J = 7.2 Hz, 2H), 1.89 (d, J = 4.6 Hz, 2H), 1.82 (d, J = 2.9 Hz, 1H), 1.58 (h, J = 7.2 Hz, 2H), 1.52–1.42 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 174.42, 172.88, 170.95, 169.11, 168.68, 168.46, 167.83, 149.49, 145.11, 141.71, 139.44, 139.22, 138.98, 136.10, 134.10, 134.03, 132.54, 131.53, 129.01, 128.55, 128.51, 127.43, 127.09, 122.49, 51.60, 47.29, 32.25, 31.93, 31.18, 28.53, 28.41, 25.03, 24.95, 24.89, 24.16, 22.39. LC-MS (M+H: 534.2): 96% purity.
The synthesis procedure of HDAC6 degraders based on HDAC6-selective inhibitor Next-A was previously reported.(Wu et al., 2019)
4-((1-butyl-3-(4-(2-(4-(((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzyl)amino)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)phenyl)ureido)methyl)-N-hydroxybenzamide 13a (21% yield).
1H NMR (400 MHz, DMSO-d6): δ 10.96 (s, 2H), 8.24 (s, 1H), 8.04 (s, 1H), 7.75–7.69 (m, 5H), 7.63 (t, J = 7.7 Hz, 1H), 7.55 (d, J = 7.5 Hz, 2H), 7.46 (d, J = 7.9 Hz, 2H), 7.35–7.29 (m, 4H), 6.82 (d, J = 8.5 Hz, 2H), 5.13 (dd, J = 13.5, 5.1 Hz, 1H), 4.82–4.70 (m, 2H), 4.64 (s, 1H), 4.59 (d, J = 8.4 Hz, 2H), 4.50-.28 (m, 3H), 3.77 (s, 4H), 3.35–3.24 (m, 2H), 3.09–2.82 (m, 1H), 2.56–2.53 (m, 1H), 2.48–2.42 (m, 1H), 2.18 (t, J = 7.3 Hz, 1H), 1.74 (s, 1H), 1.60 (d, J = 12.8 Hz, 1H), 1.47 (q, J = 7.4 Hz, 3H), 0.85 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 174.50, 172.85, 170.94, 167.91, 164.00, 155.40, 153.07, 146.30, 142.39, 140.43, 139.34, 136.69, 136.30, 134.04, 132.41, 131.56, 131.38, 128.89, 128.61, 127.83, 126.97, 123.18, 122.02, 121.87, 114.36, 66.53, 52.09, 51.74, 49.01, 47.30, 46.03, 43.45, 31.18, 29.96, 29.00, 23.97, 19.44, 13.79. LC-MS (M+H: 814.9): 99% purity.
4-((1-butyl-3-(4-(3-(4-(((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzyl)amino)methyl)-1H-1,2,3-triazol-1-yl)propoxy)phenyl)ureido)methyl)-N-hydroxybenzamide 13b (30% yield).
1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 1H), 8.23 (s, 1H), 8.02 (s, 1H), 7.72 (q, J = 8.5, 8.0 Hz, 3H), 7.63 (t, J = 7.5 Hz, 1H), 7.55 (d, J = 7.4 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 7.33 (dd, J = 13.5, 8.2 Hz, 3H), 6.82 (d, J = 8.5 Hz, 2H), 5.14 (dd, J = 13.4, 5.1 Hz, 1H), 4.72–4.30 (m, 6H), 3.92 (t, J = 6.0 Hz, 2H), 3.77 (s, 4H), 3.27 (t, J = 7.7 Hz, 2H), 2.90 (ddd, J = 18.2, 13.5, 5.4 Hz, 1H), 2.57 (d, J = 16.8 Hz, 1H), 2.43 (td, J = 13.1, 4.4 Hz, 1H), 2.30–2.15 (m, 2H), 2.04–1.94 (m, 1H), 1.91 (s, 1H), 1.73 (s, 1H), 1.65–1.36 (m, 3H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 172.86, 172.09, 170.96, 167.93, 164.02, 155.45, 153.60, 146.19, 142.44, 140.34, 139.35, 136.70, 136.33, 133.70, 132.43, 131.57, 131.39, 128.90, 128.65, 127.84, 126.99, 122.85, 122.04, 121.97, 114.19, 64.62, 51.74, 48.98, 47.31, 46.42, 46.05, 43.47, 31.19, 29.98, 29.10, 23.97, 22.40, 19.46, 13.81. LC-MS (M+H: 829.0): 97% purity.
