

Summary
Chlorcyclizine (CCZ) is a potent HCV entry inhibitor but its molecular mechanism is unknown. Here we show that CCZ directly targets the fusion peptide of HCV E1 and interferes with the fusion process. Generation of CCZ resistance-associated substitutions of HCV in vitro revealed six missense mutations in the HCV E1 protein, five being in the putative fusion peptide. A viral fusion assay demonstrated that CCZ blocked HCV entry at the membrane fusion step and that the mutant viruses acquired resistance to CCZ’s action in blocking membrane fusion. UV cross-linking of photoactivatable CCZ-diazirine-biotin in both HCV-infected cells and recombinant HCV E1/E2 protein demonstrated direct binding to HCV E1 glycoprotein. Mass spectrometry analysis revealed that CCZ cross-linked to an E1 sequence adjacent to the putative fusion peptide. Docking simulations demonstrate a putative binding model, wherein CCZ binds to a hydrophobic pocket of HCV E1 and forms extensive interactions with the fusion peptide.
Keywords: Viral infection, fusion loop, drug resistance-associated substitutions, photo-activated cross-linking, membrane fusion, molecular modelling
eTOC blurb
Hu and Rolt et al showed that anti-histamine agent chlorcyclizine inhibits HCV entry by targeting HCV E1 protein and blocking the viral fusion process. Understanding the interaction of chlorcyclizine with HCV may provide important insights into the viral fusion mechanism and development of broad-spectrum antivirals against HCV and other viruses.
Graphical Abstract
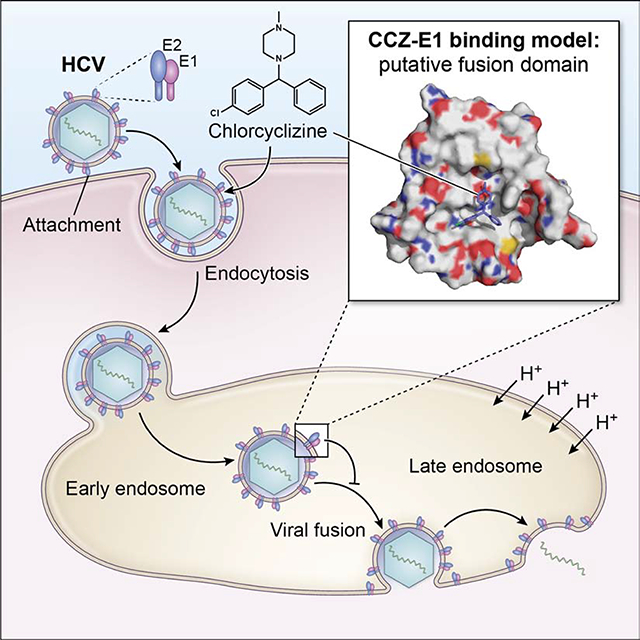
Introduction
The World Health Organization estimates that 71 million people worldwide live with hepatitis C virus (HCV) infection (WHO, 2017). While a vaccine has been developed for hepatitis B virus, no such vaccine or intervention exists for HCV. Direct-acting antivirals (DAAs) targeting the replication stage of HCV life cycle have been developed and are highly effective (Liang and Ghany, 2013). Since HCV can readily develop resistance to DAAs under monotherapy, combination therapy with multiple DAAs is now the gold standard in clinical application (Liang and Ghany, 2013). While currently approved DAAs are effective, the further development of antivirals targeting other stages of the HCV life cycle can potentially improve synergistic inhibition, further raise the evolutionary barriers for resistance, decrease the duration of treatment and improve pan-genotypic coverage.
In search of novel HCV inhibitors, we previously demonstrated that chlorcyclizine (CCZ), an approved first-generation antihistamine drug, is a potent inhibitor of HCV infection (He et al., 2015). More recently, a structure-activity relationship campaign produced analogs with increased potency and improved ADME properties that were effective against HCV in a humanized mouse model of HCV genotypes 1b and 2a (He et al., 2016; Rolt et al., 2018). Yet before further clinical development, it is necessary to define the precise molecular mechanism of action of these compounds. Using various functional assays, we showed that CCZ targets the late-entry stage of the HCV life cycle, after host cell attachment but before replication (He et al., 2015). Structurally similar scaffolds (diphenylpiperazines/piperidines/alkylamines, phenothiazines and thioxanthenes) with differing primary targets have also been identified as HCV entry inhibitors (Pietschmann, 2017). The primary targets of these scaffolds (histamine H1, dopamine receptor, Ca2+channel inhibition, etc.) do not mediate the observed anti-HCV activities of these compounds, arguing for the existence of an alternative target(s).
Since CCZ was discovered in a phenotypic high-throughput screen, the functional target and molecular mechanism remain unknown (He et al., 2015). After screening and hit optimization, target identification is required to inform further medicinal chemistry efforts and to decipher the molecular mechanism of the drug class. Viruses can acquire resistance-associated mutations when replicating in the presence of antiviral agents, particularly those targeting viral components directly. Viral mutations that impart resistance to the antivirals can either directly or indirectly inform the mechanism of action. For example, in vitro culture of HCV replicon under pressure from the DAA, daclatasvir, rapidly selected for HCV resistance-associated substitutions (RASs) in its molecular target NS5A, which prevent the binding of the drug to NS5A (Fridell et al., 2010). Serial passaging of HCV replicon in vitro with the host-acting antiviral cyclosporin A generated strains of HCV with mutations in NS5B, which was later identified as a substrate for the proline cis-trans isomerase cyclophilin A, which in turn is inhibited by cyclosporin A (Liu et al., 2009b; Robida et al., 2007).
Cellular target(s) of drugs can also be identified by affinity pull-down experiments, though this approach usually requires chemical modification of the drug of interest to include orthogonal chemical moieties such as biotin without affecting its activity significantly. This process permits enrichment and therefore identification of binding partners to that particular compound through techniques such as Western blotting and mass spectrometry. Furthermore, modifications to compound of interest such as the incorporation of a photoactivatable diazirine(Vervacke et al., 2014) enables covalent attachment to binding partners, improving pull-down performance in scenario of low binding affinity.
Here we elucidate the mechanism of action of CCZ against HCV infection. We demonstrated that following production and characterization of RASs upon serial passaging with increasing concentrations of CCZ, mutations conferring resistance to CCZ were identified in the putative fusion loop of the HCV E1 protein. Next, we synthesized a CCZ-diazirine-biotin photoaffinity probe that retains activity against HCV and binds specifically to the HCV E1 protein. Mass spectrometry localized the CCZ binding sites near the putative E1 fusion peptide (Drummer et al., 2007; Lavillette et al., 2006; Li et al., 2009a; Perez-Berna et al., 2006; Tong et al., 2018; Vieyres et al., 2010). Computational docking of CCZ to E1 showed that CCZ forms a favorable interaction with a hydrophobic pocket in the HCV E1 fusion loop. Our results depict a novel mechanism of CCZ in inhibiting HCV entry by directly targeting HCV E1 and blocking the viral fusion process.
Results
Generation of CCZ resistance-associated substitutions of HCV
To generate HCV RASs to CCZ, an infectious cell-culture drug-resistance selection system, based on HCV genotype 2a, J6/JFH1 strain (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005), was designed (Figure 1A). In this system, HCV-containing cell culture medium was serially passaged while simultaneously exposed to increasing concentrations of (S)-CCZ. After serial passages, detection of replication-competent virus at high concentrations of (S)-CCZ using HCV core immunofluorescent staining would indicate emergence of RASs (Figure 1B). Daclatasvir (DCV) was used as a control since it is known that HCV RASs to DCV occurs readily (Fridell et al., 2011). After HCV infection was established (passage 1, P1), treatment with (S)-CCZ or DCV showed progressively weaker dose-dependent inhibition of HCV infection with serial passages starting at P5 for CCZ and P4 for DCV (Figure S1A). With additional passages, viruses appeared to infect cells at higher and higher drug concentrations, reaching maximal resistance after P13 for CCZ (Figure S1A). Putative (S)-CCZ-resistant viruses generated in P13 from the wells in column 1 showed productive infection at much higher concentrations (> 5 μM) of (S)-CCZ compared to the same passage of DMSO-treated HCV-WT (Figure 1C). For DCV-treated samples, the viruses from P13 were resistant to DCV at > 1 nM with the WT virus being sensitive to DCV at < 50 pM.
Figure 1. Generation of (S)-CCZ resistance-associated substitutions (RASs) of HCV.

A. Two-part HCV infection assay to generate drug-resistant HCV. Red: infected plate; blue: uninfected plate. Drug concentrations correspond to (S)-CCZ in [μM] and daclatasvir in [nM]. B. Expected results from HCV core immunofluorescent staining before and after generating drug-resistant HCV strains. C. HCV core immunofluorescent staining for CCZ-resistant HCV strains generated above that were treated with various concentrations (S)-CCZ. Two representative (S)-CCZ-treated HCV clones (B1 and E1) and one DMSO-treated HCV clone are shown. Detailed passage data are shown in Figure S1A. D. Mutations of putative HCV RASs were identified from each individual well of the 96-well plates at passage 13. E. Sequence alignment for the putative fusion peptide (aa 264–294) in HCV E1 for each HCV genotype (genotypes 1–6 were obtained from the Virus Pathogen Resource database and genotype 7 from the NCBI). MAFFT version 7 with the G-INS-1 progressive method and Berkeley WebLogo were used to generate the alignment figure. The amino acid substitutions of the (S)-CCZ RASs are listed above the alignment. The n represents the number of HCV sequences for each genotype used for the alignment. EC50 of (S)-CCZ for each HCV genotype was shown to the left in the chimeric HCV genotype assay (complete data presented in Figure S5). F. Summary of EC50 and maximal response of HCV-WT and HCV E1 mutants against (S)-CCZ and sofosbuvir. G. (S)-CCZ dose-response curves of 3 representative HCV E1 mutants. Complete data are presented in Figure S2.
