

Abstract
Background:
Schizophrenia (SZ) is associated with increased all-cause mortality, smoking, and age-associated proteins, yet multiple previous studies found no association between SZ and biological age using Horvath’s epigenetic clock, a well-established aging biomarker based on DNA methylation (DNAm). However, numerous epigenetic clocks have been developed that may capture distinct aspects of aging. This study tested the hypothesis that altered aging in SZ manifests in these other clocks.
Methods:
We performed a comprehensive analysis of 14 epigenetic clocks, categorized according to what they were trained to predict: chronological age, mortality, mitotic divisions, or telomere length. To understand the etiology of biological age differences, we also examined DNAm predictors of smoking, alcohol, BMI, serum proteins, and cell proportions. We independently analyzed three publicly available multi-ethnic DNAm datasets from whole blood, a total of 567 SZ cases and 594 non-psychiatric controls.
Results:
All data sets showed accelerations in SZ for the three mortality clocks up to 5 years, driven by smoking and elevated levels of 6 age-associated proteins. The two mitotic clocks were decelerated in SZ related to anti-tumor NK and CD8T cells, which may help explain conflicting reports about low cancer rates in epidemiological studies of SZ. One cohort with available medication data showed clozapine is associated with male-specific decelerations up to 7 years in multiple chronological age clocks.
Conclusions:
Our study demonstrates the utility of studying the various epigenetic clocks in tandem and highlights potential mechanisms by which mental illness influences long-term outcomes including cancer and early mortality.
Keywords: schizophrenia, aging, epigenetics, smoking, cancer, clozapine
Introduction
Life expectancy in schizophrenia (SZ) is reduced by 15 years (1), and a meta-analysis of 37 studies found a standardized mortality hazard ratio of 2.5 in SZ, with excess mortality in nearly all categories of natural causes (2). This may be explained by observations that SZ is associated with changes in age-related biomarkers of inflammation, oxidative stress, and metabolism (3). Serum proteins that increase with age and predict mortality (4) are also increased in SZ including PAI-1 (5,6), TIMP-1 (6), GDF-15 (7), Cystatin C (8), beta-2 microglobulin (9,10), and adrenomedullin (11). Thus, it has been proposed that schizophrenia is a syndrome of accelerated aging (12,13).
However, multiple studies using Horvath’s DNA methylation (DNAm)-based epigenetic clock, a well-recognized and validated biomarker of chronological age, have found no evidence for the accelerated aging hypothesis of SZ in either brain or blood (14–18). A recent study observed a null association with all-cause mortality in SZ (18). The epigenetic clock was constructed using a penalized regression model to select a weighted average of 353 CpG sites, which produces very accurate chronological age predictions across all tissues and across the entire lifespan (19). Increased or decreased epigenetic age relative to chronological age is termed age acceleration or deceleration, respectively. Age acceleration predicts increased risk of age-related diseases like Alzheimer’s disease, cancer, and cardiovascular disease (20). Epigenetic clocks are influenced by substance use, diet, exercise, obesity, and socioeconomic status (20,21). Schizophrenia is associated with all these factors, especially high smoking rates and metabolic side effects of antipsychotic medications (22–27). Thus, it is surprising that epigenetic clock analyses have not identified accelerated aging in schizophrenia. Our current understanding of epigenetic aging in schizophrenia may be incomplete, and important confounding variables such as medications and smoking have not yet been sufficiently accounted for.
Numerous epigenetic clocks have recently been developed that appear to capture distinct aspects of aging. Some predict chronological age similar to Horvath’s clock (19,28–34). Others predict age-related morbidity and mortality (4,35,36), mitotic divisions (37,38), or telomere length (39). There are also DNAm biomarkers validated in blood for smoking, alcohol, BMI, and serum levels of age-associated proteins dysregulated in schizophrenia (4,40). Various clocks have age acceleration values (i.e. after adjusting for chronological age) correlated at r < 0.5, and show very little overlap in their CpG composition (41). The clocks differ in their associations with gene expression, lifestyle factors and outcomes of aging (20,41). Whether these other clocks are altered in schizophrenia is unknown. Here, we systematically investigate 14 clocks in three multi-ethnic SZ blood DNAm data sets.
Methods and Materials
Sample
Analyses utilized three Illumina 450K DNAm data sets from whole blood: UCL (University College London, GSE80417) (42), ABD (University of Aberdeen, GSE84727) (42), and CF (Chinese Famine, GSE116379) (43). We analyzed a total of 567 SZ cases and 594 non-psychiatric controls (NPC) with ages ranging from 30 to 90 (Table 1). All three data sets included information on gender and SZ status. CF consists of subjects who were conceived during or after the 1959-1961 Chinese Famine as identified by the University in Changchun. Thus, CF has a narrower age range (44 to 51) and included data on medications obtained through a structured interview, famine exposure during the first 3 months of gestation based on birth dates, and city versus rural background. See Supplementary Methods for additional information.
Table 1. Cohort characteristics.
Table S1 shows information for these data sets before filtering out individuals with age less than 30. Figure S1 shows age histograms for each data set. Table S2 (separate file) shows subject-level data for CF. Abbreviations: NPC (non-psychiatric control), SZ (schizophrenia), UCL (University of College London), ABD (University of Aberdeen), CF (Chinese Famine), AP (antipsychotics).
| Variable | UCL NPC | UCL SZ | ABD NPC | ABD SZ | CF NPC | CF SZ |
|---|---|---|---|---|---|---|
| Number | 173 | 278 | 342 | 215 | 79 | 74 |
| Sex | 83F, 90M | 80F, 198M | 78F, 264M | 72F, 143M | 48F, 31M | 29F, 45M |
| Age Range (yr) | 30-87 | 30-90 | 30-66.4 | 30-80.7 | 44.6-51.0 | 45.0-50.9 |
| Median Age (yr) | 44 | 45 | 48.6 | 47.1 | 47.5 | 47.3 |
| Mean Age (yr) | 46.0 | 47.3 | 48.4 | 48.2 | 47.9 | 47.7 |
| Famine | 25 | 23 | ||||
| City | 74 | 45 | ||||
| Antidepressants | 0 | 8 | ||||
| Typical AP | 0 | 36 | ||||
| Atypical AP | 0 | 18 | ||||
| Clozapine | 0 | 35 |
Epigenetic Clocks
Epigenetic clocks and other biomarkers (Table 2) were calculated according to published methods (4,19,28–40). Those not normalized to provide an age estimate (or other meaningful value like telomere length) were transformed into units of standard deviation. For each clock, we calculated two values of age acceleration (19,20): 1) an age residual resulting from a linear model regressing epigenetic age on chronological age and 2) a cell composition corrected age residual resulting from regressing epigenetic age on chronological age and 7 cell types as previously described (44). Both models were developed in NPC only, then applied to all subjects. Thus, average age acceleration for NPC is zero by construction. Further analyses utilized these acceleration values for biomarkers with clear age trends. We could not assume normality of distributions, so we utilized the non-parametric Kruskal-Wallis test for univariate statistics and generalized linear models for multiple regressions. See Supplementary Methods for additional information.