4-((1-butyl-3-(4-(4-(4-(((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzyl)amino)methyl)-1H-1,2,3-triazol-1-yl)butoxy)phenyl)ureido)methyl)-N-hydroxybenzamide 13c (55% yield).
1H NMR (400 MHz, DMSO-d6): δ 11.16 (s, 1H), 10.96 (s, 1H), 9.01 (s, 1H), 8.21 (s, 1H), 8.00 (s, 1H), 7.76–7.69 (m, 4H), 7.63 (t, J = 7.5 Hz, 1H), 7.55 (d, J = 7.8 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.33–7.30 (m, 4H), 6.80 (d, J = 8.6 Hz, 2H), 5.14 (dd, J = 13.2, 5.2 Hz, 1H), 4.73–4.54 (m, 3H), 4.50–4.26 (m, 3H), 3.92 (t, J = 6.4 Hz, 2H), 3.78–3.76 (m, 4H), 3.33–3.17 (m, 2H), 2.90 (ddd, J = 18.4, 13.6, 5.5 Hz, 1H), 2.63–2.52 (m, 1H), 2.49–2.33 (m, 1H), 1.95 (dq, J = 13.5, 7.5, 5.9 Hz, 3H), 1.66 (t, J = 7.4 Hz, 2H), 1.47 (p, J = 7.6 Hz, 2H), 1.24 (d, J = 8.6 Hz, 2H), 0.85 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 172.85, 170.95, 167.91, 164.03, 155.45, 153.79, 146.23, 142.44, 140.43, 139.34, 136.69, 136.31, 133.46, 132.42, 131.57, 131.38, 128.90, 128.63, 127.83, 126.98, 122.64, 122.01, 114.09, 66.90, 51.80, 51.61, 48.97, 47.29, 46.04, 43.51, 31.18, 29.97, 29.01, 26.63, 25.78, 22.38, 19.45, 13.81. LC-MS (M+H: 842.9): 98% purity.
4-((1-butyl-3-(4-((5-(4-(((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzyl)amino)methyl)-1H-1,2,3-triazol-1-yl)pentyl)oxy)phenyl)ureido)methyl)-N-hydroxybenzamide 13d (21% yield).
1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, 2H), 9.22 (s, 1H), 8.23 (s, 1H), 7.99 (s, 1H), 7.84–7.68 (m, 4H), 7.63 (t, J = 7.5 Hz, 1H), 7.55 (d, J = 7.8 Hz, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.32 (dd, J = 8.6, 4.2 Hz, 4H), 6.83–6.73 (m, 2H), 5.14 (dd, J = 13.2, 5.1 Hz, 1H), 4.68–4.51 (m, 3H), 4.49–4.31 (m, 3H), 3.89 (t, J = 6.4 Hz, 2H), 3.77 (s, 4H), 3.27 (t, J = 7.5 Hz, 2H), 2.91 (ddd, J = 17.9, 13.5, 5.3 Hz, 1H), 2.69–2.54 (m, 1H), 2.43 (td, J = 13.2, 4.4 Hz, 1H), 2.09–1.96 (m, 1H), 1.87 (p, J = 7.6 Hz, 3H), 1.71 (q, J = 6.5, 6.1 Hz, 3H), 1.51–1.42 (m, 2H), 1.43–1.35 (m, 2H), 1.26–1.22 (m, 2H), 0.85 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 173.34, 171.43, 168.40, 155.95, 154.36, 139.82, 137.18, 136.75, 133.87, 132.89, 132.04, 131.88, 131.78, 129.38, 129.07, 128.30, 127.45, 123.03, 122.50, 115.81, 114.53, 67.81, 60.51, 52.48, 52.29, 52.09, 49.62, 49.45, 49.06, 47.78, 46.50, 31.65, 30.45, 30.00, 28.61, 25.73, 24.70, 23.07, 22.85, 19.92, 14.74, 14.28. LC-MS (M+H: 857.4): 95% purity.