Emergence of CCZ resistance-associated substitutions in E1 protein of HCV
After confirming phenotypic resistance based on HCV core immunofluorescent staining, HCV RNA was isolated from cell culture medium for sequencing. A total of 5 (S)-CCZ resistant HCV clones were sequenced while 3 were excluded due to low virus titer. After excluding sequence variants observed in the respective DMSO-cultured clones, a total of 6 missense variants were identified in the HCV E1 protein (Figure 1D), 5 of them being clustered in the putative fusion peptide domain of the HCV E1 protein between HCV polyprotein amino acid 264 and 293 (Drummer et al., 2007; Lavillette et al., 2006; Li et al., 2009a; Perez-Berna et al., 2006; Tong et al., 2018): M267V, A274T, L286I, Q289H, and F291L (Figure 1D, Figure S1B). The mutations were shown in the context of the fusion peptide sequence alignment among all HCV genotypes (Figure 1E). The M267V and L286I mutations were each observed in combination with F291L. One additional missense variant, P244S, was found near the fusion loop. In general, most of the affected amino acids are conserved across all HCV genotypes except for M267, which is present only in genotype 2 (Figure 1E).
Putative RASs confer HCV resistance to CCZ
To confirm that the E1 mutations identified in these putative RASs confer HCV resistance to (S)-CCZ, each mutation was independently introduced into the HCV-WT clones. HCV constructs with double mutations of M267V/F291L or L286I/F291L were also generated. The dose-response curves of (S)-CCZ in inhibiting HCV demonstrated that all the mutant viruses had variable but clear shifts in EC50 values (except L286I containing mutants) and lower maximum responses as compared to the HCV-WT control, whereas the HCV replication inhibitor sofosbuvir inhibited the mutant viruses and HCV-WT equally (Figure 1F & 1G and Figure S2). The results indicated that the mutations identified in the HCV E1 protein are indeed responsible for viral resistance to CCZ.
To exclude the possibility that these RASs may alter viral fitness accounting for apparent CCZ resistance, we compared the replication capacity and infectivity of each mutant virus with HCV-WT. In general, all mutant viruses showed similar or slightly lower levels of replication capacity and/or infectivity comparing to HCV-WT (Figure 2), further confirming that the RASs in E1 protein are indeed CCZ-resistant mutations.
Figure 2. Viral fitness of RAS-containing HCV.

A. The scheme of viral fitness assay is shown here. The first part assesses the replication capacity of each RAS after electroporation of in vitro transcribed HCV RNA into Huh7.5.1 cells and the second part tests the infectivity of produced virus by re-infecting naïve Huh7.5.1 cells. B. Replication capacity for each HCV RAS was assessed by electroporating 3×105 Huh7.5.1 cells with 1.5 μg of RNA with the RAS-RLuc construct in a 6-well plate. Luminescence of the electroporated cells was measured 6 h after electroporation to account for background and 72 h after electroporation to assess for replication capacity. C. The infectivity of each RAS was assessed by harvesting the virus-containing medium 72 h after electroporation and re-infecting 104 naïve Huh7.5.1 cells in a 96-well plate. Luminescence was measured 48 h after re-infection. All luminescence measurements are normalized to HCV-WT. Each data point was presented as mean value ± SEM of 3 replicates. The results are representative of three separate experiments.
CCZ blocks membrane fusion of HCV during viral entry
We previously showed that CCZ inhibits HCV entry at the post-receptor binding step and follows a similar pattern as the endosomal membrane fusion inhibitor, bafilomycin A1, in the time-of-addition assay (He et al., 2015). We tested the effect of (S)-CCZ on membrane fusion of wild-type and mutant HCV in a membrane fusion assay, which is depicted in Figure 3A. NH4Cl was present in the medium throughout the viral entry period to block the endosomal membrane fusion except for a 5 min artificial pH shift to pH 5, during which endosomal acidification was artificially achieved, allowing membrane fusion and viral entry process to be completed. (S)-CCZ was added during (Figure 3A, protocol I) or after (Figure 3A, protocol II) the pH 5 shift to distinguish whether the drug would block viral fusion even when endosomal acidification was artificially restored. As shown in Figure 3B, bafilomycin A1 behaved similarly to the DMSO control in protocol I and II. As expected, the artificial lowering of the endosomal pH allowed viral entry in DMSO control and overcame the block of viral membrane fusion by bafilomycin A1. In contrast, HCV infection did not show much of an increase (1.31-fold) in cells with CCZ treatment during the pH 5 shift, which was significantly lower than that of the DMSO control (3.98-fold). Addition of CCZ after the pH 5 shift did not exert such an effect (Figure 3B, protocol II). These results indicated that CCZ blocks viral membrane fusion in the endosomes even under an acidic environment. For the mutant viruses, CCZ inhibition of HCV infection following pH 5 shift was significantly diminished (Figure 3C), suggesting that the HCV RASs are indeed resistant to the blocking of membrane fusion by CCZ.
Figure 3. (S)-CCZ inhibits HCV membrane fusion during HCV entry.

A. Scheme of the assay. In protocol I, the compound (DMSO, bafilomycin A1 or (S)-CCZ) was present during the pH 5 shift and removed after. In protocol II, the compound was added after the pH 5 shift. B. Huh7.5.1 cells were infected with HCV-WT and treated with bafilomycin (3 nM) or (S)-CCZ (5 μM) as shown in A. Cells were cultured for 72 h following the treatment before being processed for HCV core immunofluorescent staining. The number of HCV core positive foci (≥ 4 stained cells in each group) was counted, the data were normalized to the DMSO control (relative % infection) and presented as mean + SEM (n = 3). Relative percent of infection was calculated by dividing the number of HCV core-positive foci from the pH 7 by that of the pH 5 of the DMSO control (set as 100%). All data (% infection) from other conditions (Baf and CCZ) were divided by the number of HCV core-positive foci of pH 5 of the DMSO control within each protocol. The fold-change of pH 5 over pH 7 for each condition was determined by dividing the % infection of pH 5 by that of pH 7 and shown above each condition. Statistical significance of the fold-change of the Baf or CCZ condition was compared to that of the DMSO condition (Student’s t test) and shown above the graph. C. Huh7.5.1 cells were infected with (S)-CCZ-resistant HCV mutants and treated following the procedure from protocol I. Statistical significance of the fold-change for each condition (DMSO, bafilomycin A1, or CCZ) of the mutant virus was compared to the fold-change from the same condition with the wild-type HCV under protocol I (Student’s t test; * represents P < 0.05, ** P < 0.01). The results are representative of three separate experiments.
CCZ-diazirine probe cross-links HCV E1 protein by UV activation
In order to identify the molecular target for CCZ, the photoaffinity probes CCZ-diazirine and CCZ-diazirine-biotin (CCZ-DB) were synthesized and fully characterized (Figure S3 & Figure 4A). Both were stable at room temperature and in ambient light as routinely encountered in a laboratory environment, though slow decomposition (days) could be observed in natural light (determined by LCMS). Both CCZ-diazirine and CCZ-DB were found to inhibit HCV infection in the HCV infection assay in a dose-dependent manner with EC50 of 25 nM and 19.7 nM, respectively (Figure 4A). To determine the appropriate length of time for UV irradiation when performing the diazirine crosslinking in vitro, pilot studies were performed in water (10 μM) at 4°C (on ice) using a 100 W, 115 V mercury lamp with a 365 nm filter. Aliquots were removed at predetermined intervals over 2 h and analyzed by the LCMS to determine the time-dependent decomposition of the CCZ-diazirine, which readily converted to the solvent insertion products in less than 5 min without any evidence of internal rearrangement as determined by the LCMS (Figure S3).
Figure 4. Characterization of CCZ-diazirine derivatives and UV-activated crosslinking of CCZ-DB with HCV E1 protein.

A. Structures and dose-response curves of (S)-CCZ, CCZ-diazirine, CCZ-diazirine-biotin (CCZ-DB) and diazirine-biotin (DB) control. CCZ-diazirine and CCZ-diazirine-biotin were active in inhibiting HCV in the HCV infection assay, with EC50 of 25.0 nM and 19.7 nM, respectively. The DB control has the diazirine-biotin moieties but was not active against HCV. B. Cross-linking of CCZ-DB with recombinant HCV E1/E2 protein. Purified recombinant E1/E2 protein (genotype 1a) was incubated with various compounds described above at room temperature for 1 h, subjected to UV cross-linking and then purified by Neutravidin beads followed by Western blot with anti-E1 antibody. Recombinant E1/E2 protein was included on the blot as a reference. In one sample, a 100-fold higher concentration of (S)-CCZ (100 μM) was added to the CCZ-DB cross-linking condition. C. Cross-linking of CCZ-DB with E1 protein of HCV genotype 1a-infected cells. Huh7.5.1 cells were infected with high-titer HCVcc genotype 1a virus in the presence of the various compounds at 37°C for 1 h, subjected to UV cross-linking and then lysed for purification by Neutravidin beads followed by Western blot with anti-E1 antibody. The same conditions were tested as the HCV recombinant E1/E2 protein above. A high-titer stock of the HCV genotype 1a virus was also run on the blot to demonstrate the presence of E1 protein. The results are representative of three separate experiments.