Table 2.
Epigenetic clocks and other DNAm-based biomarkers
| Denotation | 1st Author, Year | Trained Phenotype | #CpGs | Tissues derived |
|---|---|---|---|---|
| GrimAge | Lu, 2019a (4) | Mortality | 1030 | Whole blood |
| PhenoAge | Levine, 2018 (35) | Mortality, phenotypic age | 513 | Whole blood |
| Zhang | Zhang, 2017 (36) | Mortality | 10 | Whole blood |
| MiAge | Youn, 2018 (37) | Mitotic divisions | 268 | 8 cancer types, adjacent tissue |
| Yang | Yang, 2016 (38) | Mitotic divisions | 385 | Whole blood |
| DNAmTL | Lu, 2019b (39) | Telomere length | 140 | DNAmTL |
| Horvath1 | Horvath, 2013 (19) | Chronological age | 353 | 51 tissues/cells |
| Horvath2 | Horvath, 2018 (33) | Chronological age | 391 | Skin, fibroblasts, buccal, cord blood |
| Vidal-Bralo | Vidal-Bralo, 2016 (31) | Chronological age | 8 | Whole blood |
| Lin | Lin, 2015 (34) | Chronological age | 99 | Whole blood |
| Hannum | Hannum, 2013 (32) | Chronological age | 71 | Whole blood |
| Weidner | Weidner, 2014 (30) | Chronological age | 3 | Whole blood |
| Garagnani | Garagnani, 2012 (29) | Chronological age | 1 | Whole blood |
| Bocklandt | Bocklandt, 2011 (28) | Chronological age | 1 | Saliva |
| GDF-15 | Lu, 2019a (4) | GDF-15 serum protein | 137 | Whole blood |
| ADM | Lu, 2019a (4) | ADM serum protein | 186 | Whole blood |
| B2M | Lu, 2019a (4) | B2M serum protein | 91 | Whole blood |
| PAI-1 | Lu, 2019a (4) | PAI-1 serum protein | 211 | Whole blood |
| TIMP-1 | Lu, 2019a (4) | TIMP-1 serum protein | 42 | Whole blood |
| Cystatin C | Lu, 2019a (4) | Cystatin C serum protein | 87 | Whole blood |
| Leptin | Lu, 2019a (4) | Leptin serum protein | 187 | Whole blood |
| Smoking1 | Lu, 2019a (4) | Smoking pack-years | 172 | Whole blood |
| Smoking2 | McCartney, 2018 (40) | Smoking | 233 | Whole blood |
| Alcohol | McCartney, 2018 (40) | Alcohol | 450 | Whole blood |
| BMI | McCartney, 2018 (40) | BMI | 1109 | Whole blood |
Results
Mortality, mitotic, and telomere length clocks are altered in SZ
Previous studies using the UCL and ABD data sets have shown no difference in the Horvath1 chronological age clock between SZ and NPC (16,17). We replicated their results (Figures 1A, S2A). We examined if altered aging manifests in clocks other than Horvath1 in same data sets, as well as in the Chinese Famine (CF) data set.
Figure 1.
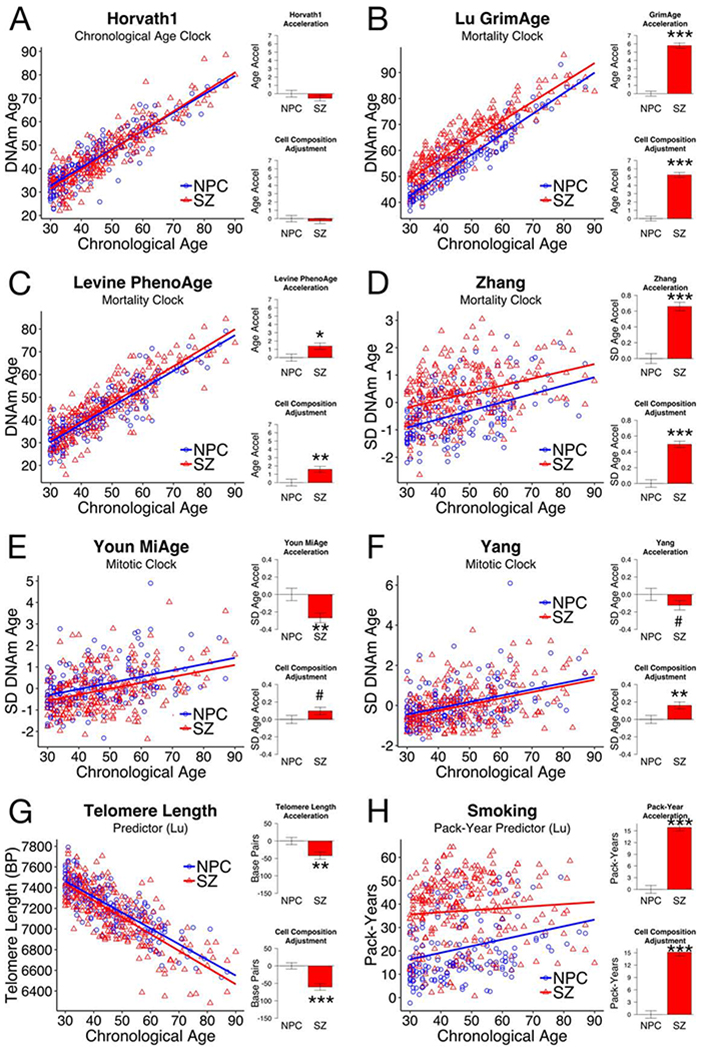
Aging clocks other than Horvath1 show changes in schizophrenia (SZ) compared to non-psychiatric controls (NPC): (A) The Horvath1 chronological age clock, (B-D) the GrimAge, PhenoAge, and Zhang mortality clocks, (E-F) the MiAge and Yang mitotic clocks, (G) predicted telomere length, (H) predicted smoking pack-years. Here we show results from UCL (NPC, n = 173; SZ, n = 278). The other two data sets showed the same patterns (Figures S2–3). Complete statistics are in Table S4. For each panel, we show chronological age compared to clock score (left), as well as average age accelerations for SZ compared to NPC (right). Age accelerations are defined as residuals from the linear model for NPC (blue line), and therefore average age acceleration for NPC is 0. The first model is adjusted only for age. The second model is adjusted for age and derived cell counts (see Supplementary Methods). Error bars are SEM. More advanced biological age is indicated by positive acceleration values for most clocks, but by negative acceleration values for telomere length. Statistical significance of difference from NPC is shown: <0.2#, <0.05*, <0.01**, <0.001***.