4-((1-butyl-3-(4-((6-(4-(((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzyl)amino)methyl)-1H-1,2,3-triazol-1-yl)hexyl)oxy)phenyl)ureido)methyl)-N-hydroxybenzamide 13e (41% yield).
1H NMR (400 MHz, DMSO-d6): δ 11.17 (s, 1H), 10.97 (s, 1H), 9.04 (s, 1H), 8.21 (s, 1H), 7.99 (s, 1H), 7.73 (q, J = 8.7, 7.8 Hz, 4H), 7.63 (d, J = 7.3 Hz, 1H), 7.56 (d, J = 7.3 Hz, 2H), 7.49 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 8.0 Hz, 4H), 6.80 (d, J = 8.7 Hz, 2H), 5.15 (dd, J = 13.6, 5.1 Hz, 1H), 4.68–4.53 (m, 3H), 4.48–4.27 (m, 3H), 3.87 (d, J = 6.7 Hz, 2H), 3.79 (s, 4H), 3.27 (d, J = 7.9 Hz, 2H), 2.91 (td, J = 15.4, 12.9, 5.1 Hz, 1H), 2.58 (d, J = 18.3 Hz, 1H), 2.43 (s, 1H), 2.03–1.78 (m, 3H), 1.67 (t, J = 7.3 Hz, 3H), 1.44 (h, J = 7.2 Hz, 4H), 1.36–1.19 (m, 6H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 173.33, 171.43, 168.39, 155.95, 154.41, 142.93, 139.83, 137.13, 136.92, 133.82, 132.90, 132.05, 131.85, 129.38, 129.22, 128.34, 127.46, 122.53, 114.52, 67.89, 52.09, 49.45, 47.77, 46.51, 31.65, 30.45, 30.15, 29.05, 26.12, 25.44, 22.86, 19.92, 14.27. LC-MS (M+H: 871.3): 96% purity.
4-((1-butyl-3-(4-((7-(4-(((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzyl)amino)methyl)-1H-1,2,3-triazol-1-yl)heptyl)oxy)phenyl)ureido)methyl)-N-hydroxybenzamide 13f (41% yield).
1H NMR (400 MHz, DMSO-d6): δ 10.95 (s, 2H), 8.25 (s, 1H), 8.03 (d, J = 5.5 Hz, 1H), 7.85–7.73 (m, 4H), 7.74–7.66 (m, 1H), 7.69–7.57 (m, 2H), 7.54 (t, J = 8.0 Hz, 2H), 7.45–7.29 (m, 2H), 7.24 (d, J = 7.6 Hz, 2H), 7.02–6.73 (m, 2H), 5.19 (dd, J = 13.2, 5.2 Hz, 1H), 4.70–4.63 (m, 3H), 4.48 (d, J = 17.3 Hz, 1H), 4.39 (t, J = 7.0 Hz, 2H), 3.94 (t, J = 6.4 Hz, 2H), 3.82 (t, J = 4.2 Hz, 3H), 3.56 (s, 1H), 3.37–3.29 (m, 2H), 3.02–2.86 (m, 1H), 2.68–2.60 (m, 1H), 2.55–2.43 (m, 1H), 2.05 (ddd, J = 13.7, 6.9, 3.1 Hz, 1H), 1.87 (p, J = 7.2 Hz, 2H), 1.71 (dd, J = 11.6, 5.0 Hz, 3H), 1.59–1.47 (m, 2H), 1.49–1.36 (m, 2H), 1.36–1.21 (m, 7H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 173.05, 171.45, 168.39, 165.19, 155.95, 154.41, 149.68, 146.80, 146.43, 141.10, 137.18, 136.74, 133.83, 132.90, 132.05, 129.38, 129.06, 128.30, 127.28, 127.17, 125.38, 122.97, 122.52, 114.52, 67.95, 52.32, 49.68, 49.07, 46.41, 31.66, 30.46, 30.42, 30.18, 30.12, 29.11, 28.60, 28.55, 26.28, 25.84, 22.87, 19.93, 14.28. LC-MS (M+H: 885.2): 95% purity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Calculation of Relative HDAC6 expression
The normalized signal (NS) was calculated by followed formula:
The relative HDAC6 expression was calculated by divide NS of compound treated well by average NS of vehicle/DMSO treated wells and marked as “relative HDAC6 expression fold to vehicle”. Normalized data were generated bar graph or dot plot representing mean relative expression (n = 3) with ± SD as error bar. For dose response, nonlinear fitting of [Inhibitor] vs. response (three parameters) was generated by GraphPad Prism with R2 over 0.95.
Calculation of Relative Cell Viability
The relative viability (RV) was measured by followed formula:
Normalized data was graphed as bar graph or dot plot representing as mean of relative viability (n = 3) with ± SD as error bar.
Statistical Analysis
All statistical analyses were done by GraphPad Prism. Statistical significance was analyzed by performing unpaired two-tailed student t-test or one-way ANOVA analysis of variance. For ANOVA, multiple group comparisons with vehicle or compound-treated group were followed Dunnett correction. Not significant (ns) P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HDAC1 (10E2) Mouse mAb | Cell Signaling Technology | #5356 |
| HDAC2 (3F3) Mouse mAb | Cell Signaling Technology | #5113 |
| HDAC4 (D15C3) Rabbit mAb | Cell Signaling Technology | #7628 |
| HDAC6 (D2E5) Rabbit mAb | Cell Signaling Technology | #7558 |
| HDAC8 Antibody (F-9) | Santa Cruz Biotechnology | sc-374180 |
| Ikaros (D6N9Y) (IKZF1) Rabbit mAb | Cell Signaling Technology | #14859s |
| Aiolos (D1C1E) (IKZF3) Rabbit mAb | Cell Signaling Technology | #15103s |
| eRF3 (GSPT1) Antibody | Cell Signaling Technology | #14980s |
| Human/Mouse/Rat beta-Actin Antibody | R&D system | MAB8929 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | #7074 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | #7076 |
| Bacterial and Virus Strains | ||
| Not Applicable | ||
| Biological Samples | ||
| Not Applicable | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| TMB Substrate Set | BioLegend | #421101 |
| Clarity ECL substrate | Bio-Rad | #1705061 |
| Thalidomide | Selleckchem | S1193 |
| Pomalidomide | Selleckchem | S1567 |
| Lenalidomide | Selleckchem | S1028 |
| CC-122 | MedChemExpress | HY-100507; CAS: 1015474-32-4 |
| CC-220 | MedChemExpress | HY-101291; CAS: 1323403-33-3 |
| CC-885 | MedChemExpress | HY-101488; CAS: 1010100-07-8 |
| Janus green B | Sigma-Aldrich | 201677; CAS: 2869-83-2 |
| Resazurin sodium salt | Sigma-Aldrich | R7017; CAS: 62758-13-8 |
| Critical Commercial Assays | ||
| Not Applicable | ||
| Deposited Data | ||
| Not Applicable | ||
| Experimental Models: Cell Lines | ||
| MM1S | ATCC | ATCC CRL-2974 |
| Jurkat, Clone E6–1 | ATCC | ATCC TIB-152 |
| Experimental Models: Organisms/Strains | ||
| Not Applicable | ||
| Oligonucleotides | ||
| Not Applicable | ||
| Recombinant DNA | ||
| Not Applicable | ||
| Software and Algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| none | ||
SIGNIFICANCE.