Using the photoaffinity probe CCZ-DB in reaction with recombinant HCV E1/E2 protein and HCV-infected cells after UV-activated cross-linking, HCV E1 was identified as the molecular target of CCZ by Western blot using a Wes ProteinSimple capillary-based immunoassay system (Figure 4B & 4C). The cross-linked E1 recombinant protein had a slightly different mobility from the native E1 (Figure 4B & 4C). Western blot with anti-E2 antibody did not identify E2 in the cross-linked complex, likely because the E2 protein is not covalently linked to E1 in the recombinant E1/E2 heterodimer and thus removed from the complex by the partially denaturing buffer for binding and washing. Enrichment of HCV E1 was only observed in the samples incubated with CCZ-DB and UV irradiation. Using various controls including DMSO, CCZ-DB without UV activation, and a biotinylated diazirine derivative with no anti-HCV properties (DB-control), the HCV E1 band was not detected. In addition, a competition experiment wherein the photoaffinity cross-linking was performed in the presence of excess concentration of (S)-CCZ (100 μM) failed to enrich HCV E1, indicating that the interaction of CCZ-DB with HCV E1 was specific.
Identification of cross-linked E1 amino acids by mass spectrometry analysis
We first validated a reporter ion/neutral loss approach based on analysis of the CCZ-DB for analysis and detection of cross-linked peptides using control peptide samples (Supplemental Methods: Mass spectrometric analysis of CCZ-diazirine-biotin cross-linked E1 glycoprotein). We then analyzed the cross-linked HCV E1 digested with an alpha lytic protease and found specific modifications of the sequence stretch YEAADAILHTPG. Multiple single cross-linked forms with the top 3 forms being Y*EAADAILHTPG (~ 46%), YEAADAIL*HTPG (~ 22%) and YE*AADAILHTPG (~ 14%) were detected (Figure S4 & Figure 5).
Figure 5. E1 sequences of (S)-CCZ cross-linking site and mutational analyses.

A. E1 Sequence comparison of various HCV genotypes near the major amino acid residues cross-linked with CCZ (marked by *) was shown as in Figure 1. B. Dose-response curves of (S)-CCZ against the HCV GT2a WT and mutants W214Y, Q215E and W214Y/Q215E. The experiment was performed in 96-well plates. Data points are expressed as mean values ± SEM from 6 samples (n = 6). The results are representative of three separate experiments.
HCV genotypes have different susceptibilities to CCZ
As shown in Figure 1E, the fusion loop sequences of the various HCV genotypes possess considerable heterogeneity. To assess the relative efficacy of CCZ against various HCV genotypes, we tested chimeric HCV viruses of various genotypes (Gottwein et al., 2011). Although CCZ showed maximal inhibition against all HCV genotypes at high doses (1–30 μM), there was a significant variation of efficacy among different genotypes. Among them, HCV GT2a was the most sensitive to CCZ (EC50 of 0.078 μM), followed by GT2b, 4a, 1b, 3a and 7a (EC50 of 1.32, 7.16, 8.18, 8.82, and 10.59 μM, respectively), and 6a, 5a and 1a being the least sensitive (EC50 of 14.06, 16.07 and 18.78 μM, respectively) (Figure 1E and Figure S5). As a control for entry inhibition, inhibitory dose responses of CD81 neutralizing antibody against the chimeric viruses were performed in a parallel plate and showed minor EC50 variations (< 5-fold) among the HCV genotypes (Figure S5).
As CCZ directly interacts with the HCV E1 protein, the variations of putative fusion peptide sequences among the genotypes may account for the genotypic difference of efficacy. Mass spectrometric analysis showed that CCZ interacts with the E1 protein of HCV genotype 1a at amino acids Y214 and E215. This region shows modest variations among various genotypes (Figure 5A). For genotype 2a, the corresponding amino acids are W214 and Q215. We, therefore, mutated the W214 and Q215 to Y214 and E215, either individually or in combination, in the HCV genotype 2a genome. Analysis of these mutant genotype 2a viruses showed a significant reduction of viral inhibition by CCZ, with EC50 shifting from 0.021 μM to 0.357 μM for W214Y mutant, 0.276 μM for Q215E mutant, and 3.37 μM for W214Y/Q215E double mutant (Figure 5B). These results indicated that sequence variations of E1 near the CCZ binding site among various HCV genotypes likely contribute to the different sensitivities of HCV genotypes to CCZ.
Molecular modeling of CCZ and HCV E1 interaction
To probe the structural basis of interaction between CCZ and E1 (particularly the putative fusion peptide), we took advantage of several published modeling reports, in which in silico models of full-length HCV E1/E2 heterodimers have been generated by using crystallographic structures of partial HCV E1 and E2 proteins, and computational tools such as co-evolution and Rosetta protocols (Cao et al., 2019; Castelli et al., 2017; Freedman et al., 2017). Using information from the above modeling and the threading program I-TASSER (Yang and Zhang, 2015), we further refined the structural model of E1 ectodomain (residues 192–310) based on experimental mutagenesis studies and molecular simulations. Since the putative fusion peptide region is highly flexible, cluster representatives of 10 structural models were generated from molecular dynamic simulations and an ensemble docking approach was applied to account for protein flexibility and induced conformational changes upon ligand binding (Figure S6A). As shown in the predicted binding model (Figure 6A), CCZ binds into a well-defined hydrophobic pocket formed by the fusion loop within the ectodomain of E1. Residue Q289 is centered at the pocket and forms a key H-bond with the N atom of the piperazine ring of CCZ. Residues M264, M267, and F291 surrounding the pocket make extensive hydrophobic interactions with CCZ. Furthermore, residues W214 and Q215 which are located at the bottom of the binding pocket, part of the previously determined alpha-helix of the E1 structure (El Omari et al., 2014), form aromatic and halogen interactions with the phenyl and chlorophenyl groups of CCZ. This model accommodates docking of both the (S) and (R) conformations of CCZ, consistent with our previous finding that they have similar anti-HCV activities (He et al., 2015). Based on the model, the (R)-CCZ can bind in either configuration (Figure S6B.a) into this pocket. Since the CCZ-DB is synthesized as mixed enantiomers and the pocket can accommodate either configuration, the diazirine can cross-link with either W214 and Q215 (Figure S6B.b & S6B.c). The Y214/E215 HCV mutant showed reduced affinity estimated to the hydrophobic pocket (ΔG = −3.14 kcal/mol) due to the potential loss of hydrogen bonding in comparison to the wild-type W214/Q215 sequence of the HCV 2a strain (ΔG = −4.26 kcal/mol).
Figure 6. Molecular modelling of CCZ binding to HCV E1 protein.

A. Predicted binding model of (S)-CCZ with E1. The (S)-CCZ fits into a hydrophobic pocket of the space-filling model of E1 on the left. On the right, interactions of (S)-CCZ with various key amino acid residues, as identified by the mutagenesis studies, are shown in a ribbon model. The hydrogen bond between Q289 and the N atom of the piperazine, and a halogen bond between Q215 and the chlorophenyl group of (S)-CCZ provide significant stabilization. B. EC50 values and predicted binding energies of CCZ analogs. The EC50 values of 10 structurally related CCZ analogs published previously were compared with their respective predicted binding energies. C. The EC50’s of 10 CCZ analogs (A) were plotted against their respective binding energies predicted based on various intermolecular interactions (e.g., hydrogen bonds, electrostatic interactions, pi-pi stacking, etc.). The anti-HCV activities of the CCZ analogs correlate well with the binding energies (R2 = 0.7815, P = 0.0007).
The predicted binding model is commensurate with the finding that Q289H/M267V/F291L mutations had a significant impact on the binding and inhibitory activity of CCZ (Figure S6B.d). It is not immediately clear how the other CCZ-resistant mutations confer resistance to CCZ, other than possibly causing a change in the secondary structure of E1 large enough to exclude CCZ from its binding pocket. The L286I mutant, which is only marginally resistant to CCZ, is observed to point away from the CCZ binding pocket in the model (Figure 6A). While it may play a stabilizing role in forming the hydrophobic pocket, it appears to have little impact on the direct interaction with CCZ binding. The resistance of the A274T mutant to CCZ may be explained by the change of the hydrophobic side chain A274 to hydrophilic side chain T274, which impairs the hydrophobic interaction of CCZ to the hydrophobic pocket. P244 is conserved and located in the ß-sheet structure upstream of the fusion loop (El Omari et al., 2014; Freedman et al., 2017); the P244S substitution may affect the conformation of the E1 ectodomain and formation of the hydrophobic pocket, resulting in reduced interaction with CCZ. Finally, it is possible that these mutations at the fusion peptide confer resistance by affecting proper E1-E2 interaction required for the fusion process (Douam et al., 2018).