The GrimAge, PhenoAge and Zhang mortality clocks were trained by regressing time-to-death on DNAm, either directly or using an intermediate measure (4,35,36). They are more sensitive to mortality than Horvath1. We found age acceleration for schizophrenia (SZ) in all mortality clocks compared to non-psychiatric controls (NPC) in all data sets (Panels B-D in Figures 1, S2, S3; Table S4). GrimAge showed accelerations in SZ of 5.8 years (UCL; p < 0.0001), 4.2 years (ABD; p < 0.0001) and 2.5 years (CF; p = 0.0003). PhenoAge was accelerated in SZ by 1.4 years (UCL; p = 0.0127), 1.9 years (ABD; p < 0.0001), and 1.5 years (CF; p = 0.0912). Zhang is not normalized to give age estimates, so we use units of standard deviation (SD). Zhang was increased in SZ by 0.66 SD (UCL), 0.61 SD (ABD) and 0.75 SD (CF), with p < 0.0001 for all.
The MiAge and Yang mitotic clocks use mathematical models of DNA methylation changes to estimate mitotic age, or the total number of lifetime cell divisions (37,38). They are accelerated in diverse cancers, high-turnover tissues, and carcinogen-exposed tissues. They were correlated to the same extent both before and after adjusting for chronological age (R2 = 0.649 (UCL) to 0.833 (CF)) (Figure S4). We found deceleration for SZ in both mitotic clocks in all three data sets (Panels E-F in Figures 1, S2, S3; Table S4). MiAge was decelerated by 0.27 SD (UCL; p = 0.0017), 0.44 SD (ABD; p < 0.0001), and 0.22 SD (CF; p = 0.1585). Yang was decelerated by 0.12 SD (UCL; p = 0.1918), 0.39 SD (ABD; p < 0.0001), and 0.42 SD (CF; p = 0.0085).
The DNAm estimator of telomere length (DNAmTL) (39) was reduced in SZ in UCL (42 base pairs, p = 0.0067) and ABD (34 base pairs, p = 0.0063) (Panel G of Figure 1, S2, Table S4). Since telomere length decreases with age, we considered DNAmTL to be accelerated in SZ, consistent with prior reports on SZ telomere length (45).
After adjusting for gender, these differences in various clocks were all maintained. In fact, SZ showed more significant effects after adjusting for gender, particularly for the mitotic clocks (Table S5). SZ males consistently showed mitotic clock deceleration in all data sets. SZ females showed significant deceleration in UCL, a deceleration trend in ABD, and no deceleration in CF (Table S5, Figure S5).
When we adjusted for cell composition, mortality clock and telomere length accelerations were slightly smaller but remained statistically significant (Figures 1, S2, S3; Tables S4, S5). However, mitotic clock deceleration in SZ was greatly reduced after correcting for cell composition, drawing attention to a relationship between SZ, mitotic age, and cell proportions.
SZ mortality clock acceleration is related to smoking and age-associated proteins
Because mortality clocks are accelerated by smoking (4,35,36), and smoking rates are much higher in SZ (25), we hypothesized smoking was a major driver of SZ mortality clocks. The DNAm-based predictor of smoking pack-years (PY) by Lu et al. (4) was increased in SZ by 15.9 PY (UCL, p < 0.0001), 10.5 PY (ABD, p < 0.0001), and 5.3 PY (CF, p = 0.007), independent of gender and cell composition (Figures 1H, S2H, S3H; Tables S4, S6). Another smoking predictor from McCartney et al. (40) shows the same pattern, and the two independently derived biomarkers are highly correlated (Figure S6).
We also examined DNAm-based predictors of alcohol and body mass index (BMI) (Tables S4, S6, Figures 2, S7–9) (40). Alcohol was positively associated with smoking. However, despite higher smoking scores in SZ, alcohol use was reduced in SZ in ABD and CF. After correcting for smoking and gender, alcohol scores were lower in SZ in all data sets, by 0.22 SD (UCL, p = 0.0468), 0.49 SD (ABD, p < 0.001), and 0.67 SD (CF, p < 0.001) (Table S6). BMI was negatively correlated with smoking. BMI was reduced in SZ only in CF, by 0.47 SD (p = 0.0034), independent of gender, smoking, and alcohol.
Figure 2.

DNA methylation biomarkers in SZ and NPC: (A-B) Alcohol and BMI, (C-I) serum protein levels, and (J-L) three cell types with altered proportions in SZ. Here we show results from UCL (NPC, n = 173; SZ, n = 278). The other two data sets showed similar patterns (Figures S7–8). Complete statistics are in Table S4.
Multiple regression analysis showed smoking was the major factor contributing to GrimAge and Zhang acceleration (Table S7). Each PY increase was associated with ~0.3 years accelerated GrimAge, and smoking alone accounted for 82% (UCL), 75% (ABD), and 47% (CF) of SZ’s influence on GrimAge. Each PY increase was associated with a 0.03-0.06 SD increase in Zhang acceleration, and smoking accounted for 70% (UCL), 97% (ABD), 13% (CF) of SZ’s influence on Zhang. Smoking’s effect on PhenoAge was less pronounced. GrimAge was positively associated with alcohol and BMI, which were lower in SZ so would not explain acceleration.
After accounting for smoking, alcohol, and BMI, SZ was still associated with a 1- to 2-year acceleration in GrimAge and PhenoAge, and a 0.02 to 0.1 SD increase in Zhang (Table S7). To investigate this excess acceleration, we examined sub-clocks of GrimAge for serum levels of 7 age-associated proteins (Figures 2, S7, S8; Table S4). Six (B2M, Cystatin C, GDF-15, TIMP-1, ADM, and PAI-1) were increased in SZ, and all except GDF-15 remained elevated after correction for smoking, alcohol, and BMI (Table S9). These proteins could fully explain excess acceleration in all three mortality clocks (see Supplemental Results for more details).
Mitotic clocks are decelerated in SZ related to NK and CD8T cells
We found mitotic clock deceleration was related to cell proportions (Figure 1E–F), so we examined the contribution of individual cell types. SZ showed increased granulocytes and plasmablasts, and reduced NK cells, CD8T, and CD4T cells (Figures 2, S7, S8; Table S4), consistent with prior literature (46). Multiple regression analysis indicated CD8T and NK cell reduction in SZ significantly explains variance in MiAge and Yang in all data sets, while increases in granulocytes and plasmablasts may also contribute but the relationship is less consistent (Table S10). Furthermore, CD8T and NK cells had a stronger relationship with mitotic clocks in SZ compared to NPC, but granulocytes and plasma cells did not (Table S11).
Smoking was an important confounder. Smoking reportedly reduces NK cell number and activity (47), and we found decreased NK cells were associated with mitotic clock deceleration. However, this conflicted with the expectation that tobacco carcinogens should accelerate mitotic clocks. Smoking in SZ explained 40-50% of NK cell reduction, but not changes in other cell types (Table S12, Figure S10). After accounting for smoking’s effects on cell types, we found smoking independently accelerated MiAge and Yang as expected (Table S10, Figure S11). Alcohol, BMI, and serum proteins could not explain MiAge and Yang acceleration (Tables S10–14, see Supplemental Results).