As an emerging and widely used technology, PROTAC degraders provide a new strategy for novel therapeutic development by controlling the cellular level of functional proteins. The development of novel E3 ligands will expand the available library of successful degraders. CRBN E3 ligase and thalidomide derivatives have been widely used in PROTAC design, but PROTAC based on them have several limitations such as off-targets. A general cell-based method that can facilitate the development of E3 ligase ligands will be valuable for more applications of PROTACs as chemical tools and therapeutics. In this article, we report a general cell-based target engagement assay for the identification of cell-permeable cereblon E3 ubiquitin ligase ligands and demonstrated the utility of the ligands identified here for the degradation of HDAC6. This strategy may provide a practical and efficient alternative to the biochemical assay for the optimization of E3 ligase ligands for many other protein targets beyond HDAC6.
Highlights.
Cell-based assay developed for evaluating the binding affinity of CRBN E3 ligands.
A small library of CRBN E3 ligands were synthesized and examined by this assay.
Selected E3 ligands were used to construct cellular active HDAC6 degraders.
ACKNOWLEDGEMENT:
The authors would like to thank the University of Wisconsin Carbone Cancer Center’s (UWCCC) Consultation Panel for support of this project. This work is also supported in part by NIH/NCI P30 CA014520-UW Comprehensive Cancer Center Support (CCSG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Figure S1-S5. Scheme S1-S2.
Supplemental Information can be found online at https://doi.org/10.1016/j.chmbiol.2020.XX.XXX
RERENCES
- An Z, Lv W, Su S, Wu W, and Rao Y (2019). Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 10, 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PL, and Vermette P (2016). Intracellular insulin quantification by cell-ELISA. Exp. Cell Res 347, 14–23. [DOI] [PubMed] [Google Scholar]
- Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, and Crews CM (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, Rychak E, Corral LG, Ren YJ, Wang M, et al. (2014). Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol 21, 803–809. [DOI] [PubMed] [Google Scholar]
- Chan K-H, Zengerle M, Testa A, and Ciulli A (2017). Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J. Med. Chem 61, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessum NEAA, Sharp SY, Caldwell JJ, Pasqua AE, Wilding B, Colombano G, Collins I, Ozer B, Richards M, Rowlands M, et al. (2018). Demonstrating In-Cell Target Engagement Using a Pirin Protein Degradation Probe (CCT367766). J. Med. Chem 61, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus K, Wehenkel M, Choi E-Y, Han H-J, Lee H, Swanson H, and Kim K-B (2011). Impact of linker length on the activity of PROTACs. Mol. BioSyst 7, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, and Fischer ES (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane radial ray syndrome. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. (2014). Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley CA, Potjewyd F, Lamb KN, James LI, and Frye SV (2020). Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay. ACS Chem. Biol 15, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan K, Chen W, Lamont DJ, Zengerle M, and Ciulli A (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goergler Annick; Norcross Roger; Schmid Philipp; Dey Fabian; Kusznir EA (2019). GLUTARIMIDE.