Our previous SAR study of CCZ had generated an extensive series of CCZ analogs with well-defined anti-HCV activities (He et al., 2015). The predicted CCZ binding model provides an opportunity to assess whether the observed anti-HCV activities can be correlated with the structural features of these compounds. We, therefore, docked a number of these analog compounds to the putative CCZ binding pocket and evaluated their binding affinities. As expected, the S and R conformations of CCZ have similar binding affinities. The most active compound (#4) from our previous SAR study (He et al., 2016; Rolt et al., 2018) has the highest binding energy, likely because the two chloro-phenyl groups and the N-ethyl group on the piperazine make a stronger interaction with the relevant amino acid residues surrounding this binding pocket. The double halogen bonds likely provide a more stabilizing interaction and the N-ethyl group fits better into the upper hydrophobic pocket (Figure S6C). The calculated binding free energies of these derivative compounds exhibit a good correlation with the experimental antiviral activities (R2 = 0.78), further supporting the binding model of CCZ with the fusion peptide in HCV E1 (Figure 6B & 6C).
Discussion
HCV entry is a complex multi-step process that involves many host factors. It starts with a well-coordinated interaction between the viral envelope proteins and multiple cell surface receptors including scavenger receptor class B type I (SR-BI), tetraspanin protein CD81, and the tight junction proteins claudin-1 and occludin, whose proper cellular distribution for HCV entry is regulated by E-cadherin (Evans et al., 2007; Flint et al., 1999; Li et al., 2016; Liu et al., 2009a; Pileri et al., 1998; Ploss et al., 2009; Scarselli et al., 2002). In addition, Niemann-Pick C1 Like 1 (NPC1-L1) facilitates HCV entry via a cholesterol-dependent step (Sainz et al., 2012). EGF receptor, transferrin receptor 1 and TGFß-1 receptor signaling pathways have also been implicated in the regulation of HCV entry process (Lupberger et al., 2011; Martin and Uprichard, 2013; Zhang et al., 2017). Following uptake, internalization and endosomal trafficking, HCV completes its entry process by releasing the viral genome via membrane fusion of viral envelope proteins in the endosome (Bartosch et al., 2003; Hsu et al., 2003; Meertens et al., 2006; Tscherne et al., 2006). Intervention in any of these steps can block viral entry and lead to inhibition of HCV infection. Neutralizing antibodies against cell surface receptor SR-BI, CD81, and claudin-1 can successfully prevent HCV infection (Xiao et al., 2015). Small molecule inhibitors, such erlotinib and ezetimibe, can block viral entry by targeting EGF receptors and NPC1-L1, respectively (Lupberger et al., 2011; Sainz et al., 2012). A nucleic acid-based compound, phosphorothioate oligonucleotide, has also been shown to prevent HCV entry (Matsumura et al., 2009).
CCZ was identified as a potent HCV entry inhibitor through a high-throughput phenotypic screen (He et al., 2015; Hu et al., 2014). Using time-of-addition and viral fusion assays, we showed here that CCZ targets the membrane fusion step of the HCV entry process in a manner that is mechanistically distinct from bafilomycin A1. Bafilomycin A1 inhibits HCV entry via preventing endosomal acidification, whereas CCZ blocks membrane fusion even in the lowered pH environment. We, therefore, reasoned that CCZ may directly interact with the HCV envelope proteins to block membrane fusion. Flunarizine, a structurally related small molecule previously reported to target the membrane fusion stage of the HCV life cycle (Perin et al., 2016), likely has the same mechanism of action as CCZ in blocking HCV entry.
Further evidence that CCZ targets the HCV envelope proteins was borne from the in vitro drug resistance selection assay, in which most of the identified and validated CCZ-resistant mutations reside in the putative fusion peptide of E1. These mutants not only showed phenotypic resistance in the HCV infection assay but also exhibited reduced susceptibility to CCZ in the viral fusion assay.
To demonstrate that HCV E1 is indeed the direct target of CCZ, we synthesized a molecular probe of CCZ with a UV-activatable diazirine group and a biotin molecule for photo-activated cross-linking and affinity purification (Rybak et al., 2004; Vervacke et al., 2014). HCV-infected cells were treated with CCZ-DB and subjected to photoaffinity cross-linking and streptavidin-mediated purification of biotinylated proteins. The CCZ-DB probe was able to specifically enrich HCV E1 from lysate of HCV-infected cells. Using recombinant HCV E1/E2 protein in the same protocol, we also specifically pulled down the E1 protein. Mass spectrometry analysis of the cross-linked E1 protein revealed that CCZ binds to a region of the E1 protein that flanks the putative fusion peptide in an E1 model. Taken together, these data demonstrate that CCZ inhibits HCV entry by directly and specifically binding to HCV E1.
The ability of CCZ to inhibit HCV is substantially different across various HCV genotypes with 2a being the most sensitive genotype following by 2b. Alignment of the fusion loop and other relevant sequences of all HCV genotypes provides a clue to the structural basis of their variable sensitivities to CCZ. The M267 residue is present only in genotype 2, which is most sensitive to CCZ, HCV genotypes 1, 3, 5 and 6 possess G267. The M267V mutation associated with CCZ resistance indicates an important role of the Met residue in binding to CCZ. The methylthio side chain of M267 may provide an important scaffold within the hydrophobic pocket to interact with CCZ, whereas the V267 residue of the CCZ RAS and the G267 residue in all other genotypes may confer a less favorable interaction. It is interesting to note that the two other CCZ RASs, L286I and F291L, represent variations between genotype 2a and 2b (Figure 1E). Our chimeric genotype study showed that the genotype 2a strain (L286/F291) is more sensitive to CCZ than genotype 2b strain J8 (I286/L291; F291 is present in a minority of genotype 2b strains). These sequence variations between 2a and 2b may also explain the difference in their different sensitivities to CCZ.
Other sequence variations may also contribute to the genotypic differences in susceptibility to CCZ. It was noted that genotypes 2 variants are much more susceptible to CCZ relative to other genotypes. Further sequence analysis of in this region highlights several other variations that are unique to genotype 2 (Figure 1E), e.g. W214 and Q215, that may contribute to the enhanced interaction of CCZ with E1. This model is also supported by the CCZ-DB cross-linking data - the bulkier aromatic side chain of W214 in genotype 2a may form a more favorable interaction with the phenyl group of CCZ via π-π stacking than the Y214 of genotype 1a and other genotypes. The neutral side chain of Q215 in genotype 2a (E215 in genotype 1a and other genotypes) may also provide a more favorable interaction with the chloro-phenyl group of CCZ. Alternatively, these amino acid variations may functionally affect certain fusion processes that are not dependent on CCZ binding directly to the fusion loop per se, but still confer resistance to CCZ, allowing the fusion process to take place despite CCZ.
The HCV envelope glycoproteins E1 and E2 form heterodimers and are the precursors for the envelopment of HCV virions (Baumert et al., 1998; Falson et al., 2015; Freedman et al., 2017). The partial structures of E1 and E2 have been determined by crystallography (El Omari et al., 2014; Khan et al., 2014; Kong et al., 2013). Based on these structures and using computational modeling tools and a variety of experimental data, models of full-length E1/E2 heterodimer have been constructed in silico (Cao et al., 2019; Castelli et al., 2017; Freedman et al., 2017). The structure of the N terminal E1 resembles the structure of phosphatidylcholine transfer protein (El Omari et al., 2014). The fusion peptide of E1 has not been structurally determined and modeling reveals a highly flexible and dynamic conformation. Based on our modeling, the fusion peptide and the surrounding sequences form a hydrophobic pocket into which CCZ can be accommodated. Docking simulation with crucial interacting amino acids based on the mutagenesis study supports this model of interaction between CCZ and the putative fusion peptide. As the E1 fusion peptide can have a highly flexible conformation based on modeling and in vitro assays (Cao et al., 2019; Castelli et al., 2017; Douam et al., 2018; Freedman et al., 2017), CCZ likely binds to this region and either stabilizes the fusion peptide in a conformation that inhibits the binding of E1 to inner endosomal membrane. Alternatively, CCZ may occlude the binding of HCV E1, competitively/sterically, to the inner endosomal membrane, thereby inhibiting the fusion process.