Chronological age clocks are decelerated in males with clozapine use
All but one clock trained to predict chronological age did not differ between NPC and SZ in UCL and ABD, both before and after correcting for sex and cell counts (Figure S12, Tables S4 and S15). Hannum showed a small acceleration in SZ in UCL (0.9 years, p = 0.0236) and ABD (0.7 years, p = 0.0219). Smoking, alcohol, and BMI had little to no effect. However, we were surprised to find SZ deceleration for Horvath1 and Lin in CF, and trends towards deceleration in other chronological age clocks (Table S4). Multiple regression analysis revealed only one factor that could explain deceleration: clozapine use (Table S15).
When we separated SZ into three medication groups (non-clozapine antipsychotics only, clozapine only, or both), both groups taking clozapine showed a significant decrease in chronological age, but strikingly this deceleration occurred only in males (Figure 3, Table S16). Clozapine-only males showed decelerations of 5.6 years in Lin (p = 0.0011), 5.0 years in Horvath1 (p = 0.0024), 2.1 years in Horvath2 (p = 0.0451), 3.7 years in Hannum (p = 0.0074), 4.1 years in Weidner (p = 0.0599). Males taking both clozapine and other antipsychotics showed decelerations of 7.9 years in Lin (p < 0.0001), 5.1 years in Horvath1 (p = 0.0053), 2.3 years in Horvath2 (p = 0.0432), 3.5 years in Hannum (p = 0.0216), 5.2 years in Weidner (p = 0.0341). SZ patients taking non-clozapine antipsychotics and females taking clozapine were not different than NPC. No clozapine effect was seen for any other clocks, or for protein, smoking, alcohol, or BMI biomarkers (Table S17–19). Although antipsychotics affect the immune system and clozapine can cause agranulocytosis (48,49), the non-clozapine antipsychotic group did not differ from the two clozapine groups for any cell type and adjusting for cell counts did not change the effect of clozapine (Tables S16, S20).
Figure 3.
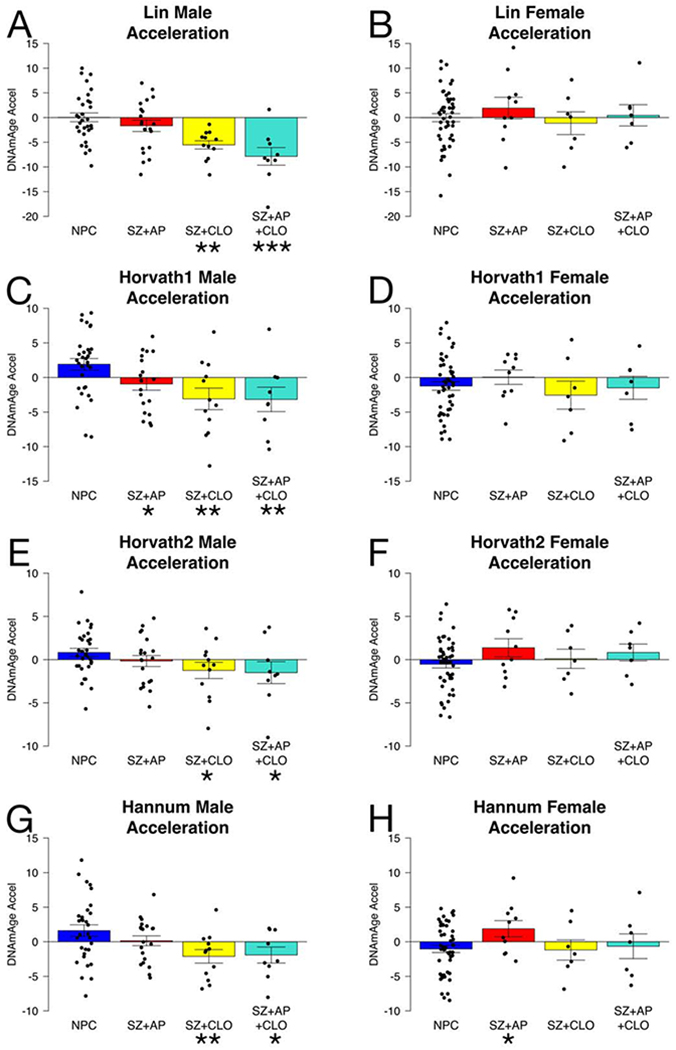
Clozapine effects on chronological age clocks in the CF data set are male-specific. Age accelerations are defined as residuals from the linear model for NPC, and therefore average age acceleration for male and female NPCs together is 0. Error bars are SEM. NPC = non-psychiatric controls, SZ = schizophrenia, AP = non-clozapine antipsychotics, CLO = clozapine. Statistical significance of difference from NPC is shown: <0.05*, <0.01**, <0.001***.
Mortality clock, mitotic clock, and clozapine effects are likely independent
Our analysis involved a large number of DNAm-based biomarkers, many of which may be correlated. Thus, it was unclear if our results concerning mortality clocks, mitotic clocks, and clozapine effects were independent, or instead reflected the same phenomenon. To address this, we sought to understand epigenetic aging on a global level rather than one biomarker at a time. Because most variables were tightly correlated due to chronological age, we utilized age residuals for biomarkers with reliable age trends.
We calculated pairwise correlations between all variables and performed hierarchical clustering (Figure S13). Because relationships between variables were broadly conserved across all data sets, we pooled the data and adjusted for differences across studies (Figures 4A, S15). There were four clusters of correlated clocks that mostly agreed with our categorization of mortality, mitotic, chronological age, and telomere length clocks. The exception was PhenoAge which clusters with chronological age clocks, though it was still highly correlated with mortality clocks. We performed principal component analysis (PCA) on the pooled data (Figures 4B–D), focusing on the first 6 PCs which explained 57.3% of the variance (Figures S16–17). PCA confirmed our findings that mortality clock acceleration is associated with smoking and age-associated proteins, while mitotic clock deceleration is associated with NK and CD8T cells. The mortality and mitotic clusters were not correlated with each other, and they loaded onto PC1 and PC2 respectively. Thus, mortality clock acceleration and mitotic clock deceleration in SZ were quantitatively independent.
Figure 4.

SZ effects on mortality, mitotic, and chronological age clocks are independent. (A) Correlation plot using biweight correlation between all measures for pooled data, adjusted based on study. Age residuals are used for variables that show robust age correlations (epigenetic clocks, proteins, smoking pack-years). Variables are ordered based on hierarchical clustering. Study-specific plots are found in Figure S13. (B-D) First six principal components, only showing variables with loadings higher than expected by chance. (E) PC correlations with SZ and sex for pooled data sets, and with SZ and antipsychotic classes for CF males. CF females are shown in Figure S18. Multiple regression analyses are shown in Table S21.
The chronological age clocks were more complex, with variance captured by shared signals from both mortality (PC1) and mitotic (PC2) clocks (Figures 4A–B), but also by PCs 3-5 (Figures 4C–D, S17). Multiple regression analysis showed clozapine affected PC3-5 specifically in males, but not PC1-2 after adjusting for SZ (Table S21, Figures 4E, S18). Thus, clozapine’s effect was independent of results concerning mortality and mitotic clocks in SZ.