- Hagner PR, Man H-W, Fontanillo C, Wang M, Couto S, Breider M, Bjorklund C, Havens CG, Lu G, Rychak E, et al. (2015). CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 126, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein SA, Hillengass J, and McCarthy PL (2018). Next-Generation Drugs Targeting the Cereblon Ubiquitin Ligase. J. Clin. Oncol 36, 2101–2104. [DOI] [PubMed] [Google Scholar]
- Ishoey M, Chorn S, Singh N, Jaeger MG, Brand M, Paulk J, Bauer S, Erb MA, Parapatics K, Müller AC, et al. (2018). Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem. Biol 13, 553–560. [DOI] [PubMed] [Google Scholar]
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, and Handa H (2010). Identification of a Primary Target of Thalidomide Teratogenicity. Science (80-.) 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, Yang C-Y, Wang M, Han X, and Wang S (2019). Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem 62, 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, Karasawa S, Carmel G, Jackson P, Abbasian M, et al. (2012). Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 26, 2326–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong K-K, Bradner JE, and Kaelin WG (2014). The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science (80-.) 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu C-C, Miller K, Fang W, Wang N-Y, Nguyen D, Houston J, et al. (2016). A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 535, 252–257. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Couto S, Zheng X, Lu G, Hui J, Stamp K, Drew C, Ren Y, Wang M, Carpenter A, et al. (2018a). SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol 14, 981–987. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Zhang W, Man H-W, Muller G, Khambatta G, Baculi F, Hickman M, LeBrun L, Pagarigan B, Carmel G, et al. (2018b). A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem 61, 535–542. [DOI] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, et al. (2018). Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A, Facon T, Sonneveld P, Bladè J, Offidani M, Gay F, Moreau P, Waage A, Spencer A, Ludwig H, et al. (2008). Thalidomide for treatment of multiple myeloma: 10 years later. Blood 111, 3968–3977. [DOI] [PubMed] [Google Scholar]
- Pettersson M, and Crews CM (2019). PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future. Drug Discov. Today Technol 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, and Prince HM (2010). Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 24, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching KM, Mahan S, Corona CR, McDougall M, Vasta JD, Robers MB, Urh M, and Daniels DL (2018). Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol 13, 2758–2770. [DOI] [PubMed] [Google Scholar]
- Riedl S, Kaiser P, Raup A, Synatschke C, Jérôme V, and Freitag R (2018). Non-Viral Transfection of Human T Lymphocytes. Processes 6, 188. [Google Scholar]
- Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Oláh J, Ovádi J, Sippl W, and Jung M (2018). Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem 61, 482–491. [DOI] [PubMed] [Google Scholar]
- Schürmann M, Janning P, Ziegler S, and Waldmann H (2016). Small-Molecule Target Engagement in Cells. Cell Chem. Biol 23, 435–441. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. (2008). IRF4 addiction in multiple myeloma. Nature 454, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers QL, Petzold G, Bunker RD, Renneville A, Słabicki M, Liddicoat BJ, Abdulrahman W, Mikkelsen T, Ebert BL, and Thomä NH (2018). Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science (80-.) 362, eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GM, Niphakis MJ, and Cravatt BF (2013). Determining target engagement in living systems. Nat. Chem. Biol 9, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, and Crews CM (2019). Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Luo T, Greenberg EF, Bradner JE, and Schreiber SL (2011). Discovery of histone deacetylase 8 selective inhibitors. Bioorganic Med. Chem. Lett 21, 2601–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wu S, Liu J, Yang K, Xie H, and Tang W (2019). Development of selective small molecule MDM2 degraders based on nutlin. Eur. J. Med. Chem 176, 476–491. [DOI] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 67, 5–18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Yang K, Zhang Z, Leisten ED, Li Z, Xie H, Liu J, Smith KA, Novakova Z, Barinka C, et al. (2019). Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem 62, 7042–7057. [DOI] [PubMed] [Google Scholar]
- Yang J, Li Y, Aguilar A, Liu Z, Yang C-Y, and Wang S (2019). Simple Structural Modifications Converting a Bona fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem 62, 9471–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Song Y, Xie H, Wu H, Wu Y-T, Leisten ED, and Tang W (2018). Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett 28, 2493–2497. [DOI] [PubMed] [Google Scholar]
- Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang CY, Zhao Y, et al. (2018). Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YX, Braggio E, Shi C-X, Bruins LA, Schmidt JE, Van Wier S, Chang X-B, Bjorklund CC, Fonseca R, Bergsagel PL, et al. (2011). Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 118, 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J, Zhu H, Farley KA, Ding W, Schiemer J, et al. (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci 115, 7285–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all biological and chemical data (1H NMR, 13CNMR and LC-MS of all compounds) generated during in this study. No unique code was generated in this study.