HCV has a distinct membrane fusion mechanism from that of class II viral fusion that is typical of flaviviruses and other arboviruses (Li and Modis, 2014). The of HCV E1/E2 heterodimer has a compact globular structure and contains a putative fusion peptide in E1, though the precise mechanism whereby this fusion peptide interacts with the host cell membrane is not clear. Recent evidence suggests that many viral class II fusion proteins have a built-in motif to accommodate glycerophospholipid head groups of host membranes and induce membrane organization by concentrating cholesterol at the site of insertion (Guardado-Calvo et al., 2017). This interaction is important for the class II fusion mechanism, in which the phosphatidylcholine (positively charged) head group fits into a recognition pocket with a conserved D residue (negatively charged) of the fusion peptides (Guardado-Calvo et al., 2017). It is conceivable that such a pocket, as partly defined by the CCZ binding domain, exists in HCV E1 and contributes to viral fusion. The binding interaction of CCZ (positively charged piperazine) with the conserved D279 in the putative hydrophobic pocket of HCV E1, together with the nearby Q289, may resemble the above recognition process of the glycerophospholipid head-group. Cholesterol flux has been implicated in HCV entry (Chamoun-Emanuelli et al., 2014), further supporting the above mechanism in viral fusion. As mentioned above, it is indeed interesting that HCV E1 has structural homology to that of phosphatidylcholine transfer protein (El Omari et al., 2014). CCZ binding to this pocket may thus interfere with this insertion mechanism and blocks subsequent fusion process of HCV. While the presented structural model based on structure-function analyses, computational modeling and docking simulations are hypothetical, the model provides an intriguing insight into the molecular mechanism of CCZ’s action on HCV entry. Complete confirmation of this hypothesis would require additional structural and biochemical studies.
In this study, we elucidate the mechanism of action of CCZ in inhibiting HCV entry and provide important virologic, biochemical and structural insights into the fusion process of HCV. Understanding this mechanism now presents an opportunity to develop broad-spectrum antivirals against HCV and other viruses. In addition, this knowledge may be useful in designing candidate immunogens based on the conserved fusion structure for HCV vaccine development.
Significance
Fusion of virus with the host cell membrane is an essential process in viral entry and infection. Little is known about the fusion mechanism of HCV. By using a previously identified potent HCV entry inhibitor, chlorcyclizine, we show that this compound binds directly to the putative fusion sequence of the HCV envelope protein and blocks the fusion process. Structure-function studies and molecular modeling reveal novel structural features of the fusion sequence, and provide important insight into the potential mechanism whereby HCV fuses with the cell endosomal membrane. Understanding this mechanism may present an opportunity to develop broad-spectrum antivirals against HCV and other viruses as well as potential immunogens for HCV vaccine development.
STAR⋆Methods
Resource Availability
Lead Contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by Lead Contact Zongyi Hu (zongyih@bdg10.niddk.nih.gov) or T. Jake Liang (jake.liang@nih.gov).
Materials Availability
All unique/stable reagents generated in this study will be provided without restriction as long as stocks remain available and reasonable compensation is provided by the requestor to cover processing and shipment.
Data and Code Availability
This study did not generate any unique datasets or code.
Experimental Model and Subject Details
Huh7.5.1 cell, which allows improved production of HCV JFH1 clone, was derived from the Huh7 cell line. Huh7 cell was originally established from a liver tumor in a 57-year-old Japanese male in 1982. The cell was cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO, USA) and 1% penicillin/streptomycin, and incubated at 37°C with 5% CO2.
CHO cells, originated from adult female Chinese hamster ovary, were cultured in Iscove’s modified Dulbecco’s medium (Thermo Fisher Scientific, Waltham, MA) containing 10% FBS, 0.1 mM/0.016 mM sodium hypoxanthine/thymidine (HT supplement; Thermo Fisher Scientific), 0.002 mM methotrexate, and 100 U/ml of penicillin and 100 μg/ml of streptomycin at 37°C with 5% CO2.
Method Details
Chemicals and Viruses
The (S)-CCZ isomer was purified from a racemic CCZ mixture (Sigma-Aldrich, St. Louis, MO, USA). Sofosbuvir (Selleckchem, Houston, TX, USA), daclatasvir (Selleckchem, Houston, TX, USA), and mouse anti-human CD81 monoclonal antibody (BD Biosciences, San Jose, CA, USA) were purchased from various sources. Cyclosporine A and bafilomycin A1 were both purchased from Sigma-Aldrich (St. Louis, MO, USA). Wild-type HCV (JFH1 and J6/JFH1 clones), J6/JFH1 clone with Renilla luciferase reporter between p7 and NS2 (HCV-Rluc) (Tscherne et al., 2006), and various reconstructed HCV mutants described below were generated based on a previously published protocol (Hu et al., 2014). Briefly, plasmids containing various HCV cDNA genome sequences were linearized with XbaI at the 3’-end. HCV sense mRNAs were transcribed in vitro with the linearized cDNA templates and T7 RNA polymerase using MEGAscript™ T7 Transcription kit (Thermo Fischer Scientific, Waltham, MA, USA). Then 20 μg of HCV RNA were electroporated into 4 × 106 Huh7.5.1 cells with the Neon Transfection System at the conditions of 1400 V, 20 ms and 1 pulse (Thermo Fischer Scientific, Waltham, MA, USA). Virus-containing media were harvested daily from day 3 and stored at −80°C. Plasmids of JFH1-based chimeric HCVs expressing core-NS2 of genotype 1 to 7 with or without the Renilla luciferase reporter gene were kindly provided by Dr. Jens Bukh (University of Copenhagen, Denmark). These recombinants are termed according to the genotype (isolate) of core-NS2: 1a(TN), 1b(J4), 2a(J6), 2b(J8), 3a(S52), 4a(ED43), 5a(SA13), 6a(HK6a), and 7a(QC69), respectively (Gottwein et al., 2011). Renilla luciferase reporter gene was inserted into C-terminal domain III of JFH1 NS5A at downstream of aa 2390. These RLuc recombinants were genetically stable after one viral passage and the genotype-specific core-NS2 sequence did not influence the response to interferon-alfa-2b (Gottwein et al., 2011).
Generation of HCV Resistance-Associated Substitutions to CCZ
A 96-well in vitro HCV infection system subjected to serial passages of increasing concentrations of (S)-CCZ was used to generate drug-resistant HCVs according to the protocol described by Bush (Bush et al., 2014). A black, clear bottom 96-well plate was seeded with Huh7.5.1 cells at 10,000 cells/well with a final volume of 100 μL/well. After overnight incubation, the cell culture medium was replaced with 100 μL/well of 1×105 focus-forming units/mL of HCV-WT and incubated for 6 hours to establish HCV infection. After establishing infection, viral medium was discarded and (S)-CCZ was added in a two-fold increasing concentration series from 10 nM to 5 μM per column (Column 10: [10 nM], 9: [20 nM], 8: [40 nM], and so on; DMSO: 0.1% v/v) with a total volume of 200 μL/well. Column 11 and 12 were treated with DMSO as a vehicle-only control (Figure 1A). After 48 hours, a new 96-well plate was seeded with uninfected Huh7.5.1 cells. After an additional 24 hours, a two-part infection was performed: during the primary (1°) infection, 100 μL of virus-containing medium was passaged well-to-well from the infected plate to the uninfected plate; the secondary infection (2°) was performed by passaging 50 μL of virus-containing medium to a two-fold higher dose in the (S)-CCZ concentration series (50 μL of column 11 from the previously infected plate to column 10 on the uninfected plate, column 10 to 9, and so on) (Figure 1A). Special care was taken to ensure that no cross-contamination occurred between each individual well in the 1° infection and that no row-to-row contamination occurred in the 2° infection. Finally, the remaining 50 μL of medium from the previously infected plate was stored at −80°C for storage. After removing all virus-containing medium, infected cells were fixed for HCV core immunofluorescent staining to test for productive infection (Figure 1B). Wells with ≥ 5 foci were considered positive for infection in this assay when compared to background controls. The two-part infection assay was repeated every 3 days until productive HCV infection was observed at 2.5 μM of (S)-CCZ. Daclatasvir was tested similarly as a control HCV drug with concentrations from 10 pM to 5 nM and a resistance threshold of 2.5 nM.
HCV Core Immunofluorescent Staining Assay
To check for productive HCV infection, HCV core immunofluorescent staining was performed as described previously (Li et al., 2009b). Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and blocked with 3% BSA in PBS (w/v) containing 0.3% Tween-20. Anti-HCV core 6G7 monoclonal antibody and Alexa Fluor 488 anti-mouse antibody (Thermo Fischer Scientific, Waltham, MA, USA) were used to detect HCV core protein. Hoechst dye (Thermo Fischer Scientific, Waltham, MA, USA) was used to stain cell nuclei. A fluorescent microscope was then used to detect HCV infection. The entire well (96-well plate) was manually scanned under the 10X objective. The number of HCV core positive foci (≥ 4 positive cells/group) of the entire well was counted for quantitation.