See Supplemental Results for further implications of clustering and PCA, including for telomere length and non-clozapine antipsychotics.
Discussion
Aging is a multifaceted process unfolding across the lifespan leading to functional decline and mortality. Patients with SZ have different long-term health outcomes compared to the general population. However, it is difficult to identify SZ patient characteristics that predict aging outcomes, often occurring decades later, due to the large number of possible mechanisms, confounders, and disease heterogeneity. Epigenetic clocks can potentially bridge the gap between risk factors and aging because they can be measured throughout the lifespan and in every individual.
Prior publications reported the surprising finding that SZ is not associated with alterations in epigenetic aging, despite elevated risk factors and poor health outcomes. Our findings reconcile these observations, demonstrating that altered aging occurs in other epigenetic clocks not previously examined in SZ, because these clocks are sensitive to different aspects of aging (see Supplemental Discussion).
Mortality clock accelerations in SZ were impressive. The 5.8 year GrimAge acceleration in UCL corresponds to a 74% increased hazard of death (HR = 1.105.8 = 1.74) (4). Mortality clocks predict cardiovascular disease, diabetes, adiposity, and comorbidity count better than chronological age clocks (4,35,36). Mortality clock acceleration was driven largely by increased smoking, reinforcing the importance of addressing smoking in SZ.
Individuals with SZ showed increased predicted levels of six age-associated proteins that contribute to mortality clock acceleration and provide molecular links to cancer, cardiovascular disease, kidney function, hypertension, and cognitive function (4). It is unlikely that these results are artifacts of the DNAm predictor, as they agree with prior direct measurements of proteins in SZ (5–11). Their associations with smoking, alcohol, and BMI are consistent with the literature (50,51). GDF-15, previously found to be inversely associated with psychosis severity (7), is highly correlated with smoking, perhaps informing the debate about the role of smoking in SZ etiology (52). The other proteins show significant smoking-independent elevation, and some were previously linked to SZ pathogenesis and cognitive symptoms (5,9,53). This suggests the underlying biology of SZ contributes to aging and raises the possibility of novel SZ treatments to improve both mental and physical health.
Mitotic clock deceleration in SZ is interesting given the debate from epidemiological studies suggesting cancer risk is reduced in SZ despite high smoking rates and obesity (54). It has been unclear if this observation represents an epiphenomenon where individuals with SZ die before the development of cancer (55). However, we found mitotic age is lower in individuals with SZ throughout the lifespan, which could provide a biological basis for reduced cancer risk. Indeed, cancer risk may be reduced even before the age of SZ diagnosis and in first-degree relatives (56). Notably, our finding that mitotic clock deceleration is less pronounced in females is consistent with prior observations that the primary cancer types not decreased in SZ are breast and endometrial cancer (56) and that socioeconomic inequalities in smoking-related mortality in many countries have decreased in men but increased in women (57). Another link with cancer incidence is the role of NK and CD8T cells, known mediators of anti-tumor immunity (58), though it seems paradoxical that reduced NK and CD8T cells are associated with slowed mitotic age. However, intrinsic NK cell activity has been reported to be elevated in SZ (59), so it is possible that overactive NK and CD8T cells in SZ protect from cancer, and negative feedback reduces the number of circulating cells. Smoking also affects other cell types, especially neutrophils, that interact with NK and CD8T cells (60). Alternatively, reduced mitotic age may simply reflect differential mitotic rates of NK and CD8T cells relative to other immune cells. Mechanistic studies are needed to untangle these complex relationships.
Clozapine, the most effective antipsychotic for SZ, was associated with a male-specific deceleration in chronological age clocks, independent of immune cell proportions. The magnitude of this effect is striking, as Lin shows a 7-year deceleration yet the standard deviation for Lin acceleration among controls is only 5 years. This result was limited by medication data only being available for one data set with 74 SZ individuals. Available long-term medication data in epigenetic aging studies is uncommon, and therefore very few studies have identified medications with any effects on epigenetic age. This highlights the need to collect detailed medication data with DNA methylation in future studies. Clozapine use rates are higher in China than most other countries (61), and 48% of SZ subjects in CF were on clozapine. Medication data is not available for UCL or ABD, but these sets collected in the UK likely have much lower clozapine use rates. This may explain why chronological clock slowing was only observed in CF.
The practical consequences of clozapine-related age deceleration are unclear. Previous long-term cohort studies found up to 60% reduced all-cause mortality and 45% reduced cardiovascular mortality associated with clozapine (62,63), though it is puzzling why this might manifest in chronological age clocks but not mortality clocks. One possible explanation is that the clocks were trained in the general population, which may miss biological mechanisms of clozapine acting in a small subset of the population. A similar phenomenon may apply to the epigenetic BMI and alcohol biomarkers (see Supplemental Discussion). It is possible clozapine impacts the aging process itself, as a recent bioinformatics study identified drugs that induce younger human gene expression profiles, and the top 30 “geroprotective” compounds included clozapine and three other antipsychotics (64). Antipsychotics alter lifespan in the nematode C. elegans by modulating the insulin-like growth factor pathway that controls aging across numerous animal species (64,65). Clozapine may influence aging through unique effects on neurotrophic factors and glutamate (66–68). A sex-specific effect on aging is plausible given sex differences in clozapine side effects, metabolism, and treatment response (49,69). However, there are alternative explanations for clozapine’s association with biological age. Individuals with lower biological age may be more resilient to the adverse effects of clozapine and hence more likely to be continued on clozapine treatment or survive to be included the study. If that is true, then DNA methylation may be a useful biomarker for identifying patients appropriate for clozapine treatment, as clozapine is currently underutilized due to concerns about serious side effects. Clozapine may also be associated with increased healthcare since clozapine requires regular laboratory monitoring. Clozapine may exert acute effects directly on clock CpGs, rather than influencing a chronic aging process. Distinguishing between these possibilities will require detailed longitudinal studies. Nevertheless, it is likely that clozapine interacts with aging, and further investigation of this clozapine effect is warranted.
This study was limited primarily by available data on subjects and its cross-sectional design. There was not data to indicate if severity of psychiatric symptoms or physical illness accelerates the clocks in SZ. Direct quantification of BMI, substance use, and cell composition may yield different results than the epigenetic biomarkers we utilized (see Supplemental Discussion), though it is notable that the smoking epigenetic proxy predicts mortality more accurately than self-reported smoking (4). We measured smoking in cumulative pack-years, but it is possible that quantifying dose and duration separately may yield different results (70). Moreover, schizophrenia is a complex, heterogeneous disorder with diverse socioeconomic ramifications, such as increased risk of poverty, unemployment, stigma, social isolation, housing instability, and poor healthcare (22–24). Stress is well-established to influence aging (71), and education and income are associated with biological age (20). The cross-sectional design has many limitations. It is unclear if survival bias played a role in our findings, though we expect that correcting for survival bias would further increase the degree of SZ mortality clock acceleration. Longitudinal studies would enable study of when altered aging occurs (e.g. rapidly during early psychosis or slowly over decades). For example, childhood trauma and lifetime PTSD severity is cross-sectionally associated with higher Hannum but not Horvath1 age (72), while a longitudinal study found alcohol use disorder and specific PTSD symptoms increases the pace of aging of Horvath1 but not Hannum (73). Our study establishing schizophrenia’s association with multiple altered epigenetic aging biomarkers should motivate future longitudinal studies using well-phenotyped populations.