Viral RNA Isolation, Construction of HCV Mutant Clones and DNA Sequencing
HCV RNA genomes were isolated from the cell culture medium using the GeneJET Viral DNA/RNA Purification Kit (Thermo Fischer Scientific, Waltham, MA, USA). cDNAs encoding the region between HCV core and NS2 were generated using the SuperScript™ III First-Strand Synthesis System (Thermo Fischer Scientific, Waltham, MA, USA). The HCV envelope gene sequence (core, E1 and E2) was PCR amplified using CloneAmp™ HiFi PCR premix (Takara Bio USA Inc., Mountain View, CA, USA) to generate amplicons for Sanger sequencing. To generate HCV mutants, an HCV cDNA fragment between EcoRI and KpnI restriction sites containing HCV core and E1 sequence was subcloned into pUC19 (pUC19.HCVnt1–1271) at the corresponding sites. Then the HCV cDNA fragments between NsiI-KpnI (nt1045–1271) containing each of the CCZ-induced resistance mutations were individually synthesized (GENEWIZ, South Plainfield, NJ, USA) and cloned into pUC19.HCVnt1–1271. Then the EcoRI and KpnI fragments of HCV containing the mutations in the HCV E1 region were cloned back into a plasmid containing the whole HCV-WT or HCV-Rluc genome using the InFusion® HD Cloning Kit (Takara Bio USA Inc., Mountain View, CA, USA) and custom primers. For HCV E1 mutants W214Y, Q215E and W214Y/Q215E, site-directed mutagenesis using Q5® Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) was performed on pUC19.HCVnt1–1271 plasmid with the primer pairs of 5’-GACAGCATTACCTATCAGCTCCAGGCTGC-3’ for W214Y and 5’-GACAGCATTACCTGGGAGCTCCAGGCTGC-3’ for Q215E as forward primers, and 5’-ATTGGTGCAGTCGTTAGTCACC-3’ for both W214Y and Q215E as reverse primer. Double mutation W214Y/Q215E was generated by a second round of site-directed mutagenesis on the basis of first round single mutation. Then the EcoRI and KpnI fragments with each single or combination of mutations were cloned into the complete HCV genome at the corresponding sites as described above. All mutant constructs were verified by sequencing after cloning.
Inhibitory Dose-Response Curves of CCZ on HCV Infection
CCZ dose responses against HCV were performed in a 96-well plate assay system. Huh7.5.1 cells were seeded at a density of 104 cells/well overnight. The cells were then infected with HCV-Rluc together with (S)-CCZ or sofosbuvir at serial concentrations of half-log increments between 0.00316 −10 μM. After 48 h culture, cells were lysed for luciferase assay according to the manufacturer’s protocol (Promega, Madison, WI, USA).
Amino Acid Sequence Alignment of HCV Fusion Loop
HCV E1 amino acid sequences for each major genotype (GT) excluding GT7 were obtained from the Virus Pathogen Resource (ViPR) database (Pickett et al., 2012). The GT7 sequences were obtained from the NCBI database. Search results were constrained to only complete genome sequences for each genotype or sub-genotype and duplicated sequences were removed. Sequences were aligned using MAFFT version 7 with the G-INS-1 progressive alignment method (Katoh and Standley, 2013). The putative fusion loop sequence was then extracted from the aligned data, and sequence logos were created using WebLogo (Crooks et al., 2004).
HCV Membrane Fusion Assay
A modified version of HCV membrane fusion assay as described by Perin et al was used to test CCZ’s mode of action in HCV inhibition (Perin et al., 2016). Huh7.5.1 cells were seeded in poly-lysine-coated 96-well plates (1.5 × 104 cells /well). Cells were treated with NH4Cl (10 mM) for 1 h at 37°C before infection of HCV-WT in the presence of NH4Cl. Cells were incubated with HCV-WT (~ 0.5 MOI) for 3 h at 4°C and then washed gently with medium containing NH4Cl. Cells were incubated in medium containing NH4Cl together with (S)-CCZ (1 μM) for 1 h. Then cells were incubated for 5 min at 37°C with freshly prepared pH 5 or 7 citrate-phosphate buffer containing 1 μM of (S)-CCZ. Cells were washed gently twice and then further incubated in medium containing NH4Cl and (S)-CCZ for 3 h. Cells were washed twice and cultured in regular DMEM for 72 h before being processed for HCV core immunofluorescent staining. As controls, DMSO or bafilomycin A1 (3 nM) was similarly treated (Figure 3). HCV core-positive foci per well were recorded for the analysis of HCV infection under various conditions.
Generation of Recombinant HCV E1/E2 Protein
The E1/E2 glycoprotein coding region from H77c (genotype 1a) (GenBank accession no. AF009606; amino acids 192 to 746), preceded by the signal peptide sequence for tissue plasminogen activator (tPA), was inserted into the SpeI/MluI site of the pTRIP lentiviral vector bearing an internal ribosome entry site (IRES)-AcGFP reporter. Lentiviral particles expressing the HCV E1E2 glycoprotein were generated by co-transfecting 4×105 293T cells in 6 well plates with pTRIP.HCVE1/E2, HIV gag-pol, and pCMV.VSV-G (the vesicular stomatitis virus glycoprotein) in a ratio of 1:0.8:0.2, respectively (Logan et al., 2017). Supernatants containing the lentiviral particles were harvested at 48 h and 72 h.
To express E1E2 glycoprotein, CHO cells were transduced with packaged lentivirus. GFP-positive CHO cells expressing recombinant E1/E2 were sorted by flow cytometry using a BD FACSAria III cell sorter (BD Biosciences). CHO cells stably expressing recombinant E1/E2 constructs from the HCV genotype 1a H77c strain were amplified and then harvested by centrifugation at 1,000 g for 10 min, washed once with cold PBS and lysed with lysis buffer (100 mM Nacl, 20 mM TrisHCl [pH7.5], 1 mM EDTA, 0.5% Triton X-100). After removing the cell debris by centrifugation at 20,000 g for 15 min, the supernatant was loaded to a 1 × 5 cm GNA-agarose column (Vector Laboratories, Burlingame, CA). The column was washed extensively with lysis buffer, and the bound proteins were eluded in 0.5 ml fractions by using 0.9 M α-D-mannopyranoside in the lysis buffer. The GNA eluate fraction was loaded on to a hydroxyapatite (HAP) column (Bio-Rad, Hercules, CA; 158–8000), and the flow through was concentrated with a 50,000-molecular-weight cutoff centrifugal filter unit (EMD Millipore, Billerica, MA) and stored at −80°C.
Synthesis and UV-Activated Crosslinking of Photoaffinity CCZ-Diazirine Probe
The chemical synthesis of CCZ-diazirine and CCZ-diazirine-biotin (CCZ-DB) are reported in Supplementary Materials and Methods including full characterization of all intermediates by H1, F19, C13 NMR and HRMS. Since the anti-E1 antibody (A4) recognizes only HCV genotype 1a E1 protein (Dubuisson et al., 1994), we performed this set of experiments with HCV genotype 1a reagents. In vitro photoaffinity cross-linking was performed by mixing 0.1 μg of recombinant HCV E1/E2 protein (genotype 1a, HCV-1 strain)(Stamataki et al., 2007, Logan et al., 2017) with 1 μM of CCZ-DB or control compound in PBS at room temperature for 2 h, followed by UV irradiation for 7 min. Irradiation was carried out with a UVP Blak-Ray™ 100 W, 115 V mercury lamp with a 365 nm filter (Thermo Fischer Scientific, Waltham, MA, USA) from a distance of approximately 5 cm from the light source, on ice. Samples were kept at 4°C for the pull-down of biotinylated proteins.
For in vivo cross-linking, Huh7.5.1 cells (3 × 106) were seeded into a 10 cm dish and cultured overnight. Subsequently, the medium was removed and replaced with 5 mL of DMEM containing high-titer HCV chimeric genotype 1a virus (Gottwein et al., 2011) at a multiplicity of infection of 0.1 together with 5 μM CCZ-DB or control compound. For competition control, 20-fold of (S)-CCZ (100 μM) was also added to the cell with CCZ-DB. Samples were incubated for 1 h at 37°C for viral entry. UV irradiation was then carried out for the photoaffinity crosslinking. The supernatant was removed and kept if necessary, and cells were washed twice with 5 mL cold PBS followed by the addition of 1 mL of lysis buffer (30 mM Tris pH 7.5, 1 mM EDTA, 150 mM NaCl, 0.3% NP-40, 0.05% SDS) with protease inhibitor cocktail. The cell lysate was pelleted by centrifugation at 4°C, 20K relative centrifugal force (RCF) for 5 min. Supernatant was isolated and kept for enrichment of CCZ-DB cross-linked proteins.
Affinity Pull-Down of CCZ-DB Cross-Linked Proteins and Western Blot Analysis
50 μL of Pierce NeutrAvidin agarose beads (Thermo Fischer Scientific, Waltham, MA, USA) were spun down, the supernatant was discarded, and the beads were washed twice with 1 mL of PBS. The cross-linked recombinant E1/E2 protein and HCV-infected cell lysate samples were added to the NeutrAvidin beads. Biotin BSA (2 μL of 2 μg/mL biotin BSA in PBS) was used as a positive control. Samples were briefly mixed and placed on a rocker for 1 h at 4°C then pelleted (Eppendorf desktop centrifuge, 5,000 RPM, 30 s). Beads were washed successively with PBS twice, lysis buffer (described above) twice, and finally PBS. Elution buffer (2% SDS, 3 mM biotin, 6 M urea, 2 M thiourea) (Rybak et al., 2004), 1:4 Laemmli buffer, and reducing agent were added to resin beads and incubated at room temperature for 10 min, then 95°C for 10 min. Samples were briefly spun and the supernatant was used for Western blot analysis with A4 anti-E1 antibody (provided by Harry Greenberg, Stanford University, Palo Alto, USA) or ALP33 anti-E2 antibody (provided by Arvind Patel of University of Glasgow, UK) using a ProteinSimple Wes capillary Western blot system (Wallingford, CT, USA).
Mass Spectrometry Analysis of UV-Crosslinked CCZ and Recombinant E1/E2 Protein
Recombinant HCV E1/E2 protein (genotype 1a, HCV-1 strain), was generated as previously described (Stamataki et al., 2007, Logan et al, 2017). 5 μg of recombinant E1/E2 protein mixed with CCZ-DB (50 μM) were cross-linked by UV irradiation as described above. Processing via mass spectrometry analysis is detailed in the Supplemental Methods (Mass spectrometric analysis of CCZ-diazirine-biotin cross-linked E1 glycoprotein, Figure S4A–C).
Structural Modeling and Molecular Dynamic Simulations of HCV E1
The 3D structure model of HCV E1 protein was constructed using the crystal structure of HCV E1 protein which consists of 79 amino acids of the N-terminal domain (PDB: 4UOI) as a template. The sequence of the HCV genotype 2a was retrieved from the UniProtKB database (access code Q99IB8). Residues 192–310 which includes the ectodomain and the putative fusion loop (264–290) were generated using the I-TASSER program (Yang and Zhang, 2015). Molecular Dynamic (MD) simulations were performed to refine the HCV E1 model in explicit solvent using the Amber 18 program (Case, 2018). The solvated protein systems were subjected to a thorough energy minimization prior to MD simulations by first minimizing the water molecules while holding the solute frozen (1,000 steps using the steepest descent algorithm), followed by 5,000 steps of conjugate gradient minimization of the whole system to remove close contacts and to relax the system. The simulated system was first subjected to a gradual temperature increase from 0 K to 300 K over 100 ps, and then equilibrated for 500 ps at 300 K, followed by a production run of 10 ns. Clustering analysis of the MD trajectories was performed using the CPPTRAJ module (Roe and Cheatham, 2013). A total of 10 clusters were generated using the hierarchical clustering from the protein simulations and the representatives of ensemble structures were extracted for the following docking study.
Ensemble Docking of CCZ Binding to HCV E1
The AutoDock 4.2 program (Morris et al., 2009) was used for the docking of CCZ and derivatives to ensembles of the E1 structure. The protein active site was defined by a grid of 70 × 70 × 70 points with a grid spacing of 0.375 Å. The Lamarckian Genetic Algorithm (LGA) (Morris, 1998) was applied with 100 runs and the maximum number of energy evaluations was set to 2 × 106. Binding mode analysis was performed using the AutoDockTool package. The top 30 poses from each docking were retained for a consensus binding mode analysis. Finally, the optimal binding model of E1 and CCZ were subjected to stepwise energy minimization and MD simulations as described above.
Quantification and Statistical Analysis
Data were analyzed with the GraphPad Prism 7.0 software for curve fitting, EC50 calculation and graphical displays. T tests were used to determine the statistical difference between the means of two groups. GraphPad Prism was used to produce Figures 1G, 2B, 2C, 3B, 3C, 4A, 5B, 6C, S2 and S5. For Figure 1G, 2C, 4A, 5B S2 and S5, data are represented as mean +/− SEM from 6 replicates (n=6, n represents the number of wells tested in a 96-well plate.) For Figure 2B, 3B and 3C, data are represented as mean +/− SEM from 3 replicates (n=3, n represents the number of wells tested in a 6-well plate.).
Supplementary Material
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HCV core monoclonal antibody (6G7) | This paper | N/A |
| Anti-HCV E1 antibody (A4) | Harry Greenberg | N/A |
| Anti-HCV E2 antibody (ALP33) | Arvind Patel | N/A |
| Mouse anti-human CD81 monoclonal antibody | BD Biosciences | Cat # 55675 |
| Alexa Fluor 488 anti-mouse antibody | Thermo Fischer Scientific | Cat # A28175 |
| Chemicals and Recombinant Proteins | ||
| Chlorcyclizine (CCZ, racemic mixture) | Sigma-Aldrich | SML1473 |
| (S)-CCZ isomer | This paper | N/A |
| Sofosbuvir | Selleckchem | S2794 |
| Daclatasvir | Selleckchem | S1482 |
| Cyclosporine A | Sigma-Aldrich | PHR1092 |
| Bafilomycin A1 | Sigma-Aldrich | 19-148 |
| CCZ-diazirine | This paper | N/A |
| CCZ-diazirine-biotin | This paper | N/A |
| Recombinant HCV E1/E2 (GT1a, HCV-1 strain) | Michael Houghton | N/A |
| Virus strains | ||
| HCV-WT (JFH1 and J6/JFH1 clones) | This paper | N/A |
| HCV-Rluc (J6/JFH1 clone) | This paper | N/A |
| HCV mutants (P224S, M267V, A274T, L286I Q289H, F291L, M267V/F291L, L286I/F291L W214Y, Q215E, W214Y/Q215E) | This paper | N/A |
| Chimeric HCV genotypes 1-7 | This paper | N/A |
| Recombinant DNA | ||
| pUC19.HCVnt1-1271 | This paper | N/A |
| pHCV.(P224S, M267V, A274T, L286I Q289H, F291L, M267V/F291L, L286I/F291L W214Y, Q215E, W214Y/Q215E) | This paper | N/A |
| pTRIP lentiviral vector | (Logan et al., 2017) | N/A |
| pTRIP.HCVE1/E2 | (Logan et al., 2017) | N/A |
| HIV gag/pol | Logan et al., 2017 | N/A |
| pCMV-VSV-G | Addgene | Cat # 8454 |
| Oligonucleotides | ||
| Forward primer: 5’-GACAGCATTACCTATCAGCTCCAGGCTGC-3’ for HCV.W214Y | ||
| This paper | N/A | |
| Forward primer: 5’-GACAGCATTACCTGGGAGCTCCAGGCTGC-3’ for HCV.Q215E | ||
| This paper | N/A | |
| Reverse primer: 5’-ATTGGTGCAGTCGTTAGTCACC-3’ for both HCV.W214Y and Q215E | ||
| This paper | N/A | |
| Critical Commercial Assays | ||
| Renilla luciferase assay kit | Promega | E2820 |
| GeneJET Viral DNA/RNA Purification Kit | Thermo Fischer Scientific | K0821 |
| SuperScript™ III First-Strand Synthesis System | Thermo Fischer Scientific | 18080051 |
| CloneAmp™ HiFi PCR premix | Takara Bio USA Inc | 639298 |
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs | E0554S |
| InFusion® HD Cloning Kit | Takara Bio USA Inc. | 638909 |
| Pierce NeutrAvidin agarose beads | Thermo Fischer Scientific | 29200 |
| ProteinSimple Wes capillary Western blot system | Wallingford | proteinsimple.com |
| MEGAscript™ T7 Transcription Kit | Thermo Fischer Scientific | AMB13345 |
| Experimental Models: Cell Line | ||
| Huh7.5.1 | Scripps Institute (UCSD) | RRID: CVCL_E049 |
| CHO | Dr. Houghton (Uni. Of Alberta) | RRID: CVCL_0213 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | Version 7 |
| I-TASSER program | Roy et al. | Nat. Protoc. 2010 |
| Amber 18 program | Amber 2018 Reference Manual | ambermd.com |
| CPPTRAJ module | Roe & Cheatham | ambermd.com |
| AutoDock4.2 program | Morris et al., 2009 | Version 4 |
| Virus Pathogen Resource (ViPR) database | Pickett et al., 2012 | viprbrc.org |
| MAFFT multiple sequence alignment software | Katoh, K. & Standley, 2013 | Version 7 |
| WebLogo | Crooks et al., 2004 | weblogoberkeley.edu |
Highlights.
CCZ blocks membrane fusion of HCV during viral entry
HCV RASs to CCZ cluster in the putative E1 fusion domain
HCV carrying CCZ RASs are resistant to the blocking of membrane fusion by CCZ
CCZ binds to E1 and forms extensive interaction with the E1 putative fusion peptide
Acknowledgments
We thank Dr. Harry Greenberg (Stanford University) for providing mouse monoclonal anti-HCV E1 antibody, Arvind Patel (University of Glasgow, UK) for providing mouse monoclonal anti-HCV E2 antibody (ALP33) and Jason Piotrowski for technical assistance. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and National Center for Advancing Translational Sciences, National Institutes of Health, USA.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Supporting Citations
The following references appear in the Supplemental Information: Rappsilber et al., 2007; Davis, et al., 2001; Cox and Mann, 2008; Meyer et al., 2014.
Supplemental Information
Supplemental Information can be found online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartosch B, Dubuisson J, and Cosset FL (2003). Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med 197, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumert TF, Ito S, Wong DT, and Liang TJ (1998). Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J. Virol 72, 3827–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush CO, Pokrovskii MV, Saito R, Morganelli P, Canales E, Clarke MO, Lazerwith SE, Golde J, Reid BG, Babaoglu K, et al. (2014). A small-molecule inhibitor of hepatitis C virus infectivity. Antimicrob. Agents Chemother 58, 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Yu B, Kong D, Cong Q, Yu T, Chen Z, Hu Z, Chang H, Zhong J, Baker D, et al. (2019). Functional expression and characterization of the envelope glycoprotein E1E2 heterodimer of hepatitis C virus. PLoS Pathog. 15, e1007759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE, Cruzeiro VWD, Darden TA, Duke RE, Ghoreishi D, Gilson MK, et al. (2018). Amber 2018 Reference Manual.