Our comprehensive epigenetic clock analysis is striking in how closely it matches prior observations about SZ: greatly increased all-cause mortality, reduced risk of cancer (except breast and endometrial), shortened telomeres, high smoking rates, derangements in immune cells and serum proteins, and unique effects of clozapine. Epigenetic clocks offer the opportunity to understand the biological basis of these observations. Importantly, this was the first epigenetic study to examine all these factors simultaneously, enabling us to dissect their relationships and suggest many of them are independent phenomena. Our work may serve as a foundation for future aging studies on the effects of genetics, developmental stressors, disorganized behavior, lifestyle factors, substance use, medications, co-morbidities, stigma, and socioeconomic factors. Given the inherent complexity of both psychiatric disease and aging, using multiple distinct epigenetic clocks simultaneously can shed new light on when, why, and how psychiatric disease influences diverse aging processes.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.ora/resources. | Include any additional information or notes if necessary. |
| Deposited Data; Public Database | GSE80417 | NCBI GEO DataSets | RRID:SCR_005012; https://www.ncbi.nlm.nih.gov/gds | |
| Deposited Data; Public Database | GSE84727 | NCBI GEO DataSets | RRID:SCR_005012; https://www.ncbi.nlm.nih.gov/gds | |
| Deposited Data; Public Database | GSE116379 | NCBI GEO DataSets | RRID:SCR_005012; https://www.ncbi.nlm.nih.gov/gds | |
| Software; Algorithm | R version 3.5.0 | R Project for Statistical Computing | RRID:SCR_001905 | |
| Software; Algorithm | New Methylation Age Calculator | PMID:24138928 | https://dnamage.genetics.ucla.edu/new | Includes Horvath1, Horvath2, Hannum, PhenoAge, GrimAge, smoking pack-years, GDF-15, ADM, B2M, PAI-1, TIMP-1, Cystatin C, Leptin, and cell composition estimates |
| Software; Algorithm | Horvath1 epigenetic clock | PMID:24138928 | N/A | |
| Software; Algorithm | Horvath2 epigenetic clock | PMID:30048243 | N/A | |
| Software; Algorithm | Vidal-Bralo epigenetic clock | PMID:27471517 | N/A | |
| Software; Algorithm | Lin epigenetic clock | PMID:26110659 | N/A | |
| Software; Algorithm | Hannum epigenetic clock | PMID:23177740 | N/A | |
| Software; Algorithm | Weidner epigenetic clock | PMID:24490752 | N/A | |
| Software; Algorithm | Garagnani epigenetic clock | PMID:23061750 | N/A | |
| Software; Algorithm | Bocklandt epigenetic clock | PMID:21731603 | N/A | EDARADD |
| Software; Algorithm | GrimAge epigenetic clock | PMID:30669119 | N/A | Includes smoking pack-years, GDF-15, ADM, B2M, PAI-1, TIMP-1, Cystatin C, Leptin |
| Software; Algorithm | Levine PhenoAge epigenetic clock | PMID:29676998 | N/A | |
| Software; Algorithm | Zhang epigenetic clock | PMID:28303888 | N/A | continuous risk score |
| Software; Algorithm | MiAge epigenetic clock | PMID:29160179 | http://www.columbia.edu/~sw2206/softwares.htm | |
| Software; Algorithm | Yang epigenetic clock | PMID:27716309 | N/A | |
| Software; Algorithm | DNAmTL epigenetic clock | PMID:31422385 | N/A | |
| Software; Algorithm | McCartney smoking, alcohol, and BMI predictors | PMID: 30257690 | N/A | |
| Software; Algorithm | WGCNA R package v1.68 | CRAN; Peter Langfelder; PMID: 19114008 | RRID:SCR_003302 | |
| Software; Algorithm | ggcorrplot R package v0.1.2 | CRAN; Alboukadel Kassambara | N/A | |
| Software; Algorithm | factoextra R package v1.0.5 | CRAN; Alboukadel Kassambara | RRID:SCR_016692 |
Acknowledgments
Dr. Higgins-Chen was supported by grants from the NIMH (2R25MH071584-11 and 2T32MH019961-21A1; Yale Neuroscience Training Program), and the Thomas P. Detre Fellowship Award in Translational Neuroscience Research (Yale University). Drs. Boks, Vinkers, and Kahn report no grants related to this work. Dr. Levine was supported by grants from the NIA (5R00AG052604-04), and the Pilot Grant from the Claude D. Pepper Older Americans Independence Center at the Yale School of Medicine (P30AG021342). Sponsors had no role in the collection, analysis, or interpretation of the data; preparation, review, approval, submission of the manuscript; or any other part of study design or conduct. We acknowledge members of the Levine lab for critical input and manuscript review. Drs. Boks, Vinkers, and Kahn previously published a manuscript describing the CF data set (GSE116379) (43). We also acknowledge Hannon et al. for the original acquisition of the UCL and ABD data sets (GSE80417, GSE84727) (42).