- Castelli M, Clementi N, Pfaff J, Sautto GA, Diotti RA, Burioni R, Doranz BJ, Dal Peraro M, Clementi M, and Mancini N (2017). A Biologically-validated HCV E1E2 Heterodimer Structural Model. Scientific Reports 7, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoun-Emanuelli AM, Pecheur EI, and Chen Z (2014). Benzhydrylpiperazine compounds inhibit cholesterol-dependent cellular entry of hepatitis C virus. Antiviral Res. 109, 141–148. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MT, Spahr CS, McGinley MD, Robinson JH, Bures EJ, Beierle J, Mort J, Yu W, Luethy R, and Patterson SD (2001). Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. II. Limitations of complex mixture analyses. Proteomics 1, 108–117. [DOI] [PubMed] [Google Scholar]
- Douam F, Fusil F, Enguehard M, Dib L, Nadalin F, Schwaller L, Hrebikova G, Mancip J, Mailly L, Montserret R, et al. (2018). A protein coevolution method uncovers critical features of the hepatitis C virus fusion mechanism. PLoS Pathog. 14, e1006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummer HE, Boo I, and Poumbourios P (2007). Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J. Gen. Virol 88, 1144–1148. [DOI] [PubMed] [Google Scholar]
- Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, and Rice CM (1994). Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol 68, 6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, and Stuart DI (2014). Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat. Commun 5, 4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, and Rice CM (2007). Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805. [DOI] [PubMed] [Google Scholar]
- Falson P, Bartosch B, Alsaleh K, Tews BA, Loquet A, Ciczora Y, Riva L, Montigny C, Montpellier C, Duverlie G, et al. (2015). Hepatitis C virus envelope glycoprotein E1 forms trimers at the surface of the virion. J. Virol 89, 10333–10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, and McKeating JA (1999). Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol 73, 6235–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman H, Logan MR, Hockman D, Koehler Leman J, Law JL, and Houghton M (2017). Computational prediction of the heterodimeric and higher-order structure of gpE1/gpE2 envelope glycoproteins encoded by hepatitis C virus. J. Virol 91, e02309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridell RA, Qiu D, Wang C, Valera L, and Gao M (2010). Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother 54, 3641–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridell RA, Wang C, Sun J-H, O’Boyle II DR, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, et al. (2011). Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in Humans: In Vitro and In Vivo Correlations. Hepatology 54, 1924–1935. [DOI] [PubMed] [Google Scholar]
- Gottwein JM, Jensen TB, Mathiesen CK, Meuleman P, Serre SBN, Lademann JB, Ghanem L, Scheel TKH, Leroux-Roels G, and Bukh J (2011). Development and application of hepatitis C reporter viruses with genotype 1 to 7 core-nonstructural protein 2 (NS2) expressing fluorescent proteins or luciferase in modified JFH1 NS5A. J. Virol 85, 8913–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardado-Calvo P, Atkovska K, Jeffers SA, Grau N, Backovic M, Perez-Vargas J, de Boer SM, Tortorici MA, Pehau-Arnaudet G, Lepault J, et al. (2017). A glycerophospholipid-specific pocket in the RVFV class II fusion protein drives target membrane insertion. Science 358, 663–667. [DOI] [PubMed] [Google Scholar]
- He S, Lin B, Chu V, Hu Z, Hu X, Xiao J, Wang AQ, Schweitzer CJ, Li Q, Imamura M, et al. (2015). Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Trans. Med 7, 282ra249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Xiao J, Dulcey AE, Lin B, Rolt A, Hu Z, Hu X, Wang AQ, Xu X, Southall N, et al. (2016). Discovery, optimization, and characterization of novel chlorcyclizine derivatives for the treatment of hepatitis C virus infection. J. Med. Chem 59, 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, and McKeating JA (2003). Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U S A 100, 7271–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Lan K-H, He S, Swaroop M, Hu X, Southall N, Zheng W, and Liang TJ (2014). Novel cell-based hepatitis C virus infection assay for quantitative high-throughput screening of anti-hepatitis C virus compounds. Antimicrob. Agents and Chemother 58, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, et al. (2014). Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509, 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, et al. (2013). Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 1090–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D, Bartosch B, Nourrisson D, Verney G, Cosset FL, Penin F, and Pecheur EI (2006). Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J. Biol. Chem 281, 3909–3917. [DOI] [PubMed] [Google Scholar]
- Li HF, Huang CH, Ai LS, Chuang CK, and Chen SS (2009a). Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. J. Biomed. Sci 16, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, and Elledge SJ (2009b). A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U S A 106, 16410–16415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sodroski C, Lowey B, Schweitzer CJ, Cha H, Zhang F, and Liang TJ (2016). Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. U S A 113, 7620–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, and Modis Y (2014). A novel membrane fusion protein family in Flaviviridae? Trends Microbiol 22, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang TJ, and Ghany MG (2013). Current and future therapies for hepatitis C virus infection. N. Engl. J. Med 368, 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. (2005). Complete replication of hepatitis C virus in cell culture. Science 309, 623–626. [DOI] [PubMed] [Google Scholar]
- Liu S, Yang W, Shen L, Turner JR, Coyne CB, and Wang T (2009a). Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol 83, 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Robida JM, Chinnaswamy S, Yi G, Robotham JM, Nelson HB, Irsigler A, Kao CC, and Tang H (2009b). Mutations in the hepatitis C virus polymerase that increase RNA binding can confer resistance to cyclosporine A. Hepatology 50, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan M, Law L, Wong J, Hockman D, Landi A, Chen C, Crawford K, Kundu J, Baldwin L, Johnson J, et al. (2017). Native folding of a recombinant gpe1/gpe2 heterodimer vaccine antigen from a precursor protein fused with Fc IgG. J Virol. 91: e01552–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, et al. (2011). EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med 17, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DN, and Uprichard SL (2013). Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. U S A 110, 10777–10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura T, Hu Z, Kato T, Dreux M, Zhang YY, Imamura M, Hiraga N, Juteau JM, Cosset FL, Chayama K, et al. (2009). Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology 137, 673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Bertaux C, and Dragic T (2006). Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol 80, 11571–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JG, Kim S, Maltby DA, Ghassemian M, Bandeira N, and Komives EA (2014). Expanding proteome coverage with orthogonal-specificity alpha-lytic proteases. Mol. Cell. Proteomics 13, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GMG, D. S.; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ (1998). Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem 19, 1639–1662. [Google Scholar]
- Perez-Berna AJ, Moreno MR, Guillen J, Bernabeu A, and Villalain J (2006). The membrane-active regions of the hepatitis C virus E1 and E2 envelope glycoproteins. Biochemistry 45, 3755–3768. [DOI] [PubMed] [Google Scholar]
- Perin PM, Haid S, Brown RJ, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, et al. (2016). Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63, 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, et al. (2012). ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 40, D593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann T (2017). Clinically approved ion channel inhibitors close gates for hepatitis C virus and open doors for drug repurposing in infectious viral diseases. J. Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. (1998). Binding of hepatitis C virus to CD81. Science 282, 938–941. [DOI] [PubMed] [Google Scholar]
- Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, and Rice CM (2009). Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robida JM, Nelson HB, Liu Z, and Tang H (2007). Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J. Virol 81, 5829–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, and Cheatham TE 3rd (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Rolt A, Le D, Hu Z, Wang AQ, Shah P, Singleton M, Hughes E, Dulcey AE, He S, Imamura M, et al. (2018). Preclinical pharmacological development of chlorcyclizine derivatives for the treatment of hepatitis C virus infection. J. Infect. Dis 217, 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak JN, Scheurer SB, Neri D, and Elia G (2004). Purification of biotinylated proteins on streptavidin resin: a protocol for quantitative elution. Proteomics 4, 2296–2299. [DOI] [PubMed] [Google Scholar]
- Sainz B Jr., Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. (2012). Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18, 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, and Vitelli A (2002). The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamataki Z, Coates S, Evans MJ, Wininger M, Crawford K, Dong C, Fong YL, Chien D, Abrignani S, Balfe P, et al. (2007). Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 25, 7773–7784. [DOI] [PubMed] [Google Scholar]
- Tong Y, Lavillette D, Li Q, and Zhong J (2018). Role of hepatitis C virus envelope glycoprotein E1 in virus entry and assembly. Front. Immunol 9, 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, and Rice CM (2006). Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol 80, 1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervacke JS, Funk AL, Wang YC, Strom M, Hrycyna CA, and Distefano MD (2014). Diazirine-containing photoactivatable isoprenoid: synthesis and application in studies with isoprenylcysteine carboxyl methyltransferase. J. Org. Chem 79, 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, and Dubuisson J (2010). Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol 84, 10159–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, et al. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med 11, 791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2017). Global hepatitis report, 2017.
- Xiao F, Fofana I, Thumann C, Mailly L, Alles R, Robinet E, Meyer N, Schaeffer M, Habersetzer F, Doffoël M, et al. (2015). Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut 64, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, and Zhang Y (2015). Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinformatics 52, 5.8.1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Sodroski C, Cha H, Li Q, and Liang TJ (2017). Infection of hepatocytes with HCV increases cell surface levels of heparan sulfate proteoglycans, uptake of cholesterol and lipoprotein, and virus entry by up-regulating smad6 and smad7. Gastroenterology 152, 257–270.e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, and Chisari FV (2005). Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U S A 102, 9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.