Disclosures: Dr. Higgins-Chen received grants from the NIMH during the conduct of the study, and consulting fees from Life Epigenetics outside the submitted work. Dr. Levine received grants from the NIA and the Glenn Foundation, and consulting fees from Elysium Health and Life Epigenetics outside the submitted work. Dr. Levine is Head of Bioinformatics for Elysium Health and holds licenses for epigenetic clocks she has developed (DNAmPhenoAge). All other authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hennekens CH, Hennekens AR, Hollar D, Casey DE (2005): Schizophrenia and increased risks of cardiovascular disease. Am Heart J 150: 1115–1121. [DOI] [PubMed] [Google Scholar]
- 2.Saha S, Chant D, McGrath J (2007): A Systematic Review of Mortality in Schizophrenia. Arch Gen Psychiatry 64: 1123. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen TT, Eyler LT, Jeste D V. (2018): Systemic biomarkers of accelerated aging in schizophrenia: A critical review and future directions. Schizophr Bull 44: 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, et al. (2019): DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 11: 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoirisch-Clapauch S, Amaral OB, Mezzasalma MAU, Panizzutti R, Nardi AE (2016): Dysfunction in the coagulation system and schizophrenia. Transl Psychiatry 6: e704–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeffries CD, Perkins DO, Fournier M, Do KQ, Cuenod M, Khadimallah I, et al. (2018): Networks of blood proteins in the neuroimmunology of schizophrenia. Transl Psychiatry 8: 0–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar P, Millischer V, Villaescusa JC, Nilsson IAK, Östenson CG, Schalling M, et al. (2017): Plasma GDF15 level is elevated in psychosis and inversely correlated with severity. Sci Rep 7: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan MK, Cooper JD, Heilmann-Heimbach S, Frank J, Witt SH, Nöthen MM, et al. (2017): Associations between SNPs and immune-related circulating proteins in schizophrenia. Sci Rep 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chittiprol S, Venkatasubramanian G, Neelakantachar N, Allha N, Shetty KT, Gangadhar BN (2009): β2-Microglobulin abnormalities in antipsychotic-naïve schizophrenia: Evidence for immune pathogenesis. Brain Behav Immun 23: 189–192. [DOI] [PubMed] [Google Scholar]
- 10.Chittiprol S, Venkatasubramanian G, Neelakantachar N, Reddy NA, Shetty KT, Gangadhar BN (2009): Longitudinal study of β2-microglobulin abnormalities in schizophrenia. Int Immunopharmacol 9: 1215–1217. [DOI] [PubMed] [Google Scholar]
- 11.Yilmaz N, Herken H, Cicek HK, Celik A, YQrekli M, Akyol Ö (2007): Increased levels of nitric oxide, cortisol and adrenomedullin in patients with chronic schizophrenia. Med Princ Pract 16: 137–141. [DOI] [PubMed] [Google Scholar]
- 12.Kirkpatrick B, Kennedy BK (2018): Accelerated aging in schizophrenia and related disorders: Future research. Schizophr Res 196: 4–8. [DOI] [PubMed] [Google Scholar]
- 13.Kirkpatrick B, Messias E, Harvey PD, Fernandez-Egea E, Bowie CR (2008): Is schizophrenia a syndrome of accelerated aging? Schizophr Bull 34: 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voisey J, Lawford BR, Morris CP, Wockner LF, Noble EP, Young RM, Mehta D (2017): Epigenetic analysis confirms no accelerated brain aging in schizophrenia. npj Schizophr 3: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKinney BC, Lin H, Ding Y, Lewis DA, Sweet RA (2017): DNA methylation evidence against the accelerated aging hypothesis of schizophrenia. npj Schizophr 3: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinney BC, Lin H, Ding Y, Lewis DA, Sweet RA (2017): DNA methylation age is not accelerated in brain or blood of subjects with schizophrenia. Schizophr Res 196: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okazaki S, Otsuka I, Numata S, Horai T, Mouri K, Boku S, et al. (2019): Epigenetic clock analysis of blood samples from Japanese schizophrenia patients. npj Schizophr 5: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowalec K, Hannon E, Mansell G, Burrage J, Ori APS, Ophoff RA, et al. (2019): Methylation age acceleration does not predict mortality in schizophrenia. Transl Psychiatry 9 10.1038/s41398-019-0489-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horvath S (2013): DNA methylation age of human tissues and cell types. Genome Biol 14: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horvath S, Raj K (2018): DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 1–14. [DOI] [PubMed] [Google Scholar]
- 21.Rosen AD, Robertson KD, Hlady RA, Muench C, Lee J, Philibert R, et al. (2018): DNA methylation age is accelerated in alcohol dependence. Transl Psychiatry 8 10.1038/s41398-018-0233-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green MF, Horan WP, Lee J, McCleery A, Reddy LF, Wynn JK (2018): Social Disconnection in Schizophrenia and the General Community. Schizophr Bull 44: 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster A, Gable J, Buckley J (2012): Homelessness in Schizophrenia. Psychiatr Clin North Am 35: 717–734. [DOI] [PubMed] [Google Scholar]
- 24.Hakulinen C, McGrath JJ, Timmerman A, Skipper N, Mortensen PB, Pedersen CB, Agerbo E (2019): The association between early-onset schizophrenia with employment, income, education, and cohabitation status: nationwide study with 35 years of follow-up. Soc Psychiatry Psychiatr Epidemiol 54: 1343–1351. [DOI] [PubMed] [Google Scholar]
- 25.De Leon J, Diaz FJ (2005): A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res 76: 135–157. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds GP, McGowan OO (2017): Mechanisms underlying metabolic disturbances associated with psychosis and antipsychotic drug treatment. J Psychopharmacol 31: 1430–1436. [DOI] [PubMed] [Google Scholar]
- 27.Barton BB, Segger F, Fischer K, Obermeier M, Musil R (2020): Update on weight-gain caused by antipsychotics: a systematic review and meta-analysis. Expert Opin Drug Saf 0: 1. [DOI] [PubMed] [Google Scholar]
- 28.Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E (2011): Epigenetic Predictor of Age. PLoS One 6: e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garagnani P, Bacalini MG, Pirazzini C, Gori D, Giuliani C, Mari D, et al. (2012): Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 11: 1132–1134. [DOI] [PubMed] [Google Scholar]
- 30.Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, et al. (2014): Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol 15 10.1186/gb-2014-15-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidal-Bralo L, Lopez-Golan Y, Gonzalez A (2016): Simplified assay for epigenetic age estimation in whole blood of adults. Front Genet 7: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda SV, et al. (2013): Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol Cell 49: 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. (2018): Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY) 10: 1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin Q, Wagner W (2015): Epigenetic Aging Signatures Are Coherently Modified in Cancer. PLoS Genet 11: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine ME, Lu AT, Quach A, Chen B, Assimes TL, Bandinelli S, et al. (2018): An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10: 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Wilson R, Heiss J, Breitling LP, Saum KU, Schöttker B, et al. (2017): DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youn A, Wang S (2018): The MiAge Calculator: a DNA methylation-based mitotic age calculator of human tissue types. Epigenetics 13: 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Wong A, Kuh D, Paul DS, Rakyan VK, Leslie RD, et al. (2016): Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol 17: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu AT, Seeboth A, Tsai P, Sun D, Quach A, Reiner AP, et al. (2019): DNA methylation-based estimator of telomere length. Aging (Albany NY) 11: 5895–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCartney DL, Stevenson AJ, Ritchie SJ, Walker RM, Zhang Q, Morris SW, et al. (2018): Epigenetic prediction of complex traits and death. Genome Biol 19: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Z, Leung D, Levine M (2019): Comparative analysis of epigenetic aging clocks from CpG characteristics to functional associations. bioRxiv. [Google Scholar]
- 42.Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. (2016): An integrated genetic-epigenetic analysis of schizophrenia: Evidence for colocalization of genetic associations and differential DNA methylation. Genome Biol 17: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boks MP, Houtepen LC, Xu Z, He Y, Ursini G, Maihofer AX, et al. (2018): Genetic vulnerability to DUSP22 promoter hypermethylation is involved in the relation between in utero famine exposure and schizophrenia. npj Schizophr 4 10.1038/s41537-018-0058-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horvath S, Levine AJ (2015): HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis 212: 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russo P, Prinzi G, Proietti S, Lamonaca P, Frustaci A, Boccia S, et al. (2018): Shorter telomere length in schizophrenia: Evidence from a real-world population and meta-analysis of most recent literature. Schizophr Res 202: 37–45. [DOI] [PubMed] [Google Scholar]
- 46.Karpiński P, Samochowiec J, Frydecka D, Sąsiadek MM, Misiak B (2018): Further evidence for depletion of peripheral blood natural killer cells in patients with schizophrenia: A computational deconvolution study. Schizophr Res 201: 243–248. [DOI] [PubMed] [Google Scholar]
- 47.Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, et al. (2017): Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 8: 268–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Røge R, Møller BK, Andersen CR, Correll CU, Nielsen J (2012): Immunomodulatory effects of clozapine and their clinical implications: What have we learned so far? Schizophrenia Research, vol. 140 Elsevier B.V., pp 204–213. [DOI] [PubMed] [Google Scholar]
- 49.Alberich S, Fernández-Sevillano J, González-Ortega I, Usall J, Sáenz M, González-Fraile E, González-Pinto A (2019): A systematic review of sex-based differences in effectiveness and adverse effects of clozapine. Psychiatry Res 280: 112506. [DOI] [PubMed] [Google Scholar]
- 50.Sasaki A, Kurisu A, Ohno M, Ikeda Y (2001): Overweight/obesity, smoking, and heavy alcohol consumption are important determinants of plasma PAI-1 levels in healthy men. Am J Med Sci 322: 19–23. [DOI] [PubMed] [Google Scholar]
- 51.Huang B, Svensson P, Ärnlöv J, Sundström J, Lind L, Ingelsson E (2016): Effects of cigarette smoking on cardiovascular-related protein profiles in two community-based cohort studies. Atherosclerosis 254: 52–58. [DOI] [PubMed] [Google Scholar]
- 52.Kendler KS, Lönn SL, Sundquist J, Sundquist K (2015): Smoking and schizophrenia in population cohorts of Swedish women and men: A prospective co-relative control study. Am J Psychiatry 172: 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salih Zoroğlu S, Herken H, Yürekli M, Uz E, Tutkun H, Savaş HA, et al. (2002): The possible pathophysiological role of plasma nitric oxide and adrenomedullin in schizophrenia. J Psychiatr Res 36: 309–315. [DOI] [PubMed] [Google Scholar]
- 54.Hodgson R, Wildgust HJ, Bushe CJ (2010): Cancer and schizophrenia: is there a paradox? J Psychopharmacol 24: 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guan NC, Termorshuizen F, Laan W, Smeets HM, Zainal NZ, Kahn RS, et al. (2013): Cancer mortality in patients with psychiatric diagnoses: A higher hazard of cancer death does not lead to a higher cumulative risk of dying from cancer. Soc Psychiatry Psychiatr Epidemiol 48: 1289–1295. [DOI] [PubMed] [Google Scholar]
- 56.Ji J, Sundquist K, Ning Y, Kendler KS, Sundquist J, Chen X (2013): Incidence of cancer in patients with schizophrenia and their first-degree relatives: A population-based study in Sweden. Schizophr Bull 39: 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gregoraci G, van Lenthe FJ, Artnik B, Bopp M, Deboosere P, Kovács K, et al. (2017): Contribution of smoking to socioeconomic inequalities in mortality: A study of 14 European countries, 1990–2004. Tob Control 26: 260–268. [DOI] [PubMed] [Google Scholar]
- 58.Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. (2006): NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood 107: 1342 LP–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yovel G, Sirota P, Mazeh D, Shakhar G, Rosenne E, Ben-Eliyahu S (2000): Higher natural killer cell activity in schizophrenic patients: The impact of serum factors, medication, and smoking. Brain Behav Immun 14: 153–169. [DOI] [PubMed] [Google Scholar]
- 60.Van Eeden SF, Hogg JC (2000): The response of human bone marrow to chronic cigarette smoking myeloperoxidase neutrophils polymorphonuclear leukocytes. Eur Respir J 15: 915–921. [DOI] [PubMed] [Google Scholar]
- 61.Xiang YT, Wang CY, Si TM, Lee EHM, He YL, Ungvari GS, et al. (2011): Clozapine use in schizophrenia: Findings of the Research on Asia Psychotropic Prescription (REAP) studies from 2001 to 2009. Aust N Z J Psychiatry 45: 968–975. [DOI] [PubMed] [Google Scholar]
- 62.Wimberley T, MacCabe JH, Laursen TM, Sørensen HJ, Astrup A, Horsdal HT, et al. (2017): Mortality and self-harm in association with clozapine in treatment-resistant schizophrenia. Am J Psychiatry 174: 990–998. [DOI] [PubMed] [Google Scholar]
- 63.Taipale H, Tanskanen A, Mehtälä J, Vattulainen P, Correll CU, Tiihonen J (2020): 20-year follow-up study of physical morbidity and mortality in relationship to antipsychotic treatment in a nationwide cohort of 62,250 patients with schizophrenia (FIN20). World Psychiatry 19: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janssens GE, Lin XX, Millan-Ariño L, Kavšek A, Sen I, Seinstra RI, et al. (2019): Transcriptomics-Based Screening Identifies Pharmacological Inhibition of Hsp90 as a Means to Defer Aging. Cell Rep 27: 467–480.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weeks KR, Dwyer DS, Aamodt EJ (2010): Antipsychotic drugs activate the C. elegans AKT pathway via the DAF-2 insulin/IGF-1 receptor. ACS Chem Neurosci 1: 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zullo JM, Drake D, Aron L, Hern PO, Dhamne SC, Davidsohn N, et al. (2019): Regulation of lifespan by neural excitation and REST. Nature 574: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Islam F, Mulsant BH, Voineskos AN, Rajji TK (2017): Brain-Derived Neurotrophic Factor Expression in Individuals With Schizophrenia and Healthy Aging: Testing the Accelerated Aging Hypothesis of Schizophrenia. Curr Psychiatry Rep 19 10.1007/s11920-017-0794-6 [DOI] [PubMed] [Google Scholar]
- 68.Krivoy A, Hochman E, Sendt KV, Hollander S, Vilner Y, Selakovic M, et al. (2018): Association between serum levels of glutamate and neurotrophic factors and response to clozapine treatment. Schizophr Res 192: 226–231. [DOI] [PubMed] [Google Scholar]
- 69.Mayerova M, Ustohal L, Jarkovsky J, Pivnicka J, Kasparek T, Ceskova E (2018): Influence of dose, gender, and cigarette smoking on clozapine plasma concentrations. Neuropsychiatr Dis Treat 14: 1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peto J (2012): That the effects of smoking should be measured in pack-years: Misconceptions 4. Br J Cancer 107: 406–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Calabrese EJ, Dhawan G, Kapoor R, lavicoli I, Calabrese V (2015): What is hormesis and its relevance to healthy aging and longevity? Biogerontology 16: 693–707. [DOI] [PubMed] [Google Scholar]
- 72.Wolf EJ, Maniates H, Nugent N, Maihofer AX, Armstrong D, Ratanatharathorn A, et al. (2018): Traumatic stress and accelerated DNA methylation age: A meta-analysis. Psychoneuroendocrinology 92: 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolf EJ, Logue MW, Morrison FG, Wilcox ES, Stone A, Schichman SA, et al. (2019): Posttraumatic psychopathology and the pace of the epigenetic clock: A longitudinal investigation. Psychol Med 49: 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.